

Abstract
Two formal syntheses and one total synthesis of fostriecin (1) have been achieved, as well as, the synthesis of its related congener dihydro-dephospho-fostriecin. All the routes use the Sharpless dihydroxylation to set the absolute stereochemistry at C-8/9 positions and a Leighton allylation to set the C-5 position of the natural product. In the formal syntheses a Noyori transfer hydrogenation of an ynone was used to set the C-11 position while the total synthesis employed a combination of asymmetric dihydroxylation and Pd-π-allyl reduction to set the C-11 position. Finally in the total synthesis, a trans-hydroboration of the C-12/13 alkyne was used in combination with a Suzuki cross coupling to establish the Z,Z,E-triene of fostriecin (1).
Introduction
The phosphorylated polyketide natural products, represented by fostriecin (1, CI-920),1 are typified by polyene-, polyol- and pyranone functionalities. Since the isolation of fostriecin (1) from Steptomyces pulveraceus in 1983, additional members of this unique class of natural products have been discovered. Not surprisingly, this group of natural products has been the subject of significant synthetic efforts.2 The synthetic studies of fostriecin (1) began with a synthesis of the C-9 diastereomer by Just,3 then the full stereochemical assignment (1997)4 and subsequent total synthesis by Boger.5 A short time after the Boger synthesis of fostriecin (1), a second total synthesis was reported by Jacobsen.6 In the subsequent years, there have been thirteen additional total or formal syntheses of fostriecin (1)7 and related approaches.8 The last of these was an effort reported by us in 20107l and then again in 2019 (Scheme 1).7m
Scheme 1.
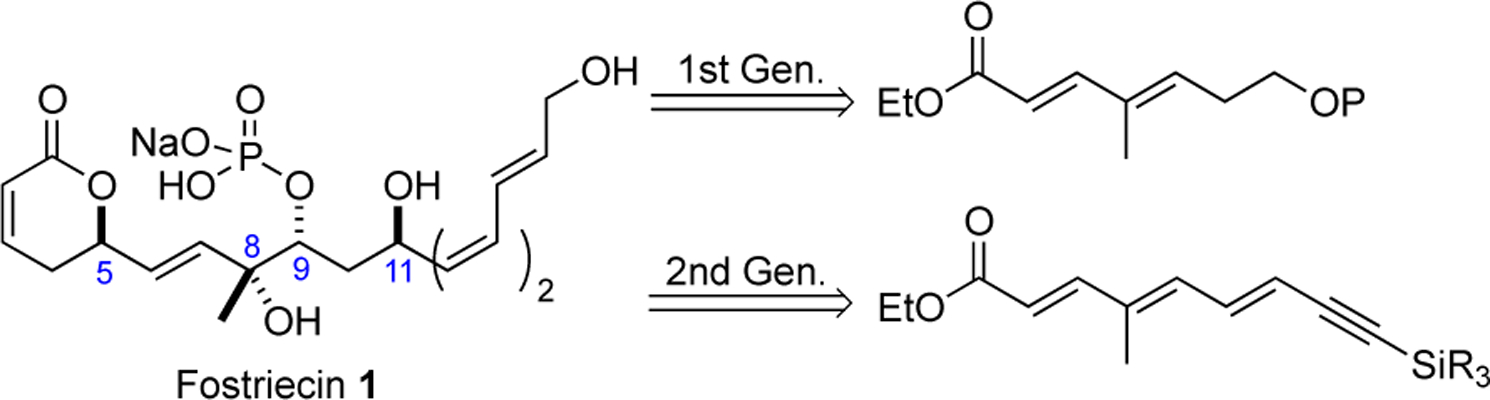
1st generation retrosynthesis of fostriecin (1)
The unique ability of fostriecin (1) to inhibit several protein phosphatases (aka, PP1, PP2A and PP4) has also inspired studies of its mechanism-of-action (MOA).9 Of particular interest is the potency and selectivity of fostriecin’s inhibition (e.g., IC50 = 45 nM; PP2A, IC50 = 1.5 nM; PP4 IC50 = 3.0 nM),10 and the resulting broad ranging cancer cell cytotoxicity (e.g., leukemia, lung cancer, breast cancer, and ovarian cancer).11,12 In fact, fostriecin (1) has been explored as a potential anti-cancer therapy, having advanced to the clinical trial stage at the National Cancer Institute.13 Our interest in a synthesis of fostriecin (1), as well as, its related congener dihydro-dephospho-fostriecin 2 arose from our general interest in the synthesis of 1,3-polyol and pyranone natural products for stereochemical-structure activity relationship (S-SAR) studies.14 Herein we disclose the full account of our synthetic efforts which involved two distinct approaches to fostriecin (1) and resulted in two formal syntheses and one total synthesis of the natural product.
Results and discussion
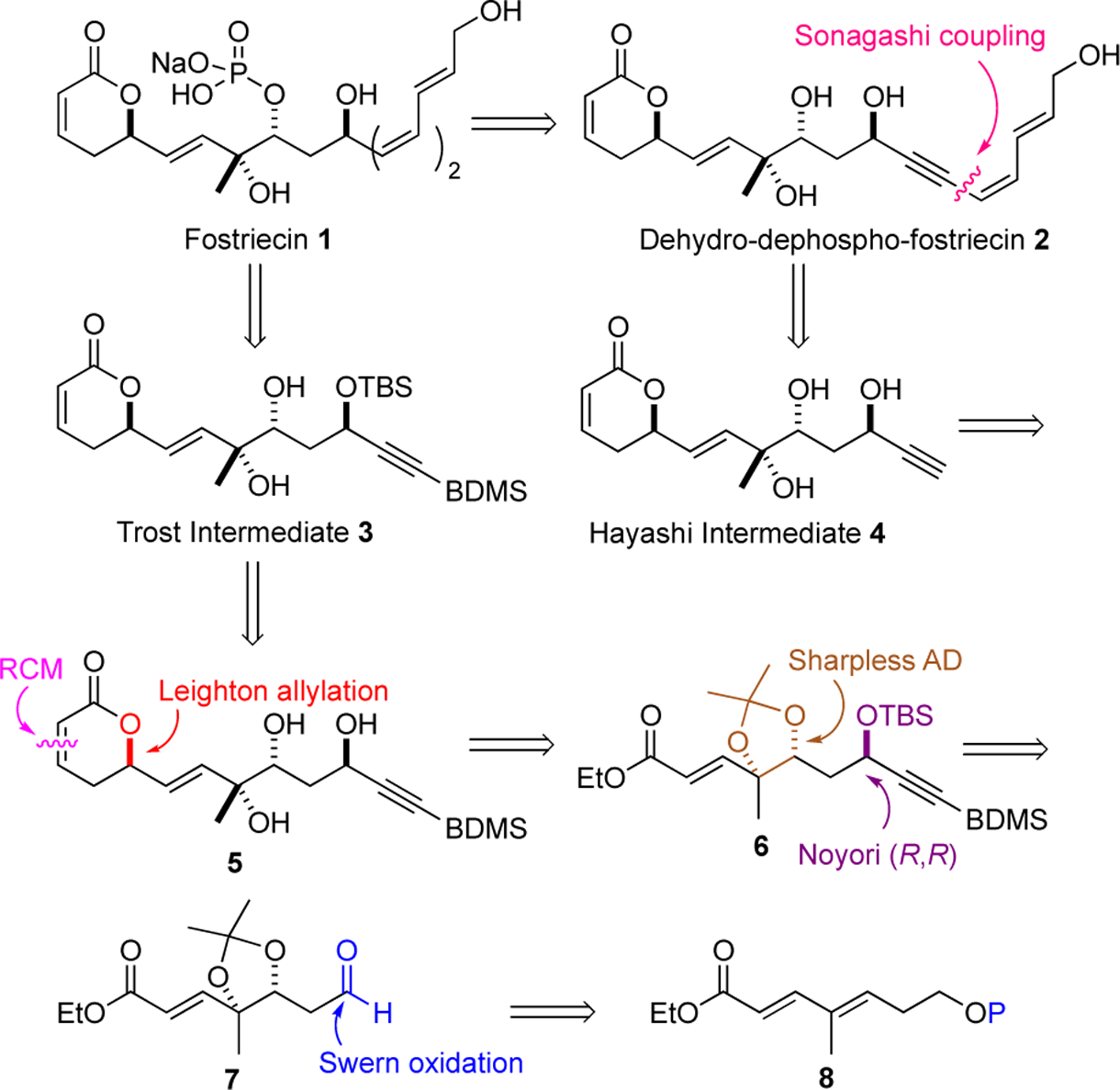
The retrosynthesis for our initial approach to fostriecin (1) was based on a desire to access both fostriecin (1) and its dehydro-fostriesin congener 2 (Scheme 1).7m At the outset, the approach was developed, fully cognizant of the approaches of Trost and Hayashi. More specifically, we envisioned that both target molecules 1 and 2 could be derived from the Trost fostriecin intermediate 3 with a terminal BDMS-protected alkyne and the Hayashi intermediate 4, respectively.7g,i Both intermediates 3 and 4 could be derived from triol 5, which could be prepared by pyranone annulation of enone 6 with the requisite C-8,9,11-triol stereochemistry installed. We envisioned the C-11-propargyl alcohol of 6 being installed by an alkyne addition to aldehyde 7. Finally, the chirality of 7 could be prepared from a suitably protected achiral enoate 8 by a Sharpless asymmetric dihydroxylation.15
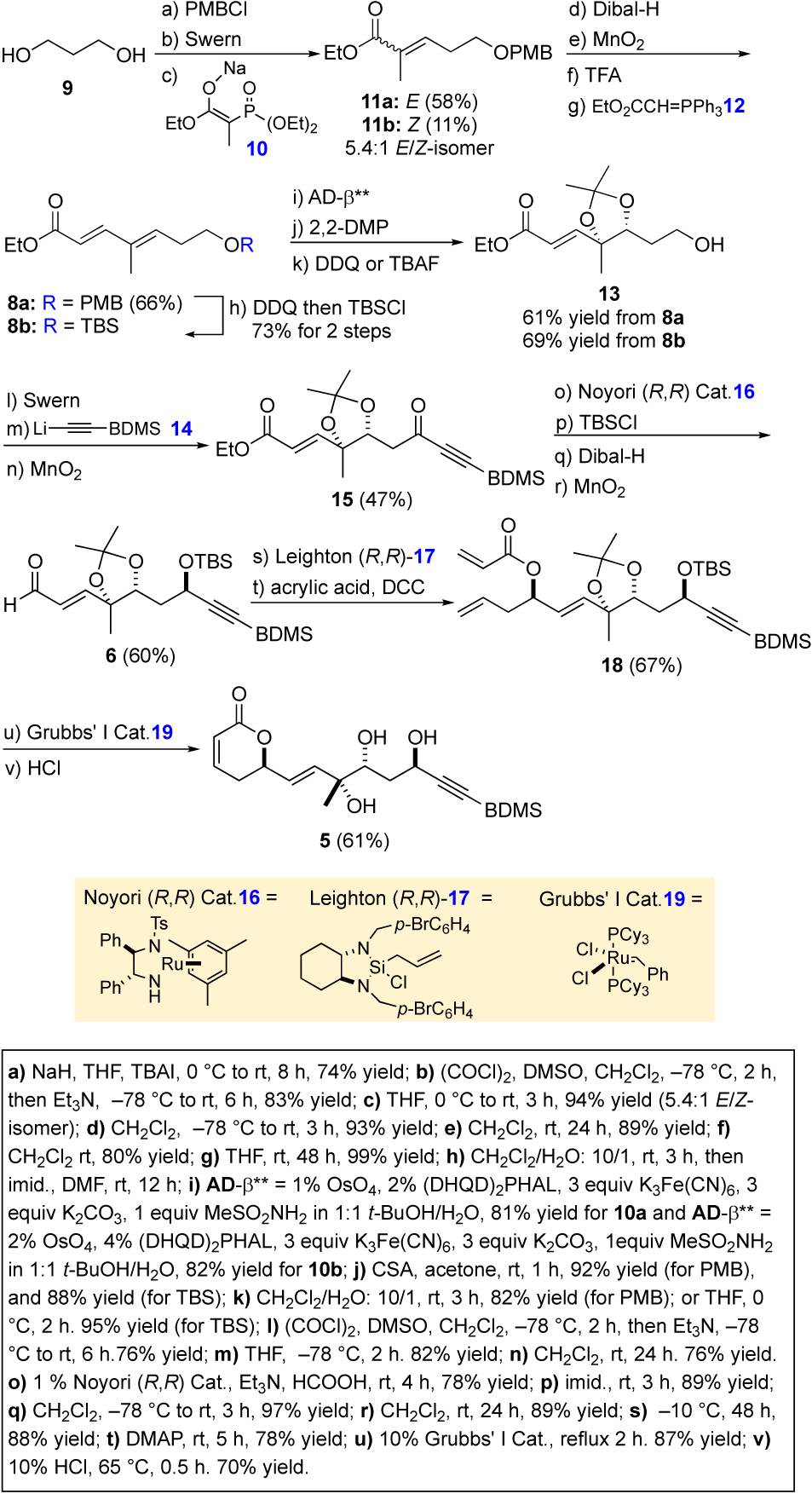
The synthesis of the key triol intermediate 5 commenced with commercially available 1,3-propane diol (9) (Scheme 2). Mono-PMB-protection, Swern oxidation and Horner-Wadsworth-Emmons olefination afforded a 5.4:1 E/Z-mixture of the PMB-protected enoates 11a/b in a 58% yield (3 steps).16,17 A Dibal-H reduction of the mixture of esters 11a/b followed by MnO2 oxidation yielded a similar E/Z-mixture of enals, with the undesired Z-isomer being easily isomerized into the E-isomer upon exposure to TFA (80%). Finally, exposure of the diastereomerically pure enal to a olefination the a stabilized Wittig reagent 12 selectively yielded the E,E-dienoate 8a (66 % yield for 4 steps).18 Because we worried about the oxidative stability of the PMB group to the dihydroxylation condition, we decided to also prepare a TBS-protected version of dienoate 8a (i.e., 8b). Thus, the PMB-protecting group was removed (DDQ) and replaced with a TBS-protecting group to give dienoate 8b in 73% yield for the 2 steps. To our delight, both dienoates 8a and 8b reacted cleanly upon being subjected to our typical dienoate variant Sharpless asymmetric dihydroxylation conditions (for 8a, 1% OsO4, 2% (DHQD)2PHAL for 8b, 2% OsO4, 4% (DHQD)2PHAL) to regioselectively form diols, which after acetonide formation and TBS-/PMB-deprotection to form primary alcohol 13 in high enantiopurity (98% ee)19 and excellent yield (61% and 69%, respectively from dienoates 8a/b).
Scheme 2.
Synthesis of pyranone 5
Oxidation of alcohol 13 via a Swern type DMSO oxidation, followed by addition of lithium acetylide 14 (BDMS-acetylene plus n-BuLi) to the resulting aldehyde afforded a 1:1 mixture of diastereomeric propargylic alcohols. A MnO2 oxidation of the crude mixture of alcohols gave ynone 15 (47%, 3 steps). The reagent controlled, diastereoselective reduction of ketone 15 was accomplished using the Noyori catalyzed (1% 16, Et3N•HCO2H) to give a single diastereomeric propargyl alcohol (dr >20:1).20 A subsequent TBS-protection, ester reduction with Dibal-H and MnO2 oxidation formed aldehyde 6. A highly diastereoselective allylation of aldehyde 6 was accomplished with the (R,R)-Leighton reagent 17 (–10 °C, 2 days) to give a homoallylic alcohol with near perfect stereocontrol (88%).21,22 A DCC-promoted esterification (4 equiv of acrylic acid, DCC) gave trienyne 18 (78%). Exposure of a refluxing CH2Cl2 solution of the trienyne 18 to the Grubbs I catalyst 19 resulted in a clean cyclization into a pyranone,23 which after acetonide deprotection gave the desired triol 5 (61%, 2 steps).
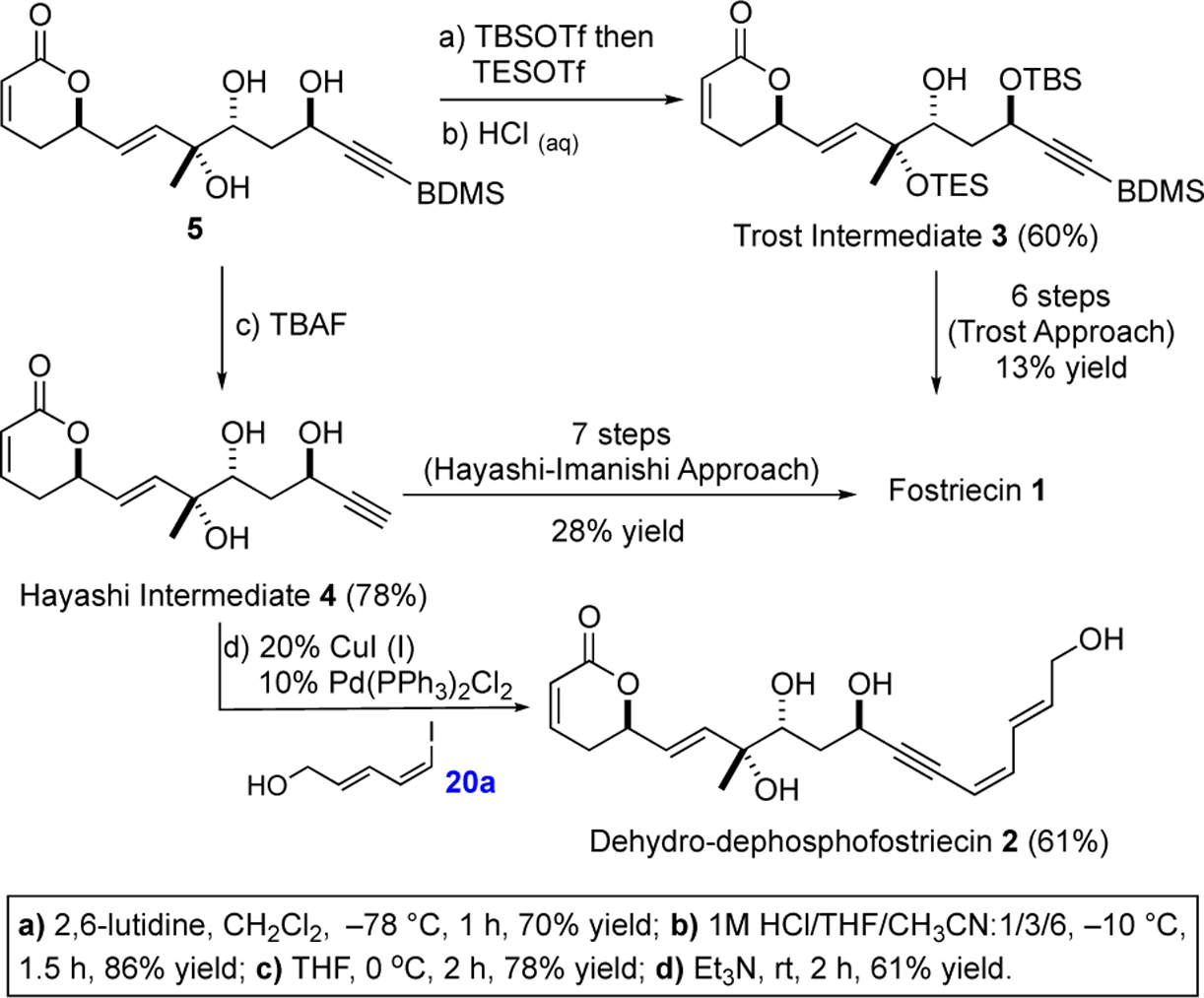
From triol 5 we were able to link it to two formal syntheses offostriecin (Scheme 3). A selective TBS-protection and TES-deprotection was used in a three-step protecting group manipulation strategy to give the Trost intermediate 3. Thus, exposure of triol 5 to one equiv of TBSOTf and excess 2,6-lutidine selectively protected the C-11 propargyl alcohol. Upon disappearance of the starting material, 2 equiv of TESOTf was added to the reaction mixture to form the persilylated material. Selective deprotection of the C-9 secondary TES-ether with aqueous HCl afforded the known Trost intermediate 3 (60%, 2 steps), which Trost et al. converted into fostriecin (1) in 6 steps with 13% yield.7g In an attempt to discover an alternative approach to fostriecin and analogues, we came interested in preparing dihydro-dephospho-fostriecin 2.7m,24 This desire led us to the synthesis of the terminal alkyne 4, an intermediate in Hayashi’s fostriecin synthesis that was completed using Imanishi’s end game.7b,25 Thus, upon treatment of BDMS-protected alkyne 5 with TBAF, the terminal alkyne 4 was prepared in 78% yield. Exposure of alkyne 4 to the Sonogashira cross coupling conditions with 5-iodo-butan-E,Z-dien-1-ol 20a afforded a 61% yield of dihydro-dephospho-fostriecin 2.26 Unfortunately, all efforts to selectively reduce the alkyne in 2 were unsuccessful (e.g., dissolving metal reductions, Red-Al, etc.). As a result, we turned our attention to an alternative route to fostriecin (1).
Scheme 3.
Two formal syntheses of fostriecin (1)
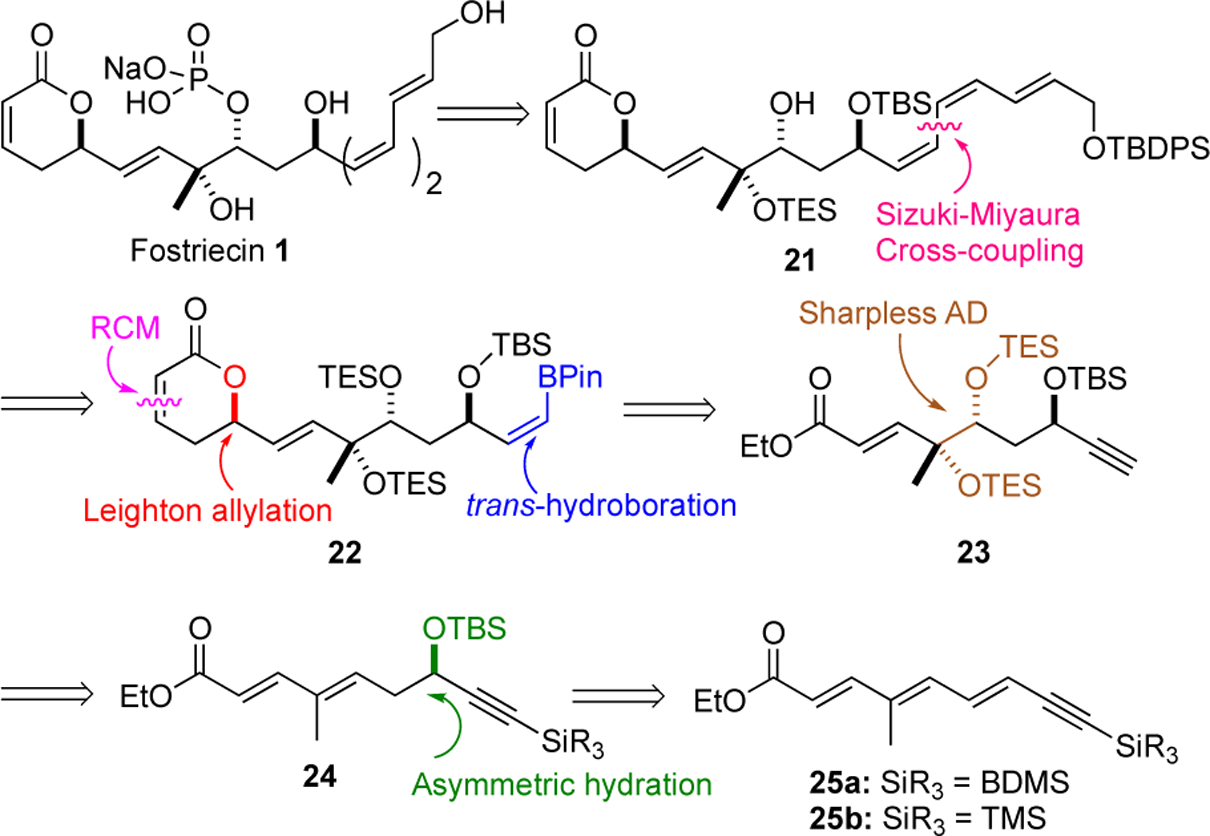
Concurrent with the approach outlined above, we explored an alternative route to fostriecin (1). Our retrosynthetic analysis for this alternative approach was focused upon our interest in using an iterative asymmetric hydration27 and dihydroxylation of achiral polyene 25 to address the C-8,9,11-triol positions (Scheme 4). Key to the approach is the reliance on the polarizing effect of ester conjugation to control the regioselectivity of two Sharpless dihydroxylation and formic acid reduction of a Pd-π-allyl intermediate (vide infra). An additional unique feature to this approach is the use of a trans-selective hydroboration/Suzuki cross-coupling reaction to install the E,Z,Z-triene functionality of fostriecine (1). As with the previous approach this approach would rely on a Leighton allylation and ring-closing metathesis to install the pyranone ring.
Scheme 4.
Our 2nd generation approach to fostriecin (1)
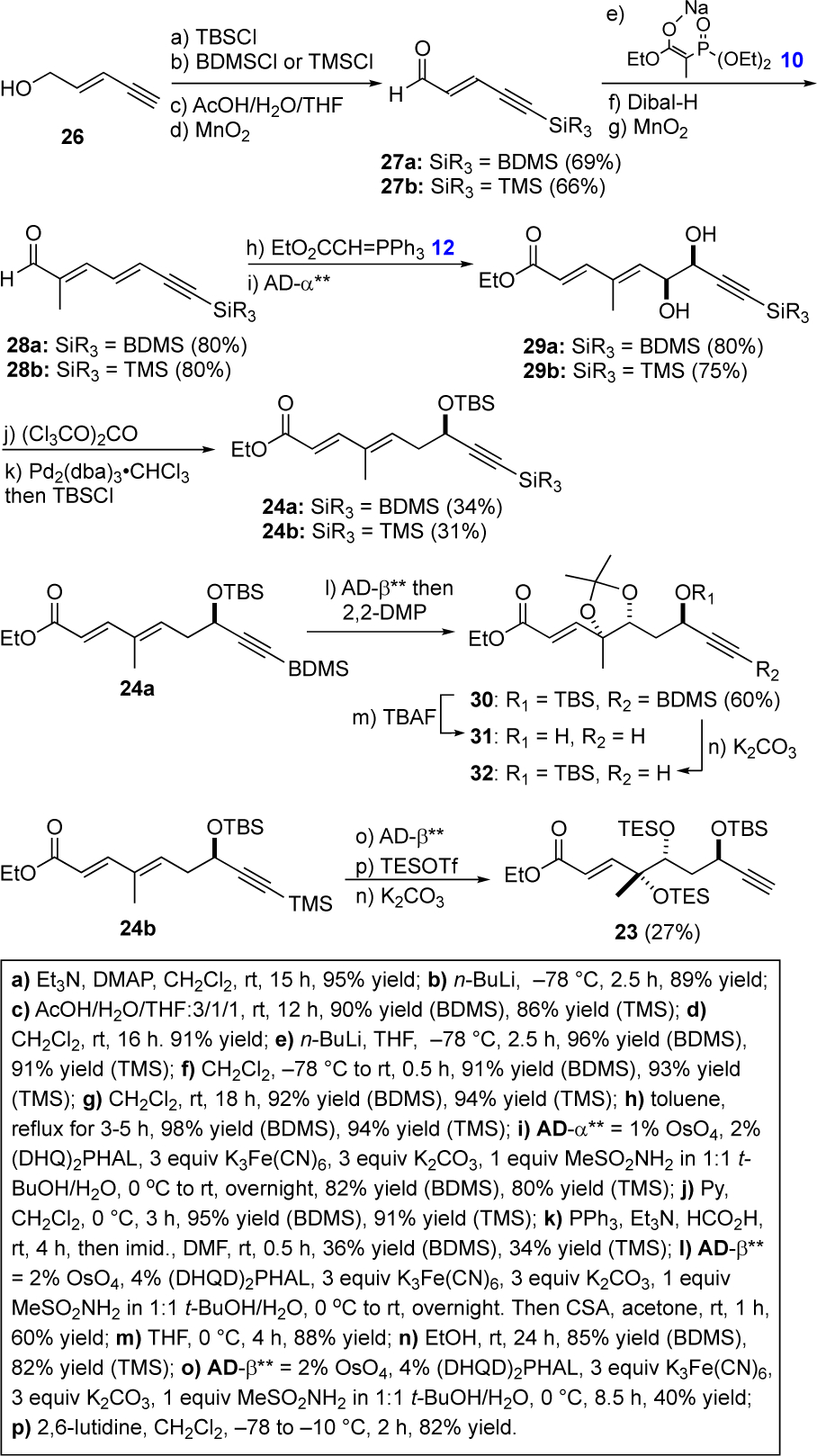
The revised approach began with the synthesis of trienynes 25a/b with a BDMS and TMS-protected alkyne, respectively, from commercially available enynol 26. A TBS-ether protection of the allylic alcohol was followed by protection of the alkyne as a TMS and BDMS alkyne. An acid catalyzed deprotection of the silyl-ether followed by a MnO2 oxidation of the allylic alcohol to form enals 27a/b (69%/66%, 4 steps). Exposure of aldehydes 27a and 27b separately to the Horner-Wadsworth-Emmons olefination gave respective enoates, which were reduced with two equiv of Dibal-H to form two allylic alcohols. A MnO2 oxidation of the allylic alcohols gave the two enals 28a/b (80%/80%, 3 steps). Wittig olefinations of 28a/b with the stabilized Wittig reagent 12 gave enynols 25a/b, which were then regio- and enantio-selectively dihydroxylated (OsO4/(DHQ)2PHAL) to give diols 29a/b (80%/75%, from 28a/b). The allylic alcohols in 29a and 29b were selectively reduced over the propargylic by a two-step cyclic carbonate formation and formic acid reduction with catalytic Pd(0)/PPh3 to form two propargyl alcohols, which after TBS-protection formed 24a/b (34%/31%, 2 steps). At this stage the routes diverged at BDMS-alkynes 24a and 24b. The BDMS-alkyne 24a was diastereoselectively dihydroxylated (OsO4/(DHQD)2PHAL) to give a crude diol, which was protected as an acetonide 30 (60%). Subsequently, the BDMS group was removed with K2CO3 in methanol to give 32 (85%). In a related sequence, the TMS-protected alkyne 24b was regioselectively oxidized under the Sharpless conditions gave a diol, which was protected as a bis-TES ether. Finally, the TMS-alkyne was deprotected with K2CO3 in methanol to give 23 (27%, 3 steps).
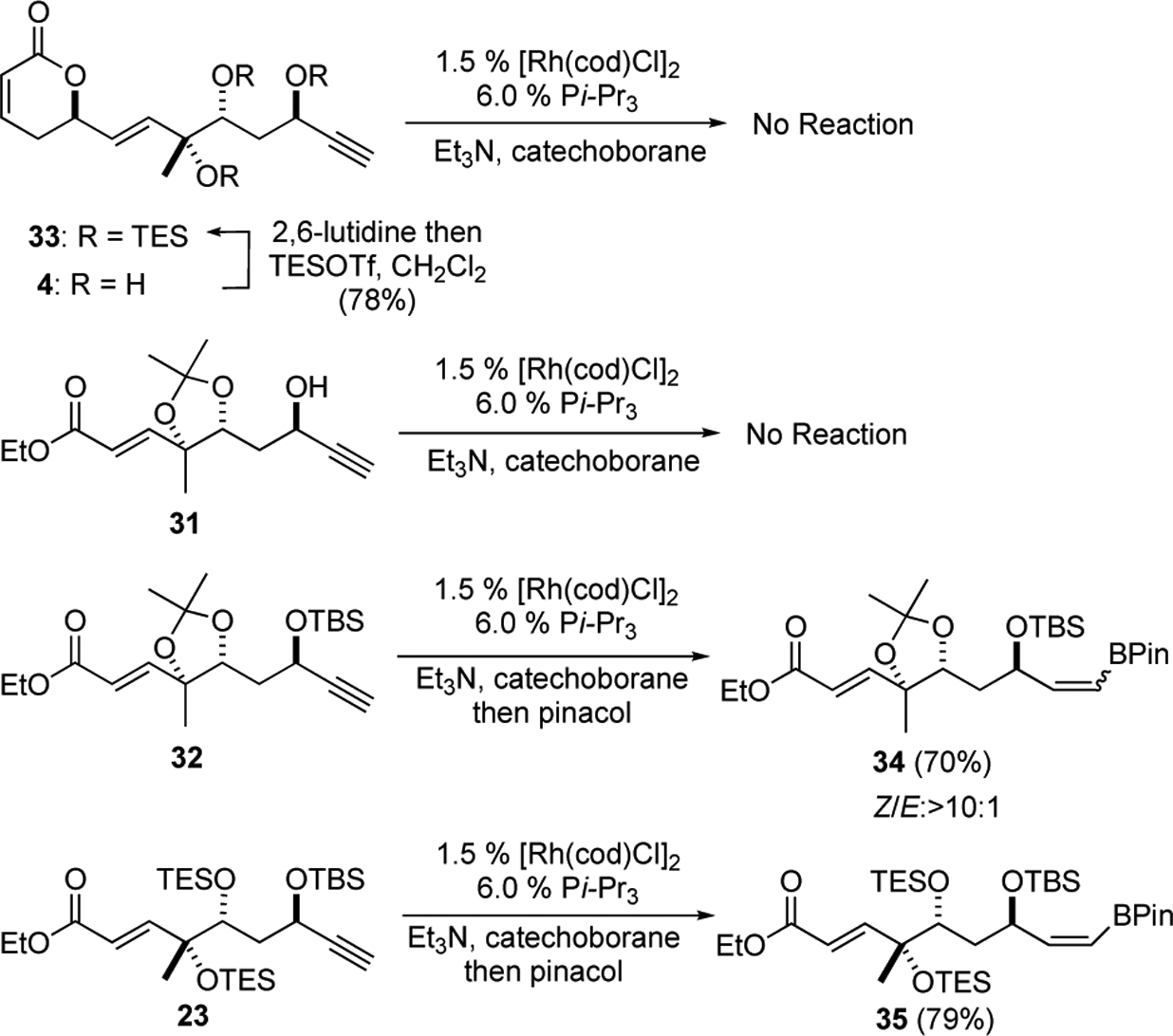
Having exhaustively explored Sonogashira type approaches, we next decided to investigate the possibility of using Miyaura’s Rh-catalyzed trans-hydroboration of alkynes28 for the generation of cross-coupling compatible organometallic species capable of delivering the requisite Z,Z,E-triene of fostriecin. Our initial efforts to explore the Miyaura’s conditions (catecholborane, 1.5% [Rh(cod)Cl2], 6.0% P(i-Pr)3 then pinacol) for the trans-hydroboration of the terminal alkyne with substrates that contained free alcohols or pyranones (e.g., 33, 4 and 31) led to less than desirable outcomes. For example, exposure of pyranone containing alkyne 33 to the reaction conditions led to no reaction. Similar negative results were seen when alkyne 31 with a free propargyl alcohol was used. This lack of reactivity was overcome by exploring substrates devoid of free alcohol and pyranone functionalities. For instance, excellent yields of pinacolboranes 34 and 35 were stereoselectivity obtained when terminal alkynes 32 and 23 were exposed to the reaction conditions.
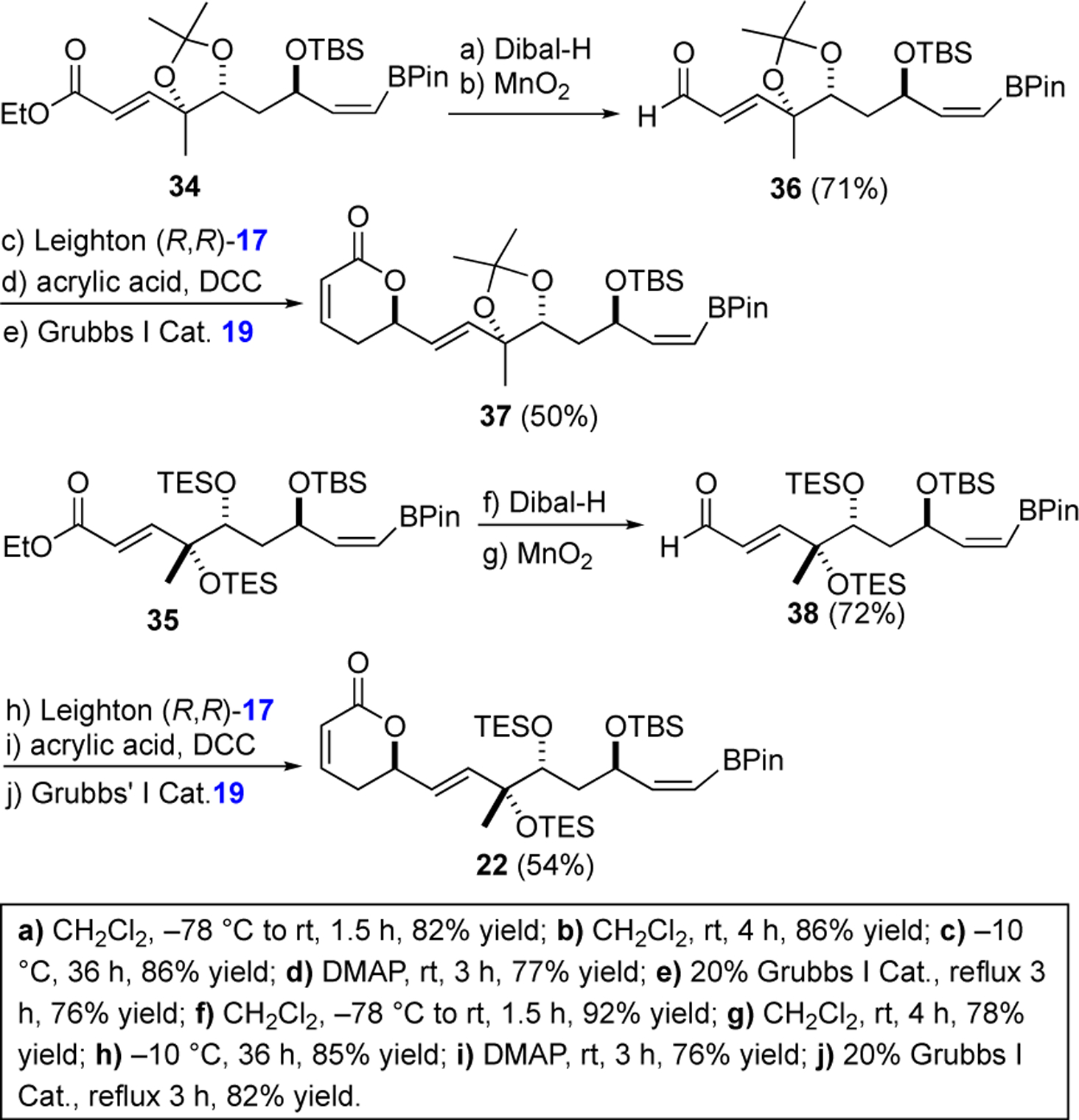
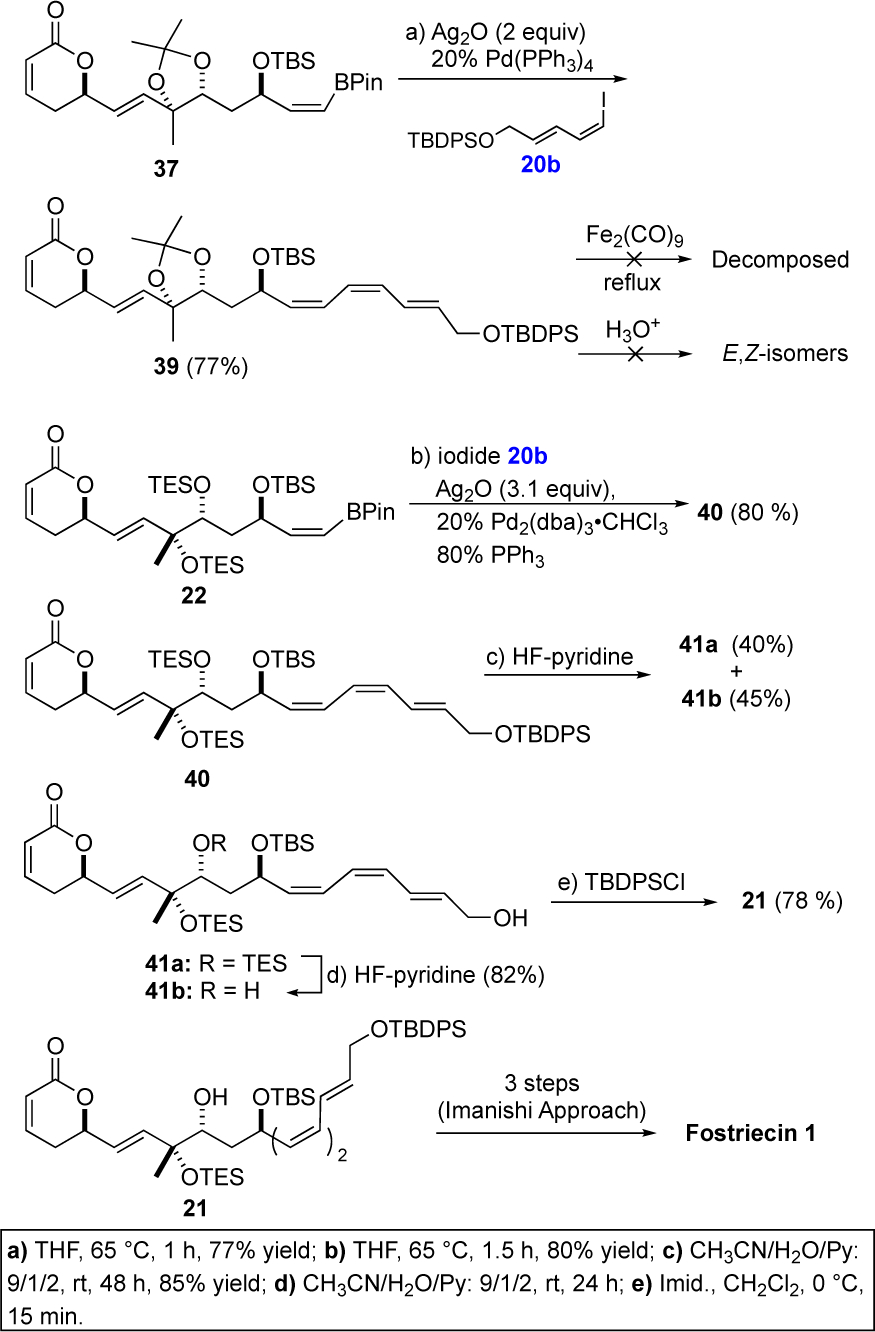
The pinacol boronate products 34 and 35 were remarkably stable to silica gel chromatography as well as to several reaction conditions. For instance, vinyl borane 34 survived exposure to excess Dibal-H and MnO2 oxidation to give aldehyde 36 (71%, 2 steps). Interestingly the stability of the vinyl borane was stereospecific as only the Z-vinyl borane was isolated after the MnO2 oxidation, as the minor E-isomer did not survive the reaction conditions. The stability to the vinyl borane unique to a Z-isomer was similarly on display as it survived the Leighton allylation of 36 and a subsequent DCC coupling reaction (DCC/acrylic acid) and ring-closing metathesis with the Grubbs I catalyst 29 to give 37 (50%, 3 steps). The Z-vinyl borane 35, with a C-8/9 bis-TES ether protection group instead of an acetonide, similarly survived the Dibal-H reduction and allylic alcohol oxidation to form 38 (72%, 2 steps). Once again, the vinyl borane functionality of 38 survived the Leighton allylation, acrylation and ring-closing metathesis to form 22 (53%, 3 steps).23,29
We first explored the Suzuki-Miyaura cross coupling Z-vinyl borane 37 with Z-vinyl iodide 20b, using Ag2O and Pd(PPh3)4 as a catalyst to form the E,E,Z-triene 39 in good overall yield and triene selectivity.30 Unfortunately, we were unable to find condition to deprotect the acetonide protecting group without isomerizing the triene. Our efforts to protect the polyene as an Fe(CO)3 complex also led to isomerization. When we turned to cross-coupling of the persilyl protected vinyl borane 22, we found the Pd(PPh3)4/Ag2O system failed to give any product. Fortunately, excellent yields of cross coupling reactions could be obtained, when we increased the catalyst loading and switched to alternative Pd(0) source while the Pd/PPh3 ratio was maintained at a 1:2 ratio (20% Pd2(dba)3•CHCl3/80% PPh3). Under these optimal conditions the Z-vinyl boranate 22 and Z-vinyl iodide 20b coupled to form the Z,Z,E-triene 40 in an 80% yield and with excellent triene E/Z-stereoselectivity (>20:1).
In triene 40 all the carbon atoms and stereocenters of fostriecin (1) have been successfully installed. To complete the synthesis all that was required was the adjustment of the silyl protecting groups to match the protecting group pattern used by Imanishi (i.e., 21). The selective deprotection of the C-9 TES-silyl ether in 40 to 21 was not straightforward, as it always occurred with concomitant deprotection of the TBDPS group. After considerable experimentation, a practical three step solution was found that began with deprotection of the TBDPS group in 40 with HF•Py to give 41a along with an equal amount of 41b with the C-9 TES-group already removed. After chromatographic separation, re-exposure of 41a to HF•Py selectively removed the C-9 TES-group to cleanly provide 41b. A subsequent re-protection of the primary hydroxyl group of 41b with TBDPSCl/imidazole afforded the desired fostriecin precursor 21. Finally, carefully following the Imanishi protocol, a three-step sequence converted precursor 21 into fostriecin 1 (~ 0.5 mg) whose 1HNMR, HRMS date matched those of the natural material.
Conclusions
In summary, three distinct enantioselective routes for the synthesis of fostriecin (1) have been developed. The two formal syntheses used a combination of Noyori ynone reduction, Sharpless dihydroxylation and a Leighton allylation to install all the stereochemistry required to converge with the syntheses established by Trost and Hiyashi. The formal approach also enabled a route to a dihydro-dephospho-congener 2. An alternative total synthesis of fostriecin was also described which used a combination of highly regioselective asymmetric dihydroxylations and Pd-π-allyl-mediated reductions of an achiral 25 trieneyne to install the C-8,9,11-triol portion of the molecule. Finally, a trans-selective hydroboration and cross coupling sequence along with a staged protection/deprotection sequence was used to complete the synthesis.
Supplementary Material
Figure 1.
Two generations of synthesizing fostriecin (1)
Scheme 5.
Synthesis and asymmetric hydration/oxidation of tri-enynoate 24
Scheme 6.
Trans-hydroboration of alkynes 23 and 30
Scheme 7.
The installation of the pyranone ring
Scheme 8.
End game of fostriecin (1)
Acknowledgements
We are grateful to NIH (GM090259) and NSF (CHE-1565788). The authors gratefully acknowledge the National Science Foundation (CHE-1565788), and the National Institutes of Health (AI146485, AI144196 and AI142040) for their support of this work.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.(a) Tunac JB; Graham BD; Dobson WE Novel Antitumor Agents CI-920, PD 113,270 and PD 113,271 – Taxonomy, Fermentation and Biological Properties. J. Antibiot 1983, 36, 1595. [DOI] [PubMed] [Google Scholar]; (b) Stampwala SS; Bunge RH; Hurley TR; Wilmer NE; Brankiewicz AJ; Steinman CE; Smitka TA; French JC Novel Antitumor Agents CI-920, PD 113,270 and PD 113,271 – Isolation and Characterization. J. Antibiot 1983, 36, 1601. [DOI] [PubMed] [Google Scholar]; (c) Hokanson GC; French JC Novel Antitumor Agents CI-920, PD 113,270 and PD 113,271 – Structure Determination. J. Org. Chem 1985, 50, 462. [Google Scholar]
- 2.Trost BM; Knopf JD; Brindle CS Synthetic Strategies Employed for the Construction of Fostriecin and Related Natural Products. Chem. Rev 2016, 116, 15035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Just G; O’Connor B Synthesis of the 5R,8R,9S,11R Dephosphorylated Derivative of CI-920, a Novel Antitumor Agent. Tetrahedron Lett. 1988, 29, 753. [Google Scholar]
- 4.(a) Boger DL; Hikota M; Lewis BM Determination of the Relative and Absolute Stereochemistry of Fostriecin (CI-920). J. Org. Chem 1997, 62, 1748. [Google Scholar]; (b) Buck SB; Hardouin C; Ichikawa S; Soenen DR; Gauss CM; Hwang I; Swingle MR; Bonness KM; Honkanen RE; Boger DL Fundamental role of the fostriecin unsaturated lactone and implications for selective protein phosphatase inhibition. J. Am. Chem. Soc 2003, 125, 15694. [DOI] [PubMed] [Google Scholar]
- 5.Boger DL; Ichikawa S; Zhong W Total Synthesis of Fostriecin (CI-920). J. Am. Chem. Soc 2001, 123, 4161. [DOI] [PubMed] [Google Scholar]
- 6.Chavez DE; Jacobsen EN Total Synthesis of Fostriecin (CI-920). Angew. Chem. Int. Ed 2001, 40, 3667. [DOI] [PubMed] [Google Scholar]
- 7.(a) Reddy YK; Falck JR Asymmetric Total Synthesis of (+)-Fostriecin. Org. Lett 2002, 4, 969. [DOI] [PubMed] [Google Scholar]; (b) Miyashita K; Ikejiri M; Kawasaki H; Maemura S; Imanishi T Total Synthesis of Fostriecin (CI-920) via a Convergent Route. Chem. Commun 2002, 7, 742. [DOI] [PubMed] [Google Scholar]; (c) Wang Y-G; Kobayashi Y Formal Total Synthesis of Fostriecin. Org. Lett 2002, 4, 4615. [DOI] [PubMed] [Google Scholar]; (d) Esumi T; Okamoto N; Hatakeyama S Versatile Enantiocontrolled Syn-thesis of (+)-Fostriecin. Chem. Commun 2002, 24, 3042. [DOI] [PubMed] [Google Scholar]; (e) Shibahara S; Fujino M; Tashiro Y; Okamoto N; Esumi T; Takahashi K; Ishihara J; Hatakeyama S Total Synthesis of (+)-Fostriecin and (+)-Phoslactomycin B. Synthesis, 2009, 17, 2935. [Google Scholar]; (f) Fujii K; Maki K; Kanai M; Shibasaki M Formal Catalytic Asymmetric Total Synthesis of Fostriecin. Org. Lett 2003, 5, 733. [DOI] [PubMed] [Google Scholar]; (g) Trost BM; Frederiksen MU; Papillon JPN; Harrington PE; Shin S; Shireman BT Dinuclear Asymmetric Zn Aldol Additions: Formal Asymmetric Synthesis of Fostriecin. J. Am. Chem. Soc 2005, 127, 3666. [DOI] [PubMed] [Google Scholar]; (h) Yadav JS; Prathap I; Tadi BP Formal Synthesis of Fostriecin by a Carbohydrate-Based Approach. Tetrahedron Lett. 2006, 47, 3773. [Google Scholar]; (i) Hayashi Y; Yamaguchi H; Toyoshima M; Okado K; Toyo T; Shoji M Formal Total Synthesis of Fostriecin via 1,4-Asymmetric Induction Using Cobalt-Alkyne Complex. Org. Lett 2008, 10, 1405. [DOI] [PubMed] [Google Scholar]; (j) Robles O; McDonald FE Conver-gent Synthesis of Fostriecin via Selective Alkene Couplings and Regioselective Asymmetric Dihydroxylation. Org. Lett 2009, 11, 5498. [DOI] [PubMed] [Google Scholar]; (k) Li D; Zhao Y; Ye L; Chen C; Zhang J A Formal Total Synthesis of Fostriecin by a Convergent Approach. Synthesis, 2010, 19, 3325. [Google Scholar]; (l) Gao D; O’Doherty GA Total Synthesis of Fostriecin: Via a Regio- and Stereoselective Polyene Hydration, Oxidation, and Hydroboration Sequence. Org. Lett 2010, 12, 3752. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Gao D; Li B; O’Doherty GA Synthesis of Dehydro-Dephospho-Fostriecin and Formal Total Synthesis of Fostriecin. Org. Lett 2019, 21, 8334. [DOI] [PubMed] [Google Scholar]
- 8.(a) Whitehead A; McReynolds MD; Moore JD; Hanson PR Multivalent Activation in Temporary Phosphate Tethers: A New Tether for Small Molecule Synthesis. Org. Lett 2005, 7, 3375. [DOI] [PubMed] [Google Scholar]; (b) Cossy J; Pradaux F; BouzBouz S Synthesis of the C1−C12 Fragment of Fostriecin. Org. Lett 2001, 3, 2233. [DOI] [PubMed] [Google Scholar]; (c) Robles O; McDonald FE Convergent Synthesis of Fostriecin via Selective Alkene Couplings and Regioselective Asymmetric Dihydroxylation. Org. Lett 2009, 11, 5498. [DOI] [PubMed] [Google Scholar]
- 9.(a) Boritzki TJ; Wolfard TS; Besserer JA; Jackson RC; Fry DW Inhibition of Type II Topoisomerase by Fostriecin. Biochem. Pharmacol 1988, 37, 4063. [DOI] [PubMed] [Google Scholar]; (b) Guo XW; Th’ng JP; Swank RA; Anderson HJ; Tudan C; Bradbury EM; Roberge M Chromosome Condensation Induced by Fostriecin Does Not Require p34cdc2 Kinase Activity and Histone H1 Hyperphosphorylation, but Is Associated with Enhanced Histone H2A and H3 Phosphorylation. EMBO J. 1995, 14, 976. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ho DT; Roberge M Cancer Biology: The Antitumor Drug Fostriecin Induces Vimentin Hyperphosphorylation and Intermediate Filament Reorganization. Carcinogenesis, 1996, 17, 967. [DOI] [PubMed] [Google Scholar]; (d) Buck SB; Hardouin C; Ichikawa S; Soenen DR; Gauss C-M; Hwang I; Swingle MR; Bonness KM; Honkanen RE; Boger DL Fundamental Role of the Fostriecin Unsaturated Lactone and Implications for Selective Protein Phosphatase Inhibition. J. Am. Chem. Soc 2003, 125, 15694. [DOI] [PubMed] [Google Scholar]; (e) Amable L; Moring KL; Swingle MR; Ratti P; Buck S; Boger DL; Honkanen R Investigations into the Structure-Activity Relationship of Fostriecin, a Potent Inhibitor of Ser/Thr Protein Phosphatases. FASEB J. 2006, 20, A924. [Google Scholar]
- 10.(a) Walsh AH; Cheng A; Honkanen RE Fostriecin, an Antitumor Antibiotic with Inhibitory Activity Against Serine/Threonine Protein Phosphatases Types 1 (PP1) and 2A (PP2A), Is Highly Selective for PP2A. FEBS Lett. 1997, 416, 230. [DOI] [PubMed] [Google Scholar]; (b) Hastie CJ; Cohen PTW Purification of Protein Phosphatase 4 Catalytic Subunit: Inhibition by the Antitumor Drug Fostriecin and Other Tumor Suppressors and Promoters. FEBS Lett. 1998, 431, 357. [DOI] [PubMed] [Google Scholar]; (c) Forbes AM; Meier GP; Haendiges S; Taylor LP Structure-Activity Relationship Studies of Flavonol Analogues on Pollen Germination. J. Agric. Food. Chem 2014, 62, 2175. [DOI] [PubMed] [Google Scholar]; (d) Amrutha K; Nanjan P; Shaji SK; Sunilkumar D; Subhalakshmi K; Rajakrishna L; Banerji A Discovery of lesser known flavones as inhibitors of NF-kB signaling in MDA-MB-231 breast cancer cells-A SAR study. Bioorg. Med. Chem. Lett 2014, 24, 4735. [DOI] [PubMed] [Google Scholar]
- 11.Reviews:; (a) Lewy DS; Gauss C-M; Soenen DR; Boger DL Fostriecin: Chemistry and Biology. Curr. Med. Chem 2002, 9, 2005. [DOI] [PubMed] [Google Scholar]; (b) De Jong RS; De Vries EGE; Mulder NH Fostriecin: a Review of the Preclinical Data. Anti-Cancer Drugs. 1997, 8, 413. [PubMed] [Google Scholar]
- 12.(a) Jackson RC; Fry DW; Boritzki TJ; Roberts BJ; Hook KE; Leopold WR The Biochemical Pharmacology of CI-920, a Structurally Novel Antibiotic with Antileukemic Activity. Adv. Enzyme Regul 1985, 23, 193. [DOI] [PubMed] [Google Scholar]; (b) Scheithauer W; Hoff DDV; Clark GM; Shillis JL; Elslager EF In Vitro Activity of the Novel Antitumor Antibiotic Fostriecin (CI-920) in a Human Tumor Cloning Assay. Eur. J. Clin. Oncol 1986, 22, 921. [DOI] [PubMed] [Google Scholar]
- 13.De Jong RS; Mulder NH; Uges DRA; Sleijfer DT; Hoppener FJP; Groen HJM; Willemse PHB; van der Graaf WT; de Vries EGE Phase I and Pharmacokinetic Study of the Topoisomerase II Catalytic Inhibitor Fostriecin. Br. J. Cancer, 1999, 79, 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Liu X; Wang Y; Duclos RI; O’Doherty GA Stereochemical Structure Activity Relationship Studies (S-SAR) of Tetrahydrolipstatin. ACS Med. Chem. Lett 2018, 9, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Goins CM; Sudasinghe TD; Liu X; Wang Y; O’Doherty GA; Ronning DR Characterization of Tetrahydrolipstatin and Stereoderivatives on the Inhibition of Essential Mycobacterium tuberculosis Lipid Esterases. Biochemistry, 2018, 57, 2383. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mulzer M; Tiegs B; Wang Y; Coates GW; O’Doherty GA Total Synthesis of Tetrahydrolipstatin, via a highly Regio- and Stereo-selective Carbonylation of Epoxyhomoallylic alcohols. J. Am. Chem. Soc 2014, 136, 10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.For Sharpless asymmetric dihydroxylation, see:; (a) Sharpless KB; Amberg W; Bennani YL; Crispino GA; Hartung J; Jeong KS; Kwong HL; Morikawa K; Wang ZM; Xu D; Zhang XL The Osmium-Catalyzed Asymmetric Dihydroxyla-tion: A New Ligand Class and a Process Improvement. J. Org. Chem 1992, 57, 2768. [Google Scholar]; (b) Kolb HC; VanNieuwenhze MS; Sharpless KB Catalytic Asymmetric Dihydroxylation. Chem. Rev 1994, 94, 2483. [Google Scholar]; For our previous approaches to C-8/9 diol installation, see:; (c) Guo G; Mortensen MS; O’Doherty GA Enantioselective Synthesis of 10-epi-Anamarine via an Iterative Dihydroxylation Sequence. Org. Lett 2008, 10, 3149. [DOI] [PubMed] [Google Scholar]; (d) Li M; O’Doherty GA De Novo Formal Synthesis of (−)-Apicularen A via an Iterative Asymmetric Hydration Sequence. Org. Lett 2006, 8, 6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Omura K; Swern D Oxidation of Alcohols by “Activated” Dimethyl Sulfoxide. A Preparative Steric and Mechanistic Study. Tetrahedron Lett. 1978, 34, 1651. [Google Scholar]
- 17.(a) Horner L; Hoffmann H; Wippel HG Phosphorus Organic Compounds. XII. Phosphine Oxides as Reagents for the Olefin Formation. Chem. Ber 1958, 91, 61. [Google Scholar]; (b) Horner L; Hoffmann H; Wippel HG; Klahre G Phosphorus Organic Compounds. XX. Phosphine Oxides as Reagents for Olefin Formation. Chem. Ber 1959, 92, 2499. [Google Scholar]; (c) Wadsworth WS Jr.; Emmons WD The Utility of Phosphonate Carbanions in Olefin Synthesis. J. Am. Chem. Soc 1961, 83, 1733. [Google Scholar]
- 18.Wittig G; llkopf U. Über. Triphenyl-Phosphin-Methylene Als Olefinbildende Reagenzien (I. Mitteil.). Chem. Ber 1954, 87, 1318. [Google Scholar]
- 19.Ahmed Md. M.; Mortensen MS; O’Doherty GA De Novo Synthesis of 2-Substituted syn-1,3-Diols via an Iterative Asymmetric Hydration Strategy. J. Org. Chem 2006, 71, 7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Noyori R; Ohkuma T Asymmetric Catalysis by Architec-tural and Functional Molecular Engineering: Practical Chemo- and Stereoselective Hydrogenation of Ketones. Angew. Chem. Int. Ed 2001, 40, 40. [PubMed] [Google Scholar]; (b) Noyori R; Yamakawa M; Hashiguchi S Metal-Ligand Bifunctional Catalysis: A Nonclassical Mechanism for Asymmetric Hydrogen Transfer between Alcohols and Carbonyl Compounds. J. Org. Chem 2001, 66, 7931. [DOI] [PubMed] [Google Scholar]; (c) Ikariya T; Murataa K; Noyori R Bifunctional transition metal-based molecular catalysts for asymmetric syntheses. Org. Biomol. Chem 2006, 4, 393. [DOI] [PubMed] [Google Scholar]
- 21.(a) Kubota K; Leighton JL A Highly Practical and Enantioselecve Reagent for the Allylation of Aldehydes. Angew. Chem. Int. Ed 2003, 42, 946. [DOI] [PubMed] [Google Scholar]; (b) Trost BM; Weiss AH The Enantioselective Addition of Alkyne Nucleophiles to Carbonyl Groups. Adv. Synth. Catal 2009, 351, 963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Kim IS; Ngai M-Y; Krische MJ Enantioselective Iridium-Catalyzed Carbonyl Allylation from the Alcohol or Alde-hyde Oxidation Level using Allyl Acetate as an Allyl Metal Surrogate. J. Am. Chem. Soc 2008, 130, 6340. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Skucas E; Bower JF; Krische MJ Carbonyl Allylation in the Absence of Preformed Allyl Metal Reagents: Reverse Prenylation via Iridium-Catalyzed Hydrogenative Coupling of Dimethylallene. J. Am. Chem. Soc 2007, 129, 12678. [DOI] [PubMed] [Google Scholar]; (c) Jadhav PK; Bhat KS; Perumal PT; Brown HC Chiral Synthesis via Organoboranes: Asymmetric Allylboration via Chiral Allyldialkylboranes. Synthesis of Homoallylic Alcohols with Exceptionally High Enantiomeric Excess. J. Org. Chem 1986, 51, 432. [Google Scholar]
- 23.(a) Grubbs RH Olefin Metathesis. Tetrahedron Lett. 2004, 60, 7117. [Google Scholar]; (b) Trnka TM; Grubbs RH The Development of L2 × 2Ru = CHR Olefin Metathesis Catalysts: An Organometallic Success Story. Acc. Chem. Res 2001, 34, 18. [DOI] [PubMed] [Google Scholar]
- 24.Hayashi Y; Yamaguchi H; Toyoshima M; Okado K; Toyo T; Shoji M Formal Total Synthesis of Fostriecin by 1,4-Asymmetric Induction with an Alkyne−Cobalt Complex. Chem. Eur. J 2010, 16, 10150. [DOI] [PubMed] [Google Scholar]
- 25.Miyashita K; Ikejiri M; Kawasaki H; Maemura S; Imanishi T Total Synthesis of an Antitumor Antibiotic, Fostriecin (CI-920). J. Am. Chem. Soc 2003, 125, 8238. [DOI] [PubMed] [Google Scholar]
- 26.Sonogashira K; Tohda Y; Hagihara N Convenient Synthesis of Acetylenes. Catalytic Substitutions of Acetylenic Hydrogen with Bromo Alkenes, Iodo Arenes, and Bromopyridines. Tetrahedron Lett. 1975, 16, 4467. [Google Scholar]
- 27.(a) Wang Y; Xing Y; Zhang Q; O’Doherty GA De Novo Synthesis of Natural Products via the Asymmetric Hydration of Polyenes. Chem. Commun 2011, 47, 8493. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li M; O’Doherty GA De Novo Asymmetric Synthesis of Milbemy-cin β3 via an Iterative Asymmetric Hydration. Org. Lett 2006, 8, 3987. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mortensen MS; Osbourn JM; O’Doherty GA De Novo Formal Synthesis of (−)-Virginiamycin M2 via the Asymmetric Hydration of Dienoates. Org. Lett 2007, 9, 3105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Guo H; Mortensen MS; O’Doherty GA Formal Total Synthesis of RK-397 via an Asymmetric Hydration and Iterative Allylation Strategy. Org. Lett 2008, 10, 3149. [DOI] [PubMed] [Google Scholar]
- 28.Ohmura T; Yamamoto Y; Miyaura N Rhodium- or Iridium-Catalyzed trans-Hydroboration of Terminal Alkynes, Giving (Z)-1-Alkenylboron Compounds. J. Am. Chem. Soc 2000, 122, 4990. [Google Scholar]
- 29.(a) Hunter TJ; O’Doherty GA An Enantioselective Synthesis of Cryptocarya Diacetate. Org. Lett 2001, 3, 2777. [DOI] [PubMed] [Google Scholar]; (b) Smith CM; O’Doherty GA Enantioselective Synthesis of Cryptocarya Triacetate, Cryptocaryolone and Cryptocaryolone Diacetate. Org. Lett 2003, 5, 1959. [DOI] [PubMed] [Google Scholar]; (c) Wang Y; O’Doherty GA Cryptocaryol A and B: Total Syntheses, Stereochemical Revision, Structure Elucidation and Structure-Activity Relationship. J. Am. Chem. Soc 2013, 135, 9334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyaura N; Suzuki A Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev 1995, 95, 2457. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.