

Abstract
Most patients with epithelial ovarian cancer (EOC) are diagnosed at advanced stage and have a poor prognosis. However, a small proportion of these patients will survive, whereas others will die very quickly. Clinicopathological factors do not allow precise identification of these subgroups. Thus, we have validated a molecular subclassification as new prognostic factor in EOC. One hundred and ninety‐four patients with Stage II–IV EOC were characterized by whole‐genome expression profiling of tumor tissues and were classified using a published 112 gene set, derived from an International Federation of Gynecology and Obstetrics (FIGO) stage‐directed supervised classification approach. The 194 tumor samples were classified into two subclasses comprising 95 (Subclass 1) and 99 (Subclass 2) tumors. All nine FIGO II tumors were grouped in Subclass 1 (P = 0.001). Subclass 2 (54% of advanced‐stage tumors) was significantly correlated with peritoneal carcinomatosis and non‐optimal debulking. Patients with Subclass 2 tumors had a worse overall survival for both serous and non‐serous histological subtypes, as revealed by univariate analysis (hazard ratios [HR] of 3.17 and 17.11, respectively; P ≤ 0.001) and in models corrected for relevant clinicopathologic parameters (HR 2.87 and 12.42, respectively; P ≤ 0.023). Significance analysis of microarrays revealed 2082 genes that were differentially expressed in advanced‐grade serous tumors of both subclasses and the focal adhesion pathway as the most deregulated pathway. In the present validation study, we have shown that, in advanced‐stage serous ovarian cancer, two approximately equally large molecular subtypes exist, independent of classical clinocopathological parameters and presenting with highly different whole‐genome expression profiles and a markedly different overall survival. Similar results were obtained in a small cohort of patients with non‐serous tumors. (Cancer Sci 2012; 103: 1334–1341)
To estimate the prognosis of malignant diseases, classical clinicopathological parameters are primarily taken into account. Molecular studies of several types of cancers describe subclasses of tumors with regard to prognosis that will hopefully allow for a more precise prognostication and reveal possible new targets for anticancer treatment. Perou et al.1 introduced microarray data for the molecular subclassification of breast cancer more than 10 years ago. Since then, researchers have published similar gene signatures for the molecular subclassification of a variety of tumor entities.2, 3, 4 Especially for breast cancer, gene signatures predicting similar tumor subclasses have been established, most of them using single sample predictors (SSPs) to define the various subclasses, such as Luminal A, Luminal B, basal like, human epidermal growth factor receptor 2 (HER2), and normal breast like.5 Nevertheless, a retrospective study evaluating three SSPs on four breast cancer data sets revealed poor consistency in predicting the subtypes using these SSPs, whereby only basal‐like tumors were consistently classified.5
Epithelial ovarian cancer (EOC) is the most lethal gynecologic malignancy, afflicting approximately 6% of women.6 It has become the fourth most frequent cause of cancer‐related deaths in women in Western countries. Approximately 75% of patients with EOC are diagnosed at advanced stage (International Federation of Gynecology and Obstetrics [FIGO] III/IV).6 Further subclassification is urgently needed because a significant proportion of these patients will fare very well, with a prognosis similar to that of patients with early stage disease, whereas others die very quickly from their disease. A better understanding of the molecular changes in ovarian cancer will improve therapy choice and guide the search for additional targeted therapeutics. Histopathologic subtyping of ovarian cancer includes the following groups: high‐grade serous, clear cell, endometrioid, mucinous, and low‐grade serous tumors.7 A recently described nine‐marker set (cyclin‐dependent kinase inhibitor 1A [CDKN1A], Dickkopf‐related protein 1 [DKK1], hepatocyte nuclear factor 1‐β [HNF1β], mouse double minute 2 [MDM2], progesterone receptor [PGR], trefoil factor 3 [intestinal] [TFF3], tumor protein p53 [TP53], vimentin [VIM] and Wilms Tumor 1 [WT1]) determined by immunohistochemistry can predict these subtypes with high sensitivity and specificity.8 Molecular signatures for the subclassification of ovarian cancer are rare, but include the following. De Cecco et al.9 published a molecular signature of significantly enriched 74 genes related to the extracellular matrix and the fibroblast growth factor (FGF) 2 pathway, dividing the samples into two approximately equally large groups, but with no significant association with any clinicopathologic or prognostic parameters. Tothill et al.10 described a k‐means clustering approach using microarray data of 285 ovarian cancer tissues yielding an optimal number of six clusters (C1–C6), two of which were associated with low malignant potential (LMP) tumors and early‐stage endometrioid tumors (C3 and C6, respectively), with considerably better prognosis. Among the remaining four subtypes, patients with a high stromal response signature (C1) had the poorest overall survival (OS) compared with patients with a higher number of CD3+ cells and lower expression of stromal response genes (C2 and C4), whereas the C5 cluster, a mesenchymal‐associated subtype, exhibited a trend for reduced OS compared with subtypes C2 and C4.10 In 2009, Yoshihara et al.11 described a gene signature comprised of 112 genes to subdivide advanced‐stage serous ovarian cancer samples into two subtypes. This signature was derived from a comparison of eight FIGO I tumors with that of 35 Stage III/IV tumors. Interestingly, a non‐negative matrix factorization analysis revealed two stable clusters, one consisting of all eight FIGO I tumors and a further 14 Stage III/IV tumors and the other consisting of the remaining 21 Stage III/IV tumors. These 21 tumors exhibited significantly reduced resectability, significantly reduced progression‐free survival (PFS), and reduced OS (P = 0.068) compared with the cluster containing the 14 other Stage III/IV tumors. Nonetheless, it is difficult to come to any conclusion on the basis of these studies because of the small sample size. None of the nine aforementioned markers for histological subclassification were among the 112 genes differently expressed in the two molecular subtypes.
To examine the role of the 112 gene signature determined by Yoshihara et al. with regard to clinical outcome, we used whole‐genome transcriptome data of 194 tumor tissues from non‐FIGO I ovarian cancer patients produced during the course of the European Union project OVCAD (Ovarian Cancer: Diagnosis of a silent killer; no. 018698) to subclassify these tumors and to correlate the subclasses obtained with clinicopathologic parameters, gene expression profiles, and outcomes in patients with serous and non‐serous tumors (excluding clear cell tumors).
Materials and Methods
Patients
The EU OVCAD project included patients from five European university hospitals (Gynecologic Oncology Clinics of the University Hospitals Berlin, Hamburg, Innsbruck, Leuven, and Vienna; OVCAD Consortium). Tissue samples were collected according to standard surgical procedures. Information on clinical and histopathological characteristics, as well as follow‐up data, was collected and documented by experienced clinicians. All histological types of EOC, except clear cell, were included. Patients with benign ovarian diseases, borderline ovarian cancer, secondary malignant tumors, or with FIGO Stage I EOC were excluded. The study protocol was approved by the ethics committees of the participating OVCAD partners. Presurgical written informed consent was obtained from each patient before their inclusion in the study. All patients received standard treatment, including debulking surgery and platinum‐based chemotherapy. Residual tumor was defined as negative if a macroscopically complete resection was accomplished. The evaluation of tumor response to chemotherapy was made according to World Health Organization (WHO)12 criteria: progression of disease after first‐line chemotherapy was defined as an at least twofold increase in the nadir serum CA‐125 level according to the Gynecological Cancer Intergroup (GCIG) criteria13 or confirmed radiologically. The response to first‐line treatment was evaluated by experienced gynecologic oncologists in the participating university clinics. At the time of analysis, the median follow‐up was 31 months, with 57 documented cases of cancer‐related deaths.
Microarray analysis
Total RNA from 213 fresh frozen tumor tissues was isolated using the ABI PRISM 6100 Nucleic Acid PrepStation (Applied Biosystems, Carlsbad, CA, USA) according to the manufacturer's instructions and quantified spectroscopically. The quality of the RNA was assessed with an Agilent 2100 Bioanalyser (Agilent, Santa Clara, CA, USA) and the RNA integrity number (RIN) in all cases was >5. Total RNA (15 μg) was labeled using the Applied Biosystems Chemiluminescent RT Labeling Kit and hybridized to the Human Genome Survey Microarray v.2.0 (Applied Biosystems), washed, and scanned according to the manufacturer's instructions. Raw microarray data were filtered using the quality flags provided with the proprietary analysis software and quantile normalized using the Bioconductor R script ABarray v.1.2 (open source software). A principal component analysis (PCA)‐guided signal‐to‐noise (S/N) cut‐off (>5400 probe IDs with S/N > 3 in each data set) filtered out nine of the 213 samples, leaving 204 arrays of acceptable quality. Because of differences between microarray batches, the normalized data were corrected as follows. First, signals were quantile normalized within each batch. Second, we subtracted the batch‐specific mean from each gene and added the overall mean. The success of this method to remove batch effects was confirmed by three‐dimensional plots of the first three principal components, in which the individual batches no longer showed any visible clustering. Finally, 10 patients were excluded during follow‐up by the stringent OVCAD exclusion criteria, mainly due to missing standard chemotherapy or fatal complications during or shortly after debulking surgery.
Subclassification
One hundred and six of the 112 genes used for subclassification by Yoshihara et al.11 could be mapped to the annotation of the Human Genome Survey Microarray v.2.0 (Applied Biosystems), yielding 126 probe Ids, including some double and triple coverages. The expression values of these 126 probe IDs were used to classify the 194 tumor tissues with non‐negative matrix factorization (NMF) implemented in the MultiExperiment Viewer (MeV) v.4.6 (open source software)14 using the parameters rank range “2–6”, number of runs “200”, maximum iterations “2000”, and cost measurement “divergence”, essentially following the classification approach by Yoshihara et al.11
Statistical analysis
Statistical correlations of the subclasses to clinicopathologic parameters were assessed by t‐tests, χ2 tests, and Fisher's exact tests, as appropriate. Results were corrected for multiple testing by the Holm–Bonferroni method.15 Impact on recurrence‐free survival and OS was determined by univariate and multiple Cox regression analyses15 and the univariate impact illustrated by Kaplan–Meier estimates.17 The proportion of explained variation (PEV) was calculated according to Heinze and Schemper.18
Significance analysis of microarrays and functional annotation
The impact of the subclass classification on whole‐genome expression changes was determined by significance analysis of microarrays (SAM) implemented in MeV v.4.614 using a false discovery rate (FDR) of 5%. Therefore 9708 probe IDs were prefiltered so that in one of both subclasses 75% of samples showed an S/N value > 2. Functional analysis of differentially expressed genes was performed using the Database for Annotation, Visualization and Integrated Discovery tool (DAVID) v.6.7.19
Results
Subclassification and correlation to clinicopathologic parameters
One hundred and ninety‐four advanced‐stage EOC tissues were classified using 126 probe IDs representing 106 of the 112 subclass‐specific genes used by Yoshihara et al.11 and the NMF method. The cophenetic correlation coefficient was highest allowing two groups for classification (0.94) compared with using more than two groups (i.e. three to six groups [cophenetic correlation coefficients always <0.69]). Ninety‐five of the 194 tissues were classified into one group, including all nine FIGO II samples (adjusted P = 0.028) and this group was therefore named Subclass 1; 99 tissues were classified into another group (Subclass 2). Age, grade, nodal status, and amount of ascites did not differ significantly between the two subclasses (data not shown) or if patients were grouped according tumor histology (Table 1). In addition to the lack of FIGO II patients in Subclass 2, patients with peritoneal carcinomatosis (adjusted P = 0.030 and 0.045 for serous and non‐serous tumors, respectively) and, in the case of non‐serous tumors only, patients with residual tumor after debulking surgery (adjusted P = 0.030) were significantly over‐represented in subclass 2. As already stated by Yoshihara et al.,11 advanced stage (i.e. FIGO III and IV) tumors were divided into approximately similar large cohorts, in our case 86 Subclass 1 and 99 Subclass 2 tumors. In Table 1, correlations are shown for subclasses and the clinicopathologic parameters of the histological serous and non‐serous tumor subtypes.
Table 1.
Clinicopathologic characteristics of patients and correlations with subclasses
| Subclass 1 | Subclass 2 | P‐value | Adjusted P‐value | |
|---|---|---|---|---|
| Serous EOC (n = 171) | ||||
| Total | 81 (47.4%) | 90 (52.6%) | 0.313* | |
| Mean (±SD) age (years) | 56.9 (12.3) | 58.7 (11.7) | ||
| FIGO stage | ||||
| FIGO II | 5 (6.2%) | 0 (0.0%) | 0.049† | |
| FIGO III | 63 (77.8%) | 72 (80.0%) | ||
| FIGO IV | 13 (16.0%) | 18 (20.0%) | ||
| Grade (one missing) | ||||
| Grade 1 | 1 (1.2%) | 5 (5.6%) | 0.322† | |
| Grade 2 | 18 (22.2%) | 21 (23.6%) | ||
| Grade 3 | 62 (76.6%) | 63 (70.8%) | ||
| Residual tumor | ||||
| No | 63 (77.8%) | 58 (64.4%) | 0.056‡ | |
| Yes | 18 (22.2%) | 32 (35.6%) | ||
| Peritoneal carcinomatosis | ||||
| No | 30 (37.0%) | 16 (17.8%) | 0.005† | 0.030 |
| Yes | 51 (63.0%) | 74 (82.2%) | ||
| Ascites | ||||
| ≤500 mL | 55 (67.9%) | 46 (51.1%) | 0.026‡ | 0.130 |
| >500 mL | 26 (32.1%) | 44 (48.9%) | ||
| Non‐serous EOC§ (n = 23) | ||||
| Total | 14 (60.9%) | 9 (39.1%) | 0.675* | |
| Mean (±SD) age (years) | 57.1 (11.5) | 55.1 (9.5) | ||
| FIGO stage | ||||
| FIGO II | 4 (28.6%) | 0 (0.0%) | 0.127† | |
| FIGO III | 63 (71.4%) | 9 (100.0%) | ||
| Grade (one missing) | ||||
| Grade 1 and 2 | 3 (21.4%) | 2 (22.2%) | 1.000† | |
| Grade 3 | 11 (78.6%) | 7 (77.8%) | ||
| Residual tumor | ||||
| No | 13 (92.9%) | 3 (33.3%) | 0.005† | 0.030 |
| Yes | 1 (7.1%) | 6 (66.7%) | ||
| Peritoneal carcinomatosis | ||||
| No | 10 (71.4%) | 1 (11.1%) | 0.009† | 0.045 |
| Yes | 4 (28.6%) | 8 (88.9%) | ||
| Ascites | ||||
| ≤500 mL | 10 (71.4%) | 6 (66.7%) | 1.000† | |
| >500 mL | 4 (28.6%) | 3 (33.3%) | ||
Unless indicated otherwise, data show the number of patients in each group, with percentages given in parentheses. *t‐test; †Fisher's exact test; ‡χ2 test. §Endometrioid n = 5; mixed epithelial n = 7; mucinous n = 3; undifferentiated carcinoma n = 8. EOC, epithelial ovarian cancer; FIGO, International Federation of Gynecology and Obstetrics.
Impact of subclassification on the prognostication of PFS and OS for patients with serous and non‐serous tumors
For analysis of PFS and OS, especially to validate the results of Yoshihara et al.,11 patients were divided into two groups, those with serous or non‐serous tumors. The nine patients with FIGO II tumors were excluded from outcome analyses because they were expected to show a generally better prognosis. Figure 1 shows the univariate impact of the subclassification on prognostication of PFS and OS of these two groups as estimated by Kaplan–Meier plots. The impact of the subclassification on FIGO III and IV high‐grade serous tumors was analyzed by multivariate Cox regression (Table 2) . In the final model for OS, only peritoneal carcinomatosis (hazard ratio [HR] 4.56; P = 0.012) and subclass (HR 2.87; P = 0.004) showed significant independent impact. For PFS, only FIGO (Stage IV vs III) and peritoneal carcinomatosis showed significant independent impact, but not the subclass. For OS, the PEV of the subclassification alone was 8.45%, already representing 60% of the PEV of the combined model using all clinicopathologic parameters except our subclassification (PEV = 14.10%). Checking the proportional hazards assumption of the Cox model by including interactions of covariates with time, we noticed that the effect of subclassification increases consistently over time, roughly doubling the risk every year (data not shown). In Table 3, results from univariate Cox regression analysis for patients with non‐serous tumors are shown, which, owing to limited event numbers, were adjusted only for peritoneal carcinomatosis by stratification. Again, subclassification can be confirmed as an independent predictor for OS with similar HRs as obtained with serous tumors. In contrast with the analysis of serous tumors, subclassification showed a significant effect on PFS in non‐serous tumors. In Figure 2, Kaplan–Meier estimates for patients with FIGO III serous and non‐serous tumors are shown, stratifying the patients in three groups using the two strongest predictive factors only, subclass and peritoneal carcinomatosis. One group of patients (Subclass 1 and no peritoneal carcinomatosis) presents with a very good prognosis (75th percentile not reached), one group (either Subclass 2 or peritoneal carcinomatosis) has a slightly worse prognosis (50th percentile not reached), and one group (Subclass 2 and peritoneal carcinomatosis) appears to have considerably worse prognosis (50th percentile reached at 33 and 18 months for patients with serous and non‐serous tumors, respectively).
Figure 1.
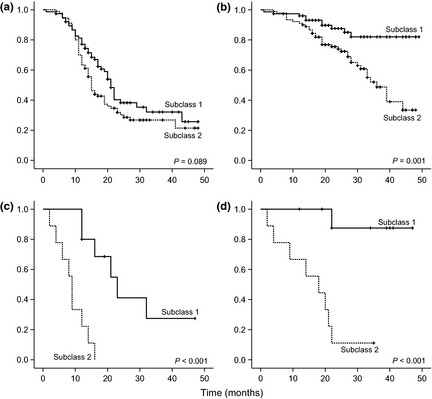
Kaplan–Meier estimates of the impact of molecular subclasses on (a,c) progression‐free survival for patients with (a) serous (P = 0.089) and (c) non‐serous (P < 0.001; log rank test) epithelial ovarian cancer (EOC) and (b,d) overall survival for patients with (b) serous (P = 0.001) and (d) non‐serous (P < 0.001; log rank test) EOC.
Table 2.
Multiple Cox regression analyses for progression‐free and overall survival, as well as the proportion of explained variations of clinicopathologic parameters and the subclassification, for late stage serous ovarian cancer patients (n = 165)
| Progression‐free survival | Overall survival | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HR | 95% CI | P | PEV alone | PEV model | Cumulative PEV | HR | 95% CI | P | PEV alone | PEV model | Cumulative PEV | |||
| Age (per decade) | 1.07 | 0.89 | 1.28 | 0.492 | 0.82% | 1.02 | 0.99 | 1.05 | 0.103 | 3.15% | ||||
| FIGO (IV vs III) | 2.17 | 1.36 | 3.46 | 0.001 | 4.84% | 1.86 | 0.92 | 3.77 | 0.083 | 3.13% | ||||
| Grade (3 vs 1&2) | 1.47 | 0.87 | 2.46 | 0.148 | 3.14% | 2.13 | 0.87 | 5.23 | 0.098 | 4.38% | ||||
| Residual tumor (yes vs no) | 1.41 | 0.93 | 2.13 | 0.108 | 3.19% | 1.07 | 0.56 | 2.02 | 0.840 | 1.04% | ||||
| Peritoneal carcinomatosis (yes vs no) | 2.69 | 1.55 | 4.67 | <0.001 | 8.20% | 14.60% | 4.56 | 1.39 | 14.99 | 0.012 | 7.90% | 14.10% | ||
| Subclass (2 vs 1) | 1.25 | 0.84 | 1.87 | 0.269 | 1.37% | 0.61% | 2.87 | 1.40 | 5.88 | 0.004 | 8.45% | 6.69% | ||
HR, hazard ratio; PEV, proportion of explained variations; 95% CI, 95% confidence interval.
Table 3.
Multiple Cox regression analyses for progression‐free survival and overall survival and proportion of explained variations (PEV) of clinicopathologic parameters and the subclassification for late stage non‐serous ovarian cancer patients (n = 19)
| Progression‐free survival | Overall survival | |||||||
|---|---|---|---|---|---|---|---|---|
| HRa | 95% CI | P | HRa | 95% CI | P | |||
| Subclass (2 vs 1) | 4.04 | 1.02 | 15.97 | 0.047 | 12.42 | 1.41 | 106.46 | 0.023 |
Adjusted for peritoneal carcinomatosis by stratification. HR, hazard ratio; 95% CI, 95% confidence interval.
Figure 2.
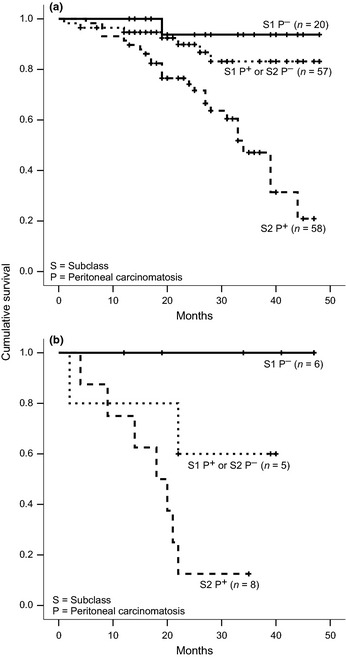
Kaplan–Meier estimates of overall survival of patients with International Federation of Gynecology and Obstetrics (FIGO) Stage III (a) serous and (b) non‐serous epithelial ovarian cancer, stratified by molecular subclasses and peritoneal carcinomatosis (both P < 0.001; log rank test).
Significance analysis of microarrays and functional analysis of the differences
To assess the difference in the transcriptome between Subclass 1 and Subclass 2 tumors from patients presenting with FIGO III and IV high‐grade serous tumors, an SAM was performed that resulted in 2082 significantly different expressed probe IDs with an FDR of 5% (see Table S1 available as Supplementary Material to this paper). These represent 21.4% (2082/9708) of all probe IDs analyzed. Of these, the expression of 1289 probe IDs was higher in Subclass 2 tumors, whereas that of 793 was lower. Functional analysis using DAVID of these 2082 probe IDs (1595 of which were annotated in DAVID) revealed highly enriched annotation clusters termed “cytoskeleton”, “cell adhesion”, “extracellular matrix”, and “cell motion” (Table 4). The most prominent over‐represented pathway was “focal adhesion”, with 42 genes involved, corresponding to a 2.1‐fold enrichment (FDR 0.5%; see Fig. S1).
Table 4.
Functional clustering of differentially expressed genes (1595 annotated) with Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.7
| Category | Term | Count | % | Fold enrichment | P‐value | FDR |
|---|---|---|---|---|---|---|
| Annotation Cluster 1 Enrichment Score: 3.46 | ||||||
| GOTERM_MF_FAT | GO:0046983–protein dimerization activity | 75 | 4.70 | 1.6 | 3.81E − 05 | 6.10E − 02 |
| GOTERM_MF_FAT | GO:0042802–identical protein binding | 82 | 5.14 | 1.5 | 2.27E − 04 | 3.63E − 01 |
| GOTERM_MF_FAT | GO:0042803–protein homodimerization activity | 44 | 2.76 | 1.5 | 4.66E − 03 | 7.21E + 00 |
| Annotation Cluster 2 Enrichment Score: 3.36 | ||||||
| GOTERM_CC_FAT | GO:0043232–intracellular non‐membrane‐bounded organelle | 294 | 18.43 | 1.3 | 1.69E − 06 | 2.47E − 03 |
| GOTERM_CC_FAT | GO:0043228–non‐membrane‐bounded organelle | 294 | 18.43 | 1.3 | 1.69E − 06 | 2.47E − 03 |
| GOTERM_MF_FAT | GO:0003779–actin binding | 53 | 3.32 | 1.9 | 8.73E − 06 | 1.40E − 02 |
| SP_PIR_KEYWORDS | Actin binding | 41 | 2.57 | 2.1 | 1.84E − 05 | 2.76E − 02 |
| GOTERM_CC_FAT | GO:0015629–actin cytoskeleton | 45 | 2.82 | 1.9 | 5.17E − 05 | 7.54E − 02 |
| SP_PIR_KEYWORDS | Cytoskeleton | 80 | 5.02 | 1.6 | 8.37E − 05 | 1.25E − 01 |
| GOTERM_MF_FAT | GO:0008092–cytoskeletal protein binding | 69 | 4.33 | 1.6 | 1.10E – 04 | 1.77E − 01 |
| GOTERM_BP_FAT | GO:0030036–actin cytoskeleton organization | 36 | 2.26 | 1.8 | 6.89E − 04 | 1.25E + 00 |
| GOTERM_CC_FAT | GO:0005856–cytoskeleton | 156 | 9.78 | 1.3 | 9.63E − 04 | 1.40E + 00 |
| GOTERM_BP_FAT | GO:0030029–actin filament‐based process | 37 | 2.32 | 1.7 | 1.14E − 03 | 2.07E + 00 |
| GOTERM_BP_FAT | GO:0007010–cytoskeleton organization | 57 | 3.57 | 1.5 | 2.50E − 03 | 4.48E + 00 |
| GOTERM_CC_FAT | GO:0015630–microtubule cytoskeleton | 62 | 3.89 | 1.3 | 3.99E − 02 | 4.48E + 01 |
| GOTERM_CC_FAT | GO:0005815–microtubule organizing center | 32 | 2.01 | 1.4 | 4.14E − 02 | 4.61E + 01 |
| GOTERM_CC_FAT | GO:0005813–centrosome | 28 | 1.76 | 1.4 | 6.38E − 02 | 6.18E + 01 |
| GOTERM_CC_FAT | GO:0044430–cytoskeletal part | 98 | 6.14 | 1.2 | 8.30E − 02 | 7.17E + 01 |
| Annotation Cluster 3 Enrichment Score: 3.29 | ||||||
| GOTERM_BP_FAT | GO:0007155–cell adhesion | 94 | 5.89 | 1.5 | 3.12E − 05 | 5.72E − 02 |
| GOTERM_BP_FAT | GO:0022610–biological adhesion | 94 | 5.89 | 1.5 | 3.30E – 05 | 6.04E − 02 |
| SP_PIR_KEYWORDS | Cell adhesion | 55 | 3.45 | 1.6 | 5.24E − 04 | 7.83E − 01 |
| GOTERM_BP_FAT | GO:0016337–cell‐cell adhesion | 31 | 1.94 | 1.3 | 1.32E − 01 | 9.26E + 01 |
| Annotation Cluster 4 Enrichment Score: 3.14 | ||||||
| SP_PIR_KEYWORDS | ECM | 55 | 3.45 | 2.8 | 4.16E − 12 | 6.24E − 09 |
| GOTERM_CC_FAT | GO:0031012–ECM | 70 | 4.39 | 2.3 | 8.15E − 11 | 1.19E − 07 |
| GOTERM_CC_FAT | GO:0005578–proteinaceous ECM | 65 | 4.08 | 2.3 | 4.01E − 10 | 5.85E − 07 |
| GOTERM_CC_FAT | GO:0005581–collagen | 16 | 1.00 | 5.1 | 9.18E − 08 | 1.34E − 04 |
| GOTERM_CC_FAT | GO:0044420–ECM part | 30 | 1.88 | 2.9 | 2.59E − 07 | 3.78E − 04 |
| SP_PIR_KEYWORDS | Hydroxylation | 21 | 1.32 | 3.7 | 5.20E − 07 | 7.79E − 04 |
| SP_PIR_KEYWORDS | Triple helix | 13 | 0.82 | 5.2 | 2.48E − 06 | 3.72E − 03 |
| SP_PIR_KEYWORDS | Hydroxylysine | 12 | 0.75 | 4.8 | 1.74E − 05 | 2.60E − 02 |
| SP_PIR_KEYWORDS | Trimer | 11 | 0.69 | 5.2 | 1.85E − 05 | 2.78E − 02 |
| GOTERM_BP_FAT | GO:0030198–ECM organization | 24 | 1.50 | 2.6 | 2.79E − 05 | 5.11E − 02 |
| SP_PIR_KEYWORDS | Collagen | 21 | 1.32 | 2.7 | 6.04E − 05 | 9.05E − 02 |
| SP_PIR_KEYWORDS | Hydroxyproline | 12 | 0.75 | 4.0 | 1.11E − 04 | 1.67E − 01 |
| GOTERM_BP_FAT | GO:0043062–extracellular structure organization | 30 | 1.88 | 2.1 | 1.89E − 04 | 3.45E − 01 |
| GOTERM_CC_FAT | GO:0005583–fibrillar collagen | 7 | 0.44 | 6.6 | 2.81E − 04 | 4.09E − 01 |
| GOTERM_CC_FAT | GO:0005604–basement membrane | 18 | 1.13 | 2.6 | 4.06E − 04 | 5.91E − 01 |
| KEGG_PATHWAY | hsa04512: ECM–receptor interaction | 20 | 1.25 | 2.4 | 4.49E − 04 | 5.49E − 01 |
| INTERPRO | IPR008160: collagen triple helix repeat | 18 | 1.13 | 2.5 | 5.57E − 04 | 9.46E − 01 |
| GOTERM_BP_FAT | GO:0030199–collagen fibril organization | 10 | 0.63 | 3.9 | 6.06E − 04 | 1.10E + 00 |
| SP_PIR_KEYWORDS | Ehlers–Danlos syndrome | 6 | 0.38 | 6.7 | 1.05E − 03 | 1.56E + 00 |
| UP_SEQ_FEATURE | Domain: fibrillar collagen NC1 | 6 | 0.38 | 6.7 | 1.08E − 03 | 1.97E + 00 |
| INTERPRO | IPR000885: fibrillar collagen, C‐terminal | 6 | 0.38 | 6.5 | 1.27E − 03 | 2.14E + 00 |
| GOTERM_MF_FAT | GO:0048407–PDGF binding | 6 | 0.38 | 6.3 | 1.38E − 03 | 2.18E + 00 |
| SMART | SM00038: COLFI | 6 | 0.38 | 6.0 | 1.73E − 03 | 2.31E + 00 |
| UP_SEQ_FEATURE | Domain:TSP N‐terminal | 8 | 0.50 | 4.3 | 1.78E − 03 | 3.24E + 00 |
| UP_SEQ_FEATURE | Region of interest:triple‐helical region | 8 | 0.50 | 4.3 | 1.78E − 03 | 3.24E + 00 |
| UP_SEQ_FEATURE | Propeptide:C‐terminal propeptide | 5 | 0.31 | 7.7 | 2.34E − 03 | 4.24E + 00 |
| GOTERM_MF_FAT | GO:0005201–ECM structural constituent | 17 | 1.07 | 2.3 | 2.43E − 03 | 3.82E + 00 |
| INTERPRO | IPR003129:Laminin G, TSP‐type, N‐terminal | 7 | 0.44 | 3.8 | 8.17E − 03 | 1.31E + 01 |
| SMART | SM00210:TSPN | 7 | 0.44 | 3.5 | 1.13E − 02 | 1.42E + 01 |
| GOTERM_MF_FAT | GO:0046332–SMAD binding | 10 | 0.63 | 2.5 | 1.49E − 02 | 2.14E + 01 |
| UP_SEQ_FEATURE | Region of interest: non‐helical region | 4 | 0.25 | 6.1 | 2.20E − 02 | 3.38E + 01 |
| GOTERM_CC_FAT | GO:0005588–collagen Type V | 3 | 0.19 | 11.2 | 2.23E − 02 | 2.80E + 01 |
| SP_PIR_KEYWORDS | Pyroglutamic acid | 9 | 0.56 | 2.4 | 2.97E − 02 | 3.64E + 01 |
| UP_SEQ_FEATURE | Glycosylation site: O‐linked (Gal…) | 4 | 0.25 | 5.5 | 3.11E − 02 | 4.42E + 01 |
| GOTERM_BP_FAT | GO:0043588–skin development | 7 | 0.44 | 2.7 | 3.75E − 02 | 5.04E + 01 |
| PIR_SUPERFAMILY | PIRSF002257: collagen α1 (V) chain | 3 | 0.19 | 8.5 | 4.08E − 02 | 4.65E + 01 |
| PIR_SUPERFAMILY | PIRSF002255: collagen α1 (I) chain | 3 | 0.19 | 6.8 | 6.41E − 02 | 6.30E + 01 |
| UP_SEQ_FEATURE | Propeptide: N‐terminal propeptide | 3 | 0.19 | 6.1 | 7.96E − 02 | 7.84E + 01 |
| PIR_SUPERFAMILY | PIRSF002258:collagen α1 (IV) chain | 3 | 0.19 | 4.9 | 1.20E − 01 | 8.53E + 01 |
| GOTERM_CC_FAT | GO:0030934–anchoring collagen | 3 | 0.19 | 3.7 | 1.87E − 01 | 9.52E + 01 |
| INTERPRO | IPR012680: Laminin G, subdomain 2 | 5 | 0.31 | 1.4 | 4.78E − 01 | 1.00E + 02 |
| INTERPRO | IPR001791: Laminin G | 5 | 0.31 | 1.3 | 5.33E − 01 | 1.00E + 02 |
| SMART | SM00282: LamG | 5 | 0.31 | 1.2 | 5.89E − 01 | 1.00E + 02 |
| INTERPRO | IPR013320: Concanavalin A‐like lectin/glucanase, subgroup | 7 | 0.44 | 1.1 | 6.40E − 01 | 1.00E + 02 |
| GOTERM_BP_FAT | GO:0008544–epidermis development | 15 | 0.94 | 0.9 | 7.54E − 01 | 1.00E + 02 |
| GOTERM_BP_FAT | GO:0007398–ectoderm development | 16 | 1.00 | 0.9 | 7.71E − 01 | 1.00E + 02 |
| Annotation Cluster 5 Enrichment Score: 3.00 | ||||||
| GOTERM_CC_FAT | GO:0060205–cytoplasmic membrane‐bounded vesicle lumen | 16 | 1.00 | 4.1 | 3.02E − 06 | 4.40E − 03 |
| GOTERM_CC_FAT | GO:0031983–vesicle lumen | 16 | 1.00 | 3.9 | 5.66E − 06 | 8.26E − 03 |
| GOTERM_CC_FAT | GO:0031093–platelet α granule lumen | 15 | 0.94 | 4.1 | 6.30E − 06 | 9.20E − 03 |
| GOTERM_CC_FAT | GO:0031091–platelet α granule | 17 | 1.07 | 3.4 | 1.86E − 05 | 2.71E − 02 |
| GOTERM_CC_FAT | GO:0044433–cytoplasmic vesicle part | 30 | 1.88 | 1.8 | 2.16E − 03 | 3.11E + 00 |
| GOTERM_CC_FAT | GO:0031410–cytoplasmic vesicle | 79 | 4.95 | 1.4 | 2.61E − 03 | 3.73E + 00 |
| GOTERM_CC_FAT | GO:0016023–cytoplasmic membrane‐bounded vesicle | 69 | 4.33 | 1.4 | 3.20E − 03 | 4.57E + 00 |
| SP_PIR_KEYWORDS | Mitogen | 10 | 0.63 | 3.2 | 3.32E − 03 | 4.86E + 00 |
| GOTERM_CC_FAT | GO:0031988–membrane‐bounded vesicle | 70 | 4.39 | 1.4 | 4.51E − 03 | 6.39E + 00 |
| GOTERM_CC_FAT | GO:0031982–vesicle | 80 | 5.02 | 1.3 | 5.29E − 03 | 7.45E + 00 |
| GOTERM_CC_FAT | GO:0042470–melanosome | 16 | 1.00 | 2.0 | 1.14E − 02 | 1.54E + 01 |
| GOTERM_CC_FAT | GO:0048770–pigment granule | 16 | 1.00 | 2.0 | 1.14E − 02 | 1.54E + 01 |
| GOTERM_CC_FAT | GO:0030141–secretory granule | 26 | 1.63 | 1.6 | 1.66E − 02 | 2.17E + 01 |
| SP_PIR_KEYWORDS | Cytoplasmic vesicle | 25 | 1.57 | 1.3 | 1.55E − 01 | 9.19E + 01 |
| Annotation Cluster 6 Enrichment Score: 2.64 | ||||||
| GOTERM_BP_FAT | GO:0030334–regulation of cell migration | 35 | 2.19 | 2.4 | 3.65E − 06 | 6.69E − 03 |
| GOTERM_BP_FAT | GO:0006928–cell motion | 72 | 4.51 | 1.7 | 5.81E − 06 | 1.06E − 02 |
| GOTERM_BP_FAT | GO:0040012–regulation of locomotion | 36 | 2.26 | 2.1 | 2.53E − 05 | 4.64E − 02 |
| GOTERM_BP_FAT | GO:0051270–regulation of cell motion | 36 | 2.26 | 2.1 | 2.84E − 05 | 5.20E − 02 |
| GOTERM_BP_FAT | GO:0001944–vasculature development | 43 | 2.70 | 1.9 | 3.54E − 05 | 6.47E − 02 |
| GOTERM_BP_FAT | GO:0001568–blood vessel development | 42 | 2.63 | 1.9 | 4.35E − 05 | 7.97E − 02 |
| GOTERM_BP_FAT | GO:0032101–regulation of response to external stimulus | 27 | 1.69 | 1.9 | 1.46E − 03 | 2.63E + 00 |
| GOTERM_BP_FAT | GO:0030335–positive regulation of cell migration | 18 | 1.13 | 2.3 | 1.74E − 03 | 3.13E + 00 |
| GOTERM_BP_FAT | GO:0040017–positive regulation of locomotion | 19 | 1.19 | 2.2 | 2.04E − 03 | 3.68E + 00 |
| GOTERM_BP_FAT | GO:0016477–cell migration | 40 | 2.51 | 1.6 | 2.08E − 03 | 3.74E + 00 |
| GOTERM_BP_FAT | GO:0001525–angiogenesis | 25 | 1.57 | 1.9 | 2.44E − 03 | 4.37E + 00 |
| GOTERM_BP_FAT | GO:0051272–positive regulation of cell motion | 18 | 1.13 | 2.1 | 4.95E − 03 | 8.69E + 00 |
| GOTERM_BP_FAT | GO:0048514–blood vessel morphogenesis | 31 | 1.94 | 1.7 | 5.86E − 03 | 1.02E + 01 |
| GOTERM_BP_FAT | GO:0051674–localization of cell | 41 | 2.57 | 1.5 | 7.62E − 03 | 1.31E + 01 |
| GOTERM_BP_FAT | GO:0048870–cell motility | 41 | 2.57 | 1.5 | 7.62E − 03 | 1.31E + 01 |
| GOTERM_BP_FAT | GO:0050921–positive regulation of chemotaxis | 8 | 0.50 | 3.1 | 1.11E − 02 | 1.86E + 01 |
| GOTERM_BP_FAT | GO:0050920–regulation of chemotaxis | 8 | 0.50 | 2.9 | 1.61E − 02 | 2.57E + 01 |
| GOTERM_BP_FAT | GO:0050927–positive regulation of positive chemotaxis | 6 | 0.38 | 3.6 | 2.14E − 02 | 3.27E + 01 |
| GOTERM_BP_FAT | GO:0050926–regulation of positive chemotaxis | 6 | 0.38 | 3.6 | 2.14E − 02 | 3.27E + 01 |
| GOTERM_BP_FAT | GO:0007169–transmembrane receptor protein tyrosine kinase signaling pathway | 30 | 1.88 | 1.5 | 2.25E − 02 | 3.41E + 01 |
| GOTERM_BP_FAT | GO:0032103–positive regulation of response to external stimulus | 12 | 0.75 | 2.1 | 2.28E − 02 | 3.44E + 01 |
| GOTERM_BP_FAT | GO:0048520–positive regulation of behavior | 8 | 0.50 | 2.7 | 2.62E − 02 | 3.85E + 01 |
| GOTERM_BP_FAT | GO:0008284–positive regulation of cell proliferation | 47 | 2.95 | 1.3 | 5.82E − 02 | 6.66E + 01 |
| GOTERM_BP_FAT | GO:0050795–regulation of behavior | 8 | 0.50 | 2.0 | 1.05E − 01 | 8.70E + 01 |
| GOTERM_BP_FAT | GO:0048584–positive regulation of response to stimulus | 27 | 1.69 | 1.3 | 1.37E − 01 | 9.32E + 01 |
COLFI, fibrillar collagens C‐terminal domain; ECM, extracellular matrix; FDR, false discovery rate; LamG, Laminin G domain; PDGF, platelet‐derived growth factor; TSP, thrombospondin; TSPN, TSPN‐terminal domain.
Discussion
The present study describes a validation analysis of a previously defined gene signature to establish its relevance as a clinically useful prognostic factor. Evaluating the prognostic outcome of women with ovarian cancer is considered a key part in the treatment planning for patients with ovarian cancer, especially those with high‐grade serous ovarian cancer. Currently, typical clinicopathologic parameters, such as age, histology, FIGO stage, grade, and residual tumor mass following primary surgery, are considered prognostic factors for patients with advanced‐stage EOC. Unfortunately, the routine use of thus far published new prognostic factors in clinical practice has had limited success.20
Histology has been described as independent predictor of prognosis. However, considering that more than two‐thirds of advanced EOC are reported to be of the serous type,21 histology cannot be used as prognostic predictor within this large group of patients. In addition, tumors classified in the past as high‐grade endometrioid carcinoma are actually mostly of the serous type.22
Evaluating the care and treatment of ovarian cancer patients is, to a large extent, related to tumor stage and grade. Considering grade, it has to be taken into account that high‐grade serous carcinoma represents the dominant subtype of ovarian carcinoma.22 In advanced ovarian carcinoma, it has been observed that grade has no strong prognostic value, or even lacks a significant impact on prognosis,23 as confirmed by our study. Addressing FIGO stage, our Cox proportional hazards model revealed a significant influence of advanced FIGO stage (IV vs III vs II) only on PFS (HR 2.03; P = 0.002) and not on OS (HR 1.60; P = 0.182), analyzed over all histological subtypes. In accordance with previous studies,24 we observed in the present study that patients with no macroscopic residual tumor mass after primary surgery have a far better PFS, but only a slightly better OS.
New and reliable prognostic factors for patients with high‐grade serous tumors, such as molecular subclassifications, could help detect patients who may benefit from different treatment regimens. Several microarray studies have been conducted to find prognostic gene sets or to subclassify different subtypes of ovarian carcinoma.25 Using a robust statistical method, Yoshihara et al.11 published such a set, consisting of 112 genes, dividing advanced‐stage serous tumors into two groups: one group with characteristics similar to early stage tumors and a better prognosis, and the other group with different characteristics and a worse prognosis. However, the sample size in that study was only 35 and therefore the survival analysis lacks power. The aim of the present study was to validate the subclassification system of Yoshihara et al.11 using a large and extensively documented patient cohort of advanced‐stage serous ovarian cancers and to assess the potential impact of this signature on the outcome of patients with non‐serous tumors. Indeed, we were able to reproduce the subclassification system of Yoshihara et al.11 and divided the advanced‐stage tumors into 86 Subclass 1 tumors with a better OS and 99 Subclass 2 tumors with a significantly worse prognosis. The prognostic effect on OS remained significant and stable in the multiple Cox regression model, corrected for all relevant known clinicopathologic parameters, for both histological serous and non‐serous types, with mainly the same HRs. Furthermore, both types of tumors showed a marked difference in their transcriptomes, with 2082 probe IDs exhibiting significantly different expression between high‐grade serous Subclass 1 and Subclass 2 tumors. A functional analysis of these genes revealed adhesion proteins and proteins involved in attachment to the extracellular matrix as the most over‐represented functional groups of genes. Focal adhesion as one of the most over‐represented pathways indicates that the differences in cell–cell and cell–extracellular matrix attachment, with corresponding signaling, play a major role in EOC biology and prognosis. Focal adhesion kinase (FAK), the central protein in the focal adhesion pathway, was shown to be overexpressed in several tumors and plays a significant role in cell survival, migration, and invasion.26 Furthermore, an FAK inhibitor was shown to increase survival in a mouse model of ovarian cancer26 and the phosphorylated (p‐) version of FAK Y397, in combination with missing deleted in liver cancer (DLC1) expression (shown to dephosphorylate p‐FAK), indicated a significant decrease in OS.27 Yoshihara et al.11 described zinc finger E‐box binding homeobox 2 (ZEB2), a regulator of E‐cadherin (CDH1), as important gene deregulated in Subclass 1 compared with Subclass 2 tumors, speculating that epithelial–mesenchymal transition (EMT) could play an important role in the worse outcome of Subclass 2 patients. Despite the fact that, in the present study, ZEB2 expression was also significantly higher (P < 0.001) in Subclass 2 patients and CDH1 expression tended to be lower (although not significantly different), typical EMT‐associated genes were not over‐represented in the differentially expressed genes (data not shown). Therefore, we would propose that classical EMT seems not to be the leading factor for stratifying advanced EOCs according to this 112 gene signature in two prognostic different subclasses.
Despite the fact that this subclassification correlates significantly with the occurrence of diffuse peritoneal carcinomatosis, both factors are significant, independent, and strong predictors for OS (peritoneal carcinomatosis HR of 4.56 and 5.36 for patient with serous and non‐serous tumors, respectively, and subclassification HR of 2.87 and 2.75, respectively).
In particular, patients presenting with the combination of Subclass 2 tumors and peritoneal carcinomatosis seem to have a exceptionally bad prognosis, regardless as to the histologically type of tumors. For PFS, only the occurrence of peritoneal carcinomatosis, and not the new subclassification, had a significant and independent prognostic value. Due to the facts that the negative influence of tumors of the subclass 2 on overall survival increases over time and the high number of differentially expressed genes obtained between these two subclasses, we suppose that tumors classified into these two subclasses render two biologically different tumor types. This also seems to be true for both types of histologies: serous and non‐serous. Recently, The Cancer Genome Atlas Research (CGAR) Network published integrated genomic analyses of high‐grade serous ovarian carcinoma.28 Using approximately 1500 intrinsically variable genes, the CGAR Network found four clusters using unsupervised non‐negative matrix factorization that they named “Differentiated”, “Immunoreactive”, “Mesenchymal”, and “Proliferative”. Nevertheless, there was no survival difference associated with these four subclusters.
To identify the biological mechanisms and pathways differentiating the two subclasses, further molecular biological research has to be conducted. Improving our understanding of the biology of EOC, especially the behavior of the tumor in the peritoneum, is of critical importance. As a next step, we plan to reduce this now independently validated 112 gene signature to a more practical and clinically applicable (i.e. smaller) gene set. Using robust linear models, followed by a validation step in a large, independent cohort of advanced‐stage EOC, we hope to answer the question whether this enhanced subclassification approach can be used in clinical practice. Furthermore, we want to validate the impact of this signature in a large cohort of patients with non‐serous tumors, including clear cell tumors.
Disclosure Statement
The authors have no conflicts of interest to declare.
Supporting information
Fig. S1. Focal adhesion pathway (KEGG pathway hsa04510) highlighting all proteins, protein families, and subpathways over‐represented in the list of differentially expressed genes.
Table S1. Differentially expressed genes between the two serous epithelial ovarian cancer subclasses.
Acknowledgment
This work was supported by the Sixth Framework Programme (FP6) Project of the European Union called “Ovarian Cancer: Diagnosis of a silent killer–OVCAD” (no. 018698) to JS, IV, SM, GH, RZ.
References
- 1. Perou CM, Sorlie T, Eisen MB et al Molecular portraits of human breast tumours. Nature 2000; 406: 747–52. [DOI] [PubMed] [Google Scholar]
- 2. Joyce T, Cantarella D, Isella C, Medico E, Pintzas A. A molecular signature for epithelial to mesenchymal transition in a human colon cancer cell system is revealed by large‐scale microarray analysis. Clin Exp Metastasis 2009; 26: 569–87. [DOI] [PubMed] [Google Scholar]
- 3. Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death‐from‐cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest 2005; 115: 1503–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Miecznikowski JC, Wang D, Liu S, Sucheston L, Gold D. Comparative survival analysis of breast cancer microarray studies identifies important prognostic genetic pathways. BMC Cancer 2010; 10: 573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weigelt B, Mackay A, A'Hern R et al Breast cancer molecular profiling with single sample predictors: a retrospective analysis. Lancet Oncol 2010; 11: 339–49. [DOI] [PubMed] [Google Scholar]
- 6. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62: 10–29. [DOI] [PubMed] [Google Scholar]
- 7. Gilks CB. Subclassification of ovarian surface epithelial tumors based on correlation of histologic and molecular pathologic data. Int J Gynecol Pathol 2004; 23: 200–5. [DOI] [PubMed] [Google Scholar]
- 8. Kalloger SE, Kobel M, Leung S et al Calculator for ovarian carcinoma subtype prediction. Mod Pathol 2010; 24: 512–21. [DOI] [PubMed] [Google Scholar]
- 9. De Cecco L, Marchionni L, Gariboldi M et al Gene expression profiling of advanced ovarian cancer: characterization of a molecular signature involving fibroblast growth factor 2. Oncogene 2004; 23: 8171–83. [DOI] [PubMed] [Google Scholar]
- 10. Tothill RW, Tinker AV, George J et al Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res 2008; 14: 5198–208. [DOI] [PubMed] [Google Scholar]
- 11. Yoshihara K, Tajima A, Komata D et al Gene expression profiling of advanced‐stage serous ovarian cancers distinguishes novel subclasses and implicates ZEB2 in tumor progression and prognosis. Cancer Sci 2009; 100: 1421–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rustin GJ, Nelstrop AE, Bentzen SM, Bond SJ, McClean P. Selection of active drugs for ovarian cancer based on CA‐125 and standard response rates in phase II trials. J Clin Oncol 2000; 18: 1733–9. [DOI] [PubMed] [Google Scholar]
- 13. Rustin GJ, Vergote I, Eisenhauer E et al Definitions for response and progression in ovarian cancer clinical trials incorporating RECIST 1.1 and CA 125 agreed by the Gynecological Cancer Intergroup (GCIG). Int J Gynecol Cancer 2011; 21: 419–23. [DOI] [PubMed] [Google Scholar]
- 14. Saeed AI, Bhagabati NK, Braisted JC et al TM4 microarray software suite. Methods Enzymol 2006; 411: 134–93. [DOI] [PubMed] [Google Scholar]
- 15. Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat 1979; 6: 65–70. [Google Scholar]
- 16. Cox DR. Regression models and life‐tables. J R Stat Soc B 1972; 34: 187–220. [Google Scholar]
- 17. Kaplan EL, Meier P. Non‐parametric estimation from incomplete observations. J Am Stat Assoc 1958; 53: 457–81. [Google Scholar]
- 18. Heinze G, Schemper M. Comparing the importance of prognostic factors in Cox and logistic regression using SAS. Comput Methods Programs Biomed 2003; 71: 155–63. [DOI] [PubMed] [Google Scholar]
- 19. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 20. Clark TG, Stewart ME, Altman DG, Gabra H, Smyth JF. A prognostic model for ovarian cancer. Br J Cancer 2001; 85: 944–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Winter WE III, Maxwell GL, Tian C et al Prognostic factors for stage III epithelial ovarian cancer: a Gynecologic Oncology Group Study. J Clin Oncol 2007; 25: 3621–7. [DOI] [PubMed] [Google Scholar]
- 22. McCluggage WG. Morphological subtypes of ovarian carcinoma: a review with emphasis on new developments and pathogenesis. Pathology 2011; 43: 420–32. [DOI] [PubMed] [Google Scholar]
- 23. Chi DS, Liao JB, Leon LF et al Identification of prognostic factors in advanced epithelial ovarian carcinoma. Gynecol Oncol 2001; 82: 532–7. [DOI] [PubMed] [Google Scholar]
- 24. Elattar A, Bryant A, Winter‐Roach BA, Hatem M, Naik R. Optimal primary surgical treatment for advanced epithelial ovarian cancer. Cochrane Database Syst Rev 2011; 10: CD007565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chon HS, Lancaster JM. Microarray‐based gene expression studies in ovarian cancer. Cancer Control 2011; 18: 8–15. [DOI] [PubMed] [Google Scholar]
- 26. Halder J, Lin YG, Merritt WM et al Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res 2007; 67: 10976–83. [DOI] [PubMed] [Google Scholar]
- 27. Fan DM, Shi HR. Pilot study: alteration of deleted in liver cancer1 and phosphorylated focal adhesion kinase y397 cytoplasmic expression and the prognostic value in advanced epithelial ovarian carcinoma. Int J Mol Sci 2011; 12: 8489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Network CGAR . Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474: 609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Focal adhesion pathway (KEGG pathway hsa04510) highlighting all proteins, protein families, and subpathways over‐represented in the list of differentially expressed genes.
Table S1. Differentially expressed genes between the two serous epithelial ovarian cancer subclasses.