

Abstract
An integrated, parallel-plate microfluidic device was engineered to interrogate and fractionate cells based on their adhesivity to a substrate surface functionalized with adhesive ligand in a tightly controlled flow environment to elucidate associated cell-intrinsic pathways. Wall shear stress levels and endothelial presentation of E-selectin were modeled after the inflamed vasculature microenvironment in order to simulate in vitro conditions under which in vivo hematogenous metastasis occurs. Based on elution time from the flow channel, the collection of separate fractions of cells – non-interacting and interacting – at high yields and viabilities enabled multiple post-perfusion analyses, including flow cytometry, in vivo metastasis modeling, and transcriptomic analysis. This platform enabled the interrogation of flow-regulated cell molecular profiles, such as (co)expression levels of natively expressed selectin ligands sLex, CD44, and carcinoembryonic antigen, and cancer stem cell marker CD24. This additionally revealed E-selectin adhesivity exhibited by metastatic human colon carcinoma cells to be a transient phenotype. Facile and rapid, this methodology for unbiased, label free sorting of large populations of cells based on their adhesion in flow represents a method of studying flow-regulated adhesion in vitro for the identification of molecular drug targets for development as anti-metastatic cancer therapeutics.
Keywords: metastasis, microfluidic, colon cancer, E-selectin, sialyl Lewis x
Graphical Abstract
1. Introduction
Over 90% of all cancer-related deaths result from metastasis, a multistep process in which cancer cells leave the primary tumor, intravasate into the blood stream, circulate in the vasculature until they are able to extravasate, and eventually take up residence in a secondary location in the body to form a metastatic tumor.[1,2] Extravasation occurs in the presence of hemodynamic force, necessitating processes to overcome its dispersive effects. Amongst multiple implicated mechanisms, adhesive interactions with endothelial presented receptors such as E-selectin amongst others, act to slow down circulating cancer cells, leading to eventual cell arrest and extravasation.[2–6] Despite the correlation between a cell’s propensity to exhibit selectin-mediated adhesion and metastatic potential, the cancer cell-intrinsic mediators facilitating the process are poorly understood.[7–11] Given that cancer is one of the largest public health burdens facing the U.S. today, afflicting over 14.5 million with nearly 1.7 million new diagnoses each year, a better understanding of the mechanisms underlying cancer metastasis and identification of relevant biomarkers or therapeutic targets is crucial for developing anti-metastatic cancer therapies and to monitor patient responses to therapy.[12,13]
To enable both the interrogation of biomechanical effects on metastasis as well as the elucidation of mechanisms of adhesion, engineered microfluidic systems are frequently implemented to model metastasis in vitro.[11,14] Despite being highly simplified systems relative to in vivo mouse models, they have the advantage of enabling experimentation under defined cellular, molecular, and/or biophysical conditions. Using video microscopy-based imaging to monitor the extent of adhesion in simulated flow fields that exhibit forces on par with those experienced in the microvasculature, many thousands of cells can be analyzed in a single experiment, enabling high sampling volumes in individual or parallelized experiments. Biochemical, biomolecular, and transcriptomic manipulations of analyzed cells can also be performed to interrogate biomolecular mechanisms contributing to metastatic adhesive phenotypes.[5,6,15–18] The capacity to probe the attributes intrinsic to cancer cells that confer adhesion in an unperturbed manner, however, is not feasible using these interventional type approaches without a priori knowledge. Imaging based analyses alone also provide limited insight into biomolecular mechanisms and also do not allow for subpopulations of cells to be collected and assayed in an unbiased manner based on their phenotypes of adhesion.
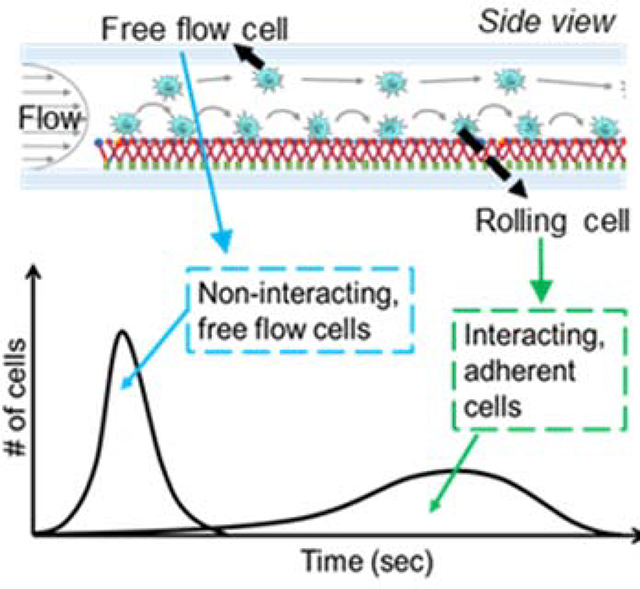
To overcome the existing limitations but leverage the advantages of such in vitro microfluidic systems in the investigation of metastasis, we engineered a flow-based device that functions as an adhesive chromatography platform. This system, based on a widely used parallel-plate flow chamber, leverages the time-averaged velocities of cells perfused as a pulse and dispersed in perfusion media in a controlled flow field as a proxy for overall adhesion propensity to a substrate with or without surface functionalization with adhesive ligands (Figure 1a). The high-throughput nature of the cell adhesion chromatography system permits the analysis of a large number of cells simultaneously in an environment that, used herein, recapitulates the physiological flow of the circulatory system. Using residence time theory, cells with lowly versus highly adhesive phenotypes can be separately enriched and collected at sufficient yields amenable to off-chip analyses, allowing further investigation of cell subpopulation expression profiles, metastatic potential, and molecular characteristics. Using this methodology, cell subpopulations exhibiting distinct adhesive behaviors within a population of genomically identical metastatic human colon carcinoma cells that nevertheless exhibit heterogeneous profiles of adhesion to E-selectin in flow were interrogated to reveal how expression and transcriptomic mechanisms associated with this transient metastatic phenotype are regulated by flow.
Figure 1. Engineered cell sorting adhesion chromatography microfluidic to investigate cancer metastasis in vitro.

(A) Elution profiles of LS174T metastatic colon carcinoma cell populations when perfused over E-selectin, demonstrating the early elution of free flow cells followed by subsequent elution of adherent cells. (B) Hemodynamic microenvironment-mimicking, parallel-plate microfluidic fabrication, into which (C, bottom) metastatic cancer cells are infused into the E-selectin functionalized channel, visualized via an integrated high speed videomicroscopy, and collected into collection tubes based on channel residence time. (D-H) Continuous perfusion experiments, (I-M) Static perfusion experiments. (D,I) Channel functionalization schematic and perfusion workflow diagram. (E) Distance of individual perfused cells from the channel bottom calculated based on measured individual cell velocity and size decreases from the inlet (channel position 1) until reaching channel position 3 after which it is uniform throughout the remaining channel length. Data represent mean ± s.e.m. (F,K) Representative relative frequency distribution of elution time of the infused cell pulse with the inset showing the mean time at which 95% of the cell pulse has eluted ± s.e.m. (G,L) Number of interacting cells along the length of the channel during the sorting phase. Data represent mean ± s.e.m. (H,M) Number of interacting cells along the width of the channel (black) and average velocity of rolling cells along the width of the channel (gray). Data represent mean ± s.e.m. (J) Location of cell pulse during static phase of Static perfusion experiments (E-H,J-M) Each point represents the mean ± s.e.m. of n≥5 independently run experiments. Cell pulse of LS174T colon carcinoma cells perfused at 1 dyn cm−2 over (E,G,I,K) 2.5 ug mL−1 E-selectin or (F,J) unfunctionalized channels. * p < 0.05 ** p<0.01 compared to unfunctionalized channel (no interacting cells) by one-sample t-test.
2. Results
2.1. Microfluidic-based adhesion chromatography
We constructed from polydimethylsiloxane (PDMS), a polystyrene tissue culture plate, and an adhesive gasket a parallel plate flow chamber device whose inlet is connected to a syringe pump to vary the wall shear stress to mimic a range of physiologically relevant shear stresses (Figure 1b,c). Easy to rapidly construct and assemble, and amenable to high speed video microscopy imaging of perfusion experimentation throughout the flow channel (Figure 1b), cells perfused as a suspension into this device in perfusion media are subjected to a roughly uniform wall shear stress level across >90% of the channel width and can be collected into an interchangeable cell reservoir at the outlet (Figure 1c). Experimentation performed under “Continuous” flow conditions (no cessation of flow during experimentation, Figure 1d) necessitated a so-called settling region within the flow channel to ensure all perfused cells were in contact with an adhesive substrate for uniform distances, given the time, which in the context of flow translates to distance, necessary for all perfused cells to settle to the inferior channel substrate (Figure 1e).[11,19] Channels were thus functionalized only within the last third of the total channel length (channel positions 7–9, Figure 1d), where cells were uniformly in contact with the substrate as defined by being within one cell radii from the interior channel wall as experimentally validated by measurement of cell velocity and size (Figure 1e). In unfunctionalized channels passivated by blocking with bovine serum albumin (BSA), the time at which 95% of the infused cell pulse had eluted through the channel was defined as commencing the sorting phase of the experiment (Figure 1f). In the presence of E-selectin, this resulted in extensive adhesion by selectin-binding LS174T cells as quantified by video image analysis that was restricted to and homogenously distributed along only the functionalized straight portion of the channel nearest the outlet (channel positions 7–9 but not 1–6, Figure 1g), extents unaffected by channel width position (Figure 1h). Adhesive LS174T cells also translated in rolling adhesion at similar velocities at various distances from the outside edge of the channel (Figure 1h), thus indicating good spatial control and uniformity of functionalization. Overall, given the exact size of this channel, the surface area of functionalization, wall shear stress level, and a minimum surface area density to prevent cell-cell interactions, adhesion chromatography experiments could be performed to a maximal loading capacity of approximately 106 cells, a number that can be tuned by parameters of channel configuration and flow.
As a point of comparison given the capacity of force to regulate the initiation of rolling adhesion, we also established a workflow for evaluating cell adhesion initiated in the absence of force but presence of flow during de-adhesion, so called “Static” experimentation. During these experiments, after loading of the cell pulse into the channel, cells were allowed to settle and interact with the substrate for 5 min, a time experimentally determined as sufficient for cells to settle in static conditions, after which time fluid flow was reinitiated to start the sorting phase of experimentation (Figure 1i).[11,20] Perfused again as a pulse, cells could be loaded into the device and distribute amongst the channel length (Figure 1j). When assessed by video microscopy, at times soon after 95% of cells had eluted from an unfunctionalized channel after reinitation of flow (Figure 1k), the extent of adhesion by LS174T cells to E-selectin was substantial throughout the channel length (Figure 1l). Notably, the highest number of interacting cells were located at the middle length of the channel (Figure 1l), implying that interacting cells roll slowly and do not have time to reach the end of the channel by the end of perfusion (10 min after the flow was restarted). The extent and velocity of rolling adhesion did not vary significantly with channel width position (Figure 1m). Approximately 5E105 cells could be loaded into the device for experimentation, a number that again could be tweaked based on channel and flow parameters.
2.2. Adhesion chromatography microfluidic enrichment of distinct and viable cell subpopulations of varying in vitro adhesivities
Experiments were performed under both Continuous and Static conditions to interrogate the effect of force on initiation of E-selectin-mediated adhesion and whether the extent and quality of experimentally observed adhesion to E-selectin differed between experimental flow types. A cell pulse of E-selectin-binding LS174T cancer cells and perfusion media was perfused through the channel surface functionalized with 2.5 μg mL−1 E-selectin at a predetermined flow rates corresponding to physiological levels of wall shear stress where selectin-mediated adhesion occurs, ranging from 0.5 to 5 dyn cm−2.[20] Unsurprisingly, the extent of LS174T cell adhesion to E-selectin decreased with increasing wall shear stress level (Figure 2a). The velocity with which cells mediated adhesion also increased as the wall shear stress increased (Figure 2b). [14,15,21] Strikingly, the velocity of rolling adhesion by LS174T cells on E-selectin did not differ between Continuous and Static experimentation at all wall shear stress levels tested, with the exception of the highest of 5 dyn cm−2, where the rolling velocity of cells of adherent cells in Continuous experiments was significantly greater than that in Static experiments (Figure 2a–b). Non-E-selectin ligand expressing B16F10 melanoma cells were also separately perfused through the channel in a cell pulse under both Static and Continuous conditions.[22,23] Expectedly, there were minimal interacting B16F10 cells at all tested wall shear stresses levels (Figure 2a).
Figure 2. Collected cell fraction analysis.

(A) Number of interacting cells during the sorting phase of perfusion. (B) Rolling velocity of interacting cells during the sorting phase of perfusion. n≥20. (A-B) Either static or continuous flow at 0.5, 1, 2, or 5 dyn cm−2. Each point represents the mean ± s.e.m. of n≥3 independently run experiments. (C) Experiment workflow diagram of static and continuous flow experiments. (D) Percent of total cells recovered from infused pulse of 250,000 cells after perfusion on either a functionalized or unfunctionalized channel. (E) Percent of cells collected in either the free flow or adherent cell fraction after either Static or Continuous flow on an unfunctionalized channel or a functionalized channel. (F) Percent of cells that are viable prior to perfusion or in each fraction after perfusion on either a functionalized or unfunctionalized channel. (G) Increase in cell number of cultured unsorted cells or collected cell fractions after 72 hr normalized to initial cell culture seeding number. FF: free flow, AD: adherent. (H) Representative brightfield images of cultured cells in a well of a 96 well plate at time of initial seeding (0 hr.) or after 72 hr. Scale bar represents 400 um. (I) Percent of LS17T or B16F10 cells collected in each fraction after perfusion under either Static or Continuous flow. (D-G, I) Perfusion experiments were at 1 dyn cm−2. Functionalized experiments were over 2.5 μg mL−1 E-selectin. Each bar represents the mean ± s.e.m. of n≥3 independently run experiments. (E, I) * p < 0.05, ** p< 0.01, **** p < 0.0001 by t-test comparing sizes of adherent cell fractions.
The difference in elution times from an flow channel that result from differential ability of cells to mediate adhesion was leveraged to quantify the fraction of total cells capable of mediating adhesion to E-selectin in flow – with non-interacting, free flow cells exiting the channel before interacting, adherent cells (Figure 1a).[11,14] Cells that eluted prior to the predetermined “non-interacting cell elution time” were collected as the “Free flow” fraction. Subsequently, the cell collection reservoir was changed and the cell subpopulation remaining within the microfluidic device removed by escalating the flow to 5 dyn cm−2 for 5 min was collected labeled as the “Adherent” fraction (Figure 2c).
Using this workflow and using a wall shear stress of at 1 dyn cm−2 and 2.5 μg mL−1 E-selectin, >70% of cells (either LS174T and B16F10) were recovered during experimentation, levels unchanged by channel functionalization (Figure 2d). Of the cells recovered, ~60% of LS174T cells eluted in the adherent population (Figure 2e), extents at levels nearing those seen for Static experimentation (Figure 2a). Conversely, nearly all of B16F10 cells eluted into the non-interacting, Free flow fraction when perfused in either Static or Continuous flow set-ups, consistent with B16F10 cells, which lack E-selectin ligands failing to mediate adhesion to E-selectin in flow (Figure 2a).[22,23] However, ~10% of perfused B16F10 cells were recovered in the Adherent fraction, a proportion similar in magnitude to the amount of LS174T cells recovered from unfunctionalized channels (Figure 2e) indicating a small extent of contamination within the enriched adherent cell subpopulation. Nevertheless, the fraction of LS174T cells comprising the adherent fraction was substantially larger than for B16F10 cells. Of the collected cells of each clone type, over 95% were viable in collected cell fractions, and viability was not affected by perfusion, channel functionalization, nor the adhesion propensity of the cell (Figure 2f). Furthermore, fractionated LS174T cell subpopulations cultured for 72 hr showed no differences in the ability to proliferate (Figure 2g) or grow (Figure 2h) in culture, indicating that the system is able to collect viable cells for further expansion and post-perfusion analysis.
When varying wall shear stress level to emulate the range of physiologically relevant flow conditions of the microenvironment a circulating metastatic cell may be exposed to, perfusion of selectin-ligand expressing LS174T cancer cells over E-selectin functionalized surfaces resulted in collection of a decreasing fraction of total perfused cells (Figure 2i). At 0.5 and 1.0 dyn cm−2, experiments performed under Continuous flow conditions resulted in slightly but significantly lower levels of cell recovery compared to Static experiments (Figure 2i). Taken together, these results indicate that this adhesion chromatography device is capable of not only separating cells based on adhesion propensity to the functionalized substrates in a manner regulated by hemodynamic force, but also that sorted cells can be retained and/or expanded for further phenotypic, genotypic, and molecular analysis post perfusion.
2.3. Sorted cell fractions exhibit different metastatic potentials in vivo
We sought to explore the relationship between propensity to mediate adhesion to E-selectin, using channel residence time as a proxy, and metastatic potential in vivo. As such, using an E-selectin functionalized channel, a cell pulse of LS174T cells were perfused through the channel at 1.0 dyn cm−2 and sorted based on elution time into free flow and adherent populations, and 105 cells from sorted fractions were subsequently injected intravenously via the tail vail into NSG mice. Markedly, the cells from the adherent subpopulation, enriched in the adhesion chromatography device based on their enhanced ability to interact with E-selectin in vitro, exhibited greater lung metastatic tumor bearing potential than the cells from the free flow subpopulation in vivo 7 and 14 d post injection as quantified by counting the total number of lung metastases in each mouse (Figure 3a–b). Further, when normalizing the number of metastasis in each lung 7 d post injection to the unsorted, parental population, the injected free flow cell group exhibited a decreased number of metastases, further elucidating the less metastatic nature of the free flow fraction (Figure 3c). Quantification of hematoxylin and eosin histology sections confirmed these results, as injection of cells from the free flow fraction resulted in a lower cell density within lung cryosections than injection of cells from either the adherent fraction or unsorted, parental population (Figure 3d). These results demonstrate the utility of this adhesion chromatography device in enriching cells with differential metastatic potentials based on their E-selectin binding capabilities.
Figure 3. LS174T cells collected using adhesion chromatography in the Free flow fraction metastasize in vivo less than Adherent fraction.

(A) Representative bright field and chromogenic images of NSG mouse whole lobes and lung sections and 7 d post intravenous (i.v.) infusion of parent (unsorted) or sorted free flow or adherent LS174T cells. Arrows indicate metastatic tumors. Scale bars, 100 μm or 1 mm in regions of interest and whole lobe images, respectively. (B) Lung metastasis count in lungs dissected from NSG mice either 7 or 14 d post i.v. infusion of collected LS174T free flow cells or adherent cells. (C) Count of lung metastases in either the free flow group or the adherent group normalized to the mean number of lung metastases per NSG mouse in the parent (unsorted) group 7 d post i.v. infusion. (D) Quantification of hematoxylin and eosin staining for each group. (A-D) Perfusion and separation of an LS174T cell pulse of 250,000 cells was over 2.5 μg mL−1 E-selectin at 1 dyn cm−2. NSG mice were injected i.v. with 105 LS174T cells. (B-C) Each point represent an individual mouse lung. Bars represent mean ± s.e.m. of n≥5 individual mice. (B) * p < 0.05 by two sample t-test. (C) * p < 0.05 by t-test Ho: μ= 1 (D) Bars represent mean ± SEM of independent samples. Analyzed with a one-way ANOVA with post hoc t-test with Bonferonni corrections for multiple comparisons * p < 0.05 ** p < 0.01
2.4. Differential selectin ligand expression by LS174T cells separated using adhesion chromatography by E-selectin adhesivity
Selectin ligand glycan epitope and glycoprotein expression levels in LS174T cell populations sorted for their enriched adhesive potential on E-selectin were measured. Sialofucosylated tetrasaccharides sialyl Lewis x (sLex) ligand expression, as measured by anti-sLex/a binding antibody HECA-452, on sLea-negative LS174T cells collected in the adherent versus free flow fractions, was assessed flow cytometrically. In both Static and Continuous flow experiments, the Free flow LS174T subpopulation (the cells that did not interact with the E-selectin during perfusion through the microfluidic device) had significantly less HECA452 immunoreactivity relative to the cells that exhibited adhesion to E-selectin (Figure 4a,b). Furthermore, when binned into high and low HECA452 stained populations based on the highest 10% and lowest 10% of the unsorted parent populations (HECAhi and HECAlo, respectively), the adherent fraction had significantly more cells HECAhi (sLex hi expressing) cells whereas the free flow cell population had significantly more HECAlo (sLex lo expressing) cells (Figure 4c). Jointly, these results substantiate the finding that increasing sLex expression is associated with enhanced LS174T cell adhesivity to E-selectin in flow.[11,14]
Figure 4. LS174T cells collected using adhesion chromatrography into Adherent subpopulation after perfusion over E-selectin exhibits enhanced HECA and CD44, but not CEA, expression levels.

(A,D,G) Representative flow cytometric histograms of sLex (HECA452), CD44, or CEA expression. (B,E,H) sLex (HECA452), CD44, or CEA expression normalized to the parent population between cells in the collected free flow fraction and adherent fraction. Each point represents the mean fluorescent intensity of analyzed cells ± s.e.m. (C,F,I) Percent of cells in each fraction that are in the top 10% or bottom 10% of sLex (HECA), CD44, or CEA expressing cells. Each bar represents the mean ± s.e.m. of n≥3 individually run experiments. (J) Representative flow cytometry gating and analysis strategy for (K,L) sLex (HECA452) expression levels normalized to the unsorted parent LS174T cell population in either high (top 10%) or low (bottom 10%) expressing subpopulations of (K) CD44 or (L) CEA. Each bar represents the mean ± s.e.m. of n≥3 individually run experiments. Perfusion and separation of an LS174T cell pulse of 250,000 cells was over 2.5 μg mL−1 E-selectin at 1 dyn cm−2. * p < 0.05 ** p < 0.01 *** p, 0.001 by paired t-test.
Relationships between LS174T cells’ capacity to mediate adhesion to E-selectin in flow and expression of CD44 and carcinoembryonic antigen (CEA), two previously identified selectin ligand glycoproteins, was also determined. Experiments performed under both Static flow and Continuous flow conditions revealed increased levels of CD44 expression on cells collected in the interacting, adherent cell fractions (Figure 4d–e).[5,11,12] Despite differences in CD44 expression levels among collected fractions in both perfusion methods, only an increase in CD44lo cells in the free flow subpopulation and an increase in CD44hi cells in the adherent subpopulation collected after continuous flow (Figure 4f) with high and low CD44 stained populations defined on the highest 10% and lowest 10% of the unsorted parent populations (CD44hi and CD44lo, respectively). Contrastingly, CEA expression levels among the two fractions did not differ (Figure 4g–h), and this lack of difference was also reflected in no change in the frequencies of the sorted populations within the high and low CEA stained populations defined on the highest 10% and lowest 10% of the unsorted parent populations (CEAhi and CEAlo, respectively) (Figure 4i). Together, these results implicate a flow-regulated role for CD44 in E-selectin mediated adhesion and suggest a redundant role of E-selectin ligand CEA, as has been previously suggested.[15]
Sorted cells were next costained to interrogate any relative co-enrichment of CD44/HECA(sLex) or CEA/HECA(sLex) expression by flow cytometry. Using mean fluorescence intensity of HECA452 as a proxy, sLex expression in CD44hi and CD44lo LS174T subpopulations (Figure 4j) was found to be enhanced in the CD44hi subpopulation in both Static and Continuous experimentation (Figure 4k). Dissimilarly, when employing the same gating strategy to analyze CEA and HECA coexpression, there was no difference found in sLex expression between the cell subpopulations with either high or low CEA expression (Figure 4l). Collectively, these findings elucidate the interrelation between the adhesion propensity of LS174T cells and their ligand expression, further demonstrating the importance of concurrent high expression of sLex and CD44 in enhancing LS174T cell interaction with E-selectin in flow. Moreover, they illustrate the utility of this platform in probing complex relations between adhesion cell adhesive phenotypes and profiles of ligand expression.
2.5. Adhesion to E-selectin in flow by LS174T cells is a transient phenotypic characteristic not associated with significant mRNA profile expression changes in adhesive ligand genes
In order to elucidate the drivers of invasive cancer metastasis, metastatic potential is often studied by probing genotypic differences among the various subpopulations of metastatic cancer cell populations.[19,24] To interrogate the ability of LS174T cells to retain their metastatic selectin adhesion characteristics for subsequent study, LS174T cells were perfused in Continuous flow experiments and collected into Adherent or Free flow fractions. The subpopulations were either reperfused immediately to confirm enrichment based on adhesivity or placed in culture for 72 hr before analysis in subsequent perfusion experiments. Expectedly, immediate reperfusion resulted in the reperfused Free flow cells showing a decreased adhesion, as indicated by significantly more cells eluting into the Free flow fraction than the other two subpopulations. Correspondingly, the reperfused Adherent cells showed a greater adhesive propensity, and the cells also had a greater expression of sLex than the reperfused free flow cells (Figure 5a–b). When the cultured subpopulations were reperfused under Continuous conditions, however, there was no difference in the relative size of the collected interacting and non-interacting fractions between subpopulations, and HECA452 staining revealed no difference in expression levels (Figure 5a–b). These results reveal E-selectin adhesivity to be a phenotypic characteristic of LS174T that is not sustained.[19]
Figure 5. Adhesivity to E-selectin is a transient phenotype by LS174T cells not caused by mRNA expression changes.

(A) Percent of LS174T cells collected in either the free flow or adherent fraction of the parent (unsorted) population or reperfused cell subpopulations either immediately after perfusion or after 2 passages. Each bar represents the mean ± s.e.m. of n≥3 individually run experiments. (B) Quantification of sLex expression of cells in each collected cell fraction. (C,D,H,I) mRNA expression heatmaps of (C) all analyzed mRNA, (D) genes regulating CEA, CD44, and sLex presentation, (H) actin-cytoskeleton structure, and (I) cell cycle stage. US0/FF0/AD0: unsorted/free flow/adherent, flash frozen at 0 hr; US48/FF84/AD48: unsorted/free flow/adherent, flash frozen at 48 hr. (E-G) Histograms of log2 fold change at 0 hr of genes regulating CEA, CD44, and sLex presentation of (E) free flow compared to adherent, (F) free flow compared to unsorted, and (G) adherent compared to unsorted. (A-C) Perfusion and separation of an LS174T cell pulse of 250,000 cells was over 2.5 μg mL−1 E-selectin at 1 dyn cm−2. * p < 0.05 *** p < 0.001 **** p< 0.0001 by (A) One way ANOVA with post-hoc t-test or (B) t-test.
To investigate the mechanisms underlying this transient metastatic phenotype of LS174T cells, mRNA sequencing was performed and gene expression profiles among various cell subpopulations were quantified and compared. Specifically, parental populations of LS174T colon carcinoma cells were perfused in Continuous flow at 1 dyn cm−2 over 2.5 μg mL−1 and sorted into Free flow and Adherent fractions based on elution time. A subset of each fraction was immediately processed for mRNA extraction and analysis, and the remaining cells from each fraction were cultured separately for 48 hr prior to transcriptome analysis. Additionally, a time-matched, perfused but unsorted population of parental LS174T cells were either immediately or cultured for 48 hr before processing for transcriptome analysis to serve as a control. Subsequently, the LS174T cells sorted in the adhesion chromatography system using E-selectin under Continuous flow conditions were analyzed and the sorted fractions were compared to each other as well as to that of the untreated parental cells.
We hypothesized that since adhesivity is not a retained characteristic of LS174T cells but rather a transient phenotype, the sorted subpopulations of cells analyzed immediately after perfusion would exhibit different expression levels of genes, particularly those related to adhesion, the actin/cytoskeleton structure, and/or cell growth phase, compared to each other, as well as compared to that of the unsorted populations and sorted populations that were analyzed after 48 hr in culture – all of which we expected to all exhibit similar expression levels. Surprisingly, mRNA expression data for all samples showed minimal differences between subpopulations, and hierarchical clustering grouped the samples not by their respective fraction, but rather by the time at which they were analyzed after perfusion (0 versus 48 hr), which is likely a growth phase related artifact (Figure 5c). Given these unexpected results, we analyzed changes in genes regulating sLex, CD44, and CEA expression but again found no significant differences between any group, and the hierarchial grouping still clustering samples by time rather than sorting (Figure 5d). Further, interrogation of fold change of the genes of interest revealed only minor fold changes between all combinations, with a majority of the differences clustering at a fold change of nearly zero (Figure 5e–g). Finally, because changes in the actin cytoskeleton structure and cell cycle stage have been implicated in cancer metastasis, we probed the mRNA profile of those pathways, which again informed similar mRNA expression among all six samples and consistent clustering as to what was seen previously (Figure 5 h–i). Taken together, these results indicate that the transient adhesive phenotypes of metastatic LS174T subpopulations is not reflected in changes mRNA profile of genes controlling adhesive ligand presentation, despite measured differences of both functional adhesive propensity on E-selectin and surface presented sLex and CD44 ligand expression. Notably, due to the lack of measurable changes in expression of pathways we hypothesized would control cell adhesivity to E-selectin, biological replicates were not conducted and transcriptome analysis was not performed.
2.6. LS174T cell E-selectin adhesive phenotype association with cancer stem cell features
The relationship between a molecular marker of cancer stem cells and adhesive phenotype on E-selectin in flow for LS174T cells were investigated through costaining sorted populations with CD24 and either HECA452, CD44, or CEA (Figure 6a–c). Upon evaluation of the expression of E-selectin ligands and cancer stem cell marker CD24, the highest 10% of HECA (sLex) and CEA expressing cells were found to exhibit higher levels of CD24 expression (Figure 6c,e).[25] Contrastingly, the lowest 10% of CD44 expressing cells correlated with higher levels of CD24 expression (Figure 6b). Using the adhesion chromatography system to enrich cells on 2.5 μg mL−1 E-selectin at 1.0 dyn cm−2, the effect of E-selectin adhesivity on the frequencies of CD24 expressing cells within adhesive versus non-adhesive (free flow) subpopulations was measured. Irrespective of perfusion type (Continuous or Static), CD24 expression levels did not differ with adhesive phenotype (Figure 6d, Figure S2a), nor was there a difference in the frequency of CD24hi and CD24lo cells in either fraction (Figure 6e, Figure S2b). When assessed as coexpression relationships via quadrant analysis, no differences in HECA452, CD44, or CEA with CD24 were observed for Static experiments (Figure S2c–e). However, under Continuous flow conditions, frequencies of HECAlo/CD24lo were increased for cells exhibiting a lowly adhesive phenotype (Figure 6f). Adherent LS174T cell subpopulations also exhibited decreased frequencies of CD44lo/CD24hi cells (Figure 6g) and increased levels of both CD44hi/CD24hi and CEAlo/CD24lo cells (Figure 6g,h).
Figure 6. CD24 expression does not differ between LS174T cells that do or do not exhibit E-selectin adhesivity.

(A-C) Representative flow cytometric histograms of sLex (HECA452), CD44, or CEA expression indicating gating for the top and bottom 10% percent of cells expressing the respective ligand and the CD24 MFI in the highest versus lowest 10% of HECA, CD44, or CEA expressing cells. (D) Average mean fluorescent intensity of cells obtained from the free flow and adherent fractions in continuous flow. (E) Percentage of cells in the top or bottom 10% of CD24 expressing cells after continuous flow. (D,E) Each bar represents the mean ± s.e.m. of n≥3 individually run experiments. (F-H) Comparisons between frequencies of cells in low versus high selectin ligand expression derived from costaining. (A-C, F-H) Each bar represents the mean ± s.e.m. of n≥3 individually run experiments. Perfusion and separation of an LS174T cell pulse of 250,000 cells was over 2.5 μg mL−1 E-selectin at 1 dyn cm−2. * p < 0.05 ** p < 0.01 by paired t-test.
3. Discussion
We demonstrate the ability to rapidly sort in an unbiased, label free manner large populations of cells based on their adhesion propensity in flow through the use of an engineered, microfluidic based adhesion chromatography platform. Using residence time as a proxy for time-averaged cell adhesive behavior to collect two distinct fractions of cells – adhesive (interacting) and free flow (non-interacting), this platform enables not only real-time visualization of cellular adhesion under hemodynamic force, but also allows post-perfusion phenotypic analysis and in vivo modeling of the fractionated cell subpopulations under defined flow conditions. Noting that increased adhesion in vitro correlated with enhanced metastatic potential in vivo by LS174T colon carcinoma cells, we demonstrate this sorting platform can be implemented to better understand the complex process of cancer metastasis by probing interactions between selectins and glycosylated ligands expressed by metastatic cells. We furthermore demonstrate the utility of this system in the interrogation of phenotypic attributes associated with E-selectin adhesion transiently exhibited by metastatic LS174T cells, noting that the observed phenotypic differences are likely not due to mRNA expression profile changes.
This cell adhesion chromatography system permitted the interrogation of attributes shared within populations of cells that exhibit similar adhesion phenotypes. Notably, sLex and CD44, but not CEA, were found to have enhanced expression on LS174T cells that were able to mediate adhesive interactions with E-selectin in flow. With respect to endogenous sLex expression, this finding is consistent with the existing literature.[19] CEA was also not found to be enriched in either collected subpopulation of cells, which is supported by knockdown of CEA expression in LS174T cells failing to alter adhesive behavior on E-selectin in flow.[29] Previous reports, on the other hand, have demonstrated a correlation between CD44 expression and selectin-mediated adhesion over P- and L-, but not E-selectin, which is in contrast with our findings of CD44 expression being enhanced with increased E-selectin adhesivity.[14] However, this noted positive correlation between adhesion propensity on E-selectin in flow and CD44 expression is in direct agreement with previous reports that also found higher levels of rolling behavior over E-selectin in cells with enhanced CD44 as well as sLex expression.[16,19] Our findings highlight the advantages of our cell-sorting microfluidic platform over systems used in previous reports for our integrated system permits the analysis of the relationship between multiple cell parameters (adhesive phenotype and ligand expression levels) of unperturbed, distinct metastatic subpopulations based on population averages without the need for cumbersome cell knockdown studies, limitations of single level cell measurements, or repeated sorting of cells as a result of the ability to permit rapid interrogation of a large number of cells in a relatively short period of time.
The apparent discrepancy between the lack of differential mRNA profiles and observed functional changes in adhesive propensity and E-selectin ligand presentation among sorted cell populations suggest that the protein expression changes result from post-transcriptional and/or post-translational regulatory processes.[30] One hypothesis is that the functional changes are a result of post-transcriptional alterations in microRNA (miRNA), which were not measured in this work. These results are consistent with previous reports that have noted only half of all protein expression changes are correlation with mRNA expression changes and post-translational repression of E-selectin ligand presentation on metastatic colon cancer cells via miRNA being implicated in regulating E-selectin mediated adhesion.[26–28] Therefore, further analysis of the genomic profile of the sorted and unsorted cell fractions transcriptomally analyzed either immediately or cultured for 48 hr by analyzing and comparing miRNA expression levels, and subsequently knocking-out any identified miRNA and quantifying adhesivity alterations using our microfluidic platform, may provide further insight into the phenotypic changes causing functional adhesive differences between subpopulations and corroborate our proposed explanation for the reported discrepancies in mRNA expression and adhesive ligand protein expression. In addition to unexplored potential post-translational differences, additional potential post-transcriptional changes include alterations in the length of proteins or untranslated regions of mRNA.[30]
Correlations between adhesive phenotype and cancer stem cell markers were assessed in LS174T cells by analyzing the cell subpopulations fractionated for adhesion to E-selectin under flow for CD24 expression, a cancer stem cell marker whose expression is implicated in increased tumor progression, motility, and metastatic potential.[16,25,31,32] We found that CD44 expression was inversely proportional to CD24 expression, noting the enrichment of CD24hi cells in a CD44lo subpopulation, generally. This observation may give further insight into reports that demonstrate in vivo CD44-knockdown experiments lead to increased lung metastasis, despite our finding that high CD44 expression is linked with increased levels of adhesion on E-selectin, as the CD44 knockdown cells are subsequently augmenting a cancer stem cell subpopulation.[15,19,29]
Together, the noted increase in a CD44hi/CD24hi subpopulation among adherent cells and increase in a CD44lo/CD24hi subpopulation among free flow cells suggest that CD44 alone is predictive of adhesive phenotype, suggesting that there is no direct correlation between CD24 expression and adhesion on E-selectin under flow. Notably, these results conflict with what we recently measured for these cells when analyzed for E-selectin adhesion potential, however, those results were at a single cell, rather than population, level and obtained at 0.5 dyn cm−1, under which conditions a significantly higher fractions of cells mediate adhesion in flow (by ~50%, Figure 2i), which may explain the discrepancy. Furthermore, despite a lack of expression level differences between CEA in sorted populations, we found an increased CEAlo/CD24lo subpopulation in the Adherent subpopulation.[19] Interestingly, although CEA expression may not be correlated with adhesive differences on E-selectin, CEAlo cells have been found to have increased cancer stem cell like properties, enhanced levels of sphere formation in vitro, and enhanced metastatic potential in vivo.[33] Therefore, using our cell sorting platform, we are inadvertently able to enrich for this population. Low adhesive potential has previously been correlated with increased cancer stem properties in melanoma and breast cancer.[34] Together our results indicate a lack of correlation between cancer stem cell marker CD24 and adhesion propensity on E-selectin in LS174T colon carcinoma cells.
Our results suggest a unique capacity for physiological force from fluid flow in influencing mechanisms of metastatic dissemination. Specifically, cell fractions collected via the Continuous perfusion method exhibited significant differences in ligand expression and coexpression between subpopulations that were not found in cell fractions collected via the Static perfusion method (Figure 4 and 6). We hypothesize that this is a result of forced contact between cell-expressed selectin ligands and the functionalized substrate in static flow experiments allowing a greater number of low affinity receptor-ligand interactions to occur in the free flow fraction that are not able to form in continuous flow experiments with a constantly applied shear stress. Thus, despite not changing the approximate frequency of adhesive cells of the total population, subtle differences in the collected cell subfractions were diluted in the absence of the dispersive effects of flow. Due to the continuous nature of fluid flow through the vasculature microenvironment in which cancer extravasation occurs, Continuous perfusion could be considered a more psychologically relevant experimental method for future investigations elaborating the mechanisms and biology of cancer metastasis.
In closing, we have engineered an integrated, parallel-plate microfluidic device to fractionate cells based on their elution time from a flow channel as a proxy for cell adhesivity to a substrate surface functionalized with adhesive ligand to investigate the pathway of cancer extravasation. Used here, wall shear stress-levels were modeled after hemodynamic flow of the vasculature microenvironment in order to simulate conditions under which in vivo metastasis occurs in vitro. By separately collecting two fractions of cancer cells - non-interacting and interacting – the cellular populations can be subsequently analyzed, reperfused, or modeled in vivo after perfusion, allowing interrogation of a large number of cells in a tightly controlled flow environment. This platform enabled the interrogation of cell molecular profiles, such as (co)expression levels of natively expressed selectin ligands (sLex, CD44, and CEA) and cancer stem cell marker CD24 on circulating cancer cells, as it relates to adhesive phenotype on a subpopulation level based on adhesive propensity, thereby probing differences that may underlie varying adhesive phenotypes of a heterogeneous metastatic cancer cell population. Using the microfluidic analysis platform to study the behavior of cancer cells in the vascular microenvironment, and therefore relating the phenotypic characteristics of a cancer cell with their adhesive potential, will inform novel biomarkers that can be used to identify cancer cells and identify molecular pathways that can be targeted with therapeutics to prevent cancer metastasis.
4. Experimental Section
Cell Culture:
Human colorectal adenocarcinoma LS174T cells and mouse melanoma B16F10 cells were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% heat inactivated fetal bovine serum (FBS) and 1% penicillin-streptomycin-amphotericin (Life Technologies). The cells were harvested via mild trypsinization with 0.25% trypsin EDTA and 0.05% trypsin EDTA respectively, centrifuged at 400 X G for 5 min, and resuspended in culture medium. Cells were either used for perfusion experiments or diluted into tissue culture treated cell flasks for subculture. Prior to use in perfusion experiments, the LS174T cells were incubated in suspension for 2 hr at 37°C to allow cell surface glycoprotein regeneration. Quantification of cell confluency was performed by analyzing the area of adherent cell cultures compared to the total surface area of the well surface bottom using ImageJ (National Institutes of Health). The confluency at various time points was normalized to that at the time of seeding the cells.
Channel Fabrication:
Microfluidic channels were fabricated as detailed previously [35]. Concisely, the microfluidic channels were made using 100 um thick double-sided adhesive tape (3M) backed with a release liner into which a U-shaped channel of two parallel 2 cm wide by 14 cm long sections connected by a 2 cm wide by 1.5 cm long section was cut using a craft cutter (Silhouette America). The adhesive channels were then affixed to PDMS (Ellsworth Adhesives), which was previously cured by mixing PDMS base with curing agent at a ratio of 9:1 and curing for 3 hr at 90°C in a Pyrex dish. To finish fabrication, an inlet hole was punched into channel using a biopsy punch, and assembly was attached to a non-tissue culture treated polystyrene plates where an outlet hole was drilled.
Channel Functionalization:
The channel was functionalized by incubating with 2.5 μg mL−1 Fc specific anti-IgG (R&D Systems) in Dulbecco’s Phosphate Buffered Saline (D-PBS) without calcium and magnesium at 4°C overnight, blocking with 1% BSA in D-PBS at room temperature for 1 hr, incubating with 2.5 μg mL−1 of E-selectin (R&D Systems) in D-PBS with calcium and magnesium at room temperature for 2 hr, and finally, blocking the entire device with 1% BSA in D-PBS at room temperature for 1 hr In between each step, the channel was washed three times with 1 mL of D-PBS with calcium and magnesium. For Static flow experiments, the channel was functionalized from the outlet up to 1 cm from the inlet. For Continuous flow experiments, only the straight portion nearest the outlet was functionalized.
Perfusion Experiments:
An inlet syringe coupled to tubing, which was filled with perfusion media (0.1 % BSA in D-PBS), were connected to a syringe pump (PhD Ultra Harvard Apparatus). The syringe pump was used to withdraw a cell pulse of 250,000 cells diluted in 200 μL of perfusion media into the inlet tubing at a rate of 0.5 mL min−1. The tubing-syringe pump was inserted into the inlet hole of the channel and a 5 mL test tube was connected to the bottom of the outlet hole as the cell collection reservoir. The entire apparatus was placed on an Eclipse TI optical microscope (Nikon) with an objective magnification of 10X and linked to NIS-Elements software (Nikon) to acquire videos at 0.281 μs and a frame rate of 25 frames per sec. To begin perfusion, the syringe pump was set to a flow rate to achieve the desired wall shear stress to initiate inflow from the syringe-tubing assembly. In Static flow experiments, perfusion was paused after two min of inflow to allow the cells to settle to the bottom of the channel and interact with the functionalized surface. After five min of stopped flow had transpired, flow was restarted for the predetermined free flow elution time of the cells (the time it takes for 95% of the cell pulse to elute out of an unfunctionalized channel), after which perfusion was stopped and the test tube containing the cell solution collected during perfusion was removed and a new test tube was attached. Finally, the syringe with inlet tubing was replaced with a new syringe-inlet tubing apparatus filled with only perfusion media and used to eject the cell solution out of the channel and into the second collection tube. In Continuous flow experiments, perfusion was stopped after the free flow elution time had been reached. At the end of the experiment, the number of cells and nonviable cells were counted, then analyzed using a BD LSRII flow cytometer (BD Biosciences) for expression of various ligands or put back into cell culture.
Cell Fraction Quantification and Cell Viability Analysis:
The collected cell fractions were centrifuged at 400 X G for 5 min, the supernatant was decanted, and the cell pellet resuspended in 200 μL of perfusion media. Trypan Blue 0.4% (Life Technologies) was diluted 1:1 with D-PBS. The 0.2% solution was mixed with collected cells in a 1:1 ratio and incubated at room temperature for 2 min. A sample of the cell-trypan solution was then loaded onto a hemocytometer and viewed under a microscope to count the total number of cells collected in each fraction as well as the fraction of non-viable cells and the viable cells.
Flow Cytometric Analysis:
The cell solution were centrifuged at 400 X G for 5 min and resuspended in diluted antibody solutions on ice for 45 min, washed with D-PBS two times, and resuspended in perfusion media for analysis on the flow cytometer. The antibodies (BD Biosciences) were diluted in D-PBS in the following ratios – 1:20 for PE CD44 and PE CEA, 1:40 for APC or PE HECA452, and 1:10 for APC CD24. Obtained flow cytometric data was analyzed using FlowJo software (Treestar Inc.).
Video Analysis:
The mean distance of the cells from the channel bottom at any given time was experimentally determined and calculated as previously described.[11] Briefly, a cell pulse followed by perfusion media was perfused into an unfunctionalized channel and five min videos were taken with the focal place set to the channel bottom at nine evenly spaced positions across the channel length. The videos were subsequently analyzed using an OpenCV-based Traffic Flow Analyzer cell tracking software that is able to detect cell edges and their x and y position, and consequently determine the cell velocity according to Equation (1). The metric yvel/0.5(rx+ry) was used to represent the distance of a cell from the channel bottom based on its horizontal velocity.
| (1) |
Cell Pulse Location Quantification:
To determine the location of the cell pulse when perfusion is paused during static flow experiments, inflow of a 200 μL cell pulse at 1.25×106 cells mL−1 followed by perfusion media into an unfunctionalized channel was started for two min, stopped, then images were taken at evenly spaced positions along the length of the channel and the number of cells at each position were counted. Alike, the number of interacting cells in functionalized channels was quantified by taking 30 sec videos across nine evenly spaced length positions in the channel and three evenly spaced width positions at each length position after steady flow had been reached, and previously described metrics were used to distinguish interacting from non-interacting cells.[14] Briefly, in perfusion experiments, a cell that paused within imaging field of view or translated substantially slower than the free flow velocity was defined as an “interacting” cell, whereas a “non-interacting” cell was defined as one that which did not mediate contact with the channel bottom during observation.[11]
Free Flow Elution Time Quantification:
The free flow elution time of the cells was determined by modifying a procedure previously used to find the residence time of cells in a microfluidic channel.[11] 30 min videos of 250,000 cells perfused as a 200 μL cell pulse at a flow rate corresponding to the desired wall shear stress were taking 0.5 cm from the channel outlet. The aforementioned cell tracker code set to threshold of 4 and a blurring factor of 7 was used to identify objects that had a diameter between 12–18 μm that had traveled more than 400 μm during the course of the video and record the time it took the object to leave the field of view after perfusion had begun. The values were put into Prism (GraphPad Software Inc) to create cumulative frequency plots. The elution time of free flow cells was defined as the time at which 95% of the tracked cells had left the field of view.
In Vivo Metastasis Model:
104 LS174T cells, either from the unsorted parental population or fractionated using the adhesion chromatography system, were suspended in 100 μL of sterile saline and each fraction was injected intravenously via the tail vein into six week old female NSG mice under isoflurane anesthesia (Jackson Labs). After 7 or 14 d, mice were sacrificed by CO2 asphyxiation, lungs dissected, and metastatic foci counted. Afterwards, the lungs were flash frozen in optimal cutting temperature (OCT) gel in a bath of 2’methylbutane cooled in a bath of liquid nitrogen then maintained in a freezer at −80°C until cryosectioning into 10μm slices, staining with hematoxylin and eosin (H&E), and imaging with a Nanozoomer microscope (Hamamatsu Photonics). Cell densities were quanitified as previously described.[35] Briefly, H&E stained histology images were imported into ImageJ using the NDPI tools plugin [36] and thresholded using identical parameters across all images to quantify the percent area above the threshholded background to quantify the cell density of each cryosection. All protocols for animal procedures were approved by the Institutional Animal Care and Use Committee at Georgia Institute of Technology.
mRNA Extraction and Sequencing
The cell samples used to for mRNA extraction were flash frozen using liquid nitrogen either immediately after perfusion or after 48 hr of culture. Total RNA was isolated by the RNeasy Micro Kit (Qiagen) according to the manufacturer’s instructions. Briefly, frozen cell pellets (35 cells per pellet) were immediately suspended in RLT Plus lysis buffer in the presence of beta-mercaptoethanol. Lysates were homogenized via centrifugation (at maximum speed for 2 min) using Qiashredder columns. One volume of 70% ethanol was added to the lysate, and mixed via pipetting. The samples were transferred to RNeasy MinElute spin columns and centrifuged for 15 sec at 8000 X G. After washing column with RW1 buffer, on column DNase digestion was performed. After on-column washing steps, columns were dried via centrifugation at full speed for 5 min. RNA was eluted in 50 μL of RNase-free water. RNA concentration and purity were determined via a NanoDrop spectrophotometer. Once the mRNA was extracted, The New England Biolabs (NEB) mRNA isolation kit was used to pull down mRNA using poly T beads. Next, the NEB Ultra II RNA directional kit was used to convert the mRNA to double stranded cDNA, utilizing illumine adapters, which were added to each fragment in order to sequence the mRNA.
mRNA Sequencing Analysis
In preparing the RNA-Seq data from 6 LS174T samples for the differential gene expression analysis, quality control was performed on 12 raw FASTQ files using FastQC (Babraham Informatics) and Trimmomatic (Usadel Lab) to ensure the quality of the data. The quantification of the gene expression levels for the 6 LS174T samples was performed using Salmon.[37] The gene counts were imported using a R package to prepare a count matrix for the downstream exploratory and gene-level analysis.[38] For the analysis involving the single replicates, GFOLD was used to observe changes in gene expression.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health R21 CA202849 and NIH T32 GM-008433.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References
- [1].Weigelt B, Peterse JL, van ‘t Veer LJ, Nat. Rev. Cancer 2005, 5, 591. [DOI] [PubMed] [Google Scholar]
- [2].Konstantopoulos K, Thomas SN, Annu. Rev. Biomed. Eng 2009, 11, 177. [DOI] [PubMed] [Google Scholar]
- [3].Steeg PS, Nat. Rev. Cancer 2003, 3, 55. [DOI] [PubMed] [Google Scholar]
- [4].Hunter KW, Crawford NP, Alsarraj J, Breast Cancer Res. BCR 2008, 10, S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lote H, Spiteri I, Ermini L, Vatsiou A, Roy A, McDonald A, Maka N, Balsitis M, Bose N, Simbolo M, Ann. Oncol 2017, 28, 1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Thomas SN, Schnaar RL, Konstantopoulos K, Am. J. Physiol. - Cell Physiol 2009, 296, C505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Borsig L, Wong R, Hynes RO, Varki NM, Varki A, Proc. Natl. Acad. Sci. U. S. A 2002, 99, 2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kim YJ, Borsig L, Varki NM, Varki A, Proc. Natl. Acad. Sci. U. S. A 1998, 95, 9325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Köhler S, Ullrich S, Richter U, Schumacher U, Br. J. Cancer 2010, 102, 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Läubli H, Borsig L, Cancer Microenviron 2010, 3, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Oh J, Edwards EE, McClatchey PM, Thomas SN, J. Cell Sci 2015, DOI 10.1242/jcs.166439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lech G, Słotwiński R, Słodkowski M, Krasnodębski IW, World J Gastroenterol 2016, 22, 1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Baumann P, Cremers N, Kroese F, Orend G, Chiquet-Ehrismann R, Uede T, Yagita H, Sleeman JP, Cancer Res 2005, 65, 10783. [DOI] [PubMed] [Google Scholar]
- [14].Edwards EE, Oh J, Anilkumar A, Birmingham KG, Thomas SN Oncotarget 2017, 8, 83585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Thomas SN, Zhu F, Schnaar RL, Alves CS, Konstantopoulos K, J. Biol. Chem 2008, 283, 15647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Napier SL, Healy ZR, Schnaar RL, Konstantopoulos K, J. Biol. Chem 2006, 282, 3433. [DOI] [PubMed] [Google Scholar]
- [17].Hanley WD, Napier SL, Burdick MM, Schnaar RL, Sackstein R, Konstantopoulos K, FASEB J 2006, 20, 337. [DOI] [PubMed] [Google Scholar]
- [18].Alves CS, Yakovlev S, Medved L, Konstantopoulos K, J. Biol. Chem 2009, 284, 1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Edwards EE, Birmingham KG, O’melia MJ, Oh J, Thomas SN, Cell Syst n.d., In print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Konstantopoulos K, Kukreti S, McIntire LV, Adv. Drug Deliv. Rev 1998, 33, 141. [DOI] [PubMed] [Google Scholar]
- [21].Chen S, Springer TA, J. Cell Biol 1999, 144, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rutter A, Hugenholtz H, Saunders JK, Smith ICP, Neurochem J. 1995, 64, 1655. [DOI] [PubMed] [Google Scholar]
- [23].J. Exp. Med 1996, 183, 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nguyen A, Yoshida M, Goodarzi H, Tavazoie SF, Nat. Commun 2016, 7, 11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Schneider M, Huber J, Hadaschik B, Siegers GM, Fiebig H-H, Schüler J, BMC Cancer 2012, 12, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang M, Matyunina LV, Walker LD, Chen W, Xiao H, Benigno BB, Wu R, McDonald JF, Sci. Rep 2017, 7, 8171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhong L, Huot J, Simard MJ, Sci. Rep 2018, 8, 2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhong L, Simoneau B, Huot J, Simard MJ, Oncotarget 2016, 8, 1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dallas MR, Liu G, Chen WC, Wirtz D, Huso DL, Konstantopoulos K, 2012, 26, 4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Vogel C, Marcotte EM, Nat. Rev. Genet 2012, 13, 3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Aigner S, Sthoeger ZM, Fogel M, Weber E, Zarn J, Ruppert M, Zeller Y, Vestweber D, Stahel R, Sammar M, Blood 1997, 89, 3385. [PubMed] [Google Scholar]
- [32].Yan C, Hu Y, Zhang B, Mu L, Huang K, Zhao H, Ma C, Li X, Tao D, Gong J, Oncotarget 2016, 7, 80700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang Y, Wu M, Han X, Wang P, Qin L, Angew. Chem. Int. Ed Engl 2015, 54, 10838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tran R, Myers DR, Denning G, Shields JE, Lytle AM, Alrowais H, Qiu Y, Sakurai Y, Li WC, Brand O, Mol. Ther 2017, 25, 2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jensen EC, Anat. Rec 2013, 296, 378. [DOI] [PubMed] [Google Scholar]
- [36].Deroulers C, Ameisen D, Badoual M, Gerin C, Granier A, Lartaud M, Diagn. Pathol 2013, 8, DOI 10.1186/1746-1596-8-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C, Nature Methods 2017, 14, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Love MI, Anders S, Kim V, Huber W, F1000Res 2015, 4, 1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.