

Summary
Experimental fibroblast growth factor 21 (FGF21) analogs can improve lipid profiles in patients with metabolic diseases. However, their effects on markers of insulin sensitivity appear to be minimal, potentially because of insufficient exposure. Systemic drug levels vary from sub-pharmacological to demonstrating pharmacodynamic effects but with dose-limiting adverse events. Here we report results from a phase 1 multiple ascending dose study of AKR-001, an Fc-FGF21 fusion protein engineered for sustained systemic pharmacologic exposure, in individuals with type 2 diabetes. With a half-life of 3–3.5 days, the peak-to-trough ratio under steady-state conditions is approximately 2 following QW dosing. AKR-001 appears to demonstrate pharmacodynamic effects on serum markers of insulin sensitivity and acceptable tolerability up to and including 70 mg QW. Positive trends in lipoprotein profile, including triglycerides, non-high-density lipoprotein (non-HDL) cholesterol, HDL-C, and apolipoproteins B and C3 are consistent with other FGF21 analogs. AKR-001’s clinical profile supports further evaluation as a treatment for metabolic diseases.
Graphical Abstract
Highlights
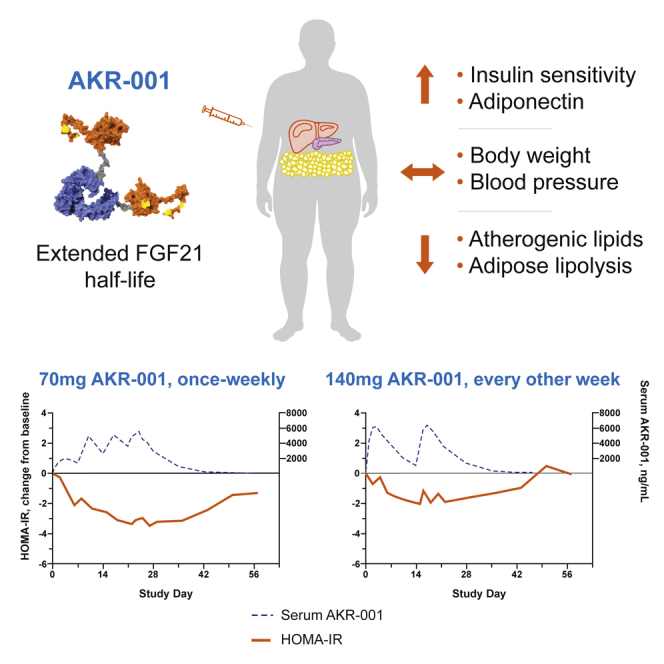
AKR-001 is an Fc-FGF21 analog with stabilized N- and C-terminal domains of FGF21
AKR-001 dosed once weekly modulates markers of insulin sensitivity in T2D patients
AKR-001 exhibits positive trends in markers of lipid metabolism
AKR-001 has acceptable tolerability, supporting ongoing clinical development
FGF21 ameliorates metabolic disease in animal models but has modest effects in humans. Kaufman et al. report the pharmacokinetic, pharmacodynamic, and safety data from a phase 1 multiple ascending dose study in T2DM of AKR-001, a long-acting FGF21 analog. They show that AKR-001 exhibits positive trends in various metabolic markers.
Introduction
Cardiovascular, kidney, and hepatic diseases are frequent co-morbidities of type 2 diabetes mellitus.1 Current treatment approaches for diabetes and other manifestations of metabolic syndrome utilize multiple agents to address each symptom or risk factor. FGF21 is an endogenous protein that regulates metabolism2 and has been studied as a treatment for metabolic disorders, with several agents tested in clinical studies and none yet approved by the US Food and Drug Administration (FDA). FGF21 activates a cell membrane co-receptor complex consisting of β-Klotho and the canonical FGF receptors (FGFRs) for FGF21—FGFR1c, FGFR2c, and FGFR3c—whose expression is tissue dependent; e.g., FGFR1c is the predominant receptor in adipose tissue.3,4 Endogenous FGF21 concentrations in healthy humans are approximately 100 pg/mL5 and are elevated as much as 10-fold in patients with obesity, nonalcoholic fatty liver disease (NAFLD), or non-alcoholic steatohepatitis (NASH),6,7 leading to the hypothesis that these may represent an FGF21-resistant state.8 Dietary changes in humans also affect circulating FGF21 levels; acute fructose or alcohol consumption robustly increases plasma FGF21,9,10 and prolonged protein restriction or carbohydrate excess similarly increase FGF21 levels.11
Exogenous administration of FGF21 and analogs of FGF21 has been shown to improve hyperglycemia and lipid profiles and reduce insulin levels and body weight in obese diabetic rodents12, 13, 14 and monkeys.15, 16, 17 Nevertheless, the promise of multi-factor improvement in metabolic disease (improved glycemic control and insulin sensitivity, weight loss, and lipid and lipoprotein effects) has not fully materialized in clinical studies of FGF21 analogs. The prototypical FGF21 analog LY2405319 improved dyslipidemia in obese patients with type 2 diabetes to a similar extent as that observed in diabetic monkeys. However, markers of insulin sensitivity were unchanged except at the highest dose levels, where the magnitude and significance of responses were variable.18 Furthermore, in placebo-controlled studies of three other FGF21 analogs (PF-05231023,19,20 pegbelfermin,21 and the FGFR1c/β-Klotho bispecific antibody BFKB8488A; C. Wong et al., 2019, AASLD, poster #2310), markers of insulin sensitivity were not significantly or consistently improved, and lipoprotein responses were variable.
A limitation of all FGF21 analogs tested in clinical studies to date has been their sub-optimal pharmacokinetic (PK) profiles. Recombinant human FGF21 has a half-life of 2 h or less when injected into monkeys or mice,14,15,22 prompting multiple approaches to engineer FGF21 to extend its duration of action. LY2405319 required daily subcutaneous (s.c.) injection, indicating a limited extension of half-life.18 Although once-weekly s.c. administration of pegbelfermin has been evaluated, daily injection appears to be required to achieve clinically meaningful improvements in high-density lipoprotein (HDL) cholesterol and triglycerides,21 consistent with an apparent half-life of 36–48 h.23 Another analog, PF-05231023, was administered intravenously twice or once weekly, respectively, in two clinical studies. However, although twice-weekly dosing was more effective than weekly dosing in reducing plasma triglyceride levels, consistent with the reported half-life of 6.5–10.3 h, more frequent dosing was associated with diarrhea, reported by 29% of patients, and one subject discontinued dosing because of ongoing diarrhea and vomiting.20,24 Diarrhea was also one of the most frequently observed adverse events (AEs) with pegbelfermin treatment.21,25
Preclinical and clinical studies have also demonstrated inconsistent effects of FGF21 analogs on bone growth, blood pressure, and heart rate. In diet-induced obese mice, administration of exogenous FGF21 led to a decrease in bone mass.26 However, a more recent preclinical study did not find evidence of bone loss in diet-induced obese mice administered recombinant human FGF21 (rhFGF21).27 Twice-weekly administration of pharmacologically effective doses of PF-05231023 in subjects with type 2 diabetes for 4 weeks was associated with increased markers of bone turnover,19 but this may have been due to the concurrent weight loss. No change in mean bone mineral density was observed following 12 weeks21 or 16 weeks25 of pegbelfermin treatment. Weekly administration of PF-05231023 increased blood pressure and heart rate over baseline compared with placebo treatment in obese people with hypertriglyceridemia.20 However, cardiovascular changes were not reported with twice-weekly administration of PF-05231023 or with the other FGF21 analogs tested. In addition, loss of FGF21 function in humans carrying a rare truncation mutant appears to be associated with increased blood pressure.28
The clinical evidence suggests that the PK characteristics of FGF21 analogs to date have prevented delivery of an acceptable balance between therapeutic benefit and mechanism-related toxicity because of high peak-to-trough exposures and inadequately sustained exposure across the inter-dose interval. Indeed, the pattern of dose-response in preclinical studies indicates that sustained exposure to FGF21 agonism will be required to demonstrate the full range of metabolic improvements. For example, in diet-induced obese mice, higher doses of FGF21 are required to reduce body weight and adiposity compared with improvements in insulin sensitivity and glucose metabolism.13 Thus, an FGF21 analog that minimizes the peak-to-trough range and maintains stable, pharmacologically active concentrations could enable delivery of the full therapeutic potential of FGF21 agonism while minimizing adverse effects.
AKR-001 (formerly Fc-FGF21(RGE), AMG 876) is a long-acting human immunoglobulin 1 (IgG1) Fc-FGF21 fusion protein that has demonstrated enhanced pharmacology across numerous preclinical animal models.29 Its C-terminal region has been modified with two amino acid substitutions (P171G and A180E). These targeted mutations substantially reduce proteolytic degradation and increase affinity of binding to β-Klotho, the obligate co-receptor of FGF21, enhancing FGFR-mediated signaling activity, functional potency, and duration of action of AKR-001 in vivo.29 The pharmaceutical stability of AKR-001 is improved by an additional substitution (L98R) to minimize aggregation when formulated as an aqueous injection. The prolonged PK and pharmacodynamics (PD) characteristics of AKR-001 may enable it to achieve the full therapeutic potential of FGF21 agonism in metabolic disease while minimizing mechanism-related unwanted side effects.
The aim of the present study was to assess the safety, tolerability, PK, and PD of AKR-001 dosed once weekly (QW) and once every 2 weeks (Q2W) in patients with type 2 diabetes over 4 weeks of treatment. PK–PD modeling of metabolic and cardiovascular endpoints was performed to understand the exposure-effect relationship and to inform dose levels and dosing frequency for future clinical studies. Post hoc exploration of biomarkers relevant to deposition and metabolism of lipids by the liver and adipose tissue was performed to determine the potential utility of AKR-001 for treatment of NASH, a metabolic disease characterized by dyslipidemia, particularly hypertriglyceridemia, as well as liver-related morbidity and mortality driven by increases in hepatic de novo lipogenesis and adipose tissue lipolysis.30
Results and Discussion
Clinical Trial Design
This was a multicenter, randomized, double-blind, placebo-controlled, ascending multiple-dose study in adult patients with type 2 diabetes (registered on https://www.clinicaltrials.gov/ as NCT01856881). The trial was conducted in full compliance with the International Council for Harmonisation (ICH) E6 Guideline for Good Clinical Practice and the principles of the Declaration of Helsinki. A total of eight cohorts were randomized to receive AKR-001 or a placebo at a ratio of 3:1. AKR-001 (7, 21, 70, or 140 mg) was administered s.c. Q2W or QW for 4 weeks (Figure 1).
Figure 1.

Summary of the Trial Design
See also Figure S1.
Patient Disposition and Baseline Characteristics
Adults 18–65 years old at the time of randomization and with a BMI of 25.0–40.0 kg/m2 at screening, a diagnosis of type 2 diabetes, glycated hemoglobin (HbA1c) level of 6.5%–10% or less, and a fasting C-peptide value of 0.8 ng/mL or more were enrolled in the study. All anti-diabetes medications were discontinued at least 14 days prior to administration of the first dose of AKR-001. Female subjects were required to be of documented non-reproductive potential. Female subjects or male subjects with female partners who were lactating or breastfeeding, pregnant, or planning to become pregnant through 4 weeks after receiving the last dose of study drug were excluded. Subjects with evidence or history of diabetic complications with significant end-organ damage, significant cardiac disease, uncontrolled hypertension (systolic blood pressure [SBP] ≥ 150 mmHg or diastolic blood pressure [DBP] ≥ 90 mmHg) on or off therapy, or an unstable medical condition (hospitalization within 28 days before study day −1, major surgery within 6 months before study day −1, or otherwise unstable as judged by a study investigator) were excluded. Subjects with triglyceride levels of 500 mg/dL or higher, hepatic liver enzymes (ALT, AST, ALP, and TBIL) 1.5 times the upper limit of normal or higher, or fasting blood glucose levels of 270 mg/dL or higher were excluded, as were patients who tested positive for HIV or hepatitis C antibodies or hepatitis B surface antigen.
Of a total of 239 screened patients, 69 were randomized to treatment, and 64 completed the study (Figure S1). Four subjects discontinued because of AEs, and one subject was lost to follow-up (Figure S1). The demographic and baseline characteristics of the 69 randomized patients are provided in Table 1.
Table 1.
Participant Demographics and Baseline Characteristics for All Treatment Groups
| Treatment Group | Placebo |
AKR-001 QW |
AKR-001 Q2W |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| QW (N = 8–9) | Q2W (N = 6–8) | 7 mg (N = 5–7) | 21 mg (N = 6) | 70 mg (N = 6) | 140mg (N = 6–9) | 7 mg (N = 5–7) | 21 mg (N = 5–6) | 70 mg (N = 5–6) | 140 mg (N = 5–9) | |
| Mean age, years (SD) | 51 (8) | 58 (6) | 60 (3) | 46 (10) | 59 (5) | 57 (6) | 58 (4) | 58 (4) | 54 (9) | 54 (7) |
| Male, N (%) | 6 (67) | 6 (75) | 3 (43) | 3 (50) | 2 (33) | 5 (56) | 3 (50) | 2 (33) | 5 (83) | 4 (67) |
| Mean weight, kg (SD) | 84 (16) | 81 (9) | 98 (18) | 83 (16) | 89 (11) | 93 (13) | 98 (18) | 73 (8) | 92 (20) | 88 (9) |
| Mean BMI, kg/m2 (SD) | 30 (3) | 29 (3) | 35 (3) | 31 (3) | 36 (3) | 32 (5) | 34 (3) | 29 (2) | 33 (6) | 32 (3) |
| Geo mean fasting glucose, mmol/L (% CV) | 9.0 (28) | 10.9 (16) | 10.7 (32) | 9.4 (26) | 10.5 (16) | 10.8 (20) | 10.6 (15) | 9.8 (15) | 10.2 (18) | 10.0 (31) |
| Geo mean fasting insulin, pmol/L (% CV) | 61 (84) | 58 (45) | 72 (44) | 56 (27) | 93 (47) | 68 (68) | 63 (87) | 64 (36) | 63 (42) | 63 (88) |
| Geo mean fasting C-peptide, nmol/L (% CV) | 0.8 (36) | 0.8 (25) | 0.8 (25) | 0.7 (31) | 0.9 (18) | 0.8 (37) | 0.7 (60) | 0.9 (13) | 0.8 (53) | 0.8 (36) |
| HOMA-IR (% CV) | 3.5 (70) | 4.0 (43) | 4.9 (40) | 3.4 (44) | 6.3 (42) | 4.7 (76) | 4.3 (97) | 4.0 (46) | 4.1 (59) | 4.0 (81) |
| Geo mean fasting glucagon, ng/L (% CV) | 88 (19) | 93 (18) | 99 (35) | 85 (32) | 93 (30) | 89 (38) | 83 (26) | 93 (22) | 97 (12) | 93 (27) |
| Geo mean fasting TGs, mmol/L (% CV) | 1.6 (20) | 1.7 (25) | 1.4 (36) | 2.1 (27) | 2.0 (30) | 1.5 (48) | 2.1 (28) | 1.9 (30) | 1.7 (33) | 1.6 (55) |
| Geo mean fasting TC, mmol/L (% CV) | 5.2 (17) | 4.7 (19) | 5.4 (20) | 5.9 (13) | 5.1 (20) | 4.5 (25) | 5.5 (23) | 5.8 (19) | 4.3 (18) | 4.8 (19) |
| Geo mean fasting HDL-C, mmol/L (% CV) | 1.2 (21) | 1.1 (30) | 1.2 (18) | 1.3 (16) | 1.1 (32) | 1.2 (24) | 1.0 (18) | 1.3 (17) | 1.0 (30) | 1.2 (28) |
| Geo mean fasting non-HDL-C, mmol/L (% CV) | 4.0 (25) | 3.6 (23) | 4.2 (28) | 4.5 (21) | 3.9 (27) | 3.2 (38) | 4.5 (25) | 4.5 (22) | 3.3 (20) | 3.5 (29) |
| Geo mean fasting FFA, μmol/L (% CV) | 440 (35) | 468 (39) | 645 (21) | 432 (24) | 541 (40) | 703 (25) | 865 (17) | 360 (23) | 539 (41) | 430 (34) |
| Geo mean fasting 3-hydoxybutyrate, μmol/L (% CV) | 114 (50) | 106 (65) | 127 (42) | 97 (39) | 97 (29) | 129 (41) | 126 (26) | 93 (8) | 142 (87) | 121 (63) |
| Geo mean fasting apoA-1, mg/dL (% CV) | 130 (18) | 120 (25) | 130 (11) | 139 (7) | 136 (28) | 138 (16) | 118 (9) | 135 (14) | 109 (24) | 138 (13) |
| Geo mean fasting apoB, mg/dL (% CV) | 100 (24) | 91 (19) | 99 (24) | 108 (20) | 101 (20) | 84 (34) | 103 (28) | 106 (20) | 85 (18) | 88 (29) |
| Geo mean fasting Adiponectin, μg/mL (% CV) | 3.3 (64) | 4.0 (55) | 4.2 (52) | 3.4 (109) | 5.2 (28) | 3.5 (52) | 4.2 (66) | 5.4 (63) | 5.2 (52) | 5.0 (63) |
| Geo mean HbA1C, % (% CV) | 8.0 (10) | 7.8 (10) | 8.0 (12) | 8.4 (13) | 8.1 (10) | 7.4 (11) | 7.6 (10) | 8.2 (9) | 7.7 (7) | 8.1 (14) |
| Geo mean fasting IGF-1, ng/mL (% CV) | 135 (47) | 155 (29) | 114 (29) | 156 (20) | 113 (33) | 147 (32) | 164 (38) | 129 (47) | 112 (53) | 183 (36) |
| Geo mean fasting C4, ng/mL (% CV) | 36 (89) | 30 (86) | 29 (162) | 59 (88) | 51 (52) | 22 (112) | 19 (323) | 52 (51) | 18 (96) | 22 (232) |
| Mean HR, bpm (SD) | 66 (6) | 62 (12) | 65 (10) | 70 (10) | 69 (7) | 63 (7) | 65 (7) | 62 (6) | 67 (9) | 63 (16) |
| Mean SBP, mmHg (SD) | 125 (12) | 118 (11) | 126 (16) | 119 (14) | 125 (13) | 121 (18) | 129 (12) | 120 (17) | 132 (19) | 126 (18) |
| Mean DBP, mmHg (SD) | 77 (5) | 74 (8) | 74 (6) | 77 (6) | 80 (7) | 73 (8) | 75 (5) | 72 (10) | 81 (6) | 75 (12) |
QW, once weekly; Q2W, once every 2 weeks; N, number of subjects per group; SD, standard deviation; BMI, body mass index; Geo, geometric; TG, triglyceride; CV, coefficient of variation; TC, total cholesterol; HDL-C, high-density lipoprotein cholesterol; FFA, free fatty acid; HOMA-IR, homeostatic model assessment of insulin resistance; apoA-1, Apolipoprotein A-1; apoB, Apolipoprotein B; HbA1c, glycated hemoglobin; IGF-1, insulin growth factor 1; C4, 7α-hydroxy-4-cholesten-3-one; HR, heart rate, SBP, systolic blood pressure; DBP, diastolic blood pressure.
PK of AKR-001
AKR-001 exhibited dose-proportional exposure across the tested doses for both dosing intervals (Figure 2). Variability in exposure was moderate, with an approximately 50% coefficient of variation for the steady-state maximum observed concentration (Cmax) and the area under the curve during an inter-dose interval (AUC0–τ) across doses (Figure 2; Table 2). The median time to maximum serum concentration (Tmax) ranged from 2 to 3.5 days, depending on dose. Approximately 2-fold accumulation was observed after four AKR-001 QW doses, with steady state achieved following the third QW dose (Figure 2; Table 2). The median peak-to-trough concentration ratio following QW dosing was 2 for all tested QW doses compared with a range of 6–11 following Q2W dosing, indicating that the steady-state concentration was more effectively maintained within a narrow range following QW compared with Q2W dosing.
Figure 2.

PK Profile of AKR-001 in Type 2 Diabetic Patients
(A and B) Median serum concentration of intact AKR-001 following (A) weekly (QW) and (B) once every 2 weeks (Q2W) dose regimens.
Table 2.
Descriptive Statistics of PK Parameter Estimates after QW and Q2W Administration of AKR-001
| PK Parameter | AKR-001 QW |
AKR-001 Q2W |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 7 mg | 21 mg | 70 mg | 140mg | 7 mg | 21 mg | 70 mg | 140 mg | ||
| Day 1 | n | 7 | 6 | 6 | 9 | 6 | 6 | 6 | 6 |
| mean Cmax, ng/mL (% CV) | 297 (71.6) | 806 (76.8) | 2,600 (67.4) | 7,590 (43.7) | 193 (16.6) | 1,010 (37.1) | 2,110 (76.6) | 6,810 (33.4) | |
| mean AUC0–τ, day⋅ng/mL (% CV) | 1,450 (72.5) | 4,060 (73.6) | 13,300 (67.8) | 36,700 (36.2) | 1,490 (17.4) | 7,170 (21.4) | 17,500 (80.1) | 50,600 (35.4) | |
| Day 15 (Q2W) or day 22 (QW) | n | 6 | 6 | 6 | 5 | 6 | 6 | 6 | 6 |
| mean Cmax, ng/mL (% CV) | 440 (47.1) | 2,290 (51.3) | 6,260 (48.3) | 11,900 (83.1) | 236 (50.2) | 881 (47.0) | 3,140 (69.5) | 7,530 (49.5) | |
| mean AUC0–τ, day⋅ng/mL (% CV) | 2,260 (43.6) | 10,700 (48.4) | 31,900 (48.5) | 57,500 (70.4) | 1,750 (44.5) | 6,110 (35.4) | 27,800 (78.5) | 55,600 (44.4) | |
| t1/2,z, days (% CV) | 3.24 (23.5) | 2.56 (12.0) | 3.30 (12.7) | 3.54 (13.8) | 2.81a (30.2) | 3.27b (13.2) | 3.29 (7.1) | 3.44 (11.3) | |
QW, once weekly; Q2W, once every 2 weeks; n, number of subjects per group; % CV, coefficient of variation expressed as percent; Cmax, maximum observed concentration; AUC0–τ, area under the concentration-time curve during the inter-dose interval, post-dose; τ, inter-dose interval (QW = 7 days, Q2W = 14 days); t1/2,z, terminal half-life.
N = 4.
N = 5.
In contrast to the approximately 2-h half-life of native human FGF21, the C-terminal modifications and the Fc scaffold of AKR-001 extend the half-life of intact C terminus AKR-001 to 3–3.5 days in humans. In comparison, the intact unmodified C-terminal domains of PF-05231023 and pegbelfermin have a reported half-life in humans of 6.5–10.3 h19 and 19–24 h,31 respectively, whereas LY2405319 appears to have a relatively short half-life given its daily administration regimen.18
Anti-drug antibodies (ADAs) were detected in 7 of 52 subjects (13.5%) who received AKR-001, but the PK of AKR-001 was unaffected by the presence of ADAs. Neutralizing antibodies were not detected in any of the ADA-positive samples.
PD Endpoints
PD endpoint data from the placebo, 21 mg, and 70 mg QW dose cohorts are plotted for all measured time points (Figure 3). The magnitude of changes in PD endpoints at 70 mg QW appeared to be maximal, with little additional effect at 140 mg QW. Tabulated data from multiple time point mixed models for repeated measures (STAR Methods) are provided for all QW cohorts on day 25, 3 days after the fourth (final) QW dose, and for all Q2W cohorts on day 18, 3 days after the second (final) Q2W dose (Table 3). The planned cohort size of 8 was selected to evaluate safety and PK, as is typical for early-phase trials. The study was not powered to demonstrate statistical significance in PD endpoints. These endpoints are presented with descriptive statistics that informed ongoing clinical development of AKR-001.
Figure 3.

Effect of AKR-001 on Markers of Glycemic Control and Lipid Metabolism
(A–H) Concentration change from baseline of fasted-state (A) glucose, (B) insulin, (C) C-peptide, (D) glucagon, (E) HOMA-IR, (F) HDL cholesterol, (G) non-HDL cholesterol, and (H) triglycerides by study day for placebo (n = 7–9), 21 mg (n = 5–6), and 70 mg (n = 5–6) QW dose cohorts. Data are presented as least-squares mean ± 95% confidence intervals.
See also Figure S2.
Table 3.
Least-Squares Mean (95% Confidence Interval) Placebo-Corrected Percentage Change from Baseline of Metabolic Markers in the Fasted State on day 18 (Q2W Cohorts) or day 25 (QW Cohorts)
| Treatment Group | AKR-001 QW |
AKR-001 Q2W |
||||||
|---|---|---|---|---|---|---|---|---|
| 7 mg (n = 6) | 21 mg (n = 6) | 70 mg (n = 6) | 140mg (n = 5) | 7 mg (n = 6) | 21 mg (n = 6) | 70 mg (n = 6) | 140 mg (n = 6) | |
| Glucose | 4 (−12, 22) | 5 (−11, 24) | −23, (−35, −9) | −20 (−32, −6) | −3 (−18, 15) | 3 (−13, 22) | −6 (−20, 12) | −1 (−16, 18) |
| Insulin | −20 (−47, 23) | −28 (−53, 12) | −49 (−67, −22) | −50 (−68, −24) | −8 (−40, 43) | −23 (−50, 19) | −15 (−45, 31) | −28 (−54, 11) |
| C-peptide | −22 (−40, 1) | −29 (−46, −7) | −39 (−54, −20) | −45 (−58, −29) | 4 (−20, 37) | −13 (−34, 14) | −4 (−27, 26) | −26 (−43, −2) |
| HOMA-IR | −18 (−49, 32) | −24 (−53, 24) | −60 (−75, −35) | −60 (−75, −36) | −12 (−46, 43) | −22 (−52, 28) | −21 (−52, 28) | −29 (−56, 16) |
| Glucagon | 36 (8, 72) | 13 (−11, 43) | 13 (−11, 43) | 24 (−2, 57) | 7 (−16, 36) | 0 (−21, 28) | 46 (15, 86) | 31 (3, 67) |
| Body weight | 0.5 (−1.4, 2.5) | 0.3 (−1.7, 2.3) | −0.7 (−2.6, 1.3) | −1.4 (−3.3, 0.6) | 1.1 (−0.9, 3.1) | −0.6 (−2.5, 1.4) | −0.6 (−2.6, 1.4) | −1.5 (−3.4, 0.5) |
| Total cholesterol | 1 (−13, 16) | 1 (−12, 17) | −8 (−21, 6) | −11 (−23, 3) | −7 (−20, 8) | 3 (−11, 19) | −1 (−15, 14) | −7 (−20, 8) |
| HDL-C | 28 (13, 46) | 38 (21, 57) | 61 (41, 84) | 37 (20, 56) | 2 (−11, 16) | 23 (8, 41) | 26 (10, 44) | 37 (20, 57) |
| Non-HDL-C | −12 (−27, 8) | −11 (−27, 8) | −30 (−43, −14) | −34 (−46, −19) | −10 (−26, 10) | −4 (−21, 18) | −9 (−26, 11) | −23 (−37, −6) |
| Triglycerides | −37 (−55, −11) | −60 (−71, −43) | −69 (−78, −57) | −60 (−71, −45) | −19 (−41, 12) | −29 (−48, −2) | −42 (−58, −20) | −55 (−67, −38) |
| FFA | −16 (−46, 30) | 15 (−27, 80) | −4 (−38, 51) | −12 (−43, 37) | −31 (−56, 9) | 30 (−17, 104) | 25 (−21, 96) | 23 (−22, 93) |
| 3-Hydroxybutyrate | 11 (−40, 106) | 53 (−18, 187) | 69 (−10, 217) | 30 (−30, 142) | −9 (−52, 72) | −13 (−54, 64) | 68 (−11, 218) | 60 (−15, 203) |
QW, once weekly; Q2W, once every 2 weeks; N, number of subjects per group; HOMA-IR, homeostatic model assessment of insulin resistance; HDL-C, high-density lipoprotein cholesterol; FFA, free fatty acid. Data are from multiple time point mixed models for repeated measures (STAR Methods); a single time point is reported for brevity. See also Figure S3 and Tables S1 and S2.
C-terminal cleavage by the prolyl endopeptidase fibroblast activation protein (FAP) inactivates FGF21, underscoring the need to measure the concentration of C terminus “intact” FGF21 to interpret physiology and pharmacology. Although endogenous intact FGF21 concentrations typically range from 100–1,000 pg/mL or approximately 5–50 pM,5, 6, 7 effective AKR-001 concentrations at doses of 21 mg QW or more appear to be much higher—around 1–2 μg/mL, which correspond to approximately 20–40 nM FGF21(RGE), accounting for two molecules of FGF21(RGE) per molecule of AKR-001. Previous work demonstrated a half-maximal effective concentration (EC50) for AKR-001 of 0.5–1 nM across multiple cell-based assays,29 about 10- to 100-fold higher than the systemic levels of native human FGF21 and about 20- to 80-fold lower than the effective concentration of AKR-001 in humans, presented here.
In part, the difference between AKR-001’s in vitro EC50 and observed systemic levels of native FGF21 in humans may be explained by AKR-001’s slightly lower in vitro potency than native human FGF21. In addition, C-terminal cleavage and inactivation by FAP may lead to a substantial concentration gradient for intact native FGF21 between its cell-surface site of action and the systemic circulation, attributable to the close proximity of membrane-bound FAP to the co-receptors mediating FGF21 signaling.32 The difference between AKR-001’s in vitro EC50 and its effective human concentration remains to be explored but may, at least in part, reflect plasma protein binding of AKR-001, a relatively hydrophobic and slightly acidic molecule.
AKR-001 Induced Dose-Dependent Effects on Metabolic Markers
Markers of Insulin Sensitivity
Measures of insulin sensitivity, such as insulin, C-peptide, and homeostatic model assessment of insulin resistance (HOMA-IR), evaluated in the fasted state at the point of maximal steady-state AKR-001 serum concentration following QW administration (3 days after the final dose, day 25), showed numerical improvement at 70 and 140 mg QW (Table 3). In contrast, there appeared to be little change in markers associated with glycemic control in Q2W cohorts, including the top dose, at the point corresponding to maximal steady-state AKR-001 serum concentration (3 days after the final dose, day 18; Table 3; Figure S2A). Comparing the 70 mg QW and 140 mg Q2W dose groups is of particular value in determining the more effective dosing regimen because the total dose and AUC0–τ were similar for these groups, but the trough and average concentrations were higher following 70 mg QW than 140 mg Q2W (Table 2). The extended half-life (3–3.5 days) combined with 2–3.5 days to reach Cmax after QW dosing appeared to result in maintenance of systemic exposure to AKR-001 above a pharmacologically effective concentration during the inter-dose interval while limiting the peak-to-trough ratio to approximately 2 under steady state conditions. In contrast, an effective concentration did not appear to be maintained with Q2W dosing of 140 mg because markers of insulin sensitivity did not appear to be improved despite similar exposure (AUC0–τ) to 70 mg QW.
Figures 3A–3E show the change from baseline in concentration of markers of glycemic control in the 21 and 70 mg QW AKR-001 and placebo cohorts at all study time points, demonstrating a trend toward dose-dependent improvement. Following the fourth QW dose, only fasting C-peptide levels appeared to be reduced at 21 mg, whereas C-peptide, insulin, HOMA-IR, and glucose were lower at 70 mg. QW doses of 21 and 70 mg were also well tolerated (Table 4), suggesting therapeutic potential across this range. From day 18, the placebo-corrected change from baseline of fasting glucose following administration of 70 mg AKR-001 QW ranged from −20% to −35% (Table 3; Figure 3A). This contrasts with the general lack of reduction for obese patients with type 2 diabetes following daily dosing of LY240531918 and pegbelfermin21 for 4 and 12 weeks, respectively, twice weekly dosing of PF-05231023,19 and weekly dosing of BFKB8488A for 12 weeks (C. Wong et al., 2019, AASLD, poster #2310). Surprisingly, a trend toward increases in fasting glucose was observed following Q2W dosing (Figure S2A), possibly because of increased hepatic glucose output, consistent with observations of elevated glucagon in the higher-dose Q2W cohorts (Table 3). The indication that AKR-001 may increase glucagon levels in humans contrasts with the unchanged glucagon levels reported for mice treated with FGF2113,14 and reduced levels for non-human primates treated with rhFGF21 or FGF21 analogs.15,29 Increasing insulin sensitivity with higher doses of AKR-001 may account for the trend of higher fasting glucose at 21 mg QW reversing to lower fasting glucose at doses of 70 mg QW or less (Table 3). Improved insulin sensitivity should increase peripheral extraction of circulating glucose, potentially offsetting any increase in glucose output by the liver in response to raised glucagon levels. Insulin has also been demonstrated to suppress the acute glucagon response to arginine in humans.33
Table 4.
Treatment-Emergent AEs by Preferred Term Reported in 2 or More Subjects in the Pooled Placebo (QW and Q2W) or across All AKR-001 Treatment Groups (7–140 mg, QW and Q2W)
| Placebo Pooled QW/Q2W |
AKR-001 QW |
AKR-001 Q2W |
AKR-001 Pooled QW/Q2W |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| (N = 17) | 7 mg (N = 7) | 21 mg (N = 6) | 70 mg (N = 6) | 140 mg (N = 9) | 7 mg (N = 6) | 21 mg (N = 6) | 70 mg (N = 6) | 140 mg (N = 6) | ≤70 mg (N = 37) | 7–140 mg (N = 52) | |
| Subjects reporting TEAEs, n (%) | 12 (70.6) | 5 (71.4) | 5 (83.3) | 5 (83.3) | 9 (100) | 4 (66.7) | 4 (66.7) | 6 (100) | 5 (83.3) | 29 (78.4) | 43 (82.7) |
| Serious AEs, n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| AE withdrawals, n (%) | 1 (5.9) | 1 (14.3) | 0 (0) | 0 (0) | 4 (44.4) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (2.7) | 5 (9.6) |
| Subjects Reporting TEAEs,aat Least 2 Subjects in Pooled Placebo or across All AKR-001 Treatment Groups, n (%) | |||||||||||
| Gastrointestinal disorders | 2 (11.8) | 4 (57.1) | 3 (50) | 3 (50) | 8 (88.9) | 2 (33.3) | 2 (33.3) | 3 (50) | 3 (50) | 17 (45.9) | 28 (53.8) |
| Nausea | 0 (0) | 2 (28.6) | 3 (50) | 2 (33.3) | 6 (66.7) | 0 (0) | 2 (33.3) | 2 (33.3) | 3 (50) | 11 (29.7) | 20 (38.5) |
| Diarrhea | 1 (5.9) | 2 (28.6) | 0 (0) | 2 (33.3) | 2 (22.2) | 0 (0) | 1 (16.7) | 2 (33.3) | 1 (16.7) | 7 (18.9) | 10 (19.2) |
| Vomiting | 0 (0) | 0 (0) | 0 (0) | 2 (33.3) | 3 (33.3) | 0 (0) | 1 (16.7) | 1 (16.7) | 2 (33.3) | 4 (10.8) | 9 (17.3) |
| Dyspepsia | 1 (5.9) | 1 (14.3) | 0 (0) | 0 (0) | 2 (22.2) | 0 (0) | 0 (0) | 1 (16.7) | 0 (0) | 2 (5.4) | 4 (7.7) |
| Abdominal pain, upper | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (16.7) | 1 (16.7) | 0 (0) | 2 (5.4) | 2 (3.8) |
| Constipation | 1 (5.9) | 0 (0) | 0 (0) | 0 (0) | 1 (11.1) | 1 (16.7) | 0 (0) | 0 (0) | 0 (0) | 1 (2.7) | 2 (3.8) |
| General disorders and administration site conditions | 2 (11.8) | 2 (28.8) | 1 (16.7) | 4 (66.7) | 2 (22.2) | 0 (0.0) | 1 (16.7) | 1 (16.7) | 2 (33.3) | 9 (24.3) | 13 (25) |
| Injection site rash | 0 (0) | 0 (0) | 1 (16.7) | 2 (33.3) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 3 (8.1) | 3 (5.8) |
| Injection site erythema | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (11.1) | 0 (0) | 0 (0) | 0 (0) | 1 (16.7) | 0 (0) | 2 (3.8) |
| Metabolism and nutrition disorders | 1 (5.9) | 2 (28.6) | 2 (33.3) | 2 (33.3) | 5 (55.6) | 0 (0) | 0 (0) | 2 (33.3) | 0 (0) | 8 (21.6) | 13 (25) |
| Increased appetite | 0 (0) | 1 (14.3) | 0 (0) | 2 (33.3) | 4 (44.4) | 0 (0) | 0 (0) | 1 (16.7) | 0 (0) | 4 (10.8) | 8 (15.4) |
| Hyperkalemia | 0 (0) | 0 (0) | 1 (16.7) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (16.7) | 0 (0) | 2 (5.4) | 2 (3.8) |
| Nervous system disorders | 5 (29.4) | 1 (14.3) | 1 (16.7) | 0 (0) | 4 (44.4) | 2 (33.3) | 2 (33.3) | 3 (50) | 0 (0) | 9 (24.3) | 13 (25) |
| Headache | 4 (23.5) | 0 (0) | 1 (16.7) | 0 (0) | 1 (11.1) | 2 (33.3) | 1 (16.7) | 2 (33.3) | 0 (0) | 6 (16.2) | 7 (13.5) |
| Tremor | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 4 (44.4) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 4 (7.7) |
| Dysgeusia | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (11.1) | 0 (0) | 1 (16.7) | 1 (16.7) | 0 (0) | 2 (5.4) | 3 (5.8) |
| Musculoskeletal and connective tissue disorders | 3 (17.6) | 0 (0) | 1 (16.7) | 1 (16.7) | 1 (11.1) | 2 (33.3) | 1 (16.7) | 0 (0) | 0 (0) | 5 (13.5) | 6 (11.5) |
| Back pain | 2 (11.8) | 0 (0) | 0 (0) | 1 (16.7) | 0 (0) | 2 (33.3) | 1 (16.7) | 0 (0) | 0 (0) | 4 (10.8) | 4 (7.7) |
| Muscle spasms | 0 (0) | 0 (0) | 1 (16.7) | 0 (0) | 1 (11.1) | 1 (16.7) | 0 (0) | 0 (0) | 0 (0) | 2 (5.4) | 3 (5.8) |
| Infections and infestations | 2 (11.8) | 0 (0) | 0 (0) | 2 (33.3) | 1 (11.1) | 0 (0) | 0 (0) | 0 (0) | 1 (16.7) | 2 (5.4) | 4 (7.7) |
| Rhinitis | 0 (0) | 0 (0) | 0 (0) | 1 (16.7) | 1 (11.1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (2.7) | 2 (3.8) |
| Upper respiratory tract infection | 0 (0) | 0 (0) | 0 (0) | 1 (16.7) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (16.7) | 1 (2.7) | 2 (3.8) |
| Cardiac disorders | 0 (0) | 0 (0) | 0 (0) | 2 (33.3) | 1 (11.1) | 0 (0) | 0 (0) | 0 (0) | 1 (16.7) | 2 (5.4) | 4 (7.7) |
| Palpitations | 0 (0) | 0 (0) | 0 (0) | 2 (33.3) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (5.4) | 2 (3.8) |
System organ class AE incidence includes preferred terms that do not themselves meet the threshold for inclusion in Table 4. All remaining AEs are reported in Table S4. Additionally, a single subject may report multiple AEs captured by multiple preferred terms. For these reasons, the sum of the preferred term-level AEs may not equal the corresponding system organ class-level AEs in this table. QW, once weekly; Q2W, once every 2 weeks; N, number of subjects per group; n, number of subjects reporting event; TEAE, treatment-emergent AE. See also Tables S3 and S4.
TEAE by system organ class and preferred term, coded using the Medical Dictionary for Regulatory Activities v.17.0.
Progressive numerical reductions in fasting insulin were observed following 70 mg AKR-001 QW repeat dosing (Figure 3B; Table 3). In contrast, insulin levels appeared to be unchanged after Q2W dosing (Figure S2B; Table 3). The substantial sustained dose-related numerical decrease in C-peptide upon weekly AKR-001 administration corroborates the observed trend toward dose-dependent reductions in circulating insulin (Figure 3C; Table 3). In contrast, the only Q2W cohort with apparent reductions in C-peptide was the 140 mg dose group (Table 3; Figure S2C). Previous reports for FGF21 analogs in placebo-controlled trials showed that only the highest daily dose of LY2405319 reduced fasting insulin,18 whereas the levels of insulin remained unchanged following all dosing regimens for PF-0523102319 and pegbelfermin.21
HOMA-IR represents an estimate of insulin resistance under fasted conditions. Following AKR-001 QW dosing, trends toward lower HOMA-IR values in the 70 mg (reduction from baseline of 54%–65% over days 23–36) and 140 mg (reduction of 32%–57% over days 23–36) groups were sustained until AKR-001 serum concentration fell from steady state (Table 3; Figure 3E). HOMA-IR numerical decreases from baseline in the Q2W dose groups were observed sporadically (Figure S2E; Table 3). Glycated hemoglobin appeared to be unchanged after QW and Q2W dosing for 4 weeks, which is to be expected given its slow turnover. There were minimal changes in body weight (Table 3).
A mixed meal tolerance test was carried out 3 days after the final dose for each cohort, following the reported fasting measurements on days 18 and 25 for the Q2W and QW groups, respectively. The mean placebo-corrected changes in AUC0–4h from the baseline test (day −1) for metabolic marker responses are provided in Table S1. Under post-prandial conditions, similar to fasting measurements, markers of glycemic control appeared to be improved more by QW than Q2W dosing. There was a trend toward reductions in glucose, insulin, and C-peptide after QW dosing, whereas after Q2W dosing, only insulin and C-peptide trended lower and to a lesser magnitude. Changes in post-prandial glucagon were variable and not dose related. It has been reported that post-prandial glucagon concentration ranges are greater than those in the fasted state, and that this range increases with age.34 This greater variability may account for the inconsistent increases in glucagon seen in the 7 mg QW, 140 mg QW, and 70 mg Q2W dose groups only. The effect of other FGF21 analogs on post-prandial measurements has not been reported.
In summary, AKR-001 demonstrated trends toward improvements in glycemic control and markers of insulin sensitivity under fasting and fed conditions, comparing favorably with previously reported clinical investigations with other FGF21 analogs (C. Wong et al., 2019, AASLD, poster #2310).18,19,21
Markers of Lipid Metabolism
As with the glycemic markers described above, trends toward improvements in the lipoprotein profile were numerically greater and appeared to be longer lasting with QW than Q2W dosing (Table 3; Figures 3F–3H; Figures S2F–S2H), confirming the need to maintain a threshold of AKR-001 exposure across the inter-dose interval to elicit the maximal therapeutic effects of FGF21 agonism. For example, the maximal placebo-corrected reductions in fasting triglyceride were 69% and 55%, whereas the increases in HDL were 61% and 37%, respectively, for QW and Q2W. Maximal numerical differences in both of these markers was achieved with QW dosing of 70 mg or more. The effect of 70 mg and 140 mg QW AKR-001 on the lipoprotein profile is directionally similar to that reported for other FGF21 analogs (C. Wong et al., 2019, AASLD, poster #2310),18,19, but the effects observed for AKR-001 appear to be quantitatively larger. For example, the highest doses of PF-05231023 and LY2405319 reduced triglyceride by up to 50% after 4 weeks,18,19 whereas the highest daily dose of pegbelfermin reduced triglyceride by only 22% after 2 weeks.21,25 For HDL, the maximal increase from baseline with other FGF21 analogs was 15%–29% (C. Wong et al., 2019, AASLD, poster #2310).18, 19, 20, 21 Similarly, non-HDL cholesterol appeared to decrease in a dose-dependent manner following AKR-001 QW administration (−34% maximum), with a lesser effect seen with Q2W dosing (Table 3; Figure 3G; Figure S2G). The corresponding maximal placebo-corrected decrease in non-HDL cholesterol following treatment with PF-05231023 was 17%.20 The maximal reduction from baseline in low-density lipoprotein (LDL) cholesterol with the highest doses of pegbelfermin evaluated (20 mg daily, QD, and 20 mg QW) was 12%.21,25
Unlike previous studies with FGF analogs, fasting serum lipoprotein levels were followed for 5–7 weeks after the last dose of AKR-001 (Figures 3F–3H). The trend toward improvement in lipoprotein profiles persisted so that, in the 70 mg QW cohort on day 56, when the AKR-001 serum concentration was below the limit of quantitation (<10 ng/mL), non-HDL cholesterol and triglycerides remained below pre-treatment levels and HDL-cholesterol above. This is consistent with a model in which AKR-001 addressed underlying dyslipidemia by inducing durable, beneficial adaptations in lipid homeostasis. Supporting this hypothesis, a recent publication has shown that FGF21 increases lipophagy in hepatocytes by inducing demethylation of specific histone residues, leading to stable modifications of gene expression.35
Other markers of lipid metabolism in the fasting state, including free fatty acid (FFA) and 3-hydroxybutyrate concentration, were little affected by administration of AKR-001 (Table 3). The range of 3-hydroxybutyrate levels following AKR-001 administration is consistent with the lack of effects reported with PF-05231023 in cynomolgus monkeys19 and the modest (2-fold) increase reported with LY2405319 in humans.18 The trends toward elevated glucagon levels following AKR-001 administration may have increased fatty acid oxidation by the liver,36 leading to a modest increase in 3-hydroxybutyrate.
In contrast to the lack of effect in the fasted state, post-prandial FFA concentration (AUC0–4h) trended lower in the AKR-001 70 mg (−31%) and 140 mg (−19%) QW groups but appeared to be unchanged by other dosing regimens (Table 3; Table S1). A post-prandial FFA reduction is consistent with the apparent insulin sensitization at the higher QW doses of AKR-001 and with possible suppression of adipose tissue lipolysis by FGF21 in vivo in the fed state37 and human adipocytes in vitro.38 The effects of pegbelfermin, BFKB8488A, PF-05231023, and LY2405319 on human post-prandial FFA concentration have not been reported.
Post Hoc Exploratory Biomarkers
The inclusion of additional post hoc exploratory biomarkers enables hypothesis generation, informing future clinical development of AKR-001 and other FGF21 analogs. Testing such hypotheses requires larger cohorts than those in this study to enable statistical inferences. Apolipoprotein A-1 (apoA-1), the major protein component of HDL particles, was numerically increased following AKR-001 QW treatment at 7, 21, and 140 mg but not at 70 mg (Table S2). Weekly administration of AKR-001 resulted in a progressive dose-related trend toward reduction of apolipoprotein B (apoB), the major protein component of very-low-density lipoprotein (VLDL) and LDL particles and an independent causal risk factor for cardiovascular disease,39 with a maximum placebo-corrected decrease of 42% from baseline following treatment with 70 mg QW by day 29 (Table S2). The effect of FGF21 analogs on apoB has only been reported for LY2405319, which elicited a maximum decrease from baseline of 21%–25%.18 The smaller quantitative decrease in apoB with LY2405319 compared with AKR-001 is consistent with the greater numerical reduction in non-HDL cholesterol by AKR-001 than LY2405319. Another independent causal risk factor for cardiovascular disease is lipoprotein(a) (Lp(a)), which forms disulfide bridges with apoB.40 The levels of Lp(a) appeared to be unchanged following QW administration of AKR-001 (Table S2).
Given the rapid and numerically large reduction of triglycerides by AKR-001, levels of proteins regulating triglyceride uptake from VLDL and chylomicrons into peripheral tissues were measured. Triglyceride uptake is mediated by the enzyme lipoprotein lipase (LPL), which is activated by apolipoprotein C-2 (apoC-2) and inhibited by apolipoprotein C-3 (apoC-3) and angiopoietin-like 4 (ANGPTL4). Weekly administration of AKR-001 was associated with approximately 50% lower circulating apoC-2 and apoC-3 across all doses equal to or greater than 70 mg (Table S2). ANGPTL4 levels appeared to be unchanged (Table S2). The magnitude of reduction of apoC-3 appeared to be greater than that reported for LY2405319, which elicited a maximum decrease from baseline of 30%–35%, consistent with a smaller maximal reduction of approximately 50% in triglyceride18 compared with 60%–69% with QW AKR-001 (Table 3).
Taken together, the changes in the levels of apoC-2, apoC-3, and ANGPTL4 may be expected to increase the activity of LPL, thereby increasing extraction of triglyceride by peripheral tissues. Heterozygous loss of function of apoC-2 in humans does not impair LPL activity and extraction of triglyceride, unlike homozygous loss of function.41 This is consistent with little to no effect on LPL activity of AKR-001’s approximately 50% reduction in apoC-2 alongside robust reductions in apoC-3 and triglycerides. The observed numerical differences in specific apolipoproteins secreted by the liver as components of VLDL (apoB) or HDL (apoA-1, apoC-2, and apoC-3)40 are consistent with a role of AKR-001 in specifically modulating protein expression in human hepatocytes. Further study is required to determine whether these changes are mediated by direct action of AKR-001 on hepatocytes, or whether they are indirect effects caused by whole-body metabolic reprogramming induced by AKR-001.
Adiponectin, an adipocyte-derived hormone, has been reported to be an insulin sensitizer that exhibits anti-diabetic, anti-inflammatory, and anti-fibrotic effects.42, 43, 44 Similar to other FGF21 analogs,18,19,21 AKR-001 appeared to increase adiponectin levels, with larger numerical increases at higher doses in general (Table S2). In mice, the beneficial effects of FGF21 depend on functional adiponectin signaling, including alleviation of obesity-associated hyperglycemia and insulin resistance.12,45 However, despite an apparently similar magnitude of increase in adiponectin in the AKR-001 QW and Q2W dose groups, larger trends toward improvements in glycemic control and insulin sensitivity were evident with QW than with Q2W dosing. This is consistent with clinical observations across FGF21 analogs, where increases in adiponectin levels were not associated with improved glycemic control.18, 19, 20 These observations call into question whether the improvements in insulin sensitivity and glycemic control in humans are primarily driven by increased secretion of adiponectin from adipose tissue mediated by activation of FGFR1c, as has been demonstrated for mice.46
AKR-001 administration did not appear to reduce bile acid synthesis because the levels of 7α-hydroxy-4-cholesten-3-one (C4) did not decrease, in contrast to the decrease in total plasma bile acid (approximately −50%) and C4 (−70% to −80%) reported following treatment with long-acting FGF21 analogs in cynomolgus monkeys.47 Further supporting unchanged bile acid metabolism upon AKR-001 administration, no discernible trend in bile acid composition was apparent. Consistent with the unchanged flux of cholesterol to bile acid, fasting levels of total and non-HDL cholesterol trended lower following AKR-001 treatment (Table 3). Other experimental treatments for metabolic diseases like NASH, such as the FGF19 analog NGM28248 and the Farnesoid X Receptor (FXR) agonist obeticholic acid,49 markedly suppress bile acid synthesis in humans, consequently raising plasma LDL cholesterol. Lower bile acid secretion into the gastrointestinal (GI) tract impairs absorption of dietary fat, resulting in steatorrhea.50 Diarrhea was reported to be a frequent (36%–41%) AE with NGM282.48 Consistent with the lack of effect on bile acid synthesis, AKR-001 either did not appear to affect (QW dosing), or numerically reduced (Q2W dosing), FGF19 levels (Table S2).
Reduced plasma levels of insulin growth factor 1 (IGF-1) correlate with insulin resistance, dyslipidemia, obesity, and metabolic syndrome.51 Although the mean change from baseline in total IGF-1 concentration trended numerically higher in all cohorts receiving AKR-001 compared with placebo treatment, particularly with QW dosing, the increases were variable and not dose dependent (Table S2). The levels of total IGF-1 increased 2- to 5-fold in obese hypertriglyceridemic subjects after 4 weeks of PF-05231023 treatment, whereas free IGF-1 and the IGF binding proteins IGFBP1, IGFBP2, and IGFBP3 were reported to be unchanged.20 These observations in humans appear to be different from those in mice. Overexpression of FGF21 in the mouse liver led to 5-fold higher circulating levels of FGF21 compared with the wild type and suppressed secretion of IGF-1 by the liver.52
Compared with reports for other FGF21 analogs, AKR-001 70 mg QW elicited the largest numerical increases in markers of insulin sensitivity, the largest apparent suppression of post-prandial lipolysis, and the largest numerical improvements in lipoprotein profile (Table S5). Lipolysis is a major source of fatty acids re-esterified into triglyceride within hepatocytes and secreted by the liver as VLDL.53,54 The apparent differences in effect on adipose tissue of AKR-001 compared with other FGF21 analogs may be due to greater potency and/or intrinsic agonist activity of AKR-001 at FGFR1c, the receptor that mediates FGF21 signaling in adipose tissue.46 AKR-001 has a similar potency as human FGF21, with potency balanced across the FGFR1c, FGFR2c, and FGFR3c proteins complexed with β-Klotho.29 LY2405319 also has similar potency as human FGF21 in a range of in vitro assays.55 However, it is not possible to make a comparison with pegbelfermin or PF-05231023 because corresponding in vitro potency data have not been reported. An alternative explanation for the numerically smaller pharmacological effects of the other FGF21 analogs on adipose tissue compared with AKR-001 is that peripheral exposure was insufficient to maximally stimulate FGFR1c in their clinical studies. PEGylation of FGF21, as in pegbelfermin, may have altered its bio-distribution, reducing exposure in the periphery relative to the liver, as shown with PEGylated insulin.56 The much shorter half-life of intact C-terminal PF-05231023 (6.5–10.3 h) dosed twice weekly,20 compared with AKR-001 (3–3.5 days), may have resulted in less sustained agonism of FGFR1c than elicited by AKR-001. Although this study was not powered to support statistical inference regarding PD endpoints, the hypotheses generated by these data and described here are the subject of a recently completed clinical study (AKR-001 phase 2a trial in histologically confirmed NASH, ClinicalTrials.gov: NCT03976401).
Tolerability Profile
There were no serious or fatal AEs at any dose during the treatment period. One unrelated serious AE occurred during washout (before randomization to treatment). Nearly all (91%) treatment-emergent AEs in subjects receiving active treatment were mild (grade 1 severity) and transient. All doses up to and including 70 mg, irrespective of dose interval, demonstrated acceptable tolerability for subsequent clinical investigation, consistent with selection of 70 mg QW as the highest dose in the recently completed phase 2a trial of AKR-001 in NASH patients (NCT03976401). Treatment-emergent AEs were reported for 43 subjects receiving any dose of AKR-001 (82.7%), 29 subjects receiving up to and including 70 mg AKR-001 (78.4%), and 12 subjects receiving a placebo (70.6%) (Table 4). The most frequent treatment-related AEs were gastrointestinal. Consistent with previous reports of FGF21 analogs,18, 19, 20, 21,25 the GI events associated with AKR-001 dosing were mild or moderate (four grade 2 severity events) and transient in nature. There were no treatment-related withdrawals at doses up to and including 70 mg QW and 140 mg Q2W AKR-001. One subject receiving 7 mg QW and one receiving placebo withdrew because of hyperglycemia considered by the investigator not to be related to the investigational product. There were four investigational product-related withdrawals from the 140 mg QW dose group: two because of GI AEs, one because of GI and tremor AEs, and one because of tremor. One subject who withdrew because of GI AEs and experienced tremor after withdrawal was administered an anti-emetic with an FDA label that included tremor among CNS side effects.57 No other neuromuscular AEs were observed at any dose level.
Although one subject in the 140 mg QW cohort experienced mild ventricular extrasystoles attributed by the investigator to AKR-001, there was no observable trend in electrocardiography measurements, including corrected QT interval. One subject in the 140 mg Q2W cohort experienced mild tachycardia, and two subjects in the 70 mg QW cohort experienced mild palpitations, none of which were considered by the investigator to be related to AKR-001. No discernible trends were observed in liver function tests or markers of renal function, including albumin, alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, bilirubin, serum electrolytes, creatinine, and blood urea nitrogen.
Of the seven subjects who developed ADAs, three were sampled 6 weeks after the end of the study, and no ADAs were detected.
PK-PD and Safety Endpoint Modeling
There was an imbalance across study cohorts of hypertension, based on medical history. Therefore, the relationship between cardiovascular measurements (heart rate, systolic and diastolic blood pressure) and AKR-001 exposure was examined by PK-PD modeling. The model included all data for treated and placebo subjects from screening to the end of the study.
The PK-PD model predicted no association between blood pressure and serum concentration of AKR-001 (Figures S3A and S3B). The predicted lack of effect is consistent with unchanged vital signs reported for pegbelfermin,25 LY2405319,18 and an initial study with PF-05231023.19 However, in a subsequent study, treatment with PF-05231023 was associated with increases in blood pressure and heart rate.20 The difference between the outcomes of the two studies with PF-05231023 could not be explained.20
The predicted lack of effect of AKR-001 on blood pressure is consistent with the observed blood pressure phenotypes of human carriers of FGF21 genetic variants. Of note, rare variants encoding putative non-functional, truncated FGF21 are associated with higher blood pressure.28 In a study of 451,099 human subjects, the minor A allele of a common variant of FGF21, rs838133, is associated with 0.29 mmHg higher systolic blood pressure per A allele.28 Although it has not been definitively demonstrated, preclinical and clinical genotype-phenotype associations indicate that the A allele may be associated with loss of FGF21 function. Congruently, preclinical studies show that increasing levels of FGF21 (by overexpression or exogenous administration) reduce blood pressure and vessel wall pathology of mice rendered hypertensive by chronic administration of fructose,58 infusion of angiotensin II,59 or induction of vascular calcification following co-administration of vitamin D and nicotine.60 Further evidence of FGF21's potential protective effects on the cardiovascular system has been demonstrated in a mouse model of deoxycorticosterone acetate and sodium chloride hypertension-induced remodeling of the myocardium, where decreasing systemic levels of FGF21 exacerbated remodeling, and restoration of FGF21 levels ameliorated symptoms.61
PK-PD modeling and simulations based on all QW and Q2W doses (Figure S3C) predicted a potential increase in heart rate of up to 2–3 beats per minute at serum concentrations of AKR-001 corresponding to doses of 70 mg QW and higher. However, another sensitivity analysis based on all data for QW and Q2W doses up to and including 70 mg (i.e., with data from 140 mg dose groups excluded) predicted no effect on heart rate (Figure S3D), implying a potential threshold exposure effect on heart rate occurring between doses of 70 and 140 mg AKR-001. To place a possible increase of 2–3 beats per minute in context, administration of the glucagon-like peptide-1 receptor agonist liraglutide (Victoza) at a dose of 1.8 mg to a type 2 diabetic population is associated with a similar magnitude of increase in heart rate.62 At this dose, liraglutide has been demonstrated to reduce the risk of cardiovascular events.63
Other PD endpoints were modeled in the same manner, including all time points measured following AKR-001 QW and Q2W administration (Figures S3E–S3L). These analyses allow comparison across the overall study for each endpoint of interest, examining in particular the different dose intervals. In general, as with an increasing AKR-001 dose, increasing AKR-001 serum concentration is predicted to improve markers of insulin sensitivity and lipid metabolism. For each endpoint examined where AKR-001 appeared to elicit a positive effect, weekly administration of AKR-001 is predicted to lead to greater maximal and sustained effects than administration every 2 weeks (Figures S3E–S3L). This likely reflects the more stable and sustained serum concentration of AKR-001 following QW versus Q2W dosing.
Overall, AKR-001 demonstrated trends toward improvement in the lipoprotein profile and in markers of insulin sensitivity in this early-phase trial in patients with type 2 diabetes. AKR-001 appeared to have acceptable toleration over the course of the study, with a maximum dose of 70 mg QW selected for future clinical development (ClinicalTrials.gov: NCT03967401). Among existing antidiabetic medications, the most widely used insulin sensitizers are metformin64 and sodium-glucose co-transporter-2 (SGLT2) inhibitors.65,66 Weight gain and perceived safety issues associated with the other class of insulin sensitizers, PPARγ agonists, have markedly reduced their use.67, 68, 69 AKR-001, with promising PD effects and acceptable tolerability at doses up to 70 mg QW, has the potential to expand the therapeutic repertoire for treating patients with metabolic syndrome-related disorders like type 2 diabetes. Notably, AKR-001 did not increase body weight, consistent with several previous FGF21 analogs18,19,25 and in contrast with an FGFR1c-selective analog.70 The superior PK profile of AKR-001 and increased affinity for β-Klotho compared with other known FGF21 analogs may result in sustained agonism of FGF21’s canonical receptors (FGFR1c, FGFR2c, or FGFR3c) and could explain the observed trend toward improvement in glycemic control.
Furthermore, AKR-001’s apparent effects on a broad range of metabolic markers after 4 weeks of administration in individuals with type 2 diabetes support its ongoing clinical development and suggest its potential for treating metabolic diseases. NAFLD, the most prevalent liver disease globally,71 is a spectrum of diseases that encapsulates progression from fatty liver without inflammation to NASH and cirrhosis. NASH is a leading cause of cirrhosis, hepatocellular carcinoma, and liver transplants.72 However, with no current FDA-approved medication for NASH, there is a substantial unmet need for efficacious therapies. The underlying cause of NASH is a surfeit of calories entering the liver, resulting in excessive deposition of fat within hepatocytes, causing lipotoxicity and ultimately leading to steatohepatitis.73,74 In the liver of patients with NAFLD, flux of fatty acid from adipose tissue to the liver contributes approximately 60% of the lipid carbon in hepatocytes. Although de novo lipogenesis by hepatocytes is up to 3-fold higher in patients with NASH compared with BMI-matched controls,75 it accounts for only ∼25% of the lipid carbon in hepatocytes, with dietary fat accounting for the remainder (∼15%).30 Therefore, to maximally reduce liver fat, inhibition of hepatic de novo lipogenesis and suppression of the flux of fatty acids from adipose tissue to the liver are required. As reported here, AKR-001 appeared to reduce post-prandial lipolysis and reduced triglycerides by more than 60%. AKR-001’s pharmacology, which mimics the agonist profile of FGF21 at FGFR1c, FGFR2c, and FGFR3c,29 would also be expected to reduce hepatic de novo lipogenesis and secretion of VLDL from the liver. FGF21 suppresses sterol-regulatory element-binding transcription factor 1 (SREBP1), resulting in lower expression of enzymes associated with de novo lipogenesis and VLDL secretion.13,76 Such a profile supports evaluating the potential of AKR-001 to reduce excessive levels of hepatic fat in NASH patients (ClinicalTrials.gov: NCT03967401). Additionally, the risk of cardiovascular disease is high in subjects with metabolic syndrome, type 2 diabetes, and NASH, the majority of whom present with dyslipidemia.1,72 In this multiple-dose 4-week study with relatively small cohorts, AKR-001 demonstrated trends toward improvements in circulating levels of four risk factors independently causal for cardiovascular disease: non-HDL cholesterol, apoB and apoC3, and triglyceride.39,77,78 If these decreases are corroborated and sustained during long-term treatment in larger studies, then AKR-001 has the potential to lower the risk of major adverse cardiovascular events.
The descriptive PD data presented in this hypothesis-generating study, as well as preclinical studies showing that FGF21 or FGF21 analogs ameliorate diet-induced hepatic steatosis, inflammation, and fibrosis,13,79, 80, 81 support further study of AKR-001 in larger trials in patients with metabolic syndrome-related disorders and, in particular, NASH. AKR-001 is currently under investigation for treatment of histologically confirmed NASH, with the primary endpoint of absolute reduction in liver fat after 12 weeks of treatment. Additional endpoints under investigation include liver histology (NAFLD activity score and fibrosis) and markers of glycemic control and lipid metabolism, testing the hypotheses generated in this phase 1 study.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Fc/linker region | Amgen, Inc. | N/A |
| Rat monoclonal anti-intact C-terminal FGF21 | Amgen, Inc. | N/A |
| Biological Samples | ||
| Human plasma and serum | Clinical Trial | NCT01856881 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fc-FGF21(RGE) | Stanislaus et al.29 | AKR-001 |
| Software and Algorithms | ||
| Prism 8 | GraphPad | https://www.graphpad.com |
| R versions 3.5.1, 3.6.0 | R | http://www.r-project.org |
| emmeans package | R | https://github.com/rvlenth/emmeans |
| lme4 package | R | https://github.com/lme4/lme4 |
| nlme package | R | https://github.com/cran/nlme |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tim Rolph (tim@akerotx.com)
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
There are restrictions to the availability of datasets analyzed in this study due to patient privacy requirements of clinical trials data. Qualified researchers may request specific data and code from the lead contact.
Experimental Model and Subject Details
This clinical trial was conducted at five centers in the United States in full conformance with the ICH E6 guideline for Good Clinical Practice and the principles of the Declaration of Helsinki. The study protocol, amendments, and informed consent received approval by the Institutional Review Board from each study center prior to study initiation. The study population comprised adult females of non-reproductive potential and males aged 18–65 years, with a body mass index of 25–40 kg/m2, and a diagnosis of type 2 diabetes. Eligible participants had an elevated level of HbA1c between 6.5% and 10%, and a fasting C-peptide value of 0.8 ng/mL or greater at screening. Key exclusions were an evidence or history of diabetic complications, significant cardiac disease, uncontrolled hypertension, fasting blood glucose of 270 mg/dL or greater at screening, and hepatic liver enzymes ALT, AST, alkaline phosphatase or total bilirubin levels greater than 1.5 times the upper limit of normal at screening. All anti-diabetic medications were discontinued two weeks before dosing with AKR-001. Subjects remained off anti-diabetic medications throughout the period of treatment and during the follow up period. Lipid-lowering medications were not discontinued during the washout period or trial. No additional lipid lowering medications were administered to any subjects. All participants provided both verbal and written informed consent prior to enrolling in the study.
Method Details
Trial design
The trial was a multicenter, randomized, double-blind, placebo-controlled, ascending multiple-dose study in patients with type 2 diabetes. A total of eight cohorts were randomized to receive AKR-001 or placebo in a ratio of 3:1. AKR-001 (7, 21, 70 or 140 mg) or placebo was administered SC Q2W, or QW for 4 weeks. The trial, NCT01856881, evaluated the safety, pharmacokinetic (PK), and pharmacodynamic (PD) profile of AKR-001 dosed up to 140 mg, based on an unacceptable tolerability profile of a single 210 mg dose in the preceding single ascending dose trial (ClinicalTrials.gov: NCT01492465). The protocol outlined a ninth cohort, with 60 subjects planned to be randomized 1:1 to placebo or AKR-001 70 mg QW for 4 weeks; however, this cohort was not enrolled as the first eight cohorts provided sufficient data relating pharmacokinetic to pharmacodynamic endpoints to inform dose selection and dosing interval for subsequent clinical trials. A summary of the trial design is provided in Figure 1.
Trial procedures
Participants discontinued all antidiabetic medications and received lifestyle counseling from two weeks prior to dosing with AKR-001 until the end of study. Participants were admitted to the research facility on the following days: (day −2 to day 6 [all cohorts]) and after the last dose of AKR-001 (day 14–20 [Q2W cohorts] and day 21–27 [QW cohorts]).
Primary endpoints
The primary endpoints were the incidence of treatment-emergent AEs, safety laboratory analytes, vital signs, electrocardiograms (ECGs) and incidence of ADAs. The incidence of ADAs was measured pre-dose, day 29 and at the end of study (day 43 [Q2W] or 57 [QW]). Samples testing positive for binding antibodies were tested for FGF21-neutralizing antibodies. Blood pressure measurements were taken in a supine position, following at least five minutes in a rested and calm state.
Secondary endpoints
Characterization of PK profile: For cohorts dosed Q2W, serum AKR-001 concentrations were measured pre-dose, 8 hours, day 2, 3, 4, 5, 6, 8, 11, 15 (pre-dose), 16, 17, 18, 19, 20, 22, 29, 36 and 43. Samples were also taken on Day 50 and 57 in the 140 mg Q2W cohort. For cohorts dosed QW, serum AKR-001 concentrations were measured pre-dose, 8 hours, day 2, 3, 4, 5, 6, 8 (pre-dose), 11, 15 (pre-dose), 18, 22 (pre-dose), 23, 24, 25, 26, 27, 29, 36, 43, 50 and 57. Measurements were also taken on day 64 and 71 in the 140 mg QW cohort. Serum AKR-001 concentrations were determined using a validated sandwich ELISA, with capture by a murine monoclonal antibody against human Fc and the linker region, and quantified using a rat monoclonal antibody which detects intact, C-terminal AKR-001. AKR-001 serum concentration–time data was used to determine PK parameters using non-compartmental methods.
Fasting PD parameters, concentration of fasting glucose, insulin, C-peptide, glucagon, total cholesterol, LDL-cholesterol, HDL-cholesterol, triglycerides, free fatty acids and β-hydroxybutyrate were measured following an overnight fast of at least 8 hours. Measurements were carried out on day −1, 2, 4, 6, 8, 11, 15, 16, 18, 20, 22, 29, 36, 43 in the cohorts dosed Q2W, and also on day 50 and 57 in the 140 mg Q2W cohort. Measurements were carried out on day −1, 2, 4, 6, 8, 11, 15, 18, 22, 23, 25, 29, 36, 43, 50 and 57 in the cohorts dosed QW and also on day 64 and 71 in the 140 mg QW cohort.
Body weight was measured during screening, on day −1, 2, 3, 4, 5, 6, 8, 11, 14, 15, 16, 17, 18, 19, 20, 22, 25, 29, 36 and 43 in the cohorts dosed Q2W, and also on day 50 and 57 in the 140 mg Q2W cohort. In the QW cohorts body weight was measured during screening, on day −1, 2, 3, 4, 5, 6, 8, 11, 15, 18, 22, 23, 24, 25, 26, 27, 29, 36, 43, 50 and 57, with measurements also taken on day 64 and 71 in the 140 mg QW cohort.
A mixed meal tolerance test measuring concentration-time profiles and AUC for metabolic parameters was carried out on day −1 (baseline) for all cohorts and day 18 (three days post second dose in cohorts receiving treatment Q2W), or day 25 (three days post fourth dose in cohorts receiving treatment QW). The test was initiated between 7 and 9 am, following an overnight fast, with the meal completed within 30 minutes of start time. The standardized meal was two bottles or cans of Ensure Plus®. Multiple blood samples were collected for measurement of fasting and post-prandial glucose, insulin, C-peptide, free fatty acids, and glucagon: at 10 and 5 minutes pre-meal and 30, 60, 90, 120, 180, and 240 minutes post-prandial.
Exploratory outcome measures
Exploratory biomarkers, including fasting C4, apoB, apoA-1, adiponectin, FGF19 and IGF-1 levels were measured on day −1 (baseline), day 4, 15 (pre-dose), 29 and at the end of study (day 43 or 57). Additional biomarkers, angiopoietin-like 4, apoC-2; apoC-3, and lipoprotein(a), were measured from samples taken from QW dose groups on day −1 (baseline), day 4, 15 (pre-dose), 29 and at the end of study (day 57).
Quantification and Statistical Analysis
A computer-generated randomization schedule was prepared prior to the start of the study. The sample size of cohorts was based on practical considerations and on typical Phase 1 study sizing when safety is the primary endpoint; however, power calculations were performed to determine predictive power. Six subjects per cohort received active, providing a 73% chance of detecting an adverse event with a true incidence of 20%. The sample size was not selected for statistical inference of pharmacodynamic endpoints.
In addition to evaluating tolerability and pharmacokinetics, the trial was intended as a hypothesis-generating study for estimation of PD effect sizes. PD endpoint data are reported as placebo-corrected percentage change from baseline of least square geometric means with 95% confidence interval and without p values. Overall, the analyses presented align with standards typically used for exploratory early-stage clinical trials to indicate trends in effect, rather than standards for pivotal registration studies to indicate replicated statistical significance.
Analysis was performed on the safety analysis set, that is all subjects who received at least one dose of investigational product. Cardiovascular safety endpoints are reported as least-squares mean percentage change from baseline. Descriptive statistics are provided for demographics, baseline characteristics, safety, and PK data. Safety data from the QW and Q2W placebo groups were combined.
Statistical modeling
Assessments presented as least-squares mean and confidence interval were fit on the log scale with a linear mixed effects model and back-transformed to the linear scale for reporting. The models had fixed effects for treatment, time, treatment by time, and a random effect on baseline by subject.
Statistical models were fit using R version 3.5.1 or 3.6.0 and the lme4 library, and least-squares means and confidence intervals were calculated using the emmeans library.
PK–PD Modeling
To help determine the optimal dose level and administration frequency for future clinical evaluation of AKR-001, PK–PD modeling and simulations were performed to understand the predicted relationship of serum drug concentration with cardiovascular and PD endpoints across the whole study duration. The complete dataset for Q2W and QW cohorts including screening and Day −2 time points, when scheduled assessments of PK and the PD endpoint occurred at the same nominal time, was used in the analysis. PK imputation only occurred for predose concentrations and placebo-subject concentrations as zero; no PD imputation occurred in the model datasets. Data for each subject were plotted as a function of serum concentration of AKR-001 versus each of: systolic blood pressure, diastolic blood pressure, heart rate, triglycerides, insulin, glucose, C-peptide, total cholesterol, HDL cholesterol, non-HDL cholesterol, and free fatty acid. The model-averaged (using Akaike information criterion weighting82), asymmetric confidence intervals around the point estimate were determined for the predicted correlation of changes in intra-subject measures. Models included in averaging were all twenty-one combinations of nonlinear mixed effect models for no effect, linear, Emax, and sigmoid Emax with potentially different slope, Emax, and EC50 parameters by dosing frequency and all combinations of normal, power, and exponential residual error; for each model, the random effect was on baseline when drug concentration was zero. Standard goodness-of-fit figures were then plotted to provide a visual predictive check on predicted means compared with observations. All pharmacometric models were fit using R version 3.5.1 and the nlme library.
Additional Resources
The trial is registered on clinicaltrials.gov with the clinical trial registry number NCT01856881: https://clinicaltrials.gov/ct2/show/NCT01856881
Acknowledgments
The authors acknowledge the assistance of the clinical trial personnel and the patients for participating in the trial. We also thank Michael Tracy for biostatistical support, Abraham Anderson for exploratory biomarker analysis, Shawna Smith for safety data collection and reporting, and afPharmative Insights for medical writing assistance. Amgen and Akero Therapeutics provided funding support for the presented study and analysis.
Author Contributions
Conceptualization, L.A. and A.K.; Methodology, L.A. and A.K.; Validation, L.A. and A.K.; Formal Analysis, L.A., A.K., W.S.D., and T.R.; Investigation, L.A. and A.K.; Data Curation, W.S.D.; Resources, L.A. and A.K.; Writing – Original Draft, E.J.T. and T.R.; Writing – Review and Editing, L.A., A.K., W.S.D., E.J.T., and T.R.; Visualization, W.S.D. and E.J.T.; Supervision, T.R.; Project Administration, T.R.
Declaration of Interests
A.K. is a shareholder of Amgen and was an employee of Amgen at the time of this study. L.A. is an employee and shareholder of Amgen. W.S.D. is a consultant to Akero Therapeutics. E.J.T. is a shareholder and employee of Akero Therapeutics. T.R. is a co-founder, shareholder, and employee of Akero Therapeutics and shareholder of Pfizer. Akero Therapeutics has licensed exclusive rights to patents relating to AKR-001’s composition and use.
Published: July 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrm.2020.100057.
Supplemental Information
References
- 1.Targher G., Chonchol M., Pichiri I., Zoppini G. Risk of cardiovascular disease and chronic kidney disease in diabetic patients with non-alcoholic fatty liver disease: just a coincidence? J. Endocrinol. Invest. 2011;34:544–551. doi: 10.3275/7614. [DOI] [PubMed] [Google Scholar]
- 2.Fisher F.M., Maratos-Flier E. Understanding the Physiology of FGF21. Annu. Rev. Physiol. 2016;78:223–241. doi: 10.1146/annurev-physiol-021115-105339. [DOI] [PubMed] [Google Scholar]
- 3.Ogawa Y., Kurosu H., Yamamoto M., Nandi A., Rosenblatt K.P., Goetz R., Eliseenkova A.V., Mohammadi M., Kuro-o M. BetaKlotho is required for metabolic activity of fibroblast growth factor 21. Proc. Natl. Acad. Sci. USA. 2007;104:7432–7437. doi: 10.1073/pnas.0701600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guan D., Zhao L., Chen D., Yu B., Yu J. Regulation of fibroblast growth factor 15/19 and 21 on metabolism: in the fed or fasted state. J. Transl. Med. 2016;14:63–67. doi: 10.1186/s12967-016-0821-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gariani K., Drifte G., Dunn-Siegrist I., Pugin J., Jornayvaz F.R. Increased FGF21 plasma levels in humans with sepsis and SIRS. Endocr. Connect. 2013;2:146–153. doi: 10.1530/EC-13-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barb D., Bril F., Kalavalapalli S., Cusi K. Plasma Fibroblast Growth Factor 21 Is Associated With Severity of Nonalcoholic Steatohepatitis in Patients With Obesity and Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2019;104:3327–3336. doi: 10.1210/jc.2018-02414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dushay J., Chui P.C., Gopalakrishnan G.S., Varela-Rey M., Crawley M., Fisher F.M., Badman M.K., Martinez-Chantar M.L., Maratos-Flier E. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology. 2010;139:456–463. doi: 10.1053/j.gastro.2010.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fisher F.M., Chui P.C., Antonellis P.J., Bina H.A., Kharitonenkov A., Flier J.S., Maratos-Flier E. Obesity is a fibroblast growth factor 21 (FGF21)-resistant state. Diabetes. 2010;59:2781–2789. doi: 10.2337/db10-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dushay J.R., Toschi E., Mitten E.K., Fisher F.M., Herman M.A., Maratos-Flier E. Fructose ingestion acutely stimulates circulating FGF21 levels in humans. Mol. Metab. 2014;4:51–57. doi: 10.1016/j.molmet.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Søberg S., Andersen E.S., Dalsgaard N.B., Jarlhelt I., Hansen N.L., Hoffmann N., Vilsbøll T., Chenchar A., Jensen M., Grevengoed T.J. FGF21, a liver hormone that inhibits alcohol intake in mice, increases in human circulation after acute alcohol ingestion and sustained binge drinking at Oktoberfest. Mol. Metab. 2018;11:96–103. doi: 10.1016/j.molmet.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maida A., Zota A., Sjøberg K.A., Schumacher J., Sijmonsma T.P., Pfenninger A., Christensen M.M., Gantert T., Fuhrmeister J., Rothermel U. A liver stress-endocrine nexus promotes metabolic integrity during dietary protein dilution. J. Clin. Invest. 2016;126:3263–3278. doi: 10.1172/JCI85946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holland W.L., Adams A.C., Brozinick J.T., Bui H.H., Miyauchi Y., Kusminski C.M., Bauer S.M., Wade M., Singhal E., Cheng C.C. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab. 2013;17:790–797. doi: 10.1016/j.cmet.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu J., Lloyd D.J., Hale C., Stanislaus S., Chen M., Sivits G., Vonderfecht S., Hecht R., Li Y.S., Lindberg R.A. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2009;58:250–259. doi: 10.2337/db08-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu J., Stanislaus S., Chinookoswong N., Lau Y.Y., Hager T., Patel J., Ge H., Weiszmann J., Lu S.C., Graham M. Acute glucose-lowering and insulin-sensitizing action of FGF21 in insulin-resistant mouse models--association with liver and adipose tissue effects. Am. J. Physiol. Endocrinol. Metab. 2009;297:E1105–E1114. doi: 10.1152/ajpendo.00348.2009. [DOI] [PubMed] [Google Scholar]
- 15.Kharitonenkov A., Wroblewski V.J., Koester A., Chen Y.F., Clutinger C.K., Tigno X.T., Hansen B.C., Shanafelt A.B., Etgen G.J. The metabolic state of diabetic monkeys is regulated by fibroblast growth factor-21. Endocrinology. 2007;148:774–781. doi: 10.1210/en.2006-1168. [DOI] [PubMed] [Google Scholar]
- 16.Véniant M.M., Komorowski R., Chen P., Stanislaus S., Winters K., Hager T., Zhou L., Wada R., Hecht R., Xu J. Long-acting FGF21 has enhanced efficacy in diet-induced obese mice and in obese rhesus monkeys. Endocrinology. 2012;153:4192–4203. doi: 10.1210/en.2012-1211. [DOI] [PubMed] [Google Scholar]
- 17.Adams A.C., Halstead C.A., Hansen B.C., Irizarry A.R., Martin J.A., Myers S.R., Reynolds V.L., Smith H.W., Wroblewski V.J., Kharitonenkov A. LY2405319, an Engineered FGF21 Variant, Improves the Metabolic Status of Diabetic Monkeys. PLoS ONE. 2013;8:e65763. doi: 10.1371/journal.pone.0065763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaich G., Chien J.Y., Fu H., Glass L.C., Deeg M.A., Holland W.L., Kharitonenkov A., Bumol T., Schilske H.K., Moller D.E. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013;18:333–340. doi: 10.1016/j.cmet.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Talukdar S., Zhou Y., Li D., Rossulek M., Dong J., Somayaji V., Weng Y., Clark R., Lanba A., Owen B.M. A Long-Acting FGF21 Molecule, PF-05231023, Decreases Body Weight and Improves Lipid Profile in Non-human Primates and Type 2 Diabetic Subjects. Cell Metab. 2016;23:427–440. doi: 10.1016/j.cmet.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 20.Kim A.M., Somayaji V.R., Dong J.Q., Rolph T.P., Weng Y., Chabot J.R., Gropp K.E., Talukdar S., Calle R.A. Once-weekly administration of a long-acting fibroblast growth factor 21 analogue modulates lipids, bone turnover markers, blood pressure and body weight differently in obese people with hypertriglyceridaemia and in non-human primates. Diabetes Obes. Metab. 2017;19:1762–1772. doi: 10.1111/dom.13023. [DOI] [PubMed] [Google Scholar]
- 21.Charles E.D., Neuschwander-Tetri B.A., Pablo Frias J., Kundu S., Luo Y., Tirucherai G.S., Christian R. Pegbelfermin (BMS-986036), PEGylated FGF21, in Patients with Obesity and Type 2 Diabetes: Results from a Randomized Phase 2 Study. Obesity (Silver Spring) 2019;27:41–49. doi: 10.1002/oby.22344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cujec T.P. United States Patent and Trademark Office; 2011. Modified FGF-21 polypeptides and their uses. US 8,012,931 B2. [Google Scholar]
- 23.Tirucherai G.S., Mora J., Revankar R., Charles E.D. Pharmacokinetics and Safety of Pegbelfermin (BMS-986036) Administered in the Abdomen and Upper Arm to Normal, Overweight, and Obese Healthy Participants. J. Hepatol. 2019;70:E798. [Google Scholar]
- 24.Talukdar S., Owen B.M., Song P., Hernandez G., Zhang Y., Zhou Y., Scott W.T., Paratala B., Turner T., Smith A. FGF21 Regulates Sweet and Alcohol Preference. Cell Metab. 2016;23:344–349. doi: 10.1016/j.cmet.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanyal A., Charles E.D., Neuschwander-Tetri B.A., Loomba R., Harrison S.A., Abdelmalek M.F., Lawitz E.J., Halegoua-DeMarzio D., Kundu S., Noviello S. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet. 2019;392:2705–2717. doi: 10.1016/S0140-6736(18)31785-9. [DOI] [PubMed] [Google Scholar]
- 26.Wei W., Dutchak P.A., Wang X., Ding X., Wang X., Bookout A.L., Goetz R., Mohammadi M., Gerard R.D., Dechow P.C. Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator-activated receptor γ. Proc. Natl. Acad. Sci. USA. 2012;109:3143–3148. doi: 10.1073/pnas.1200797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X., Stanislaus S., Asuncion F., Niu Q.T., Chinookoswong N., Villasenor K., Wang J., Wong P., Boyce R., Dwyer D. FGF21 Is Not a Major Mediator for Bone Homeostasis or Metabolic Actions of PPARα and PPARγ Agonists. J. Bone Miner. Res. 2017;32:834–845. doi: 10.1002/jbmr.2936. [DOI] [PubMed] [Google Scholar]
- 28.Frayling T.M., Beaumont R.N., Jones S.E., Yaghootkar H., Tuke M.A., Ruth K.S., Casanova F., West B., Locke J., Sharp S. A Common Allele in FGF21 Associated with Sugar Intake Is Associated with Body Shape, Lower Total Body-Fat Percentage, and Higher Blood Pressure. Cell Rep. 2018;23:327–336. doi: 10.1016/j.celrep.2018.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stanislaus S., Hecht R., Yie J., Hager T., Hall M., Spahr C., Wang W., Weiszmann J., Li Y., Deng L. A Novel Fc-FGF21 With Improved Resistance to Proteolysis, Increased Affinity Toward β-Klotho, and Enhanced Efficacy in Mice and Cynomolgus Monkeys. Endocrinology. 2017;158:1314–1327. doi: 10.1210/en.2016-1917. [DOI] [PubMed] [Google Scholar]
- 30.Donnelly K.L., Smith C.I., Schwarzenberg S.J., Jessurun J., Boldt M.D., Parks E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Charles E.D. A phase 1 study of BMS-986036 (pegylated FGF21) in healthy obese subjects. Hepatology. 2016;64:546A. [Google Scholar]
- 32.Lay A.J., Zhang H.E., McCaughan G.W., Gorrell M.D. Fibroblast activation protein in liver fibrosis. Front. Biosci. 2019;24:1–17. doi: 10.2741/4706. [DOI] [PubMed] [Google Scholar]
- 33.Ahrén B. Glucagon secretion in relation to insulin sensitivity in healthy subjects. Diabetologia. 2006;49:117–122. doi: 10.1007/s00125-005-0056-8. [DOI] [PubMed] [Google Scholar]
- 34.Melanson K.J., Greenberg A.S., Ludwig D.S., Saltzman E., Dallal G.E., Roberts S.B. Blood glucose and hormonal responses to small and large meals in healthy young and older women. J. Gerontol. A Biol. Sci. Med. Sci. 1998;53:B299–B305. doi: 10.1093/gerona/53a.4.b299. [DOI] [PubMed] [Google Scholar]
- 35.Byun S., Seok S., Kim Y.C., Zhang Y., Yau P., Iwamori N., Xu H.E., Ma J., Kemper B., Kemper J.K. Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat. Commun. 2020;11:807. doi: 10.1038/s41467-020-14384-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Longuet C., Sinclair E.M., Maida A., Baggio L.L., Maziarz M., Charron M.J., Drucker D.J. The glucagon receptor is required for the adaptive metabolic response to fasting. Cell Metab. 2008;8:359–371. doi: 10.1016/j.cmet.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hotta Y., Nakamura H., Konishi M., Murata Y., Takagi H., Matsumura S., Inoue K., Fushiki T., Itoh N. Fibroblast growth factor 21 regulates lipolysis in white adipose tissue but is not required for ketogenesis and triglyceride clearance in liver. Endocrinology. 2009;150:4625–4633. doi: 10.1210/en.2009-0119. [DOI] [PubMed] [Google Scholar]
- 38.Arner P., Pettersson A., Mitchell P.J., Dunbar J.D., Kharitonenkov A., Rydén M. FGF21 attenuates lipolysis in human adipocytes - a possible link to improved insulin sensitivity. FEBS Lett. 2008;582:1725–1730. doi: 10.1016/j.febslet.2008.04.038. [DOI] [PubMed] [Google Scholar]
- 39.Sniderman A.D., Islam S., McQueen M., Pencina M., Furberg C.D., Thanassoulis G., Yusuf S. Age and Cardiovascular Risk Attributable to Apolipoprotein B, Low-Density Lipoprotein Cholesterol or Non-High-Density Lipoprotein Cholesterol. J. Am. Heart Assoc. 2016;5:11. doi: 10.1161/JAHA.116.003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bayly G.R. Clinical Biochemistry. Elsevier; 2014. Lipids and disorders of lipoprotein metabolism; pp. 702–736. [Google Scholar]
- 41.Jiang J., Wang Y., Ling Y., Kayoumu A., Liu G., Gao X. A novel APOC2 gene mutation identified in a Chinese patient with severe hypertriglyceridemia and recurrent pancreatitis. Lipids Health Dis. 2016;15:12. doi: 10.1186/s12944-015-0171-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Achari A.E., Jain S.K. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017;18:1321. doi: 10.3390/ijms18061321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shafiei M.S., Shetty S., Scherer P.E., Rockey D.C. Adiponectin regulation of stellate cell activation via PPARγ-dependent and -independent mechanisms. Am. J. Pathol. 2011;178:2690–2699. doi: 10.1016/j.ajpath.2011.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar P., Raeman R., Chopyk D.M., Smith T., Verma K., Liu Y., Anania F.A. Adiponectin inhibits hepatic stellate cell activation by targeting the PTEN/AKT pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2018;1864:3537–3545. doi: 10.1016/j.bbadis.2018.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin Z., Tian H., Lam K.S., Lin S., Hoo R.C., Konishi M., Itoh N., Wang Y., Bornstein S.R., Xu A., Li X. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab. 2013;17:779–789. doi: 10.1016/j.cmet.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 46.Adams A.C., Yang C., Coskun T., Cheng C.C., Gimeno R.E., Luo Y., Kharitonenkov A. The breadth of FGF21's metabolic actions are governed by FGFR1 in adipose tissue. Mol. Metab. 2012;2:31–37. doi: 10.1016/j.molmet.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen M.M., Hale C., Stanislaus S., Xu J., Véniant M.M. FGF21 acts as a negative regulator of bile acid synthesis. J. Endocrinol. 2018;237:139–152. doi: 10.1530/JOE-17-0727. [DOI] [PubMed] [Google Scholar]
- 48.Harrison S.A., Rinella M.E., Abdelmalek M.F., Trotter J.F., Paredes A.H., Arnold H.L., Kugelmas M., Bashir M.R., Jaros M.J., Ling L. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2018;391:1174–1185. doi: 10.1016/S0140-6736(18)30474-4. [DOI] [PubMed] [Google Scholar]
- 49.Neuschwander-Tetri B.A., Loomba R., Sanyal A.J., Lavine J.E., Van Natta M.L., Abdelmalek M.F., Chalasani N., Dasarathy S., Diehl A.M., Hameed B., NASH Clinical Research Network Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956–965. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martinez-Augustin O., Sanchez de Medina F. Intestinal bile acid physiology and pathophysiology. World J. Gastroenterol. 2008;14:5630–5640. doi: 10.3748/wjg.14.5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Orrù S., Nigro E., Mandola A., Alfieri A., Buono P., Daniele A., Mancini A., Imperlini E. A Functional Interplay between IGF-1 and Adiponectin. Int. J. Mol. Sci. 2017;18:2145. doi: 10.3390/ijms18102145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Inagaki T., Lin V.Y., Goetz R., Mohammadi M., Mangelsdorf D.J., Kliewer S.A. Inhibition of growth hormone signaling by the fasting-induced hormone FGF21. Cell Metab. 2008;8:77–83. doi: 10.1016/j.cmet.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schlein C., Talukdar S., Heine M., Fischer A.W., Krott L.M., Nilsson S.K., Brenner M.B., Heeren J., Scheja L. FGF21 Lowers Plasma Triglycerides by Accelerating Lipoprotein Catabolism in White and Brown Adipose Tissues. Cell Metab. 2016;23:441–453. doi: 10.1016/j.cmet.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 54.Barrows B.R., Parks E.J. Contributions of different fatty acid sources to very low-density lipoprotein-triacylglycerol in the fasted and fed states. J. Clin. Endocrinol. Metab. 2006;91:1446–1452. doi: 10.1210/jc.2005-1709. [DOI] [PubMed] [Google Scholar]
- 55.Kharitonenkov A., Beals J.M., Micanovic R., Strifler B.A., Rathnachalam R., Wroblewski V.J., Li S., Koester A., Ford A.M., Coskun T. Rational design of a fibroblast growth factor 21-based clinical candidate, LY2405319. PLoS ONE. 2013;8:e58575. doi: 10.1371/journal.pone.0058575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Madsbad S. LY2605541--a preferential hepato-specific insulin analogue. Diabetes. 2014;63:390–392. doi: 10.2337/db13-1646. [DOI] [PubMed] [Google Scholar]
- 57.US Food and Drug Administration. FDA label for Phenergan. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/022341s023lbl.pdf.
- 58.He J.-L., Zhao M., Xia J.J., Guan J., Liu Y., Wang L.Q., Song D.X., Qu M.Y., Zuo M., Wen X. FGF21 ameliorates the neurocontrol of blood pressure in the high fructose-drinking rats. Sci. Rep. 2016;6:29582. doi: 10.1038/srep29582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pan X., Shao Y., Wu F., Wang Y., Xiong R., Zheng J., Tian H., Wang B., Wang Y., Zhang Y. FGF21 Prevents Angiotensin II-Induced Hypertension and Vascular Dysfunction by Activation of ACE2/Angiotensin-(1-7) Axis in Mice. Cell Metab. 2018;27:1323–1337.e5. doi: 10.1016/j.cmet.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 60.Shi Y., Wang S., Peng H., Lv Y., Li W., Cheng S., Liu J. Fibroblast Growth Factor 21 Attenuates Vascular Calcification by Alleviating Endoplasmic Reticulum Stress Mediated Apoptosis in Rats. Int. J. Biol. Sci. 2019;15:138–147. doi: 10.7150/ijbs.28873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ruan C.-C., Kong L.R., Chen X.H., Ma Y., Pan X.X., Zhang Z.B., Gao P.J. A2A Receptor Activation Attenuates Hypertensive Cardiac Remodeling via Promoting Brown Adipose Tissue-Derived FGF21. Cell Metab. 2018;28:476–489.e5. doi: 10.1016/j.cmet.2018.06.013. [DOI] [PubMed] [Google Scholar]
- 62.US Food and Drug Administration. FDA label for Victoza. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/022341s023lbl.pdf.
- 63.Marso S.P., Daniels G.H., Brown-Frandsen K., Kristensen P., Mann J.F., Nauck M.A., Nissen S.E., Pocock S., Poulter N.R., Ravn L.S., LEADER Steering Committee. LEADER Trial Investigators Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016;375:311–322. doi: 10.1056/NEJMoa1603827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wilkinson S., Douglas I., Stirnadel-Farrant H., Fogarty D., Pokrajac A., Smeeth L., Tomlinson L. Changing use of antidiabetic drugs in the UK: trends in prescribing 2000-2017. BMJ Open. 2018;8:e022768. doi: 10.1136/bmjopen-2018-022768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Merovci A., Solis-Herrera C., Daniele G., Eldor R., Fiorentino T.V., Tripathy D., Xiong J., Perez Z., Norton L., Abdul-Ghani M.A., DeFronzo R.A. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J. Clin. Invest. 2014;124:509–514. doi: 10.1172/JCI70704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.American Diabetes Association Standards of Medical Care in Diabetes-2019 Abridged for Primary Care Providers. Clin. Diabetes. 2019;37:11–34. doi: 10.2337/cd18-0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bortolini M., Wright M.B., Bopst M., Balas B. Examining the safety of PPAR agonists - current trends and future prospects. Expert Opin. Drug Saf. 2013;12:65–79. doi: 10.1517/14740338.2013.741585. [DOI] [PubMed] [Google Scholar]
- 68.Nesto R.W., Bell D., Bonow R.O., Fonseca V., Grundy S.M., Horton E.S., Le Winter M., Porte D., Semenkovich C.F., Smith S., American Heart Association. American Diabetes Association Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. October 7, 2003. Circulation. 2003;108:2941–2948. doi: 10.1161/01.CIR.0000103683.99399.7E. [DOI] [PubMed] [Google Scholar]
- 69.Lee W.-S., Kim J. Peroxisome Proliferator-Activated Receptors and the Heart: Lessons from the Past and Future Directions. PPAR Res. 2015;2015:271983. doi: 10.1155/2015/271983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.DePaoli A., Phung V., Bashir M.R., Morrow L., Beysen C., Yan A., Ling L., Baxter B., Luskey K.L., Olefsky J.M. NGM313, a Novel Activator of b-Klotho/FGFR1c, Improves Insulin Resistance and Reduces Hepatic Fat in Obese, Nondiabetic Subjects. Diabetes. 2019;68:140-LB. [Google Scholar]
- 71.Younossi Z., Anstee Q.M., Marietti M., Hardy T., Henry L., Eslam M., George J., Bugianesi E. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018;15:11–20. doi: 10.1038/nrgastro.2017.109. [DOI] [PubMed] [Google Scholar]
- 72.Liu A., Galoosian A., Kaswala D., Li A.A., Gadiparthi C., Cholankeril G., Kim D., Ahmed A. Nonalcoholic Fatty Liver Disease: Epidemiology, Liver Transplantation Trends and Outcomes, and Risk of Recurrent Disease in the Graft. J. Clin. Transl. Hepatol. 2018;6:420–424. doi: 10.14218/JCTH.2018.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ipsen D.H., Lykkesfeldt J., Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018;75:3313–3327. doi: 10.1007/s00018-018-2860-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Diehl A.M., Day C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017;377:2063–2072. doi: 10.1056/NEJMra1503519. [DOI] [PubMed] [Google Scholar]
- 75.Lambert J.E., Ramos-Roman M.A., Browning J.D., Parks E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146:726–735. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Okazaki H., Goldstein J.L., Brown M.S., Liang G. LXR-SREBP-1c-phospholipid transfer protein axis controls very low density lipoprotein (VLDL) particle size. J. Biol. Chem. 2010;285:6801–6810. doi: 10.1074/jbc.M109.079459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bowman L., Hopewell J.C., Chen F., Wallendszus K., Stevens W., Collins R., Wiviott S.D., Cannon C.P., Braunwald E., Sammons E., Landray M.J., HPS3/TIMI55–REVEAL Collaborative Group Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N. Engl. J. Med. 2017;377:1217–1227. doi: 10.1056/NEJMoa1706444. [DOI] [PubMed] [Google Scholar]
- 78.Crosby J., Peloso G.M., Auer P.L., Crosslin D.R., Stitziel N.O., Lange L.A., Lu Y., Tang Z.Z., Zhang H., Hindy G., TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N. Engl. J. Med. 2014;371:22–31. doi: 10.1056/NEJMoa1307095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fisher F.M., Chui P.C., Nasser I.A., Popov Y., Cunniff J.C., Lundasen T., Kharitonenkov A., Schuppan D., Flier J.S., Maratos-Flier E. Fibroblast growth factor 21 limits lipotoxicity by promoting hepatic fatty acid activation in mice on methionine and choline-deficient diets. Gastroenterology. 2014;147 doi: 10.1053/j.gastro.2014.07.044. 1073–83.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee J.H., Kang Y.E., Chang J.Y., Park K.C., Kim H.W., Kim J.T., Kim H.J., Yi H.S., Shong M., Chung H.K., Kim K.S. An engineered FGF21 variant, LY2405319, can prevent non-alcoholic steatohepatitis by enhancing hepatic mitochondrial function. Am. J. Transl. Res. 2016;8:4750–4763. [PMC free article] [PubMed] [Google Scholar]
- 81.Jimenez V., Jambrina C., Casana E., Sacristan V., Muñoz S., Darriba S., Rodó J., Mallol C., Garcia M., León X. FGF21 gene therapy as treatment for obesity and insulin resistance. EMBO Mol. Med. 2018;10:e8791. doi: 10.15252/emmm.201708791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wagenmakers E.-J., Farrell S. AIC model selection using Akaike weights. Psychon. Bull. Rev. 2004;11:192–196. doi: 10.3758/bf03206482. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
There are restrictions to the availability of datasets analyzed in this study due to patient privacy requirements of clinical trials data. Qualified researchers may request specific data and code from the lead contact.