

Abstract
Background
Tyrosine kinase inhibitor (TKI) resistance is a major obstacle in treatment of non-small cell lung cancer (NSCLC). MET amplification drives resistance to EGFR-TKIs in 5–20% of initially sensitive
EGFR
mutated NSCLC patients, and combined treatment with EGFR-TKIs and MET-TKIs can overcome this resistance. Yet, inevitably MET-TKI resistance will also occur. Hence, knowledge on development of this sequential resistance is important for identifying the proper next step in treatment.
Methods
To investigate sequential resistance to MET-TKI treatment, we established a two-step TKI resistance model in EGFR-mutated HCC827 cells with MET amplification-mediated erlotinib resistance. These cells were subsequently treated with increasing doses of the MET-TKIs capmatinib or crizotinib in combination with erlotinib to establish resistance.
Results
In all the MET-TKI resistant cell lines, we systematically observed epithelial-to-mesenchymal transition (EMT) evident by decreased expression of E-cadherin and increased expression of vimentin and ZEB1. Furthermore, FGFR1 expression was increased in all MET-TKI resistant cell lines and four out of the six resistant cell lines had increased sensitivity to FGFR inhibition, indicating FGFR1-mediated bypass signaling.
Conclusions
EMT is common in the development of sequential EGFR-TKI and MET-TKI resistance in NSCLC cells. Our findings contribute to the evidence of EMT as a common TKI resistance mechanism.
Keywords: Non-small cell lung cancer (NSCLC), epidermal growth factor receptor (EGFR), MET receptor tyrosine kinase, tyrosine kinase inhibitor resistance, epithelial-to-mesenchymal transition (EMT)
Introduction
Acquired drug resistance continuously causes disease progression and limits the response to targeted therapies in non-small cell lung cancer (NSCLC) patients. Bypass signaling through the MET receptor caused by gene amplification is causing drug resistance in 5–20% of EGFR-mutated NSCLC patients treated with EGFR-targeting tyrosine kinase inhibitors (TKIs) (1-3). Patients with MET-amplified tumors respond to MET-TKIs (4,5), but inevitably resistance will emerge to this treatment as well.
Epithelial-to-mesenchymal transition (EMT) is also associated with resistance to EGFR-TKIs (6) and known to drive EGFR-TKI resistance (1,7). Furthermore, increased FGFR1 expression has shown to correlate with EMT in EGFR-TKI resistance, and FGFR1-inhibition can prevent the outgrowth of resistant clones, suggesting this bypass pathway as a mediator of EMT-associated EGFR-TKI resistance (8,9). The EMT phenotype is characterized by loss of epithelial cell polarity and adhesion molecules such as E-cadherin together with acquisition of mesenchymal markers like vimentin (10). The transition depends on expression of at least a subset of the core EMT transcription factors ZEB1, ZEB2, SNAI1, SNAI2, TWIST1 and TWIST2, which are involved in repression of epithelial markers and expression of mesenchymal markers (11). Expression of the EMT transcription factors is regulated both at the transcriptional level and post-transcriptional level by microRNAs (miRNAs). The latter is exemplified by the members of the miR-200 family which regulate EMT through a double negative feedback loop with ZEB1 and ZEB2. These miRNAs are often downregulated in EMT (12,13).
Knowledge on evolution of drug resistance during TKI treatment is sparse. Resistance has been suggested to develop from selection of preexisting cells with genetic resistance mechanisms or by adaptation of subpopulations of drug-tolerant persister cells that survive the initial drug-exposure (14). These drug-persisting cells are largely quiescent cells, but can resume proliferation in the presence of drug and acquire a genetic resistance mechanism during treatment (14,15). Persister cells have been hypothesized to cause drug resistance in NSCLC to targeted therapy. One example is EGFR-TKI resistance caused by EGFR T790M. Hata and colleagues reported that both selection of T790M-positive preexisting clones or the acquisition of the T790M mutation over time in initial T790M-negative drug-tolerant cells gave rise to resistance (16).
To elucidate the resistance mechanisms to MET-TKIs in sequential exposure to EGFR inhibition, we established a cellular model in EGFR-mutated cells, where erlotinib-resistant, MET-amplified cell clones were treated with the MET-TKIs capmatinib or crizotinib combined with erlotinib to establish sequential resistant cell lines.
We present the following article in accordance with the MDAR reporting checklist (available at http://dx.doi.org/10.21037/tlcr-20-522).
Methods
Cell culture and reagents
Erlotinib-resistant HCC827ER clones were developed as described in (9) and were cultured in RPMI-1640 medium (Gibco, Thermo Fisher Scientific) supplemented with 10% FBS, penicillin and streptomycin, amphotericin, HEPES buffer, Sodium Pyruvate and 5µM erlotinib at 37 °C and 5% CO2. The original HCC827 cell line was purchased from ATCC. The authenticity of HCC827ER was verified by Short Tandem Repeat fingerprint analyses with GenePrint 10 System (Promega). All TKIs used were from Selleckchem.
Establishment of resistant cells
Erlotinib-resistant, MET-amplified HCC827ER clones were treated with increasing doses of capmatinib (0–12 nM) or crizotinib (0–1 µM) in combination with 5µM erlotinib. Resistance to the highest MET-TKI concentrations was reached after 4 months of treatment. The resistant cells were maintained in culture medium with both EGFR and MET inhibitors unless otherwise noticed.
Viability assays
Cells were seeded at a density of 5,000 cells/well in a 96-well plate and treated in four replicates with the indicated drug the following day. After 72 hours drug exposure, cell viability was measured with the CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay (Promega) according to the manufacturer's protocol.
MET copy number variation analyses
DNA was isolated from cells using the QIAamp DNA mini kit (Qiagen) according to the manufacturer’s protocol. MET copy number was determined with PrimePCR ddPCR MET Copy Number Variation Assay (Unique assay ID: dHsaCP2500321, Bio-Rad) performed using the QX200 Droplet Digital system (Bio-Rad) according to the manufacturer’s protocol. The EIF2C1 PrimePCR ddPCR assay (Unique assay ID: dHsaCP2500349, Bio-Rad) was used as copy number reference. Each sample was analyzed in technical triplicates.
RNA and microRNA extraction, cDNA and qPCR
RNA was isolated with the RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. The initial flow-through was stored and used for miRNA isolation with the RNeasy Micro Kit (Qiagen) following the manufacturer’s protocol but leaving out the steps including buffer RW1. miRNAs were eluted in a total volume of 30 µL.
cDNA was synthesized from 100 ng RNA in a 20 µL reaction mix including 1× PCR buffer, 6.25 mM MgCl2 (25 mmol/L), 50U MulV reverse transcriptase, 20U RNase inhibitor (Applied Biosystems, Thermo Fisher), 2.5 µM oligo d(T) (50 µmol/L) (DNA technology) and 1 mM of each dNTP (VWR). Reverse transcription was performed at 45 °C for 30 min, 99 °C 5min and subsequently cooled to 4 °C.
Quantitative Real-Time PCR (qPCR) was conducted on a Lightcycler 480 II PCR system (Roche) using SYBR green for quantification. The reaction mix consisted of 5 µL Lightcycler 480 SYBR Green 1 Master Mix Buffer (Roche), 250 nM of each primer (Eurofins Genomics), 1 µL cDNA and H2O to a final volume of 10 µL. SDHA was used as reference based on NormFinder analysis (17). Primer sequences and annealing temperatures are listed in Table S1. Concentrations were calculated from standard curves.
Table S1. qPCR primers.
| Gene | Forward Primer (5’-3’) | Reverse Primer (5’-3’) | Annealing temp. (°C) | Primer Conc. (pmol/µL) | Amplicon size (bp) |
|---|---|---|---|---|---|
| SDHA | TGGGAACAAGAGGGCATCTG | CCACCACTGCATCAAATTCATG | 62 | 5 | 86 |
| MET | TGGAGACACTGGATGGGAGT | CAGCGCGTTGACTTATTCAT | 60 | 5 | 193 |
| FGFR1 | TGGCCTCCAAGAAGTGCATA | AAATAATGCCTCGGGTGCCA | 60 | 5 | 179 |
| ZEB1 | AGACATGTGACGCAGTCTGGGT | TGGGCATTCATATGGCTTCTCTCCA | 58 | 5 | 129 |
| Vimentin | GACCAGCTAACCAACGACAAA | TGAAAGATTGCAGGGTGTTT | 58 | 5 | 136 |
| E-cadherin | GTCCTGGGCAGACTGAATTT | GACCAAGAAATGGATCTGTGG | 58 | 5 | 150 |
Reverse transcription of miRNA was performed on 5 µL miRNA-eluate using miRNA specific primers and TaqMan MicroRNA Reverse Transcription kit (Applied biosystems, Thermo Fisher). Afterwards, quantification was performed with specific TaqMan probes and TaqMan Universal Master Mix II (Applied Biosystems, Thermo Fisher) according to the manufacturer’s protocol on the Lightcycler 480 II PCR system (Roche). The concentration was normalized to miR-16 (Applied Biosystems, Thermo Fisher) using the delta-delta method (18).
All gene expression analyses were performed in technical triplicates.
Western blotting and phospho-receptor-tyrosine-kinase blots
Protein was harvested from cells using a NP-40 lysis buffer conditioned with 10 µg/mL aprotinin and leupeptin and 1 mM orthovanadate. Briefly, cells were scraped of in lysis buffer, incubated on ice for 15 min and then sonicated 3×15 sec at low intensity. Then samples were centrifuged at 14,000 g 10 min at 4 °C. Protein concentrations were measured using the Pierce BCA assay (Thermo Fisher) and equal amounts of protein were loaded on a NuPage 4×12% Bis-Tris gel (Thermo Fisher). After blotting, the membrane was blocked with either 5% bovine serum albumin (BSA) or 5% skimmed milk depending on the antibody as described in Table S2. The membranes were incubated with primary antibody over night at 4 °C, washed and incubated with secondary antibody 1 h at room temperature (RT) before development with ECL SuperSignal West Dura Extended Duration Substrate (Thermo Fisher) using the ImageQuant LAS 4000 system (GE Healtcare Life Sciences). Each sample was analyzed in a minimum of two replicates.
Table S2. Antibodies.
| Target | Manufacturer | Catalog number | Source | Size (kDa) | Antibody diluent | Dilution factor | Blocking buffer |
|---|---|---|---|---|---|---|---|
| Primary antibodies | |||||||
| MET | Cell Signaling Technology | 3127 | Mouse | 145, 170 | 5% milk | 1:1,000 | 5% milk |
| p-MET | Cell Signaling Technology | 3129s | Rabbit | 145, 170 | 5% BSA | 1:1,000 | 5% BSA |
| FGFR1 | Cell Signaling Technology | 9740S | Rabbit | 92, 120, 145 | 5% BSA | 1:500 | 5% BSA |
| E-cadherin | BD Biosciences | 610182 | Mouse | 120 | 5% BSA | 1:2,000 | 5% BSA |
| Vimentin | Abcam | AB20346 | Mouse | 54 | 5% milk | 1:1,000 | 5% milk |
| ZEB1 | Nordic Biosite | A301-922A | Rabbit | 200 | 5% BSA | 1:500 | 5% BSA |
| Tubulin | Sigma Aldrich | T9026 | Mouse | 50 | 5% milk | 1:1,000 | 5% milk |
| Secondary antibodies | |||||||
| Anti-rabbit | Cell Signaling Technology | 7074 | Goat | 5% milk | 1:5,000 | ||
| Anti-mouse | Cell Signaling Technology | 7076 | Horse | 5% milk | 1:2,000 | ||
Proteins were also analyzed on a Proteome Profiler Human Phospho-Receptor Tyrosine kinase array (R&D systems) screening 49 RTKs according to the manufacturer’s protocol.
DNA and RNA sequencing
DNA and RNA were submitted to sequencing with the Oncomine focus panel (Thermo Fisher) on the Ion Torrent platform (Thermo Fisher) according to the manufacturer’s protocol.
Immunofluorescence analyses
Cells were seeded in six replicates in a 96-well plate and grown to 70% confluence. Then fixed in 4% paraformaldehyde for 20 min at RT and permeabilized in 0.5% Triton X-100 (Sigma), blocked in 1% BSA 1 h and incubated with primary antibody for 1h at RT. Washed and then incubated with secondary antibody 1h at RT. The antibodies used were anti-vimentin (1:500, mouse, Abcam AB20346), anti-E-cadherin (1:1,000, mouse, BD Bioscience 61018) and secondary antibody Alexa 555 conjugated donkey anti-mouse IgG (1:2,000, Invitrogen, Thermo Fisher A32773). Nuclei were stained with DAPI (Sigma-Aldrich). Each sample was analyzed in a minimum of two replicates. Images were taken on a Zeiss Axiovert 200M, and merged using Image J (National Institutes of Health, USA).
Statistics and graphs
Data is presented as means with standard deviations (mean of technical replicates) and data analyses were performed in Graphpad prism v.8.3.0 software (Graphpad Software). Comparisons of statistical significance were performed by unpaired, t-tests with a two-sided P value. P values ≤0.05 were considered statistically significant. Multiple t-tests were corrected for multiple comparisons by the Holm-Sidak method.
Ethics statement
This study does not involve human subjects. Therefore, the authors have not obtained ethical approval or conducted the study in accordance with the Declaration of Helsinki.
Results
Establishment of MET-TKI resistant cell lines
Erlotinib-resistant HCC827ER cells were previously established by treatment of the EGFR-mutated NSCLC cell line HCC827 with increasing doses of erlotinib until resistance was reached at 5 µM erlotinib (9). MET-amplified clones were isolated from HCC827ER by limited dilution. To identify mechanisms to sequential resistance we established MET-TKI resistance in these MET-amplified, erlotinib-resistant HCC827ER cell clones.
Before initiation of resistance development, we investigated the effect of erlotinib on the response to MET-TKI treatment, since amplification of MET was acquired as a bypass mechanism to erlotinib resistance. We demonstrated that the MET-TKI in combination with erlotinib achieved the highest inhibitory effect (Figure S1). Therefore, MET-TKI resistance was established in the presence of 5 µM erlotinib. We established MET-TKI resistance in three distinct MET-amplified, erlotinib-resistant HCC827ER cell clones (3PAR, 8PAR and 12PAR) by treatment with stepwise escalating doses of the MET-TKIs capmatinib or crizotinib in combination with erlotinib. MET-TKI resistance was established in single cell derived clones of MET-amplified HCC827ER cells to exclude preexisting resistant cell populations. This resulted in a total of six sequential resistant cell lines, resistant to a combination of erlotinib and a MET-TKI. The resistant cell lines were called 3CRR, 8CRR, 12CRR, 3CAR, 8CAR and 12CAR, with the number referring to the original parental HCC827ER clone, and CRR and CAR to crizotinib or capmatinib resistant, respectively (Figure 1A). At the highest achieved MET-TKI concentrations, the resistant cell lines were insensitive to combined inhibition of MET and EGFR compared to the parental cell lines (Figure 1B). The amplified copy number of MET, acquired during establishment of erlotinib resistance, was retained in all resistant cell lines (Figure 1C). Moreover, the MET-TKI resistant cells kept the original sensitizing EGFR del19 mutation, present in the HCC827 cells before development of erlotinib resistance (data not shown). MET mRNA was expressed in all the resistant cell lines, but with decreased expression in 3CRR, 8CRR, 8CAR and 12CRR (Figure S2). MET protein expression was also detected in all the resistant cell lines, but in contrast to the parental cells, MET was not phosphorylated (Figure 2A).
Figure S1.

MTS analysis of cell viability for parental cells treated with increasing concentrations of capmatinib or crizotinib with or without 5 µM erlotinib. All values are normalized to the value of the untreated sample of each individual cell line.
Figure 1.

Development of MET-TKI resistance. (A) Schematic representation of development of MET-TKI resistance in MET-amplified clones (3PAR, 8PAR and 12PAR) isolated from the erlotinib-resistant HCC827ER cell population. Treatment of the three clones with either crizotinib or capmatinib resulted in three crizotinib resistant cell lines (3CRR, 8CRR, 12CRR) and three capmatinib resistant cell lines (3CAR, 8CAR and 12CAR). (B) MTS analysis of cell viability for parental and resistant cells treated with increasing concentrations of crizotinib and capmatinib combined with 5 µM erlotinib. All values are normalized to the value of the untreated sample of each individual cell line. Significance between parental and resistant cells is calculated for each concentration (*P≤0.05). IC50 values of capmatinib and crizotinib for each cell line are presented. (C) ddPCR analysis of MET copy number in parental and resistant cells. The MET copy number was normalized to copies of EIF2C1.
Figure S2.
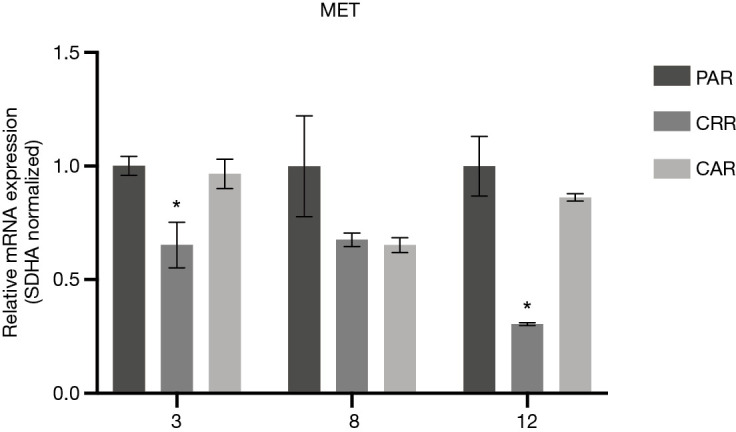
mRNA expression of MET in parental and resistant cells. Values are normalized to SDHA and subsequently to the expression in the parental cell line. Significance between the resistant cells compared to the parental cells is calculated and denoted by an asterisk (*P≤0.05).
Figure 2.

Characterization of MET-TKI resistant cells. (A) Western blot analysis of MET, pMET and EMT markers. Tubulin was used as loading control. (B) Expression profile of EMT markers in parental and resistant cells. Values are normalized to SDHA and subsequently to the parental cell line. miR-200c expression is normalized the level of miR-16 and subsequently to the parental cell line. Significance between the resistant cells compared to the parental cells is calculated and denoted by an asterisk (*P≤0.05). (C) Immunofluorescence staining of vimentin and E-cadherin (red) in parental and resistant cells (×40, scale bar =20 µm). Nuclear staining with DAPI (blue). (D) mRNA expression profile of EMT markers in 12CRR and 12CAR during development of resistance. Values are normalized to SDHA and subsequently to the expression in 12PAR. Significance between the resistant cells from each concentration step compared to the parental cells is calculated and denoted by an asterisk (*P≤0.05).
Sequential MET-TKI resistance is associated with EMT
In all six cases of MET-TKI resistance the cells presented with an EMT phenotype. The MET-TKI resistant cells changed morphology to an elongated spindle-like phenotype compared to the parental cells, together with simultaneous increased expression of the mesenchymal markers vimentin and ZEB1 and decreased expression of E-cadherin (Figure 2A,B,C, Figure S3). 12CAR and 12CRR presented the most pronounced EMT gene-expression phenotype, while EMT-associated gene expression changes in 3CRR, 3CAR, 8CRR and 8CAR were less pronounced and occurred at a later time point in the resistance development (Figure 2D, Figure S4). Expression of another EMT epithelial marker, miR-200c, was substantially decreased in 12CAR and 12CRR, and to a minor degree in 3CAR and 3CRR (Figure 2B). The decrease in mir-200c expression occurred at a later stage in the resistance development than the increase in ZEB1 expression (Figure 2D). This is in accordance with the previously described transcriptional repression of miR-200c by ZEB1 in an auto-regulatory loop (12). In support of ZEB1 as a key factor in transcriptional downregulation of epithelial genes in this model, E-cadherin expression decrease also occurred at a later stage than the increase of ZEB1 expression in 12CAR and 12CRR (Figure 2D).
Figure S3.

Morphology of parental and resistant cells (×10, scale bar =100 µm).
Figure S4.

Expression profile of EMT markers and FGFR1 in 3CAR and 3CRR (A) and 8CAR and 8CRR (B) during development of resistance. Values are normalized to SDHA and subsequently to the corresponding parental cell line. Significance between the resistant cells from each concentration step compared to the parental cells is calculated and denoted by an asterisk (*P≤0.05).
FGFR1 is involved in MET-TKI resistance
Increased FGFR1 expression and signaling have previously been associated with EMT (8,9,19). Here, we observed a substantial increase in FGFR1 expression in all resistant cell lines (Figure 3A,B) that occurred simultaneously with increase in expression of vimentin and ZEB1 (Figure 3C, Figure 2D, Figure S4). Additionally, 4 out of the 6 resistant cell lines, 3CAR, 3CRR, 8CRR and 12CRR, showed significantly decreased survival in response to treatment with the FGFR inhibitor AZD4547. Treatment with AZD4547 only had minimal effect on the parental cells. 8PAR and 12PAR showed no change in survival, whereas a significant reduction in survival was observed in 3PAR. However, this reduction was less pronounced than the effect of AZD4547 on the corresponding resistant cell lines 3CRR and 3CAR (Figure 3D). Phospho-receptor-tyrosine-kinase arrays, detecting phosphorylation of 49 human receptor tyrosine kinases (RTKs), showed no activation of RTKs in the resistant cells (data not shown). We have previously noted that FGFR1 phosphorylation is not detected on these RTK arrays despite examining HCC827 cells with active FGFR1 signaling (20).
Figure 3.

FGFR1 expression and AZD4547 sensitivity. (A) Expression of FGFR1 mRNA in parental and resistant cells. Values are normalized to SDHA and subsequently to the expression in the parental cell line. Significance between the resistant cells compared to the parental cells is calculated and denoted by an asterisk (*P≤0.05). (B) Western blot analysis of FGFR1. Tubulin was used as loading control. (C) Expression of FGFR1 in 12CRR and 12CAR during development of resistance. Values are normalized to SDHA and subsequently to the expression in 12PAR. Significance between the resistant cells from each concentration step compared to the parental cells is calculated and denoted by an asterisk (*P≤0.05). (D) MTS analysis of cell viability for parental and resistant cells treated with 5 µM AZD4547, an FGFR inhibitor. All values are normalized to the value of the untreated sample of each individual cell line (*P≤0.05, ns: not significant).
Targeted sequencing with the Oncomine Focus Panel, covering SNVs, indels, CNVs and fusions in 52 cancer-associated genes did not reveal evidence of alterations previously described to confer TKI resistance. We noted a minor HER2 copy number gain in 3CAR, 8CRR and 8CAR compared to the parental cells (https://cdn.amegroups.cn/static/application/f77d9bd5fdf9777716519cebfc4ab1cc/tlcr-20-522-1.pdf), but this was not associated with HER2 activation in the phospho-RTK array analysis (data not shown). The obtained sequencing results are more in alignment with MET-TKI resistance being epigenetically mediated through EMT and the associated upregulation of FGFR1 than being mediated by a common genetic alteration.
Discussion
MET bypass signaling caused by amplification of the MET gene is the second leading cause of EGFR-TKI resistance in NSCLC second to EGFR T790M mutations. Combining EGFR-TKIs and MET-TKIs can overcome this resistance, but inevitably MET-TKI resistance develops and causes sequential resistance. With the persisting obstacle of acquired resistance and emergence of sequential TKI resistance, there is a need to understand the resistance mechanism and dynamics of resistance development to improve treatment strategies. In this study, we treated erlotinib-resistant, MET-amplified HCC827ER clones with increasing doses of crizotinib or capmatinib in combination with erlotinib to investigate the mechanisms causing MET-TKI resistance in a sequential resistance cell model. MET-TKI resistance was developed in combination with erlotinib to achieve the highest inhibitory effect, which is in line with previous research (3). We found that all replicates of resistance presented with EMT independent of the type of MET-TKI. EMT is a known mechanism of resistance to TKIs in NSCLC primarily documented in cellular studies exploring resistance to EGFR-TKIs, but also in resistance to first-line MET-TKIs and ALK-TKIs (6,8,9,21,22). Furthermore, EMT has been verified in a limited number of clinical studies of progression samples (1,22-24). To our knowledge, this is the first evidence of EMT in resistance to crizotinib or capmatinib in NSCLC and in sequential EGFR-TKI and MET-TKI resistance. The resistant cells presented with varying expression of EMT markers. This can be explained by expressional heterogeneity of EMT markers at the mesenchymal endpoint determined by differences in the acquired expression of the core EMT transcription factors. Moreover, EMT is a spectrum of intermediary stages with varying expression of epithelial and mesenchymal characteristics, rather than separate epithelial or mesenchymal stages (10,25).
FGFR1 signaling has previously been reported as a mediator of EMT-caused resistance (8,9,26), and in accordance FGFR1 expression was substantially increased in all resistant cells. The MET-amplified HCC827ER clones used in this study existed in parallel with EMT clones in the total erlotinib-resistant HCC827ER cell population (9). These EMT clones showed increased FGFR1 expression as an early event in resistance development similar to our MET-TKI resistant EMT cells. This suggests that FGFR1 bypass signaling is a preferred mechanism to obtain an immediate survival benefit upon TKI-mediated inhibition of the primarily used RTK signaling pathways. Increased FGFR1 expression has also been associated with EMT markers in clinical samples, but further research is needed to determine the significance of FGFR1 in clinical development of TKI resistance (19,27). Furthermore, four out of the six MET-TKI resistant cell lines showed increased sensitivity to treatment with the pan-FGFR inhibitor AZD4547 compared to the parental cells. Recently, AZD4547 was investigated in clinical trials in treatment of NSCLC patients with FGFR-activated cancer. Only modest responses were observed, indicating that other factors in the heterogenous tumor affect the response (28,29).
Similar to FGFR1 bypass signaling, activation of IGF1R was found as a resistance mechanism in a previous in vitro study of sequential EGFR-TKI and MET-TKI treatment (30). In contrast, MET Y1248H and D1246N mutations and EGFR gene amplification have been observed in clinical samples from patients with acquired sequential resistance (31). Sequencing of the resistant cell lines did not show any new alterations with known potential to confer resistance in the 52 investigated genes. We note that MET and EGFR were included in the sequencing panel. We acknowledge the limitation examining a panel of preselected cancer-associated genes. However, the result is in alignment with MET-TKI resistance being epigenetically mediated through EMT and the associated up-regulation of FGFR1.
Resistance can develop either by selection of preexisting resistant cells or from drug-tolerant persister cells. Persister cells can emerge in initial sensitive cells cell populations as an adaptive response to drug-exposure (14). In this study, we observed EMT in cell clones with the original sensitizing EGFR mutation and the acquired MET-amplification demonstrating that EMT can evolve in the original drug-sensitive, epithelial cells in response to MET-TKI exposure. Transcriptionally induced rewiring and switch in pathway dependency of cells exposed to TKIs, has also been suggested as the mechanism behind survival of drug-tolerant persister cells (32), and mesenchymal markers have been observed in persister cells (16,27). This leads to the question if the EMT observed in MET-TKI resistant cells could be a drug-tolerant state in the resistance development conferring sufficient cell survival to allow later development of a genetically based resistance mechanism.
Conclusions
In conclusion the main finding of EMT as a common MET-TKI resistant mechanism in HCC827 erlotinib-resistant cells, and the possibility that EMT can evolve in original drug-sensitive cells contribute to the evidence of EMT as a resistance mechanism to TKIs and emphasizes the need to investigate EMT in clinical samples. Additionally, this study underlines the role of FGFR1 signaling in EMT-associated resistance, and that FGFR inhibitors can inhibit growth of these cells. These findings provide important insight into the development of resistance to MET-TKI treatment in sequential resistance and raise awareness of therapeutic strategies to prevent resistance development.
Supplementary
The article’s supplementary files as
Acknowledgments
Thanks to Birgit Westh Mortensen for skillful technical assistance.
Funding: None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. This study does not involve human subjects. Therefore, the authors have not obtained ethical approval or conducted the study in accordance with the Declaration of Helsinki.
Footnotes
Reporting Checklist: The authors have completed the MDAR reporting checklist. Available at http://dx.doi.org/10.21037/tlcr-20-522
Peer Review File: Available at http://dx.doi.org/10.21037/tlcr-20-522
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/tlcr-20-522). The authors have no conflicts of interest to declare.
References
- 1.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011;3:75ra26. 10.1126/scitranslmed.3002003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu HA, Arcila ME, Rekhtman N, et al. Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR-TKI Therapy in 155 Patients with EGFR-Mutant Lung Cancers. Clin Cancer Res 2013;19:2240-7. 10.1158/1078-0432.CCR-12-2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316:1039-43. 10.1126/science.1141478 [DOI] [PubMed] [Google Scholar]
- 4.Caparica R, Yen CT, Coudry R, et al. Responses to Crizotinib Can Occur in High-Level MET-Amplified Non–Small Cell Lung Cancer Independent of MET Exon 14 Alterations. J Thorac Oncol 2017;12:141-4. 10.1016/j.jtho.2016.09.116 [DOI] [PubMed] [Google Scholar]
- 5.Gainor JF, Niederst MJ, Lennerz JK, et al. Dramatic response to combination erlotinib and crizotinib in a patient with advanced, EGFR-mutant lung cancer harboring de Novo MET amplification. J Thorac Oncol 2016;11:e83-5. 10.1016/j.jtho.2016.02.021 [DOI] [PubMed] [Google Scholar]
- 6.Thomson S, Buck E, Petti F, et al. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res 2005;65:9455-62. 10.1158/0008-5472.CAN-05-1058 [DOI] [PubMed] [Google Scholar]
- 7.Jakobsen KR, Demuth C, Sorensen BS, et al. The role of epithelial to mesenchymal transition in resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Transl Lung Cancer Res 2016;5:172-82. 10.21037/tlcr.2016.04.07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ware KE, Hinz TK, Kleczko E, et al. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogenesis 2013;2:e39. 10.1038/oncsis.2013.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jakobsen KR, Demuth C, Madsen AT, et al. MET amplification and epithelial-to-mesenchymal transition exist as parallel resistance mechanisms in erlotinib-resistant, EGFR-mutated, NSCLC HCC827 cells. Oncogenesis 2017;6:e307. 10.1038/oncsis.2017.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nieto MA, Huang RYJ, Jackson RA, et al. EMT: 2016. Cell 2016;166:21-45. 10.1016/j.cell.2016.06.028 [DOI] [PubMed] [Google Scholar]
- 11.Stemmler MP, Eccles RL, Brabletz S, et al. Non-redundant functions of EMT transcription factors. Vol. 21, Nat. Cell Biol. Nature Publishing Group; 2019:102-12. [DOI] [PubMed] [Google Scholar]
- 12.Burk U, Schubert J, Wellner U, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep 2008;9:582-9. 10.1038/embor.2008.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bracken CP, Gregory PA, Kolesnikoff N, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res 2008;68:7846-54. 10.1158/0008-5472.CAN-08-1942 [DOI] [PubMed] [Google Scholar]
- 14.Sharma SV, Lee DY, Li B, et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010;141:69-80. 10.1016/j.cell.2010.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramirez M, Rajaram S, Steininger RJ, et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat Commun 2016;7:10690. 10.1038/ncomms10690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hata AN, Niederst MJ, Archibald HL, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med 2016;22:262-9. 10.1038/nm.4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersen CL, Jensen J, Orntoft T. Normalization of Real Time Quantitative Reverse Transcription PCR Data: A Model Based Variance Estimation Approach to Identify Genes Suited for Normalization, Applied to Bladder and Colon Cancer Data Sets. Cancer Res 2004;64:5245. 10.1158/0008-5472.CAN-04-0496 [DOI] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402-8. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 19.Vad-Nielsen J, Gammelgaard KR, Daugaard TF, et al. Cause-and-Effect relationship between FGFR1 expression and epithelial-mesenchymal transition in EGFR-mutated non-small cell lung cancer cells. Lung Cancer 2019;132:132-40. 10.1016/j.lungcan.2019.04.023 [DOI] [PubMed] [Google Scholar]
- 20.Gammelgaard KR, Vad-Nielsen J, Clement MS, et al. Up-Regulated FGFR1 Expression as a Mediator of Intrinsic TKI Resistance in EGFR-Mutated NSCLC. Transl Oncol 2019;12:432-40. 10.1016/j.tranon.2018.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rastogi I, Rajanna S, Webb A, et al. Mechanism of c-Met and EGFR tyrosine kinase inhibitor resistance through epithelial mesenchymal transition in non-small cell lung cancer. Biochem Biophys Res Commun 2016;477:937-44. 10.1016/j.bbrc.2016.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fukuda K, Takeuchi S, Arai S, et al. Epithelial-to-mesenchymal transition is a mechanism of ALK inhibitor resistance in lung cancer independent of ALK mutation status. Cancer Res 2019;79:1658-70. 10.1158/0008-5472.CAN-18-2052 [DOI] [PubMed] [Google Scholar]
- 23.Uramoto H, Shimokawa H, Hanagiri T, et al. Expression of selected gene for acquired drug resistance to EGFR-TKI in lung adenocarcinoma. Lung Cancer 2011;73:361-5. 10.1016/j.lungcan.2011.01.008 [DOI] [PubMed] [Google Scholar]
- 24.Gainor JF, Dardaei L, Yoda S, et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov 2016;6:1118-33. 10.1158/2159-8290.CD-16-0596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karacosta LG, Anchang B, Ignatiadis N, et al. Mapping lung cancer epithelial-mesenchymal transition states and trajectories with single-cell resolution. Nat Commun 2019;10:5587. 10.1038/s41467-019-13441-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Terai H, Soejima K, Yasuda H, et al. Activation of the FGF2-FGFR1 Autocrine Pathway: A Novel Mechanism of Acquired Resistance to Gefitinib in NSCLC. Mol Cancer Res 2013;11:759-67. 10.1158/1541-7786.MCR-12-0652 [DOI] [PubMed] [Google Scholar]
- 27.Raoof S, Mulford IJ, Frisco-Cabanos H, et al. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene 2019;38:6399-413. 10.1038/s41388-019-0887-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aggarwal C, Redman MW, Lara PN, et al. SWOG S1400D (NCT02965378), a Phase II Study of the Fibroblast Growth Factor Receptor Inhibitor AZD4547 in Previously Treated Patients With Fibroblast Growth Factor Pathway–Activated Stage IV Squamous Cell Lung Cancer (Lung-MAP Substudy). J Thorac Oncol 2019;14:1847-52. 10.1016/j.jtho.2019.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paik PK, Shen R, Berger MF, et al. A Phase Ib Open-Label Multicenter Study of AZD4547 in Patients with Advanced Squamous Cell Lung Cancers. Clin Cancer Res 2017;23:5366-73. 10.1158/1078-0432.CCR-17-0645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamaoka T, Ohmori T, Ohba M, et al. Acquired Resistance Mechanisms to Combination Met-TKI/EGFR-TKI Exposure in Met-Amplified EGFR-TKI–Resistant Lung Adenocarcinoma Harboring an Activating EGFR Mutation. Mol Cancer Ther 2016;15:3040-54. 10.1158/1535-7163.MCT-16-0313 [DOI] [PubMed] [Google Scholar]
- 31.Li A, Yang J, Zhang XC, et al. Acquired MET Y1248H and D1246N mutations mediate resistance to MET inhibitors in non-small cell lung cancer. Clin Cancer Res 2017;23:4929-37. 10.1158/1078-0432.CCR-16-3273 [DOI] [PubMed] [Google Scholar]
- 32.Kleczko EK, Heasley LE. Mechanisms of rapid cancer cell reprogramming initiated by targeted receptor tyrosine kinase inhibitors and inherent therapeutic vulnerabilities. Mol Cancer 2018;17:60. 10.1186/s12943-018-0816-y [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as