

Abstract

The synthesis of shape-persistent organic cage compounds by the formation of imine bonds opens the possibility to realize cages of different sizes, geometries, topologies, and functions. It is generally assumed that the imine bond is rather chemically labile allowing a self-correction mechanism until thermodynamic equilibrium is reached, which is often the case if a cage is formed. However, there are some contradictory experimental data to this assumption. To get a deeper insight into the imine bond dynamics of covalent organic cages, we studied the formation and exchange of both dialdehydes and triamines of two different [2 + 3] imine cages with the aid of a deuterated dialdehyde molecular building block.
Introduction
The interest in shape-persistent organic cages has increased significantly since a few years.1 This is mainly attributed to the relatively easy access to even larger structures from rather simple molecular building blocks through the formation of imine bonds.1d,1g,2 By this reaction, a large variety of cages with different sizes and geometries have meanwhile been reported.1a,3,4 Based on the assumption that the bond formation and opening of the imine units is dynamic,5 it is generally proposed that the imine cages are thermodynamically controlled products rather than kinetically controlled ones. It is worth mentioning that (almost) all theoretical calculations predicting cage structures are based on this assumption.6,7
Indeed, a number of experimental findings somehow support this hypothesis. Some pioneering work was reported by Warmuth et al., investigating cage formations based on a resorcinarene tetraaldehyde with various diamines and triamines.8 In different solvents, the same molecular building blocks gave cages of different geometries. Although this is still not fully understood, it demonstrates the synthetic potential of dynamic covalent chemistry. Unfortunately, no clear evidence for the dynamic nature of the imine bonds was given, for example, by switching between the cages by switching or adding solvent in situ. Mukherjee and co-workers have investigated a four-component system (two bis-aldehydes plus two triamines) and observed a highly selective self-sorting, giving two products exclusively.9 Based on experimental data on cage stabilities and formation, possible exchange of molecular building blocks was elaborated and later on studied in more detail:10 a thermodynamic, less stable cage was transferred to a more stable one by adding either another triamine or dialdehyde. A similar observation was made by the Li group.11 Cram and Stoddart suggested an imine exchange of a carcerand by an acid-catalyzed bar-opening/bar-closing mechanism by exchanging diamine units.12 Very recently, Kołodziejski et al. presented a very detailed study of the self-sorting and exchange of aldehyde precursor units of TREN-based cages.13 We and other researchers studied chiral self-sorting, which also relies on a dynamic interconversion of imine bonds.4g,14 Most convincing experiments to demonstrate the dynamics of the imine bond formation were done by the Li group by investigating the interconversion of a pillared [2 + 3] cage into a tetrahedral [4 + 4] cage under different conditions. Depending on temperature or concentration, a clear shift toward one of these two cages was observed.15 No exchange of other building blocks was needed.
Despite the large number of examples supporting a thermodynamically controlled formation of imine cages, there are also observations made, proposing that imine cages are rather kinetically controlled products. For instance, the formation of a cubic [4 + 4] salicylimine cage was monitored by time with matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry (MS).1a,4h At no time any oligomeric byproducts with m/z-values larger than the cage have been found, suggesting that the formation of the cage is a kinetically controlled reaction. Here, the cage formation is driven by precipitation, similar to that for most salicylimine cages described previously.4e,4g,16 One clear example of a kinetically controlled product is the formation of truncated tetrahedral [4 + 4] imine cages that have been made in acetonitrile.17 As soon as trifluoroacetic acid is added, the cages decompose to give an insoluble polymeric material, which turned out to be a crystalline covalent-organic framework.17 As mentioned above, computational screening of large combinatorial libraries of possible cages relies on the assumption that these are thermodynamically favored, and exactly these truncated [4 + 4] cages were predicted not to form.6b Besides the “lost hit” of the truncated tetrahedral [4 + 4] cages, there are a few more examples, where such calculations were not able to explain the experimentally found preferred product. In this respect, Cooper and co-workers observed the cage enlargement of a [4 + 6] to an [8 + 12] imine cage, despite the theoretical calculated preference for the smaller one.6c The same group observed the formation of open-pot cages preferably to a social self-sorted mixture of [4 + 6] and [4 + 4] imine cages despite the calculated thermodynamic favor toward the mixture.7
Because of all these “contradictory” findings, we were interested in investigating the dynamics of the imine bonds of organic cages in more detail, once these have formed. To exclude any driving forces by electronic differences of the used amines or aldehydes or different solubility of either reactants, intermediates, or cages, we decided to look at scrambling of [2 + 3] imine cages and their deuterated congeners under different conditions, which we describe herein.
Results and Discussion
Synthesis and Characterization of Authentic Material
According to previous protocols,13,18,19 the imine cages 4, 4-d27, 5, and 5-d27 were synthesized by stirring the corresponding aldehydes 1 or 1-d9 with the triamines 2 or 3 in methanol for 2 days to give cages in isolated yields between 53 and 99%, respectively (Scheme 1). All cages were fully characterized by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy, MS, IR, and elemental analysis (see the Supporting Information). Furthermore, the cages were characterized by single-crystal X-ray diffraction (Figure 2).
Scheme 1. Synthesis of [2 + 3] Imine Cages 4 and 5 and Their Deuterated Congeners.

Figure 2.
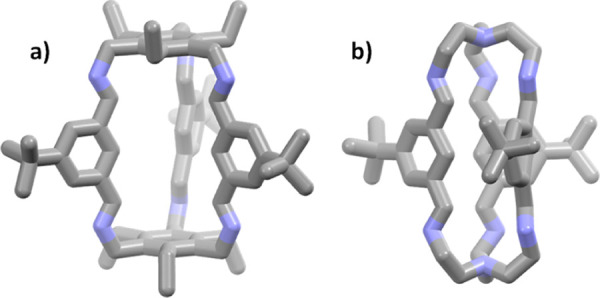
X-ray crystal structures of cages 4 (a) and 5 (b). Hydrogen (deuterium) atoms and solvent have been omitted for clarity.
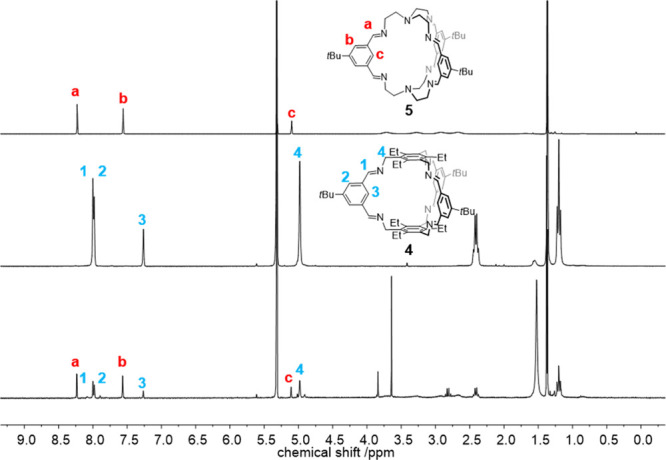
The incorporation of the deuterated tert-butyl groups (tBu-d9) into cage 4-d27 was unambiguously proven by missing the signal of the tert-butyl protons at δ = 1.37 ppm in the 1H NMR spectrum, which was previously observed for cage 4 (see Figure 1). Similar observation was made for the pair 5 and 5-d27 (here the tert-butyl protons resonate at 1.39 ppm). More important for our studies is the clear differentiation by MALDI-TOF MS. The molecular isotopic peak for cage 4 ([M + H]+) appears at m/z = 961.684 and is shifted by 27 mass units to m/z = 988.853 for cage 4-d27 ([M + H]+). For cage 5 and its deuterated congener 5-d27, the peaks are found at m/z = 755.549 ([M + H]+) and 782.718 ([M + H]+), respectively.
Figure 1.

1H NMR spectra (CD2Cl2, 600 MHz) of cage 4 and 4-d27 (a,b) and 5 and 5-d27 (c,d) for comparison.
Solvent Effect on Cage Formation
As mentioned above, several conditions (different solvents, presence or absence of catalytic amounts of acid) have been reported for the synthesis of cages. For [2 + 3] imine cages of the general structure of 4, typically EtOH, MeOH, CH2Cl2, CHCl3, or acetonitrile are reported.18a,18b,18d,18e,19a,19c Since we found for [4 + 4] cages that these are formed exclusively in acetonitrile without acid, a solvent screening (with and without acid) was done for the cage synthesis of 4 and 5 to study its influence (see Schemes 2 and 3 and Tables 1 and 2 for NMR analytics, see the Supporting Information, Figures S46–S49). It is worth mentioning that although all reactions were performed at least twice, we were not interested in determination of exact yields, but in estimating the number of formed cages in solution and in the solid state of a formed precipitate. If a precipitate was formed, it was collected by filtration, dried, weighed, and analyzed by 1H NMR spectroscopy. The remaining mother liquor was dried, a defined amount of 1,3,5-trimethoxybenzene added as internal standard, and investigated by 1H NMR spectroscopy to estimate the amount of formed cages by integration of a characteristic proton signal (4: δ = 7.18 ppm, 5: δ = 7.59 ppm) and 1,3,5-trimethoxybenzene (δ = 7.09 ppm).
Scheme 2. Formation of Cage 4 under Various Conditions.

Scheme 3. Formation of Cage 5 under Various Conditions.

Table 1. Formation Experiments of Cage 4 in Different Solvents with and without Addition of TFAa.

Reaction time: 2 days; reaction temperature: room temperature.
Calculated from 1H NMR spectrum with the addition of 1,3,5-trimethoxabenzene as standard.
Sum of yield of precipitate and mother liquor.
No precipitate formation observed after reaction.
Table 2. Formation Experiments of Cage 5 in Different Solvents with and without Addition of TFAa.

Reaction time: 2 days; reaction temperature: room temperature.
Calculated from 1H NMR spectrum with the addition of 1,3,5-trimethoxabenzene as standard.
Sum of amount of cage 5 of precipitate and mother liquor.
No precipitate observed after reaction.
First, we investigated the formation of cage 4 in different solvents without acid. With the exception of dichloromethane (DCM) and toluene (entries 4 and 8), in all other solvents the pure cage precipitated in yields between 40 and 80%. The highest yield (80%) was observed in MeOH (entry 1), the lowest in EtOH (40%, entry 2). It is worth mentioning that in MeOH, MeCN, and tetrahydrofuran (THF), no cage was found in the residual mother liquor, suggesting that it is little soluble herein and precipitation is the driving force. As mentioned above, in DCM and toluene (entries 4 and 8), no precipitate was formed, but about 40% of cage formation was detected in solution. The same reactions were performed with the addition of 2 mol % of trifluoroacetic acid (TFA). In general, the observed trends were comparable to the experiments without acid, with the only difference being that in all solvents (with the exception of methanol) the yields increased.
The formation of TREN-cage 5 was investigated under the same conditions as cage 4 (see Scheme 3 and Table 2, for NMR analytics see the Supporting Information, Figures S50–S53). In contrast to cage 4, cage 5 was much more soluble and stayed in solution in most solvents, except in MeCN, THF, and dioxane (entries 3, 6, and 7). The yields were close to quantitative in the protic solvents MeOH and EtOH (entries 1 and 2), and NMR spectra were as clean as those of the precipitates mentioned above. The remaining solutions of the reactions in MeCN, dioxane, and THF contained besides cage 5 significant amounts of other species as can be seen by additional signals in the related 1H NMR spectra that cannot be assigned to any cage protons. In DCM, toluene, and CHCl3 (entries 4, 5, and 8), no precipitate was observed and cage 5 was formed in solution; however, especially in toluene and DCM, the formation was not nearly as clean as from EtOH. Comparable to the experiments for cage 4, here the addition of a catalytic amount of TFA has the same effect and cage 5 was formed in substantially higher yields, in MeOH even quantitatively. It is worth mentioning that for these solvents, the formation was less clean (toluene, DCM, and MeCN); only in DCM, the appearance of byproducts vanished upon addition of TFA.
Exchange Experiments
To investigate the dynamic nature of the imine bond, cage 4 and cage 4-d27 were mixed in approx. equal amounts, treated under the same conditions as described above, and the slurry or solution was investigated by MALDI-TOF mass spectroscopy (MS) (Scheme 4 and Table 3 see also Figure 3). The only difference in the conditions above is the elongation of reaction time to 7 days to allow the system to adjust thermodynamic equilibrium.
Scheme 4. Scrambling of Cage 4 and Cage 4-d27 at Various Conditions (See Table 3).

Table 3. Scrambling Experiments of Cage 4 and 4-d27 in Different Solvents with and without Addition of TFA.

Observed in MALDI MS.
After addition of water.
Please note that the intensity of the found signals for 4-d9 and 4-d18 is very low.
Figure 3.

MALDI mass spectra of scrambling reactions of cage 4 and cage 4-d27 according to Scheme 4 and Table 3 without TFA and with water (entries 1–8).
First, we screened the exchange without the addition of acid. Only in DCM very small peaks of 4-d18 and 4-d9 were observed by MALDI-TOF MS (see the Supporting Information, Figures S54–S57). Although the solvents were not dried to ensure that water is present in reasonable amounts to allow the possibility of imine bond hydrolysis, water was added and the mixtures stirred for another 7 days. Basically, the results were the same. Even the addition of catalytic amounts of acid did not foster exchange in other solvents than DCM, where small peaks of scrambled cages are observed by MS, concluding that at least reasonable solubility is necessary to allow dynamic exchange.
TREN-based cages 5 and 5-d27 were treated the same (Scheme 5 and Table 4, see the Supporting Information, Figures S58–S61). Although cage 5 is soluble in a larger number of solvents than cage 4, under no conditions exchange was observed, concluding that the aliphatic imine bond of the TREN precursor is even more stable than the one of the benzylic imine in cage 4.
Scheme 5. Scrambling of Cage 5 and Cage 5-d27 at Various Conditions (See Table 4).

Table 4. Scrambling Experiments of Cage 5 and 5-d27 in Different Solvents with and without Addition of TFA.

Observed in MALDI MS.
After addition of water.
Next, we reacted cage 4 with three equivalents of deuterated aldehyde 1-d9 in various solvents with and without acid (Scheme 6 and Table 5 for NMR analytics, see the Supporting Information, Figures S63–S66 and S68–S71). Whereas without acid, scrambling was observed only in THF (entry 6), with TFA, scrambling was observed in all aprotic solvents except dioxane, most pronounced in toluene and THF (entries 14 and 16). All scrambled products are observed by MALDI MS (see the Supporting Information, Figures S62, S68), and for MeCN, DCM, and CHCl3, the intensity of observed signals is decreasing with higher degree of deuteration. The 4-d0 signal is still the most intense one in all spectra, whereas in toluene and THF, the distribution is more of a Gaussian form with the highest peaks for 4-d9 and 4-d18, suggesting that scrambling is still kinetically hampered in DCM, MeCN, and CHCl3, especially for DCM, where the cage is soluble; this clearly shows that it is not solubility exclusively which is responsible for scrambling to occur or not.
Scheme 6. Scrambling Experiments of Cage 4 with Dialdehyde 1-d9 under Various Conditions (See Table 5).

Table 5. Scrambling Experiments of Cage 4 with Aldehyde 1-d9 in Different Solvents with and without Addition of TFAb.

Observed by MALDI MS.
( ): very weak signal intensity.
): very weak signal intensity.
The TREN cage 5 behaved very differently from cage 4 (Scheme 7 and Table 6 for NMR analytics, see the Supporting Information, Figures S63–S66 and S68–S71. For MS MALDI, see the Supporting Information, Figures S72 and S77). Here without acid, weak scrambling occurred in methanol and toluene with the non-deuterated signal for cage 5 still being the most prominent one besides some very small peaks of all partially deuterated cages 5-d9, 5-d18, and 5-d27, being little higher than background noise. With the addition of acid, the situation changes. Now scrambling was observed additionally in MeCN and EtOH, which can be explained by facilitated hydrolysis and condensation reactions due to a lowered energy barrier of transition states.
Scheme 7. Scrambling Experiments of Cage 5 with Dialdehyde 1-d9 under Various Conditions (See Table 6).

Table 6. Scrambling Experiments of Cage 5 with Aldehyde 1-d9 in Different Solvents with and without Addition of TFAb.

Observed by MALDI MS.
( ): very weak signal intensity.
): very weak signal intensity.
Finally, we investigated the addition of the opposite amines to the cages and the precipitates as well as the mother liquor by 1H NMR spectroscopy (see the Supporting Information, Tables S5–S7, Figures S82–S93). First, we treated cage 4 under different conditions with 3 equiv of TREN 3 (Scheme 8 and Figure 4 see also the Supporting Information). It is worth mentioning that mass spectra showed both cages 4 and 5, which is somewhat contradictory to 1H NMR spectra. However, we conclude that cage 5 with its TREN unit and the containing tertiary amine is much more sensitive to be ionized than cage 4 and can be seen even in traces. Therefore, we discuss NMR results preferably. Without acid, transformation to cage 5 was observed in DCM and MeOH but only in liquid phases. The solid phases were clean cage 4 in all experiments. This changed somewhat with the addition of acid. Now the precipitate from acetonitrile and DCM contained TREN cage 5 too. The corresponding solutions showed major peaks of TREN cage 5 in DCM, CHCl3, MeCN, toluene, and MeOH. It is worth mentioning that in none of the measured mass spectra nor by 1H NMR spectroscopy was any formation of mixed cage 6 observed, suggesting that it does not need two different aldehydes and two different amines to observe self-sorting effects such as those observed in the molecular marriage experiment by Mukherjee et al.1d,9
Scheme 8. Exchange of Cages 4 to 5 and Vice Versa by the Addition of Amine 3 or 2, Respectively.

For conditions, see the Supporting Information.
Figure 4.
1H NMR spectra (300 MHz, CD2Cl2) of cage 5 (top) and 4 (middle) and of the isolated precipitate (bottom) after reaction of cage 4 with 2 equiv of amine 3 in CH2Cl2 with catalytic amounts of TFA.
The opposite reaction of TREN cage 5 with stoichiometric amount of triamine 2 showed no substantial formation of cage 4 without acid, neither by 1H NMR spectroscopy nor by MS, suggesting that the TREN cage 5 is thermodynamically significantly more stable than cage 4. An exception is methanol; here cage 4 was observed by MS and in the solid state by 1H NMR. With the addition of acid, the trend was basically the same, with the difference that cage 4 was seen now aside from cage 5 in the solid from MeCN; in the solid from MeOH, it was cage 4 exclusively. By concentration-dependent 1H NMR analysis of a reaction of amines 2 and 3 plus aldehyde 1 in odd stoichiometric ratios in CD2Cl2 at room temperature, we calculated a reaction constant of approx. Keq ≈ 11 in favor of TREN cage 5, which corresponds to a ΔGeq of approx. −6 kJ/mol. In toluene-d8 where both cages are soluble, is comparable.
Discussion
To summarize, we had a closer and more detailed look on the imine cage formation and their exchange of building blocks (aldehydes or amines) once formed. For this initial study, [2 + 3] imine cages based on the two most frequently used triamines were investigated. A large solvent screening with or without the addition of TFA as the catalyst revealed that the two cages behave already differently. While cage 4 based on the benzylic amine 2 is precipitating from almost every solvent (except DCM and toluene), this seems to be the driving force in agreement with Le Chatelier’s principle of removing cage 4 from the system (here the solution) and hamper its reaction. Indeed, TFA does not change the situation significantly; only the yields have risen a bit. It is worth mentioning that those systems (toluene and dichloromethane) where cage 4 was not precipitating, the amounts of formed cage were always much lower than in the cases where it solidified, once more underlining the suggestion that the formation is mainly kinetically driven by precipitation.
TREN-based cage 5 stays in solution in most solvents and is formed in high to quantitative yields. Here, yields are higher when it does not precipitate, suggesting that it is more thermodynamically stable in comparison to cage 4.
Both cages once formed do not scramble with their deuterated congeners in any solvent, neither with or without the addition of acid nor the addition of water or both. Only for cage 4, very small peaks in the MS, not substantially higher than the noise of the baseline for scrambled cages, are observed in dichloromethane. Even when deuterated isopthalaldehyde 1-d9 is added to the reaction mixtures of cage 4, no scrambling is observed in all solvents, except in THF. Here, peaks of scrambled cages can be observed in the MS but with low intensity, again not much higher than the noise. With TFA, a clear scrambling was observed in toluene, where cage 4 is soluble, and some scrambling in acetonitrile, DCM, CHCl3, and THF occurred. No scrambling was observed in EtOH or MeOH—solvents most often used to synthesize these [2 + 3]-cages. Again, cage 5 is different. No scrambling was observed with aldehyde 1-d9, when no acid is present, but scrambling is detected in toluene, MeCN, MeOH, and EtOH with acid. Cage 5 is thermodynamically favored over cage 4 by ΔGeq = −6 kJ/mol. Nevertheless, cage 4 was not converted to cage 5 upon the addition of TREN when the reaction mixture was kept in the solid state—once more emphasizing how important the solvent and solubility of cages and intermediates therein are, to allow the system to reach thermodynamic equilibrium.
To demonstrate the role of kinetics in imine cage chemistry, we were able to convert thermodynamically more stable cage 5 to cage 4 nearly quantitatively based on the knowledge generated herein. If a solution of cage 5 in methanol is treated with amine 2 (20 equiv), cage 4 precipitates in pure form and 87% yields (Scheme 9).
Scheme 9. Quantitative Transformation of Cage 5 to Cage 4.

Conclusions
This study emphasizes how important the role of solvent for imine cage formation is and that once the cage has formed, under most conditions it does not undergo a dynamic exchange with an aldehyde of the same solubility and reactivity as the one already used for the cage-forming reaction. This does not necessarily mean that it is (but it can be!) the kinetic product, especially when it precipitates out of the reaction mixture as has already been stated by De Rycke et al. earlier: “The literature emphasizes the thermodynamic stability of such assemblies [cages] but in precipitation processes one might consider the reaction to be kinetically controlled.”18b This may explain why computational screening of thermodynamic data (such as heat or energy of formation) of final compounds exclusively can lead to contradictory experimental observations, such as has been reported in the case of the formation of a (thermodynamically unfavored) cage pot7 or a (thermodynamically unfavored) larger “doubled” cage.6c Although by high throughput screening of thermodynamic heat of formations of cages, a large number of interesting new cage structures were found to form, but purely relying on these calculations can lead to the missing of very interesting structures, such as the tetrahedral [4 + 4] imine cages from our group,17 which have successfully been synthesized and studied further.18c,20 As Jelfs and co-workers recently pointed out,7 computations of kinetic pathways of cage formations are much more complicated and time-consuming than those for calculating relative energies of formation but may need to be considered in cases where theoretical and experimental data do not give the same picture.
Experimental Section
General Remarks
Melting points (not corrected) were measured with a Büchi melting point B-545. IR-spectra were recorded on a Bruker TENSOR 27 spectrometer on a ZnSe ATR crystal. NMR spectra were taken on Bruker AVANCE III 300 (300 MHz), Bruker AVANCE DRX 300 (300 MHz), Bruker AVANCE III 400 (400 MHz), Bruker AVANCE III 500 (500 MHz), and Bruker AVANCE III 600 (600 MHz) spectrometers. Chemical shifts (δ) are reported in parts per million (ppm) relative to traces of the non-deuterated solvent in the corresponding deuterated solvent. Structural assignments were made with additional information from gCOSY, gHSQC, and gHMBC experiments. HRMS experiments were carried out on a Fourier transform ion cyclotron resonance mass spectrometer solariX (Bruker Daltonik GmbH, Bremen, Germany) equipped with a 7.0 T superconducting magnet and interfaced to an Apollo II Dual ESI/MALDI source. MALDI-TOF MS experiments were carried out on a Bruker Daltonik Reflex III, Bruker ApexQe, or Bruker AutoFlex Speed TOF with DCTB (trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile) as matrix. Elemental analysis was performed by the Microanalytical Laboratory of the University of Heidelberg using an Elementar Vario EL machine. Crystal structure analysis was accomplished on a STOE Stadivari diffractometer with a copper source (Cu Kα = 1.54178 Å). All crystallographic information files (2016234 (4-d27), 2016637 (4), and 2016235 (5)) have been deposited in the Cambridge Crystallographic Data Centre and can be downloaded free of charge via www.ccdc.camac.uk/data_request/cif.
Synthesis of (2,4,6-Triethylbenzene-1,3,5-triyl)trimethanamine (2)
1,3,5-Tri(bromomethyl)-2,4,6-triethylbenzene21 and (2,4,6-triethylbenzene-1,3,5-triyl)-trimethanamine (2)22 were obtained by following known procedures from the literature. All obtained analytical data were in accordance with the literature.21,22
Synthesis of 1-(tert-Butyl)-3,5-dimethylbenzene
To an ice bath cooled solution of m-xylene (5.00 mL, 40.5 mmol) and tert-butylchloride (2.50 mL, 22.7 mmol) in dichloromethane (10 mL), aluminum chloride (250 mg, 187 mmol) was added. After stirring the reaction mixture for 1 h, it was allowed to warm up to room temperature and stirred for another 1.5 h at room temperature. Under ice bath, cooling water (150 mL) was added and the aqueous phase was separated and extracted with dichloromethane (3 × 50 mL). The organic layers were combined and dried over magnesium sulfate. After filtration, the solvent was removed under reduced pressure (10 mbar, 50 °C) to give a pale-yellow oil (2.54 g, 15.7 mmol, 70%). [n]D25 = 1.493, 1H NMR (300 MHz, CDCl3): δ (ppm) 7.02 (s, 2H, Ar–2/6-H), 6.84 (s, 1H, Ar–4-H), 2.33 (s, −CH3), and 1.31 (s, 9H, −C(CH3)3). The analytical data are in accordance with the literature.23,24
Synthesis of 5-(tert-Butyl)isophthalic Acid
Potassium permanganate (33.0 g, 207 mmol), 1-(tert-butyl)-3,5-dimethylbenzene (5.60 g, 34.5 mmol), and sodium carbonate (9.65 g, 91.1 mmol) were suspended in a mixture of water/tert-butanol [60 mL, 1:1 (v/v)] and stirred under reflux (oil bath) for 1 h. After cooling the reaction mixture to room temperature, the solid was separated by filtration, washed with water (200 mL), and disposed. The mother liquor was diluted with a saturated solution of sodium thiosulfate (400 mL) and the resulting solid separated by filtration. The mother liquor was washed with methyl-tert-butylether (3 × 100 mL) and acidified with 5 M hydrochloride acid (100 mL). After cooling the aqueous phase with an ice bath, a colorless solid precipitated, which was isolated by filtration. (4.73 g, 21.3 mmol, 62%) mp > 300 °C, 1H NMR (300 MHz, MeOD): δ (ppm) 8.44 (s, 1H, Ar–4-H), 8.30 (s, 2H, Ar–2/4-H), and 1.39 (s, 9H, −C(CH3)3). The analytical data are in accordance with the literature.23
Synthesis of (5-(tert-Butyl)-1,3-dihydroxymethylenebenzene
Under argon atmosphere and ice bath cooling, a solution of 5-(tert-butyl)isophthalic acid (4.00 g, 18.0 mmol) in THF (dry, 70 mL) was added to a suspension of lithium aluminum hydride (4.10 g, 108 mmol) in THF (dry, 100 mL). The ice bath cooling was removed and the reaction mixture stirred for 12 h. Water (50 mL) was slowly added under ice bath cooling and a saturated solution of sodium tartate (50 mL) was added. The reaction mixture was extracted with ethyl acetate (3 × 200 mL), the organic layers were combined and dried over magnesium sulfate. After filtration, the solvent was evaporated under reduced pressure (8 mbar, 50 °C) to give the diol as a colorless solid (4.00 g, 20.6 mmol, quant.). mp > 76 °C,11H NMR (300 MHz, CDCl3): δ (ppm) 7.32 (s, 2H, Ar–2/4-H), 7.19 (s, 1H, Ar–4-H), 4.68 (s, 4H, −CH2OH), and 1.33 (s, 9H, −C(CH3)3). The analytical data are in accordance with the literature.25
Synthesis of 5-(tert-Butyl)isophthalaldehyde (1)
To 1,3-bis(hydroxymethyl)-5-tert-butylbenzene (4.00 g, 20.6 mmol) and manganese dioxide (18.0 g, 206 mmol), chloroform (90 mL) was added. The reaction mixture was stirred under reflux (oil bath) for 12 h and then cooled to room temperature. After filtration over Celite, the solvent was removed under reduced pressure (10 mbar, 50 °C) to give the dialdehyde as a colorless solid (2.33 g, 12.3 mmol, 60%). mp > 64 °C,11H NMR (300 MHz, CDCl3): δ (ppm) 10.1 (s, 2H, CHO), 8.18 (s, 3H, Ar–H), and 1.41 (s, 9H, −C(CH3)3). The analytical data are in accordance with the literature.25
Synthesis of 1-(tert-Butyl)-3,5-dimethylbenzene-d9
To a solution of m-xylene (2.50 mL, 18.6 mmol) and tert-butylchloride (1.19 mL, 9.90 mmol) in dichloromethane (5 mL), aluminum chloride (115 mg, 86.0 mmol) was added under ice bath cooling. After stirring the reaction mixture for 1 h, the cooling was removed and the reaction mixture was stirred for another 1.5 h at room temperature. Water (30 mL) was added under ice bath cooling; the aqueous phase was separated and extracted with dichloromethane (3 × 20 mL). The organic layers were combined and dried over magnesium sulfate. After filtration, the solvent was removed under reduced pressure (6 mbar, 50 °C) to give a colorless oil (1.15 g, 7.10 mmol, 72%). [n]D25 = 1.493, 1H NMR (400 MHz, CDCl3): δ (ppm) 7.02 (s, 2H, Ar–2/6-H), 6.84 (s, 1H, Ar–4-H), 2.35 (s, −CH3). 13C{1H} NMR (100 MHz, CDCl3): δ (ppm) 151.6 (C1), 137.5 (C3, C5), 127.2 (C4), 123.3 (C2, C6), 34.8 (C(CD3)3) 30.46 (“octett”, JCD = 16 Hz, C(CD3)3), and 22.1 (CH3). FT-IR (ATR) ν̃ (cm–1): 3023 (m), 2918 (m), 2864 (m), 2731 (m), 2215 (s), 2160 (m), 2124 (m), 2073 (m), 2051 (m), 1767 (w), 1738 (w), 1603 (s), 1466 (m), 1376 (m), 1205 (m), 1170 (m), 1126 (m), 1060 (s), 1033 (s), 891 (w), 850 (s), 814 (m), 750 (m), 698 (s), and 632 (w). HRMS-EI (pos): m/z = 46.0697 (4), 66.1262 (13), 109.0982 (10), 121.0990 (15), 153.1546 (100), and 171.1963 (30). [M]+ calcd for C12H9D9, 171.1968; found, 171.1963. Elemental Anal. Calcd for C12H9D9O2: C, 84.12; H, 10.60. Found: C, 84.02; H, 10.71.
Synthesis of 5-(tert-Butyl)isophthalic Acid-d9
Potassium permanganate (6.47 g, 40.6 mmol), 1-(nonadeutero-tert-butyl)-3,5-dimethylbenzene-d9 (1.15 g, 6.73 mmol), and sodium carbonate (1.88 g, 17.7 mmol) were suspended in a mixture of water/tert-butanol [30 mL, 1:1 (v/v)] and stirred under reflux (oil bath) for 1 h. After cooling the reaction mixture to room temperature, the solid was separated by filtration, washed with water (50 mL), and disposed. The mother liquor was diluted with a saturated solution of sodium thiosulfate (100 mL), and the formed solid was separated by filtration. The mother liquor was washed with methyl-tert-butylether (3 × 50 mL) and acidified with 5 M hydrochloride acid (50 mL). After cooling the aqueous phase with an ice bath, a colorless solid precipitated, which was collected by filtration (911 mg, 3.94 mmol, 59%). mp > 300 °C. 1H NMR (300 MHz, MeOD): δ (ppm) 8.47 (s, 1H, Ar–4-H), and 8.29 (s, 2H, Ar–2/4-H). 13C{1H} NMR (150 MHz, MeOD): δ (ppm) 169.2 (COOH), 153.4 (C1, C3), 132.3 (C5), 131.9 (C4, C6), 129.3 (C2), 35.1 (C(CD3)3), and 30.4 (“octett”, JCD = 19 Hz, C(CD3)3). FT-IR (ATR) ν̃ (cm–1): 2823 (w), 2552 (w), 2219 (w), 1684 (s), 1602 (m), 1451 (m), 1409 (m), 1331 (m), 1283 (s), 1242 (m), 1153 (m), 1115 (m), 1058 (m) 949 (m), 917 (m), 851 (m), 760 (m), 750 (m), 737 (m), 695 (m), and 680 (m). HRMS-EI (pos): m/z = 94.0733 (5), 119.0798 (3), 181.0478 (34), 213.1030 (100), and 231.1449 (5). [M]+ calcd for C12H5D9O4, 231.1452; found, 231.1449. Elemental Anal. Calcd for C12H5D9O4: C, 62.32; H, 6.10. Found: C, 62.54; H, 6.24.
Synthesis of (5-(tert-Butyl)-1,3-dihydroxymethylenebenzene-d9
Under argon atmosphere and ice bath cooling, a solution of 5-(nonadeutero-tert-butyl)isophthalic acid (911 mg, 3.94 mmol) in THF (dry, 20 mL) was added to a suspension of lithium aluminum hydride (895 mg, 23.5 mmol) in THF (dry, 20 mL). The ice bath cooling was removed, and the reaction mixture stirred for 12 h. Water (25 mL) was slowly added under ice bath cooling, and a saturated solution of sodium tartate (25 mL) was added. The reaction mixture was extracted with ethyl acetate (3 × 100 mL); the organic layers were combined and dried over magnesium sulfate. After filtration, the solvent was evaporated under reduced pressure (10 mbar, 50 °C) to give the diol as a colorless solid (699 mg, 3.43 mmol, 87%). mp > 75–76 °C, 1H NMR (300 MHz, CDCl3): δ (ppm) 7.31 (s, 2H, Ar–2/4-H), 7.19 (s, 1H, Ar–4-H), 4.69 (s, 4H, −CH2OH), and 1.76 (s, 2H, −OH). 13C{1H} NMR (100 MHz, CDCl3): δ (ppm) 152.2 (C1, C3), 141.1 (C5), 123.6 (C4, C6), 123.0 (C2), 65.7 (CH2OH), and 34.3 (C(CD3)3) 30.4 (“octett”, JCD = 16 Hz, C(CD3)3). FT-IR (ATR) ν̃ (cm–1): 3286 (m), 2945 (m), 2922 (m), 2867 (m), 2215 (m), 2123 (m), 2071 (m), 2050 (m), 1603 (m), 1466 (m), 1443 (m), 1421 (m), 1360 (m), 1321 (m), 1306 (m), 1264 (m), 1237 (m), 1202 (m), 1163 (m), 1050 (s), 1016 (s), 973 (s), 893 (m), 879 (m), 853 (m), 815 (m), 750 (m), 715 (m), 700 (m), and 659 (m). HRMS-ESI (neg): m/z [M – H]− calcd for C12H8D9O2, 202.1799; found, 202.1802. Elemental Anal. Calcd for C12H8D9O2: C, 70.89; H, 8.83. Found: C, 70.86; H, 9.06.
Synthesis of 5-(tert-Butyl)isophthalaldehyde-d9 (1-d9)
To 1,3-bis(hydroxymethyl)-5-(nonadeutero-tert-butyl)benzene (699 mg, 3.44 mmol) and manganese dioxide (3.00 g, 3.44 mmol), chloroform (15 mL) was added. The reaction mixture was stirred under reflux for 12 h and then cooled to room temperature. After filtration over Celite the solvent was removed under reduced pressure (10 mbar, 50 °C) to give the product a colorless solid (459 mg, 2.30 mmol, 67%). mp > 60–61 °C, 1H NMR (300 MHz, CDCl3): δ (ppm) 10.1 (s, 2H, CHO), 8.19 (s, 1H, Ar–H), and 8.18 (s, 2H, Ar–H). 13C{1H} NMR (150 MHz, CDCl3): δ (ppm) 191.7 (CHO), 153.9 (C5), 137.1 (C1, C3), 131.8 (C4, C6), 129.2 (C2), and 34.7 (C(CD3)3) 30.0 (o, J = 16 Hz, C(CD3)3). FT-IR (ATR) ν̃ (cm–1): 3369 (w), 3253 (w), 3052 (w), 2933 (w), 2839 (w), 2737 (w), 2218 (m), 2126 (w), 2056 (w), 1943 (w), 1846 (w), 1701 (s), 1687 (s), 1596 (m), 1462 (m), 1397 (m), 1382 (m), 1294 (m), 1197 (m), 1146 (m), 1055 (m), 1010 (m), 967 (m), 891 (m), 823 (m), 736 (m), 686 (m), and 652 (m). HRMS-EI (pos): m/z = 79.0512 (1), 94.0728 (4), 149.0570 (16), 181.1122 (100), and 199.1543 (4). [M]+ calcd for C12H5D9O2, 199.1553; found, 199.1543. Elemental Anal. Calcd. for C12H5D9O2: C, 72.32; H, 7.09. Found: C, 72.46; H, 7.14.
Synthesis of Imine Cage 4
To a solution of dialdehyde 1 (119 mg, 0.63 mmol) in methanol (25 mL), a solution of (2,4,6-triethylbenzene-1,3,5-triyltrimethanamine 2 (100 mg, 0.40 mmol) in methanol (25 mL) was added dropwise over 10 min. After stirring the reaction mixture for 2 days at room temperature, the precipitate was collected by filtration and washed with methanol (50 mL). The solid was dried in vacuum (10 mbar, 50 °C) to give 4 as a colorless solid (134 mg, 0.14 mmol, 70%). mp > 300 °C. 1H NMR (300 MHz, CD2Cl2): δ (ppm) 8.00 (s, 6H, H–C(Ar′)=N−), 7.98 (s, 6H, Ar′–4/6-H), 7.27 (s, 3H, Ar′–2-H), 4.98 (s, 12H, Ar–CH2–N), 2.42 (q, J = 7.4 Hz, 12 H, Ar–CH2CH3), 1.37 (s, 27H, −C(CH3)3), and 1.20 (t, J = 7.5 Hz, 18H, Ar–CH2CH3). 13C{1H} NMR (150 MHz, CD2Cl2): δ (ppm) 159.7 (HC(Ar)=N−), 152.5 (Ar′C-1/3), 144.5 (ArC-2/4/6), 137.2 (Ar′C-5), 132.4 (ArC-1/3/5), 127.5 (Ar′C-2), 126.6 (Ar′C-4/6), 56.0 (Ar–CH2), 35.4 (Ar′–C(CH3)), 31.6 (Ar′–C(CH3)), 23.8 (Ar–CH2CH3), and 16.2 (Ar–CH2CH3). FT-IR (ATR) ν̃ (cm–1): 2964 (s), 2902 (s), 2871 (s), 1643 (s), 1593 (m), 1479 (m), 1452 (m), 1365 (m), 1322 (m), 1264 (m), 1228 (m), 1158 (m), 1043 (m), 975 (m), 698 (m), and 657 (m). HRMS-MALDI (DCTB): m/z [M + H]+ calcd for C66H85N6, 961.6836; found, 961.6835. Elemental Anal. Calcd. for C66H84N6·MeOH: C, 81.00; H, 8.93; N, 8.46. Found: C, 81.38; H, 8.73; N, 8.48.
Synthesis of Imine Cage 4-d27
To a solution of dialdehyde 1-d9 (50 mg, 251 μmol) in methanol (10 mL), a solution of (2,4,6-triethylbenzene-1,3,5-triyltrimethanamine 2 (45 mg, 181 μmol) in methanol (10 mL) was added dropwise over 10 min. After stirring the reaction mixture for 2 days at room temperature, the precipitate was collected by filtration and washed with methanol (20 mL). The solid was dried in vacuum (10 mbar, 50 °C) to give 4-d27 as a colorless solid (47 mg, 47.6 μmol, 53%). mp > 300 °C. 1H NMR (600 MHz, CD2Cl2): δ (ppm) 8.33 (br, 12H, HC(Ar)=N–, Ar′–4/6-H), 7.27 (s, 3H, Ar′–2-H), 4.98 (s, 12H, Ar–CH2–N), 2.41 (q, J = 7.6 Hz, 12 H, Ar–CH2CH3), and 1.20 (t, J = 7.5 Hz, 18H, Ar–CH2CH3). 13C{1H} NMR (150 MHz, CD2Cl2): δ (ppm) 159.6 (HC(Ar′)=N−), 152.5 (Ar′C-1/3), 144.4 (ArC-2/4/6), 137.2 (Ar′C-5), 132.4 (ArC-1/3/5), 127.5 (Ar′C-2), 126.5 (Ar′C-4/6), 55.9 (Ar–CH2), 34.7 (Ar′–C(CD3)), 30.3 (“octett”, JCD = 19 Hz, Ar′–C(CD3)), 23.7 (Ar–CH2CH3), and 16.2 (Ar–CH2CH3). FT-IR (ATR) ν̃ (cm–1): 2967 (m), 2874 (m), 2830 (m), 2214 (m), 1642 (s), 1592 (m), 1487 (m), 1452 (m), 1376 (m), 1312 (m), 1265 (m), 1235 (m), 1200 (m), 1158 (m), 1060 (m), 1044 (m), 975 (s), 868 (m), 786 (w), 750 (m), 693 (m), 656 (s), and 610 (m). HRMS-MALDI (DCTB): m/z [M + H]+ calcd for C66H58D27N6, 988.8530; found, 988.8529. Elemental Anal. Calcd. for C66H57D27N6·MeOH: C, 79.79; H, 8.61; N, 8.42. Found: C, 79.65; H, 8.55; N, 8.35.
Synthesis of Imine Cage 5
To a solution of dialdehyde 1 (50 mg, 0.26 mmol) in methanol (10 mL), a solution of tris(2-aminoethyl)amine 3 (25 mg, 0.17 mmol) in methanol (10 mL) was added dropwise over 10 min. After stirring the reaction mixture for 2 days at room temperature, the solvent was removed under reduced pressure (10 mbar, 50 °C) to give 5 as a colorless solid (74 mg, 0.10 mmol, >99%). mp = 200 °C (dec.). 1H NMR (400 MHz, CDCl3): δ (ppm) 8.25 (s, 6H, Ar–4/6-H), 7.59 (s, 6H, HC(Ar)=N−), 5.13 (s, 3H, Ar–2-H), 3.80 (br, 6H, CH2–CH2–N=CH), 3.28 (br, 6H, CH2–CH2–N=CH), 2.96 (br, 6H, CH2–CH2–N=CH), 2.71 (br, 6H, H2–CH2–N=CH), and 1.40 (s, 27H, C(CH3)3). 13C{1H} NMR (150 MHz, CDCl3): δ (ppm) 161.1 (HC=N−), 152.3 (ArC-1/3), 136.8 (ArC-5), 130.3 (ArC-2), 124.6 (ArC-4/6), 60.3 (CH2–CH2–N=CH), 56.4 (CH2–CH2–N=CH), 35.3 (C(CH3)3), and 31.6 (C(CH3)3). FT-IR (ATR) ν̃ (cm–1): 3423 (w), 2961 (m), 2869 (m), 2835 (m), 1642 (s), 1592 (m), 1476 (m), 1434 (m), 1381 (m), 1364 (m), 1337 (m), 1298 (m), 1266 (m), 1227 (m), 1163 (m), 1067 (m), 1036 (m), 953 (m), 921 (s), 880 (m), 810 (m), 733 (m), 703 (m), and 665 (m). MS-MALDI (DCTB): m/z [M + H]+ calcd for C48H67N8, 775.5489; found, 755.549. Elemental Anal. Calcd for C66H66N8·0.5 H2O: C, 75.45; H, 8.84; N, 14.66. Found: C, 75.44; H, 8.64; N, 14.67.
Synthesis of Imine Cage 5-d27
To a solution of dialdehyde 1-d9 (20 mg, 101 μmol) in methanol (4 mL), a solution of tris(2-aminoethyl)amine 3 (10 mg, 65.5 μmol) in methanol (4 mL) was added dropwise over 2 min. After stirring the reaction mixture for 2 days at room temperature, the solvent was removed under reduced pressure (6 mbar, 50 °C) and the colorless residue was suspended in acetone (25 mL). The insoluble solid was disposed after filtration, and 5-d27 was obtained as a colorless solid (20 mg, 34.3 μmol, >99%) after removal of the solvent under reduced pressure (6 mbar, 50 °C). mp = 210 °C (dec.). 1H NMR (600 MHz, CD2Cl2): δ (ppm) 8.23 (s, 6H, Ar′–4/6-H), 7.57 (s, 6H, HC(Ar)=N−), 5.10 (s, 3H, Ar–2-H), 3.74 (br, 6H, CH2–CH2–N=CH), 3.27 br, 6H, CH2–CH2–N=CH), 2.95 (br, 6H, CH2–CH2–N=CH), and 2.65 (br, 6H, CH2–CH2–N=CH). 13C{1H} NMR (150 MHz, CDCl3): δ (ppm) 161.1 (HC=N−), 152.4 (ArC-1/3), 137.3 (ArC-5), 130.5 (ArC-2), 124.8 (ArC-4/6), 60.6 (CH2–CH2–N=CH), 56.6 (CH2–CH2–N=CH), 34.8 (C(CD3)3), and 30.6 (“octett”, JCD = 18 Hz, C(CD3)3). FT-IR (ATR) ν̃ (cm–1): 2945 (m), 2909 (m), 2873 (m), 2833 (m), 2214 (m), 2122 (m), 2073 (m), 2052 (m), 1642 (s), 1558 (m), 1433 (m), 1379 (m), 1363 (s), 1337 (m), 1295 (m), 1266 (m), 1238 (m), 1204 (m), 1162 (m), 1063 (s), 1034 (s), 929 (m), 902 (m), 878 (m), 795 (m), 749 (m), 717 (m), 697 (s), and 664 (m). HRMS-MALDI (DCTB): m/z [M + H]+ calcd for C48H39D27N8, 782.7183; found, 782.7183. Elemental Anal. Calcd for C48H39D27N68·H2O: C, 72.07; H, 8.57; N, 14.01. Found: C, 72.18; H, 8.31; N, 13.91.
Cage Formation Experiment of Compound 4 without TFA
After dissolving aldehyde 1 (9 mg, 47.4 μmol) in 2 mL of solvent (a: methanol, b: ethanol, c: acetonitrile, d: dichloromethane, e: chloroform, f: THF, g: 1,4-dioxane, h: toluene), a solution of amine 2 (8 mg, 32.4 μmol) in 2 mL of solvent (a–h) was added dropwise. After stirring for 2 days at room temperature, an analytical aliquot of the reaction mixture was taken for analysis by MALDI MS. The precipitate (a, b, c, e, f, g) was separated from the mother liquor by filtration, and the solvent of the mother liquor was removed under reduced pressure (10 mbar, 50 °C). The precipitate and the residue were investigated independently by 1H NMR spectroscopy. For reactions (d) and (h), no precipitate formation was observed and the solvent was removed under reduced pressure (10 mbar, 50 °C), and the residue was investigated by 1H NMR spectroscopy. Since the precipitates did not contain side-products, after isolation the yield could be calculated by mass of the precipitate. If solvent signals were visible in the 1H NMR, the yield was corrected by subtraction of the amount of solvent in the sample. The amount for the cage left in the solution was estimated by 1H NMR by using 1,3,5-trimethoxybenzene (>99%) as internal standard (ca. 50 mass % of crude). See figures S46 and S47 in the Supporting Information for 1H NMR spectra.
Cage Formation Experiment of Compound 4 with TFA
After dissolving aldehyde 1 (10 mg, 52.6 μmol) in 2 mL of solvent which contained 2 mol % TFA per amine unit (a: methanol, b: ethanol, c: acetonitrile, d: dichloromethane, e: chloroform, f: THF, g: 1,4-dioxane, and h: toluene), a solution of amine 2 (9 mg, 36.1 μmol) in 2 mL of solvent (a–h) was added dropwise. After 2 days stirring at room temperature, an analytical aliquot of the reaction mixture was taken for analysis by MALDI MS. The precipitates (a, b, c, e, f, and g) were separated from the mother liquors by filtration, and the solvent of the mother liquor was removed under reduced pressure (10 mbar, 50 °C). Since the precipitate did not contain side products after isolation, the yield could be calculated by mass of the precipitate. If solvent signals were visible in the 1H NMR, the yield was calculated by subtraction of the amount of solvent in the sample. For reaction (d) and (h), the solvent was removed under reduced pressure (10 mbar, 50 °C), and the residue was investigated by 1H NMR. The amount of cage left in the solution was estimated by 1H NMR by using 1,3,5-trimethoxybenzene (>99%) as standard (ca. 50 mass % of crude). See figures S48 and S49 in the Supporting Information for 1H NMR spectra.
Cage Formation Experiment of Compound 5 without TFA
After dissolving aldehyde 1 (11 mg, 57.9 μmol) in 2 mL of solvent (a: methanol, b: ethanol, c: acetonitrile, d: dichloromethane, e: chloroform, f: THF, g: 1,4-dioxane, and h: toluene), a solution of amine 3 (6 mg, 41.0 μmol) in 2 mL of solvent (a–h) was added dropwise. After 2 days stirring at room temperature, an analytical aliquot of the reaction solution was taken for analysis by MALDI MS. The precipitates (c, f, and g) were separated from the mother liquors by filtration, and the solvent of the mother liquor was removed under reduced pressure (10 mbar, 50 °C). Since the precipitate did not contain side-products after isolation, the yield could be calculated by mass of the precipitate. If solvent signals were visible in the 1H NMR, the yield was calculated by subtraction of the amount of solvent in the sample. For reaction a, b, d, e, and h, the solvent was removed under reduced pressure (10 mbar, 50 °C) after the reaction, and the residue was investigated by 1H NMR. The amount of the cage left in the solution was estimated by1H NMR by using 1,3,5-trimethoxybenzene (>99%) as standard (ca. 50 mass % of crude). See figures S50 and S51 in the Supporting Information for 1H NMR spectra.
Cage Formation Experiment of Compound 5 with TFA
After dissolving aldehyde 1 (11 mg, 57.9 μmol) in 2 mL of solvent which contained 2 mol % TFA per amine (a: methanol, b: ethanol, c: acetonitrile, d: dichloromethane, e: chloroform, f: THF, g: 1,4-dioxane, and h: toluene), a solution of amine 2 (7 mg, 47.9 μmol) in 2 mL of solvent (a–h) was added dropwise. After 2 days stirring at room temperature, an analytical aliquot of the reaction solution was taken for analysis by MALDI MS. The precipitates (c, f, and g) were separated from the mother liquors by filtration, and the solvent of the mother liquor was removed under reduced pressure (10 mbar, 50 °C). For reactions a, b, d, e, and h, the solvent was removed under reduced pressure (10 mbar, 50 °C), and the residue was investigated by 1H NMR spectroscopy. Since the precipitate did not contain side-products after isolation, the yield could be calculated by mass of the precipitate. If solvent signals were visible in the 1H NMR, the yield was calculated by subtraction of the amount of solvent in the sample. The yield for the cage left in the solution was estimated by 1H NMR by using 1,3,5-trimethoxybenzene (>99%) as standard (ca. 50 mass % of crude). See figures S52 and S53 in the Supporting Information for 1H NMR spectra.
Cage-to-Cage Scrambling Experiment of Cages 4 and 4-d27 without TFA
Cage compounds 4 (1 mg, 1 μmol) and 4-d27 (1 mg, 1 μmol) were immersed/dissolved in 250 μL of solvent (a: methanol, b: ethanol, c: acetonitrile, d: dichloromethane, e: chloroform, f: THF, g: 1,4-dioxane, and h: toluene) and stirred at room temperature. After 7 days, an analytical sample was taken out of the reaction solution. After removing the solvent of this sample, the residue was mixed with a pre-prepared DCTB solution in THF and investigated by MALDI MS. After 7 days, water (1 μL) was added to the reaction mixture and stirred for another 7 days at room temperature. The reaction mixture was investigated again by MALDI MS as described above. See figures S54 and S55 in the Supporting Information for mass spectra.
Cage-to-Cage Scrambling Experiment of Cages 4 and 4-d27 with TFA
Cage compounds 4 (1 mg, 1 μmol) and 4-d27 (1 mg, 1 μmol) were immersed/dissolved in 250 μL of solvent containing 2 mol % TFA (a: methanol, b: ethanol, c: acetonitrile, d: dichloromethane, e: chloroform, f: THF, g: 1,4-dioxane, and h: toluene) and stirred at room temperature. After 7 days, an analytical sample was taken out of the reaction solution. After removing the solvent of this analytical sample, the residue was mixed with a pre-prepared solution of DCTB in THF and investigated by MALDI MS. To the reaction mixture, water (1 μL) was added and stirred for another 7 days at room temperature. The reaction mixture was investigated by MALDI MS as described above. See figures S56 and S57 in the Supporting Information for mass spectra.
Cage-to-Cage Scrambling Experiment of Cages 5 and 5-d27 without TFA
Cage compounds 5 (1 mg, 1.33 μmol) and 5-d27 (1 mg, 1.28 μmol) were dissolved/immersed in 332 μL of solvent (a: methanol, b: ethanol, c: acetonitrile, d: dichloromethane, e: chloroform, f: THF, g: 1,4-dioxane, and h: toluene) and stirred at room temperature. After 7 days, an analytical sample was taken out of the reaction solution. After removing the solvent from this analytical sample, the residue was mixed with a pre-prepared DCTB solution in THF and investigated by MALDI MS. To the reaction mixture, water (1 μL) was added and stirred for another 7 days at room temperature. The reaction mixture was investigated again by MALDI MS as described above. See figures S58 and S59 in the Supporting Information for mass spectra.
Cage-to-Cage Scrambling Experiment of Cages 5 and 5-d27 with TFA
Cage compounds 5 (1 mg, 1.33 μmol) and 5-d27 (1 mg, 1.28 μmol) were dissolved in 332 μL of solvent containing 2 mol % TFA (a: methanol, b: ethanol, c: acetonitrile, d: dichloromethane, e: chloroform, f: THF, g: 1,4-dioxane, and h: toluene) and stirred at room temperature. After 7 days, an analytical sample was taken out of the reaction solution. After removing the solvent from this analytical sample, the residue was mixed with a pre-prepared DCTB solution in THF and investigated by MALDI MS. To the reaction mixture, water (1 μL) was added and stirred for another 7 days at room temperature. The reaction mixture was again investigated by MALDI MS as described above. See figures S60 and S61 in the Supporting Information for mass spectra.
Aldehyde Exchange Experiment with Cage 4 and Aldehyde 1-d9 without TFA
To cage 4 (8 mg, 8.33 μmol) and aldehyde 1-d9 (7 mg, 35.2 μmol) was added 2 mL of solvent (a: methanol, b: ethanol, c: acetonitrile, d: dichloromethane, e: chloroform, f: THF, and g: toluene), and the reaction mixture was stirred at room temperature. After 7 days, an analytical sample was taken from the reaction solution and investigated by MALDI MS. From reaction a, b, c, e, f, and g, the precipitate was separated by filtration and investigated by 1H NMR. The solvent of the mother liquor was removed under reduced pressure (10 mbar, 50 °C), and the residue was analyzed by 1H NMR. For reactions d and h, no precipitate was observed, and the solvent was removed under reduced pressure (10 mbar, 50 °C). The residue was investigated by 1H NMR. See figures S62–S66 in the Supporting Information for mass and 1H NMR spectra.
Aldehyde Exchange Experiment with Cage 4 and Aldehyde 1-d9 with TFA
To cage 4 (8 mg, 8.33 μmol) and aldehyde 1-d9 (7 mg, 35.2 μmol) 2 mL of solvent was added with 2 mol % TFA per imine bond (a: methanol, b: ethanol, c: acetonitrile, d: dichloromethane, e: chloroform, f: THF, and g: toluene), and the reaction mixture was stirred at room temperature. After 7 days, an analytical sample was taken out of the reaction mixture and investigated by MALDI TOF MS. From reactions a, b, c, e, f, and g, the precipitate was separated by filtration and investigated by 1H NMR. The solvent of the mother liquor was removed under reduced pressure (10 mbar, 50 °C), and the residue was analyzed in 1H NMR. For reactions d and h, no precipitate was observed, and the solvent was removed under reduced pressure (10 mbar, 50 °C). The residue was investigated by 1H NMR. See figures S67–S71 in the Supporting Information for mass and 1H NMR spectra.
Aldehyde Exchange Experiment with Cage 5 and Aldehyde 1-d9 without TFA
To cage 5 (7 mg, 9.28 μmol) and aldehyde 1-d9 (6 mg, 30.2 μmol) was added 2 mL of solvent (a: methanol, b: ethanol, c: acetonitrile, d: dichloromethane, e: chloroform, f: THF, g: 1,4-dioxane, and h: toluene), and the reaction mixture was stirred at room temperature. After 7 days, an analytical sample was taken out of the reaction solution and investigated by MALDI MS. From reactions b, c, f, and g, the precipitate was separated by filtration and investigated by 1H NMR. The solvent of the mother liquor was removed under reduced pressure (10 mbar, 50 °C), and the residue was analyzed in 1H NMR. For reactions a, d, e, and h, no precipitate was observed, and the solvent was removed under reduced pressure (10 mbar, 50 °C). The residue was investigated in 1H NMR. See figures S72–S76 in the Supporting Information for mass and 1H NMR spectra.
Aldehyde Exchange Experiment with Cage 5 and Aldehyde 1-d9 with TFA
To cage 5 (7 mg, 9.28 μmol) and aldehyde 1-d9 (6 mg, 30.1 μmol) was added 2 mL of solvent with 2 mol % TFA per imine bond (a: methanol, b: ethanol, c: acetonitrile, d: dichloromethane, e: chloroform, f: THF, g: 1,4-dioxane, and h: toluene), and the reaction was stirred at room temperature. After 7 days, an analytical sample was taken out of the reaction solution and investigated by MALDI MS. From reactions b, c, f, and g, the precipitate was separated by filtration and investigated by 1H NMR. The solvent of the mother liquor was removed under reduced pressure (10 mbar, 50 °C), and the residue was analyzed by 1H NMR. For reactions a, d, e, and h, no precipitate was observed, and the solvent was removed under reduced pressure (10 mbar, 50 °C). The residue was investigated by 1H NMR. See figures S77–S81 in the Supporting Information for mass and 1H NMR spectra.
Amine Exchange Experiment with Cage 4 and Amine 3 without TFA
Cage compound 4 (10 mg, 10.4 μmol) and amine 3 (6 mg, 31.5 μmol) were immersed/dissolved in 2 mL of solvent (a: methanol, b: ethanol, c: acetonitrile, d: dichloromethane, e: chloroform, f: THF, g: 1,4-dioxane, and h: toluene) and stirred at room temperature. After 7 days, an analytical sample was taken out of the reaction solution and investigated by MALDI MS. From reactions a, b, c, e, f, and g, the precipitate was separated by filtration and investigated by 1H NMR. The solvent of the mother liquor was removed under reduced pressure (10 mbar, 50 °C), and the residue was analyzed by 1H NMR. For reactions d and h, no precipitate was observed, and the solvent was removed under reduced pressure (10 mbar, 50 °C). The residue was investigated by 1H NMR. See table S5 for summary and figures S82–S84 in the Supporting Information for mass and 1H NMR spectra.
Amine Exchange Experiment with Cage 4 and Amine 3 with TFA
Cage compound 4 (10 mg, 10.4 μmol) and amine 3 (6 mg, 31.5 μmol) were immersed/dissolved in 2 mL of solvent which contained 2 mol % TFA per imine bond (a: methanol, b: ethanol, c: acetonitrile, d: dichloromethane, e: chloroform, f: THF, g: 1,4-dioxane, and h: toluene) and stirred at room temperature. After 7 days, an analytical sample was taken out of the reaction solution and investigated by MALDI MS. From reaction a–g, the precipitate was separated by filtration and investigated by 1H NMR. The solvent of the mother liquor was removed under reduced pressure (10 mbar, 50 °C), and the residue was analyzed in 1H NMR. For reaction h, no precipitate was observed, and the solvent was removed under reduced pressure (10 mbar, 50 °C). The residue was investigated by 1H NMR. See table S6 for summary and figures S85–S87 in the Supporting Information for mass and 1H NMR spectra.
Amine Exchange Experiment with Cage 5 and Amine 2 without TFA
Cage compound 5 (7 mg, 9.28 μmol) and amine 2 (5 mg, 20.1 μmol) were immersed/dissolved in 2 mL of solvent (a: methanol, b: ethanol, c: acetonitrile, d: dichloromethane, e: chloroform, f: THF, g: 1,4-dioxane, and h: toluene) and stirred at room temperature. After 7 days, an analytical sample was taken out of the reaction solution and investigated by MALDI MS. From reactions a, c, f, and g, the precipitate was separated by filtration and investigated by 1H NMR. The solvent of the mother liquor was removed under reduced pressure (10 mbar, 50 °C), and the residue was analyzed by 1H NMR. For reactions b, d, e, and h, no precipitate was observed, and the solvent was removed under reduced pressure (10 mbar, 50 °C). The residue was investigated by 1H NMR. See table S7 for summary and figures S88–S90 in the Supporting Information for mass and 1H NMR spectra.
Amine Exchange Experiment with Cage 5 and Amine 2 with TFA
Cage compound 5 (7 mg, 9.28 μmol) and amine 2 (5 mg, 20.1 μmol) were immersed/dissolved in 2 mL of solvent which contained 2 mol % TFA (per imine bond) (a: methanol, b: ethanol, c: acetonitrile, d: dichloromethane, e: chloroform, f: THF, g: 1,4-dioxane, and h: toluene) and stirred at room temperature. After 7 days, an analytical sample was taken out of the reaction solution and investigated by MALDI MS. From reaction a, c, f, and g, the precipitate was separated by filtration and investigated by 1H NMR. The solvent of the mother liquor was removed under reduced pressure (10 mbar, 50 °C), and the residue was analyzed by 1H NMR. For reactions b, d, e, and h, no precipitate was observed, and the solvent was removed under reduced pressure (10 mbar, 50 °C). The residue was investigated by 1H NMR. See table S8 for summary and figures S91–S93 in the Supporting Information for mass and 1H NMR spectra.
Estimating Thermodynamic Data for Amine Exchange in Dichloromethane-d2
Aldehyde 1 with amines 2 and 3 with different ratios of the amines 3 and 2 were stirred at room temperature in dichloromethane (1.00 mL) with a catalytic amount of TFA. After 7 days, a 1H NMR spectrum was recorded and diagnostic signals picked (table S9). These were for cage 5 (δ = 7.57 ppm; 6H, Ar′–4,6-H), cage 4 (δ = 7.33 ppm (3H, Ar′–2-H), amine 2 (δ = 3.84 ppm (6H, Ar–CH2NH2), and amine 3 (δ = 2.71 ppm (6H, NCH2−). Since the signals of amine 3 are overlapping with signals of cage 5, the integral was estimated by subtraction of the integral of signal at δ = 3.29 ppm (6H, NCH2–, cage 5). Since the molar amounts of the starting material for the synthesis of cage 4 and 5 are known, the molar amounts of compounds 2, 3, 4, and 5 in the thermodynamic equilibrium could be calculated by integration of diagnostic signals from the 1H NMR spectrum (table S10). The equilibrium constant was calculated by using equation I (see the Supporting Information) with the calculated concentrations (table S11). The average equilibrium constant for transformation of cage 4 to cage 5 is Keq,av = 11.0, and thus ΔGeq,av = −5.83 kJ mol–1. See tables S9–S11 for data evaluation and figure S94 in the Supporting Information for 1H NMR spectra.
Estimating Thermodynamic Data for Amine Exchange in Toluene-d8
Aldehyde 1 with amines 2 and 3 with different ratios of the amines 3 and 2 were stirred at room temperature in dichloromethane (1.00 mL) with catalytic amount of TFA. After 5 days, a 1H NMR spectrum was recorded and diagnostic signals picked (table S12). These were for cage 5 (δ = 7.86 ppm; 6H, Ar′–4,6-H), cage 4 (δ = 8.741 ppm (36H, Ar′–4,6-H), amine 2 (δ = 3.74 ppm (6H, Ar–CH2NH2), and amine 3 (δ = 2.18 ppm (6H, NCH2−). Since the signals of amine 3 are overlapping with signals of cage 5, the integral was estimated by subtraction of the integral of signal at δ = 3.29 ppm (6H, NCH2–, cage 5). Since the molar amounts of the starting material for the synthesis of cage 4 and 5 are known, the molar amounts of compounds 2, 3, 4, and 5 in the thermodynamic equilibrium could be calculated by integration of diagnostic signals from the 1H NMR spectrum (table S13). The equilibrium constant was calculated by using equation I (see the Supporting Information) with the calculated concentrations (table S14). The average equilibrium constant for transformation of cage 4 to cage 5 in toluene is Keq,av = 31.4, and thus ΔGeq,av = −8.35 kJ mol–1. See tables S12–S14 for data evaluation and figure S95 in the Supporting Information for 1H NMR spectra.
Transformation of Cage 5 to Cage 4
Imine cage 5 (9 mg, 11.9 μmol) was dissolved in methanol (1.00 mL) and mixed with a solution of amine 2 (59 mg, 238 μmol) in methanol (0.50 mL). After stirring the reaction at room temperature with a catalytic amount of TFA overnight, the formed precipitate was isolated by filtration, washed with methanol (3 × 2 mL), and dried under vacuum (12 mbar, 60 °C) to obtain a colorless solid (10 mg, 87%). The solid was characterized by 1H NMR spectroscopy and identified as cage 4.
Acknowledgments
The authors thank the European Research Council (ERC) for funding this project in the frame of the consolidators grant CaTs n DOCs (grant agreement no. 725765).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c01887.
Crystallographic information files (4-d27, 4, and 5) (CIF)
NMR Spectra; DOSY experiments; mass spectra; infrared spectra; single-crystal X-ray diffraction data; NMR and MS data of cage formation experiments, cage-to-cage scrambling, aldehyde exchange, amine exchange; and estimating thermodynamic data for amine exchange (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Mastalerz M. Porous Shape-Persistent Organic Cage Compounds of Different Size, Geometry, and Function. Acc. Chem. Res. 2018, 51, 2411–2422. 10.1021/acs.accounts.8b00298. [DOI] [PubMed] [Google Scholar]; b Beuerle F.; Gole B. Covalent Organic Frameworks and Cage Compounds: Design and Applications of Polymeric and Discrete Organic Scaffolds. Angew. Chem., Int. Ed. 2018, 57, 4850–4878. 10.1002/anie.201710190. [DOI] [PubMed] [Google Scholar]; c Mukhopadhyay R. D.; Kim Y.; Koo J.; Kim K. Porphyrin Boxes. Acc. Chem. Res. 2018, 51, 2730–2738. 10.1021/acs.accounts.8b00302. [DOI] [PubMed] [Google Scholar]; d Acharyya K.; Mukherjee P. S. Organic Imine Cages: Molecular Marriage and Applications. Angew. Chem., Int. Ed. 2019, 58, 8640–8653. 10.1002/anie.201900163. [DOI] [PubMed] [Google Scholar]; e Hasell T.; Cooper A. I. Porous organic cages: soluble, modular and molecular pores. Nat. Rev. Mater. 2016, 1, 16053. 10.1038/natrevmats.2016.53. [DOI] [Google Scholar]; f Zhang G.; Mastalerz M. Organic cage compounds - from shape-persistency to function. Chem. Soc. Rev. 2014, 43, 1934–1947. 10.1039/c3cs60358j. [DOI] [PubMed] [Google Scholar]; g Mastalerz M. Shape-Persistent Organic Cage Compounds by Dynamic Covalent Bond Formation. Angew. Chem., Int. Ed. 2010, 49, 5042–5053. 10.1002/anie.201000443. [DOI] [PubMed] [Google Scholar]
- Rue N. M.; Sun J.; Warmuth R. Polyimine Container Molecules and Nanocapsules. Isr. J. Chem. 2011, 51, 743–768. 10.1002/ijch.201100064. [DOI] [Google Scholar]
- Santolini V.; Miklitz M.; Berardo E.; Jelfs K. E. Topological landscapes of porous organic cages. Nanoscale 2017, 9, 5280–5298. 10.1039/c7nr00703e. [DOI] [PubMed] [Google Scholar]
- a Skowronek P.; Gawronski J. Chiral Iminospherand of a Tetrahedral Symmetry Spontaneously Assembled in a [6 + 4] Cyclocondensation. Org. Lett. 2008, 10, 4755–4758. 10.1021/ol801702j. [DOI] [PubMed] [Google Scholar]; b Skowronek P.; Warżajtis B.; Rychlewska U.; Gawroński J. Self-assembly of a covalent organic cage with exceptionally large and symmetrical interior cavity: the role of entropy of symmetry. Chem. Commun. 2013, 49, 2524–2526. 10.1039/c3cc39098e. [DOI] [PubMed] [Google Scholar]; c Ding H.; Wu X.; Zeller M.; Xie Y.; Wang C. Controllable Synthesis of Covalent Porphyrinic Cages with Varying Sizes via Template-Directed Imine Condensation Reactions. J. Org. Chem. 2015, 80, 9360–9364. 10.1021/acs.joc.5b01781. [DOI] [PubMed] [Google Scholar]; d Ding H.; Yang Y.; Li B.; Pan F.; Zhu G.; Zeller M.; Yuan D.; Wang C. Targeted synthesis of a large triazine-based [4+6] organic molecular cage: structure, porosity and gas separation. Chem. Commun. 2015, 51, 1976–1979. 10.1039/c4cc08883b. [DOI] [PubMed] [Google Scholar]; e Mastalerz M. One-pot synthesis of a shape-persistent endo-functionalised nano-sized adamantoid compound. Chem. Commun. 2008, 4756–4758. 10.1039/b808990f. [DOI] [PubMed] [Google Scholar]; f Schneider M. W.; Oppel I. M.; Mastalerz M. Exo-Functionalized Shape-Persistent [2+3] Cage Compounds: Influence of Molecular Rigidity on Formation and Permanent Porosity. Chem.—Eur. J. 2012, 18, 4156–4160. 10.1002/chem.201200032. [DOI] [PubMed] [Google Scholar]; g Slater A. G.; Little M. A.; Briggs M. E.; Jelfs K. E.; Cooper A. I. A solution-processable dissymmetric porous organic cage. Mol. Syst. Des. Eng. 2018, 3, 223–227. 10.1039/c7me00090a. [DOI] [Google Scholar]; h Elbert S. M.; Rominger F.; Mastalerz M. Synthesis of a Rigid C3v-Symmetric Tris-salicylaldehyde as a Precursor for a Highly Porous Molecular Cube. Chem.—Eur. J. 2014, 20, 16707–16720. 10.1002/chem.201404829. [DOI] [PubMed] [Google Scholar]
- Rowan S. J.; Cantrill S. J.; Cousins G. R. L.; Sanders J. K. M.; Stoddart J. F. Dynamic Covalent Chemistry. Angew. Chem., Int. Ed. 2002, 41, 898–952. 10.1002/1521-3773(20020315)41:6<898::aid-anie898>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- a Jelfs K. E.; Eden E. G. B.; Culshaw J. L.; Shakespeare S.; Pyzer-Knapp E. O.; Thompson H. P. G.; Bacsa J.; Day G. M.; Adams D. J.; Cooper A. I. In silico Design of Supramolecules from Their Precursors: Odd–Even Effects in Cage-Forming Reactions. J. Am. Chem. Soc. 2013, 135, 9307–9310. 10.1021/ja404253j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Greenaway R. L.; Santolini V.; Bennison M. J.; Alston B. M.; Pugh C. J.; Little M. A.; Miklitz M.; Eden-Rump E. G. B.; Clowes R.; Shakil A.; Cuthbertson H. J.; Armstrong H.; Briggs M. E.; Jelfs K. E.; Cooper A. I. High-throughput discovery of organic cages and catenanes using computational screening fused with robotic synthesis. Nat. Commun. 2018, 9, 2849. 10.1038/s41467-018-05271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Pugh C. J.; Santolini V.; Greenaway R. L.; Little M. A.; Briggs M. E.; Jelfs K. E.; Cooper A. I. Cage Doubling: Solvent-Mediated Re-equilibration of a [3 + 6] Prismatic Organic Cage to a Large [6 + 12] Truncated Tetrahedron. Cryst. Growth Des. 2018, 18, 2759–2764. 10.1021/acs.cgd.7b01422. [DOI] [Google Scholar]
- Greenaway R. L.; Santolini V.; Pulido A.; Little M. A.; Alston B. M.; Briggs M. E.; Day G. M.; Cooper A. I.; Jelfs K. E. From Concept to Crystals via Prediction: Multi-Component Organic Cage Pots by Social Self-Sorting. Angew. Chem., Int. Ed. 2019, 58, 16275–16281. 10.1002/anie.201909237. [DOI] [PubMed] [Google Scholar]
- a Liu X.; Liu Y.; Warmuth R. Multi-Component Synthesis of Tetracavitand Nanocapsules. Supramol. Chem. 2008, 20, 41–50. 10.1080/10610270701742546. [DOI] [Google Scholar]; b Liu Y.; Liu X.; Warmuth R. Multicomponent Dynamic Covalent Assembly of a Rhombicuboctahedral Nanocapsule. Chem.—Eur. J. 2007, 13, 8953–8959. 10.1002/chem.200701067. [DOI] [PubMed] [Google Scholar]; c Liu X.; Warmuth R. Solvent Effects in Thermodynamically Controlled Multicomponent Nanocage Syntheses. J. Am. Chem. Soc. 2006, 128, 14120–14127. 10.1021/ja0644733. [DOI] [PubMed] [Google Scholar]; d Liu X.; Liu Y.; Li G.; Warmuth R. One-Pot, 18-Component Synthesis of an Octahedral Nanocontainer Molecule. Angew. Chem., Int. Ed. 2006, 45, 901–904. 10.1002/anie.200504049. [DOI] [PubMed] [Google Scholar]
- Acharyya K.; Mukherjee S.; Mukherjee P. S. Molecular Marriage through Partner Preferences in Covalent Cage Formation and Cage-to-Cage Transformation. J. Am. Chem. Soc. 2013, 135, 554–557. 10.1021/ja310083p. [DOI] [PubMed] [Google Scholar]
- a Acharyya K.; Mukherjee P. S. Hydrogen-Bond-Driven Controlled Molecular Marriage in Covalent Cages. Chem.—Eur. J. 2014, 20, 1646–1657. 10.1002/chem.201303397. [DOI] [PubMed] [Google Scholar]; b Acharyya K.; Mukherjee P. S. Shape and size directed self-selection in organic cage formation. Chem. Commun. 2015, 51, 4241–4244. 10.1039/c5cc00075k. [DOI] [PubMed] [Google Scholar]
- Jiao T.; Wu G.; Chen L.; Wang C.-Y.; Li H. Precursor Control over the Self-Assembly of Organic Cages via Imine Condensation. J. Org. Chem. 2018, 83, 12404–12410. 10.1021/acs.joc.8b01421. [DOI] [PubMed] [Google Scholar]
- Ro S.; Rowan S. J.; Pease A. R.; Cram D. J.; Stoddart J. F. Dynamic Hemicarcerands and Hemicarceplexes. Org. Lett. 2000, 2, 2411–2414. 10.1021/ol005962p. [DOI] [PubMed] [Google Scholar]
- Kołodziejski M.; Stefankiewicz A. R.; Lehn J. M. Dynamic polyimine macrobicyclic cryptands – self-sorting with component selection. Chem. Sci. 2019, 10, 1836–1843. 10.1039/c8sc04598d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Beaudoin D.; Rominger F.; Mastalerz M. Chiral Self-Sorting of [2+3] Salicylimine Cage Compounds. Angew. Chem. 2017, 129, 1264–1268. 10.1002/ange.201610782. [DOI] [PubMed] [Google Scholar]; b Wang X.; Wang Y.; Yang H.; Fang H.; Chen R.; Sun Y.; Zheng N.; Tan K.; Lu X.; Tian Z.; Cao X. Assembled molecular face-rotating polyhedra to transfer chirality from two to three dimensions. Nat. Commun. 2016, 7, 12469. 10.1038/ncomms12469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao N.; Wang Y.; Zheng X.; Jiao T.; Li H. Controllable Self-Assembly of Pills and Cages via Imine Condensation for Silver Cation Detection. Org. Lett. 2018, 20, 7447–7450. 10.1021/acs.orglett.8b03174. [DOI] [PubMed] [Google Scholar]
- Schneider M. W.; Siegfried Hauswald H.-J.; Stoll R.; Mastalerz M. A shape-persistent exo-functionalized [4 + 6] imine cage compound with a very high specific surface area. Chem. Commun. 2012, 48, 9861–9863. 10.1039/c2cc35002e. [DOI] [PubMed] [Google Scholar]
- Lauer J. C.; Zhang W.-S.; Rominger F.; Schröder R. R.; Mastalerz M. Shape-Persistent [4+4] Imine Cages with a Truncated Tetrahedral Geometry. Chem.—Eur. J. 2018, 24, 1816–1820. 10.1002/chem.201705713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mateus P.; Delgado R.; Brandão P.; Carvalho S.; Félix V. Selective recognition of tetrahedral dianions by a hexaaza cryptand receptor. Org. Biomol. Chem. 2009, 7, 4661–4673. 10.1039/b912940e. [DOI] [PubMed] [Google Scholar]; b De Rycke N.; Marrot J.; Couty F.; David O. R. P. Synthesis and characterization of hexasubstituted azacryptands. Tetrahedron Lett. 2010, 51, 6521–6525. 10.1016/j.tetlet.2010.10.019. [DOI] [Google Scholar]; c Schick T. H. G.; Lauer J. C.; Rominger F.; Mastalerz M. Transformation of Imine Cages into Hydrocarbon Cages. Angew. Chem., Int. Ed. 2019, 58, 1768–1773. 10.1002/anie.201814243. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Francesconi O.; Ienco A.; Moneti G.; Nativi C.; Roelens S. A Self-Assembled Pyrrolic Cage Receptor Specifically Recognizes β-Glucopyranosides. Angew. Chem., Int. Ed. 2006, 45, 6693–6696. 10.1002/anie.200602412. [DOI] [PubMed] [Google Scholar]; e Arunachalam M.; Ravikumar I.; Ghosh P. A New Hexaaza Bicyclic Cyclophane with Dual Binding Sites. J. Org. Chem. 2008, 73, 9144–9147. 10.1021/jo801762b. [DOI] [PubMed] [Google Scholar]
- a MacDowell D.; Nelson J. Facile synthesis of a new family of cage molecules. Tetrahedron Lett. 1988, 29, 385–386. 10.1016/s0040-4039(00)80103-3. [DOI] [Google Scholar]; b Wang F.; Sikma E.; Duan Z.; Sarma T.; Lei C.; Zhang Z.; Humphrey S. M.; Sessler J. L. Shape-persistent pyrrole-based covalent organic cages: synthesis, structure and selective gas adsorption properties. Chem. Commun. 2019, 55, 6185–6188. 10.1039/c9cc02490e. [DOI] [PubMed] [Google Scholar]; c Jazwinski J.; Lehn J.-M.; Lilienbaum D.; Ziessel R.; Guilhem J.; Pascard C. Polyaza macrobicyclic cryptands: synthesis, crystal structures of a cyclophane type macrobicyclic cryptand and of its dinuclear copper(I) cryptate, and anion binding features. J. Chem. Soc., Chem. Commun. 1987, 1691–1694. 10.1039/c39870001691. [DOI] [Google Scholar]; d Drew M. G. B.; McDowell D.; Nelson J. A new ditopic polyaza macrobicyclic ligand: X-ray crystallographic structure determination. Polyhedron 1988, 7, 2229–2232. 10.1016/s0277-5387(00)81811-2. [DOI] [Google Scholar]
- Lauer J. C.; Pang Z.; Janßen P.; Rominger F.; Kirschbaum T.; Elstner M.; Mastalerz M. Host-Guest Chemistry of Truncated Tetrahedral Imine Cages with Ammonium Ions. ChemistryOpen 2020, 9, 183–190. 10.1002/open.201900357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacca A.; Nativi C.; Cacciarini M.; Pergoli R.; Roelens S. A New Tripodal Receptor for Molecular Recognition of Monosaccharides. A Paradigm for Assessing Glycoside Binding Affinities and Selectivities by 1H NMR Spectroscopy. J. Am. Chem. Soc. 2004, 126, 16456–16465. 10.1021/ja045813s. [DOI] [PubMed] [Google Scholar]
- Ryu E.-H.; Yan J.; Zhong Z.; Zhao Y. J. Org. Chem. 2006, 71, 7205–7213. 10.1021/jo0607663. [DOI] [PubMed] [Google Scholar]
- Nightingale D.; Smith L. I. The Action of Aluminum Chloride on Aromatic Hydrocarbons. I.1 The 1,3-Dimethyl-4-butylbenzenes. J. Am. Chem. Soc. 1939, 61, 101–104. 10.1021/ja01870a033. [DOI] [Google Scholar]
- Joshi-Pangu A.; Wang C.-Y.; Biscoe M. R. Nickel-Catalyzed Kumada Cross-Coupling Reactions of Tertiary Alkylmagnesium Halides and Aryl Bromides/Triflates. J. Am. Chem. Soc. 2011, 133, 8478–8481. 10.1021/ja202769t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S.-H.; Moorefield C. N.; Dai L.; Newkome G. R. Functional Nanohybrids Constructed via Complexation of Multiwalled Carbon Nanotubes with Novel Hexameric Metallomacrocyles. Chem. Mater. 2006, 18, 4019–4024. 10.1021/cm060510d. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.