

Abstract
A short, scalable total synthesis of meayamycin is described by an approach that entails a longest linear sequence of 12 steps (22 steps overall) from commercially available chiral pool materials (ethyl L-lactate, BocNH-Thr-OH, and D-ribose) and introduces the most straightforward preparation of the right-hand subunit detailed to date. The use of the approach in the divergent synthesis of a representative series of O-acyl analogues is exemplified.
Graphical abstract
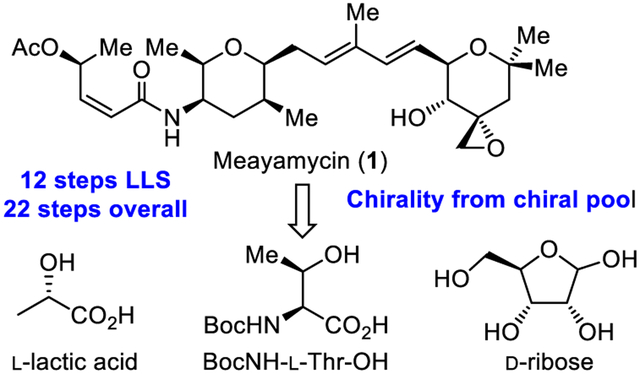
FR901464, isolated by Fujisawa Pharmaceutical Company from pseudomonas sp. No. 2663,1 is the first member of a growing class of potent antitumor antibiotics that includes the spliceostatins2 and thailanstatins.3 Initial total syntheses of FR901464 confirmed the assigned structure and relative stereochemistry within each noncontiguous subunit and permitted assignment of absolute stereochemistry. These were disclosed first by Jacobsen and co-workers (40 steps),4 utilizing a powerful asymmetric hetero Diels–Alder reaction to form the two tetrahydropyrans, and shortly thereafter by Kitahara (41 steps),5 enlisting the chiral pool to access the two tetrahydropyrans. Subsequent total syntheses by Koide6 (28 steps) and later by Ghosh7 (22 steps) provided more expedient assembly of the two highly substituted and functionalized tetrahydropyran rings.8 In addition to demonstrating the critical role of the epoxide,4 Jacobsen defined the instability of FR901464 and further showed that removal of the right-hand subunit tertiary alcohol not only abrogated this instability, but also enhanced biological potency.4 In an extension of these observations, Koide latter found that replacement of the tertiary alcohol with a methyl group (vs H)4 not only avoided the rapid compound degradation but also improved potency as much as 100-fold.6 Such synthetic analogues, named the meayamycins (Figure 1), represented an early contribution to the growing number of natural products and analogues in the class accessed by total synthesis.4-13 In 2007, Yoshida disclosed that FR901464 binds a subunit of the spliceosome disrupting conversion of pre-mRNA to mRNA.14 Shortly thereafter, Koide established that the meayamycins act in an analogous manner.15 The mechanism by which spliceosome inhibition is achieved has been shown to be effective in selectively controlling both cancer cell proliferation and metastasis with selected members exhibiting efficacious in vivo antitumor activity.16
Figure 1.
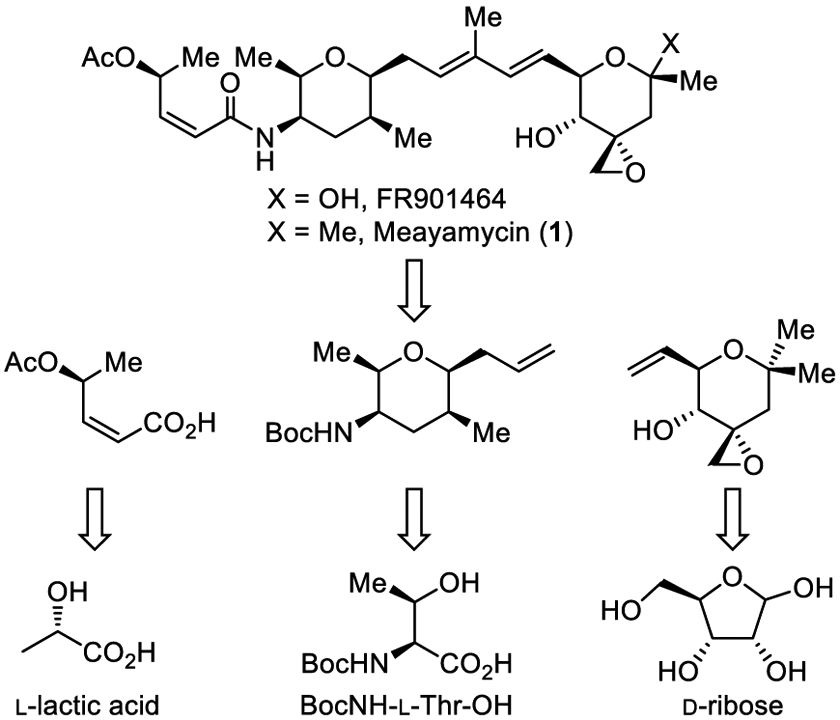
Target structures, key intermediates, and chiral pool starting materials.
In a program that explores natural product analogues as therapeutics17 or components of antibody-drug conjugates, we targeted the potent meayamycins with the objective of developing a short, scalable total synthesis. After several iterations on the approach, each subunit used to assemble 1 herein is derived from chiral pool starting materials with all 8 chiral centers introduced or controlled by chirality found in inexpensive commercial starting materials (Figure 1). The approach provided 1 in a longest linear sequence of 12 steps (22 steps overall),18 complementary to an improved approach recently disclosed by Koide (11 longest linear sequence, 24 steps overall).19
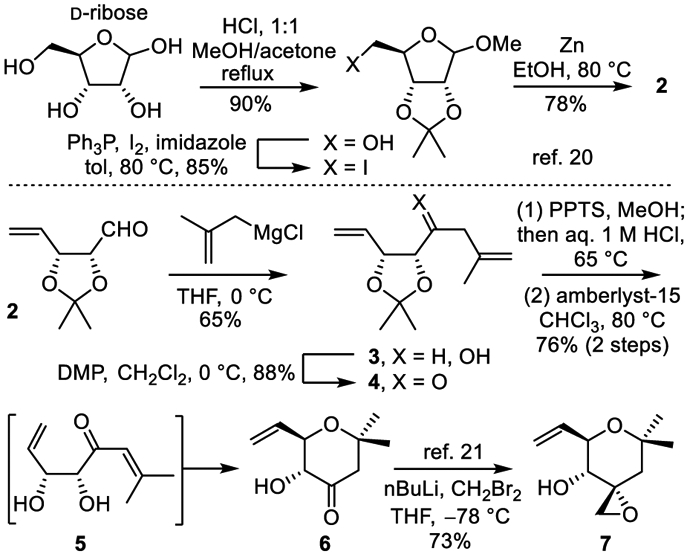
The most challenging subunit is the right-hand tetrahydropyran 7, bearing three of the stereocenters and the essential epoxide. Intermediate 7 was accessed in four steps from known aldehyde 2,20 itself prepared from D-ribose in three steps (Scheme 1). Grignard addition of 2-methylallylmagnesium chloride (2 equiv) to aldehyde 2 (THF, 5 h, 0–25 °C, 5 h, 61–65%) provided 3 as an inconsequential diastereomeric mixture. Dess–Martin periodinane (DMP) oxidation (1.5 equiv DMP, CH2Cl2, 0 °C, 3 h, 78–88%) and subsequent acid-catalyzed acetonide deprotection, alkene isomerization, and 6-endo-trig cyclization of the distal alcohol provided cyclic ketone 6. Initial optimization of this reaction found that treatment of 3 with pyridinium p-toluenesulfonate (PPTS) in MeOH (0.2 equiv PPTS, 60 °C, 4 h) afforded principally the corresponding diol and subsequent addition of aqueous 1 M HCl (MeOH/H2O 9:1, 60 °C, 2–4 h) completed the isomerization of the double bond into conjugation with the ketone to provide 5. Without purification, treatment of crude 5, already containing substantial amounts of 6, with Amberlyst-15 (CHCl3, 12 h, 80 °C, 12 h) completed the distal alkoxy conjugate addition to provide 6 (76% overall). Conversion of 6 to 7 was achieved by diastereoselective epoxide introduction (73%) following Koide’s protocol.21 In addition to the concise nature of the synthesis of 7 (4–5 steps, 32% from 2; 7–8 steps, 20% from D-ribose), the approach avoids the generation of diastereomers and provides full control of the absolute stereochemistry.
Scheme 1.
The central tetrahydropyran, bearing four chiral centers, was accessed by an approach that relied on the chiral pool to set the absolute stereochemistry (Scheme 2). Although inspired by Koide’s original synthesis,6 it is substantially shorter (5 vs 8 steps) and enlists further optimized protocols for overlapping steps. Analogous improvements were independently reported by Koide earlier this year for synthesis of intermediate lactone 12.19 Additional subtle improvements are also highlighted herein for the original conversion6 of 12 to 15.18 Central to the synthesis of 15 was use of the known starting material 9,22 the BocNH-L-Thr derived variant of Garner’s aldehyde, available in three steps from BocNH-L-Thr (1 equiv MeONHMe, 1.2 equiv EDCI, 1.2 equiv HOBt, 2 equiv (iPr2)NEt, CH2Cl2, 25 °C, 22 h; 0.2 equiv PPTS, 10 equiv MeC(OMe)2Me, THF, reflux, 18 h, 85-88% for two steps), including the reported DIBAL-H reduction of the Weinreb amide (2 equiv DIBAL-H, CH2Cl2, −78 °C, 3 h).22 A Z-selective modified Wadsworth–Horner–Emmons reaction of 1023 with aldehyde 9 provided the α,β-unsaturated ester 11 (86% for two steps, 4.6:1 Z:E) where preferential generation of the Z-isomer facilitates but may not be required for an ensuing lactonization. Acid-catalyzed N,O-ketal cleavage effected with 10-camphorsulfonic acid (CSA) and in situ lactonization (0.05 equiv CSA, MeOH, 23 °C, 74%) provided 12 in a single step. The subsequent alkene reduction of 12 proceeded with lower diastereoselectivity (6:1 vs 10:1) than reported and provided 13 contaminated with lactone ethanolysis product in our hands under conditions reported (2 mol % PtO2, H2, EtOH, 23 °C, 2 h, 98%).6 For our purposes, this was reoptimized to provide 13 in high yield (quantitative) and excellent diastereoselectivity (10:1, THF > EtOH, iPrOH, EtOAc) through use of THF as solvent (23 °C, 15 h) without competitive solvent lactone ring opening.
Scheme 2.
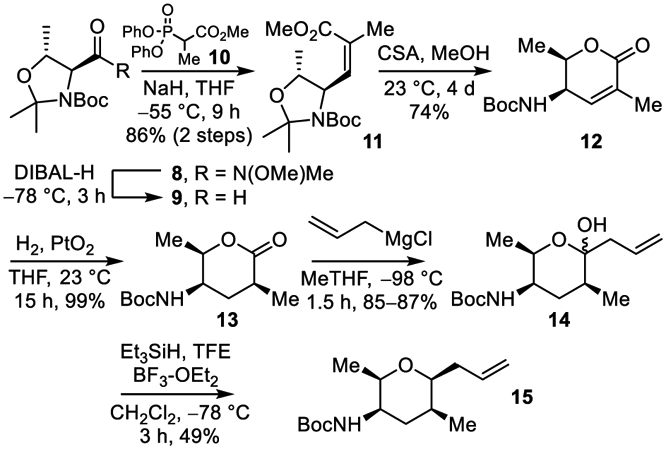
More substantial optimizations were required for implementation of the Koide6 two-step conversion of 13 to 15. Significant amounts of double addition product were observed when the reaction of allylmagnesium chloride (1.9 equiv) with 13 was conducted as detailed, likely accounting for the lower overall conversion of 13 to 15 than reported herein. Although a reduction in the amount of allylmagnesium chloride (1.3–1.6 equiv) attenuated the over addition, increasing amounts of recovered starting 13 offset any improvement in this selectivity. However, by lowering the reaction temperature (−98 vs −78 °C) and adjusting the reaction solvent (2-MeTHF vs THF), 14 was obtained in excellent yield (85-87%) with minimal over addition (6%) or recovered starting lactone (7%). Final diastereoselective reduction of the lactol provided 15 in improved conversions (49%) provided triethylsilane (10 equiv) and trifluoroethanol (TFE, 8 equiv) were also added at −78 °C and stirred for 10 min prior to addition of BF3-OEt2 (4 equiv). The entire sequence and latter reaction were conducted on gram scales, completing the synthesis of the central subunit (24% overall, 8 steps from BocNH-L-Thr).
The left-hand subunit was assembled by several approaches, two of which are illustrated in Scheme 3 that were employed depending on the target structure (e.g., 1 vs O-acyl analogues). For meayamycin (1) itself, we first implemented a straightforward approach starting with the commercially available optically active alcohol 16. Its acetal protection with ethyl vinyl ether (96%), alkyne carboxylation with Boc2O and acetal deprotection without intermediate purification of 18 (80-89%, 2 steps) provided 19. Alcohol acetylation (82%) followed by stereoselective reduction of the alkyne to the cis alkene with Lindlar’s catalyst and subsequent t-butyl ester deprotection (96%, 2 steps) provided 22. The conversion of 16 to 20 could be accomplished without purification of intermediates to provide 20 in yields as high as 81% (>7 g scale) and 22 in 78% overall yield (6 steps). A more versatile subunit 27, permitting late-stage divergent24 functionalization and avoiding a coupling isomerization, was also employed. Compound 27 was prepared with small variations on the approach described by Kitahara5 and itself adopted from earlier unrelated studies.25 Commercial ethyl L-lactate protected as its TBDPS ether25d (98%) was reduced with DIBAL-H (Et2O, −78 °C, 3 h) to the aldehyde 2425d and subjected to Z-selective Wadsworth–Horner–Emmons olefination with 2523 (THF, −78 to −55 °C, 10 h), affording 26 in high yield (95%, two steps) and stereoselectivity (>99:1 Z:E). Methyl ester hydrolysis (LiOH, 90%) completed the synthesis of 27, which was conducted on gram scales and required 4 steps (84% overall) from ethyl L-lactate.
Scheme 3.
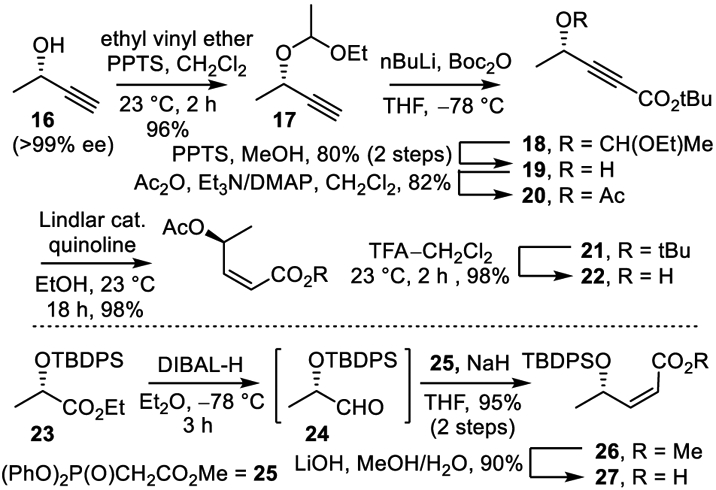
The initial assemblage of the subunits for meayamycin paralleled earlier approaches and is summarized in Scheme 4.6 Deprotection of 15 (10% TFA–CH2Cl2, 23 °C) followed by acylation of the liberated amine with 22 (1.2 equiv HATU, 4 equiv iPr2NEt, MeCN, 23 °C, 3–6 h, 69%) provided 28. Cross metathesis of 28 with methacrolein (20 equiv) provided the α,β-unsaturated aldehyde 29 (0.2 equiv Grubbs II26 cat., CH2Cl2, 23 °C, 36–48 h, 60–80%) or 0.01 equiv Grela cat.,27 CH2Cl2, 23 °C, 12 h, 60%) followed by Wittig olefination afforded 30 (1.5 equiv Ph3P+MeBr−, 1.4 equiv tBuOK, THF, 0– 23 °C, 4 h, 57–65%). Final cross metathesis of 30 with the right-hand subunit 7 (0.2 equiv Grela cat., 0.3 equiv benzoquinone (p-BQ), ClCH2CH2Cl, 45 °C, 12 h) notably without the protection of the secondary alcohol provided meayamycin (1) in yields as high as 54% and on scales up to 130 mg. This completed a synthesis of 1 by an approach that required a longest linear sequence of 12 steps (22 steps overall) from commercial materials and introduced what we suggest is the most straightforward preparation of a right-hand subunit detailed to date.
Scheme 4.
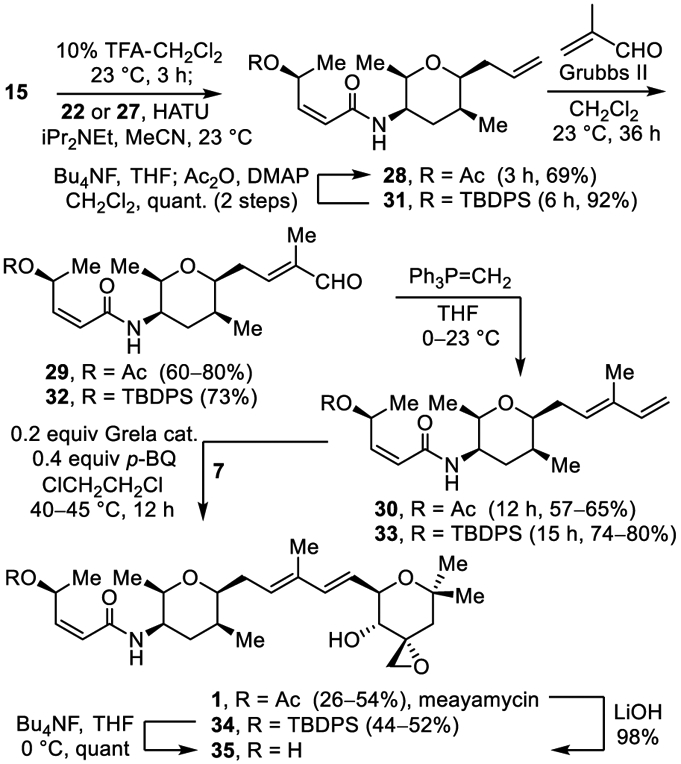
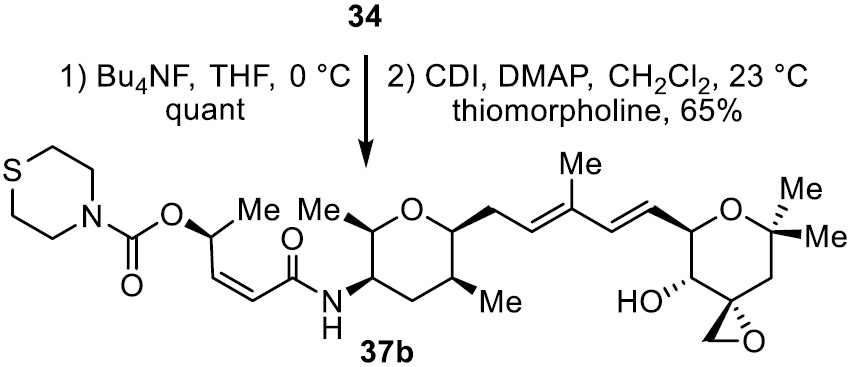
A further improved approach is also summarized in Scheme 4 and enlisted 27 in place of 22. Introduction of the meayamycin O-acetyl group may be accomplished at any stage in this alternative sequence and is exemplified with the conversion of 31 to 28 by silyl ether deprotection and acetylation. It uses the left-hand subunit 27 available in four steps, proceeds in higher overall yields because of the more robust stability of the alcohol substituent, avoids a problematic Z to E isomerization of the left-hand subunit that we observed during the coupling introduction of 22 to provide 28, proceeds with a more reproducible cross metathesis reaction in the conversion of 33 to 34 (0.3 equiv Grela cat., 0.4 equiv p-BQ, ClCH2CH2Cl, 40 °C, 12 h, 44–52%), and permits a late stage diversification of the O-acyl substituent. Without optimization, this latter feature was exemplified by the preparation of carbamate 37b from 34 (Bu4NF, THF, 0 °C, quant.; 3 equiv carbonyldimidazole (CDI), 0.2 equiv 4-dimethylaminopyridine (DMAP), CH2Cl2, 23 °C then thiomorpholine), eq 1.
 |
(1) |
As this latter approach was in development, an initial series of meayamycin O-acyl analogues, including meayamycin B,12 was prepared from the alcohol derived from acetate hydrolysis of 30 (K2CO3, MeOH, 95%) or later by TBDPS deprotection of 33 (Bu4NF, THF, 23 °C, 4 h, quant), its acylation to access alternative and representative more stable carbamates,12 and final cross-metathesis of the resulting 37a–c with 7, Figure 2.
Figure 2.

O-Acyl analogues, synthesis and activity.
The results of their initial evaluation, along with that of 1 and intermediate alcohol 35 are summarized in Figure 2. Whereas the removal of the O-acyl substituent reduced potency (35) as previously disclosed,12 the more stable carbamate replacements for the acetate (37a–c) matched the activity of 1. These observations, and those not reported herein,18 complement those disclosed initially by Koide12 and Webb11 and more recently by Nicolaou10 on the thailanstatins and help define a site and functionality available for productive modification on the stand-alone drugs themselves or for linkage as antibody-drug conjugates.
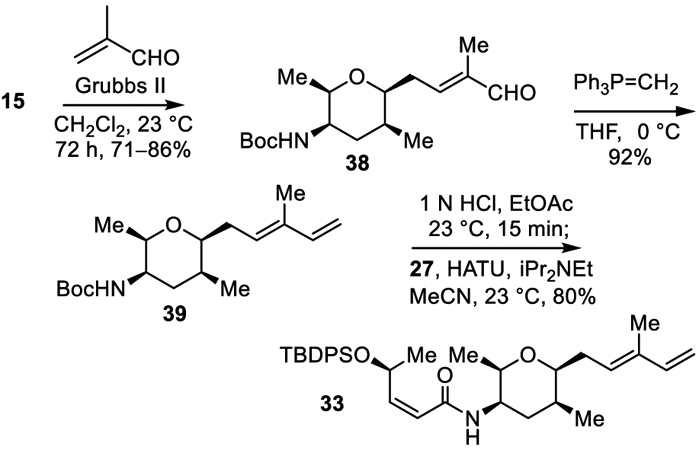
Finally, and as these studies were concluding, we reexamined an alternative sequence for assembling 33 from (Scheme 5). This entailed first elaborating 15 to 39 through cross metathesis with acrolein and subsequent Wittig olefination prior to introduction of the left-hand amide. In the absence of the labile acetate and Z-alkene found in the left-hand subunit, the transformations proved easier to implement and proceeded in higher conversions. In prior studies, acid-catalyzed Boc deprotection (10% TFA, CH2Cl2, 23 °C) conducted on 39 was reported to result in diene isomerization6 and we confirmed these observations. However, we found that Boc deprotection under alternative conditions (1 N HCl, EtOAc, 23 °C, 15 min) did not suffer competitive isomerization and the liberated free amine could be coupled with 27 (1.2 equiv HATU, iPr2NEt, MeCN, 25 °C, 15 h) to provide 33 in excellent yields, providing a viable alternative in future studies.
Scheme 5.
Herein, a short scalable total synthesis of meayamycin is described by an approach that entails a longest linear sequence of 12 steps (22 steps overall) from inexpensive commercially available chiral pool materials (ethyl L-lactate, BocNH-Thr-OH, and D-ribose). It introduces the most straightforward preparation of the ornate right-hand subunit that contains the essential epoxide by an approach amenable to structural modifications. Its use in the divergent synthesis of O-acyl analogues is exemplified that helps define a site for productive modification on the stand-alone drugs or for linkage as antibody-drug conjugates.
Supplementary Material
ACKNOWLEDGMENT
We gratefully acknowledge the financial support of the NIH (CA042056, D.L.B.) and Bristol Myers Squibb.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Full experimental details and 1H and 13C NMR spectra (PDF).
REFERENCES
- (1).(a) Nakajima H; Sato B; Fujita T; Takase S; Terano H; Okuhara M New antitumor substances, FR901463, FR901464 and FR901465. 1. Taxonomy, fermentation, isolation, physicochemical properties and biological activities. J. Antibiot 1996, 49, 1196–1203. [DOI] [PubMed] [Google Scholar]; (b) Nakajima H; Hori Y; Terano H; Okuhara M; Manda T; Matsumoto S; Shimomura K New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. J. Antibiot 1996, 49, 1204–1211. [DOI] [PubMed] [Google Scholar]; (c) Nakajima H; Takase S; Terano H; Tanaka H New antitumor substances, FR901463, FR901464 and FR901465. III. Structures of FR901463, FR901464 and FR901465. J. Antibiot 1997, 50, 96–99. [DOI] [PubMed] [Google Scholar]
- (2).(a) Liu X; Biswas S; Tang GL; Cheng YQ Isolation and characterization of spliceostatin B, a new analogue of FR901464, from Pseudomonas sp. No. 2663. J. Antibiot. 2013, 66, 555–558. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) He H; Ratnayake AS; Janso JE; He M; Yang HY; Loganzo F; Shor B; O’Donnell CJ; Koehn FE Cytotoxic spliceostatins from Burkholderia sp. and their semisynthetic analogues. J. Nat. Prod 2014, 77, 1864–1870. [DOI] [PubMed] [Google Scholar]
- (3).Liu X; Biswas S; Berg MG;Antapli CM; Xie F; Wang Q; Tang M-C; Tang G-L; Zhang L; Dreyfuss G; Cheng Y-Q Genomics-guided discovery of thailanstatins A, B and C as pre-mRNA splicing inhibitors and anti-proliferative agents from Burkholderia thailandensis MSMB43. J. Nat. Prod 2013, 76, 685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Thompson CF; Jamison TF; Jacobsen EN Total synthesis of FR901464. Convergent assembly of chiral components prepared by asymmetric catalysis. J. Am. Chem. Soc 2000, 122, 10482–10483. [Google Scholar]; (b) Thompson CF; Jamison TF; Jacobsen EN FR901464: Total synthesis, proof of structure, and evaluation of synthetic analogues. J. Am. Chem. Soc 2001, 123, 9974–9983. [DOI] [PubMed] [Google Scholar]
- (5).(a) Horigome M; Motoyoshi H; Watanabe H; Kitahara T A synthesis of FR901464. Tetrahedron Lett. 2001, 42, 8207–8210. [Google Scholar]; (b) Motoyoshi H; Horigome M; Watanabe H; Kitahara T Total synthesis of FR901464: Second generation. Tetrahedron 2006, 62, 1378–1389. [Google Scholar]
- (6).(a) Albert BJ; Sivaramakrishnan A; Naka T; Koide K Total synthesis of FR901464, an antitumor agent that regulates the transcription of oncogenes and tumor suppressor genes. J. Am. Chem. Soc 2006, 128, 2792–2793. [DOI] [PubMed] [Google Scholar]; (b) Albert BJ; Sivaramakrishnan A; Naka T; Czaicki NL; Koide K Total syntheses, fragmentation studies, and antitumor/antiproliferative activities of FR901464 and its low picomolar analogue. J. Am. Chem. Soc 2007, 129, 2648–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Ghosh AK; Chen Z-H Enantioselective syntheses of FR901464 and spliceostatin A: Potent inhibitors of spliceosome. Org. Lett 2013, 15, 5088–5091. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghosh AK; Chen Z-H; Effenberger KA; Jurica MS Enantioselective total syntheses of FR901464 and spliceostatin A and evaluation of splicing activity of key derivatives. J. Org. Chem 2014, 79, 5697–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Review: Koide K; Albert BJ Total syntheses of FR901464. J. Synth. Org. Chem., Jpn 2007, 65, 119–126. [Google Scholar]
- (9).(a) Ghosh AK; Veitschegger AM; Nie S; Relitti N; MacRae AJ; Jurica MS Enantioselective synthesis of thailanstatin A methyl ester and evaluation of in vitro splicing activity. J. Org. Chem 2018, 83, 5187–5198. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghosh AK; Reddy GC; MacRae AJ; Jurica MS Enantioselective synthesis of spliceostatin G and evaluation of bioactivity of spliceostatin G and its methyl ester. Org. Lett. 2018, 20, 96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]; See also: (c) Ghosh AK; Born JR; Veitschegger AM; Jurica MS Copper-catalyzed Stille cross-coupling and application in the synthesis of the splicesome core structure. J. Org. Chem 2020, 85, 8111–8120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Nicolaou KC; Rhoades D; Kuma SM Total syntheses of thailanstatins A–C, spliceostatin D, and analogues thereof. Stereodivergent synthesis of tetrasubstituted dihydro- and tetrahydropyrans and design, synthesis, biological evaluation, and discovery of potent antitumor agents. J. Am. Chem. Soc 2018, 140, 8303–8320. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC; Rhoades D; Lamani M; Pattanayak Manas R.; Kumar SM Total synthesis of thailanstatin A. J. Am. Chem. Soc 2016, 138, 7532–7535. [DOI] [PubMed] [Google Scholar]
- (11).(a) Lagisetti C; Pourpak A; Goronga T; Jiang Q; Cui X; Hyle J; Lahti JM; Morris SW; Webb TR Synthetic mRNA splicing modulator compounds with in vivo antitumor activity. J. Med. Chem 2009, 52, 6979–6990. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lagisetti C; Palacios G; Goronga T; Freeman B; Caufield W; Webb TR Optimization of antitumor modulators of pre-mRNA splicing. J. Med. Chem 2013, 56, 10033–10044. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Makowski K; Vigevani L; Albericio F; Valcarcel J; Alvarez M Sudemycin K: A synthetic antitumor splicing inhibitor variant with improved activity and versatile chemistry. ACS Chem. Biol 2017, 12, 163–173. [DOI] [PubMed] [Google Scholar]
- (12).Osman S; Albert BJ; Wang Y; Li M; Czaicki NL; Koide K Structural requirements for the antiproliferative activity of pre-mRNA splicing inhibitor FR901464. Chem. – Eur. J 2011, 17, 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ghosh AK; Reddy GC; Kovela S; Relitti N; Urabe VK; Prichard BE; Jurica MS Enantioselective synthesis of a cyclopropane derivative of spliceostatin A and evaluation of bioactivity. Org. Lett 2018, 20, 7293–7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Kaida D; Motoyoshi H; Tashiro E; Nojima T; Hagiwara M; Ishigami K; Watanabe H; Kitahara T; Yoshida T; Nakajima H; Tani T; Horinouchi S; Yoshida M Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat. Chem. Biol 2007, 3, 576–578. [DOI] [PubMed] [Google Scholar]; (b) Lo C-W; Kaida D; Nishimura S; Matsuyama A; Yashiroda Y; Taoka H; Ishigami K; Watanabe H; Nakajima H; Tani T; Horinouchi S; Yoshida M Inhibition of splicing and nuclear retention of pre-mRNA by spliceostatin A in fission yeast. Biochem. Biophys. Res. Commun 2007, 364, 573–577. [DOI] [PubMed] [Google Scholar]
- (15).Albert BJ; McPherson PA; O'Brien K; Czaicki NL; DeStefino V; Osman S; Li M; Billy W Day BW; Grabowski PJ; Moore MM; Vogt A; Koide K Meayamycin inhibits pre-mRNA splicing and exhibits picomolar activity against multidrug resistant cells. Mol. Cancer Ther 2009, 8, 2308–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).(a) Bonnal S; Vigevani L; Valcarcel J The spliceosome as a target of novel antitumour drugs. Nat. Rev. Drug Discov 2012, 11, 847–859. [DOI] [PubMed] [Google Scholar]; (b) Desterro J; Bak-Gordon P; Carmo-Fonseca M Targeting mRNA processing as an anticancer strategy. Nat. Rev. Drug Discov 2020, 19, 112–129. [DOI] [PubMed] [Google Scholar]
- (17).(a) Boger DL The difference a single atom can make: Synthesis and design at the chemistry–biology interface. J. Org. Chem 2017, 82, 11961–11980. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Boger DL; Johnson DS CC-1065 and the duocarmycins: Unraveling the keys to a new class of naturally derived DNA alkylating agents. Proc. Natl. Acad. Sci. U.S.A 1995, 92, 3642–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Boger DL; Johnson DS CC-1065 and the duocarmycins: Understanding their biological function through mechanistic studies. Angew. Chem. Int. Ed. Engl 1996, 35, 1438–1474. [Google Scholar]; (d) Boger DL; Boyce CW; Garbaccio RM; Goldberg JA CC-1065 and the duocarmycins: Synthetic studies. Chem. Rev 1997, 97, 787–828. [DOI] [PubMed] [Google Scholar]; (e) Boger DL; Cai H Bleomycin: Synthetic and mechanistic studies. Angew. Chem. Int. Ed 1999, 38, 448–476. [DOI] [PubMed] [Google Scholar]; (f) MacMillan KS; Boger DL Fundamental relationships between structure, reactivity, and biological activity for the duocarmycins and CC-1065. J. Med. Chem 2009, 52, 5771–5780. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Sears JE; Boger DL Total synthesis of vinblastine, related natural products, and key analogues and development of inspired methodology suitable for the systematic study of their structure–function properties. Acc. Chem. Res 2015, 48, 653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) James RC; Pierce JG; Okano A; Xie J; Boger DL Redesign of glycopeptide antibiotics: Back to the future. ACS Chem. Biol 2012, 7, 797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Okano A; Isley NA; Boger DL Total synthesis of vancomycin related glycopeptide antibiotics and key analogues. Chem. Rev 2017, 117, 11952–11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Boger DL; Chowdari NS; Gangwar S Meayamycin and its analogues, methods for their preparation, and methods of use. US Patent App. 63/007,564, Filed 4/September/20.
- (19).Bressin RK; Osman S; Pohorilets I; Basu U; Koide K Total synthesis of meayamycin B. J. Org. Chem 2020, 85, 4637–4647. [DOI] [PubMed] [Google Scholar]
- (20).(a) Smith AB III; Han Q,; Breslin PAS; Beauchamp GK Synthesis and assignment of absolute configuration of (−)-oleocanthol: A potent, naturally occurring non-steroidal antiinflammatory and anti-oxidant agent derived from extra virgin olive oils. Org. Lett 2005, 7, 5075–5078. [DOI] [PubMed] [Google Scholar]; (b) Prabhakar P; Rajaram S; Reddy DK; Shekar V; Venkateswarlu Y Total synthesis of the phytotoxic stagonolides A and B. Tetrahedron: Asymmetry 2010, 21, 216–221. [Google Scholar]
- (21).Koide K; Osman S Synthesis of FR901464 and analogs with antitumor activity. US Patent App. 2015, US 20150307512A1.
- (22).(a) Wilson ZE; Fenner S; Ley SV Total syntheses of linear polythiazole/oxazole plantazolicin A and its biosynthetic precursor plantazolicin B. Angew. Chem. Int. Ed 2015, 54, 1284–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Garner P; Park JM The synthesis and configurational stability of differentially protected β-hydroxy-α-amino aldehydes. J. Org. Chem 1987, 52, 2361–2364. [Google Scholar]; (c) Sawamura M Nakayama Y; Kato T; Ito Y Gold(I)-catalyzed asymmetric aldol reaction of N-methoxy-N-methyl-α-isocyanoacetamide (α-isocyano Weinreb amide). An efficient synthesis of optically active β-hydroxy α-amino aldehydes and ketones. J. Org. Chem 1995, 60, 1727–1732. [Google Scholar]
- (23).Akwaboah DC; Wu D; Forsyth C Stereoselective synthesis of the C1–C9 and C11–C25 fragments of amphidinolides C, C2, C3, and F. Org. Lett 2017, 19, 1180–1183. [DOI] [PubMed] [Google Scholar]
- (24).Boger DL; Brotherton CE Total synthesis of azafluoranthene alkaloids: Rufescine and imeluteine. J. Org. Chem 1984, 49, 4050–4055. [Google Scholar]
- (25).(a) Massad SK; Hawkins LD; Baker DC A series of (2S)-2-O-protected-2-hydroxypropanals (L-lactaldehydes) suitable for use as optically active intermediates. J. Org. Chem 1983, 48, 5180–5182. [Google Scholar]; (b) Annunziata R; Cinquini M; Cozzi F; Dondio G; Raimondi L Intramolecular nitrile oxide cycloaddition on chiral olefins: A stereocontrolled approach to β-ketol precursors. Tetrahedron 1987, 43, 2369–2380. [Google Scholar]; (c) Crimmins MT; Jacobs DL Asymmetric total synthesis of pyranicin. Org. Lett 2009, 11, 2695–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Srinivas B; Sridhar R; Rao KR Stereoselective total synthesis of (+)-varitriol. Tetrahedron 2010, 66, 8527–8535. [Google Scholar]
- (26).Scholl M; Ding S; Lee CW; Grubbs RH Synthesis and activity of a new generation of ruthenium-based olefin metathesis catalysts coordinated with 1,3-dimesityl-4,5-dihydroimidazol-2-ylidene ligands. Org. Lett 1999, 1, 953–956. [DOI] [PubMed] [Google Scholar]
- (27).(a) Grela K; Harutyunyan S; Michrowska A A highly efficient ruthenium catalyst for metathesis reactions. Angew. Chem., Int. Ed 2002, 41, 4038–4040. [DOI] [PubMed] [Google Scholar]; (b) Michrowska A; Bujok R; Harutyunyan S; Sashuk V; Dolgonos G; Grela K Nitrosubstituted Hoveyda–Grubbs ruthenium carbenes: enhancement of catalyst activity through electronic activation. J. Am. Chem. Soc 2004, 126, 9318–9325. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.