

Abstract
Sulfur trioxide (SO3) is a crucial compound for atmospheric sulfuric acid (H2SO4) formation, acid rain formation, and other atmospheric physicochemical processes. During the daytime, SO3 is mainly produced from the photo-oxidation of SO2 by OH radicals. However, the sources of SO3 during the early morning and night, when OH radicals are scarce, are not fully understood. We report results from two field measurements in urban Beijing during winter and summer 2019, using a nitrate-CI-APi-LTOF (chemical ionization-atmospheric pressure interface-long-time-of-flight) mass spectrometer to detect atmospheric SO3 and H2SO4. Our results show the level of SO3 was higher during the winter than during the summer, with high SO3 levels observed especially during the early morning (∼05:00 to ∼08:30) and night (∼18:00 to ∼05:00 the next day). On the basis of analysis of SO2, NOx, black carbon, traffic flow, and atmospheric ions, we suggest SO3 could be formed from the catalytic oxidation of SO2 on the surface of traffic-related black carbon. This previously unidentified SO3 source results in significant H2SO4 formation in the early morning and thus promotes sub-2.5 nm particle formation. These findings will help in understanding urban SO3 and formulating policies to mitigate secondary particle formation in Chinese megacities.
Introduction
Atmospheric SO3 is a vital intermediate in gaseous H2SO4 formation, which in turn is a crucial compound in acid rain formation, new particle formation, secondary aerosol formation, and other atmospheric physicochemical processes.1−8 During the daytime, when the intensity of UVB (ultraviolet radiation B) radiation that leads to the formation of OH radicals is high, SO3 is mainly formed by atmospheric photo-oxidation of SO2 by OH radicals. The mechanism of formation of SO3 from photo-oxidation can be written as
| R1 |
| R2 |
However, during the early morning and at night, when OH radicals are scarce, the sources of atmospheric SO3 remain unclear. Stabilized Criegee intermediates (sCI; general formula of R1R2COO) generated from the ozonolysis of alkenes have been found to be an important gas-phase oxidant for SO2 in addition to OH (eq R3):9,10
| R3 |
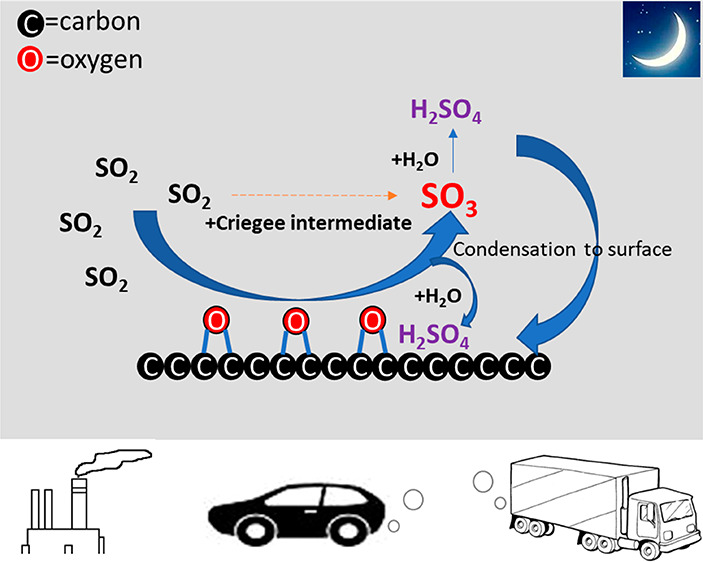
Besides gas-phase oxidation of SO2 by OH radicals and sCI, SO3 may also be formed from the heterogeneous oxidation of SO2 on oxygen-functionalized graphene and soot surfaces.11−13 He et al. have proposed that SO2 molecules can react with surface epoxide groups of carbonaceous (or soot) aerosols, leading to SO3 formation. These surface epoxy groups are considerably enhanced by atmospheric aging of soot particles in the presence of O2.12,14 Additionally, a recent theoretical study revealed high atmospheric water content could promote the oxidation of SO2 to SO3 by O2 on carbonaceous aerosol surfaces.15 Although extensive laboratory studies have reported that persistent particulate H2SO4 and sulfate can be formed from catalytic heterogeneous reaction between SO2 and carbon (soot) particles in the presence of O2, O3, NOx, NH3, and water,16−19 the intermediate precursors of particulate sulfate, and the molecular-level details of this process, remain unclear. In addition to atmospheric formation of SO3 via the oxidation of SO2, a large amount of SO3 can also be emitted to the air from coal-fired power plants and other industrial processes related to coal combustion and then be rapidly converted to H2SO4.20−22
In the atmosphere, the water-catalyzed hydration of SO3 plays an essential role in H2SO4 formation (eq R4).
| R4 |
In reaction R4, the second water molecule acts as a catalyst and significantly reduces the energy barrier of SO3 hydration.23,24 The rate coefficient of the reaction of SO3 with the water dimer [(H2O)2], or H2O-catalyzed hydrolysis, is between 10–12 and 10–10 cm3 molecule–1 s–1, resulting in a very short lifetime (<1 s) of SO3.23−26 In the atmosphere, besides water-catalyzed hydration, ammonia, sulfuric acid, formic acid, nitric acid, and oxalic acid (among others) can also act as catalysts for the SO3 hydration process and thus facilitate H2SO4 formation.25,27−29
The detection of SO3 is challenging owing to its high reactivity with water in ambient air. Various measurement technologies have been utilized for SO3 detection in flue gases, including the controlled condensation method, absorption by isopropyl alcohol, selective reaction method with calcium oxalate, and spectroscopy and mass spectrometry methods.22,30 The detection limits of these methods are unfortunately too high to measure trace-level SO3 in ambient air. In some laboratory studies, the ions SiF5–, SF6–, NO3–·HNO3, and n-C3H7NH3+ have been used as reagent ions to detect SO3.31−35
In this study, we deployed a nitrate-chemical ionization-atmospheric pressure interface-long-time-of-flight (nitrate-CI-APi-LTOF) mass spectrometer in two field measurements for atmospheric SO3 and H2SO4 during the winter from January 20 to March 31 and during the summer from June 1 to July 10, 2019, in urban Beijing. This paper presents, for the first time, the trace-level measurement of gaseous SO3 by a nitrate-CI-APi-LTOF mass spectrometer in an urban atmosphere. Additionally, atmospheric naturally charged ions were also measured from November 9 to 22, 2018. Combining the SO3 measurements with data on trace gases, black carbon (BC), traffic flow, and atmospheric ions, we suggest a possible mechanism of formation of SO3 in urban Beijing. We also probe the effects of SO3 on atmospheric H2SO4 and sub-2.5 nm particle formation.
Materials and Methods
Detection of SO3 and H2SO4 by a Nitrate-CI-APi-LTOF Mass Spectrometer
The working principle of the nitrate-CI-APi-LTOF mass spectrometer is described in Text S2 and many other studies.5,36 The high-resolution peak fit of SO3·(NO3–) and its isotope peak [34SO3·(NO3–)] are depicted in Figure S1. The nitrate-CI-APi-LTOF mass spectrometer was calibrated by in situ-generated SO3 and H2SO437 (Text S4). The calibration coefficients for SO3 and H2SO4 are determined to be 1.7 × 1010 and 6.1 × 109 cm–3, respectively (Figure S4). The ratio between the calibration coefficients of SO3 and H2SO4 is 2.8, which is consistent with the theoretical prediction of a difference of a factor of 3 in the charging efficiency (Text S5). On the basis of repeat experiments, the 1σ of the SO3 calibration coefficient was ±10%.
According to our quantum chemical calculation for the binding thermodynamics of SO3·(NO3–) and HNO3·(NO3–) ion–molecule clusters, the electronic binding energy of SO3·(NO3–) is −44.4 kcal/mol, which is substantially higher than that of HNO3·(NO3–) (−29.2 kcal/mol) (Texts S6 and S7 and Table S1). Thus, SO3 molecules can be efficiently charged by nitrate ions. The highly favorable ligand exchange reaction between neutral SO3 molecules and nitrate ions can be written as
| R5 |
Furthermore, on the basis of the quantum chemical calculation, reaction R6 is exothermal by 8.7 kcal/mol in free energy (Text S6). The lowest-free energy structure for SO3·(NO3–)·H2O is depicted in Figure S6. Therefore, the hydrate complex intermediate (SO3·H2O) also can be detected as SO3·(NO3–).
| R6 |
Besides the ligand exchange reaction, the reaction between SO3 and NO3– can also lead to SO4– formation (eq R7).32 However, SO4– can also be produced by the reaction of SO2 with O2–·(H2O)n.34,38 From our ambient data, the averaged ratios of SO4– to SO3·(NO3–) were 0.26 ± 0.07 (winter) and 2.51 ± 2.60 (summer). During the summer, a large abundance of O2–·(H2O)n favored SO4– formation. Hence, only the signal of SO3·(NO3–) was taken into account in the SO3 quantification, and it could cause a slight underestimation of SO3 concentrations.
| R7 |
Atmospheric ions were also measured with the CI-APi-LTOF mass spectrometer by turning off the chemical ionization unit. Details of other auxiliary measuring instruments for trace gases, BC, meteorological parameters, and sub-3 nm particles can be found in Texts S8 and S9. Also, the calculation of condensation sink (CS) and quantification of SO3 and H2SO4 were introduced in Texts S10 and S3.
Results and Discussion
Abundance and Diurnal Behavior of SO3 in the Winter and Summer
As shown in Figure 1A, the abundance of SO3 was significantly higher in the winter than in the summer. During the winter, the mixing ratios of SO3 varied from ∼4.0 × 104 to 1.9 × 106 molecules cm–3. In comparison, during the summer, SO3 concentrations ranged from ∼5.0 × 103 to 1.4 × 105 molecules cm–3. Under 298 K and 1 atm, the atmospheric SO3 concentration has been proposed to reach 105 molecules cm–3 around noon.25 In this study, because the influence of ambient ions (i.e., SO3·NO3–) was not excluded and the hydrate complex intermediate (SO3·H2O) also could be detected as SO3·NO3– (eq R6), SO3 concentrations could be overestimated. Median diurnal variations of SO3 and SO2 concentrations, water dimer concentrations [(H2O)2; computed from the relative humidity and temperature],26,39 and intensities of UVB (280–315 nm) on all measurement days during the winter and summer are illustrated in Figure 1B. The water dimer concentration during the summer was notably higher than during the winter. In contrast, the mixing ratio of SO2 during the summer was lower than that during the winter. In the atmosphere, due to the large abundance of water, it is generally accepted that hydration to H2SO4 is the main fate of SO3.24 As a second water molecule is needed to lower the barrier of the SO3 hydration reaction, the reaction rate effectively depends on the water dimer concentration.23,40 Therefore, the low SO3 concentrations during the summer likely result from the large abundance of the water dimer and low SO2 concentration.
Figure 1.

(A) Time series of SO3 during the winter (January 20 to March 31, 2019) and summer (June 1 to July 10, 2019), (B) median diurnal patterns of SO3, UVB, and atmospheric water dimer concentrations during the winter and summer, and (C) median normalized intensities of the atmospheric ion SO3·NO3–. Rainy and snowy days were excluded. The shadows show the values from the 25th to 75th percentile. In panel B, the dashed lines show diurnal variations of SO3 during haze and nonhaze days. The water dimer concentration was calculated on the basis of temperature and relative humidity.26,39 In panel C, the signals of atmospheric ion SO3·NO3– were normalized by the sum of NO3– and HNO3·NO3– that are dominant natural charged ions in urban Beijing (Figure S7).
During the winter and summer, SO3 showed similar diurnal patterns (Figure 1B). During the winter, SO3 levels increased from ∼05:00 and reached their peak at ∼08:30. The abundance of SO3 was higher during the early morning from ∼06:00 to ∼09:00 and night from ∼18:00 to ∼03:00 (the next day) than around noon. Similarly, during the summer, SO3 concentrations increased from ∼4:00 and reached their peak values at ∼07:00. A peak of SO3 in the early morning (∼04:00 to ∼08:00) was also observed. During both the winter and the summer, lower SO3 concentrations were observed around noon when the water dimer concentration reached its minimum level (Figure 1B). Besides water-catalyzed hydration, around noon, a large abundance of other atmospheric components (e.g., H2SO4, HCOOH, HNO3, and oxalic acid) also promoted the conversion of SO3, leading to relatively short lifetimes. In addition, diel patterns of SO3 on nonhaze and haze days in winter are also shown in Figure 1B. The abundance of SO3 exhibited a similar daily pattern during both haze and nonhaze days (Text S8), though the actual values were higher on haze days.
The averaged mass spectra of atmospheric naturally charged ions for a day are shown in Figure S7. The dominant anions in urban Beijing are nitrate ions (NO3– and HNO3·NO3–), similar to the case in Shanghai.5 Therefore, the normalized intensity of SO3·NO3– by the sum of nitrate ions also can represent the abundance of SO3 in the air. Figure 1C shows the diel variation of the normalized signals of SO3·NO3– in November 2018. The normalized signals also exhibited two peaks, in the early morning (∼6:00 to ∼9:00) and at night (∼17:00 to ∼20:00).
Potential Source Identification for SO3
To investigate the possible sources of SO3 molecules during the early morning and night, we monitored other trace gases (SO2 and NOx), BC, and traffic flow of the main road nearby.41 During the summer, the average SO3 concentration was 2.9 × 104 molecules cm–3, which is likely close to the detection limit of the instrument. Hence, the possible source identification for SO3 was focused on winter data. During the winter, the median concentrations of SO2, NOx, and BC in PM2.5 exhibited diurnal trends similar to those of SO3 (Figure 2A). On the basis of previous studies of SO2 and together with stable weather conditions (low wind speeds and shallow mixing layer) (Text S11 and Figure S8), the elevated SO2 concentration during the early morning could mainly be attributed to local emissions (e.g., residential and industry emission) and transportation.42−50 BC concentrations were tightly linked with traffic emission dominated by diesel vehicles (Figure 2A).
Figure 2.

(A) Median diurnal variations in the concentrations of SO3, SO2, black carbon (BC), NOx, the approximate abundance term {([BC][SO2])/[(H2O)2]} of SO3, and gasoline and diesel vehicle flow of the “West Third Ring Road”, which was ∼550 m to the west of the sampling station in 2017,41 and (B) correlation between the SO3 concentration and its approximate abundance term during the night and the early morning (from 18:00 to 08:00 the next day) for the whole field measurement during the winter. In panel A, the units of BC, SO2, and water dimer [(H2O)2] in approximate source terms were micrograms per cubic meter, molecules per cubic meter, and molecules per cubic meter, respectively. In panel B, the SO3 concentrations were divided into logarithmic bins, and the median values in each bin are shown as squares. The orange shadow represents the values from the 25th to 75th percentile.
During the early morning, the UVB intensity was low, which meant that OH radical concentrations from photochemistry were also low, because of the linear correlation between OH concentrations and the ozone photolysis frequency [j(O1D)] in Beijing during daytime.51 During a summer night in the suburban area of Beijing, OH radical concentrations from ∼0.5 to 3 × 106 cm–3 still can be observed.52,53 Nevertheless, during the winter in suburban Beijing, the mean observed OH concentrations were <3 × 105 cm–3 at night and <1 × 106 cm–3 in the early morning (06:00–09:00).54,55 Hence, the oxidation of SO2 by OH radicals was not the main source for SO3 during the early morning and night. In addition, during the early morning, the median ozone concentration was only ∼5 ppb, which was low in comparison to the values during the rest of the day, resulting in a minor contribution to SO3 production from the reaction between sCI and SO2. Although SO3 molecules can also be co-emitted with SO2,20−22,56,57 transportation is not feasible for SO3 owing to its short lifetime (<1s) in the air. Previous studies based on quantum chemical calculations have suggested that the heterogeneous reaction between SO2 and soot can lead to SO3 formation.11,12 If this mechanism were to dominate SO3 formation, and further assuming that the SO3 is removed by reaction with the water dimer, the concentration of SO3 would be proportional to ([BC][SO2])/[(H2O)2]. Figure 2A shows that during the early morning (06:00–09:00) and night (from 18:00 to 6:00 the next day), the median diurnal variations of SO3 and ([BC][SO2])/[(H2O)2] were consistent with each other. Also, for the whole measurement period during the winter from 18:00 to 08:00 the next day, the correlation between SO3 and this approximate abundance term was positive [r = 0.7; P < 0.0001 (Figure 2B)]. Furthermore, on nonhaze days, SO3 was tightly linked to particulate sulfate, implying SO3 may originate from heterogeneous reaction (Figure S9). Therefore, our results together suggested the heterogeneous reaction between SO2 and BC could be the possible source of SO3 during the early morning and at night. However, other formation mechanisms (e.g., Criegee intermediate and SO2) may also contribute.9
Enhancement of Sulfuric Acid and Sub-2.5 nm Particle Formation
Median diurnal variations of the concentrations or intensities of H2SO4, the sulfuric acid dimer (H2SO4·HSO4–), SO3, sub-2.5 nm particles, CS, SO2, ozone, and UVB during the winter and summer are shown in panels A and B of Figure 3A and B, respectively. During the winter and summer, elevated levels of SO3 and H2SO4 were simultaneously observed in the early morning. During the winter, the median concentrations of H2SO4 during the early morning were comparable with that around noon. In addition, it showed a good correlation (r = 0.7) between atmospheric ions of HSO4– and SO3·NO3– during the early morning (5:00–8:00) and night (from 18:00 to 5:00 the following day) (Figure S10). Thus, the SO3 formed from nonphotochemical processes enhanced H2SO4 formation during the early morning and night. In a recent study in urban Beijing, under clean conditions, oxidation of SO2 by oxidants produced in the ozonolysis of alkenes (i.e., sCI and “dark” OH) was suggested as a source of H2SO4 during the night.58 This study also pointed out that, under polluted conditions, additional sources of H2SO4 exist. Hence, during the night in urban Beijing, H2SO4 can be enhanced both by SO3 molecules produced by the heterogeneous reaction between BC and SO2 and by the oxidation of SO2 by oxidants from ozonolysis of alkenes.
Figure 3.

Median diurnal variations in the concentrations and intensities of SO3, the sulfuric acid (SA) monomer (H2SO4) and dimer, sub-2.5 nm particles, SO2, O3, UVB, and condensation sink (CS) during the (A) winter and (B) summer.
As shown in Figure 3, during the winter or summer, the median concentrations of H2SO4, H2SO4·HSO4–, and sub-2.5 nm particles followed the same diurnal behavior. The concentrations of H2SO4·HSO4– and sub-2.5 nm particles started to increase at ∼5:00. During the early morning during the winter, the median concentrations of H2SO4·HSO4– and sub-2.5 nm particles were comparable with that around noon. On the basis of the studies in urban Shanghai, if atmospheric bases were abundant, the sulfuric acid dimer could be treated as an indicator of nanocluster formation.5,59 In urban Beijing, the concentrations of atmospheric bases (e.g., amines and ammonia) may also be sufficient for efficient clustering. Thus, the increased level of formation of SO3, possibly produced by the oxidation of SO2 on top of soot particles, can intensify the production of secondary particles and enhance gas to particle conversion during the early morning and night. We note that the contribution to nanoclusters of traffic, or the reaction of SO3 with ammonia/methanol, cannot be wholly excluded.25,60−62 Our results together point toward the need to control the emission of SO2 and soot to mitigate secondary aerosol formation in urban Beijing.
Acknowledgments
The work is supported by Academy of Finland (Center of Excellence in Atmospheric Sciences, project no. 307331, and PROFI3 funding, 311932), the European Research Council via ATM-GTP (742206) and via CHAPAs (850614) and the EMME-CARE project which has received funding from the European Union’s Horizon 2020 Research and Innovation Programme, under Grant Agreement No. 856612.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.estlett.0c00615.
Description of the sampling site (Text S1), the nitrate-CI-APi-LTOF mass spectrometer (Text S2), detection of sulfuric acid with nitrate reagent ions (Text S3), detailed calibration experiment for SO3 (Text S4), quantum chemical calculations (Text S6), computational details (Text S7), PM2.5, black carbon, particulate sulfate, trace gases, meteorological parameters, and UVB measurements (Text S8), sub-3 nm particle measurements (Text S9), calculation of condensation sink (Text S10), source identification of SO2 during the winter (Text S11), high-resolution peak fitting of the 32SO3·NO3– peak and its 34SO3·NO3– main isotope peak (Figure S1), schematic of the calibration experiment setup (Figure S2), time series of normalized signals of H2SO4 and SO3, and [H2O] in the calibration experiment (Figure S3), correlation between normalized SO3 signals measured by the CI-APi-LTOF mass spectrometer and SO3 concentrations formed by photo-oxidation of SO2 by OH radicals (Figure S4), optimized structure of the SO3·(NO3–) cluster (Figure S5), lowest-free energy (at 298 K) structure found for SO3·(NO3–)·H2O (Figure S6), averaged mass spectra of atmospheric naturally charged ions for one whole day (November 10, 2018) (Figure S7), median diurnal variation of the concentrations of SO3 and SO2, the mixing layer heights (MLH), intensities of UVB, and wind speeds during the winter (Figure S8), time profile of the SO3 concentration and mass concentration of sulfate in PM2.5 and median diel variation of SO3 and sulfate for all nonhaze days during the winter measurement period (Figure S9), relationship between the atmospheric ion signals of HSO4– and SO3·NO3– during the night (from 18:00 to 5:00 next day) and early morning (5:00–8:00) from November 9 to 22, 2018 (Figure S10), and comparison of the binding thermodynamics of HNO3·(NO3–) and SO3·(NO3–) ion–molecule clusters (Table S1) (PDF)
Author Contributions
L.Y. and X.F. contributed equally to this work. L.Y. and F.B. designed the research and analyzed the data. L.Y., C.Y., Y.G., C.L., X.F., Y.Z., K.R.D., F.Z., Y.W., and T.C. performed the measurements for this study. T.K. provided quantum calculation results. L.Y., X.F., B.C., F.B., T.P., M.P.R., L.W., Y.L., J.J., D.R.W., V.-M.K., T.K., and M.K. interpreted the results and revised the manuscript. M.K., F.B., and H.H. supported and supervised this research. L.Y. wrote the manuscript with contributions from all co-authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Larssen T.; Lydersen E.; Tang D. G.; He Y.; Gao J. X.; Liu H. Y.; Duan L.; Seip H. M.; Vogt R. D.; Mulder J.; Shao M.; Wang Y. H.; Shang H.; Zhang X. S.; Solberg S.; Aas W.; Okland T.; Eilertsen O.; Angell V.; Li Q. R.; Zhao D. W.; Xiang R. J.; Xiao J. S.; Luo J. H. Acid rain in China. Environ. Sci. Technol. 2006, 40 (2), 418–425. 10.1021/es0626133. [DOI] [PubMed] [Google Scholar]
- Sipilä M.; Berndt T.; Petäjä T.; Brus D.; Vanhanen J.; Stratmann F.; Patokoski J.; Mauldin R. L.; Hyvarinen A. P.; Lihavainen H.; Kulmala M. The Role of Sulfuric Acid in Atmospheric Nucleation. Science 2010, 327 (5970), 1243–1246. 10.1126/science.1180315. [DOI] [PubMed] [Google Scholar]
- Kulmala M.; Kontkanen J.; Junninen H.; Lehtipalo K.; Manninen H. E.; Nieminen T.; Petäjä T.; Sipilä M.; Schobesberger S.; Rantala P.; Franchin A.; Jokinen T.; Jarvinen E.; Aijala M.; Kangasluoma J.; Hakala J.; Aalto P. P.; Paasonen P.; Mikkila J.; Vanhanen J.; Aalto J.; Hakola H.; Makkonen U.; Ruuskanen T.; Mauldin R. L.; Duplissy J.; Vehkamaki H.; Back J.; Kortelainen A.; Riipinen I.; Kurtén T.; Johnston M. V.; Smith J. N.; Ehn M.; Mentel T. F.; Lehtinen K. E.; Laaksonen A.; Kerminen V. M.; Worsnop D. R. Direct observations of atmospheric aerosol nucleation. Science 2013, 339 (6122), 943–6. 10.1126/science.1227385. [DOI] [PubMed] [Google Scholar]
- Bianchi F.; Tröstl J.; Junninen H.; Frege C.; Henne S.; Hoyle C. R.; Molteni U.; Herrmann E.; Adamov A.; Bukowiecki N.; Chen X.; Duplissy J.; Gysel M.; Hutterli M.; Kangasluoma J.; Kontkanen J.; Kurten A.; Manninen H. E.; Munch S.; Perakyla O.; Petäjä T.; Rondo L.; Williamson C.; Weingartner E.; Curtius J.; Worsnop D. R.; Kulmala M.; Dommen J.; Baltensperger U. New particle formation in the free troposphere: A question of chemistry and timing. Science 2016, 352 (6289), 1109–1112. 10.1126/science.aad5456. [DOI] [PubMed] [Google Scholar]
- Yao L.; Garmash O.; Bianchi F.; Zheng J.; Yan C.; Kontkanen J.; Junninen H.; Mazon S. B.; Ehn M.; Paasonen P.; Sipilä M.; Wang M.; Wang X.; Xiao S.; Chen H.; Lu Y.; Zhang B.; Wang D.; Fu Q.; Geng F.; Li L.; Wang H.; Qiao L.; Yang X.; Chen J.; Kerminen V. M.; Petäjä T.; Worsnop D. R.; Kulmala M.; Wang L. Atmospheric new particle formation from sulfuric acid and amines in a Chinese megacity. Science 2018, 361 (6399), 278–281. 10.1126/science.aao4839. [DOI] [PubMed] [Google Scholar]
- Chu B. W.; Kerminen V. M.; Bianchi F.; Yan C.; Petäjä T.; Kulmala M. Atmospheric new particle formation in China. Atmos. Chem. Phys. 2019, 19 (1), 115–138. 10.5194/acp-19-115-2019. [DOI] [Google Scholar]
- Chen H.; Wang M.; Yao L.; Chen J.; Wang L. Uptake of Gaseous Alkylamides by Suspended Sulfuric Acid Particles: Formation of Ammonium/Aminium Salts. Environ. Sci. Technol. 2017, 51 (20), 11710–11717. 10.1021/acs.est.7b03175. [DOI] [PubMed] [Google Scholar]
- Kerminen V. M.; Chen X. M.; Vakkari V.; Petäjä T.; Kulmala M.; Bianchi F. Atmospheric new particle formation and growth: review of field observations. Environ. Res. Lett. 2018, 13 (10), 103003. 10.1088/1748-9326/aadf3c. [DOI] [Google Scholar]
- Mauldin R. L.; Berndt T.; Sipilä M.; Paasonen P.; Petäjä T.; Kim S.; Kurtén T.; Stratmann F.; Kerminen V. M.; Kulmala M. A new atmospherically relevant oxidant of sulphur dioxide. Nature 2012, 488 (7410), 193–196. 10.1038/nature11278. [DOI] [PubMed] [Google Scholar]
- Welz O.; Savee J. D.; Osborn D. L.; Vasu S. S.; Percival C. J.; Shallcross D. E.; Taatjes C. A. Direct Kinetic Measurements of Criegee Intermediate (CH2OO) Formed by Reaction of CH2I with O2. Science 2012, 335 (6065), 204–207. 10.1126/science.1213229. [DOI] [PubMed] [Google Scholar]
- He G. Z.; He H. DFT studies on the heterogeneous oxidation of SO2 by oxygen functional groups on graphene. Phys. Chem. Chem. Phys. 2016, 18 (46), 31691–31697. 10.1039/C6CP06665H. [DOI] [PubMed] [Google Scholar]
- He G. Z.; Ma J. Z.; He H. Role of Carbonaceous Aerosols in Catalyzing Sulfate Formation. ACS Catal. 2018, 8 (5), 3825–3832. 10.1021/acscatal.7b04195. [DOI] [Google Scholar]
- Lizzio A. A.; DeBarr J. A. Mechanism of SO2 removal by carbon. Energy Fuels 1997, 11 (2), 284–291. 10.1021/ef960197+. [DOI] [Google Scholar]
- Han C.; Liu Y. C.; Ma J. Z.; He H. Key role of organic carbon in the sunlight-enhanced atmospheric aging of soot by O2. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (52), 21250–21255. 10.1073/pnas.1212690110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G.; He H. Water Promotes the Oxidation of SO2 by O2 over Carbonaceous Aerosols. Environ. Sci. Technol. 2020, 54 (12), 7070–7077. 10.1021/acs.est.0c00021. [DOI] [PubMed] [Google Scholar]
- Novakov T.; Chang S. G.; Harker A. B. Sulfates as Pollution Particulates - Catalytic Formation on Carbon (soot) Particles. Science 1974, 186 (4160), 259–261. 10.1126/science.186.4160.259. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Liu Y. C.; Ma J. Z.; Ma Q. X.; He H. Heterogeneous reaction of SO2 with soot: The roles of relative humidity and surface composition of soot in surface sulfate formation. Atmos. Environ. 2017, 152, 465–476. 10.1016/j.atmosenv.2017.01.005. [DOI] [Google Scholar]
- He X.; Pang S. F.; Ma J. B.; Zhang Y. H. Influence of relative humidity on heterogeneous reactions of O3 and O3/SO2 with soot particles: Potential for environmental and health effects. Atmos. Environ. 2017, 165, 198–206. 10.1016/j.atmosenv.2017.06.049. [DOI] [Google Scholar]
- Zhang F.; Wang Y.; Peng J.; Chen L.; Sun Y.; Duan L.; Ge X.; Li Y.; Zhao J.; Liu C.; Zhang X.; Zhang G.; Pan Y.; Wang Y.; Zhang A. L.; Ji Y.; Wang G.; Hu M.; Molina M. J.; Zhang R. An unexpected catalyst dominates formation and radiative forcing of regional haze. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (8), 3960–3966. 10.1073/pnas.1919343117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J. L.; Zheng C. H.; Xu L. J.; Zhang Y.; Zhang Y. X.; Liu S. J.; Gao X. Atmospheric emission inventory of SO3 from coal-fired power plants in China in the period 2009–2014. Atmos. Environ. 2019, 197, 14–21. 10.1016/j.atmosenv.2018.10.008. [DOI] [Google Scholar]
- Yang Z.; Ji P.; Li Q.; Jiang Y.; Zheng C.; Wang Y.; Gao X.; Lin R. Comprehensive understanding of SO3 effects on synergies among air pollution control devices in ultra-low emission power plants burning high-sulfur coal. J. Cleaner Prod. 2019, 239, 118096. 10.1016/j.jclepro.2019.118096. [DOI] [Google Scholar]
- Roy B.; Chen L. G.; Bhattacharya S. Nitrogen Oxides, Sulfur Trioxide, and Mercury Emissions during Oxyfuel Fluidized Bed Combustion of Victorian Brown Coal. Environ. Sci. Technol. 2014, 48 (24), 14844–14850. 10.1021/es504667r. [DOI] [PubMed] [Google Scholar]
- Morokuma K.; Muguruma C. Ab-Initio Molecular-Orbital Study of the Mechanism of the Gas-Phase Reaction SO3+H2O - Importance of the 2nd Water Molecule. J. Am. Chem. Soc. 1994, 116 (22), 10316–10317. 10.1021/ja00101a068. [DOI] [Google Scholar]
- Kolb C. E.; Jayne J. T.; Worsnop D. R.; Molina M. J.; Meads R. F.; Viggiano A. A. Gas-Phase Reaction of Sulfur-Trioxide with Water-Vapor. J. Am. Chem. Soc. 1994, 116 (22), 10314–10315. 10.1021/ja00101a067. [DOI] [Google Scholar]
- Li H.; Zhong J.; Vehkamaki H.; Kurten T.; Wang W. G.; Ge M. F.; Zhang S. W.; Li Z. S.; Zhang X. H.; Francisco J. S.; Zeng X. C. Self-Catalytic Reaction of SO3 and NH3 To Produce Sulfamic Acid and Its Implication to Atmospheric Particle Formation. J. Am. Chem. Soc. 2018, 140 (35), 11020–11028. 10.1021/jacs.8b04928. [DOI] [PubMed] [Google Scholar]
- Sarkar S.; Oram B. K.; Bandyopadhyay B. Influence of Ammonia and Water on the Fate of Sulfur Trioxide in the Troposphere: Theoretical Investigation of Sulfamic Acid and Sulfuric Acid Formation Pathways. J. Phys. Chem. A 2019, 123 (14), 3131–3141. 10.1021/acs.jpca.8b09306. [DOI] [PubMed] [Google Scholar]
- Lv G. C.; Sun X. M.; Zhang C. X.; Li M. Understanding the catalytic role of oxalic acid in SO3 hydration to form H2SO4 in the atmosphere. Atmos. Chem. Phys. 2019, 19 (5), 2833–2844. 10.5194/acp-19-2833-2019. [DOI] [Google Scholar]
- Bandyopadhyay B.; Kumar P.; Biswas P. Ammonia Catalyzed Formation of Sulfuric Acid in Troposphere: The Curious Case of a Base Promoting Acid Rain. J. Phys. Chem. A 2017, 121 (16), 3101–3108. 10.1021/acs.jpca.7b01172. [DOI] [PubMed] [Google Scholar]
- Long B.; Chang C. R.; Long Z. W.; Wang Y. B.; Tan X. F.; Zhang W. J. Nitric acid catalyzed hydrolysis of SO3 in the formation of sulfuric acid: A theoretical study. Chem. Phys. Lett. 2013, 581, 26–29. 10.1016/j.cplett.2013.07.012. [DOI] [Google Scholar]
- Fleig D.; Vainio E.; Andersson K.; Brink A.; Johnsson F.; Hupa M. Evaluation of SO3Measurement Techniques in Air and Oxy-Fuel Combustion. Energy Fuels 2012, 26 (9), 5537–5549. 10.1021/ef301127x. [DOI] [Google Scholar]
- Lovejoy E. R.; Hanson D. R.; Huey L. G. Kinetics and products of the gas-phase reaction of SO3 with water. J. Phys. Chem. 1996, 100 (51), 19911–19916. 10.1021/jp962414d. [DOI] [Google Scholar]
- Arnold S. T.; Morris R. A.; Viggiano A. A.; Jayne J. T. Ion Chemistry Relevant for Chemical-Ionization Detection of SO3. J. Geophys. Res. 1995, 100 (D7), 14141–14146. 10.1029/95JD01004. [DOI] [Google Scholar]
- Jayne J. T.; Poschl U.; Chen Y. M.; Dai D.; Molina L. T.; Worsnop D. R.; Kolb C. E.; Molina M. J. Pressure and temperature dependence of the gas-phase reaction of SO3 with H2O and the heterogeneous reaction of SO3 with H2O/H2SO4 surfaces. J. Phys. Chem. A 1997, 101 (51), 10000–10011. 10.1021/jp972549z. [DOI] [Google Scholar]
- Sorokin A.; Katragkou E.; Arnold F.; Busen R.; Schumann U. Gaseous SO3 and H2SO4 in the exhaust of an aircraft gas turbine engine: measurements by CIMS and implications for fuel sulfur conversion to sulfur (VI) and conversion of SO3 to H2SO4. Atmos. Environ. 2004, 38 (3), 449–456. 10.1016/j.atmosenv.2003.09.069. [DOI] [Google Scholar]
- Berndt T.; Scholz W.; Mentler B.; Fischer L.; Hoffmann E. H.; Tilgner A.; Hyttinen N.; Prisle N. L.; Hansel A.; Herrmann H. Fast Peroxy Radical Isomerization and OH Recycling in the Reaction of OH Radicals with Dimethyl Sulfide. J. Phys. Chem. Lett. 2019, 10 (21), 6478–6483. 10.1021/acs.jpclett.9b02567. [DOI] [PubMed] [Google Scholar]
- Jokinen T.; Sipilä M.; Junninen H.; Ehn M.; Lönn G.; Hakala J.; Petäjä T.; Mauldin R. L.; Kulmala M.; Worsnop D. R. Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF. Atmos. Chem. Phys. 2012, 12 (9), 4117–4125. 10.5194/acp-12-4117-2012. [DOI] [Google Scholar]
- Kürten A.; Rondo L.; Ehrhart S.; Curtius J. Calibration of a chemical ionization mass spectrometer for the measurement of gaseous sulfuric acid. J. Phys. Chem. A 2012, 116 (24), 6375–86. 10.1021/jp212123n. [DOI] [PubMed] [Google Scholar]
- Mohler O.; Reiner T.; Arnold F. The Formation of SO5– by Gas-Phase Ion–Molecule Reactions. J. Chem. Phys. 1992, 97 (11), 8233–8239. 10.1063/1.463394. [DOI] [Google Scholar]
- Anglada J. M.; Hoffman G. J.; Slipchenko L. V.; Costa M. M.; Ruiz-Lopez M. F.; Francisco J. S. Atmospheric Significance of Water Clusters and Ozone-Water Complexes. J. Phys. Chem. A 2013, 117 (40), 10381–10396. 10.1021/jp407282c. [DOI] [PubMed] [Google Scholar]
- Larson L. J.; Kuno M.; Tao F. M. Hydrolysis of sulfur trioxide to form sulfuric acid in small water clusters. J. Chem. Phys. 2000, 112 (20), 8830–8838. 10.1063/1.481532. [DOI] [Google Scholar]
- Yang D. Y.; Zhang S. J.; Niu T. L.; Wang Y. J.; Xu H. L.; Zhang K. M.; Wu Y. High-resolution mapping of vehicle emissions of atmospheric pollutants based on large-scale, real-world traffic datasets. Atmos. Chem. Phys. 2019, 19 (13), 8831–8843. 10.5194/acp-19-8831-2019. [DOI] [Google Scholar]
- Xu W. Y.; Zhao C. S.; Ran L.; Lin W. L.; Yan P.; Xu X. B. SO2 noontime-peak phenomenon in the North China Plain. Atmos. Chem. Phys. 2014, 14 (15), 7757–7768. 10.5194/acp-14-7757-2014. [DOI] [Google Scholar]
- Liu J.; Mauzerall D. L.; Chen Q.; Zhang Q.; Song Y.; Peng W.; Klimont Z.; Qiu X. H.; Zhang S. Q.; Hu M.; Lin W. L.; Smith K. R.; Zhu T. Air pollutant emissions from Chinese households: A major and underappreciated ambient pollution source. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (28), 7756–7761. 10.1073/pnas.1604537113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R.; Fu H. B.; Cui L. L.; Li J. L.; Wu Y.; Meng Y.; Wang Y. T.; Chen J. M. The spatiotemporal variation and key factors of SO2 in 336 cities across China. J. Cleaner Prod. 2019, 210, 602–611. 10.1016/j.jclepro.2018.11.062. [DOI] [Google Scholar]
- Huang Q.; Cheng S. Y.; Perozzi R. E.; Perozzi E. F. Use of a MM5-CAMx-PSAT Modeling System to Study SO2 Source Apportionment in the Beijing Metropolitan Region. Environ. Model. Assess. 2012, 17 (5), 527–538. 10.1007/s10666-012-9312-8. [DOI] [Google Scholar]
- Kampa M.; Castanas E. Human health effects of air pollution. Environ. Pollut. 2008, 151 (2), 362–367. 10.1016/j.envpol.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Zhong Q. R.; Shen H. Z.; Yun X.; Chen Y. L.; Ren Y. A.; Xu H. R.; Shen G. F.; Ma J. M.; Tao S. Effects of International Fuel Trade on Global Sulfur Dioxide Emissions. Environ. Sci. Technol. Lett. 2019, 6 (12), 727–731. 10.1021/acs.estlett.9b00617. [DOI] [Google Scholar]
- Klimont Z.; Smith S. J.; Cofala J. The last decade of global anthropogenic sulfur dioxide: 2000–2011 emissions. Environ. Res. Lett. 2013, 8 (1), 014003. 10.1088/1748-9326/8/1/014003. [DOI] [Google Scholar]
- Su S. S.; Li B. G.; Cui S. Y.; Tao S. Sulfur Dioxide Emissions from Combustion in China: From 1990 to 2007. Environ. Sci. Technol. 2011, 45 (19), 8403–8410. 10.1021/es201656f. [DOI] [PubMed] [Google Scholar]
- Zheng H. T.; Cai S. Y.; Wang S. X.; Zhao B.; Chang X.; Hao J. M. Development of a unit-based industrial emission inventory in the Beijing-Tianjin-Hebei region and resulting improvement in air quality modeling. Atmos. Chem. Phys. 2019, 19 (6), 3447–3462. 10.5194/acp-19-3447-2019. [DOI] [Google Scholar]
- Rohrer F.; Lu K. D.; Hofzumahaus A.; Bohn B.; Brauers T.; Chang C. C.; Fuchs H.; Haseler R.; Holland F.; Hu M.; Kita K.; Kondo Y.; Li X.; Lou S. R.; Oebel A.; Shao M.; Zeng L. M.; Zhu T.; Zhang Y. H.; Wahner A. Maximum efficiency in the hydroxyl-radical-based self-cleansing of the troposphere. Nat. Geosci. 2014, 7 (8), 559–563. 10.1038/ngeo2199. [DOI] [Google Scholar]
- Lu K. D.; Rohrer F.; Holland F.; Fuchs H.; Brauers T.; Oebel A.; Dlugi R.; Hu M.; Li X.; Lou S. R.; Shao M.; Zhu T.; Wahner A.; Zhang Y. H.; Hofzumahaus A. Nighttime observation and chemistry of HOx in the Pearl River Delta and Beijing in summer 2006. Atmos. Chem. Phys. 2014, 14 (10), 4979–4999. 10.5194/acp-14-4979-2014. [DOI] [Google Scholar]
- Tan Z.; Fuchs H.; Lu K.; Hofzumahaus A.; Bohn B.; Broch S.; Dong H.; Gomm S.; Häseler R.; He L.; Holland F.; Li X.; Liu Y.; Lu S.; Rohrer F.; Shao M.; Wang B.; Wang M.; Wu Y.; Zeng L.; Zhang Y.; Wahner A.; Zhang Y. Radical chemistry at a rural site (Wangdu) in the North China Plain: observation and model calculations of OH, HO2 and RO2 radicals. Atmos. Chem. Phys. 2017, 17 (1), 663–690. 10.5194/acp-17-663-2017. [DOI] [Google Scholar]
- Tan Z. F.; Rohrer F.; Lu K. D.; Ma X. F.; Bohn B.; Broch S.; Dong H. B.; Fuchs H.; Gkatzelis G. I.; Hofzumahaus A.; Holland F.; Li X.; Liu Y.; Liu Y. H.; Novelli A.; Shao M.; Wang H. C.; Wu Y. S.; Zeng L. M.; Hu M.; Kiendler-Scharr A.; Wahner A.; Zhang Y. H. Wintertime photochemistry in Beijing: observations of ROx radical concentrations in the North China Plain during the BEST-ONE campaign. Atmos. Chem. Phys. 2018, 18 (16), 12391–12411. 10.5194/acp-18-12391-2018. [DOI] [Google Scholar]
- Lu K.; Fuchs H.; Hofzumahaus A.; Tan Z.; Wang H.; Zhang L.; Schmitt S. H.; Rohrer F.; Bohn B.; Broch S.; Dong H.; Gkatzelis G. I.; Hohaus T.; Holland F.; Li X.; Liu Y.; Liu Y.; Ma X.; Novelli A.; Schlag P.; Shao M.; Wu Y.; Wu Z.; Zeng L.; Hu M.; Kiendler-Scharr A.; Wahner A.; Zhang Y. Fast Photochemistry in Wintertime Haze: Consequences for Pollution Mitigation Strategies. Environ. Sci. Technol. 2019, 53 (18), 10676–10684. 10.1021/acs.est.9b02422. [DOI] [PubMed] [Google Scholar]
- Srivastava R. K.; Miller C. A.; Erickson C.; Jambhekar R. Emissions of sulfur trioxide from coal-fired power plants. J. Air Waste Manage. Assoc. 2004, 54 (6), 750–762. 10.1080/10473289.2004.10470943. [DOI] [PubMed] [Google Scholar]
- Yang Z. D.; Zheng C. H.; Zhang X. F.; Zhou H.; Silva A. A.; Liu C. Y.; Snyder B.; Wang Y.; Gao X. Challenge of SO3 removal by wet electrostatic precipitator under simulated flue gas with high SO3 concentration. Fuel 2018, 217, 597–604. 10.1016/j.fuel.2017.12.125. [DOI] [Google Scholar]
- Guo Y.; Yan C.; Li C.; Feng Z.; Zhou Y.; Lin Z.; Dada L.; Stolzenburg D.; Yin R.; Kontkanen J.; Daellenbach K. R.; Kangasluoma J.; Yao L.; Chu B.; Wang Y.; Cai R.; Bianchi F.; Liu Y.; Kulmala M. Atmos. Chem. Phys. Discuss. 2020, 10.5194/acp-2019-1111. [DOI] [Google Scholar]
- Yao L.; Wang M.-Y.; Wang X.-K.; Liu Y.-J.; Chen H.-F.; Zheng J.; Nie W.; Ding A.-J.; Geng F.-H.; Wang D.-F.; Chen J.-M.; Worsnop D. R.; Wang L. Detection of atmospheric gaseous amines and amides by a high-resolution time-of-flight chemical ionization mass spectrometer with protonated ethanol reagent ions. Atmos. Chem. Phys. 2016, 16 (22), 14527–14543. 10.5194/acp-16-14527-2016. [DOI] [Google Scholar]
- Liu L.; Zhong J.; Vehkamaki H.; Kurten T.; Du L.; Zhang X.; Francisco J. S.; Zeng X. C. Unexpected quenching effect on new particle formation from the atmospheric reaction of methanol with SO3. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (50), 24966–24971. 10.1073/pnas.1915459116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olin M.; Kuuluvainen H.; Aurela M.; Kalliokoski J.; Kuittinen N.; Isotalo M.; Timonen H. J.; Niemi J. V.; Rönkkö T.; Dal Maso M. Traffic-originated nanocluster emission exceeds H2SO4-driven photochemical new particle formation in an urban area. Atmos. Chem. Phys. 2020, 20 (1), 1–13. 10.5194/acp-20-1-2020. [DOI] [Google Scholar]
- Ronkko T.; Kuuluvainen H.; Karjalainen P.; Keskinen J.; Hillamo R.; Niemi J. V.; Pirjola L.; Timonen H. J.; Saarikoski S.; Saukko E.; Jarvinen A.; Silvennoinen H.; Rostedt A.; Olin M.; Yli-Ojanpera J.; Nousiainen P.; Kousa A.; Dal Maso M. Traffic is a major source of atmospheric nanocluster aerosol. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (29), 7549–7554. 10.1073/pnas.1700830114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.