

Abstract
Aging is the major predictor for developing multiple neurodegenerative diseases, including Alzheimer’s disease (AD) other dementias, and Parkinson’s disease (PD). Senescent cells, which can drive aging phenotypes, accumulate at etiological sites of many age-related chronic diseases. These cells are resistant to apoptosis and can cause local and systemic dysfunction. Decreasing senescent cell abundance using senolytic drugs, agents that selectively target these cells, alleviates neurodegenerative diseases in preclinical models. In this review, we consider roles of senescent cells in neurodegenerative diseases and potential implications of senolytic agents as an innovative treatment.
1. Introduction
1.1. Aging and neurodegenerative diseases
Great strides have been made in extending human life expectancy. However, age-related neurodegenerative diseases pose an increasing risk in the ever-growing, aging population. In 2010, approximately 4.7 million Americans were living with Alzheimer’s disease (AD), the most common neurodegenerative condition. By 2050, this number is projected to reach 16 million (Hebert et al., 2013). Of note, this epidemiological trajectory is expected to impose an immense burden on healthcare utilization and expenditures (Yang et al., 2013).
The foremost predictor of many chronic diseases and disabilities, including most neurodegenerative diseases, diabetes, strokes, frailty, atherosclerosis, blindness, cancers, and musculoskeletal diseases, is aging (Kirkland, 2013; Miller, 2002). Age-associated cognitive impairment and neurodegenerative diseases, such as AD and PD, are characterized by progressive degeneration of the structure and function of the nervous system. With aging, the central nervous system (CNS) undergoes significant changes, including brain and spinal cord atrophy, decreases in gray matter, and accumulation of Amyloid beta (Aβ) plaques, neurofibrillary tangles, and senescent cells (Wyss-Coray, 2016).
There is much work to be done on deciphering the underlying causes of these changes. Because both natural aging and neurodegeneration share some structural and functionally deleterious features that are accentuated as neurodegenerative diseases progress, it is important to explore mechanisms and precursors of these changes to pinpoint predisposing factors that drive neurodegeneration (Wyss-Coray, 2016). Currently there is no cure available for most neurodegenerative diseases. Lack of detailed understanding about upstream neurodegenerative disease mechanisms hinders the development of effective therapeutic strategies. Recent evidence suggests that AD pathophysiology begins decades prior to manifestation of symptoms (Cummings et al., 2016). This limited understanding of mediators that drive disease onset and progression during the long prodromal, asymptomatic stages is a barrier to developing effective therapeutic strategies. Therefore, identification of early molecular mechanisms of neurodegeneration and novel targets for intervention to delay, prevent, or alleviate these diseases is a priority.
Mild cognitive impairment (MCI), AD, brain atrophy, and brain hypoperfusion are all more likely to occur with aging (Albert et al., 2011). Given that ageing is a major risk factor for many neurodegenerative and other diseases, there is growing interest in targeting fundamental aging mechanisms in order to treat multiple, age-related comorbidities (Kirkland, 2013, 2016; Kirkland and Peterson, 2009). One of the central mechanisms being explored includes cellular senescence, a cell fate in which cells lose replicative capability upon damage or stress. Accumulation of senescent cells increasingly is recognized as a critical step in age-dependent tissue dysfunction and is implicated in the pathogenesis of many degenerative diseases, including AD. There are many cell types in the CNS, including neurons, oligodendrocytes, microglia, and astrocytes. Each of these cell types can acquire a senescent phenotype. Senescent cells in the CNS are associated to neuroinflammation, impaired neurogenesis, and degenerative changes in tissue structure and integrity (Baker and Petersen, 2018).
1.2. Fundamental aging mechanisms and the geroscience hypothesis
Lopez-Otin et al. (2013) defines the nine hallmarks of aging as the following: genomic instability; telomere attrition; epigenetic alterations; loss of proteostasis; dysregulated nutrient-sensing; mitochondrial dysfunction; stem cell exhaustion; altered intercellular communication; and cellular senescence. These processes are highly interlinked and associated with deterioration of biological functions during natural aging. The Unitary Theory of Fundamental Aging Processes holds that disrupting development of one, such as clearing senescent cells, could alleviate multiple hallmarks of aging and therefore age-related dysfunction. Interestingly, these processes are operative in the pathogenesis of many age-related diseases, including diabetes, cardiovascular disease, frailty, and dementias. The Geroscience Hypothesis holds that targeting these fundamental aging processes, including cellular senescence, as root cause contributors to diseases and disorders might not only delay aging, but simultaneously prevent, delay or alleviate the onset of multiple chronic diseases (Kennedy et al., 2014; Lopez-Otin et al., 2013).
1.3. Aging and risk for multiple disabilities and diseases, including neurodegenerative diseases
Cellular senescence plays a direct role in chronic and age-related diseases and conditions, such as diabetes, atherosclerosis, neurovascular dysfunction, frailty, and dementias. Accumulation of senescent cells with aging contributes to multiple, age-related comorbidities that are frequently accompanied by neurodegenerative diseases, especially AD.
Frailty is a geriatric syndrome characterized by decreased strength and reduced capacity to respond to stress (Michaud et al., 2013). Its prevalence increases with chronological aging and dementia (Kojima et al., 2017; Ness et al., 2018). Frailty is associated with considerable morbidity, including decreased mobility, increased burden of age-related chronic diseases, loss of independence, nursing home admission, and mortality. Epidemiological evidence also suggests that physical frailty may be associated with decline in capacity for activities of daily living (ADLs) later in life and increased incidence of AD, MCI, vascular dementia (VaD), and other dementias in addition to AD, as well as AD pathology in older subjects with and without a diagnosis of dementia (Panza et al., 2015).
Cellular senescence is associated with increased systemic chronic, low grade, sterile inflammation, as is chronological aging (Xu et al., 2015b). Senescent cell abundance is higher in adipose tissue of frail than non-frail women in their 80s (Justice et al., 2017). Transplanting small numbers of senescent cells into middle-aged mice causes frailty while transplanting non-senescent cells does not (Xu et al., 2016). Thus, frailty is linked to both senescent cell burden and neurodegenerative diseases.
A key step in working toward alleviating age-related diseases and disorders, such as cognitive dysfunction and neurodegenerative diseases, may be to focus interventions on the biological mechanisms of aging, particularly cellular senescence (Kritchevsky and Justice, 2020).
2. Cellular senescence and aging
Cellular senescence is a cell fate that involves essentially irreversible replicative arrest, apoptosis resistance, and frequently increased protein synthesis, metabolic shifts with increased glycolysis, decreased fatty acid oxidation, increased generation of reactive oxygen species (ROS), and, sometimes, acquisition of a senescence-associated secretory phenotype (SASP). Cellular senescence was initially discovered in vitro in human fibroblasts in 1961 (Hayflick and Moorehead, 1961). These cells enter an irreversible, non-dividing state, “replicative senescence,” yet remain viable. Cellular senescence has come to be associated primarily with aging and age-related diseases. However, cellular senescence is also an important part of several biological processes including embryogenesis (Rajagopalan and Long, 2012), pregnancy (Velarde and Menon, 2016), wound healing (Jun and Lau, 2010), and tissue remodeling (Krizhanovsky et al., 2008).
Cellular senescence can be induced by internal and external stimuli. It is a complex stress response, whereby accumulation of DNA damage and/or other cellular stressors (e.g., oncogenic insults, reactive metabolites, proteotoxic stress) (Kirkland and Tchkonia, 2017; Kirkland et al., 2017; LeBrasseur et al., 2015; Swanson et al., 2013; Tchkonia et al., 2013; Zhu et al., 2014) cause proliferating (Campisi, 2005; Campisi and d’Adda di Fagagna, 2007; Hayflick and Moorehead, 1961), as well as terminally differentiated, non-dividing cells (Anderson et al., 2019; Farr et al., 2016; Jurk et al., 2012, 2014) to undergo profound chromatin, transcriptomic, and secretome changes, replicative arrest, increased expression of cell cycle inhibitors such as p16INK4A and/or p21CIP1, metabolic dysfunction, cellular remodeling, and resistance to apoptosis (Tchkonia et al., 2013; Wang, 1995).
Accumulation of senescent cells can cause significant tissue dysfunction. The detrimental effects of senescent cells are in part due to SASP factors. The SASP can include pro-inflammatory cytokines, such as Tumor Necrosis Factor-α (TNF-α), viral FK506-Binding Protein (vFKBP), and interleukin- (IL-) 6, chemokines, and extracellular matrix proteases, as well as bioactive lipids (bradykines, ceramides, prostenoids), micro-RNA’s (miRNAs), other non-coding nucleotides, and extracellular vesicles that induce spread of senescence to non-senescent cells, tissue destruction, protein aggregation, and apoptotic and necrotic cell death in local and distant tissues (Acosta et al., 2013; Coppé et al., 2008, 2010; Kuilman and Peeper, 2009; Nelson et al., 2012; Tchkonia et al., 2007; Xu et al., 2015a, 2015b, 2016, 2018). Even a relatively low abundance of senescent cells is sufficient to cause tissue dysfunction. We found that transplanting 106 senescent syngeneic adipocyte progenitors or autologous ear fibroblasts into the abdomen of middle-aged mice so that only 1/10,000 cells in the transplanted mice was senescent, was sufficient to cause frailty, early onset of age-related phenotypes, and premature death (Xu et al., 2018). Transplanting the same number of non-senescent cells did not do so. Senescent cell burden is low in young individuals, but increases with aging in all tissues, especially in the brain, adipose tissue, skeletal muscle, kidney, skin, and ovaries (Herbig et al., 2006; Tchkonia et al., 2010).
2.1. Senescent astrocytes
Increasing evidence suggests an important role for astrocytes in the initiation and progression of neurodegenerative disease and cognitive decline with aging. Astrocytes exert many essential complex functions in the healthy CNS and respond to multiple types of CNS insults through a process referred to as reactive astrogliosis. This appears to be a pathological hallmark of CNS structural lesions in neurodegenerative diseases. In astrogliosis, astrocytes become hypertrophic, with enlargement of both the cell body and processes. There is also an increase in secretion of pro-inflammatory cytokines, chemokines, and growth factors, characteristics in common with other types of senescent cells. Secretion of these factors has potentially detrimental effects, including exacerbation of neuroinflammation. “Astrosenescence” (see Fig. 1) has an impact not only on astrocytes, but also on surrounding neurons (Kawano et al., 2012) and neural progenitor cell (NPC) proliferation (Miranda et al., 2012). It has been reported that astrocytes isolated from post-mortem brains of subjects with AD display increased levels of the cellular senescence-associated factors p16INK4a and p21CIP1 (Blasko et al., 2004). In the frontal cortex, astrocytes express high levels of p16INK4a and increase with aging. Furthermore, there is also a larger age-associated increase of senescent astrocytes in AD patients compared to age-matched controls. There is a parallel increase of the SASP component matrix metalloproteinase-1 (MMP-1) in the cortex. There is little astrocyte senescence in the cerebellum, which exhibits little AD pathology. Thus, astrocyte senescence may be a contributor to AD via effects of senescent astrocytes on their microenvironment. A recent study in mice with tau-dependent neurodegeneration suggested that clearing senescent astrocytes with senolytic drugs could alleviate age-associated dysfunction (Bussian et al., 2018).
Fig. 1.
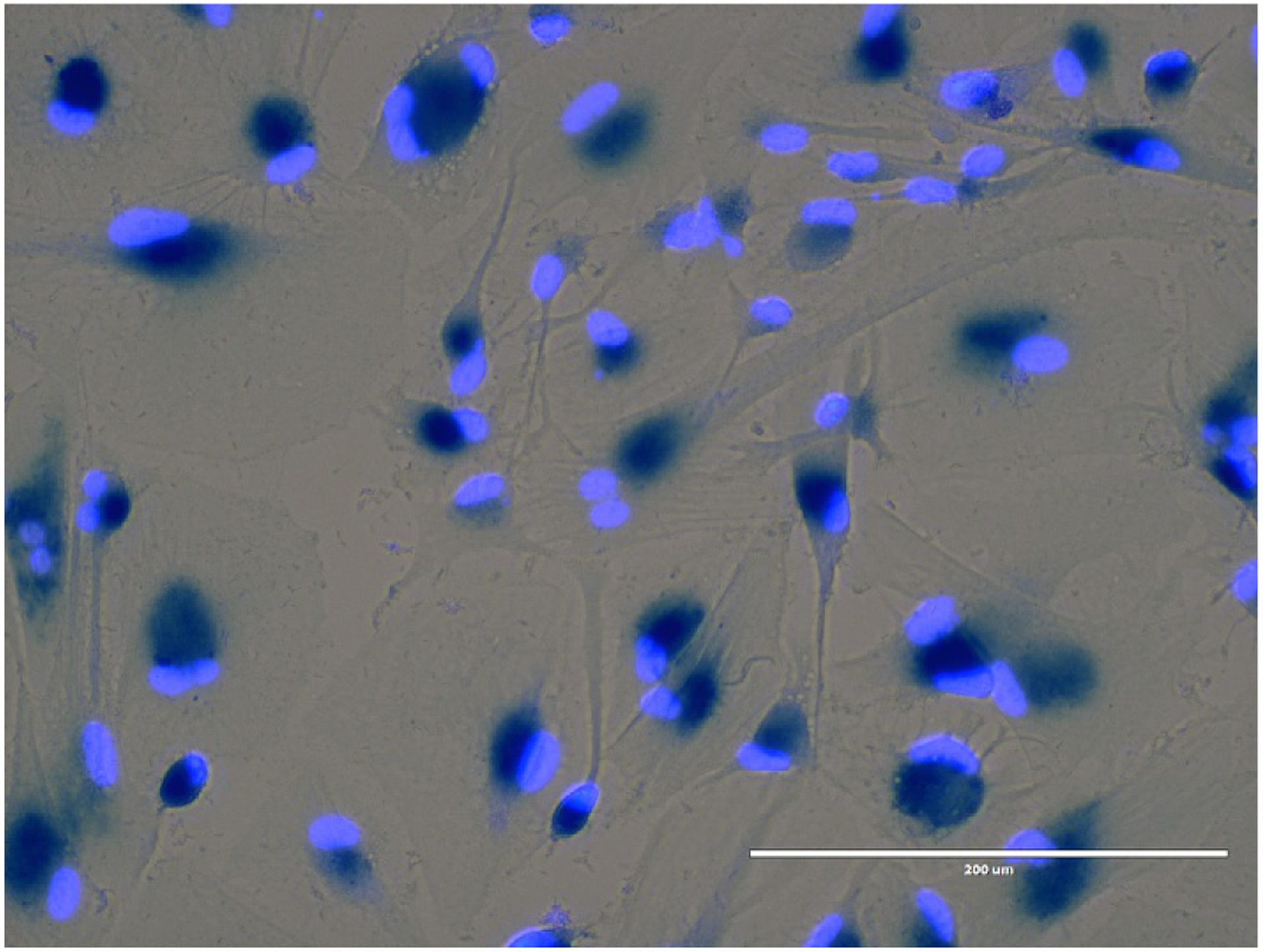
Senescent human astrocytes. Primary human astrocytes were irradiated for 10Gy and studied 20days after irradiation. All of the astrocytes in this field exhibit increased SA-β-gal (blue).
2.2. Senescent oligodendrocyte progenitor cells
As with other types of stem and progenitor cells, cell cycle regulation is crucial for Oligodendrocyte Progenitor Cells (OPCs) to fulfill their developmental role, as well as to sustain their population in the adult and aging CNS in order to maintain homeostasis and respond effectively to degenerative insults. Cognitive decline during aging, especially in the context of pathologies, such as late-onset AD, might be related to failure to maintain myelin homeostasis (Bartzokis, 2011; Lu et al., 2013, 2014). To what extent this involves defects in the mitotic capacity of OPCs that could lead to the occurrence of OPC-poor areas in the brain remains controversial. A fraction of OPCs enters senescence in the aging brain (Kujuro et al., 2010). Also, decreased proliferation of OPCs in response to demyelination has been observed in the aged mouse brain (Ruckh et al., 2012). According to a recent study, Aβ induces senescence in OPC cells (Zhang et al., 2019). Clearance of senescent OPCs alleviated Aβ plaque-associated inflammation and cognitive deficits in AD mice.
2.3. Senescent microglia
As the innate immune effector of the brain, microglia are involved in several functions. These include: regulation of inflammation, synaptic connectivity, programmed cell death, wiring and circuitry formation, phagocytosis of cell debris, synaptic pruning, and sculpting of postnatal neural circuits (Mecca et al., 2018). Age-related dystrophic microglial cells have been reported throughout the human brain (Streit, 2006). Microglial cells are efficient sensors of changes in the CNS microenvironment, and their neuroprotective role has been hypothesized to be impaired during aging (Soto et al., 2015). Recent work in the aged human brain has provided morphological evidence of structural deterioration of microglia, and work in rodents suggests that microglial cells are subject to replicative senescence, with loss of mitotic ability after repeated rounds of replication. In a tau-based model of neurodegeneration, destruction of senescent (non-dividing) microglial cells greatly reduced observed pathology (Bussian et al., 2018). This implies that destruction of senescent microglia may be of benefit in neurodegenerative diseases. However, conclusive demonstration of a role for senescent microglia in neurodegenerative disease remains to be proven. Such evidence would require the ability to specifically identify and target these cells.
2.4. Senescent-like post-mitotic neuronal cells
Neurons are post-mitotic cells that supposedly cannot enter a senescent state. However, it has been shown that neurons in old mice develop a senescent-like phenotype. In vivo, pronounced induction of senescence-associated β-galactosidase activity (SA-β-gal) in the hippocampus of 24-month-old mice was observed (Piechota et al., 2016). This is consistent with other findings (Fielder et al., 2017) that neurons surviving with a persistently activated DNA damage response have multiple features consistent with cellular senescence, including metabolic dysregulation, mitochondrial dysfunction, and the hyper-production of pro-oxidant, pro-inflammatory, and matrix-remodeling factors. These cells, termed senescent-like neurons, can negatively influence the extracellular environment and may promote induction of the same phenotype in surrounding cells, as well as drive aging and age-related neurodegenerative diseases.
3. Implications of senescent cell accumulation for neurological diseases (in preclinical animal models and patients)
3.1. Alzheimer’s disease
Advanced chronological age is the greatest risk factor for developing AD, which suggests that aging and AD may share cellular and molecular pathways. Recent evidence suggests that AD pathophysiology begins decades prior to symptoms (Cummings et al., 2016). Effects of a toxic insult, such as cellular senescence, might lay dormant for years or decades. Consistent with this, we found a lag period between transplanting senescent cells into mice and development of phenotypes (Xu et al., 2018).
Using gene expression in post-mortem human brain tissue from individuals with AD, our collaborators found that neurons with neurofibrillary tangles (NFTs) have profiles consistent with cellular senescence (Musi et al., 2018). These include upregulation of pro-survival and inflammatory pathways and down-regulation of cell death pathways. Predicted upstream regulators of these gene expression profiles were also consistent with senescence in human AD and transgenic AD mouse models. In brains from humans with AD, senescent oligodendrocyte precursor cells were found in the inferior parietal lobes near Aβ plaques, while in subjects without dementia, these senescent cells were not observed (Zhang et al., 2019). In transgenic mice with neurodegeneration due to expression of Aβ and presenilin-1, senescent oligodendrocyte lineage cells were found near plaques.
Preclinical studies conclusively demonstrate that senescent cells are associated with tau accumulation and senescent cell clearance reduces neurodegeneration, brain atrophy, white matter hyperintensities (WMH), ventricular enlargement, and cognitive impairment (Bussian et al., 2018; Musi et al., 2018; Zhang et al., 2019). Tau accumulation is the most common pathology and closest clinical correlate with dementia and neurodegeneration in AD (Arriagada et al., 1992; Ossenkoppele et al., 2016). Thus, tau-related cellular senescence may represent a novel target for intervention.
3.2. Mild cognitive impairment
Mild cognitive impairment can be a transitional stage between normal aging and AD, representing the first stage in which clinical symptoms become evident (DeCarli, 2003). Cellular senescence as a central aging mechanism is potentially involved in the progression of brain aging to MCI and AD. Although the preclinical animal models to study the mechanism of this progression are lacking and there is limited access to human biopsies to examine directly the etiology of MCI, mounting evidence suggests that cellular senescence contributes to MCI. For example, Ott et al. (2018) assessed cerebrospinal fluid (CSF) and blood collected from individuals with MCI, and found that certain pro-inflammatory cytokines, including IL2, TNF-α, and IL-17a, correlate with severity of impaired cognition. These cytokines are also frequently components of the SASP, suggesting that senescent cell burden is associated with impaired cognition in individuals with MCI. Furthermore, another group (Sheinerman et al., 2012) developed a non-invasive assay for detection of MCI based on analysis of levels of brain-enriched miRNA, including neurite- and synapse-enriched miRNA, in plasma and identification of miRNA biomarker pairs capable of successfully differentiating MCI patients from aged-matched controls. Many of these miRNAs are also secreted by senescent cells (Suh, 2018). Cognitive frailty, defined as the coexistence of MCI symptoms and the physical frailty phenotype in older persons, is increasingly considered the main geriatric condition predisposing to dementia. It appears that cellular senescence plays a causal role in impairment of physical activity, at least in mice, which also suggests that high senescent cell burden potentially contributes to MCI.
3.3. Vascular dementia
Vascular dementia is the second most common form of dementia after AD. Atherosclerosis is one of the most commonly observed cerebrovascular diseases that can result in cerebrovascular lesions. Aging is a primary risk factor for cardiovascular disease. Arteries undergo remodeling with the aging process regardless of presence of cardiovascular disease (Katsuumi et al., 2018). Increasing evidence indicates that several pathogenic mechanisms promoting atherosclerosis are also involved in neurodegenerative diseases, and that insight into the factors determining the susceptibility to, and long-term progression of, atherosclerosis may be of relevance for the etiology of neurodegenerative diseases such as dementia. One mechanism that underlies age-related atherosclerosis may be cellular senescence (Roos et al., 2016). The accumulation of senescent cells near early atherosclerotic lesions may drive the progression of atherosclerosis. Consequently, senescent cell removal approaches might block effects of aging on cardiovascular performance, attenuate atherosclerosis, and hold promising potential as a therapy. Preclinical studies have demonstrated that the alleviation of senescent cell burden in naturally aged mice and mice with established atherosclerosis reduced intimal plaque calcification (Roos et al., 2016).
Neurovascular dysfunction is also considered to be a risk factor for vAD. Within the past decade, there has been increasing interest in the potential vascular etiology of neurodegeneration and dementia. The neurovascular unit is a functional unit with multiple components in the CNS. Pathological dysfunction of the neurovascular unit has emerged as an important element in neurodegenerative diseases, in which neuronal cell death, glial cell activation, blood-brain barrier (BBB) disruption, and penetration of peripheral immune cells may ensue with disease progression (Baker and Petersen, 2018; Cai et al., 2017; Lo and Rosenberg, 2009). Also, in epidemiological, clinical, and experimental studies, there is evidence of a complex functional impairment of cerebral microvessels and astrocytes, which likely contributes to neurovascular dysfunction and cognitive decline with aging and in age-related neurodegenerative diseases. Endothelial cells, pericytes, and astrocytes in the aging brain can become senescent and express characteristic SASP proteins, including various species of potentially neurotoxic factors, which may disrupt functional neurovascular coupling and induce BBB damage.
Aside from the hallmark pathological lesions, there is evidence to suggest a role for immunological and inflammatory mechanisms in the pathophysiology of vAD. Neuroinflammation encompasses local endothelial activation, leading to the extravasation of fluid via a dysfunctional BBB, resulting in tissue damage in the surrounding parenchyma and eventually leading to the activation of perivascular macrophages, microglia, and other glial subtypes. This neuroinflammation is most likely mediated by senescent cells, including senescent endothelial cells or pericytes and senescent astrocytes, which constantly produce mediators such as MCP-1 and IL-6 (Mulugeta et al., 2008).
3.4. Stroke
The incidence of strokes increases with age, and strokes are the second leading cause of death worldwide (Feigin et al., 2014). Stroke and other cerebrovascular diseases are linked with older age at onset of dementia (Schneider et al., 2007; Sonnen et al., 2007). Lo Coco et al. (2016) theorize that the process of neurodegenerative changes in the brain begins decades before the onset of clinical dementia and cerebrovascular disease, causing neuroanatomical lesions in strategic areas of the brain. The onset of a stroke, such as the rupture of an atherosclerotic plaque, the embolization of a blood clot, or damage to a vessel and subsequent bleeding, likely initiates a series of overlapping pathophysiological changes that lead to neuronal death within and beyond the initial area of damage. Also, senescence of vascular endothelial and smooth muscle cells impairs BBB integrity and contributes to pro-inflammatory milieu in diseases, such as stroke and AD (Yamazaki et al., 2016). Neurodegeneration is often considered a secondary outcome of vascular dysfunction caused by stroke. In the context of senescence hallmarks, human astrocytes, endothelial cells, pericytes, and microglia are highly sensitive to oxidative stress and trigger a senescence program when faced with multiple types of stressors induced by a series of pathological changes after stroke. However, the mechanistic role of senescent cells in these processes requires extensive investigation.
3.5. Parkinson’s disease
Parkinson’s disease (PD) is the second, most commonly occurring neurodegenerative disease, in which patients have affected motor movement (bradykinesia) and rigidity and/or tremor (Kalia and Lang, 2015; Postuma et al., 2018). Individuals with PD often experience non-motor symptoms, such as impaired psychiatric and cognitive behavior, disrupted sleep patterns, increased pain sensation, and fatigue (Jellinger, 2015; Kalia and Lang, 2015; Schapira et al., 2017). At a pathological and histological level, PD brains have loss of dopaminergic neurons in the substantia nigra pars compacta and the presence of insoluble α-synuclein aggregates in intracytoplasmic and intraneurite eosinophilic inclusions (Lewy bodies) (Braak and Braak, 2000; Pakkenberg et al., 1991).
As with AD, PD pathology begins before clinical manifestations of disease. Non-motor symptoms like GI tract disturbances, depression, sleep disorders or rapid eye movement are sometimes the first clinical signs that a patient will present (Schapira et al., 2017), whereas motor symptoms only become evident when more than 50% of dopaminergic neurons have died (Braak and Braak, 2000; Braak et al., 2003; Ross et al., 2004). Furthermore, accurate diagnosis of early stage PD is difficult partly due to its overlapping non-motor symptoms with other neurodegenerative diseases, among them dementia with Lewy bodies and vascular parkinsonism (Tolosa et al., 2006). To this end, PD is primarily diagnosed on its clinical late-stage motor symptoms and confirmed depending on its response to levodopa, which targets motor symptoms, or DaTscan imaging.
There are two types of PD: the familial form, which can begin at around 50 years of age and the more common (approximately 85% of cases) sporadic form, generally occurring after 65years of age. Genetic mutations involved in mitochondrial respiration and function, leucine-rich repeat kinase 2 (LRRK2), PTEN-induced kinase 1 (PINK1), and Parkin RBR E3 ubiquitin protein ligases 2 and 7 (PRKN2 and 7), as well as α-synuclein (SNCA) have been implicated, as have toxic environmental compounds (Hawkes et al., 2007; Singh et al., 2019; Singleton et al., 2013).
The most widely used pharmacological treatments for PD, such as levodopa, modulate brain dopamine (Barbeau, 1974; Connolly and Lang, 2014). However, the efficacy of levodopa in reducing motor symptoms decreases over time, not all patients respond to it, and it has little effect on the non-motor symptoms of PD (Baker et al., 2009). Therefore, there has been a need for the development of new drugs for treating individuals with PD. New treatments, including gene therapies, are in ongoing clinical trials. Currently, there are no clinically-available therapies targeting the neurodegenerative aspects of this disease (Harris et al., 2020; Simmnacher et al., 2019).
Parkinson’s disease is associated with many negative changes at the molecular level, including mitochondrial and lysosomal dysfunction, misfolded protein accumulation, perturbed calcium homeostatis, and neuroinflammation (Chan et al., 2009; Tansey and Goldberg, 2010; Winklhofer and Haass, 2010). Mitochondrial dysfunction has long been considered to be a key driver of PD (Lill, 2016; Park et al., 2018). Mutations in genes involved in mitochondrial function (LRRK2, PINK1, and PRKN2 and 7) have detrimental effects on dopaminergic neurons and increase oxidative stress (Lill, 2016; Park et al., 2018). Dysfunctional mitochondria play a key role in PD, although their exact role is yet to be elucidated (Park et al., 2018). Senescent cells are present at sites of pathology and might play a key role in PD. Senescent cells have been identified in CSF, neurons, and astrocytes in PD (Chinta et al., 2018; van Dijk et al., 2013; Wan et al., 2014). In CSF, SA-β-gal activity increases during early stages of idiopathic PD (van Dijk et al., 2013). Furthermore, an important regulator of cell cycle progression, retinoblastoma protein (Rb), is down-regulated in neurons in the substantia nigra, mid-frontal cortex, and hippocampus of PD patients. Interestingly, within the substantia nigra, the inactive hyperphosphorylated isoform of Rb co-localizes with Lewy bodies (Jordan-Sciutto et al., 2003).
Astrocytes have recently been noted to play a role in many neurodegenerative diseases and neuroinflammation (Bussian et al., 2018; Musi et al., 2018; Yabluchanskiy et al., 2020; Zhang et al., 2019), including PD (Chinta et al., 2018; Wan et al., 2014). Senescent astrocytes in PD were found following exposure to an environmental toxic compound known to cause PD, i.e., 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) (Gonzalez-Barbosa et al., 2017; Wan et al., 2014). This induced astrocytes to become prematurely senescent, with increased p16INK4a, p21CIP1, and SA-β-gal expression (Wan et al., 2014). Senescent cell markers are present within astrocytes of post-mortem PD brain tissues (Chinta et al., 2018). These tissues also show increases in pro-inflammatory SASP factors, including IL-6, IL-8, and IL1α. Also consistent with being senescent, astrocytes in these tissues exhibit loss of lamin B1. Lamin B1 is a nuclear lamina marker. Down-regulation of lamin B1 has been suggested to be a marker of cellular senescence and is believed to be down-regulated in senescent cells due to changes in chromatin organization (Freund et al., 2012).
In vitro studies showed that paraquat, a toxic, environmental compound that leads to PD, induced cellular senescence and stimulated production of SASP factors in astrocytes and this in turn, led to neuronal cell death. Removal of senescent astrocytes reversed the toxic effects of paraquat by restoring neurogenesis, motor function, and neuronal cell numbers (Chinta et al., 2018).
3.6. Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a progressive motor neuron disease with no known cure (Robberecht and Philips, 2013). It is the third most common neurodegenerative disease after AD and PD (Renton et al., 2014). The neuropathology of ALS involves death of both upper and lower motor neurons in the brain and spinal cord, leading to paralysis of voluntary muscles. Studies in ALS mouse models have shown that neuron death in ALS is non-cell autonomous, as the astrocytes and microglial cells that surround motor neurons contribute to disease onset and progression (Wang et al., 2009, 2011; Yamanaka et al., 2008). Trias et al. (2019) found microglia, astrocytes, and motor neurons displaying phenotypic markers of senescence, including SASP factors, in a rat model of ALS. Mitochondrial ROS generation is frequently increased in ALS. Lipids accumulate in the nervous tissue, further contributing to cell damage, as often occurs in senescent cells with impaired mitochondrial function and increased glycolysis. Thus, astrocytes and microglia can acquire senescent phenotypes and have toxic effects in both animal models of ALS and individuals with ALS (Haidet-Phillips et al., 2011).
Neuroinflammation has been considered to be a secondary, non-specific reaction to the degeneration of motor neurons in ALS (Kjaeldgaard et al., 2018; Liu and Wang, 2017). Senescent cells, acting to promote inflammation, likely play a role in the neuroinflammation that is commonly present in ALS. Taking these points together, persistence of senescent cells might alter the cellular microenvironment through their SASP, exacerbating progressive neuroinflammation and motor neuron death in ALS.
3.7. Obesity/diabetes-related neuropsychiatric dysfunction
There is accumulation of senescent cells with obesity (Minamino et al., 2009; Ogrodnik et al., 2017; Schafer et al., 2016). Obesity has been connected to several neurodegenerative and neuropsychiatric disorders, including anxiety and depression (Gariepy et al., 2010; Hryhorczuk et al., 2013; Stunkard and Wadden, 1992). Ogrodnik et al. (2019) showed that clearance of senescent cells in obese mice alleviates anxiety-like behavior, highlighting cellular senescence as a target pathway for treating obesity-related neuropsychiatric dysfunction.
Aging is a leading risk factor for type 2 diabetes mellitus (T2DM), a condition involving peripheral insulin resistance that progresses to pancreatic failure related to increased demand for insulin secretion (Palmer et al., 2015). Prevalence of T2DM is increased in AD (Shinohara and Sato, 2017). Peripheral insulin resistance, detectible among other features by inappropriately high insulin levels for a given blood glucose, increases with aging. This peripheral insulin resistance can, in turn, be caused by fundamental aging processes, including the chronic, low grade inflammation in multiple tissues that is associated with senescent cell accumulation (Palmer et al., 2019; Xu et al., 2015a). More and more studies confirm that T2DM and AD are not independent disorders, since they share many common pathophysiological mechanisms (Klimova et al., 2018). Senescent cells accumulate in obese, insulin-resistant humans and rodents (Tchkonia et al., 2010; Villaret et al., 2010). We found that reducing senescent cell burden alleviates metabolic and adipose tissue dysfunction in obese mice (Palmer et al., 2019). Consequently, senescent cell burden in key target tissues are likely shared with the development and clinical phenotypes of T2DM and neurodegenerative diseases, and have potential as therapeutic targets for treating these two, age-related diseases.
3.8. Brain tumors
Senescence can mitigate risk of neoplastic transformation by removing compromised cells from the cell cycle (Bartkova et al., 2006). Oncogene-induced senescence may act variably through p38 mitogen-activated protein (MAP) kinase, forkhead box O (FoxO), phosphoinositide 3-kinase (PI3K), protein kinase B (AKT), and mechanistic target of rapamycin (mTOR) pathways converging on cyclin-dependent kinase inhibitor 2A (p16INK4a)/Retinoblastoma (Rb) and protein (p)14/p53/p21 to induce cell cycle arrest and induce the SASP (Xu et al., 2014). That some cells ultimately become malignant, however, is evidence that oncogene-induced senescence is an imperfect protection against tumorigenesis (Marumoto et al., 2009). Glioblastoma is the most common and most deadly adult brain tumor, and most glioblastomas exhibit mutations in p16INK4a/Rb and/or p53 signaling pathways (Ohgaki, 2005). Over half have homozygous deletion of p16INK4a, loss of which portends poor prognosis in older patients (Batchelor et al., 2004). Mutations in p53 facilitate resistance to standard treatments for glioma (Squatrito et al., 2010), which comprise alkylating chemotherapy, and radiation (Stupp et al., 2005). Unfortunately, these standard genotoxic therapies are insufficient to eliminate tumor, but can temporarily push residual cells into a senescent state often enabling a brief respite of tumor dormancy prior to inevitable recurrence. Even with the most aggressive management by surgical resection, chemotherapy and radiation, most patients with glioblastoma live less than 18months (Johnson and O’Neill, 2012).
The SASP has been shown to have pro-tumorigenic effects in some contexts by promoting infiltrative growth (Lecot et al., 2016). Whether or not this is true for glioblastoma, there is at least a strong overlap of pathways involved in senescence and the mesenchymal glioblastoma subtype, which portends therapeutic resistance and poor prognosis (Bhat et al., 2013). Recent evidence suggests that the process of passing through senescence after chemotherapy prior to recurrence induces chromatin remodeling that exacerbates aggressiveness and resilience of re-emergent cancer stem cells (Milanovic et al., 2018). Whether the state of transient therapy-induced senescence could be harnessed as an avenue to eliminate latent tumor cells prior to recurrence is an important area for future investigation (Samaraweera et al., 2017).
In contrast to glioblastoma, some CNS malignancies respond more favorably to chemotherapy and radiation, making long-term survivorship a meaningful possibility. Pediatric brain tumors are increasingly curable with aggressive therapy. However, the senescence-inducing actions of chemoradiation can yield devastating impacts on the CNS (Reimers et al., 2003).
3.9. Radiation-induced brain dysfunction
The long-term sequelae of brain radiation are severe and currently irreversible, with pathologic hallmarks persisting even decades after radiation in human subjects (Monje et al., 2007). Such hallmarks include vascular injury (Lyubimova and Hopewell, 2004), microglial activation, loss of neurogenesis (Monje et al., 2002), mitochondrial dysfunction, white matter atrophy, and compromised synaptic density (Kempf et al., 2015). Given the relevance of hippocampal neurogenesis to memory function and the sensitivity of this structure to radiation, hippocampal-avoidance dosimetry plans are now increasingly utilized when feasible (Gondi et al., 2014). Severity of cognitive outcomes are inversely proportional to the age of radiation, with youngest patients suffering a neurotoxic combination of arrested brain development/myelination and premature degeneration (Mabbott et al., 2006). Secondary malignancies, including radiation-induced glioblastoma (Donson et al., 2007), may sometimes yield a tragically premature end to a life, marked by profound cognitive and psychosocial impairment, with double-digit loss of IQ potential and inability to live independently.
Given the degree of chronic, radiation-induced neuroinflammation, microglia and astrocytes have long been under scrutiny as potential pathogenic mediators (Kyrkanides et al., 1999). Indeed, within a month after radiation, the microglial transcriptome closely mirrors that of aged microglia, with p21CIP1 being among the most highly upregulated genes (Li et al., 2015). Microglial ablation through colony stimulating factor 1 receptor (CSF1R) blockade has yielded promise in preclinical models of radiation-induced brain injury (Acharya et al., 2016). However, considering the importance of microglia for normal cognitive function (Kettenmann et al., 2013), one wonders if a more selective ablation of senescent microglia could be feasible and preferable. Navitoclax is a senolytic compound that targets the anti-apoptotic pathways of BCL2 and BCL-XL which are upregulated in senescent cells. Use of navitoclax as a senolytic agent (Zhu et al., 2015, 2016) was recently suggested to provide functional benefit in mice after brain radiation. Investigators reported clearance of senescent astrocytes and preservation of neurovascular coupling (Yabluchanskiy et al., 2020). The direct pathogenic role of senescence after radiation in other CNS cell types, including OPCs (Zhang et al., 2019), myelinating oligodendrocytes, and neurons themselves (Jurk et al., 2012), is tempting to speculate, though warranting further specific evaluation after in vivo radiation.
3.10. Chemotherapy-induced brain dysfunction
Cognitive sequelae stemming from dysfunction of multiple CNS cell types is well established following chemotherapy (Gibson et al., 2019). Like radiation, systemic chemotherapeutic agents induce cellular senescence (Aasland et al., 2019), with potential impacts on the CNS—either directly in the case of CNS-penetrant chemotherapeutics, or indirectly for non-penetrant agents. To what extent systemic release of SASP factors could contribute to “chemobrain” symptoms of cognitive impairment following chemotherapy remains to be directly determined (Mandelblatt et al., 2013).
3.11. Post-traumatic brain injury
Many traumatic brain injuries lead to some level of cognitive impairment. There are limited treatment options for post-traumatic brain injury (TBI) cognitive problems. When running a search for post-TBI and senescence in PubMed, we found very little information about physiological mechanisms related to aging. However, one group found that levels of neurofilaments in the blood could be an indicator of axonal injury after TBI (Khalil et al., 2018). This is an area of research that should be further explored (Fig. 2).
Fig. 2.
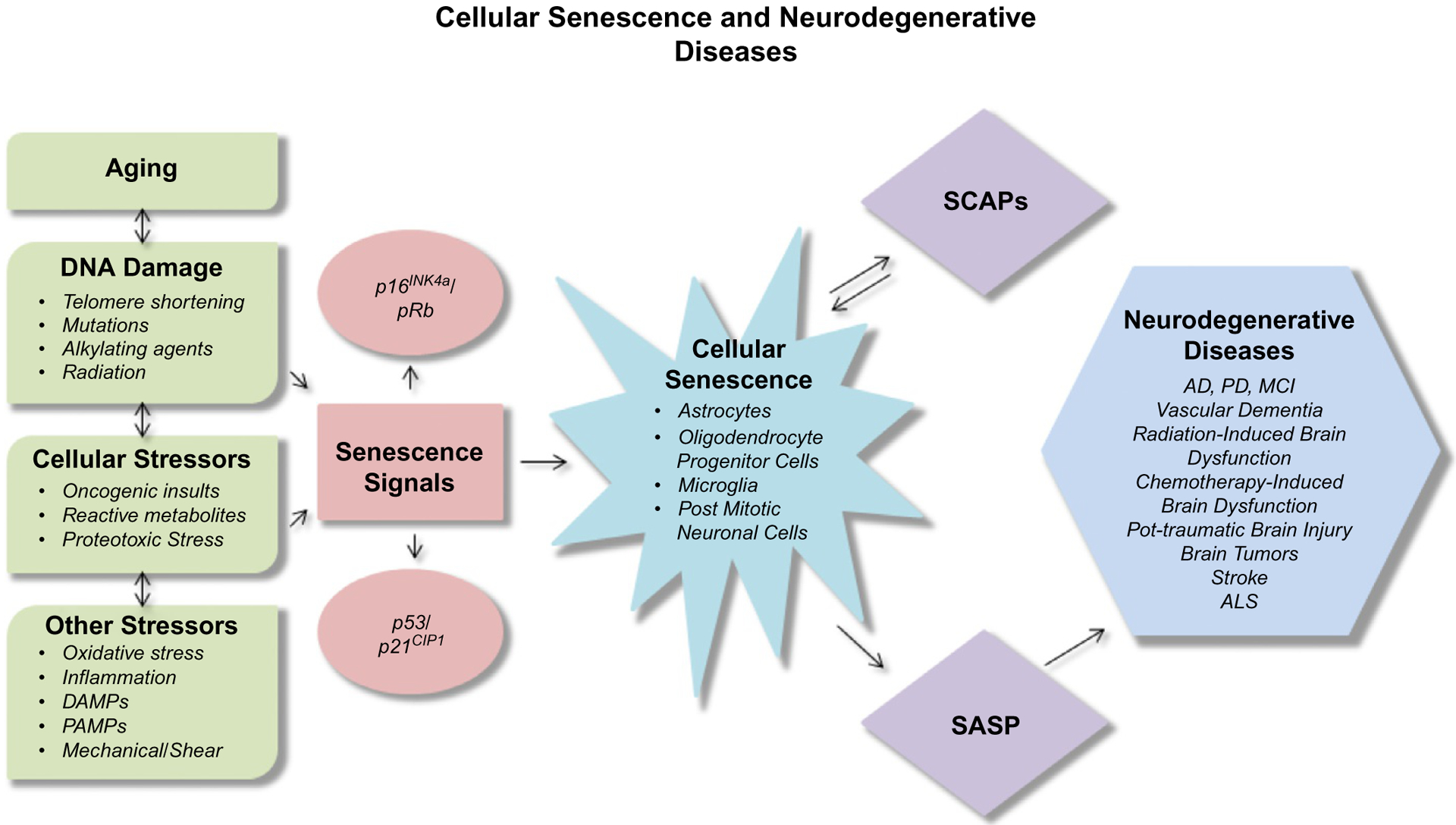
Cellular senescence and neurodegenerative diseases.
4. Senolytics
Using a hypothesis-driven, mechanism-based, drug discovery approach, we developed senolytics, agents that selectively eliminate senescent cells (Kirkland and Tchkonia 2017; Zhu et al., 2015, 2016, 2017). One of the best-characterized senolytic treatments is Dasatinib plus Quercetin (D+Q). Dasatinib, a tyrosine kinase inhibitor with BBB penetrance, decreased microgliosis and exerted neuroprotective effects in AD preclinical models (Farr et al., 2016; Jurk et al., 2014; Wang, 1995). Quercetin, a naturally occurring flavonoid, reduced AD-associated protein accumulation and cognitive and emotional impairments in transgenic AD mice (Sabogal-Guaqueta et al., 2015). Dasatinib and Quercetin have favorable side effect profiles, which makes them attractive for in-human trials. Dasatinib plus Quercetin alleviates frailty and muscle weakness in progeroid and chronologically aged mice, delays onset of age-related diseases, and extends median and maximum remaining lifespan in old mice (Yousefzadeh et al., 2018). Fisetin (F), a subsequently discovered senolytic, is a flavonoid occurring in many plants, fruits, and vegetables and has been widely available as a nutritional supplement for many years (Yousefzadeh et al., 2018; Zhu et al., 2017). In old mice, we found that intermittent F administration enhanced cognitive function (unpublished observations).
We found drugs that inhibit the SASP alleviate frailty even in very old mice, as shown by increases in activity, grip strength, and treadmill running endurance (Xu et al., 2015b). Dasatinib plus Quercetin reduces frailty in progeroid (accelerated aging) and chronologically aged mice, with improvements in an index that includes activity, strength, appearance, coat density, weight, and continence (Xu et al., 2018; Yousefzadeh et al., 2018; Zhu et al., 2015, 2016). Furthermore, D+Q increased healthspan and lifespan in old naturally aged mice. We and others found that senolytics delay, prevent, or alleviate multiple age-related diseases and disorders in murine models (Kirkland and Tchkonia, 2017; Kirkland et al., 2017; Tchkonia and Kirkland, 2018).
Using intermittent treatment with senolytic agents, we found improved metabolic parameters in obese mice, manifested by improved glucose tolerance, lower hemoglobin A1c (HbA1c), and enhanced insulin sensitivity, without affecting body weight or food intake. Complications of diabetes, including microalbuminuria and renal dysfunction, were alleviated. Mediators of insulin resistance, including inflammatory cytokines [TNF-α, Interferon gamma (IFNγ), osteopontin], the anti-adipogenic protein activin A, and immune cell chemokines in adipose tissue [Regulated on Activation, Normal T Cell Expressed and Secreted (RANTES), Macrophage Inflammatory Protein-1β (MIP-1β), and Monocyte Chemoattractant Protein-1 (MCP-1)], were reduced by eliminating senescent cells.
5. Senolytics in preclinical animal models of neurodegenerative diseases
In mice, senolytic drugs, including D+Q, have been shown to reduce NFT density, neuron loss, and ventricular enlargement, and enhance neurogenesis and memory (Musi et al., 2018; Ogrodnik et al., 2019; Zhang et al., 2019), as well as to improve carotid vascular reactivity and exercise capacity, reduce osteoporosis and loss of intervertebral disk proteoglycans, alleviate metabolic dysfunction, improve renal function, and provide many other benefits (Kirkland et al., 2017; Tchkonia and Kirkland, 2018; Xu et al., 2018; Zhu et al., 2015, 2016).
Recently, our collaborators treated AD mice with D+Q combination therapy and found a significant improvement in multiple AD-relevant phenotypes, including reduced ventricular volume, aberrant cerebral blood flow, and WMH pathology (Musi et al., 2018). In a tau+ mouse line with mild tau overexpression resulting in neurocognitive symptoms only in later life (after age 22months, equivalent to age 70years in humans), administering senolytics intermittently decreased brain senescent cells, reduced neuroinflammation and gliosis, enhanced neuron density, partially restored lost cortical brain tissue, and decreased ventricular enlargement (Musi et al., 2018). These significant improvements occurred concomitantly with clearing 35% of pro-inflammatory senescent NFT-containing neurons. In these old mice, clinically relevant outcomes were improved after only six single oral doses of D+Q spanning 12weeks. These findings were confirmed in a recent study by Zhang et al. (2019), in which D+Q also cleared brain senescent cells (senescent oligodendrocytes), decreased neuroinflammation, and improved memory demonstrated by novel object recognition and Stone maze performance. Senolytics caused apoptosis of senescent, but not normal oligodendrocyte lineage cells derived from human brains and also in the transgenic mice. Treating the mice intermittently with senolytics decreased neuroinflammation and improved memory acquisition and retention. The results of the preclinical studies suggest the efficacy of intermittent administration of senolytics for alleviating AD-associated deficits in brain structure and function.
6. Clinical trials underway or planned of senolytics for diseases, including neurodegenerative diseases
To our knowledge, there are no published studies utilizing D+Q to alleviate AD or other neurodegenerative diseases in humans. Based on our preclinical experience with 46 agents/drugs examined and recent studies with D and F, these senolytics induce apoptosis of senescent cells in vitro with minimal effects on non-senescent or healthy cells (Xu et al., 2015a; Zhu et al., 2017).
Early phase clinical trials are aimed at moving these novel findings in mice to humans. In an upcoming Phase II pilot study (NCT04063124), men and women over age 65 with mild to moderate symptomatic AD [clinical dementia rating scale (CDR) stage 1.0], but who are still mobile and living in the community, will be screened for tau and Aβ protein and presence of senescent cells in CSF or p16INK4A+ CD3+ T cells and other blood assays. Subjects with AD by brain magnetic resonance imaging (MRI), fluorodeoxyglucose (FDG) positron emission tomography (PET), amyloid PET, or tau PET imaging and evidence of senescent cells by CSF or blood assays will be administered D+Q orally for six intermittent cycles over the course of their 2-year participation. Cerebrospinal fluid, questionnaires, performance-based measures of cognition and physical performance, urine, and blood tests for markers of senescent cell burden, SASP factors, inflammation, fasting glucose and insulin, and bone turnover, will be assayed before and periodically after initiating treatment with D+Q.
7. Conclusions
Physiological mechanisms that connect aging with age-related and neurodegenerative diseases are still largely unknown. Cellular senescence is linked to age-related and neurodegenerative diseases. Cellular senescence is driven by many fundamental aging processes, including genomic instability, telomere dysfunction/shortening, metabolic dysregulation, protein aggregation and misfolding, and nuclear membrane and mitochondrial dysfunction.
While senolytics provide a promising avenue for exploration as a treatment for age-related and neurodegenerative diseases, the safety and effectiveness of these agents has not been clearly established. Senolytic drugs, including those discussed in this review, should not be used for humans outside of clinical trials where there is sufficient monitoring for potential adverse effects.
Acknowledgments
The authors are supported by NIH Grants R37AG013925, P01AG062413, and the Translational Geroscience Network (R33AG061456), the Alzheimer’s Association Part the Cloud Program, the Alzheimer’s Drug Discovery Foundation, Robert and Arlene Kogod, the Connor Group, Robert J. and Theresa W. Ryan, and the Noaber Foundation.
Abbreviations
- Aβ
amyloid beta
- AD
Alzheimer’s disease
- AKT
protein kinase B
- BBB
blood-brain barrier
- CDKN2A or p16INK4a
cyclin-dependent kinase inhibitor 2A
- CDR
clinical dementia rating scale
- CNS
central nervous system
- CSF
cerebrospinal fluid
- CSF1R
colony stimulating factor 1 receptor
- D
dasatinib
- F
fisetin
- FDG
fluorodeoxyglucose
- FoxO
forkhead box O
- IFNγ
interferon gamma
- IL
interleukin
- LRRK2
leucine-rich repeat kinase 2
- MAP
mitogen-activated protein
- MCP-1
monocyte chemoattractant protein-1
- MCI
mild cognitive impairment
- MIP-1β
macrophage inflammatory protein-1β
- MMP-1
matrix metalloproteinase
- MRI
magnetic resonance imaging
- mTOR
mechanistic target of rapamycin
- NFT
neurofibrillary tangle
- OPCS
oligodendrocyte progenitor cells
- P
protein
- PD
Parkinson’s disease
- PET
positron emission tomography
- PI3K
phosphoinositide 3-kinase
- PINK1
PTEN-induced kinase 1
- PRKN2 and 7
Parkin RBR E3 ubiquitin protein ligases 2 and 7
- Q
quercetin
- RANTES
regulated on activation normal T cell expressed and secreted
- Rb
retinoblastoma
- ROS
reactive oxygen species
- SASP
senescence-associated secretory phenotype
- SCAP
senescent cell anti-apoptotic pathway
- SNCA
α-synuclein
- T2DM
type 2 diabetes mellitus
- TBI
traumatic brain injury
- TNF-α
tumor necrosis factor-α
- WMH
white matter hyperintensities
- vAD
vascular dementia
- vFKBP
viral FK506-binding protein
Footnotes
Competing financial interests
J.L.K., T.T., and Y.Z. have a financial interest related to this research. Patents on senolytic drugs are held by Mayo Clinic. This research has been reviewed by the Mayo Clinic Conflict of Interest Review Board and was conducted in compliance with Mayo Clinic Conflict of Interest policies. No conflicts of interest, financial or otherwise, are declared by E.O.W.G., B.M.W., U.T., or T.C.B.
References
- Aasland D, Gotzinger L, Hauck L, Berte N, Meyer J, Effenberger M, et al. (2019). Temozolomide induces senescence and repression of DNA repair pathways in glioblastoma cells via activation of ATR-CHK1, p21, and NF-kappaB. Cancer Research, 79(1), 99–113. [DOI] [PubMed] [Google Scholar]
- Acharya MM, Green KN, Allen BD, Najafi AR, Syage A, Minasyan H, et al. (2016). Elimination of microglia improves cognitive function following cranial irradiation. Scientific Reports, 6, 31545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. (2013). A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nature Cell Biology, 15(8), 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. (2011). The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association work-groups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement, 7(3), 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson R, Lagnado A, Maggiorani D, Walaszczyk A, Dookun E, Chapman J, et al. (2019). Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. The EMBO Journal, 38(5), 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, & Hyman BT (1992). Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology, 42(3 Pt 1), 631–639. [DOI] [PubMed] [Google Scholar]
- Baker DJ, & Petersen RC (2018). Cellular senescence in brain aging and neurodegenerative diseases: Evidence and perspectives. The Journal of Clinical Investigation, 128(4), 1208–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker WL, Silver D, White CM, Kluger J, Aberle J, Patel AA, et al. (2009). Dopamine agonists in the treatment of early Parkinson’s disease: A meta-analysis. Parkinsonism & Related Disorders, 15(4), 287–294. [DOI] [PubMed] [Google Scholar]
- Barbeau A (1974). The clinical physiology of side effects in long-term L-DOPA therapy. Advances in Neurology, 5, 347–365. [PubMed] [Google Scholar]
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. (2006). Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature, 444(7119), 633–637. [DOI] [PubMed] [Google Scholar]
- Bartzokis G (2011). Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiology of Aging, 32(8), 1341–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor TT, Betensky RA, Esposito JM, Pham LD, Dorfman MV, Piscatelli N, et al. (2004). Age-dependent prognostic effects of genetic alterations in glioblastoma. Clinical Cancer Research, 10(1 Pt 1), 228–233. [DOI] [PubMed] [Google Scholar]
- Bhat KPL, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, et al. (2013). Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell, 24(3), 331–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasko I, Stampfer-Kountchev M, Robatscher P, Veerhuis R, Eikelenboom P, & Grubeck-Loebenstein B (2004). How chronic inflammation can affect the brain and support the development of Alzheimer’s disease in old age: The role of microglia and astrocytes. Aging Cell, 3(4), 169–176. [DOI] [PubMed] [Google Scholar]
- Braak H, & Braak E (2000). Pathoanatomy of Parkinson’s disease. Journal of Neurology, 247(Suppl 2), II3–10. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RAI, Steur ENHJ, & Braak E (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of Aging, 24(2), 197–211. [DOI] [PubMed] [Google Scholar]
- Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, & Baker DJ (2018). Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature, 562, 578–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Zhang K, Li P, Zhu L, Xu J, Yang B, et al. (2017). Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative diseases: An aging effect. Ageing Research Reviews, 34, 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J (2005). Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell, 120, 513–522. [DOI] [PubMed] [Google Scholar]
- Campisi J, & d’Adda di Fagagna F (2007). Cellular senescence: When bad things happen to good cells. Nature Reviews Molecular Cell Biology, 8(9), 729–740. [DOI] [PubMed] [Google Scholar]
- Chan CS, Gertler TS, & Surmeier DJ (2009). Calcium homeostasis, selective vulnerability and Parkinson’s disease. Trends in Neurosciences, 32(5), 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta SJ, Woods G, Demaria M, Rane A, Zou Y, McQuade A, et al. (2018). Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Reports, 22(4), 930–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly BS, & Lang AE (2014). Pharmacological treatment of Parkinson disease:A review. JAMA, 311(16), 1670–1683. [DOI] [PubMed] [Google Scholar]
- Coppé JP, Desprez PY, Krtolica A, & Campisi J (2010). The senescence-associated secretory phenotype: The dark side of tumor suppression. Annual Review of Pathology, 5, 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé JP, Patil C, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. (2008).Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biology, 6, 2853–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings J, Aisen PS, DuBois B, Frolich L, Jack CR Jr., Jones RW, et al. (2016). Drug development in Alzheimer’s disease: The path to 2025. Alzheimer’s Research & Therapy, 8, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCarli C (2003). Mild cognitive impairment: Prevalence, prognosis, aetiology, and treatment. Lancet Neurology, 2(1), 15–21. [DOI] [PubMed] [Google Scholar]
- Donson AM, Erwin NS, Kleinschmidt-DeMasters BK, Madden JR, Addo-Yobo SO, & Foreman NK (2007). Unique molecular characteristics of radiation-induced glioblastoma. Journal of Neuropathology and Experimental Neurology, 66(8), 740–749. [DOI] [PubMed] [Google Scholar]
- Farr JN, Fraser DG, Wang H, Jaehn K, Ogrodnik MB, Weivoda MM, et al. (2016). Identification of senescent cells in the bone microenvironment. Journal of Bone and Mineral Research: The Official Journal of the American Society for Bone and Mineral Research, 31(11), 1920–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigin VL, Forouzanfar MH, Krishnamurthi R, Mensah GA, Connor M, Bennett DA, et al. (2014). Global and regional burden of stroke during 1990–2010: Findings from the global burden of disease study 2010. Lancet, 383(9913), 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielder E, von Zglinicki T, & Jurk D (2017). The DNA damage response in neurons: Die by apoptosis or survive in a senescence-like state? Journal of Alzheimer’s Disease, 60(s1), S107–S131. [DOI] [PubMed] [Google Scholar]
- Freund A, Laberge RM, Demaria M, & Campisi J (2012). Lamin B1 loss is a senescence-associated biomarker. Molecular Biology of the Cell, 23(11), 2066–2075. 10.1091/mbc.E11-10-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gariepy G, Nitka D, & Schmitz N (2010). The association between obesity and anxiety disorders in the population: A systematic review and meta-analysis. International Journal of Obesity, 34(3), 407–419. [DOI] [PubMed] [Google Scholar]
- Gibson EM, Nagaraja S, Ocampo A, Tam LT, Wood LS, Pallegar PN, et al. (2019). Methotrexate chemotherapy induces persistent tri-glial dysregulation that underlies chemotherapy-related cognitive impairment. Cell, 176(1–2), 43–55. (e13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondi V, Pugh SL, Tome WA, Caine C, Corn B, Kanner A, et al. (2014).Preservation of memory with conformal avoidance of the hippocampal neural stem-cell compartment during whole-brain radiotherapy for brain metastases (RTOG 0933): A phase II multi-institutional trial. Journal of Clinical Oncology, 32(34), 3810–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Barbosa E, Mejia-Garcia A, Bautista E, Gonzalez FJ, Segovia J, & Elizondo G (2017). TCDD induces UbcH7 expression and synphilin-1 protein degradation in the mouse ventral midbrain. Journal of Biochemical and Molecular Toxicology, 31(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, et al. (2011). Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nature Biotechnology, 29(9), 824–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JP, Burrell JC, Struzyna LA, Chen HI, Serruya MD, Wolf JA, et al. (2020). Emerging regenerative medicine and tissue engineering strategies for Parkinson’s disease. NPJ Parkinson’s Disease, 6, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes CH, Del Tredici K, & Braak H (2007). Parkinson’s disease: A dual-hit hypothesis. Neuropathology and Applied Neurobiology, 33(6), 599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L, & Moorehead P (1961). The serial cultivation of human diploid strains.Experimental Cell Research, 25, 585–621. [DOI] [PubMed] [Google Scholar]
- Hebert LE, Weuve J, Scherr PA, & Evans DA (2013). Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology, 80(19), 1778–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbig U, Ferreira M, Condel L, Carey D, & Sedivy JM (2006). Cellular senescence in aging primates. Science, 311, 1257. [DOI] [PubMed] [Google Scholar]
- Hryhorczuk C, Sharma S, & Fulton SE (2013). Metabolic disturbances connecting obesity and depression. Frontiers in Neuroscience, 7, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA (2015). Neuropathobiology of non-motor symptoms in Parkinson disease.Journal of Neural Transmission (Vienna), 122(10), 1429–1440. [DOI] [PubMed] [Google Scholar]
- Johnson DR, & O’Neill BP (2012). Glioblastoma survival in the United States before and during the temozolomide era. Journal of Neuro-Oncology, 107(2), 359–364. [DOI] [PubMed] [Google Scholar]
- Jordan-Sciutto KL, Dorsey R, Chalovich EM, Hammond RR, & Achim CL(2003). Expression patterns of retinoblastoma protein in Parkinson disease. Journal of Neuropathology and Experimental Neurology, 62(1), 68–74. [DOI] [PubMed] [Google Scholar]
- Jun JI, & Lau LF (2010). The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nature Cell Biology, 12(7), 676–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, et al. (2012). Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell, 11(6), 996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurk D, Wilson C, Passos JF, Oakley F, Correia-Melo C, Greaves L, et al. (2014). Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nature Communications, 2, 4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice JN, Gregory H, Tchkonia T, LeBrasseur NK, Kirkland JL, Kritchevsky SB, et al. (2017). Cellular senescence biomarker p16INK4a+ cell burden in thigh adipose is associated with poor physical function in older women. The Journals of Gerontology Series A, Biological sciences and medical sciences, 73(7), 939–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia LV, & Lang AE (2015). Parkinson’s disease. Lancet, 386(9996), 896–912. [DOI] [PubMed] [Google Scholar]
- Katsuumi G, Shimizu I, Yoshida Y, & Minamino T (2018). Vascular senescence in cardiovascular and metabolic diseases. Frontiers in Cardiovascular Medicine, 5, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano H, Katsurabayashi S, Kakazu Y, Yamashita Y, Kubo N, Kubo M, et al. (2012). Long-term culture of astrocytes attenuates the readily releasable pool of synaptic vesicles. PLoS One, 7(10), e48034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempf SJ, Moertl S, Sepe S, von Toerne C, Hauck SM, Atkinson MJ, et al. (2015). Low-dose ionizing radiation rapidly affects mitochondrial and synaptic signaling pathways in murine hippocampus and cortex. Journal of Proteome Research, 14(5), 2055–2064. [DOI] [PubMed] [Google Scholar]
- Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, et al. (2014). Geroscience: Linking aging to chronic disease. Cell, 159(4), 709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Kirchhoff F, & Verkhratsky A (2013). Microglia: New roles for the synaptic stripper. Neuron, 77(1), 10–18. [DOI] [PubMed] [Google Scholar]
- Khalil M, Teunissen CE, Otto M, Piehl F, Sormani MP, Gattringer T, et al. (2018). Neurofilaments as biomarkers in neurological disorders. Nature Reviews Neurology, 14(10), 577–589. [DOI] [PubMed] [Google Scholar]
- Kirkland JL (2013). Translating advances from the basic biology of aging into clinical application. Experimental Gerontology, 48(1), 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland JL (2016). Translating the science of aging into therapeutic interventions. ColdSpring Harbor Perspectives in Medicine, 6(3), a025908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland JL, & Peterson C (2009). Healthspan, translation, and new outcomes for animal studies of aging. The Journals of Gerontology Series A, Biological Sciences and Medical Sciences, 64(2), 209–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland JL, & Tchkonia T (2017). Cellular senescence: A translational perspective. eBioMedicine, 21, 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, & Robbins PD (2017). The clinical potential of senolytic drugs. Journal of the American Geriatrics Society, 65(10), 2297–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjaeldgaard AL, Pilely K, Olsen KS, Pedersen SW, Lauritsen AO, Moller K, et al. (2018). Amyotrophic lateral sclerosis: The complement and inflammatory hypothesis. Molecular Immunology, 102, 14–25. [DOI] [PubMed] [Google Scholar]
- Klimova B, Kuca K, & Maresova P (2018). Global view on Alzheimer’s disease and diabetes mellitus: Threats, risks and treatment Alzheimer’s disease and diabetes mellitus. Current Alzheimer Research, 15(14), 1277–1282. [DOI] [PubMed] [Google Scholar]
- Kojima G, Liljas A, Iliffe S, & Walters K (2017). Prevalence of frailty in mild to moderate Alzheimer’s disease: A systematic review and meta-analysis. Current Alzheimer Research, 14(12), 1256–1263. [DOI] [PubMed] [Google Scholar]
- Kritchevsky SB, & Justice JN (2020). Testing the geroscience hypothesis: Early days. The Journals of Gerontology Series A, Biological Sciences and Medical Sciences, 75(1), 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. (2008).Senescence of activated stellate cells limits liver fibrosis. Cell, 134, 657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, & Peeper DS (2009). Senescence-messaging secretome: SMS-ing cellular stress. Nature Reviews Cancer, 9, 81–94. [DOI] [PubMed] [Google Scholar]
- Kujuro Y, Suzuki N, & Kondo T (2010). Esophageal cancer-related gene 4 is a secreted inducer of cell senescence expressed by aged CNS precursor cells. Proceedings of the National Academy of Sciences of the United States of America, 107(18), 8259–8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrkanides S, Olschowka JA, Williams JP, Hansen JT, & O’Banion MK (1999).TNF alpha and IL-1beta mediate intercellular adhesion molecule-1 induction via microglia-astrocyte interaction in CNS radiation injury. Journal of Neuroimmunology, 95(1–2), 95–106. [DOI] [PubMed] [Google Scholar]
- LeBrasseur NK, Tchkonia T, & Kirkland JL (2015). Cellular senescence and the biology of aging, disease, and frailty. Nestle Nutrition Institute Workshop Series, 83, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecot P, Alimirah F, Desprez PY, Campisi J, & Wiley C (2016). Context-dependent effects of cellular senescence in cancer development. British Journal of Cancer, 114(11), 1180–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MD, Burns TC, Kumar S, Morgan AA, Sloan SA, & Palmer TD (2015).Aging-like changes in the transcriptome of irradiated microglia. Glia, 63(5), 754–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill CM (2016). Genetics of Parkinson’s disease. Molecular and Cellular Probes, 30(6), 386–396. [DOI] [PubMed] [Google Scholar]
- Liu J, & Wang F (2017). Role of neuroinflammation in amyotrophic lateral sclerosis:Cellular mechanisms and therapeutic implications. Frontiers in Immunology, 8, 1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Coco D, Lopez G, & Corrao S (2016). Cognitive impairment and stroke in elderly patients. Vascular Health and Risk Management, 12, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo EH, & Rosenberg GA (2009). The neurovascular unit in health and disease:Introduction. Stroke, 40(3 Suppl), S2–S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, & Kroemer G (2013). The hallmarks of aging. Cell, 153(6), 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PH, Lee GJ, Shapira J, Jimenez E, Mather MJ, Thompson PM, et al. (2014). Regional differences in white matter breakdown between frontotemporal dementia and early-onset Alzheimer’s disease. Journal of Alzheimer’s Disease, 39(2), 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PH, Lee GJ, Tishler TA, Meghpara M, Thompson PM, & Bartzokis G (2013). Myelin breakdown mediates age-related slowing in cognitive processing speed in healthy elderly men. Brain and Cognition, 81(1), 131–138. [DOI] [PubMed] [Google Scholar]
- Lyubimova N, & Hopewell JW (2004). Experimental evidence to support the hypothesis that damage to vascular endothelium plays the primary role in the development of late radiation-induced CNS injury. The British Journal of Radiology, 77(918), 488–492. [DOI] [PubMed] [Google Scholar]
- Mabbott DJ, Noseworthy MD, Bouffet E, Rockel C, & Laughlin S (2006).Diffusion tensor imaging of white matter after cranial radiation in children for medulloblastoma: Correlation with IQ. Neuro-Oncology, 8(3), 244–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelblatt JS, Hurria A, McDonald BC, Saykin AJ, Stern RA, VanMeter JW, et al. (2013). Cognitive effects of cancer and its treatments at the intersection of aging: What do we know; what do we need to know? Seminars in Oncology, 40(6), 709–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marumoto T, Tashiro A, Friedmann-Morvinski D, Scadeng M, Soda Y, Gage FH, et al. (2009). Development of a novel mouse glioma model using lentiviral vectors. Nature Medicine, 15(1), 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecca C, Giambanco I, Donato R, & Arcuri C (2018). Microglia and aging: The role of the TREM2-DAP12 and CX3CL1-CX3CR1 axes. International Journal of Molecular Sciences, 19(1), 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud M, Balardy L, Moulis G, Gaudin C, Peyrot C, Vellas B, et al. (2013). Proinflammatory cytokines, aging, and age-related diseases. Journal of the American Medical Directors Association, 14(12), 877–882. [DOI] [PubMed] [Google Scholar]
- Milanovic M, Fan DNY, Belenki D, Dabritz JHM, Zhao Z, Yu Y, et al. (2018). Senescence-associated reprogramming promotes cancer stemness. Nature, 553(7686), 96–100. [DOI] [PubMed] [Google Scholar]
- Miller RA (2002). Extending life: Scientific prospects and political obstacles. The MilbankQuarterly, 80(1), 155–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, et al. (2009). A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nature Medicine, 15(9), 1082–1087. [DOI] [PubMed] [Google Scholar]
- Miranda CJ, Braun L, Jiang Y, Hester ME, Zhang L, Riolo M, et al. (2012). Aging brain microenvironment decreases hippocampal neurogenesis through Wnt-mediated survivin signaling. Aging Cell, 11(3), 542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje ML, Mizumatsu S, Fike JR, & Palmer TD (2002). Irradiation induces neural precursor-cell dysfunction. Nature Medicine, 8(9), 955–962. [DOI] [PubMed] [Google Scholar]
- Monje ML, Vogel H, Masek M, Ligon KL, Fisher PG, & Palmer TD (2007). Impaired human hippocampal neurogenesis after treatment for central nervous system malignancies. Annals of Neurology, 62(5), 515–520. [DOI] [PubMed] [Google Scholar]
- Mulugeta E, Molina-Holgado F, Elliott MS, Hortobagyi T, Perry R, Kalaria RN, et al. (2008). Inflammatory mediators in the frontal lobe of patients with mixed and vascular dementia. Dementia and Geriatric Cognitive Disorders, 25(3), 278–286. [DOI] [PubMed] [Google Scholar]
- Musi N, Valentine JM, Sickora KR, Baeuerle E, Thompson CS, Shen Q, et al. (2018). Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell, 17(6), e12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, et al. (2012). A senescent cell bystander effect: Senescence-induced senescence. Aging Cell, 11(2), 345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness KK, Kirkland JL, Gramatges MM, Wang Z, Kundu M, McCastlain K, et al. (2018). Premature physiologic aging as a paradigm for understanding increased risk of adverse health across the lifespan of survivors of childhood Cancer. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 36(21), 2206–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, et al. (2017).Cellular senescence drives age-dependent hepatic steatosis. Nature Communications, 8, 15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogrodnik M, Zhu Y, Langhi LGP, Tchkonia T, Kruger P, Fielder E, et al. (2019). Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell: Metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgaki H (2005). Genetic pathways to glioblastomas. Neuropathology, 25(1), 1–7. [DOI] [PubMed] [Google Scholar]
- Ossenkoppele R, Schonhaut DR, Scholl M, Lockhart SN, Ayakta N, Baker SL, et al. (2016). Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain, 139(Pt 5), 1551–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott BR, Jones RN, Daiello LA, de la Monte SM, Stopa EG, Johanson CE, et al. (2018). Blood-cerebrospinal fluid barrier gradients in mild cognitive impairment and Alzheimer’s disease: Relationship to inflammatory cytokines and chemokines. Frontiers in Aging Neuroscience, 10, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakkenberg B, Moller A, Gundersen HJ, Mouritzen Dam A, & Pakkenberg H (1991). The absolute number of nerve cells in substantia nigra in normal subjects and in patients with Parkinson’s disease estimated with an unbiased stereological method. Journal of Neurology, Neurosurgery, and Psychiatry, 54(1), 30–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AK, Gustafson B, Kirkland JL, & Smith U (2019). Cellular senescence: At the nexus between ageing and diabetes. Diabetologia, 62(10), 1835–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AK, Tchkonia T, LeBrasseur NK, Chini EN, Xu M, & Kirkland JL (2015). Cellular senescence in type 2 diabetes: A therapeutic opportunity. Diabetes, 64(7), 2289–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panza F, Seripa D, Solfrizzi V, Tortelli R, Greco A, Pilotto A, et al. (2015). Targeting cognitive frailty: Clinical and neurobiological roadmap for a single complex phenotype. Journal of Alzheimer’s Disease, 47(4), 793–813. [DOI] [PubMed] [Google Scholar]
- Park JS, Davis RL, & Sue CM (2018). Mitochondrial dysfunction in Parkinson’s disease: New mechanistic insights and therapeutic perspectives. Current Neurology and Neuroscience Reports, 18(5), 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piechota M, Sunderland P, Wysocka A, Nalberczak M, Sliwinska MA, Radwanska K, et al. (2016). Is senescence-associated beta-galactosidase a marker of neuronal senescence? Oncotarget, 7(49), 81099–81109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postuma RB, Poewe W, Litvan I, Lewis S, Lang AE, Halliday G, et al. (2018). Validation of the MDS clinical diagnostic criteria for Parkinson’s disease. Movement Disorders, 33(10), 1601–1608. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, & Long EO (2012). Cellular senescence induced by CD158d reprograms natural killer cells to promote vascular remodeling. Proceedings of the National Academy of Sciences of the United States of America, 109(50), 20596–20601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimers TS, Ehrenfels S, Mortensen EL, Schmiegelow M, Sonderkaer S,Carstensen H, et al. (2003). Cognitive deficits in long-term survivors of childhood brain tumors: Identification of predictive factors. Medical and Pediatric Oncology, 40(1), 26–34. [DOI] [PubMed] [Google Scholar]
- Renton AE, Chio A, & Traynor BJ (2014). State of play in amyotrophic lateral sclerosis genetics. Nature Neuroscience, 17(1), 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberecht W, & Philips T (2013). The changing scene of amyotrophic lateral sclerosis.Nature Reviews Neuroscience, 14(4), 248–264. [DOI] [PubMed] [Google Scholar]
- Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, Hagler M, Jurk D, Smith LA, Casaclang-Verzosa G, Zhu Y, Schafer MJ, Tchkonia T, Kirkland JL, & Miller JD (2016). Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell, 15(5), 973–977. 10.1111/acel.12458. (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross GW, Petrovitch H, Abbott RD, Nelson J, Markesbery W, Davis D, et al. (2004). Parkinsonian signs and substantia nigra neuron density in decendents elders without PD. Annals of Neurology, 56(4), 532–539. [DOI] [PubMed] [Google Scholar]
- Ruckh JM, Zhao JW, Shadrach JL, van Wijngaarden P, Rao TN, Wagers AJ, et al. (2012). Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell, 10(1), 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabogal-Guaqueta AM, Munoz-Manco JI, Ramirez-Pineda JR, Lamprea-Rodriguez M, Osorio E, & Cardona-Gomez GP (2015). The flavonoid quercetin ameliorates Alzheimer’s disease pathology and protects cognitive and emotional function in aged triple transgenic Alzheimer’s disease model mice. Neuropharmacology, 93, 134–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaraweera L, Adomako A, Rodriguez-Gabin A, & McDaid HM (2017). A novel indication for panobinostat as a senolytic drug in NSCLC and HNSCC. Scientific Reports, 7(1), 1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer MJ, White TA, Evans G, Tonne JM, Verzosa GC, Stout MB, et al. (2016). Exercise prevents diet-induced cellular senescence in adipose tissue. Diabetes, 65(6), 1606–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AHV, Chaudhuri KR, & Jenner P (2017). Non-motor features of Parkinson disease. Nature Reviews Neuroscience, 18(8), 509. [DOI] [PubMed] [Google Scholar]
- Schneider JA, Arvanitakis Z, Bang W, & Bennett DA (2007). Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology, 69(24), 2197–2204. [DOI] [PubMed] [Google Scholar]
- Sheinerman KS, Tsivinsky VG, Crawford F, Mullan MJ, Abdullah L, & Umansky SR (2012). Plasma microRNA biomarkers for detection of mild cognitive impairment. Aging (Albany NY), 4(9), 590–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M, & Sato N (2017). Bidirectional interactions between diabetes and Alzheimer’s disease. Neurochemistry International, 108, 296–302. [DOI] [PubMed] [Google Scholar]
- Simmnacher K, Lanfer J, Rizo T, Kaindl J, & Winner B (2019). Modeling cell-cell interactions in Parkinson’s disease using human stem cell-based models. Frontiers in Cellular Neuroscience, 13, 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Zhi L, & Zhang H (2019). LRRK2 and mitochondria: Recent advances and current views. Brain Research, 1702, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer MJ, & Bonifati V (2013). The genetics of Parkinson’s disease:Progress and therapeutic implications. Movement Disorders, 28(1), 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnen JA, Larson EB, Crane PK, Haneuse S, Li G, Schellenberg GD, et al. (2007). Pathological correlates of dementia in a longitudinal, population-based sample of aging. Annals of Neurology, 62(4), 406–413. [DOI] [PubMed] [Google Scholar]
- Soto I, Graham LC, Richter HJ, Simeone SN, Radell JE, Grabowska W, et al. (2015). APOE stabilization by exercise prevents aging neurovascular dysfunction and complement induction. PLoS Biology, 13(10), e1002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squatrito M, Brennan CW, Helmy K, Huse JT, Petrini JH, & Holland EC (2010). Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell, 18(6), 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ (2006). Microglial senescence: Does the brain’s immune system have an expiration date? Trends in Neurosciences, 29(9), 506–510. [DOI] [PubMed] [Google Scholar]
- Stunkard AJ, & Wadden TA (1992). Psychological aspects of severe obesity.The American Journal of Clinical Nutrition, 55(2 Suppl), 524S–532S. [DOI] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. (2005). Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England Journal of Medicine, 352(10), 987–996. [DOI] [PubMed] [Google Scholar]
- Suh N (2018). MicroRNA controls of cellular senescence. BMB Reports, 51(10), 493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson EC, Manning B, Zhang H, & Lawrence JB (2013). Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. The Journal of Cell Biology, 203(6), 929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey MG, & Goldberg MS (2010). Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiology of Disease, 37(3), 510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchkonia T, Cartwright M, Wise B, Pirtskhalava T, Karagiannides I, Shpilman A, et al. (2007). Increased TNFα and CCAAT/enhancer binding protein homologous protein (CHOP) with aging predispose preadipocytes to resist adipogenesis. American Journal of Physiology, 293, E1810–E1819. [DOI] [PubMed] [Google Scholar]
- Tchkonia T, & Kirkland JL (2018). Aging, cell senescence, and chronic disease: Emerging therapeutic strategies. Journal of the American Medical Association, 320(13), 1319–1320. [DOI] [PubMed] [Google Scholar]
- Tchkonia T, Morbeck DE, von Zglinicki T, van Deursen J, Lustgarten J, Scrable H, et al. (2010). Fat tissue, aging, and cellular senescence. Aging Cell, 9, 667–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchkonia T, Zhu Y, van Deursen J, Campisi J, & Kirkland JL (2013). Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. The Journal of Clinical Investigation, 123(3), 966–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolosa E, Wenning G, & Poewe W (2006). The diagnosis of Parkinson’s disease. LancetNeurology, 5(1), 75–86. [DOI] [PubMed] [Google Scholar]
- Trias E, Beilby PR, Kovacs M, Ibarburu S, Varela V, Barreto-Nunez R, et al. (2019). Emergence of microglia bearing senescence markers during paralysis progression in a rat model of inherited ALS. Frontiers in Aging Neuroscience, 11, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk KD, Persichetti E, Chiasserini D, Eusebi P, Beccari T, Calabresi P, et al. (2013). Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson’s disease. Movement Disorders, 28(6), 747–754. [DOI] [PubMed] [Google Scholar]
- Velarde MC, & Menon R (2016). Positive and negative effects of cellular senescence during female reproductive aging and pregnancy. The Journal of Endocrinology, 230(2), R59–R76. [DOI] [PubMed] [Google Scholar]
- Villaret A, Galitzky J, Decaunes P, Esteve D, Marques MA, Sengenes C, et al. (2010). Adipose tissue endothelial cells from obese human subjects: Differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes, 59, 2755–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan C, Liu J, Nie X, Zhao J, Zhou S, Duan Z, et al. (2014). 2, 3, 7,8-Tetrachlorodibenzo-P-dioxin (TCDD) induces premature senescence in human and rodent neuronal cells via ROS-dependent mechanisms. PLoS One, 9(2), e89811. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wang E (1995). Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Research, 55(11), 2284–2292. [PubMed] [Google Scholar]
- Wang L, Gutmann DH, & Roos RP (2011). Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Human Molecular Genetics, 20(2), 286–293. [DOI] [PubMed] [Google Scholar]
- Wang L, Sharma K, Grisotti G, & Roos RP (2009). The effect of mutant SOD1 dismutase activity on non-cell autonomous degeneration in familial amyotrophic lateral sclerosis. Neurobiology of Disease, 35(2), 234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winklhofer KF, & Haass C (2010). Mitochondrial dysfunction in Parkinson’s disease.Biochimica et Biophysica Acta, 1802(1), 29–44. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T (2016). Ageing, neurodegeneration and brain rejuvenation. Nature,539(7628), 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Bradley EW, Weivoda MM, Hwang SM, Pirtskhalava T, Decklever T, et al. (2016). Transplanted senescent cells induce an osteoarthritis-like condition in mice. The Journals of Gerontology Series A, Biological Sciences and Medical Sciences, 72, 780–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Li N, Xiang R, & Sun P (2014). Emerging roles of the p38 MAPK and PI3K/AKT/mTOR pathways in oncogene-induced senescence. Trends in Biochemical Sciences, 39(6), 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, White TA, et al. (2015a). Targeting senescent cells enhances adipogenesis and metabolic function in old age. eLife. 10.7554/eLife.12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. (2018). Senolytics improve physical function and increase lifespan in old age. Nature Medicine, 24, 1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, et al. (2015b). JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proceedings of the National Academy of Sciences of the United States of America, 112(46), E6301–E6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabluchanskiy A, Tarantini S, Balasubramanian P, Kiss T, Csipo T, Fulop GA, et al. (2020). Pharmacological or genetic depletion of senescent astrocytes prevents whole brain irradiation-induced impairment of neurovascular coupling responses protecting cognitive function in mice. Geroscience. (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H,Gutmann DH, et al. (2008). Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nature Neuroscience, 11(3), 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Baker DJ, Tachibana M, Liu CC, van Deursen JM, Brott TG, et al. (2016). Vascular cell senescence contributes to blood-brain barrier breakdown. Stroke, 47(4), 1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Lin PJ, & Levey A (2013). Monetary costs of dementia in the United States. TheNew England Journal of Medicine, 369(5), 489. [DOI] [PubMed] [Google Scholar]
- Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, et al. (2018). Fisetin is a senotherapeutic that extends health and lifespan. eBioMedicine, 36, 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S, et al. (2019). Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nature Neuroscience, 22, 719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Armstrong JL, Tchkonia T, & Kirkland JL (2014). Cellular senescence and the senescent secretory phenotype in age-related chronic diseases. Current Opinion in Clinical Nutrition and Metabolic Care, 17(4), 324–328. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, et al. (2017). New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging, 9, 955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, et al. (2016). Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell, 15(3), 428–435. 10.1111/acel.124452016, (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Tchkonia T, Pirtskhalava T, Gower A, Ding H, Giorgadze N, et al. (2015). The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell, 14, 644–658. [DOI] [PMC free article] [PubMed] [Google Scholar]