

Abstract
Extracellular fluid (ECF) potassium concentration ([K+]) is maintained by adaptations of kidney and skeletal muscle, responses heretofore studied separately. We aimed to determine how these organ systems work in concert to preserve ECF [K+] in male C57BL/6J mice fed a K+-deficient diet (0K) versus 1% K+ diet (1K) for 10 days (n = 5–6/group). During 0K feeding, plasma [K+] fell from 4.5 to 2 mM; hindlimb muscle (gastrocnemius and soleus) lost 28 mM K+ (from 115 ± 2 to 87 ± 2 mM) and gained 27 mM Na+ (from 27 ± 0.4 to 54 ± 2 mM). Doubling of muscle tissue [Na+] was not associated with inflammation, cytokine production or hypertension as reported by others. Muscle transporter adaptations in 0K- versus 1K-fed mice, assessed by immunoblot, included decreased sodium pump α2-β2 subunits, decreased K+-Cl− cotransporter isoform 3, and increased phosphorylated (p) Na+,K+,2Cl− cotransporter isoform 1 (NKCC1p), Ste20/SPS-1-related proline-alanine rich kinase (SPAKp), and oxidative stress-responsive kinase 1 (OSR1p) consistent with intracellular fluid (ICF) K+ loss and Na+ gain. Renal transporters’ adaptations, effecting a 98% reduction in K+ excretion, included two- to threefold increased phosphorylated Na+-Cl− cotransporter (NCCp), SPAKp, and OSR1p abundance, limiting Na+ delivery to epithelial Na+ channels where Na+ reabsorption drives K+ secretion; and renal K sensor Kir 4.1 abundance fell 25%. Mass balance estimations indicate that over 10 days of 0K feeding, mice lose ~48 μmol K+ into the urine and muscle shifts ~47 μmol K+ from ICF to ECF, illustrating the importance of the concerted responses during K+ deficiency.
Keywords: KCC3, NCC, NKCC1, NKCC2, sodium pump α2-β2
INTRODUCTION
Transmembrane potassium concentration ([K+]) gradients impact membrane potential, which fuels nervous and muscle cell excitability. Extracellular fluid (ECF) [K+] is tightly regulated between 3.5 and 5.0 mEq/L (73). Lower and higher values of serum [K+] promote cardiac arrhythmias due to altered cellular balances of K+, Na+, and Ca2+ that are interlinked through Na+,K+-ATPase and Na+-Ca2+ exchange (4, 73). Thus potassium homeostasis has a high homeostatic priority and multiple systems interact to control ECF [K+] during the challenges of feast and famine. Most of the body’s potassium is sequestered in intracellular fluid (ICF), ~80% in skeletal muscle, with only 2% in the ECF. This small ECF pool is regulated by input from the gut, output via the kidneys and stool, and redistribution between the ECF and the ICF (19, 36, 81). Previous studies in rats have shown that skeletal muscle Na+,K+-ATPase abundance, especially α-catalytic isoform 2, increases during high K+ intake to shift excess ECF K+ to ICF (7) and decreases when ECF [K+] falls to shift ICF K+ to ECF, which is accompanied by reciprocal increases in cellular [Na+] (46, 64, 65). Accumulation of tissue Na+ has been associated with inflammation, specifically, greater circulating and renal proinflammatory cytokines and less production of anti-inflammatory cytokines (21, 75, 80, 84). Whether the pronounced accumulation of muscle sodium that occurs during K+ deprivation is associated with inflammation has not been previously explored.
Fine control of ECF [K+] is determined by renal regulation of K+ excretion. Various renal adaptations reduce renal K+ excretion to near zero during dietary K+ deficiency (36): 1) reducing glomerular filtration rate (GFR) (18, 71); 2) increasing active K+ reabsorption via collecting duct (CD) intercalated cell H,K-ATPase (70); 3) retracting CD principal cell renal outer medullary K+ channels (ROMK) from apical membranes (28); and 4) detection of lower ECF [K+] by distal convoluted tubule (DCT) basolateral membrane sensor, Kir 4.1/Kir 5.1, hyperpolarizes the cells and decreases cell [Cl−], leading to the release of with-no-lysine kinase (WNK) inhibition by Cl−. This, in turn, activates Ste20/SPS-1-related proline-alanine-rich kinase (SPAK), which phosphorylates Na-Cl cotransporter (NCC). NCCp raises fractional Na+ reabsorption in the DCT and reduces Na+ delivery downstream to the connecting tubule and collecting duct (CNT/CD) epithelial Na+ channel (ENaC), where Na+ reabsorption drives K+ channel-mediated K+ secretion and excretion (48, 61).
The array of adaptations activated as ECF [K+] falls exemplifies the critical importance of maintaining K+ homeostasis when K+ is limited, e.g., during periods of famine, illnesses associated with low food intake, or K+-poor diets. Most previous studies, including our own, have focused on defining adaptations in muscle or kidney separately. In the current study, we aimed to assess the coordinated adaptation of skeletal muscles and kidneys in mice on K+-deficient diet. We investigated the shift of K+ from muscle to ECF along with the change in urinary K+ excretion and plasma K+, the impact of the rise in tissue Na+ on inflammatory cytokines, and how K+ deprivation alters the protein abundance of muscle and renal sodium pumps, cotransporters, channels, and kinases that regulate cotransporters. The results demonstrate the importance of concerted efforts of both organ systems in blunting the fall of ECF [K+] during reduced K+ intake.
METHODS
Animal protocol.
All animal procedures were approved by the Institutional Animal Care and Use Committee of the Keck School of Medicine of the University of Southern California or the Institutional Animal Care and Use Committee of the Vanderbilt University Medical Center and were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Experiments were performed on male C57BL/6J mice, 7–9 wk of age, purchased from Jackson Laboratories (Bar Harbor, ME) in three consecutive sets. All mice were fed one of two gel-based diets for 10 days with ad libitum access to both food and water: without added K+ (Envigo TD.88239: 0% K and 0.74% NaCl) or the same diet supplemented with KCl to 1% K+. Gel-based diet was made as previously described (45) and stored as frozen cubes. In set 1 (n = 3/group), at day 6 of the K+-deficient (0K) or 1% K+ (1K) feeding, body weights were recorded, spot urines were collected to verify K+ adaptation, and blood was collected via submandibular vein under brief anesthesia (1.5% isoflurane). In set 2 (n = 5/group), on day 6 of 0K or 1K feeding, mice were placed in metabolic cages for overnight urine collection (16 h) with ad libitum access to food and water. The following morning, body weights were recorded, and blood was collected via submandibular vein under brief anesthesia (1.5% isoflurane). Mice in both sets 1 and 2 were terminated after 10 days of feeding: mice were anesthetized intraperitoneally with ketamine-xylazine (“K-X”) in a 1:1 volume ratio of ketamine (80 mg/kg, Phoenix Pharmaceuticals, St. Joseph, MO):xylazine (8 mg/kg, Lloyd Laboratories, Shenandoah, IA), and then renal arteries were clamped and kidneys removed and quick frozen in liquid nitrogen. Terminal blood was collected by cardiac puncture. Mixed gastrocnemius plus soleus muscles were removed and quick frozen in liquid nitrogen. Tissues, plasma, and urines were stored at −80°C until assayed. In set 3 (n = 5/group), mice were fed the same diets ad libitum for 10 days and blood pressure, albuminuria, and tissue inflammation were assessed as described below. Protocol notes: kidneys from two 0K and two 1K mice in set 2 were not processed for immunoblot and terminal blood was not successfully collected in one 1K mouse. Overnight urine electrolytes are reported from set 2 alone (n = 5) since only spot urines were collected in sets 1 and 3. No unexpected deaths were recorded. The n values for combined set 1 and set 2 samples are provided in each figure legend for immunoblots.
Muscle tissue preparation for electrolyte analysis.
After fascia, tendons, and other connective tissue were picked off, mixed gastrocnemius plus soleus muscles were blotted to remove excess adherent fluid and then repeatedly minced on ice with a razor blade and pressed between two Pyrex Glass Tissue Culture Plates until tissue was homogenous. Tissue was diluted 1:25 in 5% trichloroacetic acid (TCA) and homogenized on ice for 5 min at 20,000 rpm using an Ulta-Turrax. The tube was placed on ice for 30 min with vortexing every 10 min, then centrifuged at 2,000 g for 20 min. Supernatants were retained and analyzed for electrolytes, pellets discarded.
Electrolyte analyses.
Urinary volumes were measured with pipettes. [Na+], [K+], and [Li+] in plasma, urine, and muscle TCA extract were measured by flame photometry (Cole-Parmer, model 02655-10. IL). Clearance of lithium (CLi) was calculated as (urinary [Li+] × urine volume)/plasma [Li+] and normalized to body weight. Nernst potential across muscle plasma membrane was estimated assuming muscle extract [Na+] and [K+] was equivalent to intracellular values, and that extracellular values were equivalent to plasma values of [Na+] and [K+] using the equation: VEQ = [RT/zF]ln([X]Plasma/[X]Muscle), where R is ideal gas constant, 8.314 J/mol-K; T is temperature, 298.25 K; z is valence of the ion, +1 for Na+ and K+; F is Faraday’s constant, 95,484.56 C/mol; and X is [Na+] or [K+]. Urine osmolality was measured using the Precision Systems μ-Osmette. Urinary albumin and creatinine concentrations were measured by ELISA (Exocell, cat. no.1011 and no.1012).
Blood pressure measurement.
BP was measured noninvasively by tail cuff (Hatteras Instruments), as previously described (47). Mice were acclimated and trained in the machine for 2 days, BP was measured on two adjacent days, and values were averaged for each timepoint.
Flow cytometry of aortic and renal leukocytes.
Single cell suspensions of thoracic aorta and kidney were prepared, as previously described (56). Briefly, isolated thoracic aortas with perivascular fat were digested in RPMI 1640 media containing 10% FBS, 1 mg/mL collagenase A, 1 mg/mL collagenase B, and 100 µg/mL DNAse I for 30 min at 37°. Kidneys were digested in RPMI 1640 media containing 10% FBS, 2 mg/mL collagenase D and 100 µg/mL DNAse I for 20 min at 37°. Digested tissues were filtered through 40-μm cell strainers. Viability staining was performed using LIVE/DEAD Fixable Violet Dead Cell Stain (Life Technologies). Antibodies used to stain for total leukocytes are as follows: CD45+: anti-CD45, fluorophore BV510, from BioLegend clone 30-F11; total T cells: anti-CD3, fluorophore PerCPCy5.5, from BioLegend clone 17A2; monocytes/macrophages: anti-F4/80, fluorophore AF488, from BioLegend clone BM8. We performed flow cytometry on BD FACSCanto II and analyzed data using Flowjo software.
Splenic T cell isolation, culture, and cytokine quantification.
Spleens were homogenized and filtered through a 40-μm cell strainer to get single cell suspensions. After depletion of red blood cells (RBCs) using RBC lysis buffer (eBioscience), CD4+ and CD8+ T cells were isolated using Miltenyi negative selection cell separation kits and following the manufacturer’s instructions. Splenic CD4+ and CD8+ T cells were then cultured in nontissue culture treated 96-well plates coated with mouse anti-CD3 (2 mg/mL) and mouse anti-CD28 (2 mg/mL) antibodies (BD Biosciences) for 72 h. Cell culture supernatants were collected to measure mouse interleukin 17A (IL-17A) and interferon-γ (IFNγ) using ELISA kits (Invitrogen, cat. no. 88-7371-88 and no. 88-7314-88).
Sample preparation for Immunoblot. Skeletal muscle homogenate preparation.
Total homogenate was prepared from hindlimb mixed gastrocnemius and soleus after hair, major connective tissue, tendon, and fascia were removed. Muscles were weighed, minced in a small amount of ice-cold muscle homogenizing buffer (5% sorbitol, 5 mM histidine-imidazole, 0.5 mM Na2EDTA, 0.5 mM PMSF, 1 µg/mL leupeptin, and 1 mM pABAD per 1 mL) and then homogenized 1:25 (wt/vol) on ice with the Ulta-Turrax T25 using 4 × 15 s bursts (to limit heating) at the maximum setting (20,000 rpm). Samples were vortexed and quick frozen in liquid N2 and stored at −80°C as single use aliquots. SPAK knockout muscle tissue (mixed gastrocnemius plus soleus provided by E. Delpire, Vanderbilt) and mouse brain were prepared as outlined above and used as a negative control for SPAK and a positive control for Na+,K+-ATPase subunits, respectively.
Renal homogenate preparation.
As previously described (66), kidneys were thawed in ice cold PBS, decapsulated, cortex and medulla dissected on an ice-pack and homogenized separately for 5 min on ice with an Ultra-Turrax T25 at a low-speed setting in 1 mL of isolation buffer (5% sorbitol, 0.5 mM disodium EDTA, and 5 mM histidine-imidazole buffer, pH 7.5, with the addition of 0.2 mM phenylmethylsulfonyl fluoride, 9 μg/mL aprotinin, and 5 μl/mL of a phosphatase inhibitor cocktail; Sigma P0044) and then centrifuged at 2,000 g for 10 min. Supernatants were retained and the cortex (not medulla) pellet was rehomogenized in another 1 mL of isolation buffer, recentrifuged, and pooled with the first supernatant. Samples were quick frozen in liquid N2 and stored at −80°C as single-use aliquots.
Semiquantitative immunoblotting.
After protein concentration was determined with the bicinchoninic acid (BCA) assay, homogenates were denatured in Laemmli sample buffer for 20 min at 60°C (kidneys) or 30 min at 37°C (muscles). Uniform sample loading was confirmed by densitometry of Coomassie stained gels loaded with same amount of protein as described previously (35) (Supplemental Fig. 1A: all Supplemental material is available at https://doi.org/10.6084/m9.figshare.12115863). To assess the linearity of the detection system, 1× and ½× the amounts of sample were loaded in parallel and quantitated (Supplemental Fig. S1B). Table 1 provides loading amounts, antibody information, incubation times, dilutions, and vendors. Blots were never stripped and reprobed. Signals were detected with the Odyssey Infrared Imaging System (LI-COR, Lincoln, NE) and quantified by accompanying software. Protocol exception: abundance of potassium chloride cotransporter isoform 3 (KCC3) and KCC3 phosphorylated at Thr1049 (KCC3p) were detected by chemiluminescence (Luminata Forte; Millipore) bands visualized and quantified using a C-DiGit Blot Scanner (LI-COR, Lincoln, NE). Arbitrary density units of both 1× and ½× loading amounts were normalized to mean intensity of 1K group, defined as 1.0. Since recent studies shed light on a five amino acid deletion in NKCC2 (ΔT96-N100) in C57BL/6J mice, encompassing a key phosphorylation site (41), NKCCpT96pT101 detection was not included for kidney but was included for muscle detection of NKCC1pT212pT217.
Table 1.
Antibody and immunoblot protocol details
| Antibody Target | ~kDa | Cortex, μg/lane | Medulla, μg/lane | Primary Ab Supplier | Ab Host | Dilution | Time | Secondary Ab Supplier | Host and Target | Dilution | Time | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Muscle immunoblot details | ||||||||||||
| KCC3 | 150 100 |
40, 20 | NA | A. Mercado (Mexico) | Sh | 2 μg/mL | O/N | Invitrogen | a-sheep* | 1:5,000 | 1 h | (38, 40) |
| KCC3p-T1048 | 150 | 20, 10 | NA | A. Mercado (Mexico) | Sh | 2 μg/mL | O/N | Invitrogen | a-sheep* | 1:5,000 | 1 h | (38, 40) |
| NKAα1 | 100 | 1, 0.5 | NA | Kashgarian (Yale) | Mu | 1:2,000 | O/N | Invitrogen | GAM680 | 1:5,000 | 1 h | (20) |
| NKAα2 | 100 | 5, 2.5 | NA | McDonough | Rb | 1:2,000 | O/N | Invitrogen | GAR680 | 1:5,000 | 1 h | (54) |
| NKAβ1 | 37–50 | 5, 2.5 | NA | P. Martin-Vasallo | Rb | 1:2,000 | O/N | Invitrogen | GAR680 | 1:5,000 | 1 h | (49) |
| NKAβ2 | 37–50 | 5,2.5 | NA | P. Martin-Vasallo | Rb | 1:2,000 | O/N | Invitrogen | GAR680 | 1:5,000 | 1 h | (49) |
| NKCC1 | 150 | 20, 10 | NA | DSHB (Iowa) T4 | Mu | 1:6,000 | O/N | Invitrogen | GAM680 | 1:5,000 | 1 h | (30) |
| NKCC1p-T212pT217 | 150 | 20, 10 | NA | Forbush (Yale) | Rb | 1:2,000 | O/N | Invitrogen | GAR680 | 1:5,000 | 1 h | (13) |
| OSR1 | 60–70 | 10 | NA | DSTT, Dundee | Sh | 6 μg/25 mL | 2 h | Invitrogen | DAS680 | 1:5,000 | 1 h | (55) |
| SPAK | 60–70 | 20, 10 | NA | Delpire (Vanderbilt) | Rb | 1:3,000 | O/N | Invitrogen | GAR680 | 1:5,000 | 1 h | (15, 51) |
| SPAKp-S373 OSR1p-S325 |
60–70 | 40 | NA | DSTT, Dundee | Sh | 6 μg/25 mL | 2 h | Invitrogen | DAS680 | 1:5,000 | 1 h | (15, 55) |
| Kidney immunoblot details | ||||||||||||
| AQP1 | 35 24 |
10, 5 | NA | Alpha Diagnostic Intl Inc. | Rb | 1:5,000 | O/N | Invitrogen | GAR680 | 1:5,000 | 1 h | (3) |
| AQP2 | 37 23 |
20, 10 | NA | Santa Cruz (sc. 9882) | Go | 1:500 | O/N | Invitrogen | DAG680 | 1:5,000 | 1 h | (43) |
| γENaC | 80 60 |
40, 20 | 15, 7.5 | Loffing (Zurich) | Rb | 1:10,000 | 2 h | Invitrogen | GAR680 | 1:5,000 | 1 h | (69) |
| Kir 4.1 | 40 | 40, 20 | NA | Alomone APC-035 |
Rb | 1:2,000 | O/N | Invitrogen | GAR680 | 1:5,000 | 1 h | (42) |
| NaPi2 | 85 | 40, 20 | NA | McDonough | Rb | 1:1000 | 2 h | Invitrogen | GAR680 | 1:5,000 | 1 h | (27) |
| NCC | 150 | 40, 20 | NA | McDonough | Rb | 1:5,000 | 2 h | Invitrogen | GAR680 | 1:5,000 | 1 h | (44) |
| NCCpT53 | 150 | 40, 20 | NA | Loffing (Zurich) | Rb | 1:5,000 | 2 h | Invitrogen | GAR680 | 1:5,000 | 1 h | (60) |
| NCCpS71 | 150 | 40, 20 | NA | Loffing (Zurich) | Rb | 1:5,000 | 2 h | Invitrogen | GAR680 | 1:5,000 | 1 h | (60) |
| NHE3 | 83 | 40, 20 | 10, 5 | McDonough | Rb | 1:2,000 | 2 h | Invitrogen | GAR680 | 1:5,000 | 1 h | (79) |
| NHE3-pS552 | 83 | 5, 2.5 | 10, 5 | Santa Cruz (sc. 53962) | Mu | 1:1,000 | 2 h | Invitrogen | GAM680 | 1:5,000 | O/N | (22) |
| NKAα1 | 100 | 1, 0.5 | 1, 0.5 | Kashgarian (Yale) | Mu | 1:2,000 | O/N | Invitrogen | GAM680 | 1:5,000 | 1 h | (20) |
| NKCC | 150 | 20, 10 | 10, 5 | DSHB T4-c (Iowa) | Mu | 1:6,000 | 2 h | Invitrogen | GAM680 | 1:5,000 | O/N | (30) |
| ROMK | 50 37 |
40, 20 | NA | O. Staub (Lausanne) | Gp | 1:1,000 | 2 h | Invitrogen | GAGP 680 |
1:5,000 | 1 h | (2) |
| SPAK | 60–70 | 20, 10 | NA | Delpire (Vanderbilt) | Rb | 1:3,000 | O/N | Invitrogen | GAR680 | 1:5,000 | 1 h | (51) |
| SPAKp-S373 OSR1p-S325 |
60–70 | 40, 20 | NA | DSTT, Dundee | Sh | 6 μg/25 mL | 2 h | Invitrogen | DAS680 | 1:5,000 | 1 h | (55) |
| Villin | 100 | 5, 2.5 | NA | Santa Cruz (sc. 58897) | Mu | 1:2,000 | 2 h | Invitrogen | GAM680 | 1:5,000 | 1 h | (68) |
KCC3, potassium chloride cotransporter isoform 3; NKA, Na+,K+-ATPase; NKCC, Na+,K+,2Cl− cotransporter1; SPAK, Ste20/SPS-1-related proline-alanine-rich kinase; OSR1, oxidative stress-responsive kinase 1; AQP, aquaporin 1; ROMK, renal outer medullary K+ channel; ENaC, epithelial Na+ channel; NaPi2, Na+-Pi cotransporter isoform 2; NCC, Na+-Cl− cotransporter; NHE, Na+/H+ exchanger; ~kDa, apparent molecular mass determined by Bio-Rad Precision Plus Protein Dual Color Standards; Ab, antibody; Mu, mouse; Rb, rabbit; O/N, overnight; a-sheep*, anti-sheep imaged using chemiluminescence (clarity maximum: 5 min); DAS, donkey anti-sheep, DAG, donkey anti-goat; GAM, goat anti-mouse; GAR, goat anti-rabbit; GAGP, goat anti-guinea pig; NA, not assayed.
Statistical analysis.
Data are represented as individual values and reported as means ± SE. Statistical analyses were performed using GraphPad Prism 8.2 (San Diego, CA). Significance was assessed using paired parametric Student’s t test for body weight and plasma [Na+] and [K+], two-way ANOVA for blood pressure, and other physiological parameters, transporter abundance, and immune cell analysis were assessed using unpaired parametric Student’s t test. Individual P values are provided.
RESULTS
Effects of potassium-deficient feeding on physiological parameters.
Male C57BL/6J mice were fed chow with no added K+ (0K) or 1% K+ (1K) for 10 days. By day 6, plasma [K+] was 25% lower in 0K versus 1K (3.30 ± 0.2 versus 4.32 ± 0.2 mM); by day 10, plasma [K+ ] fell 50% to 2.13 ± 0.2 mM in 0K (P = 0.0001) and was unchanged in 1K mice (Fig. 1A). Urine K+ loss, assessed at day 6 by 16 h overnight collection in metabolic cages, was 187 ± 30 μmol in 1K and fell 98% to just 3.2 ± 0.5 μmol in 0K fed (P = 0.0002). Plasma [Na+] remained unchanged in both groups: 144.9 ± 0.3 mM in 1K and 149 ± 1 mM in 0K at day 10; likewise, urine Na+ excretion and urine volume (UV) were not significantly changed by 0K diet (Fig. 1A). Although food intake was not quantified, Na+ content was the same in both diets, and urinary Na+ excretion was not different in 0K versus 1K fed, suggesting similar amounts of chow were consumed. Ten days of K+-deficient feeding reduced body weight from 29.1 ± 0.1 g (1K) to 26.9 ± 0.3 g (0K; P = 0.04) while body weight did not change in the 1K mice (Fig. 1B). Urine osmole excretion (UosmV) tended to fall in 0K versus 1K mice (753 ± 125 versus 1,302 ± 227 mosM/16 h UV, P = 0.07), as did urine osmolality (1,111 ± 122 versus 1,389 ± 130 mosmol/kgH2O, P = 0.16; Fig. 1B), potential evidence of diabetes insipidus (Fig. 1B).
Fig. 1.

Effects of zero K+ feeding on physiological parameters in male C57BL/6J mice. Mice (n = 5–6/group) were fed gel-based diet made without K+ (0K, ○) or the same diet supplemented with 1% K+ (1K, ●) for 10 days. At day 6, animals were placed in metabolic cages for urine collection with access to food and water ad libitum overnight for 16 h; afterwards, body weight was measured and blood collected via submandibular vein. At day 10, mice were anesthetized, tissues were removed for electrolyte and transporter assays, and terminal blood was collected by cardiac puncture. A: Na+ and K+ concentrations ([Na+] and [K+]) in plasma, urine, and muscle tissue extracts were assessed by flame photometry. Na+ and K+ excretion (UNaV and UKV, respectively; µmol) were calculated using overnight (16 h) urine volume. Nernst potentials across muscle plasma membrane was estimated using the equation: VEQ = [RT/zF]ln([X]Plasma/[X]Muscle) assuming muscle extract [Na+] and [K+] were equivalent to intracellular (ICF) values and that extracellular (ECF) values were equivalent to plasma values of [Na+] and [K+]. B: body weight (g) measured at day 2 on diet and before termination (day 10); urine volume (UV), urine osmolality (Uosm; mosmol/kgH2O), and urine osmole excretion (UosmV; mosml/16 h UV) from overnight urine (16 h). Comparisons for plasma [Na+] and [K+] and body weight were performed with GraphPad Prism parametric paired Student’s t test. Unpaired Student’s t test was used for 1K to 0K comparisons in UNaV, UKV, UV, muscle extract [Na+] and [K+], and Nernst potentials UosmV and Uosm. P values are provided.
To determine whether skeletal muscle donates K+ from ICF to the ECF as plasma [K+] falls in mouse muscle, tissue [Na+] and [K+] were measured in hindlimb muscle (mixed gastrocnemius and soleus). Muscle tissue concentrations in this study reflect combined ICF and ECF compartments [in a previous study in rats we determined (with l-[3H]glucose) that extracellular fluid in the muscle averaged ~10% of wet weight (64)]. Concentrations in muscle tissue are displayed as mM, assuming 1 g tissue wet weight is equivalent to 1 mL. By day 10, hindlimb muscle [K+] dropped 25% in 0K fed versus 1K controls to 87 ± 2 mM versus 115 ± 2 mM (P < 0.001). Reciprocally, hindlimb muscle [Na+] doubled during K+ deficiency in 0K versus 1K to 54.3 ± 2.5 mM versus 27.1 ± 0.4 (P < 0.001). The 29-mM drop in muscle [K+] was equivalent to the 27 mM gain in muscle [Na+] (Fig. 1A). Nernst reversal potentials (E) were estimated, assuming muscle extract [Na+] and [K+] were equivalent to ICF values and that plasma [Na+] and [K+] were equivalent to muscle ECF. When comparing 1K to 0K feeding, EK fell from −83 ± 1 to −95 ± 3 mV (P = 0.002), and ENa fell from 43.3 ± 0.4 to 26 ± 1 mV (P < 0.0001; Fig. 1A) with the diminution of transmembrane gradients.
Responses of skeletal muscle transporters to potassium-deficient diet.
To investigate regulation of key membrane transporters during the shift of K+ from skeletal muscle ICF to ECF, we analyzed hindlimb muscle homogenates after 10 days 0K versus 1K feeding (Fig. 2, A and B). Sodium pumps [Na+,K+-ATPase (NKA)] are heterodimers of α- catalytic and β glycoprotein subunits (10). During 0K feeding, pool sizes of the ubiquitous α1- and β1-NKA subunits did not change, while the abundance of muscle-enriched α2 and β2 NKA subunits decreased 20% (P = 0.0007) and 50% (P = 0.01), respectively. The electroneutral potassium chloride cotransporter isoform 3 (KCC3) is detected as core and mature glycosylated forms at 100 and 150 kDa, respectively. After 10 days 0K diet, abundance of the mature form decreased 40% (P = 0.003), while changes in the core form and KCCpT1048 abundance were not detected. In contrast, abundance of skeletal muscle Na+,K+,2Cl− cotransporter isoform 1 (NKCC1), as well as the activated form NKCC1pThr212,Thr217, was 9% (P = 0.03) and 18% (P = 0.04) more abundant, respectively, after 0K feeding.
Fig. 2.

Response of skeletal muscle transporters to K+-deficient state assayed by immunoblot of muscle homogenates. 1× and ½× amounts were assayed to confirm linearity of detection system (one amount is shown); assay and antibody details are provided in Table 1. A: individual values plotted as relative abundance normalized to mean of the 1% K (1K) group = 1; data are displayed as means ± SE; n = 6/group were assayed for all but NKAβ2 isoform (n = 3) because this noncommercial antibody degraded upon reuse and we did not identify a replacement for detecting mouse NKAβ2. Na+,K+-ATPase αβ subunits (NKAα1, α2, β1, β2); Na+,K+,2Cl− cotransporter isoform 1 (NKCC1); NKCC1 phosphorylated at Thr212 and Thr 217 (NKCC1p); K+-Cl− cotransporter isoform 3 (KCC3) glycosylated at 150 kDa and core at 100 kDa; KCC3 phosphorylated at Thr 1048 (KCC3p); Ste20/SPS-1-related proline-alanine-rich kinase (SPAK); SPAK phosphorylated at Ser 373 (SPAKp); oxidative stress-responsive kinase 1 phosphorylated at Ser 325 (OSR1p). B: typical immunoblots and normalized values (1K mean = 1), means ± SE calculated for n = 6/group (n = 3 for NKAβ2) and n = 3/group shown for simplicity. Molecular weights (MW; kDa), –br and –kid indicate mouse brain and kidney homogenate positive controls, respectively, and KO indicates a lane loaded with skeletal muscle homogenate from a SPAK-KO mouse (E. Delpire, Vanderbilt). Comparisons were performed using GraphPad Prism parametric unpaired Student’s t test and P values are provided.
The activated kinases responsible for phosphorylating these cotransporters, SPAKpS373 and OSR1pS325, were 1.7- and 2-fold more abundant during 0K feeding, respectively, (P = 0.001 for both; Fig. 2, A and B) while the total pool sizes of these kinases were reduced by 15–20% (P = 0.01 for SPAK).
K+ deficiency does not affect blood pressure or inflammation.
Previous studies showed that potassium depletion was associated with lower blood pressure in normotensive animals and lowered blood pressure in hypertensive animals and humans (62). However, meta-analysis revealed that low potassium intake may play an important role in the genesis of high blood pressure in humans and that K+ supplementation can lower blood pressure (74). Additionally, studies have reported that accumulation of tissue Na+ is associated with inflammation and hypertension (23). We investigated whether the Na+ accumulation with K+ depletion affects blood pressure, albuminuria, or end-organ inflammation in mice fed 0K versus 1K diet for 10 days. There was no difference in BP between groups (Fig. 3A). K+ depletion with Na+ accumulation did not affect the number of aortic total leukocytes, T cells, or macrophages (Fig. 3B). To investigate whether K+ depletion/Na+ accumulation affects glomerular injury, we measured urinary albumin:creatinine ratio and did not detect an increase (Fig. 3C). Renal total leukocytes, T cells, and macrophages were, likewise, not different between 0K- and 1K-fed mice (Fig. 3D). Finally, to determine if K+ depletion/Na+ accumulation was associated with increased systemic inflammation, we examined splenic CD4+ T cell production of IL-17A, and CD8+ T-cell production of interferon-γ (IFNγ). Interestingly, despite a doubling in muscle cell [Na+], these were unchanged between 0K and 1K-fed mice (Fig. 3E).
Fig. 3.

K+-deficient state with elevated muscle cell Na+ does not affect blood pressure or provoke inflammation. Mice were fed diet supplemented with 1% K+ (1K, ●) or diet without K+ (0K, ○) for 10 days. A: systolic blood pressure (SBP) was measured at baseline and day 5 and day 10 (n = 5/group). B: flow cytometric quantification of total leukocytes (CD45+), T cells (CD3+), and macrophages (F4/80+) in the thoracic aorta from both groups (n = 5/group). C: albumin:creatinine ratio was measured in both groups by ELISA (n = 4/group). D: flow cytometric quantification of total leukocytes (CD45+), T cells (CD3+), and macrophages (F4/80+) in the kidney from both groups (n = 5/group). E: splenic CD4+ T-cell production of IL-17A and CD8+ T cell production of IFNγ were quantified in both groups by ELISA (n = 4–5/group). Data are expressed as means ± SE. Comparisons were performed using GraphPad Prism parametric unpaired Student’s t test and P values are provided.
Responses of renal cortical proximal nephron and medullary thick ascending limb Na+ transporters to potassium-deficient diet.
K+ conservation mechanisms are evident in specific regions of the nephron. Along the mouse proximal tubule (PT), we did not detect significant changes in pool sizes of NHE3, NHE3pS552 (a marker for NHE3 localization to the base of the microvilli where it is less active), and NaPi2, during 0K feeding, although there was a tendency for lower NHE3; cortical AQP1 and villin also remained unchanged (Supplemental Fig. S2). Renal KCC3 abundance was not investigated despite expression of KCC3 in the PT because a previous study detected no change in KCC3 abundance with low- or high-K+ diets (37). Along the medullary thick ascending limb (mTAL), the abundance of NHE3pS552 increased 1.8-fold (P = 0.001), while the mNKCC2 abundance decreased 25% (P = 0.047) in 0K relative to 1K mice (Fig. 4, A and B). The renal clearance of endogenous lithium (CLi), a marker for volume flow from the PT and mTAL, was measured to assess physiological impact of 0K diet in this region: CLi fell 8-fold from 192 ± 26 to 23 ± 5 mL·16 h−1·kg body wt−1 by day 6 of 0K feeding. In contrast, clearance of sodium, reflecting transport along the entire nephron, was similar in 1K versus 0K-fed mice: 24 ± 5 versus 18 ± 5 mL·16 h−1·kg body wt−1 (Fig. 4C).
Fig. 4.

Response of renal cortical proximal nephron and medullary thick ascending limb Na+ transporters to K+-deficient (0K) state. Na+ transporters were assayed by immunoblot. 1× and ½× loading amounts were assayed to confirm linearity of detection system (one amount is shown above); assay and antibody details are provided in Table 1. A: individual values are plotted as relative abundance normalized to mean of the 1% K (1K) group = 1; displayed as means ± SE (n = 6/group). Shown are Na+/H+ exchanger isoform 3 (NHE3); NHE3 phosphorylated at Ser 552 (NHE3p); Na+-Pi cotransporter isoform 2 (NaPi2); Na+,K+-ATPase α1-subunit (NKAα1); medullary NHE3 (mNHE3); mNHE3 phosphorylated at Ser 552 (mNHE3p); and Na+,K+,2Cl− cotransporter isoform 2 (mNKCC2). B: typical immunoblots with means ± SE calculated for n = 6/group and n = 3/group shown for simplicity. MW indicates molecular weight in kDa. C: clearance of Li+, an estimate of volume flow leaving the proximal tubule and medullary thick ascending limb, calculated classically as CLi = ([U/P]Li × UV])/body wt. Clearance of Na+ calculated by CNa = ([U/P]Na × UV])/body wt. Comparisons were performed using GraphPad Prism parametric unpaired Student’s t test. P values are provided; *P < 0.05 in B.
Distal nephron adaptations to conserve potassium during 0K diet are facilitated by activation of regulatory kinases and cotransporters.
The kidney balances K+ output to K+ intake, at least in part, by sensing a fall in ECF [K+] (36); Previous studies implicate the basolateral K+-sensing channel Kir 4.1/5.1 in this NKCC2 and NCC regulation (11, 82, 83). Abundance of the basolateral Kir 4.1 was depressed 25% in 0K mice in which plasma [K+] had fallen to 2 mM (P = 0.004; Fig. 5, A and B). Distal convoluted tubule nephron cells, in response to sensing low ECF K+, activate Na+ reabsorption to reduce Na+ delivery downstream to ENaC where Na+ reabsorption drives K+ secretion (36, 63, 67). 0K feeding had no effect on cTAL NKCC2 abundance (NKCC2p detection excluded from study - see methods) but increased the Na-Cl cotransporter (NCC), NCCpT53, and NCCpS71 1.6-, 3.4-, and 3.1-fold, respectively (all P < 0.03; Fig. 5, A and B). Kinases that phosphorylate these cotransporters are regulated during K+ deprivation in rat (45); in agreement herein, mouse SPAKp and OSR1p abundance increased 1.6- and 1.9-fold (P < 0.004) in response to 0K diet (Fig. 5, A and B). At the locus of K+ secretion (late DCT to cortical collecting duct) there was no evidence of γ-epithelial sodium channel activation by cleavage (γENaC-Cl) in 0K or 1K mice, and there was 1.6-fold accumulation of the full-length unactivated form (γENaC-Fl, P < 0.0001) with 0K feeding. Likewise, the renal outer medullary potassium channel (ROMK) abundance was unchanged. The abundance of aquaporin-2 glycosylated (AQP2-37kDa) and core (AQP2-23kDa) forms was 25–33% lower in 0K animals compared with 1K control animals (Fig. 5, A and B), as previously reported (32).
Fig. 5.

Response of distal nephron to K+-deficient state assayed by immunoblot. 1× and ½× loading amounts were assayed to confirm linearity of detection system (one amount is shown above); assay and antibody details are provided in Table 1. A: individual values plotted as relative abundance normalized to mean of the 1% K (1K) group = 1; data displayed as means ± SE (n = 6/group). Cortical NKCC2 (NKCC2); inwardly rectifying K+ channel 4.1 (Kir 4.1); Na+-Cl− cotransporter (NCC); NCC phosphorylated at Thr 53 (NCCpT53) and Ser 71 (NCCpS71); renal outer medullary K+ channel (ROMK); epithelial Na+ channel (ENaC) full length (-Fl) and cleaved (-Cl); Ste20/SPS-1-related proline-alanine-rich kinase (SPAK); SPAK phosphorylated at Ser 373 (SPAKp); oxidative stress-responsive kinase 1 phosphorylated at Ser 325 (OSR1p); aquaporin 2 (AQP2). B: typical immunoblots with means ± SE calculated for n = 6/group and n = 3/group shown for simplicity. MW indicates molecular weight in kDa. Comparisons were performed using GraphPad Prism parametric unpaired Student’s t test. P values are provided; *P < 0.05 in B.
Figure 6 summarizes the impact of 0K diet on the pool sizes of transporters, channels, and kinases in skeletal muscle and kidney. Phosphorylation of SPAK and OSR1 is increased in both organs associated with higher phosphorylation of NKCC1 in muscle and NCC in kidney, connecting sodium transporter regulation to potassium adaptation in muscle and kidney.
Fig. 6.

Impact of K+-deficient diet (0K) diet on abundance of muscle and kidney transporters, channels, and kinases relative to values on baseline 1% K+ diet (1K). Data plotted as box-whisker plots of 0K means ± SE (n = 6/group), normalized to 1K = 1 (dotted line). Analyzed by parametric unpaired Student’s t test. *P < 0.05.
DISCUSSION
Extracellular potassium concentration is regulated within a narrow range (3.5–5 mM) and is a key determinant of cellular membrane potential (73). Thus proper ECF [K+] has a high physiologic priority because alterations below or above this range can lead to life-threatening cardiac arrythmias. As animals transition between “feast and famine,” their systems must adapt to maintain ECF [K+]. We previously investigated the response of rats to chronic K+ deprivation and confirmed that skeletal muscles, contributed ICF K+ to the ECF and reciprocally accumulated ICF Na+ (34, 64, 65). That is, muscle cells do not adapt to retain their ICF [K+] (e.g., by increasing active K+ uptake via sodium pumps); rather, muscles activate mechanisms that donate K+ (net K+ efflux response) to the whole body’s ECF, which buffers the fall in plasma [K+] in the absence of K+ intake. The response is associated with a posttranslational decrease in abundance of sodium pump α2-β2 isoform, an isoform that is enriched in skeletal muscles, especially fast-twitch white fibers (34, 64, 65).
In the current study, we report that mice exhibit an analogous altruistic donation of K+ from skeletal muscle ICF to ECF. Specifically, plasma [K+] fell during K+ deprivation from ~4.5 mM (baseline) to ~3 mM (day 6) to ~2 mM (day 10). This drop occurred despite kidney adaptations to conserve K+ loss via urine, from ~200 µmol/16 h at baseline to 3 µmol/16 h at day 6 of 0K diet. Although only 1.5% of baseline excretion persists, it is still a sizeable loss considering the small pool of ECF K+ in a mouse. Assuming that 1) baseline ECF volume in a mouse is 0.2 × total body weight [based on a report that ECF in a 29 g mouse is equal to 5.8 mL (9)]; and 2) ECF [K+] is 4.5 mM (Fig. 1), the baseline ECF K+ pool is ~26 µmol. Thus, it is unsustainable to lose 4.5 µmol K+/day. In fact, ECF K+ pool falls only ~15 µmol to 11 µmol by day 10 of K+ deprivation (Fig. 7), illustrating the importance of the K+ shift from muscle ICF to ECF. While the adaptations are not perfect (as plasma [K+] continues to gradually fall to 2 mM), enough K+ remains in the ECF to sustain organ function: the Nernst potential for K+ across muscle membranes falls from −83 to −95 mV (Fig. 1).
Fig. 7.
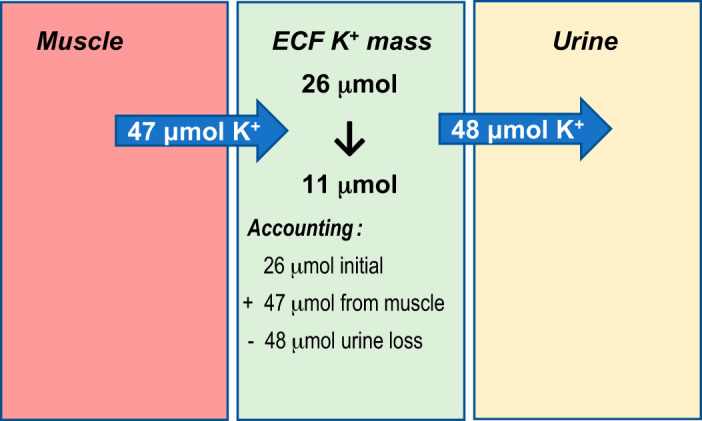
Muscle and kidney both contribute to buffering the fall in extracellular fluid (ECF) K+ concentration ([K+]). 1) How much does the ECF K+ mass decrease during 10 days of dietary K+ deprivation? K+ mass in ECF = ECF volume in a mouse [0.2 × body weight (9)] × K+ concentration in ECF: (1K) K+ mass = (29 g × 0.2) mL × 4.5 mM = 26 μmol. During K+ deprivation (0K), K+ mass = (27 × 0.2) mL × 2 mM = 11 μmol. Answer is that ECF loses ~15 μmol K+ over 10 days, which is more than half of the initial ECF K+ mass. 2) How many μmol K+ are lost into the urine over 10 days? Assuming K+ is lost over 10 days at the same rate as K+ is lost overnight during 16 h collection, the cumulative amount lost will be 15-fold more than lost overnight: (24 h/day × 10 days)/16 h = 15-fold more than that measured during 16 h overnight collection. The 1K mice K+ loss in urine over 10 days = 187 μmol/16 h × 15 = 2,807 μmol. The 0K mice K+ loss in urine over 10 days = 3.2 μmol/16 h × 15 = 48 μmol. Answer is that urinary K+ excretion drops by ~99.9% to 48 μmol over 10 days. 3) How many μmol K+ shift from muscle ICF to ECF? To estimate this value, first we need to assume all skeletal muscles lose about the same K+ as hindlimb, and second, assume a value for the fraction of mouse body weight that is skeletal muscle. First, normalizing [K+] measured/wet weight to the combined wet weights of left and right hindlimbs (0.26 g for 1K and 0.24 g for 0K) provides ~30 μmol K+ in 1K-fed hindlimbs and 21 μmol K+ in 0K-fed hindlimbs at day 10. Second, the fraction of body weight that is skeletal muscle in mouse was estimated from a report of wet weight of mouse gastrocnemius + gluteus + triceps at 0.65 g, which is 1.3 g for both left + right (52), an underestimate but reasonable. From these values we calculate that if hindlimbs lose 30–21 = 9 μmol K+/0.25 g over 10 days, then 1.3 g skeletal muscle would shift 47 μmol to the ECF, quite similar in amount to the urinary K+ loss. ECF K+ falls, nonetheless, likely due to unaccounted-for routes of K+ loss, such as from feces and saliva.
After 10 days K+ deprivation, [K+] in mouse hindlimb muscle (ICF and adherent ECF) fell 25% from 115 to 87 mM. Muscle can lose K+ by either reducing active influx (via Na+,K+-ATPase) or increasing passive gradient driven efflux by channels or transporters. As in rat (34), mouse muscle Na+,K+-ATPase α2-catalytic isoform-β2 glycoprotein subunit abundance fell 25 and 50% respectively. The greater fall in β2 is significant because the glycoprotein subunit is rate limiting for assembly and transit of the active α-β heteromers to the plasma membrane (10, 33). This response was isoform specific in mouse hindlimb as the ubiquitous α1-β1 isoform subunits’ abundance was unchanged during K+ deprivation (Fig. 6).
In addition to decreases in the α2-β2 Na+,K+-ATPase active influx pathway, the abundance of muscle cotransporters (that can operate in influx or efflux modes) was impacted by the 0K diet. Using a candidate approach, we detected KCC3, phosphorylated (inactive) KCC3, NKCC1, phosphorylated (activated) NKCC1, and the kinases that phosphorylate the cotransporters SPAK and OSR1 (Fig. 2, 6). An unbiased assessment by proteomics could likely identify other relevant K+ transporters. Evidence for expression of these cotransporters in muscle has been previously reported (25) and a nonadaptive role for KCC3 in the K+ efflux that accompanies cardiac ischemia has been reported (40, 59, 78). Classic thought is that KCC3 and NKCC1, driven by gradients established by the Na+,K+-ATPase(s), are inactive at baseline and KCC3 is activated (through dephosphorylation) during cell volume decreases to increase influx of Na+ and Cl− (14, 40). In this study, we also observe reciprocal patterns of regulation: abundance of mature glycosylated KCC3 falls 40%, while NKCC1p increases 18% (Figs. 2 and 6). No change in KCC3p was detected, suggesting KCC3 remains inactive. We can speculate that this pattern is a response to depressed muscle cell volume during hypokalemia: body weight falls 10% (Fig. 1), and Dôrup and Clausen (12) have highlighted the inhibitory effects of K+ deficiency on body weight in rodents. A fall in KCC3 abundance with no change in KCC3p abundance, coupled with greater SPAKp and OSR1p, suggests that the inhibitory KCC3p/total ratio may increase. Thus, KCC3 is unlikely to play a direct role in K+ efflux or the fall in muscle cell [K+] during K+ deprivation. More likely, activation of NKCC1—consistent with greater SPAKp and OSR1p abundance—may play a role in ICF Na+ accumulation as cell K+ falls and muscles shrink; a number of studies have implicated NKCC1 in muscle cellular Na+ uptake (24, 29, 77). We are unaware of previous studies of these muscle cotransporters or their regulatory kinases during K+ deficiency, and our speculation suggests they are adapting in a classical manner.
We estimated the net K+ donation from all of the muscles’ ICF to the ECF during 0K feeding by making assumptions (Fig. 7): 1) normalizing [K+] in mixed gastrocnemius + soleus to the combined wet weight of the left and right side hindlimb muscles (0.26 ± 0.008 g for 1K fed; 0.24 ± 0.007 g for 0K fed) provides an estimate of the µmol K+ in these dissected muscles at ~30 and 21 µmol K+ in 1K- and 0K-fed mice, respectively, at the end of the 10-day feeding; 2) to estimate total skeletal muscle weight in a mouse, we refer to a study in which the weight of gastrocnemius + gluteus + triceps is reported (0.65 ± 0.06 g for a 30 g mouse (52)). Accounting for both left and right muscles, weight can be estimated as 1.3 g (likely an underestimate but reasonable); and 3) if soleus + gastrocnemius in this study donates 30 (1K) –21 µmol K+ (0K) = ~9 µmol K+/0.25 g muscle, then 1.3 g muscle is estimated to donate 47 µmol K+ from ICF to ECF over the course of 10 days in 0K mice (Fig. 7). While based on many assumptions, 47 µmol K+ is in the same range as the 48 µmol K+ estimated to be lost in the urine over the same time period, illustrating the importance of skeletal muscle to the maintenance of ECF K+ during dietary K+ restriction (Fig. 7). However, despite this concordance, ECF [K+] still falls to 2 mM during 10 days 0K feeding, indicating there are unaccounted-for routes of potassium loss such as feces and saliva (39). Under basal conditions ~90% of K+ excretion is through renal loss and ~10% through fecal loss (1). Other potential adaptations include intestinal and salivary conservation. Investigation of intestinal K+ handling indicates significant reductions in fecal K+ loss during hypokalemia (1, 39). Less is known about the role of salivary adaptation. Potassium concentration in saliva can be two- to fivefold greater than in ECF (8) and there is a study suggesting a fall in salivary K+ secretion in K+-depleted rats (57). Thus continued leakage of K+ via these routes may contribute to the fall in ECF [K+] when intake is near zero.
In response to the fall in plasma [K+] during 10 days of K+-deficient feeding, skeletal muscle ICF [K+] falls with equivalent accumulation of muscle [Na+], likely to maintain intracellular volume, as illustrated for cotransporter regulation. We, and others, have reported the same reciprocal changes in Na+ and K+ in K+-deficient rats (6, 45). Tissue sodium accumulation, attributed to hypertonic interstitial sodium, has been linked to immune activation (76). However, it is not known whether high intracellular muscle sodium secondary to K+ deficiency leads to immune activation and hypertension. In our study, despite a significant doubling in muscle tissue sodium, we did not detect any change in blood pressure or an increase in renal, vascular, or systemic inflammation. Thus it appears that increased muscle sodium alone is not proinflammatory.
Reducing urinary excretion of K+ is key to maintaining ECF K+ when intake is low. In the 1960s, reports indicated K+ deprivation in rats suppressed glomerular filtration rate (GFR) ~20%, presumed to be an adaptation to limit K+ filtration and excretion (18, 71). While not measured in this study, we assume GFR is similarly reduced in mice during hypokalemia. In 1964, Malnic et al. (31) showed, by micropuncture, that rat kidneys match K+ excretion to K+ intake by specific adjustments in K+ reabsorption and secretion along the nephron. Significant molecular details have been described during the subsequent 60 years, summarized in numerous reviews (17, 36, 50, 61, 63). We measured a pronounced reduction in endogenous lithium clearance, CLi, during K+ depletion, which is a measure of volume flow leaving the proximal tubule and loop of Henle, without a change in sodium clearance, CNa, a measure of whole nephron reabsorption. These findings in mice are consistent with the earlier studies of Shirley and Walter (58) and Walter et al. (71) in rats, which reported by micropuncture 70% reduction in CLi during K+ depletion, associated with reduced GFR, similar Li+ handling along the proximal tubule, and increased Li+ reabsorption along the distal tubule and amiloride-sensitive collecting duct. While others report fourfold increases in NHE3 abundance in potassium-deficient animals (5), we observed no significant change in transporters’ abundance (Fig. 6) in the proximal tubule despite the 85% reduction in CLi. We can speculate that decreased filtered loads of Na+ and K+ during K+ deficiency, coupled to maintained basal levels of Na+ and K+ reabsorption in the PT (31) may account for greater fractional reabsorption of Na+ and K+. Additionally, along the distal nephron, the 1.6- to 3-fold increases in abundance of NCCp [an indicator of active NCC in the apical plasma membrane (26)] and associated activated kinases SPAKp and OSR1p, may also contributes to the fall in CLi, consistent with previously reported increase in Li+ reabsorption along the distal tubule in K+ depleted rats (58).
It is generally believed that the advantage of cotransporter activation is to limit delivery of Na+ downstream to ENaC where Na+ reabsorption creates a potential gradient favorable for K+ secretion by ROMK- and flow-activated BK channels (36). In our previous report in rats fed K+-deficient diets, we detected increased SPAKp and OSR1p but did not detect increase in NCCp (45), as reported by many other laboratories (16, 28, 72). In retrospect, we suppose this is protocol related: rats were anesthetized for an hour and infused with maintenance saline to measure blood pressure via arterial cannulation before termination, so NCC may have been dephosphorylated secondary to saline infusion. In this study, by contrast, kidneys were removed and quick frozen within minutes of termination.
The abundance of Kir4.1 in 0K-fed mice was 25% lower than in 1K-fed mice. Kir4.1, detected in cTAL, DCT, connecting tubule, and CD cells, is a key component of the DCT sensing of ECF [K+] through Kir4.1/5.1 heterotetramer (11, 61, 82, 83): when ECF [K+] falls, activity of basolateral Kir4.1/5.1 increases, hyperpolarizing the cell and decreasing cytoplasmic [Cl−], which diminishes its inhibition of with-no-lysine (WNK) allowing it to phosphorylate (activate) SPAK and activate NCC (61). The decrease in Kir4.1 during 0K feeding suggests an adaptation that would self-limit the hyperpolarization, as the ECF [K+] falls to 2 mM.
In the cortical collecting duct, 0K-fed mice exhibited increased abundance of full-length γENaC with no detectable cleaved (activated) ENaC, providing evidence for appropriate suppression of ENaC-mediated Na+ reabsorption. Abundance of ROMK, shown to be retracted to subapical pools during low K+ feeding (28), was unchanged. Cortical AQP2 abundance was also suppressed in mice during 0K feeding, consistent with previous studies in rats (32, 53).
In summary, 10 days of K+-deficient feeding in mice led to coordinated adaptations of the renal and muscle systems (Fig. 7) to blunt the fall in plasma K+. 1) The renal system responds by reducing K+ secretion to near zero by first reducing GFR and K+ filtration (mediating signal unknown) and then by activating distal Na+ cotransporters to reduce Na+ delivery downstream to ENaC, where Na+ reabsorption drives K+ secretion (Fig. 6). This response is mediated by sensing of ECF [K+] in the DCT, which initiates the WNK-SPAK cascade that activates cotransporters. 2) Skeletal muscle participates in adaptation during 0K feeding by reducing NKA α2-β2 isoform abundance (reducing ICF K+ entry) and increasing NKCC1 abundance and activation (increasing ICF Na+ entry) (Fig. 6). These adaptations shift K+ from ICF to ECF and increase intracellular Na+, which does not provoke inflammation. While the muscle signaling mechanisms are unexplored, the fall in ECF [K+] is most likely sensed by a skeletal muscle specific sensor similar to that seen in the kidney. In the end, the persistent urinary loss of potassium prevents ECF [K+] correction until K+ intake is increased.
GRANTS
This study was supported by American Heart Association (AHA) Grant 15GRNT23160003 (to A.A.McD.), NIH National Institute of Diabetes and Digestive and Kidney Diseases Grant 2R01DK083785 (to A.A.McD.), NIH National Heart, Lung, and Blood Institute Grant DP2HL137166 (to M.S.M), AHA Grants EIA34480023 and IPLOI34760558 (to M.S.M.), Conacyt, Mexico Grant S-8290 (to G.G.), and the China Scholarship Council (to Y.C.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
B.E.M., Y.C., A.M., G.G., M.S.M., and A.A.M. conceived and designed research; B.E.M., Y.C., T.S.P., D.L.R., and A.M. performed experiments; B.E.M., Y.C., T.S.P., A.M., G.G., M.S.M., and A.A.M. analyzed data; B.E.M., Y.C., A.M., G.G., M.S.M., and A.A.M. interpreted results of experiments; B.E.M., Y.C., T.S.P., M.S.M., and A.A.M. prepared figures; B.E.M., Y.C., and M.S.M. drafted manuscript; B.E.M., Y.C., T.S.P., D.L.R., A.M., G.G., M.S.M., and A.A.M. approved final version of manuscript; D.L.R., A.M., G.G., M.S.M., and A.A.M. edited and revised manuscript.
ACKNOWLEDGMENTS
The authors thank E. Delpire (Vanderbilt University) for providing SPAK KO muscle tissue and SPAK primary antibody, and J. Loffing for antibodies to NCC and ENaC. We also thank Timothy Reilly for critical review of the manuscript.
REFERENCES
- 1.Agarwal R, Afzalpurkar R, Fordtran JS. Pathophysiology of potassium absorption and secretion by the human intestine. Gastroenterology 107: 548–571, 1994. doi: 10.1016/0016-5085(94)90184-8. [DOI] [PubMed] [Google Scholar]
- 2.Al-Qusairi L, Basquin D, Roy A, Stifanelli M, Rajaram RD, Debonneville A, Nita I, Maillard M, Loffing J, Subramanya AR, Staub O. Renal tubular SGK1 deficiency causes impaired K+ excretion via loss of regulation of NEDD4-2/WNK1 and ENaC. Am J Physiol Renal Physiol 311: F330–F342, 2016. doi: 10.1152/ajprenal.00002.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bachmann S, Mutig K, Bates J, Welker P, Geist B, Gross V, Luft FC, Alenina N, Bader M, Thiele BJ, Prasadan K, Raffi HS, Kumar S. Renal effects of Tamm-Horsfall protein (uromodulin) deficiency in mice. Am J Physiol Renal Physiol 288: F559–F567, 2005. doi: 10.1152/ajprenal.00143.2004. [DOI] [PubMed] [Google Scholar]
- 4.Blaustein MP. The pump, the exchanger, and the holy spirit: origins and 40-year evolution of ideas about the ouabain-Na+ pump endocrine system. Am J Physiol Cell Physiol 314: C3–C26, 2018. doi: 10.1152/ajpcell.00196.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyd-Shiwarski CR, Weaver CJ, Beacham RT, Shiwarski DJ, Connolly KA, Nkashama LJ, Mutchler SM, Griffiths SE, Knoell SA, Sebastiani RS, Ray EC, Marciszyn AL, Subramanya AR. Effects of extreme potassium stress on blood pressure and renal tubular sodium transport. Am J Physiol Renal Physiol 318: F1341–F1356, 2020. doi: 10.1152/ajprenal.00527.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bundgaard H, Kjeldsen K. Potassium depletion increases potassium clearance capacity in skeletal muscles in vivo during acute repletion. Am J Physiol Cell Physiol 283: C1163–C1170, 2002. doi: 10.1152/ajpcell.00588.2001. [DOI] [PubMed] [Google Scholar]
- 7.Bundgaard H, Schmidt TA, Larsen JS, Kjeldsen K. K+ supplementation increases muscle [Na+-K+-ATPase] and improves extrarenal K+ homeostasis in rats. J Appl Physiol (1985) 82: 1136–1144, 1997. doi: 10.1152/jappl.1997.82.4.1136. [DOI] [PubMed] [Google Scholar]
- 8.Burgen AS. The secretion of potassium in saliva. J Physiol 132: 20–39, 1956. doi: 10.1113/jphysiol.1956.sp005500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chapman ME, Hu L, Plato CF, Kohan DE. Bioimpedance spectroscopy for the estimation of body fluid volumes in mice. Am J Physiol Renal Physiol 299: F280–F283, 2010. doi: 10.1152/ajprenal.00113.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clausen MV, Hilbers F, Poulsen H. The structure and function of the Na,K-ATPase isoforms in health and disease. Front Physiol 8: 371, 2017. doi: 10.3389/fphys.2017.00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuevas CA, Su XT, Wang MX, Terker AS, Lin DH, McCormick JA, Yang CL, Ellison DH, Wang WH. Potassium sensing by renal distal tubules requires Kir4.1. J Am Soc Nephrol 28: 1814–1825, 2017. doi: 10.1681/ASN.2016090935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dôrup I, Clausen T. Effects of potassium deficiency on growth and protein synthesis in skeletal muscle and the heart of rats. Br J Nutr 62: 269–284, 1989. doi: 10.1079/BJN19890029. [DOI] [PubMed] [Google Scholar]
- 13.Flemmer AW, Gimenez I, Dowd BF, Darman RB, Forbush B. Activation of the Na-K-Cl cotransporter NKCC1 detected with a phospho-specific antibody. J Biol Chem 277: 37551–37558, 2002. doi: 10.1074/jbc.M206294200. [DOI] [PubMed] [Google Scholar]
- 14.Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev 85: 423–493, 2005. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- 15.Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, Giani JF, Nguyen MT, Riquier-Brison AD, Seth DM, Fuchs S, Eladari D, Picard N, Bachmann S, Delpire E, Peti-Peterdi J, Navar LG, Bernstein KE, McDonough AA. The absence of intrarenal ACE protects against hypertension. J Clin Invest 123: 2011–2023, 2013. doi: 10.1172/JCI65460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grimm PR, Coleman R, Delpire E, Welling PA. Constitutively active SPAK causes hyperkalemia by activating NCC and remodeling distal tubules. J Am Soc Nephrol 28: 2597–2606, 2017. doi: 10.1681/ASN.2016090948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gritter M, Rotmans JI, Hoorn EJ. Role of dietary K+ in natriuresis, blood pressure reduction, cardiovascular protection, and renoprotection. Hypertension 73: 15–23, 2019. doi: 10.1161/HYPERTENSIONAHA.118.11209. [DOI] [PubMed] [Google Scholar]
- 18.Holliday MA, Egan TJ. Changes in GFR and C-H2O before and after repair of K deficiency in rats. Am J Physiol 202: 773–776, 1962. doi: 10.1152/ajplegacy.1962.202.4.773. [DOI] [PubMed] [Google Scholar]
- 19.Hoorn EJ, Gritter M, Cuevas CA, Fenton RA. Regulation of the renal NaCl cotransporter and its role in potassium homeostasis. Physiol Rev 100: 321–356, 2020. doi: 10.1152/physrev.00044.2018. [DOI] [PubMed] [Google Scholar]
- 20.Kashgarian M, Biemesderfer D, Caplan M, Forbush B 3rd. Monoclonal antibody to Na,K-ATPase: immunocytochemical localization along nephron segments. Kidney Int 28: 899–913, 1985. doi: 10.1038/ki.1985.216. [DOI] [PubMed] [Google Scholar]
- 21.Kirabo A. A new paradigm of sodium regulation in inflammation and hypertension. Am J Physiol Regul Integr Comp Physiol 313: R706–R710, 2017. doi: 10.1152/ajpregu.00250.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kocinsky HS, Girardi AC, Biemesderfer D, Nguyen T, Mentone S, Orlowski J, Aronson PS. Use of phospho-specific antibodies to determine the phosphorylation of endogenous Na+/H+ exchanger NHE3 at PKA consensus sites. Am J Physiol Renal Physiol 289: F249–F258, 2005. doi: 10.1152/ajprenal.00082.2004. [DOI] [PubMed] [Google Scholar]
- 23.Kopp C, Linz P, Wachsmuth L, Dahlmann A, Horbach T, Schöfl C, Renz W, Santoro D, Niendorf T, Müller DN, Neininger M, Cavallaro A, Eckardt KU, Schmieder RE, Luft FC, Uder M, Titze J. (23)Na magnetic resonance imaging of tissue sodium. Hypertension 59: 167–172, 2012. doi: 10.1161/HYPERTENSIONAHA.111.183517. [DOI] [PubMed] [Google Scholar]
- 24.Kristensen M, Hansen T, Juel C. Membrane proteins involved in potassium shifts during muscle activity and fatigue. Am J Physiol Regul Integr Comp Physiol 290: R766–R772, 2006. doi: 10.1152/ajpregu.00534.2004. [DOI] [PubMed] [Google Scholar]
- 25.Kristensen M, Juel C. Potassium-transporting proteins in skeletal muscle: cellular location and fibre-type differences. Acta Physiol (Oxf) 198: 105–123, 2010. doi: 10.1111/j.1748-1716.2009.02043.x. [DOI] [PubMed] [Google Scholar]
- 26.Lee DH, Maunsbach AB, Riquier-Brison AD, Nguyen MT, Fenton RA, Bachmann S, Yu AS, McDonough AA. Effects of ACE inhibition and ANG II stimulation on renal Na-Cl cotransporter distribution, phosphorylation, and membrane complex properties. Am J Physiol Cell Physiol 304: C147–C163, 2013. doi: 10.1152/ajpcell.00287.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leong PK, Devillez A, Sandberg MB, Yang LE, Yip DK, Klein JB, McDonough AA. Effects of ACE inhibition on proximal tubule sodium transport. Am J Physiol Renal Physiol 290: F854–F863, 2006. doi: 10.1152/ajprenal.00353.2005. [DOI] [PubMed] [Google Scholar]
- 28.Lin DH, Sterling H, Wang WH. The protein tyrosine kinase-dependent pathway mediates the effect of K intake on renal K secretion. Physiology (Bethesda) 20: 140–146, 2005. doi: 10.1152/physiol.00044.2004. [DOI] [PubMed] [Google Scholar]
- 29.Lindinger MI, Hawke TJ, Vickery L, Bradford L, Lipskie SL. An integrative, in situ approach to examining K+ flux in resting skeletal muscle. Can J Physiol Pharmacol 79: 996–1006, 2001. doi: 10.1139/y01-083. [DOI] [PubMed] [Google Scholar]
- 30.Lytle C, Xu JC, Biemesderfer D, Forbush B 3rd. Distribution and diversity of Na-K-Cl cotransport proteins: a study with monoclonal antibodies. Am J Physiol Cell Physiol 269: C1496–C1505, 1995. doi: 10.1152/ajpcell.1995.269.6.C1496. [DOI] [PubMed] [Google Scholar]
- 31.Malnic G, Klose RM, Giebisch G. Micropuncture study of renal potassium excretion in the rat. Am J Physiol 206: 674–686, 1964. doi: 10.1152/ajplegacy.1964.206.4.674. [DOI] [PubMed] [Google Scholar]
- 32.Marples D, Frøkiaer J, Dørup J, Knepper MA, Nielsen S. Hypokalemia-induced downregulation of aquaporin-2 water channel expression in rat kidney medulla and cortex. J Clin Invest 97: 1960–1968, 1996. doi: 10.1172/JCI118628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McDonough AA, Geering K, Farley RA. The sodium pump needs its beta subunit. FASEB J 4: 1598–1605, 1990. doi: 10.1096/fasebj.4.6.2156741. [DOI] [PubMed] [Google Scholar]
- 34.McDonough AA, Thompson CB, Youn JH. Skeletal muscle regulates extracellular potassium. Am J Physiol Renal Physiol 282: F967–F974, 2002. doi: 10.1152/ajprenal.00360.2001. [DOI] [PubMed] [Google Scholar]
- 35.McDonough AA, Veiras LC, Minas JN, Ralph DL. Considerations when quantitating protein abundance by immunoblot. Am J Physiol Cell Physiol 308: C426–C433, 2015. doi: 10.1152/ajpcell.00400.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McDonough AA, Youn JH. Potassium homeostasis: the knowns, the unknowns, and the health benefits. Physiology (Bethesda) 32: 100–111, 2017. doi: 10.1152/physiol.00022.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melo Z, Cruz-Rangel S, Bautista R, Vázquez N, Castañeda-Bueno M, Mount DB, Pasantes-Morales H, Mercado A, Gamba G. Molecular evidence for a role for K(+)-Cl(-) cotransporters in the kidney. Am J Physiol Renal Physiol 305: F1402–F1411, 2013. doi: 10.1152/ajprenal.00390.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melo Z, de los Heros P, Cruz-Rangel S, Vázquez N, Bobadilla NA, Pasantes-Morales H, Alessi DR, Mercado A, Gamba G. N-terminal serine dephosphorylation is required for KCC3 cotransporter full activation by cell swelling. J Biol Chem 288: 31468–31476, 2013. doi: 10.1074/jbc.M113.475574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meneton P, Schultheis PJ, Greeb J, Nieman ML, Liu LH, Clarke LL, Duffy JJ, Doetschman T, Lorenz JN, Shull GE. Increased sensitivity to K+ deprivation in colonic H,K-ATPase-deficient mice. J Clin Invest 101: 536–542, 1998. doi: 10.1172/JCI1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mercado A, Vázquez N, Song L, Cortés R, Enck AH, Welch R, Delpire E, Gamba G, Mount DB. NH2-terminal heterogeneity in the KCC3 K+-Cl- cotransporter. Am J Physiol Renal Physiol 289: F1246–F1261, 2005. doi: 10.1152/ajprenal.00464.2004. [DOI] [PubMed] [Google Scholar]
- 41.Moser S, Loffing J. Different NKCC2 amino acid sequences between 129/Sv and C57BL/6 mice affect analysis of NKCC2 phosphorylation with phosphoform-specific antibodies (Abstract). American Society of Nephrology ASN Kidney Week Washington, D.C. November 5–10, 2019. Abstr. no. FR-PO614, https://www.asn-online.org/education/kidneyweek/2019/program-abstract.aspx?controlId=3236906. [Google Scholar]
- 42.Nakajima M, Kawamura T, Tokui R, Furuta K, Sugino M, Nakanishi M, Okuyama S, Furukawa Y. Enhanced accumulation of Kir4.1 protein, but not mRNA, in a murine model of cuprizone-induced demyelination. Brain Res 1537: 340–349, 2013. doi: 10.1016/j.brainres.2013.09.024. [DOI] [PubMed] [Google Scholar]
- 43.Nedvetsky PI, Tabor V, Tamma G, Beulshausen S, Skroblin P, Kirschner A, Mutig K, Boltzen M, Petrucci O, Vossenkämper A, Wiesner B, Bachmann S, Rosenthal W, Klussmann E. Reciprocal regulation of aquaporin-2 abundance and degradation by protein kinase A and p38-MAP kinase. J Am Soc Nephrol 21: 1645–1656, 2010. doi: 10.1681/ASN.2009111190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen MT, Lee DH, Delpire E, McDonough AA. Differential regulation of Na+ transporters along nephron during ANG II-dependent hypertension: distal stimulation counteracted by proximal inhibition. Am J Physiol Renal Physiol 305: F510–F519, 2013. doi: 10.1152/ajprenal.00183.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nguyen MT, Yang LE, Fletcher NK, Lee DH, Kocinsky H, Bachmann S, Delpire E, McDonough AA. Effects of K+-deficient diets with and without NaCl supplementation on Na+, K+, and H2O transporters’ abundance along the nephron. Am J Physiol Renal Physiol 303: F92–F104, 2012. doi: 10.1152/ajprenal.00032.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nørgaard A, Kjeldsen K, Clausen T. Potassium depletion decreases the number of 3H-ouabain binding sites and the active Na-K transport in skeletal muscle. Nature 293: 739–741, 1981. doi: 10.1038/293739a0. [DOI] [PubMed] [Google Scholar]
- 47.Norlander AE, Saleh MA, Pandey AK, Itani HA, Wu J, Xiao L, Kang J, Dale BL, Goleva SB, Laroumanie F, Du L, Harrison DG, Madhur MS. A salt-sensing kinase in T lymphocytes, SGK1, drives hypertension and hypertensive end-organ damage. JCI Insight 2: e92801, 2017. doi: 10.1172/jci.insight.92801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palygin O, Pochynyuk O, Staruschenko A. Distal tubule basolateral potassium channels: cellular and molecular mechanisms of regulation. Curr Opin Nephrol Hypertens 27: 373–378, 2018. doi: 10.1097/MNH.0000000000000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peng L, Martin-Vasallo P, Sweadner KJ. Isoforms of Na,K-ATPase alpha and beta subunits in the rat cerebellum and in granule cell cultures. J Neurosci 17: 3488–3502, 1997. doi: 10.1523/JNEUROSCI.17-10-03488.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Penton D, Czogalla J, Loffing J. Dietary potassium and the renal control of salt balance and blood pressure. Pflugers Arch 467: 513–530, 2015. doi: 10.1007/s00424-014-1673-1. [DOI] [PubMed] [Google Scholar]
- 51.Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J Biol Chem 277: 50812–50819, 2002. doi: 10.1074/jbc.M208108200. [DOI] [PubMed] [Google Scholar]
- 52.Qiu S, Mintz JD, Salet CD, Han W, Giannis A, Chen F, Yu Y, Su Y, Fulton DJ, Stepp DW. Increasing muscle mass improves vascular function in obese (db/db) mice. J Am Heart Assoc 3: e000854, 2014. doi: 10.1161/JAHA.114.000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Radin MJ, Yu MJ, Stoedkilde L, Miller RL, Hoffert JD, Frokiaer J, Pisitkun T, Knepper MA. Aquaporin-2 regulation in health and disease. Vet Clin Pathol 41: 455–470, 2012. doi: 10.1111/j.1939-165x.2012.00488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rhee MS, Perianayagam A, Chen P, Youn JH, McDonough AA. Dexamethasone treatment causes resistance to insulin-stimulated cellular potassium uptake in the rat. Am J Physiol Cell Physiol 287: C1229–C1237, 2004. doi: 10.1152/ajpcell.00111.2004. [DOI] [PubMed] [Google Scholar]
- 55.Richardson C, Rafiqi FH, Karlsson HK, Moleleki N, Vandewalle A, Campbell DG, Morrice NA, Alessi DR. Activation of the thiazide-sensitive Na+-Cl- cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci 121: 675–684, 2008. doi: 10.1242/jcs.025312. [DOI] [PubMed] [Google Scholar]
- 56.Saleh MA, McMaster WG, Wu J, Norlander AE, Funt SA, Thabet SR, Kirabo A, Xiao L, Chen W, Itani HA, Michell D, Huan T, Zhang Y, Takaki S, Titze J, Levy D, Harrison DG, Madhur MS. Lymphocyte adaptor protein LNK deficiency exacerbates hypertension and end-organ inflammation. J Clin Invest 125: 1189–1202, 2015. doi: 10.1172/JCI76327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schneyer CA, Schneyer LH. Salivary gland function and electrolyte composition in potassium-deficient rats. Proc Soc Exp Biol Med 108: 584–586, 1961. doi: 10.3181/00379727-108-27003. [DOI] [PubMed] [Google Scholar]
- 58.Shirley DG, Walter SJ. Renal tubular lithium reabsorption in potassium-depleted rats. J Physiol 501: 663–670, 1997. doi: 10.1111/j.1469-7793.1997.663bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shivkumar K, Deutsch NA, Lamp ST, Khuu K, Goldhaber JI, Weiss JN. Mechanism of hypoxic K loss in rabbit ventricle. J Clin Invest 100: 1782–1788, 1997. doi: 10.1172/JCI119705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int 83: 811–824, 2013. doi: 10.1038/ki.2013.14. [DOI] [PubMed] [Google Scholar]
- 61.Su XT, Ellison DH, Wang WH. Kir4.1/Kir5.1 in the DCT plays a role in the regulation of renal K+ excretion. Am J Physiol Renal Physiol 316: F582–F586, 2019. doi: 10.1152/ajprenal.00412.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tannen RL. Effects of potassium on blood pressure control. Ann Intern Med 98: 773–780, 1983. doi: 10.7326/0003-4819-98-5-773. [DOI] [PubMed] [Google Scholar]
- 63.Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, Weinstein AM, Wang WH, Yang CL, Ellison DH. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 21: 39–50, 2015. doi: 10.1016/j.cmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thompson CB, Choi C, Youn JH, McDonough AA. Temporal responses of oxidative vs. glycolytic skeletal muscles to K+ deprivation: Na+ pumps and cell cations. Am J Physiol Cell Physiol 276: C1411–C1419, 1999. doi: 10.1152/ajpcell.1999.276.6.C1411. [DOI] [PubMed] [Google Scholar]
- 65.Thompson CB, McDonough AA. Skeletal muscle Na,K-ATPase alpha and beta subunit protein levels respond to hypokalemic challenge with isoform and muscle type specificity. J Biol Chem 271: 32653–32658, 1996. doi: 10.1074/jbc.271.51.32653. [DOI] [PubMed] [Google Scholar]
- 66.Veiras LC, Girardi ACC, Curry J, Pei L, Ralph DL, Tran A, Castelo-Branco RC, Pastor-Soler N, Arranz CT, Yu ASL, McDonough AA. Sexual dimorphic pattern of renal transporters and electrolyte homeostasis. J Am Soc Nephrol 28: 3504–3517, 2017. doi: 10.1681/ASN.2017030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Veiras LC, Han J, Ralph DL, McDonough AA. Potassium supplementation prevents sodium chloride cotransporter stimulation during angiotensin II hypertension. Hypertension 68: 904–912, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Veiras LC, Pei L, Yu ASL, McDonough AA. Sexual dimorphic expression of renal claudins, water channels and transporters accounts for the downstream shift in salt and volume reabsorption along the nephron in female vs. male rats. The FASEB J 30: 967.29, 2016. [Google Scholar]
- 69.Wagner CA, Loffing-Cueni D, Yan Q, Schulz N, Fakitsas P, Carrel M, Wang T, Verrey F, Geibel JP, Giebisch G, Hebert SC, Loffing J. Mouse model of type II Bartter’s syndrome. II. Altered expression of renal sodium- and water-transporting proteins. Am J Physiol Renal Physiol 294: F1373–F1380, 2008. doi: 10.1152/ajprenal.00613.2007. [DOI] [PubMed] [Google Scholar]
- 70.Walter C, Tanfous MB, Igoudjil K, Salhi A, Escher G, Crambert G. H,K-ATPase type 2 contributes to salt-sensitive hypertension induced by K+ restriction. Pflugers Arch 468: 1673–1683, 2016. doi: 10.1007/s00424-016-1872-z. [DOI] [PubMed] [Google Scholar]
- 71.Walter SJ, Shore AC, Shirley DG. Effect of potassium depletion on renal tubular function in the rat. Clin Sci (Lond) 75: 621–628, 1988. doi: 10.1042/cs0750621. [DOI] [PubMed] [Google Scholar]
- 72.Wang MX, Cuevas CA, Su XT, Wu P, Gao ZX, Lin DH, McCormick JA, Yang CL, Wang WH, Ellison DH. Potassium intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the Kir4.1 potassium channel. Kidney Int 93: 893–902, 2018. doi: 10.1016/j.kint.2017.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weiss JN, Qu Z, Shivkumar K. Electrophysiology of hypokalemia and hyperkalemia. Circ Arrhythm Electrophysiol 10: e004667, 2017. doi: 10.1161/CIRCEP.116.004667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Whelton PK, He J, Cutler JA, Brancati FL, Appel LJ, Follmann D, Klag MJ. Effects of oral potassium on blood pressure. Meta-analysis of randomized controlled clinical trials. JAMA 277: 1624–1632, 1997. doi: 10.1001/jama.1997.03540440058033. [DOI] [PubMed] [Google Scholar]
- 75.Wiig H, Luft FC, Titze JM. The interstitium conducts extrarenal storage of sodium and represents a third compartment essential for extracellular volume and blood pressure homeostasis. Acta Physiol (Oxf) 222: e13006, 2018. doi: 10.1111/apha.13006. [DOI] [PubMed] [Google Scholar]
- 76.Wilck N, Balogh A, Markó L, Bartolomaeus H, Müller DN. The role of sodium in modulating immune cell function. Nat Rev Nephrol 15: 546–558, 2019. doi: 10.1038/s41581-019-0167-y. [DOI] [PubMed] [Google Scholar]
- 77.Wong JA, Fu L, Schneider EG, Thomason DB. Molecular and functional evidence for Na+-K+-2Cl− cotransporter expression in rat skeletal muscle. Am J Physiol Regul Integr Comp Physiol 277: R154–R161, 1999. doi: 10.1152/ajpregu.1999.277.1.R154. [DOI] [PubMed] [Google Scholar]
- 78.Yan GX, Chen J, Yamada KA, Kléber AG, Corr PB. Contribution of shrinkage of extracellular space to extracellular K+ accumulation in myocardial ischaemia of the rabbit. J Physiol 490: 215–228, 1996. doi: 10.1113/jphysiol.1996.sp021137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang L, Leong PK, Chen JO, Patel N, Hamm-Alvarez SF, McDonough AA. Acute hypertension provokes internalization of proximal tubule NHE3 without inhibition of transport activity. Am J Physiol Renal Physiol 282: F730–F740, 2002. doi: 10.1152/ajprenal.00298.2001. [DOI] [PubMed] [Google Scholar]
- 80.Yi B, Titze J, Rykova M, Feuerecker M, Vassilieva G, Nichiporuk I, Schelling G, Morukov B, Choukèr A. Effects of dietary salt levels on monocytic cells and immune responses in healthy human subjects: a longitudinal study. Transl Res 166: 103–110, 2015. doi: 10.1016/j.trsl.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Youn JH, McDonough AA. Recent advances in understanding integrative control of potassium homeostasis. Annu Rev Physiol 71: 381–401, 2009. doi: 10.1146/annurev.physiol.010908.163241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang C, Wang L, Su XT, Lin DH, Wang WH. KCNJ10 (Kir4.1) is expressed in the basolateral membrane of the cortical thick ascending limb. Am J Physiol Renal Physiol 308: F1288–F1296, 2015. doi: 10.1152/ajprenal.00687.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang C, Wang L, Zhang J, Su XT, Lin DH, Scholl UI, Giebisch G, Lifton RP, Wang WH. KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1). Proc Natl Acad Sci USA 111: 11864–11869, 2014. doi: 10.1073/pnas.1411705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang J, Rudemiller NP, Patel MB, Karlovich NS, Wu M, McDonough AA, Griffiths R, Sparks MA, Jeffs AD, Crowley SD. Interleukin-1 receptor activation potentiates salt reabsorption in angiotensin II-induced hypertension via the NKCC2 co-transporter in the Nephron. Cell Metab 23: 360–368, 2016. doi: 10.1016/j.cmet.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]