

Abstract
A novel coronavirus (SARS-CoV-2) identified in Wuhan state of China in 2019 is the causative agent of deadly disease COVID-19. It has spread across the globe (more than 210 countries) within a short period. Coronaviruses pose serious health threats to both humans and animals. A recent publication reported an experimental 3D complex structure of the S protein of SARS-CoV-2 showed that the ectodomain of the SARS-CoV-2 S protein binds to the peptidase domain (PD) of human ACE2 with a dissociation constant (Kd) of ~ 15 nM. In this study, we focused on inhibitors for ACE2: S protein complex using virtual screening and inhibition studies through molecular docking for over 200,000 natural compounds. Toxicity analysis was also performed for the best hits, and the final complex structures for four complexes were subjected to 400 ns molecular dynamics simulations for stability testing. We found two natural origin inhibitors for the S protein: human ACE2 complex (Andrographolide and Pterostilbene) which displayed better inhibition potential for ACE2 receptor and its binding with the S protein of SARS-CoV-2. Comparative studies were also performed to test and verify that these two drug candidates are also better than hydroxychloroquine which is known to inhibit this complex. However, we needed better potential drug candidates to overcome the side effects of hydroxychloroquine. Supplementary experimental studies need to be carried forward to corroborate the viability of these two new inhibitors for ACE2: S protein complex so as to curb down COVID-19.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00894-020-04599-8.
Keywords: COVID-19, SARS-CoV-2, Molecular dynamics, Andrographolide, S protein: human ACE2 complex
Introduction
A novel coronavirus SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) was subsequently detected in the Wuhan state of China in the last quarter of 2019 as the causative pathogen for the COVID-19 (coronavirus disease 2019), a lethal respiratory tract infection. Genetically this virus closely resembles the SARS virus. It has spread across the globe (more than 210 countries) within a short period. Coronaviruses pose serious health threats to both humans and animals. The mortality rate for 2019-nCoV (novel coronavirus) is not as high (approximately 2–3%), in comparison to SARS-CoV (severe acute respiratory syndrome coronavirus) having fatality rate of ∼ 10% and MERS-CoV (Middle East respiratory syndrome coronavirus) having fatality rate of ∼ 36%, but its rapid propagation has resulted in the activation of protocols to stop its spread [1]. The genomic RNA of coronaviruses is the largest among RNA viruses, approximately 27 to 30 kb [2]. The genome of 2019-nCoV is reported to have a 79.6% sequence identity to SARS-CoV [3]. Phylogenetic analysis have revealed that SARS-CoV-2 comes under genus Beta coronavirus and falls under the subgenus of Sarbecovirus, a relatively close family member to SARS like coronaviruses which have been derived from bats. These viruses have been identified as bat-SL-CoVZXC21 and bat-SL-CoVZC45 having 96% sequence similarity.
The homology modeling analyses have revealed that SARS-CoV-2 and SARS-CoV have a similar receptor-binding domain structure, despite amino acid variation at some key residues [4]. The S protein (spike protein) of SARS-CoV-2 also called 2019-nCov may also interact with human ACE2 as like SARS-CoV for host infection [5, 6]. Trimer of the S protein is known to be cleaved making them into S1 and S2 units of which the S1 subunit is released to the post-fusion conformation in this transition during viral infection [7–10]. S1 subunit is known to contain RBD (receptor-binding domain), known to directly bind the PD (peptidase domain) of human ACE2 (angiotensin-converting enzyme 2), also known as type 1 transmembrane metallocarboxypeptidase in homology to ACE receptor [1, 11]. On the other hand, S2 subunit is studied to be responsible for the fusion of membranes. When the former S1 subunit interacts with the host receptor ACE2, an additional site for cleavage is exposed on the S2 subunit to be cleaved by host proteases, known to be critical for viral infection [7, 12, 13]. This deadly disease has resulted in a lot of publications recently of which one reported the 3D structure of the S protein of SARS-CoV-2 displayed that the ectodomain of the SARS-CoV-2 binds to the PD (peptidase domain) of human ACE2 with a strong association displaying a Kd (dissociation constant) of 15 nM [14]. ACE2 expression is detected on the type I and type II alveolar epithelium, upper respiratory system, heart, kidney tubular epithelium, pancreas, endothelial cells, and enterocytes. Thus, the external spike protein determines the infectious nature and host specificity of 2019-nCoV that produce new spikes and glycoproteins that are assembled into numerous copies of the virus. After replication of the genetic material, the Golgi bodies exocytose the viruses. As an immune response, CD4+ T helper cells develop immunity against 2019-nCoV by producing IFN-γ and IL-17. 2019-nCoV also targets these circulating immune cells and induces apoptosis of CD3, CD8, and CD4 cells, causing lymphocytopenia, overproduction of cytokines, and failure of multiple organs [15].
Angiotensin 2 is known to be a major substrate for human ACE2. It is well-studied that the human ACE2 is known to degrade angiotensin 2, in order to generate angiotensin’s 1–7 which is negatively known to regulate RAS pathway. Different organs including the cardiovascular system are found to be affected by the human ACE2 receptor as a protective function (https://www.rndsystems.com/resources/articles/ace-2-sars-receptor-identified). The virus upon entry into human being is known to perform its first step in the form of the trimeric viral spike protein interacting with the human ACE2 (angiotensin-converting enzyme 2) receptor. Human ACE2 is known to be in a dimeric form; therefore, researchers have suggested the possibility of two trimeric viral spike proteins binding to a human ACE2 dimer, identifying it as 1:1 interaction [16]. The structural biology approach with experimental 3D structures of the complex provides a strong basis for drug development and therapeutics approach to target this deadly interaction [16]. The structural analysis, therefore, has been important to provide insights into the mechanism of action for virus recognition leading to infection [16]. To date, there is no specific treatment available to treat COVID-19 infection [17, 18]. This study is a vital step to find novel compounds which can act as remedy for COVID-19.
Plants produce certain natural phenolic compounds known as stilbenes. These compounds are phytoalexins because they are synthesized in response to UV radiation and other abiotic stresses [19, 20]. The stilbene compounds have backbone similar to each other with varying functional groups on the rings. Stilbene compounds are widely studied because of their varied biological roles in individuals [21]. The reason for research is these compounds are acting as anti-inflammatory [22], antitumoral [23], antiviral [24], antioxidative [25], and life-span extension [26]. Many other compounds being structurally related to resveratrol also carry biological activities. Piceatannol is a metabolite of resveratrol frequently found in berries, grapes, white tea, and rhubarb [27]. These compounds have also been found to have antioxidant and anticancer activities [27–29]. Additional dimethyl ether analog of resveratrol is Pterostilbene. Pterostilbene has numerous pharmacological resemblances with other stilbenes including antidiabetic [30], antiaging [31], and anti-inflammatory [32] effects. These compounds are known to develop effects on many vital cellular processes such as apoptosis [33] cell proliferation and antioxidant activity [34, 35].
Material and methods
Protein retrieval and preparation
The published structure of the 2019-nCoV RBD/ACE2-B0AT1 complex (PDB-ID: 6M17) was used for virtual screening. As a starting protein structure, we used a 10-μs simulated structure of the human ACE2 ectodomain in a complex state with the receptor-binding domain of a spike protein from SARS-CoV-2 (PDB 6M17) with frames saved every 1.2 ns by DE Shaw Research [36] for our study. The reason behind using an already simulated structure for 10 μs would help us to achieve the lowest energy conformation of the protein. The simulation also helped us know the important junctions of interaction between the S protein: human ACE2 complex.
Virtual screening
Initial virtual screening through blind docking of 203,458 compounds from natural ZINC library [37] was performed using PyRx [38] with default settings. The 4 best scoring compounds with lowest energy of binding or binding affinity were extracted and aligned with receptor structure for further analysis.
Ligand preparation
We then prepared the best hit ligands by downloading the 3D structure of the ligands. Hydroxychloroquine as said to be a potential inhibitor against SARS-CoV-2 is known to subject heavy side effects such as cough or hoarseness to difficulty breathing reaching loss of hearing. Therefore, need of safe natural ligands with minute or no side effects is of high importance. We used hydroxychloroquine as a positive control for comparative studies. The ligands were energy minimized using Avogadro 4.0 [39].
Toxicity prediction
We then used an online webserver ProTox-II [40] for calculating and predicting toxicity reports which would suggest if the natural ligands used in this research do not have any toxic effects on the body. These studies were also important to compare with hydroxychloroquine as we are in search of a better alternative. ProTox-II identified drug toxicity by separating the input molecules into 6 classes for toxicity (from 1 to 6). Class 1 has LD50 ≤ 5 which is fatal in nature; on the other hand, class 6 shows LD50 > 5000 which means the compound is non-toxic.
Inhibition studies through molecular docking
Molecular docking was carried out using a well-known software AutoDock 4.0 suite [41]. The inhibition mechanism for all the suggested drugs was performed with pre-defined protocols [42]. Flexible docking approach was followed to achieve best results. Steps for generating PDBQT files for the S protein: human ACE2 complex and drug candidates, preparation, and creation of the grid box were completed using GUI version of the same program AutoDock Tools 4.0 [41, 43–45]. ADT successfully assigned hydrogens (polar), Kollman charges for united atoms, parameters for solvation, and fragmental volumes. ADT then saved the prepared files in the readable PDBQT format. The grid map was then prepared using a grid box in AutoGrid. The size for the grid box was set to 62 × 70 × 88 xyz points with a spacing in the grid at 0.375 Å. The centers for the grid were designated at X = 190.404, Y = 101.754, and Z = − 0.753 dimensions.
In addition, the exhaustiveness of 100 was used to get the best output which takes more computational power and time for the analyses. The LGA algorithm (Lamarckian Genetic) was picked to search for the conformers which fit best. Ten different conformers were considered for each drug molecule during the docking experiment. Random seed was generated with a population size of 150. Energy evaluation was set to a maximum of 2,500,000, and generations were set to a maximum of 27,000. The mutation rate was set to 0.02, while the automatically survived top individuals were set to 1 with a crossover rate of 0.8. Rest of all docking parameters was set to be default with 10 LGA runs.
Most favored binding was detected by calculating the least binding energy in the form of kcal/mol, and at the same time, the results were to be less than 1.0 Å in RMSD in position. Further structural analyses were done by extraction of the best results and aligned to the reference structure. Ligand interaction diagram was analyzed using PoseView of Protein Plus server [46, 47], and figures were prepared using LigPlot module in Accelrys Discovery Studio 4.0 (http://www.accelrys.com).
Molecular dynamics simulations
System setup
The complex structures for S protein and human ACE2 with the inhibited drugs were prepared for molecular dynamics simulations. Four hundred nanosecond accumulative simulations were performed with the CHARMM36 force field [48]. We prepared the solvated systems using TCL scripts in VMD [49] and performed MD simulations using NAMD [50, 51]. The system consisted of the protein complex, TIP3P water, counter ions Na+/Cl-, and 150 mM NaCl.
Simulation setup
The system setup was then subjected to energy minimization which lasted for around 3200 steps following 1000-ps equilibration. The molecular dynamics production run was set up for 100 ns each, totally to accumulative 400 ns MD production procedure. NPT ensemble was used (1 bar) with a time step of 2 fs. The temperature was set up at 300 K with low damping coefficient, while pressure was controlled using Nose-Hoover Langevin piston. Electrostatics were calculated using particle mesh Ewald (PME). A total cut-off at 12 Å was given for short range and van der Walls electrostatics. All simulations were replicated thrice with initialized random seed to get average.
Data analyses
Data analysis for the produced trajectories was performed using TCL-scripts previously implemented in VMD [49] and data were plotted using gnuplot (http://gnuplot.info). We have also calculated RMSF α alignments for carbons for all residues and structural changes by RMSD throughout the simulation run. Calculation between the hydrogen donor and acceptor was set with a cut-off at 3.6 Å, which included the backbone as well as side-chain. Other analysis such as radius of gyration (ROG), solvent accessible surface area (SASA), secondary structure content (DSSP), and H-bond formations upon ligand binding were calculated using TCL bash scripts. RMSD, RMSF, total energy, SASA, radius of gyration, and H-bonds were plotted using prism.
Analysis for binding free energy (MMPBSA) from MD simulations
MMPBSA.py module was used to calculate the free energy and interaction energy of the ligand. The mathematical formula used to calculate the energies was:
The solvation energy for all the states was calculated using Generalized Born (GB) and Poisson Boltzman (OB). This analysis revealed the electrostatic contribution of the solvation state. The final data was plotted using prism.
Results and discussion
Protein selection and preparation
SARS-CoV-2 when gets into the human body and tries to find the host cell, the receptor-binding domain (RBD) of the CoV-2 spike protein binds to the human ACE2 receptor. This is the first point of contact between the human body cells and SARS-CoV-2. Therefore, the protein structure used in the study is the complex between the RBD of SARS-CoV-2 and the human ACE2. The protein used for this study is an experimentally solved structure of ACE2 and S protein complex (PDB-ID: 6M17), which was previously subjected to a 10-μs molecular dynamics simulation to have the most stable structure in the least energy conformation.
Virtual screening and ligand selection
Natural ligands were acquired using the ZINC natural library with a total of ~ 203,458 drug molecules. These molecules were tested through blind docking against the S protein: human ACE2 complex to shortlist best candidates. Primary virtual screening gave optimum hits for 20 compounds mentioned in Supplementary Table 2. Final 4 drugs—Andrographolide, Artemisinin, Pterostilbene, and Resveratrol—were then selected on the basis of multiple criteria such as binding score, hydrophobic, electrostatic, and pi-pi cationic interactions with the protein. Therefore, we continued further studies using the mentioned drug candidates. People have reported that hydroxychloroquine abolishes this interaction and binds between the interface of these two proteins [52]. Focus of the study is to find alternative drugs which can inhibit this particular region and at the same time have lesser side effects than hydroxychloroquine. Hydroxychloroquine was used as a positive control to confirm and compare the interaction. The final list of ligands tested thoroughly is mentioned with PubChem ID and 2D structures (Fig. 1).
Fig. 1.

Shortlisted drugs from natural library of over 200,000 compounds with their respective 2D structures and PubChem identification numbers
Toxicity prediction
Finalized inhibitors were then tested for compare their toxic effects using online tool ProTox-II. The results for toxicity prediction suggested that these shortlisted natural ligands used in this research are identified as less toxic than previously used hydroxychloroquine. The toxicity values suggest that Andrographolide was being put in the class 5 with Artemisinin categorizing them as the least toxic, while Pterostilbene, Resveratrol, and hydroxychloroquine were categorized in class 4 (Table 1). Toxicity radar charts for all the ligands explaining toxicity effects are shown in Supplementary Fig. 1.
Table 1.
Toxicity prediction results for the selected compounds as calculated using online tool ProTox-II [40]
| Natural drug Candidates |
Predicted LD50 (mg/kg) | Predicted toxicity class | Average similarity (%) | Prediction accuracy (%) | Toxicity |
|---|---|---|---|---|---|
| Andrographolide | 5000 | 5 | 68.61 | 68.07 | Immunotoxicity |
| Artemisinin | 4228 | 5 | 100 | 100 | Immunotoxicity |
| Pterostilbene | 1560 | 4 | 80.11 | 70.97 | Immunotoxicity, estrogen receptor alpha |
| Resveratrol | 1560 | 4 | 69.97 | 68.07 | Androgen receptor, estrogen receptor alpha, estrogen receptor ligand binding domain, mitochondrial membrane potential, ATPase family AAA domain-containing protein 5 |
| Hydroxychloroquine | 1240 | 4 | 100 | 100 | Immunotoxicity, mutagenicity |
Molecular docking results
Molecular docking studies were performed using flexible docking module of AutoDock 4 further strengthened this research to find an alternate natural inhibitor for S protein: human ACE2 complex. The molecular docking was performed using AutoDock 4 using default settings.
Final inhibition scores in the form of binding energies and major interacting residues for all the drug candidates are mentioned in Table 2. With the binding energy of − 9.1 kcal/mol, Andrographolide shows the best binding with the receptor.
Table 2.
Binding energy of protein-ligand complex obtained after performing molecular docking using AutoDock 4.2 [41]
| Drug | Binding energy (Kcal/mol) |
Interacting residues |
|---|---|---|
| Andrographolide | − 9.1 | Asp-30, Asn-33, His-34, Pro-389, Arg-393, Tyr-505 |
| Artemisinin | − 6.2 | His-34, Ala-387, Pro-389, Tyr-505 |
| Pterostilbene | − 8.9 | His-34, Ser-494, Gly-496 |
| Resveratrol | − 8.7 | Gly-496 |
| Hydroxychloroquine | − 7.1 | Glu-37, Arg-393, Gln-388, Pro-389, Gly-496, Tyr-505, His-34, Tyr-495, Lys-403, Ser-494 |
Further structural analyses were carried out using PyMOL (www.pymol.org) and PoseView module of Protein Plus. We found that the best inhibitor for binding Andrographolide fits perfectly between the interface of S protein: human ACE2 complex. The binding of Andrographolide with the protein complex showed interactions with residues Asn-33, Arg-393, and Tyr-505 in the form of H-bonds with the drug candidate. His-34 and Pro-389 formed alkyl and pi-alkyl interactions (Fig. 2).
Fig. 2.

Docking pose (left) for Andrographolide docked with ACE2: S protein complex in the interface between both proteins. (Right) Ligand interaction diagram showing important interactions involved in the complex
Structural analyses for the second drug candidate—Artemisinin—also showed binding between the interface of S protein: human ACE2 complex. However, the docking score was lower than what we achieved for other candidates. Artemisinin showed the formation 1 H-bond with Tyr-505 residue of the ACE2 receptor. His-34 and Ala-387 again formed alkyl and pi-alkyl contacts with the receptor. Pro-389 forms a carbon H-bond. The docking pose and ligand interaction diagram for Artemisinin inhibiting the protein complex is shown in Supplementary Fig. 2.
We then moved on to analyze the third drug candidate “Pterostilbene,” which was the candidate with second best drug inhibition score in terms of binding energy (− 8.9 kcal/mol). Structural analyses showed Pterostilbene forming a pi-pi stack with His-34 of ACE2 along-with two H-bonds (Gly-496 and Ser-494) (Fig. 3).
Fig. 3.

Docking pose (left) for Pterostilbene docked with human ACE2: S protein complex in the interface between both proteins. (Right) Ligand interaction diagram showing important interactions involved in the complex
Resveratrol, which is from the same Stilbene family as Pterostilbene, also showed similar interaction as Pterostilbene with a docking score of − 8.7 kcal/mol. However, when the structural analyses of the complex were performed, it was surprising that Resveratrol only showed one H-bond with Gly-496 in this interaction (Fig. 4).
Fig. 4.

Docking pose (left) for Resveratrol docked with human ACE2: S protein complex in the interface between both proteins. (Right) Ligand interaction diagram showing important interactions involved in the complex
Hydroxychloroquine which is a known inhibitor and was used as a positive control to compare and confirm this interaction also showed expected binding at the same interface as other ligands. Structural analyses of the known inhibitor showed wide interactions with the ACE2 receptor including one H-bond with Gly-496, Tyr-505, Tyr-495, and Lys-403 as alkyl and pi-alkyl contacts; Gln-388 is contacted as the amide-pi stacked residue. One pi-cation interaction was also observed with Arg-393 (Supplementary Fig. 3).
Molecular dynamics simulations
To confirm the stability of the complex structures in combination with the drug candidates, we performed an accumulative 400-ns molecular dynamics simulation on all the 4 complexes. All the simulations are performed in triplicates for more concrete data analysis. This production run was post 1 ns equilibration using NAMD. We found that the RMSD fluctuations between structures are not too high which explains why the structures with complexed ligand are very stable. Overall trajectory analyses for all the compounds are more or less equilibrated with an average change of approx. 2 Å in the RMSD (Fig. 5). The most deviation observed (2.80 Å) as average RMSD change for around steps 40,000 and 45,000 ps for Artemisinin (shown in green) (Fig. 5). Artemisinin also had the least docking score, and this deviation may be because of the hydrophobic interactions of the cyclic groups with the receptor residues. Trajectories for Andrographolide (in red) and Pterostilbene (shown in violet) after 36,000 ps show equilibration, suggesting stable binding with the ACE2 receptor macromolecule (Fig. 5).
Fig. 5.

RMSD analysis (for backbone and C-alpha) for the production run of Andrographolide (red), Pterostilbene (violet), Resveratrol (blue), and Artemisinin (green) inhibiting the S protein of SARS-CoV-2 in complex with human ACE2
Similarly, RMSF plot for the trajectories shows approx. same per-residue fluctuation in the case of Andrographolide as of Artemisinin (Fig. 6). Pterostilbene and Resveratrol showed slight fluctuations. Artemisinin as expected from the RMSD plot showed more local residue-based fluctuation. Residue number 240 to 260 shows the highest fluctuation in all the 4 cases. This cluster of residue could be the functional site of the ligand binding phenomenon. Table 3 recorded the average values for all three individual simulation runs. Average RMSD (for backbone and c-alpha), RMSF, and number of H-bonds formed are recorded. Supplementary Fig. 4 demonstrated the hydrogen-bonding pattern observed during 100 ns simulation in all 4 protein-ligand complexes. Approximately near 50 ns, no bonds were observed in any complex. Average numbers of H-bonds formed in case of Andrographolide, Pterostilbene, Resveratrol, and Artemisinin are 3, 3, 2, and 2, respectively (Table 3).
Fig. 6.
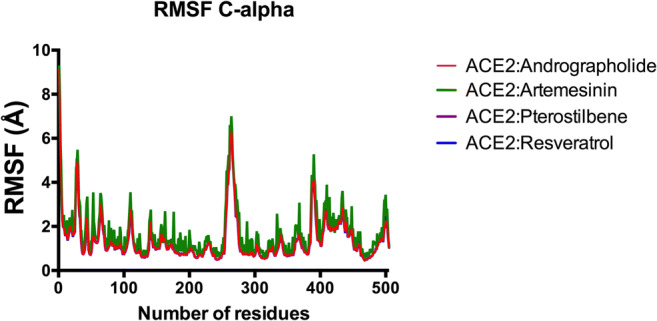
RMSF graph (for alpha carbon) for all 4 protein-ligand complexes during 100-ns simulation run. RMSF for Pterostilbene and Resveratrol are exactly overlapped by Artemisinin
Table 3.
The average RMSD for backbone and C-alpha trace, RMSF, and average H-bonds formed between protein and compound across simulations for all the complexes over 3 replicates of 100-ns molecular dynamics simulations
| Complex | RMSD Backbone (Å) | RMSD C-alpha (Å) | RMSF (Å) | Average H-bonds formed |
|---|---|---|---|---|
| ACE2:Andrographolide | 1.95 ± 0.19 | 1.99 ± 0.21 | 1.39 ± 0.11 | 2.97 ± 0.15 |
| ACE2:Artemesinin | 2.80 ± 0.31 | 2.89 ± 0.34 | 1.72 ± 0.20 | 1.62 ± 0.48 |
| ACE2:Pterostilbene | 1.96 ± 0.18 | 2.04 ± 0.14 | 1.41 ± 0.15 | 2.93 ± 0.14 |
| ACE2:Resveratrol | 2.45 ± 0.24 | 2.49 ± 0.28 | 1.53 ± 0.19 | 2.24 ± 0.31 |
These results suggest that Andrographolide and Pterostilbene can be good inhibitors for the S protein: human ACE2 complex interface which will inhibit the binding of S protein of SARS-CoV-2 to the ACE2 receptor without showing any side effect.
Apart from RMSD, RMSF, and number of hydrogen bonds formed between protein and ligand, radius of gyration is also calculated. Supplementary Fig. 5 depicted the radius of gyration plots for all the 4 complexes over 100 ns of simulation time. As we can observe that Rg is decreasing in all cases over the time, it suggests that binding of ligands helps in the stabilization and compactness of the protein. The radius of gyration (Rg) of a particle is the root-mean-square distance of all electrons from their center of gravity. It is an important parameter and is often useful as an indicator for structural changes of a substance. Changes studied through the use of the radius of gyration are, for instance, association and dissociation effects, conformational changes by denaturation, binding of coenzymes, and temperature effects (O. Kratky, P. Laggner, in Encyclopedia of Physical Science and Technology (Third Edition), 2003).
Solvent accessible surface area for all the proteins was also calculated to check the effect of ligand binding on the residue profiling of the surface of the protein. Supplementary Fig. 6 shows solvent accessible surface area (SASA) plot for all 4 complexes as obtained using gmx sas command in GROMACS [53, 54] for 100-ns simulation run. Pterostilbene and Resveratrol plots are exactly overlapped by Artemisinin. This suggests that there is no major change in the structure of the protein on binding with the different ligand. The total energy of the complexes and individual energy components are depicted in Supplementary Figs. 7 and main text Fig. 7. Individual energy components like van der Waals forces, coulomb, and H-bond are calculated using MM-PBSA/MM-GBSA tool in GROMACS. A table (Table 4) representing these values is also included in the text. The complex of ACE2 and Andrographolide shows the highest Gibbs free energy (− 48.164 kJ/mol). It suggests that Andrographolide is the best lead molecule which shows good interaction with ACE2 receptor exhibiting it as the potential target for human ACE2 binding protein. Also, Supplementary Fig. 8 depicted the secondary structure change plot for all 4 complexes as obtained using do_dssp command in GROMACS [53, 54] for 100-ns simulation run. Pterostilbene and Resveratrol plots are exactly overlapped by Artemisinin again referring Andrographolide and Pterostilbene as best leads for further drug development process.
Fig. 7.
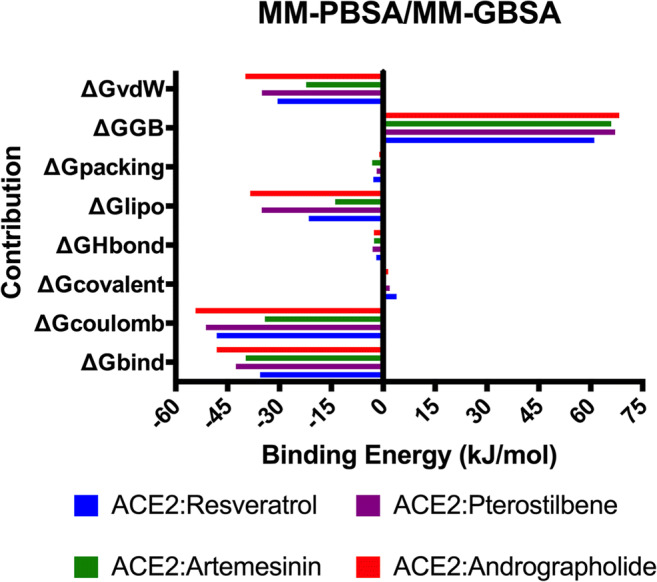
MM-PBSA/MM-GBSA graph of all 4 complexes providing the distribution of energy components during the course of simulation
Table 4.
MMPBSA/MMGBSA analysis performed using the script MMPBSA.py module showing different energy contributions during the 100-ns molecular dynamics simulation for each of the four complexes
| Contribution | ACE2 Andrographolide |
ACE2 Artemisinin |
ACE2 Pterostilbene |
ACE2 Resveratrol |
|---|---|---|---|---|
| ∆Gbind | − 48.164 ± 3.56 | − 39.776 | − 42.625 | − 35.619 |
| ∆Gcoulomb | − 54.2546 ± 4.18 | − 34.216 | − 51.247 | − 48.154 |
| ∆Gcovalent | 1.456 | 0.472 | 1.895 | 3.843 |
| ∆GHbond | − 2.708 | − 2.621 | − 3.085 | − 2.011 |
| ∆Glipo | − 38.462 | − 13.884 | − 35.125 | − 21.493 |
| ∆Gpacking | − 1.135 | − 3.173 | − 1.875 | − 2.856 |
| ∆GGB | 68.272 | 65.941 | 67.054 | 61.048 |
| ∆GvdW | − 39.881 | − 22.294 | − 35.051 | − 30.548 |
Conclusions
Initial molecular dynamics, primary screening, molecular docking, and post-complex molecular dynamics simulations for 100 ns each (in triplicates) in this research suggested that the interaction between the S protein: human ACE2 complex is very important. The interactions are strongly on the helices of the human ACE 2 protein, which are important in the interaction with the receptor-binding domain of the S protein of SARS-CoV-2. This was shown in the initial 10-μs simulation by DE Shaw Research [36]. This interface interaction also explains why it is important to abolish this interaction. The most important residue which we see from all the ligand interaction diagrams is His-34 of human ACE2 receptor which lies on the surface and hence a very important in terms of interaction with the S protein. We compare and show that our positive control as well all suggested drug candidates have shown interaction with His-34 with utilizing non-polar binding. This interaction will be an important factor in abolishing the connection between the S-protein and human ACE2, further stopping the spread by this first point of contact. Andrographolide and Pterostilbene have shown promising binding and stability results by molecular dynamics indicating their usefulness in the form of inhibiting this important complex. Experimental in vitro studies are suggested with the use of Andrographolide and Pterostilbene for further analysis and corroboration.
Supplementary information
(DOCX 2309 kb)
Data availability
Not applicable.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Code availability
Not applicable
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science (80- ) 2005;309(5742):1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 2.Spaan W, Cavanagh D, Horzinek MC. Coronaviruses: structure and genome expression. J Gen Virol. 1988;69(Pt 12):2939–2952. doi: 10.1099/0022-1317-69-12-2939. [DOI] [PubMed] [Google Scholar]
- 3.Li F. Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol. 2016;3(1):237–261. doi: 10.1146/annurev-virology-110615-042301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11(8):875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belouzard S, Chu VC, Whittaker GR. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci U S A. 2009;106(14):5871–5876. doi: 10.1073/pnas.0809524106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simmons G, Reeves JD, Rennekamp AJ, Amberg SM, Piefer AJ, Bates P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc Natl Acad Sci U S A. 2004;101(12):4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simmons G, Zmora P, Gierer S, Heurich A, Pöhlmann S. Proteolytic activation of the SARS-coronavirus spike protein: cutting enzymes at the cutting edge of antiviral research. Antivir Res. 2013;100(3):605–614. doi: 10.1016/j.antiviral.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song W, Gui M, Wang X, Xiang Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018;14(8):e1007236. doi: 10.1371/journal.ppat.1007236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jarcho JA, Ingelfinger JR, Hamel MB, D’Agostino RB, Harrington DP. Inhibitors of the renin-angiotensin-aldosterone system and Covid-19. N Engl J Med. 2020;382(25):2462–2464. doi: 10.1056/NEJMe2012924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Millet JK, Whittaker GR. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015;202:120–134. doi: 10.1016/j.virusres.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci U S A. 2005;102(33):11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science (80- ) 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dariya B, Nagaraju GP. Understanding novel COVID-19: its impact on organ failure and risk assessment for diabetic and cancer patients. Cytokine Growth Factor Rev. 2020;53:43–52. doi: 10.1016/j.cytogfr.2020.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science (80- ) 2020;367(6485):1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pawar AY. Combating devastating COVID −19 by drug repurposing. Int J Antimicrob Agents. 2020;17:105984. doi: 10.1016/j.ijantimicag.2020.105984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rane JS, Chatterjee A, Kumar A, Ray S (2020) Targeting SARS-CoV-2 spike protein of COVID-19 with naturally occurring phytochemicals: an in silico study for drug development [DOI] [PMC free article] [PubMed]
- 19.Langcake P, Cornford CA, Pryce RJ. Identification of pterostilbene as a phytoalexin from Vitis vinifera leaves. Phytochemistry. 1979;18(6):1025–1027. [Google Scholar]
- 20.Langcake P, Pryce RJ. The production of resveratrol by Vitis vinifera and other members of the Vitaceae as a response to infection or injury. Physiol Plant Pathol. 1976;9(1):77–86. [Google Scholar]
- 21.Weiskirchen S, Weiskirchen R. Resveratrol: how much wine do you have to drink to stay healthy? Adv Nutr. 2016;7(4):706–718. doi: 10.3945/an.115.011627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tili E, Michaille J-J, Adair B, Alder H, Limagne E, Taccioli C, et al. Resveratrol decreases the levels of miR-155 by upregulating miR-663, a microRNA targeting JunB and JunD. Carcinogenesis. 2010;31(9):1561–1566. doi: 10.1093/carcin/bgq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khan OS, Bhat AA, Krishnankutty R, Mohammad RM, Uddin S. Therapeutic potential of resveratrol in lymphoid malignancies. Nutr Cancer. 2016;68(3):365–373. doi: 10.1080/01635581.2016.1152386. [DOI] [PubMed] [Google Scholar]
- 24.Yiu C-Y, Chen S-Y, Chang L-K, Chiu Y-F, Lin T-P. Inhibitory effects of resveratrol on the Epstein-Barr virus lytic cycle. Molecules. 2010;15(10):7115–7124. doi: 10.3390/molecules15107115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gülçin İ. Antioxidant properties of resveratrol: a structure–activity insight. Innovative Food Sci Emerg Technol. 2010;11(1):210–218. [Google Scholar]
- 26.Khan M, Park S, Kim H-J, Lee K-J, Kim D-H, Baek S-H, et al (2019) The resveratrol rice DJ526 callus significantly increases the lifespan of drosophila (resveratrol rice DJ526 callus for longevity). Nutrients 11(5) [DOI] [PMC free article] [PubMed]
- 27.Seyed MA, Jantan I, Bukhari SNA, Vijayaraghavan K. A comprehensive review on the chemotherapeutic potential of piceatannol for cancer treatment, with mechanistic insights. J Agric Food Chem. 2016;64(4):725–737. doi: 10.1021/acs.jafc.5b05993. [DOI] [PubMed] [Google Scholar]
- 28.Piotrowska H, Kucinska M, Murias M. Biological activity of piceatannol: leaving the shadow of resveratrol. Mutat Res. 2012;750(1):60–82. doi: 10.1016/j.mrrev.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 29.Murias M, Jäger W, Handler N, Erker T, Horvath Z, Szekeres T, et al. Antioxidant, prooxidant and cytotoxic activity of hydroxylated resveratrol analogues: structure-activity relationship. Biochem Pharmacol. 2005;69(6):903–912. doi: 10.1016/j.bcp.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 30.Bhakkiyalakshmi E, Sireesh D, Sakthivadivel M, Sivasubramanian S, Gunasekaran P, Ramkumar KM. Anti-hyperlipidemic and anti-peroxidative role of pterostilbene via Nrf2 signaling in experimental diabetes. Eur J Pharmacol. 2016;777:9–16. doi: 10.1016/j.ejphar.2016.02.054. [DOI] [PubMed] [Google Scholar]
- 31.Kasiotis KM, Pratsinis H, Kletsas D, Haroutounian SA. Resveratrol and related stilbenes: their anti-aging and anti-angiogenic properties. Food Chem Toxicol. 2013;61:112–120. doi: 10.1016/j.fct.2013.03.038. [DOI] [PubMed] [Google Scholar]
- 32.Paul S, Rimando AM, Lee HJ, Ji Y, Reddy BS, Suh N. Anti-inflammatory action of pterostilbene is mediated through the p38 mitogen-activated protein kinase pathway in colon cancer cells. Cancer Prev Res (Phila) 2009;2(7):650–657. doi: 10.1158/1940-6207.CAPR-08-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwon O, Seo Y, Park H. Pinosylvin exacerbates LPS-induced apoptosis via ALOX 15 upregulation in leukocytes. BMB Rep. 2018;51(6):302–307. doi: 10.5483/BMBRep.2018.51.6.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jeong E, Lee H-R, Pyee J, Park H. Pinosylvin induces cell survival, migration and anti-adhesiveness of endothelial cells via nitric oxide production. Phytother Res. 2013;27(4):610–617. doi: 10.1002/ptr.4770. [DOI] [PubMed] [Google Scholar]
- 35.Koskela A, Reinisalo M, Hyttinen JMT, Kaarniranta K, Karjalainen RO. Pinosylvin-mediated protection against oxidative stress in human retinal pigment epithelial cells. Mol Vis. 2014;20:760–769. [PMC free article] [PubMed] [Google Scholar]
- 36.Shaw DE (2020) Molecular dynamics simulations related to SARS-CoV-2. Molecular dynamics simulations related to SARS-CoV-2. [cited 2020 Aug 23]. Available from: https://www.deshawresearch.com/downloads/download_trajectory_sarscov2.cgi/
- 37.Sterling T, Irwin JJ. ZINC 15--ligand discovery for everyone. J Chem Inf Model. 2015;55(11):2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Biol. 2015;1263:243–250. doi: 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- 39.Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. Aust J Chem. 2012;4(1):17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46(W1):W257–W263. doi: 10.1093/nar/gky318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bikadi Z, Hazai E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J Cheminform. 2009;1:15. doi: 10.1186/1758-2946-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alazmi M (2019) Molecular modeling and docking of aquaporin inhibitors to reveal new insights into schistosomiasis treatment. Curr Comput Aided Drug Des [DOI] [PubMed]
- 43.Illingworth CJR, Morris GM, Parkes KEB, Snell CR, Reynolds CA. Assessing the role of polarization in docking. J Phys Chem A. 2008;112(47):12157–12163. doi: 10.1021/jp710169m. [DOI] [PubMed] [Google Scholar]
- 44.Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. J Comput Chem. 2007;28(6):1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 45.Danish S. A simple click by click protocol to perform docking: AutoDock 4.2 made easy [PMC free article] [PubMed]
- 46.Stierand K, Rarey M. From modeling to medicinal chemistry: automatic generation of two-dimensional complex diagrams. ChemMedChem. 2007;2(6):853–860. doi: 10.1002/cmdc.200700010. [DOI] [PubMed] [Google Scholar]
- 47.Stierand K, Rarey M. Drawing the PDB: protein-ligand complexes in two dimensions. ACS Med Chem Lett. 2010;1(9):540–545. doi: 10.1021/ml100164p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang J, MacKerell AD. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem. 2013;34(25):2135–2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):27. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 50.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Acun B, Hardy DJ, Kale LV, Li K, Phillips JC, Stone JE. Scalable molecular dynamics with NAMD on the summit system. IBM J Res Dev. 2018;62(6):1–9. doi: 10.1147/jrd.2018.2888986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu J, Cao R, Xu M, Wang X, Zhang H, Hu H, et al. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020;6:16. doi: 10.1038/s41421-020-0156-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;91(1–3):43–56. [Google Scholar]
- 54.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 2309 kb)
Data Availability Statement
Not applicable.