

Abstract
In Gram-negative bacteria, the folding and insertion of β-barrel outer membrane proteins (OMPs) to the outer membrane are mediated by the β-barrel assembly machinery (BAM) complex. Two leading models of this process have been put forth: the hybrid barrel model, which claims that a lateral gate in BamA’s β-barrel can serve as a template for incoming OMPs, and the passive model, which claims that a thinned membrane near the lateral gate of BamA accelerates spontaneous OMP insertion. To examine the key elements of these two models, we have carried out 45.5 μs of equilibrium molecular dynamics simulations of BamA with and without POTRA domains from Escherichia coli, Salmonella enterica, Haemophilus ducreyi and Neisseria gonorrhoeae, together with BamA’s homolog, TamA from E. coli, in their native, species-specific outer membranes. In these equilibrium simulations, we consistently observe membrane thinning near the lateral gate for all proteins. We also see occasional spontaneous lateral gate opening and sliding of the β-strands at the gate interface for N. gonorrhoeae, indicating that the gate is dynamic. An additional 14 μs of free-energy calculations shows that the energy necessary to open the lateral gate in BamA/TamA varies by species, but is always lower than the Omp85 homolog, FhaC. Our combined results suggest OMP insertion utilizes aspects of both the hybrid barrel and passive models.
Author summary
Gram-negative bacteria such as Escherichia coli have a second, outer membrane surrounding them. This outer membrane provides an additional layer of protection, but also presents an additional challenge in its construction, exacerbated by the lack of chemical energy in this region of the bacterial cell. For example, proteins in the outer membrane are inserted via BamA, itself an integral membrane protein. The precise mechanisms by which BamA assists in the insertion process are still unclear. Here, we use extensive simulations in atomistic detail of BamA from multiple species in its native outer membrane environment to shed light on this process. We find that the lateral gate of BamA, a proposed pathway into the membrane, is dynamic, although to a degree varying by species. On the other hand, thinning of the outer membrane near BamA’s lateral gate is observed consistently across all simulations. We conclude that multiple features of BamA contribute to protein insertion into the outer membrane.
Introduction
Gram-negative bacteria are enveloped by two membranes, an inner membrane and an outer membrane (OM) [1]. This OM serves as a strong barrier against drugs targeting Gram-negative bacteria [2] with lipopolysaccharides/lipooligosaccharides (LPS/LOS) on the outer leaflet and a mixture of phospholipids on the inner leaflet [3, 4]. The outer membrane proteins (OMPs) in the asymmetric OM play important roles in nutrient transport [5, 6], waste export [7, 8], cell signaling [9, 10] and membrane biogenesis [11, 12] and are almost exclusively β-barrel structures [13]. The folding and insertion of these β-barrel OMPs are mediated by the β-barrel assembly machinery (BAM) complex without ATP or ion gradients [14–16]. Although multiple structures have been solved by X-ray crystallography or cryo-electron microscopy, the exact mechanisms of the process remain unknown [17–20]. In E. coli, the BAM complex consists of five members, named BamA-E [16, 21]. BamA contains a 16-strand transmembrane β-barrel and five periplasmic polypeptide-transport-associated (POTRA) domains [22]. BamB-E, which interact with BamA primarily through its POTRA domains, are lipoproteins with their N-termini embedded in the OM [23–25]. Experiments have shown that although BamA itself is able to accelerate OMP folding in vitro [26], BamD is also required for cell viability in vivo [27], and all BAM components are required to achieve maximum efficiency [28, 29].
The central component of the BAM complex, BamA, is a member of the Omp85 family of proteins that is essential for OM biogenesis in mitochondria, chloroplasts and bacteria [30, 31]. Several structural features of BamA have been recognized and highlighted, leading to two corresponding mechanistic models of insertion [32]. The seam of the BamA barrel between β1 and β16 strand (Fig 1A) is dynamic [33], and it can open laterally [22, 34]. This opening may serve as a template for nascent proteins to initiate β-barrel formation through β-augmentation, referred to as the hybrid barrel model [35]. The hybrid barrel model is supported by a series of experiments [36], including a recent structure of two BamAs, one joined to the other at their lateral gates [37]. Alternatively, the passive model focuses on the narrowed hydrophobic surface at the lateral gate, which is predicted to disorder the membrane and attenuate its thickness [22, 34]. The thinner, less stable membrane would allow OMPs to fold into it directly [38, 39]. Additionally, a homolog of BamA, TamA also consists of a 16-stranded β-barrel at its C-terminus and three POTRA domains at its N-terminus [40]. TamA’s POTRA3 is similar to BamA POTRA domains, and it has been suggested that the function of the β-barrels of BamA and TamA is conserved [41].
Fig 1. BamA structure.

(A) BamA of N. gonorrhoeae in its OM with protein in white. β1 is highlighted in red and β16 is highlighted in purple. (B) Five POTRA domains of BamA of N. gonorrhoeae in tandem with different colors.
With the exact mechanisms of OMP insertion by BamA and TamA unknown, we investigated the structural features of BamAs of different species together with TamA of E. coli using molecular dynamics (MD) simulations with each embedded in its respective native OM. We constructed 12 systems with and without POTRA domains and ran 45.5 μs of equilibrium simulations as well as over 14 μs of replica-exchange umbrella sampling (REUS) to probe the lateral gating of BamA (see Table 1 for the full list). Our simulations show that the frequency of lateral gating for BamA is species dependent while membrane thinning for BamA is species independent, thus shedding light on potential insertion mechanisms.
Table 1. A summary of equilibrium simulations and REUS simulations performed for this study.
| Label | Equilibrium Simulations | REUS Simulations | ||
|---|---|---|---|---|
| Time × Replica | Sum | Replica × Time | Sum | |
| EcBamA | 2000 ns × 2 | 4000 ns | 18 × 70 ns | 1260 ns |
| EcBamAΔP | 2000 ns × 2 | 4000 ns | 18 × 60 ns | 1080 ns |
| EcBamAcw | 1500 ns × 1 | 1500 ns | ||
| SeBamAΔP | 2000 ns × 2 | 4000 ns | 17 × 50 ns | 850 ns |
| HdBamA | 2000 ns × 2 | 4000 ns | 17 × 100 ns | 1700 ns |
| HdBamAΔP | 2000 ns × 2 | 4000 ns | 17 × 65 ns | 1105 ns |
| NgBamA | 2000 ns × 2 | 4000 ns | 17 × 100 ns | 1700 ns |
| NgBamAΔP | 2000 ns × 2 | 4000 ns | 18 × 50 ns | 900 ns |
| EcTamA | 2000 ns × 2 | 4000 ns | 16 × 90 ns | 1440 ns |
| EcTamAΔP | 2000 ns × 2 | 4000 ns | 17 × 90 ns | 1530 ns |
| BpFhaC | 2000 ns × 2 | 4000 ns | 19 × 70 ns | 1330 ns |
| BpFhaCΔP | 2000 ns × 2 | 4000 ns | 20 × 75 ns | 1500 ns |
Materials and methods
System construction
We constructed 12 membrane-protein systems in total with and without POTRA domains (ΔP) for this study using the CHARMM-GUI membrane builder [42, 43]. All proteins were placed in asymmetric outer membranes (OM) consisting of a phospholipid (PL) inner leaflet and a lipopolysaccharide (LPS) outer leaflet with O-antigen excluded. The composition of the inner leaflet is decided by species. We assigned lipid tails with different degrees of unsaturation and different numbers of carbons to different types of phospholipid (phosphatidylethanolamine, phosphatidylglycerol, phosphatidylcholine and cardiolipin are considered in this study) to satisfy the composition of lipid tails as well as the composition of lipid types [44–49]. We modelled LPS with their respective lipid A and core oligosaccharides for E. coli [50], S. enterica [50], H. ducreyi [51, 52], N. gonorrhoeae [53] and Bordetella cepacia [54, 55]. All systems are solvated with TIP3P water [56]. Magnesium ions were used to neutralize the negative charges of LPS while 0.15 M KCl was used to neutralize the systems. See S1 Table for detailed information of all systems.
An additional system starting from EcBamA was also constructed. In this system, a model of the E. coli peptidoglycan (PG) cell wall [57, 58] was added in the periplasm. The PG was anchored to the OM via five Lpp trimers (PDB 1EQ7 [59]), which are tri-acylated at their N-termini (inserted into the inner leaflet of the OM) and covalently bonded to PG for one out of every three copies at their C-terminal end [60]. The number of Lpp was selected to be commensurate with the roughly 500,000 copies per cell [1].
MD Protocol
We ran simulations in both NAMD [61] and Amber [62]. We used the CHARMM36m force field for proteins [63] and CHARMM36 for lipids [64]. Langevin dynamics (damping constant γ = 1.0 ps−1) was used to keep the temperature (310 K) constant, and an anisotropic Langevin piston barostat (in NAMD) or a Monte Carlo barostat (in Amber) was used to enforce constant pressure (1 atm) [65]. The time step of all simulations without hydrogen mass repartitioning (HMR) was 2 fs. Bonded interactions and short-range nonbonded interactions (less than the 12-Å cutoff) are calculated at every time step. The Particle-mesh Ewald (PME) method was used for long-rang interactions, updated every other time step [66]. VMD was used to analyze all results [67].
Equilibrium Simulations
All systems were equilibrated first in NAMD by releasing system components sequentially (lipid tails for 1 ns, everything except protein for 10 ns, everything except protein backbone for 10 ns, everything for 10 ns). The systems were minimized for 2000 steps before each step. After equilibration, all systems but the PG-containing one were run twice in Amber for 2000 ns each using HMR and a time step of 4 fs [68, 69]. Because systems with bonds crossing periodic boundaries cannot be run in Amber, the PG-containing system was run in NAMD for 1500 ns using HMR.
Replica Exchange Umbrella Sampling (REUS) simulations
We used REUS to calculate the potential of mean force (PMF) for lateral gate opening [70]. Targeted Molecular Dynamics (TMD) was used to generate starting states for REUS [71] (see S1 Fig for more details about TMD). The collective variable for BamA of N. gonorrhoeae is defined as the distance between N and O atoms that potentially form hydrogen bonds at the lateral gate projected to the direction that lateral gates open (see S2 Fig). The colvars module of NAMD was used to construct all collective variables [72]. A total of 17-20 unevenly distributed windows were used for REUS, covering a range from 2-12 Å. Different force constants were used for different windows during REUS: see S2 Table for centers and force constants. The sampling data of REUS was used to calculate the PMF using the weighted histogram analysis method (WHAM) [73]. Each REUS simulation was run until an additional 5 ns changed the PMF endpoint by less than 0.2 kcal/mol at which point it was considered to be converged (see S3 Fig).
Results
Membrane hydrophobic thickness of BamA and TamA systems
BamAs exhibit narrowed hydrophobic surfaces at their lateral gates, which is predicted to thin the membrane and create a disordered lipid region [22]. Previous MD simulations of BamA of E. coli, H. ducreyi and N. gonorrhoeae supported this membrane thinning [22, 34], which can accelerate nascent proteins folding and inserting into the OM [39, 74]. In our 2-μs equilibrium simulations, we observed membrane thinning near different strands of the barrel in different systems (Fig 2A). For BamA of E. coli, S. enterica, H. ducreyi and N. gonorrhoeae, membranes near the lateral gate are thinned by 5.3±1.7 Å, demonstrating that the membrane thinning at the lateral gate is a universal feature of BamA (Fig 2B and S4 Fig). We also quantified the extent of membrane thinning by measuring the area of the membrane for which the thickness is below 20 Å (S5 Fig). The results indicate that the areas can be very different, ranging from 4 Å2 (in one replica of HdBamAΔP) to 768 Å2 (in one replica of EcBamAΔP), with the average at ∼350 Å2. This average area is slightly more than the cross-sectional area of an eight-stranded β-barrel, suggesting that it is of sufficient extent to accelerate passive, concerted insertion of at least small OMPs.
Fig 2. Membrane thickness in equilibrium simulations.

(A) Average thickness around the β-barrel for the first run of each system (See S6 Fig for all systems). Labeled concentric circles represent the membrane thickness in Å. (B) Average thickness of the whole membrane (in red) as well as average thickness near β8 and β9 at the back of the β-barrel in grey and near the lateral gate in blue.
Some systems also exhibit minor thinning near β8-β10 strands (Fig 2B). Although TamA’s structure is broadly similar to BamA’s, TamA systems have the most obviously thinned membrane near the β8-β10 strands (See S6 Fig for the average thickness near each strand for all replicas). For EcTamA, membranes near β8 and β9 are thinned by 3.7±1.7 Å. When we inspect the structure of TamA, we find a series of charged residues, R394, R396, R398, D407 and R437 on the periplasmic side of β8, β9 and β10 resulting in membrane thinning. Additionally, three hydrophilic residues, Q409, Y411 and N433, in the middle of β8, β9 and β10 contribute to the membrane defect (S7 Fig) [75].
Spontaneous lateral gate opening and sliding
Previously, spontaneous lateral gate opening of the barrel of N. gonorrhoeae BamA in MD simulations was reported to take place in DMPE at 310 K [22], in DLPC at 310 K and 340 K, and in OM at 340 K [34]. Here we observed lateral gate opening of NgBamA in its native OM at 310K in our equilibrium simulations (Fig 3A). The β1 and β16 strands separate spontaneously with the number of hydrogen bonds formed between β1 and β16 dropping at the beginning of both replicas (Fig 3C). We also observed fluctuations of the lateral gates in HdBamAΔP, HdBamA, and NgBamAΔP systems (S8 Fig). Strand separation stays at 3 Å in SeBamAΔP, EcBamAΔP and EcBamA systems (S8 Fig), which matches the length of a hydrogen bond. TamA’s lateral gate also opens in equilibrium simulations, and it stays open due to a lipid that enters it (S8 Fig).
Fig 3. Lateral gate opening and sliding in equilibrium simulations.

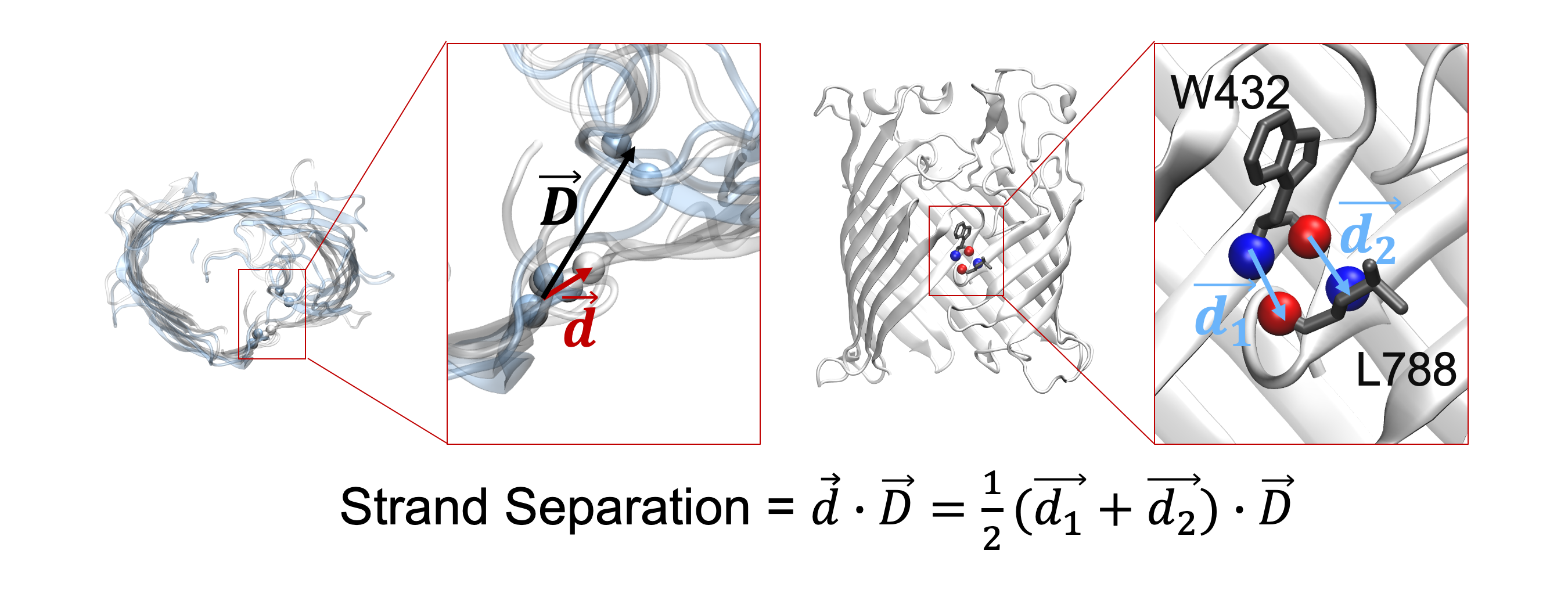
(A) Snapshots of a closed state of NgBamA (top) and the maximum strand separation in NgBamA (bottom) with β1 in red and β16 in purple. (B) Snapshots of β-barrel of NgBamA before (left) and after (right) the sliding. (C) Strand separation (top) and number of hydrogen bonds between backbones of β1 and β16 (bottom) of NgBamA system over time. Strand separation is defined by the average of the distance between O of W432 and N of L788 and the distance between N of W432 and O of L788 in NgBamA. A cutoff distance of 3.5 Å and an angle of 30 degrees are used to define a hydrogen bond.
One of the most unexpected observations from our equilibrium simulations is sliding of the lateral gate (Fig 3B). In our NgBamA equilibrium simulation, W432 and L788 form hydrogen bonds initially. As the lateral gate opens, the hydrogen bonds break and the distance between W432 and L788 fluctuates. Then the distance sharply increases and stays above 8 Å while the number of hydrogen bonds rises simultaneously (Fig 3C), which results from F786 sliding down and forming hydrogen bonds with W432. Additionally, we observed sliding of the lateral gate in HdBamAΔP, which explains the apparent sharp increase in strand separation (S8 Fig). This observed sliding agrees with experimental results in which EcBamA can form disulfide cross-links between lateral gate residues (G433C-T809C and G431C-Q803C) that are 14 Å apart in the crystal structure [33], demonstrating its flexibility.
Energetics of lateral gate opening
To further investigate lateral gate opening, we ran replica-exchange umbrella sampling (REUS) simulations to calculate the potential of mean force (PMF) for opening of all systems with and without POTRA domains. In order to avoid conflating the sliding conformation with the lateral gate opening conformation, we projected the distance of atoms forming hydrogen bonds at the lateral gate on the direction it opens. For example, we projected the vector that the N/O atoms of W432 and O/N atoms of L788 form in NgBamA (and the corresponding residues for other proteins) on their direction at maximum separation, defined as the lateral gate opening coordinate (S2 Fig).
We use FhaC as a control since it is also a member of the Omp85 family but does not assist with OMP insertion [76]. As expected, with or without POTRA domains, the PMFs of BamA and TamA are lower than those of FhaC [34]. Unexpectedly, although all PMFs tend to increase as the strands separate, a well in the PMF appears from 3.5 Å to 5.5 Å. This small drop in the PMFs is due to water molecules going between β1 and β16 and forming hydrogen bonds with the backbone, stabilizing the open state slightly (S9 Fig).
The energy required to open the lateral gate for NgBamAΔP and EcTamAΔP is about 4 kcal/mol lower than that of EcBamAΔP, SeBamAΔP and HdBamAΔP (Fig 4B). The PMFs for systems with POTRA domains are within a relatively larger range from 8 to 20 kcal/mol compared to barrel-only systems (Fig 4A). Interestingly, the PMF of lateral gate opening for NgBamA is sufficiently low to happen spontaneously on the microsecond timescale [34], as observed in our equilibrium simulations. Opposite from NgBamA, whose PMF is lower than NgBamAΔP’s, the PMFs for EcBamA and EcTamA are both higher than those of their respective barrel-only systems. Finally, the PMFs for HdBamA and HdBamAΔP are similar to each other.
Fig 4. PMFs for lateral gate opening.

(A) PMFs of lateral gate opening for systems with POTRA domains. (B) PMFs of lateral gate opening for barrel-only systems. In both panels, the region over which hydrogen bonds between the N- and C-terminal strands are ruptured is indicated.
Interactions between β-barrels and their environments
Based on our equilibrium simulations, we quantified the interaction propensity of each β-barrel residue with lipids, LPS, water, and other protein residues. For each residue, the number of atoms within 4 Å of its side chain for each type of molecule is accumulated over the two 2-μs runs. We plot the percentage for each type in S10 Fig. Unsurprisingly, the 16 β-stands primarily interact with lipids while loops and turns interact with water, as previously reported for another OMP, OmpLA [77]. Additionally, it is evident that the β-strand residues whose side chains are inside the barrel are interacting more with water, while the β-strand residues whose side chains are outside the barrel are interacting with lipid A head group, lipid A tail, PL tail and PL head group periodically.
We developed a metric to quantify the overall difference in the interaction propensities between pairs of proteins (Table 2 and S11 Fig). Looking at EcBamA, for example, we find that SeBamA has the most similar interaction pattern, followed by (in order) HdBamA, NgBamA, EcTamA, and BpFhaC. These differences are consistent with the evolutionary relationships between these bacteria and/or proteins [78], further demonstrating that evolution operates within constraints presented by each protein’s local environment.
Table 2. Measure of the difference of interaction propensities between the proteins.
Briefly, this difference is a Euclidean distance between each set of interaction propensities, averaged over all aligned residues between two proteins. See S11 Fig for further details of the calculation.
| EcBamA | SeBamAΔP | HdBamA | NgBamA | EcTamA | BpFhaC | |
|---|---|---|---|---|---|---|
| EcBamA | 0 | 0.079 | 0.120 | 0.156 | 0.172 | 0.218 |
| SeBamAΔP | 0.079 | 0 | 0.118 | 0.149 | 0.176 | 0.221 |
| HdBamA | 0.120 | 0.118 | 0 | 0.152 | 0.184 | 0.226 |
| NgBamA | 0.156 | 0.149 | 0.152 | 0 | 0.178 | 0.234 |
| EcTamA | 0.172 | 0.176 | 0.184 | 0.178 | 0 | 0.219 |
| BpFhaC | 0.218 | 0.221 | 0.226 | 0.234 | 0.219 | 0 |
One of the most prominent features of the interaction pattern is that some loop residues interact predominantly with water, making them attractive targets of novel antibiotics [79–81]. To better quantify their exposure, we calculated the ratio of each loop residue’s contact area with water to its surface area (S12 Fig). A series of residues on L4, L6 and L7 of EcBamA and SeBamAΔP, which are on the surface of the barrel lid, stand out. The results are consistent with the recent finding that N534, L536, V543, E554, Q561, D562, D568, D569, K644, E645 and L699 of EcBamA interact with a new peptide antibiotic [81]. Notably, loop residues of E. coli and S. enterica could be buried by O-antigen of the bacteria, which is not included in our E. coli and S. enterica systems [82]. In contrast, since the LPS in N. gonorrhoeae and H. ducreyi is a lower-weight form lipooligosaccharides (LOS), which lacks O-antigen [83], the residues on L4 and L7 of NgBamA as well as on L4 and L6 of HdBamA would always be exposed to water.
POTRA domains interact with the OM and with Lpp
We observed extensive interactions between the POTRA domains of all BamAs and TamA with periplasmic leaflet of the OM, as was previously reported for EcBamA [84]. As the contact areas with the OM surface indicate (S13 Fig), the POTRA domains have a strong tendency to touch the membrane. Interestingly, the P1-P3 domains of TamA present levels of contact area similar to the corresponding POTRA domains of BamA of E. coli. For example, P2 of TamA has almost no contact with the OM, similar to P4 of EcBamA. All other POTRA domains of TamA and of EcBamA form extensive interactions with the periplasmic leaflet of the OM; their average contact areas over time ranges from 100 to 2000 Å2, which is considerable compared to the ∼5785 Å2 surface area on average for a POTRA domain (S3 Table).
Peptidoglycan (PG) and its highly abundant protein anchor, Lpp, which positions the PG ∼8-10 nm from the bottom of the OM in the periplasm [57, 60], may affect the conformational dynamics of the POTRA domains. To examine this possibility, we ran a 1.5-μs simulation of EcBamA with PG anchored by Lpp below (Fig 5). Indeed, we observed a number of hydrogen bonds between the POTRA domains and Lpp (∼7 on average). Nonetheless, we still observed significant interactions between the POTRA domains and the OM as well (S13 Fig).
Fig 5. EcBamA with PG anchored by Lpp below.

Glycan strands are in blue and peptides in green, viewed for the membrane plane (A) and periplasm (B). Lpp trimers are shown in purple; BamA is in white.
Discussion
To interrogate the hybrid barrel model of OMP insertion and probe the dynamics of the lateral gate in BamA from different species, we ran a series of equilibrium simulations at 310 K with or without POTRA domains to examine the gate separation. In two 2-μs simulations for each of the nine BamA/TamA systems, we observed spontaneous lateral gate opening for N. gonorrhoeae BamA and E. coli TamA. No lateral gate opening is observed for E. coli, S. enterica, and H. ducreyi BamA, nor for our control protein, FhaC, on the 2-μs time scale. We next calculated the energetic landscape of lateral gate opening, which supports the observations from the equilibrium simulations. NgBamA requires the least energy to open its gate, explaining why it was observed in 2 μs. E. coli and S. enterica systems, on the other hand, require the most energy, consistent with no lateral gate opening observed in equilibrium simulations. Also, after 2-4 Å of separation, all the PMFs of gate opening in BamA and TamA are lower than the respective FhaC systems (with or without POTRA domains; Fig 3). Additional evidence for a functional role of lateral gating in BamA comes from a recent structure in which two BamAs, both with open gates, are in contact [37].
We also analyzed the thickness of membranes around the BamA/TamA barrels to evaluate the passive model of OMP insertion, in which a destabilized membrane region near BamA accelerates the process. Our results clearly show that all BamAs and TamA exhibit a narrowed hydrophobic region in their native outer membranes near the lateral gate. The consistency of this observation across multiple species and membrane compositions strongly supports a key tenet of the passive model.
Overall, our results provide evidence supporting both the hybrid barrel and passive models of BamA-mediated insertion of OMPs. However, these models do not necessarily contradict each other, but rather each may reflect an aspect of the insertion process. Our PMFs indicate that the lateral gate of BamA from some species may not open spontaneously, at least in the absence of some stimulating factor(s). This factor could be, for example, interaction with a nascent OMP, aided by the thinned membrane. Additionally, the positions of accessory proteins may alter the gate structure [17, 18, 20]. For example, BamB may stabilize the open lateral gate through its interactions with the POTRA domains. Although BamB is absent in the BAM complex of N. gonorrhoeae, our results demonstrate that its BamA is able to perform spontaneous lateral gate opening. Other accessory proteins, such as the essential BamD, may serve similar roles in regulating the conformation of BamA.
Supporting information
(XLSX)
(PDF)
(PDF)
(PDF)
For BamA of N. gonorrhoeae and TamA of E. coli, we selected an open state observed in equilibrium simulations to use as a target in Targeted Molecular Dynamics (TMD) for generation of starting states for REUS. For other BamAs for which an open state was not observed in their own equilibrium simulations, we set the target to the backbone of the β-strands of the NgBamA target according to the alignment, and we use TamA as a target for FhaC (See S1 File. for the alignment).
(PNG)
{kind=link}
is the direction that lateral gates open and is the vector formed by N and O atoms that potentially form hydrogen bonds at the lateral gate. For example, the collective variable for BamA of N. gonorrhoeae is defined as the average of the distance between 432 N and 788 O and the distance between 432 O and 788 N projected to the vector between them in the target. N and O of Y432 and I806 are used for BamA of E. coli, Y432 and I800 for S. enterica, Y429 and V789 for H. ducreyi, W432 and L788 for N. gonorrhoeae, Y274 and I571 for TamA of E. coli, and N219 and I548 for FhaC of B. pertussis. The maximum separation and a close state are overlapped on the left with the maximum separation shown in blue and the close state in white. The atoms forming hydrogen bonds at the lateral gate are shown as spheres and colored by name on the right.
(PNG)
{kind=link}
We use 45-100 ns as the final sampling interval for NgBamA. For each umbrella sampling simulation, we calculated PMFs using different sampling intervals. When an additional 5-ns of sampling at the endpoint does not change the PMF by more than 0.2 kcal/mol, the simulation is considered converged.
(PNG)
{kind=link}
(PNG)
{kind=link}
(PNG)
{kind=link}
(PNG)
{kind=link}
(PNG)
{kind=link}
(PNG)
{kind=link}
(PNG)
{kind=link}
For each residue, we add up the number of atoms of water, core, lipid A head group, lipid A tails, PL tails, PL head group, and protein (backbones of the nearby residues are excluded) within 4 Å of the side chain, respectively, over the 4 μs of combined equilibrium simulations. The graph shows the ratio of each category.
(PNG)
{kind=link}
(PNG)
{kind=link}
For each residue, the fraction of surface area in contact with water is determined for each frame; the resulting list over the entire 2×2-μs trajectories is binned into 10 deciles and plotted. The darker the bar for a given residue, the more it is exposed to water.
(PNG)
{kind=link}
(PNG)
{kind=link}
Acknowledgments
Computational resources were provided through the Extreme Science and Engineering Discovery Environment (XSEDE; TG-MCB130173), which is supported by National Science Foundation (NSF; ACI-1548562). This work also used the Hive cluster, which is supported by the NSF under grant number 1828187 and is managed by the Partnership for an Advanced Computing Environment (PACE) at the Georgia Institute of Technology.
Data Availability
All relevant data are within the manuscript and its Supporting information files.
Funding Statement
This work was supported by the National Institutes of Health (R01-GM123169). JL was supported by Office of Undergraduate Education, funded by Tang Aoqing Honors Program in Science. JL is grateful to College of Chemistry of Jilin University for sustaining research training abroad. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Silhavy TJ, Kahne D, Walker S. The bacterial cell envelope. Cold Spring Harb Perspect Biol. 2010;2(5):a000414 10.1101/cshperspect.a000414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fair RJ, Tor Y. Antibiotics and bacterial resistance in the 21st century. Perspect Medicin Chem. 2014;6:2564 10.4137/PMC.S14459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mühlradt PF, Golecki JR. Asymmetrical distribution and artifactual reorientation of lipopolysaccharide in the outer membrane bilayer of Salmonella typhimurium. Eur J Biochem. 1975;51(2):343–352. 10.1111/j.1432-1033.1975.tb03934.x [DOI] [PubMed] [Google Scholar]
- 4. Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003;67:593–656. 10.1128/MMBR.67.4.593-656.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Black PN, Said B, Ghosn C, Beach J, Nunn W. Purification and characterization of an outer membrane-bound protein involved in long-chain fatty acid transport in Escherichia coli. J Biol Chem. 1987;262(3):1412–1419. [PubMed] [Google Scholar]
- 6. Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003;67(4):593–656. 10.1128/MMBR.67.4.593-656.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pagès JM, Amaral L. Mechanisms of drug efflux and strategies to combat them: challenging the efflux pump of Gram-negative bacteria. Biochim Biophys Acta—Proteins and Proteomics. 2009;1794(5):826–833. 10.1016/j.bbapap.2008.12.011 [DOI] [PubMed] [Google Scholar]
- 8. Li XZ, Plésiat P, Nikaido H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin Microbiol Rev. 2015;28(2):337–418. 10.1128/CMR.00117-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Khan NA, Shin S, Chung JW, Kim KJ, Elliott S, Wang Y, et al. Outer membrane protein A and cytotoxic necrotizing factor-1 use diverse signaling mechanisms for Escherichia coli K1 invasion of human brain microvascular endothelial cells. Microb Pathog. 2003;35(1):35–42. 10.1016/S0882-4010(03)00090-1 [DOI] [PubMed] [Google Scholar]
- 10. Castanié-Cornet MP, Cam K, Jacq A. RcsF is an outer membrane lipoprotein involved in the RcsCDB phosphorelay signaling pathway in Escherichia coli. J Bacteriol. 2006;188(12):4264–4270. 10.1128/JB.00004-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rollauer SE, Sooreshjani MA, Noinaj N, Buchanan SK. Outer membrane protein biogenesis in Gram-negative bacteria. Phil Trans R Soc Lond B. 2015;370(1679):20150023 10.1098/rstb.2015.0023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Konovalova A, Kahne DE, Silhavy TJ. Outer membrane biogenesis. Annu Rev Microbiol. 2017;71:539–556. 10.1146/annurev-micro-090816-093754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Koebnik R, Locher KP, Van Gelder P. Structure and function of bacterial outer membrane proteins: barrels in a nutshell. Mol Microbiol. 2000;37(2):239–253. 10.1046/j.1365-2958.2000.01983.x [DOI] [PubMed] [Google Scholar]
- 14. Voulhoux R, Tommassen J. Omp85, an evolutionarily conserved bacterial protein involved in outer-membrane-protein assembly. Res Microbiol. 2004;155(3):129–135. 10.1016/j.resmic.2003.11.007 [DOI] [PubMed] [Google Scholar]
- 15. Walther DM, Rapaport D, Tommassen J. Biogenesis of β-barrel membrane proteins in bacteria and eukaryotes: evolutionary conservation and divergence. Cell Mol Life Sci. 2009;66(17):2789–2804. 10.1007/s00018-009-0029-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hagan CL, Silhavy TJ, Kahne D. β-Barrel membrane protein assembly by the Bam complex. Annu Rev Biochem. 2011;80:189–210. 10.1146/annurev-biochem-061408-144611 [DOI] [PubMed] [Google Scholar]
- 17. Gu Y, Li H, Dong H, Zeng Y, Zhang Z, Paterson NG, et al. Structural basis of outer membrane protein insertion by the BAM complex. Nature. 2016;531(7592):64–69. 10.1038/nature17199 [DOI] [PubMed] [Google Scholar]
- 18. Bakelar J, Buchanan SK, Noinaj N. The structure of the β-barrel assembly machinery complex. Science. 2016;351(6269):180–186. 10.1126/science.aad3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Han L, Zheng J, Wang Y, Yang X, Liu Y, Sun C, et al. Structure of the BAM complex and its implications for biogenesis of outer-membrane proteins. Nat Struct Mol Biol. 2016;23(3):192 10.1038/nsmb.3181 [DOI] [PubMed] [Google Scholar]
- 20. Iadanza MG, Higgins AJ, Schiffrin B, Calabrese AN, Brockwell DJ, Ashcroft AE, et al. Lateral opening in the intact β-barrel assembly machinery captured by cryo-EM. Nat Commun. 2016;7:12865 10.1038/ncomms12865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ricci DP, Silhavy TJ. The Bam machine: a molecular cooper. Biochim Biophys Acta—Bioener. 2012;1818(4):1067–1084. 10.1016/j.bbamem.2011.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Noinaj N, Kuszak AJ, Gumbart JC, Lukacik P, Chang H, Easley NC, et al. Structural insight into the biogenesis of β-barrel membrane proteins. Nature. 2013;501(7467):385–390. 10.1038/nature12521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Noinaj N, Fairman JW, Buchanan SK. The crystal structure of BamB suggests interactions with BamA and its role within the BAM complex. J Mol Biol. 2011;407(2):248–260. 10.1016/j.jmb.2011.01.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim KH, Aulakh S, Paetzel M. Crystal structure of β-barrel assembly machinery BamCD protein complex. J Biol Chem. 2011;286(45):39116–39121. 10.1074/jbc.M111.298166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Knowles TJ, Browning DF, Jeeves M, Maderbocus R, Rajesh S, Sridhar P, et al. Structure and function of BamE within the outer membrane and the β-barrel assembly machine. EMBO Rep. 2011;12(2):123–128. 10.1038/embor.2010.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Plummer AM, Fleming KG. BamA alone accelerates outer membrane protein folding in vitro through a catalytic mechanism. Biochemistry. 2015;54(39):6009–6011. 10.1021/acs.biochem.5b00950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, Kahne D. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell. 2005;121(2):235–245. 10.1016/j.cell.2005.02.015 [DOI] [PubMed] [Google Scholar]
- 28. Hagan CL, Kim S, Kahne D. Reconstitution of outer membrane protein assembly from purified components. Science. 2010;328(5980):890–892. 10.1126/science.1188919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roman-Hernandez G, Peterson JH, Bernstein HD. Reconstitution of bacterial autotransporter assembly using purified components. eLife. 2014;3:e04234 10.7554/eLife.04234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gentle I, Gabriel K, Beech P, Waller R, Lithgow T. The Omp85 family of proteins is essential for outer membrane biogenesis in mitochondria and bacteria. J Cell Biol. 2004;164(1):19–24. 10.1083/jcb.200310092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heinz E, Lithgow T. A comprehensive analysis of the Omp85/TpsB protein superfamily structural diversity, taxonomic occurrence, and evolution. Front Microbiol. 2014;5:370 10.3389/fmicb.2014.00370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Noinaj N, Gumbart JC, Buchanan SK. The β-barrel assembly machinery in motion. Nat Rev Microbiol. 2017;15(4):197–204. 10.1038/nrmicro.2016.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Doerner PA, Sousa MC. Extreme dynamics in the BamA β-barrel seam. Biochemistry. 2017;56(24):3142–3149. 10.1021/acs.biochem.7b00281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lundquist K, Bakelar J, Noinaj N, Gumbart JC. C-terminal kink formation is required for lateral gating in BamA. Proc Natl Acad Sci USA. 2018;115(34):E7942–E7949. 10.1073/pnas.1722530115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Noinaj N, Kuszak AJ, Balusek C, Gumbart JC, Buchanan SK. Lateral opening and exit pore formation are required for BamA function. Structure. 2014;22(7):1055–1062. 10.1016/j.str.2014.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Doyle MT, Bernstein HD. Bacterial outer membrane proteins assemble via asymmetric interactions with the BamA β-barrel. Nat Commun. 2019;10(1):1–13. 10.1038/s41467-019-11230-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tomasek D, Rawson S, Lee J, Wzorek JS, Harrison SC, Li Z, et al. Structure of a nascent membrane protein as it folds on the BAM complex. Nature. 2020; p. 1–6. 10.1038/s41586-020-2370-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ricci DP, Silhavy TJ. Outer Membrane Protein Insertion by the β-barrel Assembly Machine. Protein Secretion in Bacteria. 2019; p. 91–101. 10.1128/ecosalplus.ESP-0035-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kleinschmidt JH. Folding of β-barrel membrane proteins in lipid bilayersUnassisted and assisted folding and insertion. Biochim Biophys Acta—Bioener. 2015;1848(9):1927–1943. 10.1016/j.bbamem.2015.05.004 [DOI] [PubMed] [Google Scholar]
- 40. Gruss F, Zähringer F, Jakob RP, Burmann BM, Hiller S, Maier T. The structural basis of autotransporter translocation by TamA. Nat Struct Mol Biol. 2013;20(11):1318 10.1038/nsmb.2689 [DOI] [PubMed] [Google Scholar]
- 41. Selkrig J, Belousoff MJ, Headey SJ, Heinz E, Shiota T, Shen HH, et al. Conserved features in TamA enable interaction with TamB to drive the activity of the translocation and assembly module. Sci Rep. 2015;5:12905 10.1038/srep12905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J Comput Chem. 2008;29(11):1859–1865. 10.1002/jcc.20945 [DOI] [PubMed] [Google Scholar]
- 43. Wu EL, Cheng X, Jo S, Rui H, Song KC, Dvila-Contreras EM, et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J Comput Chem. 2014;35(27):1997–2004. 10.1002/jcc.23702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vance JE, Vance DE. Biochemistry of lipids, lipoproteins and membranes. Elsevier; 2008. [Google Scholar]
- 45. Olsen RW, Ballou CE. Acyl phosphatidylglycerol a new phospholipid from Salmonella typhimurium. J Biol Chem. 1971;246(10):3305–3313. [PubMed] [Google Scholar]
- 46. Sud IJ, Feingold DS. Phospholipids and fatty acids of Neisseria gonorrhoeae. J Bacteriol. 1975;124(2):713–717. 10.1128/JB.124.2.713-717.1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jantzen E, Berdal BP, Omland T. Cellular fatty acid composition of Haemophilus species, Pasteurella multocida, Actinobacillus Actinomycetemcomitans and Haemophilus vaginalis (Corynebacterium vaginale). Acta Pathol Microbiol Scand B. 1980;88(1–6):89–93. [DOI] [PubMed] [Google Scholar]
- 48. White DC. Lipid composition of the electron transport membrane of Haemophilus parainfluenzae. J Bacteriol. 1968;96(4):1159–1170. 10.1128/JB.96.4.1159-1170.1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kawai Y, Moribayashi A. Characteristic lipids of Bordetella pertussis: simple fatty acid composition, hydroxy fatty acids, and an ornithine-containing lipid. J Bacteriol. 1982;151(2):996–1005. 10.1128/JB.151.2.996-1005.1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Raetz CR. Biochemistry of endotoxins. Annu Rev Biochem. 1990;59(1):129–170. 10.1146/annurev.bi.59.070190.001021 [DOI] [PubMed] [Google Scholar]
- 51. Trees DL, Morse SA. Chancroid and Haemophilus ducreyi: an update. Clin Microbiol Rev. 1995;8(3):357–375. 10.1128/CMR.8.3.357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gibson BW, Melaugh W, Phillips NJ, Apicella MA, Campagnari AA, Griffiss J. Investigation of the structural heterogeneity of lipooligosaccharides from pathogenic Haemophilus and Neisseria species and of R-type lipopolysaccharides from Salmonella typhimurium by electrospray mass spectrometry. J Bacteriol. 1993;175(9):2702–2712. 10.1128/jb.175.9.2702-2712.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jenning HJ, Johnson KG, Kenne L. The structure of an R-type oligosaccharide core obtained from some lipopolysaccharides of Neisseria meningitidis. Carbohydr Res. 1983;121:233–241. 10.1016/0008-6215(83)84020-8 [DOI] [PubMed] [Google Scholar]
- 54. Geurtsen J, Dzieciatkowska M, Steeghs L, Hamstra HJ, Boleij J, Broen K, et al. Identification of a novel lipopolysaccharide core biosynthesis gene cluster in Bordetella pertussis, and influence of core structure and lipid A glucosamine substitution on endotoxic activity. Infect Immun. 2009;77(7):2602–2611. 10.1128/IAI.00033-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. AlBitar-Nehme S, Basheer SM, Njamkepo E, Brisson JR, Guiso N, Caroff M. Comparison of lipopolysaccharide structures of Bordetella pertussis clinical isolates from pre-and post-vaccine era. Carbohydr Res. 2013;378:56–62. 10.1016/j.carres.2013.05.002 [DOI] [PubMed] [Google Scholar]
- 56. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. 10.1063/1.445869 [DOI] [Google Scholar]
- 57. Gumbart JC, Beeby M, Jensen GJ, Roux B. Escherichia coli peptidoglycan structure and mechanics as predicted by atomic-scale simulations. PLoS Comput Biol. 2014;10:e1003475 10.1371/journal.pcbi.1003475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hwang H, Paracini N, Parks JM, Lakey JH, Gumbart JC. Distribution of mechanical stress in the Escherichia coli cell envelope. Biochim Biophys Acta Biomembr. 2018;1860:2566–2575. 10.1016/j.bbamem.2018.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shu W, Liu J, Ji H, Lu M. Core structure of the outer membrane lipoprotein from Escherichia coli at 1.9 Å resolution. J Mol Biol. 2000;299:1101–1112. 10.1006/jmbi.2000.3776 [DOI] [PubMed] [Google Scholar]
- 60. Asmar AT, Collet JF. Lpp, the Braun lipoprotein, turns 50 major achievements and remaining issues. FEMS Microbiol Lett. 2018;365(18):fny199 10.1093/femsle/fny199 [DOI] [PubMed] [Google Scholar]
- 61. Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802. 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Case DA, Cheatham TE III, Darden T, Gohlke H, Luo R, Merz KM Jr, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–1688. 10.1002/jcc.20290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods. 2017;14(1):71–73. 10.1038/nmeth.4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B. 2010;114:7830–7843. 10.1021/jp101759q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys. 1994;101(5):4177–4189. 10.1063/1.467468 [DOI] [Google Scholar]
- 66. Darden T, York D, Pedersen L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092. 10.1063/1.464397 [DOI] [Google Scholar]
- 67. Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graphics. 1996;14(1):33–38. 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- 68. Hopkins CW, Le Grand S, Walker RC, Roitberg AE. Long-time-step molecular dynamics through hydrogen mass repartitioning. J Chem Theory Comput. 2015;11(4):1864–1874. 10.1021/ct5010406 [DOI] [PubMed] [Google Scholar]
- 69. Balusek C, Hwang H, Lau CH, Lundquist K, Hazel A, Pavlova A, et al. Accelerating membrane simulations with Hydrogen Mass Repartitioning. J Chem Theory Comput. 2019;15:4673–4686. 10.1021/acs.jctc.9b00160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sugita Y, Kitao A, Okamoto Y. Multidimensional replica-exchange method for free-energy calculations. J Chem Phys. 2000;113(15):6042–6051. 10.1063/1.1308516 [DOI] [Google Scholar]
- 71. Schlitter J, Engels M, Krüger P. Targeted molecular dynamics: a new approach for searching pathways of conformational transitions. J Mol Graphics. 1994;12(2):84–89. 10.1016/0263-7855(94)80072-3 [DOI] [PubMed] [Google Scholar]
- 72. Fiorin G, Klein ML, Hénin J. Using collective variables to drive molecular dynamics simulations. Mol Phys. 2013;111(22–23):3345–3362. 10.1080/00268976.2013.813594 [DOI] [Google Scholar]
- 73. Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J Comput Chem. 1992;13(8):1011–1021. [Google Scholar]
- 74. Danoff EJ, Fleming KG. Membrane defects accelerate outer membrane β-barrel protein folding. Biochemistry. 2015;54(2):97–99. 10.1021/bi501443p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157(1):105–132. 10.1016/0022-2836(82)90515-0 [DOI] [PubMed] [Google Scholar]
- 76. Maier T, Clantin B, Gruss F, Dewitte F, Delattre AS, Jacob-Dubuisson F, et al. Conserved Omp85 lid-lock structure and substrate recognition in FhaC. Nat Commun. 2015;6:7452 10.1038/ncomms8452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wu EL, Fleming PJ, Yeom MS, Widmalm G, Klauda JB, Fleming KG, et al. E. coli outer membrane and interactions with OmpLA. Biophys J. 2014;106(11):2493–2502. 10.1016/j.bpj.2014.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ciccarelli FD, Doerks T, Von Mering C, Creevey CJ, Snel B, Bork P. Toward automatic reconstruction of a highly resolved tree of life. Science. 2006;311(5765):1283–1287. 10.1126/science.1123061 [DOI] [PubMed] [Google Scholar]
- 79. Imai Y, Meyer KJ, Iinishi A, Favre-Godal Q, Green R, Manuse S, et al. A new antibiotic selectively kills Gram-negative pathogens. Nature. 2019;576(7787):459–464. 10.1038/s41586-019-1791-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Urfer M, Bogdanovic J, Monte FL, Moehle K, Zerbe K, Omasits U, et al. A peptidomimetic antibiotic targets outer membrane proteins and disrupts selectively the outer membrane in Escherichia coli. J Biol Chem. 2016;291(4):1921–1932. 10.1074/jbc.M115.691725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Luther A, Urfer M, Zahn M, Müller M, Wang SY, Mondal M, et al. Chimeric peptidomimetic antibiotics against Gram-negative bacteria. Nature. 2019;576(7787):452–458. 10.1038/s41586-019-1665-6 [DOI] [PubMed] [Google Scholar]
- 82. Domínguez-Medina CC, Pérez-Toledo M, Schager AE, Marshall JL, Cook CN, Bobat S, et al. Outer membrane protein size and LPS O-antigen define protective antibody targeting to the Salmonella surface. Nat Commun. 2020;11(1):1–11. 10.1038/s41467-020-14655-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Preston A, Mandrell RE, Gibson BW, Apicella MA. The lipooligosaccharides of pathogenic gram-negative bacteria. Crit Rev Microbiol. 1996;22(3):139–180. 10.3109/10408419609106458 [DOI] [PubMed] [Google Scholar]
- 84. Fleming PJ, Patel DS, Wu EL, Qi Y, Yeom MS, Sousa MC, et al. BamA POTRA domain interacts with a native lipid membrane surface. Biophys J. 2016;110(12):2698–2709. 10.1016/j.bpj.2016.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
(PDF)
(PDF)
(PDF)
For BamA of N. gonorrhoeae and TamA of E. coli, we selected an open state observed in equilibrium simulations to use as a target in Targeted Molecular Dynamics (TMD) for generation of starting states for REUS. For other BamAs for which an open state was not observed in their own equilibrium simulations, we set the target to the backbone of the β-strands of the NgBamA target according to the alignment, and we use TamA as a target for FhaC (See S1 File. for the alignment).
(PNG)
is the direction that lateral gates open and is the vector formed by N and O atoms that potentially form hydrogen bonds at the lateral gate. For example, the collective variable for BamA of N. gonorrhoeae is defined as the average of the distance between 432 N and 788 O and the distance between 432 O and 788 N projected to the vector between them in the target. N and O of Y432 and I806 are used for BamA of E. coli, Y432 and I800 for S. enterica, Y429 and V789 for H. ducreyi, W432 and L788 for N. gonorrhoeae, Y274 and I571 for TamA of E. coli, and N219 and I548 for FhaC of B. pertussis. The maximum separation and a close state are overlapped on the left with the maximum separation shown in blue and the close state in white. The atoms forming hydrogen bonds at the lateral gate are shown as spheres and colored by name on the right.
(PNG)
We use 45-100 ns as the final sampling interval for NgBamA. For each umbrella sampling simulation, we calculated PMFs using different sampling intervals. When an additional 5-ns of sampling at the endpoint does not change the PMF by more than 0.2 kcal/mol, the simulation is considered converged.
(PNG)
(PNG)
(PNG)
(PNG)
(PNG)
(PNG)
(PNG)
For each residue, we add up the number of atoms of water, core, lipid A head group, lipid A tails, PL tails, PL head group, and protein (backbones of the nearby residues are excluded) within 4 Å of the side chain, respectively, over the 4 μs of combined equilibrium simulations. The graph shows the ratio of each category.
(PNG)
(PNG)
For each residue, the fraction of surface area in contact with water is determined for each frame; the resulting list over the entire 2×2-μs trajectories is binned into 10 deciles and plotted. The darker the bar for a given residue, the more it is exposed to water.
(PNG)
(PNG)
Data Availability Statement
All relevant data are within the manuscript and its Supporting information files.