

Abstract
Global histone modification patterns correlate with tumor phenotypes and prognostic factors in multiple tumor types. Recent studies suggest that aberrant histone modifications play an important role in cancer. However, the effects of global epigenetic rearrangements on cell functions remain poorly understood. In this study, we show that the histone H3 lysine 9 (H3K9) methyltransferase SUV39H1 is clearly involved in regulating cell migration in vitro. Overexpression of wild‐type SUV39H1, but not enzymatically inactive SUV39H1, activated migration in breast and colorectal cancer cells. Inversely, migration was reduced by knockdown of SUV39H1 or chemical inhibition by chaetocin. In addition, H3K9 trimethylation (H3K9me3) was specifically increased in invasive regions of colorectal cancer tissues. Moreover, the presence of H3K9me3 positively correlated with lymph node metastasis in colorectal cancer patients. Furthermore, overexpression of SUV39H1 drove tumorigenesis in mouse, resulting in a considerable decrease in survival rate. These data indicate that H3K9 trimethylation plays an important role in human colorectal cancer progression, possibly by promoting collective cell invasion.
Global changes in the epigenetic landscape are a hallmark of cancer.1, 2 For example, genome‐wide loss of lysine 16 acetylation and lysine 20 trimethylation of histone H4 (H4K16ac and H4K20me3, respectively) is observed in multiple types of human cancers.3, 4 Moreover, global histone modification patterns predict clinical outcome, and correlate with tumor phenotypes and prognostic factors, in tumors including breast, prostate, lung, and gastric cancer.5, 6, 7, 8, 9, 10 Although somatic mutation is widely accepted as the origin of cancer, recent studies suggest that epigenetic alterations may be key initiating events, and that genetic and epigenetic alterations interact at all stages of cancer development and cancer progression.11, 12 Importantly, epigenetic aberrations, unlike genetic mutations, are potentially reversible and can potentially be restored to their normal state by epigenetic therapy.13, 14 Thus, the reversible nature of epigenetic aberrations has led to the promising field of epigenetic therapy, which includes anticancer drugs targeting histone deacetylases and histone methyltransferase.15, 16
Cell migration is a critical step in tumor invasion and metastasis.17 Recent studies suggest that chromatin structure may play a role in cell migration.18 Global changes in chromatin organization are induced by cell migration cues, particularly elevation in trimethylation of histone H3 at lysine 9.19 It has also been reported that cell migration requires chromatin condensation.20 Consistently, we have shown that the activation of breast cancer cell migration occurs concomitantly with a slight increase in global histone H3 lysine 9 trimethylation (H3K9me3).21
Histone H3 lysine 9 methylation is a repressive mark that is related to heterochromatin formation.22 Despite a clear correlation among the global H3K9 methylation patterns in several cancers such as gastric cancer,6, 7, 8, 10 the underlying molecular link between H3K9 methylation and cancer progression remains unknown. Therefore, in this study we investigated the role of global H3K9me3 in cancer progression in vivo and in vitro.
Materials and Methods
Cell culture and transfection
Maintenance of the human breast cancer cell lines MDA‐MB‐231 and ZR75‐30 cells (ATCC, Manassas, VA, USA) was carried out as described previously.21 The human colorectal adenocarcinoma cell line DLD‐1 (ATCC) was cultured in RPMI‐1640 supplemented with 10% (w/v) FBS (Biowest, Miami, FL, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) or Polyethylenimine “Max” (Polysciences, Warrington, PA, USA).
Antibodies and reagents
Mouse anti‐H3K9me3 and a rat anti‐histone H3 antibody were described previously.23 Mouse anti‐β‐tubulin mAb and chaetocin were from Sigma‐Aldrich (St. Louis, MO, USA). Small interfering RNAs against the SUV39H1 coding region (#1 GAACCTCTATGACTTTGAA, #2 CCAACTACCTGGTGCAGAA, #3 CTAAGAAGCGGGTCCGTAT) and negative control siRNA were obtained from Nippon Gene (Tokyo, Japan) and transfected into cells using RNAi Max (Invitrogen).
Immunohistochemistry
Formalin‐fixed, paraffin‐embedded specimens of human colorectal cancer were obtained from surgical cases from Osaka University Hospital (Osaka, Japan) following approval by the ethics committee. Sections (4 μm) were deparaffinized in xylene, dehydrated in a graded series of ethanol, and processed for antigen retrieval by autoclaving in 0.01 M citrate buffer. Endogenous peroxidase was blocked by incubating in 3% H2O2‐methanol at room temperature for 15 min. After washing with 0.05% Tween 20‐supplemented Tris‐buffered saline (TBS‐T) and blocking with 5% BSA in TBS‐T at room temperature for 30 min, sections were incubated with the primary antibodies indicated in figure legends and then with appropriate biotinylated secondary antibodies. Sections were further incubated with peroxidase‐conjugated streptavidin (Dako, Glostrup, Denmark) and visualized with 3,3‐diaminobenzidine solution (Sigma‐Aldrich). The sections were then counterstained with hematoxylin.
Staining scores
The immunohistochemical staining of tissues was evaluated based on the percentage of the cancer cells stained.8 First, each cell was defined as negative or positive (either intermediate or strong nuclear staining) in whole specimens. The percentage of positive cells in each specimen was then graded using the following categories: score 1, <10% cancer cells positively stained; score 2, 10–30% cancer cells positively stained; score 3, 31–70% cancer cells positively stained; and score 4, >70% cancer cells positively stained. The results were further classified into two groups, low expression (score 1,2) and high expression (score 3,4).
Immunofluorescence microscopy
Immunofluorescence micro‐scopy was carried out as described previously.24 In this study, image acquisition was carried out using an IX81 (Olympus) or Eclipse E600 (Nikon, Tokyo, Japan) microscope equipped with a Plan Apo 60 × /NA 1.4 lens.
Construction of plasmids
Human SUV39H1 cDNA was amplified and inserted into the vector pEGFP‐C1. Point‐ mutated SUV39H1 (SUVH1‐C326A) was generated by site‐ directed mutagenesis using appropriate oligo‐DNA primer sets and inserted into pEGFP‐C1. All cDNA constructs were verified by DNA sequencing.
Western blot analysis
Cell lysates were sonicated and centrifuged. Supernatants were resolved by SDS‐PAGE and transferred to a nitrocellulose membrane (Millipore, Billerica, MA, USA). Membranes were probed with the indicated antibodies and detected with using the ECL Plus kit (GE Healthcare, Little Chalfont, UK).
Wound healing assay
Confluent MDA‐MB‐231 cells were scratched using a pipette tip (time = 0), incubated for the indicated times, and fixed. Phase‐contrast images were obtained every 5 min. For quantitative analysis, the distance of the wound was measured and the ratio to the original wound distance was shown (n = 7). Results are expressed as the mean distance (n = 7) ± SD.
Cell motility assay
Cell migration assays were carried out as described previously, using Boyden chambers containing polycarbonate membrane inserts (8‐μm pore size; Neuro Probe, Gaithersburg, MD, USA) coated with Cellmatrix Type I‐C (Nitta Gelatin, Osaka, Japan),21 with some modification. Briefly, plasmids indicated in figure legends were transiently transfected into the cells, and 1 × 104 cells were seeded into the upper chamber in medium containing 0.1% BSA without FBS. Then FBS was added to the lower chamber as a chemoattractant. The number of cells migrating to the lower surface in 3 h was counted. These experiments were repeated a minimum of four times.
Real‐time cell motility assay
Real‐time cell motility assays were carried out as described previously, using an xCELLigence real‐time cell analyzer (Roche, Basel, Switzerland).21 Briefly, the CIM‐ plate was coated with Cellmatrix Type I‐C. Then, 17 h after transfection, 5 × 104 cells were seeded into the upper chamber in medium containing 0.1% BSA but no FBS. The lower chamber was filled with medium containing FBS, which served as a chemoattractant. The top chamber was sealed at the bottom with a microporous membrane containing gold microelectrodes. Cells were permitted to migrate to the bottom chamber, and the impedance (indicated as the “cell index”) was measured. The cell index increased due to cell migration through membrane pores and adhesion to the sensor side of the membrane. These experiments were repeated a minimum of three times and the results were recorded as the cell index value after 9 h.
Matrigel invasion assay
Transwell inserts (Chemotaxicells with 8‐μm pore size; Kurabo, Osaka, Japan) were coated with 100 μg/mL Matrigel (Becton Dickinson Biosciences, Franklin Lakes, NJ, USA), which includes laminin, type IV collagen, and perlecan, and then inserted into a 24‐well chamber. Next, 1 × 105 DLD‐1 cells transiently transfected with the indicated plasmid were seeded in the upper chamber in medium containing 0.1% BSA without FBS. The lower chamber was filled with medium containing FBS as a chemoattractant. Cells were permitted to migrate for 24 h then fixed and stained with hematoxylin. The number of cells that invaded to the lower surface was counted. Each assay was carried out in triplicate.
Cell proliferation assay
Cells were seeded into 96‐well plates, and cell growth was measured using WST‐1 reagent (Dojindo Laboratories, Kumamoto, Japan) as described previously.21
In vivo tumorigenesis
Before experiments were initiated, all protocols were approved by the Institutional Animal Care and Use Committee of the Osaka University Graduate School of Medicine and Health Science. DLD‐1 cells were transfected with pEGFP‐C1‐SUV39H1 or empty vector (pEGFP‐C1). On the next day, cells were suspended in culture medium (1 × 106/50 μL) and injected s.c. into the backs of 4‐week‐old female BALB/c nude mice (n = 4 for each cell line; two injection points per mouse). Mice were obtained from Oriental Yeast Co. (Tokyo, Japan) and maintained under specific pathogen‐free conditions in animal facilities in accordance with institutional guidelines. The sizes of the implanted tumors were measured using calipers three times per week for 3 months. Tumor volumes were estimated using the formula V = LW 2 × (π/6), where V is volume (mm3), L is longest diameter (mm), and W is smallest diameter (mm). For immunohistochemistry, 1 month after injection, implanted tumors were isolated, fixed, and embedded into paraffin. Sections were immunostained with anti‐H3K9me3 mAb. Survival rate was calculated by Kaplan–Meier analysis.
Correlation between H3K9 trimethylation and clinicopathologic variables
Tissues were obtained from stage II and III patients (51–87 years old, mean 69.3 years; 28 male and 24 female) and stained with anti‐H3K9me3 mAbs.
For statistical analyses, Student's t‐test and Fisher's test were applied. P‐values <0.05 were considered statistically significant.
Anchorage‐independent colony formation assay
Two milliliters of culture medium with 0.6% agar was first plated into each 6‐well plate. After the bottom agar was solidified, another 1.5 mL of 0.3% agar in culture medium carrying 1–5 × 104 cells was poured. After 2 weeks' incubation, colonies were directly photographed and counted.
Results
Histone H3K9 methyltransferase SUV39H1 promotes breast cancer cell migration
Cell migration is a critical step in tumor invasion and metastasis. Recently, we showed that the activation of breast cancer cell migration occurs concomitantly with a slight increase in global H3K9me3.21 Therefore, to directly assess the requirement of H3K9me3 for directional migration, we focused on the ubiquitously expressed histone H3 lysine 9 methyltransferase SUV39H1,25 and blocked its enzymatic activity using the chemical inhibitor chaetocin.26 First, we studied the effect of chaetocin on a wound healing assay using the breast cancer cell line MDA‐MB‐231. Control cells closed wounds, generated by scratching, within 10 h (Fig. 1a, upper). In contrast, in the presence of chaetocin, the wounds remained unclosed over the same time period (Fig. 1a, lower). In addition we found a subtle, but reproducible, decrease in cell migration in another chaetocin‐treated breast cancer cell line, ZR75‐30 (Fig. 1b).
Figure 1.
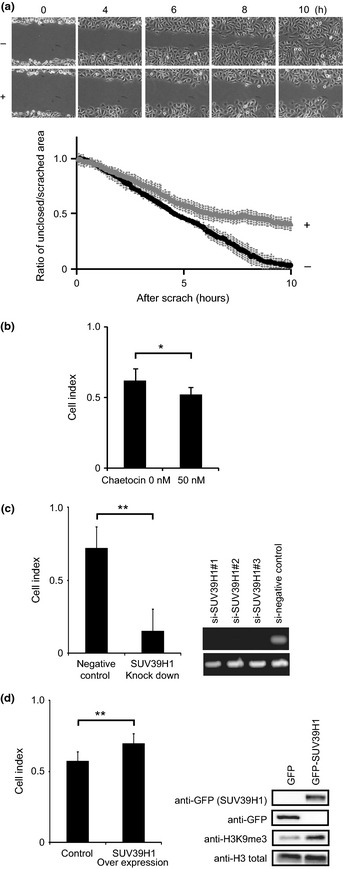
Histone H3 lysine 9 (H3K9) methylation is associated with breast cancer cell migration. (a) For this experiment we used an aggressive human breast cancer cell line, MDA‐MB‐231. Confluent cells were incubated with (lower) or without (upper) 200 nM chaetocin for 2 h then scratched to create a model wound. Phase‐contrast images were obtained every 5 min. Snapshots taken every 2 h are shown (upper). Unclosed area was quantified (lower). (b) Cell migration of another human breast cancer cell line, ZR75‐30, was analyzed using an xCELLigence instrument in the presence or absence of chaetocin. (c) ZR75‐30 cells were transfected with either siRNA against SUV39H1 or a negative control siRNA, and cell migration was measured (left). Knockdown efficiency was confirmed by RT‐PCR (right). (d) ZR75‐30 cells were transfected with plasmids expressing either GFP or GFP‐ SUV39H1, and cell migration was measured (left). The transfectant with empty vector (pEGFPC1) was used as the control for all following experiments. The expression level of GFP‐SUV39H1 and H3K9me3 level was analyzed in Western blotting using anti‐GFP mAb and anti‐H3K9 trimethylation (anti‐H3K9me3) (right). *P < 0.05, **P < 0.01.
To confirm the specificity of chaetocin, we next knocked down SUV39H1 using siRNA. Consistent with the results obtained with the drug, knockdown of SUV39H1 led to decreased cancer cell migration (Fig. 1c).
In contrast, cell migration activity increased in cells overexpressing SUV39H1 (Fig. 1d, left), in which the level of SUV39H1 and H3K9me3 increased as expected (Fig. 1d, right). These findings clearly indicate that histone H3K9me3 is involved in promoting cell migration. It is to be noted that the addition of chaetocin suppressed cell motility within 3 h in the wound healing assay (Fig. S1), suggesting that transcriptional regulation is not required for H3K9me3‐dependent cell migration.
Enzymatic activity of SUV39H1 is required for activation of cell migration and invasion
In order to determine whether upregulation of H3K9me3 promotes migration of other types of cancer cells, we investigated the colorectal cancer cell line DLD‐1. We chose this line because it shows moderately lower levels of H3K9me3, but similar total histone H3 levels, relative to other cell lines, as revealed by Western blotting (Fig. 2a). As expected, SUV39H1 overexpression increased the level of H3K9me3 (Fig. 2b, right). The expression of wild‐ type SUV39H1 significantly promoted colorectal cancer cell migration relative to cells transfected with an empty vector (Fig. 2b, left). SUV39H1 is a methyltransferase but is also known to accumulate at the heterochromatin,27 suggesting that it may play other roles, as in the case of other methyl‐ modifying enzymes that have functions independent of their enzymatic activities.28 We therefore asked whether activation of cell migration by SUV39H1 is dependent on its enzymatic activity. To this end, we introduced a point mutation (cysteine 326 to alanine) in a cysteine‐rich region of the SET domain, yielding SUV39H1‐C326A, which lacks enzymatic activity.29 Expression of the C326A mutant did not promote cell motility, but instead decreased motility relative to GFP‐transfected cells (Fig. 2b), suggesting that trimethylation of H3K9 by SUV39H1 is likely to be required for promotion of directional migration. Moreover, cell invasion was also increased in DLD‐1 cells overexpressing wild‐type SUV39H1, but not in cells expressing the methyltransferase‐deficient C326A mutant (Fig. 2c). Because we observed cell invasion after 24 h, it is possible that differences in cell growth might have affected the measurement of invasion activity. To rule this out, we analyzed proliferation of DLD‐1 cells transfected with SUV39H1, SUV39H1‐C326A, or empty vector. All of these cells showed almost the same growth rate over 3 days (Fig. 2d). From these findings, we conclude that the enzymatic activity of SUV39H1 is required for activation of cell migration. In addition, SUV39H1 overexpression activated anchorage‐independent colony formation, which is a key parameter for cells to acquire a metastatic potential (Fig. 2e,f).
Figure 2.
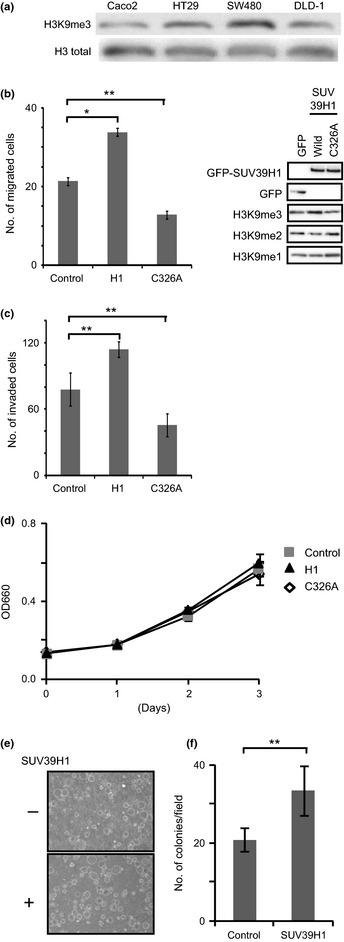
Histone H3 lysine 9 (H3K9) methylation by SUV39H1 activates colorectal cancer cell migration and invasion. (a) Levels of H3K9 trimethylation (H3K9me3) and histone H3 in several human colorectal cancer cells were analyzed using Western blotting, with anti‐H3K9me3 and anti‐histone H3 mAbs. Total H3 protein level was used as an internal control. (b) DLD‐1 cells were transfected with GFP, GFP‐SUV39H1, or GFP‐SUV39H1‐C326A, and cell migration activity was examined using a Boyden chamber. Data collected from the Boyden chamber and xCELLigence system yielded consistent results.21 (c) DLD‐1 cells were transfected with plasmids expressing GFP‐SUV39H1 or GFP‐SUV39H1‐C326A, and cell invasion was measured using Chemotaxicell coated with Matrigel basement membrane. (d) DLD‐1 cells were transfected with plasmids expressing GFP, GFP‐SUV39H1, or GFP‐SUV39H1‐C326A. After 24 h, cells were seeded in 96‐well plates (time = 0), and cell proliferation was monitored using the WST assay. (e,f) Anchorage‐independent colony formation of SUV39H1 forced expressing DLD‐1 and their parental cells was assessed on 0.6% agar. After 14 days' incubation, colonies were directly photographed (e) and numbers of colonies/field were indicated (f). *P < 0.05, **P < 0.01, asterisk shows significant difference relative to GFP‐expressing cells.
Histone H3 lysine 9 trimethylation specifically increases in the invasive region of colorectal cancer tissue
To investigate the clinical significance of H3K9 trimethylation, we carried out immunohistochemistry on colorectal cancer tissue, using anti‐ H3K9me3 mAb and a series of mAbs specific for particular modified histones.23 We observed strong H3K9me3 staining at the invasive front (Fig. 3a, right, Fig. 3c), whereas H3K9me3 staining was moderate next to the surface of the tumor (Fig. 3a left, Fig. 3b). It should be noted that this differential staining pattern could be observed even within a single specimen (Fig. S2). We observed no detectable difference in other histone modifications, including H3K9me2, H3K36me2, and H3K36me3, between cancer and non‐cancerous tissue (Fig. S3).
Figure 3.
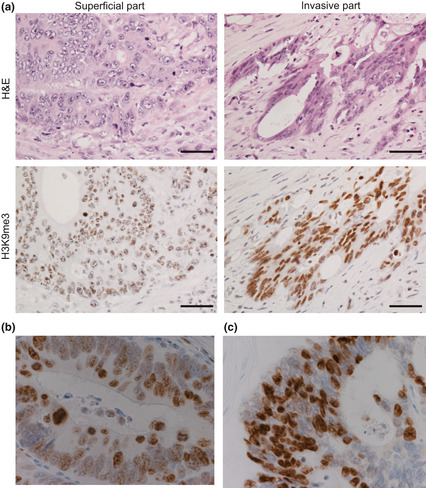
Increased histone H3 lysine 9 trimethy‐lation (H3K9me3) levels in the invasive region of colorectal cancer tissue. H3K9me3 levels in colorectal cancer tissue was examined by immunohisto‐chemistry. (a–c) Paraffin‐embedded colorectal cancer tissues were stained using anti‐H3K9me3 mAb. Representative overall staining patterns of colorectal cancer tissue are shown. Note the high level of H3K9me3 staining in malignant cells in comparison with the surrounding normal cells. Bar = 50 μm. (a) Representative cases of non‐invasive (left) and invasive areas (right) are shown (n = 52). (b,c) Higher magnification photographs (×80) of non‐invasive (b) and invasive (c) areas.
Staining intensity of H3K9me3 correlates with lymph node metastasis
To evaluate the pathological significance of H3K9me3 in colorectal tumors, we analyzed the correlation between histone H3K9me3 levels and various clinicopathological data in 52 patients with colorectal cancer. The H3K9me3 staining scores were divided into two groups, low (specimens containing <30% H3K9me3‐positive cells) and high (specimens containing >30% H3K9me3‐positive cells); details described in Figure S4 and “Materials and Methods”. As shown in Table 1, the H3K9me3 staining status positively correlated with lymph node metastasis (P = 0.018), whereas H3K9me3 was not correlated with other clinicopathological data such as lymphovascular or vascular invasion.
Table 1.
Correlation between histone H3 lysine 9 trimethylation (H3K9me3) and clinicopathologic variables in patients with colorectal cancer (n = 52)
| Overall | K9me3‐low | K9me3‐high | P‐valuea | |
|---|---|---|---|---|
| WHO classification | ||||
| Well | 11 | 2 | 9 | 0.728 |
| Moderate | 37 | 8 | 29 | |
| Poor | 1 | 0 | 1 | |
| Others | 2 | 1 | 1 | |
| Tumor stage | ||||
| T1 | 2 | 1 | 1 | 0.556 |
| T2 | 3 | 0 | 3 | |
| T3 | 46 | 10 | 36 | |
| T4 | 1 | 0 | 1 | |
| Lymph node metastasis | ||||
| Present | 31 | 3 | 28 | 0.018 |
| Absent | 21 | 8 | 13 | |
| Vascular invasion | ||||
| Present | 24 | 4 | 20 | 0.324 |
| Absent | 27 | 7 | 20 | |
| Lymphovascular invasion | ||||
| Present | 48 | 10 | 38 | 0.886 |
| Absent | 3 | 1 | 2 | |
Fisher's exact test. K9me3‐high, specimens containing >30% H3K9me3‐positive cells; K9me3‐low, specimens containing <30% H3K9me3‐positive cells. The total number of analyzed patients varies between 51 and 52 because some of the clinical data was not available.
Upregulated histone lysine 9 methylation drives tumor formation
In order to investigate the effect of global H3K9 methylation on tumorigenesis, DLD‐1 cells expressing SUV39H1 were injected s.c. into the backs of nude mice, and tumor volume was followed for 3 months (Fig. 4a). One month after injection, we began to observe differences in tumor sizes between mice implanted with GFP‐SUV39H1‐expressing DLD‐1 cells and those implanted with GFP‐expressing control cells. Subsequently, the size differences increased significantly (Fig. 4a), and the survival rate of mice implanted with SUV39H1‐expressing cells decreased considerably (Fig. 4b). To confirm the H3K9 methylation level in vivo, tumors were isolated from mice and immunostained with anti‐H3K9me3 mAb. Interestingly, the tumor tissue from control mice showed H3K9me3 staining only at the tumor border (Fig. 4c). In contrast, the tumor tissue obtained from mice implanted with SUV39H1‐expressing cells showed strong H3K9me3 staining in most cells.
Figure 4.
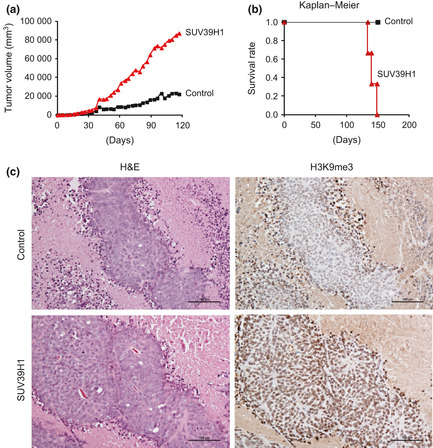
Tumor formation driven by induced histone H3 lysine 9 (H3K9) methylation. (a) GFP‐, or GFP‐SUV39H1‐expressing DLD‐1 cells were implanted into the backs of nude mice. Tumor sizes were measured at the indicated time points, and the tumor volumes were calculated. (b) Survival rate was calculated by Kaplan–Meier analysis. (c). One month after implantation, tumors were isolated, fixed in formalin, and embedded in paraffin. Tissue sections were stained with anti‐H3K9me3 mAb and H&E. Representative pictures are shown. Bar = 100 μm.
Discussion
Correlations between H3K9 trimethylation and tumor progression have been reported in multiple cancers. However, the pathological effects of H3K9me3 have remained unknown. Here, we demonstrated that the level of global H3K9me3 is especially elevated in the invasive area in colorectal cancer tissue, and that H3K9 trimethylation correlates with lymph node metastasis. Through overexpression of H3K9 methyltransferase, we revealed that SUV39H1 positively regulates cell migration in vitro through its enzymatic activity. Furthermore, global H3K9 methylation upregulates tumorigenesis in vivo.
How does the overexpression of SUV39H1 activate directional cell migration in vitro?
Figures 1 and 2 suggest that trimethylation of H3K9 by SUV39H1 is required for directional cell migration. However, the molecular mechanism underlying SUV39H1‐induced cell migration remains unknown. H3K9me3 is widely accepted as a transcriptional repression mark; therefore, one plausible explanation is that H3K9me3 at specific chromatin loci may regulate cell migration through transcriptional regulation, as has been reported for other methyl‐modifying enzymes.29 However, our result suggests that transcriptional regulation is not required for H3K9me3‐dependent cell migration (Fig. S1). Thus, another attractive possibility is that H3K9me3 could serve as a platform for a nuclear envelope‐spanning protein complex, the linker of nucleoskeleton and cytoskeleton (LINC) complex, which associates with and coordinates both chromatin and the centrosome.30, 31, 32 In migrating cells, the centrosome is positioned between the nucleus and the leading edge, and proper positioning of centrosome is essential for directional cell movement. Thus, regulation of the centrosome by H3K9me3 through the LINC complex may be required for cell migration. Along these lines, it is noteworthy that the H3K9 mono‐ and dimethyltransferase G9a is essential for proper centrosome function.33 H3K9 mono‐ and dimethylation is an indispensable step on the pathway to trimethylation; therefore, disrupting G9a might block SUV39H1‐induced H3K9 trimethylation.
How does induced H3K9 methylation drive tumorigenesis in vivo?
Some reports have indicated the importance of SUV39H1 in tumorigenesis: suppression of SUV39H1 attenuates the anchorage‐independent growth of transformed NHBZ cells,34 and the mRNA of SUV39H1 is elevated in primary colorectal cancer,35 whereas the detection of SUV39H1 in immunohistochemistry is difficult in colorectal cancer tissue probably because SUV39H1 protein is expressed below detection level. In this study, we showed that SUV39H1 expression drives tumorigenesis in vivo. Importantly, we revealed that strong H3K9me3 staining is observed only at the surface of tumors in mice implanted with control DLD‐1 cells (Fig. 4c). Overexpression of SUV39H1 activated both cell invasion and migration (Fig. 2b), but did not affect cell growth in vitro (Fig. 2d). Moreover, we did not observe significant differences in the staining pattern for vascular formation markers between tumor samples derived from GFP‐ and GFP‐SUV39H1‐expressing cells (data not shown). Recently it has been reported that collective cell invasion is required for expansive tumor growth.36, 37 Based on these findings, we propose that upregulation of H3K9me3 by SUV39H1 activates cell invasion and migration, resulting in an expansion of the tumor volume.
The analysis of the mechanisms underlying the roles of epigenetic alteration in tumor progression is still an early stage. However, based on the early data, inhibition of H3K9 methylation is a rational target for cancer therapy.
Disclosure Statement
A part of this study was carried out as a joint research program with Wako Pure Chemical Industries. This work was supported by: Leave a Nest (http://lne.st/, to YY); Grants‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science (to NM and MH); and a Grant‐in‐Aid for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to MH). The authors have no conflict of interest.
Supporting information
Fig. S1. Addition of chaetocin immediately affected the cell migration suggesting that the impaired cell motility was not a result of transcriptional regulations.
Fig. S2. Histone H3 lysine 9 trimethylation level is higher at invasive area than surface area in colorectal cancer tissue.
Fig. S3. There is no detectable differences in H3K9me2, H3K36me2, and H3K36me3, between cancer and non‐cancerous tissue.
Fig. S4. Histone H3 lysine 9 trimethylation level in colorectal cancer tissue was analyzed in immunohistochemistry.
Acknowledgments
We gratefully acknowledge Ms Junko Imada (Osaka University, Osaka, Japan), Ms Kumiyo Hirai (Osaka University) and Ms Yuka Kaneko (Wako Pure Chemical Industries, Osaka, Japan) for technical assistance. We also thank Dr Tsutomu Kurokawa and Dr Susumu Honda (Wako Pure Chemical Industries) and Dr Yumi Kinugasa (Osaka University) for valuable discussions.
(Cancer Sci 2013; 104: 889–895)
References
- 1. Füllgrabe J, Kavanagh E, Joseph B. Histone onco‐modifications. Oncogene 2011; 30: 3391–403. [DOI] [PubMed] [Google Scholar]
- 2. Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 2012; 13: 343–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fraga MF, Ballestar E, Villar‐Garea A et al Loss of acetylation at Lys16 and tri‐methylation at Lys20 ofhistone H4 is a common hallmark of human cancer. Nat Genet 2005; 37: 391–400. [DOI] [PubMed] [Google Scholar]
- 4. Behbahani TE, Kahl P, von der Gathen J et al Alterations of global histone H4K20 methylation during prostate carcinogenesis. BMC Urol 2012; 12: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Seligson DB, Horvath S, Shi T et al Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005; 435: 1262–6. [DOI] [PubMed] [Google Scholar]
- 6. Song JS, Kim YS, Kim DK, Park SI, Jang SJ. Global histone modification pattern associated with recurrence and disease‐free survival in non‐small cell lung cancer patients. Pathol Int 2012; 62: 182–90. [DOI] [PubMed] [Google Scholar]
- 7. Park YS, Jin MY, Kim YJ, Yook JH, Kim BS, Jang SJ. The global histone modification pattern correlates with cancer recurrence and overall survival in gastric adenocarcinoma. Ann Surg Oncol 2008; 15: 1968–76. [DOI] [PubMed] [Google Scholar]
- 8. Nguyen CT, Weisenberger DJ, Velicescu M et al H3K9: Histone H3‐lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5‐aza‐2'‐deoxycytidine. Cancer Res 2002; 62: 6456–61. [PubMed] [Google Scholar]
- 9. Elsheikh SE, Green AR, Rakha EA et al Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res 2009; 69: 3802–9. [DOI] [PubMed] [Google Scholar]
- 10. Nakazawa T, Kondo T, Ma D et al Global histone modification of histone H3 in colorectal cancer and its precursor lesions. Hum Pathol 2012; 43: 834–42. [DOI] [PubMed] [Google Scholar]
- 11. Chi P, Allis CD, Wang GG. Covalent histone modifications—miswritten, misinterpreted and mis‐erased in human cancers. Nat Rev Cancer 2010; 10: 457–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Albert M, Helin K. Histone methyltransferases in cancer. Semin Cell Dev Biol 2010; 21: 209–20. [DOI] [PubMed] [Google Scholar]
- 13. Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov 2006; 5: 37–50. [DOI] [PubMed] [Google Scholar]
- 14. Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol 2010; 28: 1069–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 2006; 5: 769–84. [DOI] [PubMed] [Google Scholar]
- 16. Wagner T, Jung M. New lysine methyltransferase drug targets in cancer. Nat Biotechnol 2012; 30: 622–3. [DOI] [PubMed] [Google Scholar]
- 17. Yamaguchi H, Wyckoff J, Condeelis J. Cell migration in tumors. Curr Opin Cell Biol 2005; 17: 559–64. [DOI] [PubMed] [Google Scholar]
- 18. Gerlitz G, Bustin M. The role of chromatin structure in cell migration. Trends Cell Biol 2011; 21: 6–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gerlitz G, Livnat I, Ziv C, Yarden O, Bustin M, Reiner O. Migration cues induce chromatin alterations. Traffic 2007; 8: 1521–9. [DOI] [PubMed] [Google Scholar]
- 20. Gerlitz G, Bustin M. Efficient cell migration requires global chromatin condensation. J Cell Sci 2010; 123: 2207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tanaka H, Nishioka Y, Yokoyama Y et al Nuclear envelope‐localized EGF family protein amphiregulin activates breast cancer cell migration in an EGF‐like domain independent manner. Biochem Biophys Res Commun 2012; 420: 721–6. [DOI] [PubMed] [Google Scholar]
- 22. Schotta G, Lachner M, Sarma K et al A silencing pathway to induce H3–K9 and H4–K20 tri‐methylation at constitutive heterochromatin. Genes Dev 2004; 18: 1251–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kimura H, Hayashi‐Takanaka Y, Goto Y et al The organization of histone H3 modifications as revealed by a panel of specific monoclonal antibodies. Cell Struct Funct 2008; 33: 61–73. [DOI] [PubMed] [Google Scholar]
- 24. Hieda M, Isokane M, Koizumi M et al Membrane‐anchored growth factor HB‐EGF, on the cell surface targeted to the inner nuclear membrane. J Cell Biol 2008; 180: 763–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rea S, Eisenhaber F, O'Carroll D et al Regulation of chromatin structure by site‐specific histone H3 methyltransferases. Nature 2000; 406: 593–9. [DOI] [PubMed] [Google Scholar]
- 26. Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3–9. Nat Chem Biol 2005; 3: 143–5. [DOI] [PubMed] [Google Scholar]
- 27. Grewal SIS, Jia S. Heterochromatin revisited. Nat Rev Genet 2007; 8: 35–46. [DOI] [PubMed] [Google Scholar]
- 28. Miller SA, Mohn SE, Weinmann AS. JMJD3 and UTX play a demethylase‐ independent role in chromatin remodeling to regulate T‐box family member‐dependent gene expression. Mol Cell 2010; 40: 594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen MW, Hua KT, Kao HJ et al H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep‐CAM. Cancer Res 2010; 70: 7830–40. [DOI] [PubMed] [Google Scholar]
- 30. Crisp M, Liu Q, Roux K et al Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol 2005; 172: 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Méjat A, Misteli T. LINC complexes in health and disease. Nucleus 2010; 1: 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Malone CJ, Misner L, Le Bot N et al The C. elegans hook protein, ZYG‐12, mediates the essential attachment between the centrosome and nucleus. Cell 2003; 115: 825–36. [DOI] [PubMed] [Google Scholar]
- 33. Kondo Y, Shen L, Ahmed S et al Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS ONE 2008; 3: e2037. doi: 10.1371/journal.pone.0002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Watanabe H, Soejima K, Yasuda H et al Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell Int 2008; 8: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kang MY, Lee BB, Kim YH et al Association of the SUV39H1 histone methyltransferase with the DNA methyltransferase 1 at mRNA expression level in primary colorectal cancer. Int J Cancer 2007; 121: 2192–7. [DOI] [PubMed] [Google Scholar]
- 36. Friedl P, Alexander S. Cancer invasion and metastasis: plasticity and reciprocity. Cell 2011; 174: 992–1009. [DOI] [PubMed] [Google Scholar]
- 37. Friedl M, Locker J, Sahai E, Jeffrey E. Segall Classifying collective cancer cell invasion. Nat Cell Biol 2012; 14: 777–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Addition of chaetocin immediately affected the cell migration suggesting that the impaired cell motility was not a result of transcriptional regulations.
Fig. S2. Histone H3 lysine 9 trimethylation level is higher at invasive area than surface area in colorectal cancer tissue.
Fig. S3. There is no detectable differences in H3K9me2, H3K36me2, and H3K36me3, between cancer and non‐cancerous tissue.
Fig. S4. Histone H3 lysine 9 trimethylation level in colorectal cancer tissue was analyzed in immunohistochemistry.