

Summary
Accumulation of CD103+CD8+ resident memory T (TRM) cells in human lung tumors has been associated with a favorable prognosis. However, the contribution of TRM to anti-tumor immunity and to the response to immune checkpoint blockade has not been clearly established. Using quantitative multiplex immunofluorescence on cohorts of non-small cell lung cancer patients treated with anti-PD-(L)1, we show that an increased density of CD103+CD8+ lymphocytes in immunotherapy-naive tumors is associated with greatly improved outcomes. The density of CD103+CD8+ cells increases during immunotherapy in most responder, but not in non-responder, patients. CD103+CD8+ cells co-express CD49a and CD69 and display a molecular profile characterized by the expression of PD-1 and CD39. CD103+CD8+ tumor TRM, but not CD103−CD8+ tumor-infiltrating counterparts, express Aiolos, phosphorylated STAT-3, and IL-17; demonstrate enhanced proliferation and cytotoxicity toward autologous cancer cells; and frequently display oligoclonal expansion of TCR-β clonotypes. These results explain why CD103+CD8+ TRM are associated with better outcomes in anti-PD-(L)1-treated patients.
Keywords: CD8 TRM cells; CTL; Tc17; lung cancer; anti-PD-1 immunotherapy; tumor-infiltrating lymphocytes; ICB response biomarkers; Aiolos, AhR, and T-bet transcription factors; CD103 integrin; TCR repertoire
Graphical Abstract
Highlights
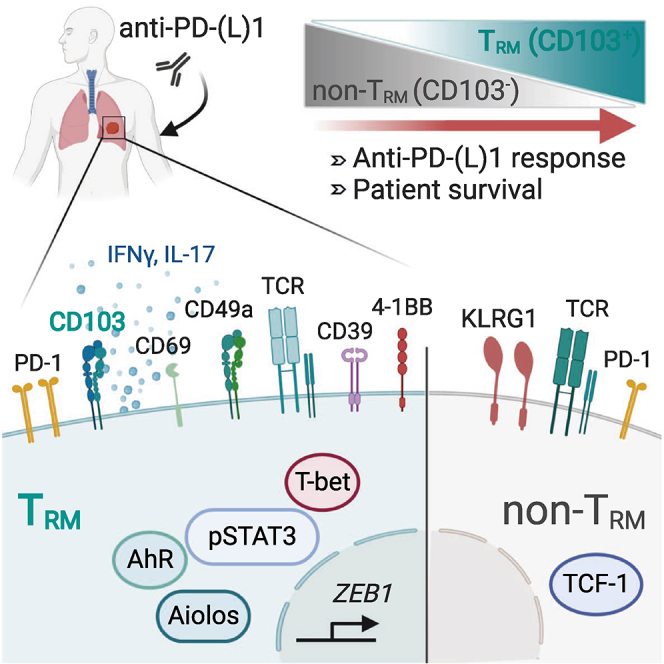
A high density of CD103+CD8+ cells in tumors correlates with response to anti-PD-(L)1
The density of CD103+CD8+ cells increases after anti-PD-1 in most responder patients
CD103+CD8+ TRM cells are enriched with tumor-specific T cells
A subset of CD103+CD8+ TRM cells display a Tc17 differentiation program
Using retrospective cohorts of anti-PD-(L)1-treated NSCLC, Corgnac et al. show that the density of CD103+CD8+ cells in tumors is associated with better progression-free survival. CD103+CD8+ tumor-infiltrating lymphocytes are tumor-specific resident memory T cells (TRM) enriched with a subset displaying a unique Tc1/Tc17 differentiation program that differs from CD103−CD8+ T cells (non-TRM).
Introduction
Effective cancer immunotherapy requires the generation of persistent tumor-reactive cytotoxic T lymphocytes (CTLs) that continuously eliminate primary malignant cells and micro-metastases. Immune checkpoint blockade (ICB), in particular via anti-programmed cell death-1 (PD-1) and anti-PD-L1 neutralizing monoclonal antibodies (mAbs), has led to improved outcomes in non-small cell lung cancer (NSCLC) patients by blocking the interaction of PD-1 inhibitory receptor on tumor-specific T cells with its ligand PD-L1 on target cells, thereby reactivating their anti-tumor effector functions. ICB, as a single immunotherapy agent, has demonstrated responses in up to 20% of patients with advanced NSCLC who failed platinum-based chemotherapy. In this context, the clinical benefits of anti-PD-(L)1 therapy require that tumors be infiltrated by CD8+ T lymphocytes expressing high levels of PD-1 and mediating tumor mutation-derived neoantigen-specific responses.1,2 However, little is known of the nature of these CD8+ T lymphocytes, whether they belong to the memory pool of T cells, and whether they patrol through blood or are confined to tumor tissues, where they are permanently retained. Recently, a lineage of CD8+ memory T cells, called resident memory T (TRM) cells, was localized in several human solid tumors, including NSCLC, and was associated with a favorable prognosis.3, 4, 5, 6, 7, 8
TRM cells were initially identified in infectious diseases, where they were located at sites of pathogen entry so as to trigger optimized protection against viral re-infections.9 They are distinct from central memory T (TCM) and effector memory T (TEM) cells due to their non-recirculating behavior and restricted localization in non-lymphoid peripheral tissues. This memory T cell subset lacks molecules such as S1pr1, Klf2, CCR7, and CD62L, enabling egress from tissue and migration to lymph nodes, and has a unique surface phenotype characterized by the expression of the CD69 activation marker and CD103 (αE(CD103) β7) and CD49a (α1(CD49a) β1) integrins, which likely participate in their residency feature by binding to E-cadherin and type IV collagen on epithelial and endothelial cells, respectively.10,11 TRM cells also upregulate lymphocyte function-associated antigen-1 (LFA-1 or αL(CD11a) β2) integrin, which sustains their residence in liver tissue by binding to intercellular adhesion molecule-1 (ICAM-1) on endothelial cells.12 Formation and persistence of antiviral TRM are regulated by a specific repertoire of transcription factors, including Runx3, Blimp-1, Hobit (homolog of Blimp-1 in T cells), and Notch, and involve a specific set of cytokines, including transforming growth factor β (TGF-β), interleukin-15 (IL-15), type I interferon (IFN), and IL-12.13, 14, 15 TRM have been shown to produce pro-inflammatory cytokines such as IFN-γ and tumor necrosis factor (TNF) and cytotoxic serine protease granzyme B, which participate in recruiting other immune cells at the sites of infection and mediating cytotoxic function, respectively. With regard to cancer diseases, much less is known about the generation and maintenance of TRM at the tumor site and their role in anti-tumor immune responses. Thus far, the phenotypic and functional features of human tumor TRM cells and their contribution to T cell immunity to cancer and responses to ICB are poorly understood. In this report, we show that human lung tumor CD8+ TRM cells are a homogeneous CD103+CD49+CD69+ population characterized by the expression of T-bet, phosphorylated (p)STAT-3, and Aiolos transcription factors, and that a subset of them produces IFN-γ and IL-17, specific to Tc17 cells. NSCLC CD8+ TRM cells also produce granzyme B, form stable conjugates with autologous tumor cells, and mediate specific cytotoxic activity toward the cognate target. This tumor TRM subset, but not its CD103−CD8+ tumor-infiltrating counterpart, proliferate and clonally expand in some tumors, suggesting that it is enriched in tumor-specific CD8+ T cells. Remarkably, an enhanced CD103+CD8+ subset within the tumor microenvironment (TME) is associated with better progression-free survival (PFS) of anti-PD-(L)1-treated NSCLC patients. This subset is increased upon anti-PD-1 administration in tumors from most responder patients, but not in tumors from non-responder patients, and represents the major CD8+ tumor-infiltrating lymphocytes (TILs) population in intraepithelial tumor regions. Thus, CD103+CD8+ TRM cells could be considered potential biomarkers when selecting patients who may benefit from ICB.
Results
Accumulation of CD103+CD8+ Cells in Tumors Is Associated with Better Outcomes in Anti-PD-(L)1-Treated NSCLC Patients
To determine the contribution of TRM cells to the response to ICB, we established a retrospective tumor cohort (discovery cohort) of 111 patients with advanced NSCLC treated with single-agent anti-PD-(L)1 mAbs (Table S1). Tumor sections from formalin-fixed, paraffin-embedded (FFPE) samples were stained simultaneously with anti-CD8, anti-CD103, and anti-cytokeratin mAbs, and the density and distribution of CD103+CD8+ TIL and CD103−CD8+ TIL in epithelial and stromal regions were assessed using InForm software (Figure 1A). Among the 111 analyzed tumor samples, conclusive staining for the quantification of CD103+CD8+ TIL was obtained for 86 patients. Multiplex fluorescent immunohistochemistry (IHC) revealed that the density of the CD103+CD8+ TIL subset varied between tumors, ranging from 0 to 1,320 cells/mm2, with a median of 103 cells/mm2. Most CD103+CD8+ TILs were located within the stroma, with a median density of 153 cells/mm2. In epithelial tumor regions, there were fewer lymphocytes than in the stroma (median density of 68 cells/mm2); however, 76% of these CD8+ TILs were CD103+ (median density of 45 cells/mm2), whereas only 48% in the stroma (Table S1). Increased intraepithelial lymphocyte infiltration was particularly observed in tumors with a high density of CD103+CD8+ cells in the stromal compartment (Figure S1A).
Figure 1.

Increased CD103+CD8+ TILs Are Associated with Improved Outcomes of Anti-PD-1-Treated NSCLC
(A) Fluorescent IHC image of CD8, CD103, cytokeratin, and DAPI staining in lung tumor section (top). Digital markup image shows epithelial and stromal zones of the tumor section defined by cytokeratin staining (center). Digital markup image shows CD103+CD8+ (yellow), CD103−CD8+ (green), CD103+CD8− (red), and cytokeratin+ (pink) cells in lung tumors (bottom). Scale bar, 2 cm.
(B) Kaplan-Meier curve shows iPFS of anti-PD-1-treated patients with tumors harboring high (>252/mm2) (n = 19) or low (<252/mm2) (n = 65) densities of CD103+CD8+ cells (n = 84).
(C) Density of total CD103+CD8+ cells in tumors depending on iORRs of non-responders (NR) (n = 65) and responders (R) (n = 17) patients to PD-1 blockade.
(D) Kaplan-Meier curve shows iPFS of anti-PD-1-treated patients with tumor epithelial regions harboring a high (>48/mm2) (n = 40) or low (<48/mm2) (n = 44) density of CD103+CD8+ cells (n = 84).
(E) Density of CD103+CD8+ cells in epithelial tumor regions depending on iORRs of NR (n = 65) and R (n = 17) patients to anti-PD-1.
(F) Kaplan-Meier curve shows iPFS of PD-1 blockade-treated patients with epithelial regions of tumors harboring a high (>17/mm2) (n = 43) or low (<17/mm2) (n = 41) density of CD103−CD8+ cells.
(G) Density of CD103−CD8+ cells in epithelial tumor regions depending on iORRs of NR (n = 65) and R patients (n = 17) to PD-1 blockade.
(H) Kaplan-Meier curve shows iPFS of anti-PD-1-treated patients with tumors harboring a satisfactory (CD103+CD8+high/CD103−CD8+low, red, n = 12), intermediate (CD103+CD8+high or CD103−CD8+low, blue, n = 57), or poor (CD103+CD8+low/CD103−CD8+high, black, n = 15) CD103/CD8 score.
(I) Percentages of CD103+ cells among CD8+ TILs in epithelial tumor areas of anti-PD-1-treated patients undergoing a long-response (PFS > 6 months and OS > 12 months; n = 13) or a fast-progression (OS < 12 weeks; n = 17). ∗∗p < 0.01.
(J) Fluorescent IHC images show CD103+CD8+ (orange), CD103−CD8+ (green), cytokeratin+ (pink), and DAPI+ (blue) cells in tumors from responder and non-responder patients before and after administration of anti-PD-1. Scale bar, 1 cm. Right, density of CD103+CD8+ cells in epithelial tumor regions of tumors before and after the administration of anti-PD-(L)1 in responder (n = 5) or non-responder (n = 4) patients.
Each symbol represents an individual patient; horizontal lines correspond to mean ± standard deviation (SD) (C, E, and G). p value was determined by log-rank test (B, D, F, and H), Chi-square test (C, E, and G) or unpaired t test (I).
See also Figures S1 and S2, and Tables S1–S6.
We then correlated the survival of patients from the date of the first immunotherapy administration with the score of tumor infiltration by CD8+ TILs. In this cohort, the objective response rate (ORR) was 20% (n = 17/86), corresponding to responders. As expected, patients with strong CD8 cell (CD8high) tumor infiltration had increased survival compared to patients with CD8low tumors (hazard ratio [HR] = 0.40, 95% confidence interval [CI] 0.20–0.77, p = 0.006), and median immunotherapy progression-free survival (iPFS) of 9.92 months (95% CI 2.73–not reached) (Figure S1B; Table S2). The density of CD8+ TIL was also higher in responder patients than in non-responders (Figure S1C). Remarkably, when patients were stratified by their tumor CD103+CD8+ cell infiltration, iPFS dramatically increased in the patient population harboring tumors that were strongly infiltrated by CD103+CD8+ cells (CD103+CD8+high), with median iPFS reaching 30 months (95% CI 2.73–not-reached) (Figure 1B and Table S2). The density of total CD103+CD8+ TILs also correlated with the ORR (Figure 1C). In contrast, patients with tumors slightly infiltrated by CD103+CD8+ cells had only a 2.3-month (95% CI 1.68–5.03) median iPFS. Multivariate analyses showed that tumor infiltration by CD103+CD8+ cells was an independent factor in satisfactory prognosis, based on iPFS, with a HR of 0.39 (95% CI 0.18–0.85, p = 0.01) (Table S3). More important, median iPFS in the CD103+CD8+high population was 20 months longer than in the total CD8+high population (Table S2). As expected, PD-L1 expression by tumor cells correlates with a better PFS (Table S1). These results highlight the strong benefit of an enhanced CD103+CD8+ cell subset in tumors compared to the heterogeneous CD8+ subset in the response to anti-PD-(L)1.
Correlation between Intraepithelial CD103+CD8+ TILs, Tumor E-Cadherin, and ICAM-1 Expression and Response to ICB
Next, we examined CD8+ TILs in epithelial tumor regions where immune cells are in close contact with tumor cells. Patients with high intraepithelial CD103+CD8+ TILs responded more effectively to anti-PD-(L)1 than patients with low intraepithelial CD103+CD8+ infiltration (HR = 0.52, 95% CI 0.31–0.88, p = 0.01) (Figure 1D). The density of intraepithelial CD103+CD8+ TILs also correlated with the ORR (Figure 1E). Similar results were observed when analyzing total CD8+ cell infiltrate in the epithelial compartment, predominantly composed of CD103+CD8+ TILs (Figures S1D and S1E). Correlation between an enhanced total (Figures S1F and S1G), but to a lesser extent epithelial (Figures S1H and S1I), CD103+CD8+ subset with better PFS of anti-PD-(L)1-treated patients was confirmed in a second cohort (validation cohort) of 41 NSCLC, including 10 (26%) responder patients (Table S4). In contrast, patients with increased intraepithelial CD103−CD8+ TIL did not respond efficiently to anti-PD-1, with a HR of 1.74 (95% CI 1.05–2.88, p = 0.03), and no correlation was observed between the density of intraepithelial CD103−CD8+ cells and the ORR (Figures 1F and 1G). Since epithelial tumor region infiltration by either high numbers of CD103+CD8+ cells or low numbers of CD103−CD8+ cells was associated with better outcomes, we stratified the cohort into 3 groups based on these 2 parameters. Using a CD103/CD8 score, we considered a group satisfactory if both parameters were present (CD103+CD8+high and CD103−CD8+low), intermediate if only 1 parameter was present (CD103+CD8+high or CD103−CD8+low), and poor if neither was present (CD103+CD8+low and CD103−CD8+high). Interestingly, the group with a satisfactory CD103/CD8 score had a median PFS of 26.2 months (95% CI 15.3–not-reached) versus 2.7 months (95% CI 1.7–7.3) for the group with an intermediate score and 1.38 months for the group with a poor score (95% CI 0.72–1.97, p < 0.0001) (Figure 1H; Table S2). In addition, anti-PD-1-long-responder patients (PFS > 6 months and overall survival [OS] > 12 months) displayed a higher frequency of CD103+ cells among CD8+ TILs than fast-progressor patients (defined by an “early death” occurring during the first 12 weeks after the beginning of ICB) (Figure 1I). A higher density of intraepithelial CD103+CD8+ TILs in anti-PD-1-long-responder patients than in fast-progressor patients was also obtained in the validation cohort (Figure S1J). More important, quantitative multiplex fluorescent IHC staining of tumors collected from 9 patients (5 from the validation cohort and 4 additional patients) before (2–25 weeks) and after anti-PD-1 administration showed an increase in the density of epithelial CD103+CD8+ TILs in 4 of 5 responder patients. In contrast, no expansion of this subset was observed in 4 of 4 non-responder patients (Figure 1J; Table S5). An increase in total CD103+CD8+ and total CD8+ TILs was also observed in responder patients (Table S5). These results highlight the importance of enhanced intratumoral CD103+CD8+ cells in response to anti-PD-(L)1 and support the conclusion of a role of this subset in patient outcomes.
We then assessed the expression of E-cadherin and ICAM-1, the respective ligands of CD103 and LFA-1 integrins, known to play a major role in strengthening the interaction between CTLs and tumor cells following T cell receptor (TCR) recognition of major histocompatibility complex (MHC) class I-peptide complexes.16,17 The expression of E-cadherin on cancer cells from 105 evaluable tumor sections of the 111 analyzed NSCLCs was scored into 4 groups: scores 0 and 1 were considered low (12%, n = 13/105) and scores 3 and 4 were high (88%, n = 92/105) (Figure S2A; Table S6). Data analyses indicated that E-cadherin expression does not affect PFS, and no effect on the density of intraepithelial CD103+CD8+ TILs was observed (Figures S2B and S2C). Similarly, according to ICAM-1 expression on tumor cells from 101 evaluable tumor sections of the 111 analyzed tumors, the population was classified into low and high groups (Figure S2D), with no impact observed on survival or infiltration by CD103+CD8+ cells in epithelial regions (Figures S2E and S2F). Nevertheless, for tumors combining an epithelial CD103+CD8+high plus ICAM-1high profile, median PFS increased up to 15.3 months (95% CI 9.63–not reached) versus a median PFS of 3.8 months (95% CI 2.56–not reached) observed for the epithelial CD103+CD8+high plus ICAM-1low profile (Figure S2G; Table S2). These results support the observation that CD103 promoted CD8 T cell infiltration within epithelial tumor regions, and suggest that these CD103+CD8+ lymphocytes are tumor-reactive TRM cells.
CD8+ TILs from NSCLC Are Highly Enriched in CD103+CD49a+CD69+ TRM
Experiments were next performed to delineate the phenotypic and molecular features of CD103+CD8+ NSCLC TILs, and to determine whether they are a homogeneous population or whether heterogeneity exists in the expression of conventional TRM surface markers. With this aim, CD8+ TILs from 39 freshly resected treatment-naive early-stage NSCLC tumors were isolated and stained for CD3, CD8, CD103, CD49a, and CD69, as well as the effector T cell (TEff) surface marker KLRG1 (Figure S3A). The results indicated that the percentage of CD8+ T cells displaying a CD103+KLRG1− phenotype ranged from 18% to 81%, with a mean of 51%, while the percentage of CD8+ T cells displaying a CD103−KLRG1+ TEff phenotype ranged from 7% to 52%, with a mean percentage of 26% (Figure 2A). The mean percentages of CD103+KLRG1+ and CD103−KLRG1− T cell subpopulations were only ~10% and 12% of CD8+ TILs, respectively. Notably, the expression of CD103 inversely correlated with that of KLRG1 on CD8+ TILs and appeared mutually exclusive (Figure 2B).
Figure 2.

Expression of CD103, CD49a, and CD69 on CD8 T Cells Infiltrating Treatment-Naive NSCLC and Healthy Lung Tissues
(A) Freshly resected NSCLC tumors were dissociated and the CD8+ T cell subset was analyzed for CD103 and KLRG1 expression. Representative dot plots from 4 different TIL samples are included (bi-exponential scale). Right, percentages of CD103+KLRG1−, CD103−KLRG1+, CD103+KLRG1+, and CD103−KLRG1− TIL subsets among CD3+CD8+ cells (n = 39).
(B) Inverse correlation between percentage of CD103+ and KLRG1+ cells among CD8+ TILs (n = 40). The r value indicates Pearson correlation coefficient.
(C) Expression of CD69 and CD49a on CD103+ and KLRG1+ CD8+ TILs. Representative dot plot from 1 TIL sample is shown. Right, percentages of CD69+ and CD49a+ cells among CD103+ and KLRG1+ CD8 TILs are presented (n = 17 and n = 16, respectively).
(D) Correlation between percentage of CD103+ and CD49a+ cells among CD8+ TIL (n = 27). The r value indicates Pearson correlation coefficient.
(E) Percentages of KLRG1+ (n = 16) and CD103+CD49+ cells (n = 19) among CD8+ T cells infiltrating tumors or autologous healthy lung tissues.
(F) Percentages of CD69+ cells among CD103+CD49a+ CD8 T cells from tumor or healthy lung tissues (n = 19).
(G) Total numbers of CD103+CD69+CD49a+ CD8 (TRM) cells per milligram of tumor tissues and paired normal lung tissues (n = 18).
Each symbol represents individual TILs or lung sample; horizontal lines correspond to means ± standard errors of the mean (SEM). ∗∗p < 0.01, ∗∗∗p < 0.001 (paired t test).
We then further characterized the CD103+CD8+ TIL subset by evaluating the expression of CD69 and CD49a TRM markers. The results showed that >90% of CD103+CD8+ T cells expressed CD69 and CD49a (Figures 2C, S3A, and S3B), and that the expression of CD49a correlated with that of CD103 (Figure 2D). In addition, most CD103+ TILs displayed a CCR7−CD45RA− profile, which is characteristic of TEM cells (Figure S3C). In contrast, only 50% and 35% of KLRG1+CD8+ TILs expressed CD69 and CD49a, respectively (Figure 2C). This T cell subset is more heterogeneous in CD45A expression and is mainly composed of CCR7-CD45RA− TEM and CCR7−CD45RAhigh terminally differentiated (EMRA) T cells (Figure S3C). CD103+CD49a+CD69+ CD8 T cells, hereafter called TRM cells are also present in adjacent healthy lung tissues, but at a lower frequency and lower expression level of CD69 than in autologous tumor tissues (Figures 2E and 2F). In contrast, the KLRG1+ CD8 T cell subset, hereafter referred to as non-TRM cells, is more abundant in healthy lung tissue than in the cognate tumor (Figure 2E). In addition, the number of TRM cells per milligram of tissue is much higher in the tumor than in the cognate normal lung, with mean values of ~1,000 TRM/mg of tumor versus 100 TRM/mg of healthy lung (Figure 2G), with no correlation between the 2 tissue compartments within each patient sample (Table S7). These data support the conclusion that NSCLCs are strongly infiltrated by a homogeneous CD103+CD49a+CD69+ CD8 TRM subset, which is readily distinguishable from the KLRG1+ CD8 TEff subset.
Molecular Features of NSCLC CD8 TRM Cells
To determine the transcriptional profile of the tumor TRM subset, we performed RNA-based next-generation sequencing (RNA-seq) of CD103+CD8+ and matched KLRG1+CD8+ T cells freshly isolated from the TILs of 7 treatment-naive early-stage NSCLCs. Gene expression analyses performed with p ≤ 0.05 revealed a large number of transcripts (n = 2,958) differentially expressed in the 2 T cell subsets, supporting the notion that they correspond to 2 divergent populations (Figure S3D). Among these transcripts, we identified a gene signature that is characteristic of antiviral TRM cells and NSCLC T cells,5,13,18,19 including the upregulation of CXCR6 and SIRPG transcripts (Figure S3E; Table S8). A gene signature with the upregulation of GPR34, ITGAE, and CXCR6 genes and the downregulation of SIPR5 and SIPR1, encoding sphingosine phosphate receptors, KLF2, encoding Krüppel-like transcription factors that regulate T cell trafficking, and SELL, encoding L-selectin CD62L, important in lymphocyte homing to lymphoid organs, was identified in tumor TRM cells (Figure S3F; Table S8). The downregulation of S1pr1 in tumor CD103+CD8+ TRM cells was confirmed by multiparametric flow cytometry, with levels similar to those of healthy lung TRM cells (Figure S3G). In addition, gene set enrichment analysis (GSEA) showed that several hallmark gene sets, such as inflammation, cell cycle, TGF-β signaling pathways, mammalian target of rapamycin (mTOR), and hypoxia, were enriched in TRM cells (Figures S4A and S4B; Table S9).
Among TRM signature genes, a panel of genes involved in T cell exhaustion, including CTLA-4, BTLA, HAVCR2, PDCD1, LAG3, and TIGIT, were more strongly expressed in CD103+ than in KLRG1+ TIL subsets (Figures 3A and S4C; Table S8). The upregulation of SPRY1 (Sprouty), ENTPD1 (CD39), LAYN (Layilin), and TOX genes was also observed in TRM cells (Figure S4C). Flow cytometry analyses confirmed enhanced expression of PD-1 on TRM cells from tumors, but not on non-TRM cells and TRM cells from cognate healthy lung tissue (Figure 3B). Notably, ectonucleotidase CD39 was specifically expressed by TRM cells, and its expression was associated with PD-1 (Figures 3C and S4D). Moreover, t-distributed stochastic neighbor embedding (t-SNE) analysis highlighted the strong correlation of expression of CD39 and PD-1 with the TRM cluster, whereas the non-TRM cluster showed a weak association with these markers (Figure 3D). The expression levels of CD103 on CD8+ TILs correlated with CD39 and 4-1BB (CD137) levels, and CD103high T cells also displayed CD39high (Figure 3E) and 4-1BBhigh (Figure 3F) expression profiles characteristic of antigen-experienced T lymphocytes.20,21 As expected, TRM cells from adjacent normal lung expressed only low levels of 4-1BB, excluding the recent engagement of TCR with specific antigen (Figure 3F). These results support the hypothesis that NSCLC CD103+CD8+ TILs were enriched with tumor-reactive T cells harboring all of the features of activated TRM cells.
Figure 3.

Expression of T Cell Exhaustion Hallmark in TRM Cells from NSCLC Tumors
(A) Heatmap of transcripts involved in T cell exhaustion differentially expressed in CD103+ and KLRG1+ CD8+ TILs (n = 7). Different expression patterns correspond to different isoforms of the same gene.
(B) Expression of PD-1 on CD103+ and KLRG1+ CD8+ TILs. Dot plots of 1 representative patient. Right, percentages of PD-1+ cells among TRM and non-TRM (n = 21) and paired TRM from healthy lung (n = 13).
(C) Percentages of CD39+ cells in paired TRM and non-TRM from NSCLCs (n = 13).
(D) t-SNE map of CD103+CD49a+ (blue) and KLRG1+ (pink) cells among CD8+ TILs. Right, t-SNE analysis of CD39 and PD-1 expression on CD103+CD49a+ (TRM) and KLRG1+ (non-TRM). The data are from 2 representative TIL samples (patients 3 and 4).
(E) Dot plots of CD39 expression on CD103+CD8+ TRM, displaying high (CD103high), intermediate (CD103int), and low (CD103low) CD103 phenotypes, and CD103−CD8+ TIL from 1 representative tumor. Right, percentages of CD39+ cells among TRM expressing high, intermediate, and low levels of CD103 and CD103−CD8+ TIL (n = 16).
(F) Dot plot of 4-1BB expression on CD103+CD8+ TILs from 1 representative tumor. Right, percentages of 4-1BB+ cells among TRM displaying high, intermediate, and low CD103 profiles. CD103−CD8+ TIL (n = 7) and CD103+CD8+ TRM from autologous normal lungs (n = 4) are included. CD103 intensity is shown by a gradient color code.
Symbols represent individual TILs or lung samples; horizontal lines correspond to means ± SEMs. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 (paired t test or ANOVA with Bonferroni post hoc test); ns, not significant.
See also Figures S3 and S4 and Table S8.
Lung Tumor TRM Cells Express Transcription Factors Involved in Th17 Differentiation
To explore potential pathways involved in TRM formation in tumors, we studied genes encoding transcription factors differentially expressed in CD103+CD8+ and KLRG1+CD8+ TILs. RNA-seq analyses indicated that CD103+CD8+ T cells displayed a specific signature characterized by the upregulation of ZEB1 encoding the zinc-finger E-box binding homeobox-1, ZNF683 (zinc finger 683) encoding the Blimp1 homolog Hobit, and PRDM1 (BLIMP1), and the downregulation of EOMES, IKZF2 (HELIOS), and TCF7 genes (Figures 4A, 4B, and S5A; Table S8). Remarkably, a set of genes encoding transcription factors involved in Th17 differentiation, such as IKZF3 encoding the Ikaros transcription factor family member Aiolos, and AHR encoding an aryl hydrocarbon receptor, were more strongly expressed in TRM cells than in non-TRM cells (Figure 4B and S5A). A panel of genes associated with the Aiolos pathway, including SMARCA4, UBE2I, and GRAP2, were also upregulated in TRM cells (Figure S5B).
Figure 4.

Transcription Factor Profiles of TRM and non-TRM from NSCLCs
(A) Heatmap of transcripts encoding transcription factors differentially expressed in CD103+ and KLRG1+ CD8+ TIL (n = 7) (p < 0.05).
(B) Expression of ZEB1, ZNF683 (HOBIT), PRDM1 (BLIMP1), IKZF3 (AIOLOS), EOMES, and AHR genes in TRM and non-TRM from NSCLCs (n = 7). Values are transcripts per milion (TPM).
(C) Percentages of Hobit+ cells among TRM and non-TRM cells from NSCLCs (n = 21) and among healthy lung TRM cells (n = 15). Right, expression of Hobit (gMFI) in TRM and non-TRM cells from NSCLCs (n = 21) and in healthy lung TRM cells (n = 15).
(D) Expression of TCF-1 (gMFI) in tumor TRM and non-TRM cells (n = 7).
(E) Percentages of T-bet+ cells among tumor TRM and non-TRM cells (n = 21) and among healthy lung TRM cells (n = 11). Right, expression of Aiolos (gMFI) among tumor TRM and non-TRM and paired healthy lung TRM (n = 13). Heathy donor Th17 control cells are included (n = 2).
(F) Expression of AhR (gMFI) in tumor TRM and non-TRM (n = 10), and in Th17+ control cells (n = 2).
(G) Expression of pSTAT3 (gMFI) in tumor TRM and non-TRM cells (n = 10), and in Th17+ controls (n = 2). Right, expression of pSTAT3 (gMFI) in tumor TRM cells displaying CD103high (High), CD103int (Int), and CD103low (Low) phenotype, and in CD103−CD8+ TILs (Neg) (n = 10).
Each symbol represents an individual donor; horizontal lines are means ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 (paired t test or ANOVA with Bonferroni post hoc test). gMFI, geometric mean fluorescence intensity; ns: not significant.
Flow cytometry analyses showed that tumor TRM cells expressed lower levels of Hobit protein than non-TRM cells, but levels higher than in healthy lung TRM cells (Figure 4C). The downregulation of the TCF7 gene-encoding protein TCF-1 in tumor TRM as compared to non-TRM cells was confirmed at the protein level (Figure 4D). In contrast, T-bet and Aiolos transcription factors were more strongly expressed in tumor TRM than in non-TRM cells, at levels similar to or higher than those in healthy lung TRM cells (Figure 4E). Moreover, AhR protein and phosphorylated-STAT3 (pSTAT3), also known as the master regulators of Th17 differentiation, were more strongly expressed in TRM than in non-TRM cells, and at levels similar to those of healthy lung TRM cells (Figures 4F, 4G, and S5C). Notably, tumor TRM cells displaying high levels of CD103 (CD103high) expressed higher levels of pSTAT3 than TRM cells with intermediate (CD103Int) and low (CD103low) CD103 expression (Figure 4G). In addition, tumor TRM cells expressed similar levels of pSTAT3 and Aiolos than healthy donor Th17+ control cells (Figures 4E and 4G). In contrast, AhR was more strongly expressed in Th17+ controls than in tumor TRM cells (Figure 4F). The transcription factor RORγt was equally expressed by TRM and non-TRM cells, and the chemokine receptor CCR6 was more strongly expressed in TRM than in non-TRM cells (Figure S5D; Table S8). These results demonstrate that, together with T-bet, human lung tumor CD8+ TRM cells express Aiolos, AhR, and pSTAT3, three master regulators of the Th17 pathway, and ultimately suggest that they are enriched with Tc17 lymphocytes.
Lung Tumor CD8+ TRM Cells Contain Tc17 and are Tumor-Reactive CTL
To gain further insight into the molecular characteristics of human lung tumor CD8+ TRM cells, we studied the expression profiles of genes involved in T cell activation. The results showed that TRM displayed a signature of more activated lymphocytes than non-TRM cells (Figure 5A). Notably, GSEA highlighted the upregulation of several genes belonging to the Th17/Tc17 cell lineage in TRM cells (Figure 5B; Table S8). Among these genes, IL17A and IL17RA, encoding IL-17 and the IL-17 receptor (IL-17R), were more strongly expressed in TRM than in non-TRM cells (Figure 5C; Table S8). The upregulation of IL17A transcript in some TRM, but not in non-TRM cells, was further confirmed by qRT-PCR using paired CD103+CD8+ and KLRG1+CD8+ T cells from 6 additional lung TILs (Figure S5E). Moreover, intracellular flow cytometry analysis of fresh TILs stimulated ex vivo with phorbol myristate acetate (PMA) plus ionomycin showed production of IL-17 by TRM cells and healthy donor Th17+ control cells, but not by non-TRM cells (Figure 5D). Remarkably, higher percentages of IL-17-producing cells were observed in CD103high TRM cells than in CD103int and CD103low cells (Figure S5F), and the expression of CD39 and 4-1BB was frequently higher in IL-17+ than in IL-17− TRM cells (Figure S5G). ELISA also confirmed the restricted secretion of IL-17 in supernatants from TRM, but not from non-TRM cells (Figure 5E). In contrast, both non-TRM and TRM cells from tumors were able to produce high levels of IFN-γ and TNF-α following ex vivo stimulation with PMA plus ionomycin (Figure 5F). Moreover, a subset of TRM cells from lung tumors produced both IL-17 and IFN-γ (Figure 5G). These IL-17-producing CD8+ T cells were not TCR-γδ lymphocytes, which often display a CD4−CD8− profile and represent ~3.6% of CD3+ TILs, and only 2.2% of them produced IL-17 (Figures S5H and S5I).
Figure 5.

A Subset of Tumor CD103+CD8+ TRM Cells Displays a Tc17-Polarized Pattern
(A) Heatmap of activation gene signature differentially expressed in paired CD103+ and KLRG1+ CD8+ TIL (n = 7) (p < 0.05).
(B) GSEA of gene set from the Th17 signature in the transcriptome of CD103+CD8+ TILs relative to KLRG1+CD8+ TILs (n = 7). Running enrichment score (RES) for the gene set as the analysis “walks down” the ranked list of genes; the position of gene set members (black vertical lines) in the ranked list of genes and the value of ranking metric are shown.
(C) Expression of IL17A and IL17RA transcripts encoding IL-17 and IL-17R, respectively, in tumor TRM and non-TRM cells (n = 7). TPMs are shown.
(D) Intracellular expression of IL-17 in CD8+ TILs stimulated for 4 h with PMA plus ionomycin. One representative dot plot is shown. Right, percentages of IL-17+ cells among TRM and non-TRM cells from tumors (n = 12). Healthy donor Th17+ control cells are included (n = 2).
(E) Production of IL-17 measured by ELISA in supernatant of tumor TRM and non-TRM cells stimulated overnight with PMA plus ionomycin. The data are concentrations of IL-17 in picograms per milliliter per 100,000 cells (n = 5).
(F) Intracellular expression of IFN-γ and TNF-α in CD8+ TILs stimulated with PMA plus ionomycin. The dot plots from 1 representative patient are shown. Right, percentages of IFN-γ+ (n = 13) and TNF-α+ (n = 7) cells among TRM and non-TRM cells from tumors.
(G) Intracellular co-expression of IL-17 and IFN-γ in tumor TRM cells stimulated with PMA plus ionomycin. Two representative patients are shown.
Symbols represent individual TILs or lung samples; horizontal lines are means ± SEMs (C, D, E, and F). ∗p < 0.05 and ∗∗∗ p < 0.001 (paired t test or ANOVA with Bonferroni post hoc test).
Flow cytometry analyses showed that tumor TRM cells expressed higher levels of proliferation marker protein Ki67 compared to tumor non-TRM and TRM cells from adjacent healthy lung (Figure 6A). Intracellular immunofluorescence also revealed that tumor TRM cells expressed higher levels of granzyme B than non-TRM and healthy lung TRM cells, supporting the hypothesis that they are tumor-reactive CTLs (Figure 6B). Consistently, further functional studies by confocal microscopy showed that tumor TRM cells formed more stable conjugates with autologous cancer cells than non-TRM cells, as evaluated by tyrosine phosphorylation (pTyr) at the contact zone between T cells and target cells (Figure 6C). Moreover, tumor TRM cells were able to kill autologous tumor cells much more efficiently than non-TRM cells (Figure 6D). These results further support the conclusion that TRM cells infiltrating NSCLC tumors are activated tumor-specific cytotoxic effectors.
Figure 6.

TRM Cells Are Tumor-Specific CD8+ T Cells
(A) Intracellular expression of Ki67 in TRM and non-TRM cells from NSCLCs. The dot plot of 1 representative patient is shown. Right, percentages of Ki67+ cells in paired TRM and non-TRM cells from tumors and paired TRM cells from healthy lung (n = 18).
(B) Intracellular expression of granzyme B in tumor TRM and non-TRM cells. The dot plot of 1 representative patient is shown. Right, percentages of granzyme B+ cells in TRM and non-TRM cells from tumors and paired TRM cells from healthy lung (n = 14).
(C) Conjugates formed between freshly isolated CD8+ tumor TRM or non-TRM cells and autologous tumor cells were analyzed by confocal microscopy for p-Tyr (green fluorescence) accumulation in the contact area. T cells were labeled with anti-CD8 mAb (red). Scale bars, 10 μm. Right, quantification of pTyr fluorescence intensity fold increase in CD8 T cells at contact zone with cognate target cells. Four representative patients are included. Each symbol represents an individual synapse from 15 to 37 analyzed conjugates.
(D) Cytotoxic activity of freshly isolated tumor TRM and non-TRM cells toward autologous cancer cells determined by a conventional 4 h chromium release assay at a 20:1 E:T ratio. The data correspond to 4 independent experiments with 4 different tumor samples. Horizontal lines are means ± SEMs from triplicate.
Symbols represent individual TILs or lung samples; horizontal lines are means ± SEMs (A and B). ∗p < 0.05 and ∗∗∗p < 0.001 (paired t test or ANOVA with Bonferroni post hoc test).
Lung Tumor CD8+ TRM Cells Express a More Clonal TCR Repertoire Than Non-TRM Cells
To assess the clonality of NSCLC CD8+ TRM cells, we investigated the TCR-β repertoire of 9 freshly sorted CD103+CD8+ TILs and autologous KLRG1+CD8+ TILs by deep TCR sequencing (TCR-seq). Results indicated that tumor TRM cells were more oligoclonal than paired non-TRM cells (Figures 7A and S6A). The 10 most frequent (top 10) TCR-β clonotypes, defined by hypervariable complementarity-determining region 3 (CDR3) nucleotide sequences, accounted for >60% of the TRM population and <45% of the non-TRM population (Figure 7B). In contrast, only a marginal difference was observed between TCR-β repertoires of TRM cells from NSCLC tumors and proximal healthy lung (Figure S6B). TCRV-β (TCR variable β chain) family distribution within each T cell subset was variable across patients, but it was different between TRM and non-TRM cells from a given patient (Figure 7C). However, TRM and non-TRM populations shared some clonotypes, the proportions of which varied from one patient to another and ranged from 4% to 28% (Figures S6C–S6E). Next, we questioned whether the restricted TCR-β repertoire observed in TRM cells correlated with the expression of PD-1, reported to identify the CD8+ tumor-specific repertoire infiltrating human tumors.22 The data revealed a correlation between the productive frequency of the top 10 clonotypes and the percentage of PD-1+CD103+CD8+ TILs (Figure 7D), further supporting the conclusion that NSCLC CD8+ TRM cells are an oligoclonal subpopulation enriched with activated tumor-specific T cells that underwent antigen-driven expansion at the tumor site. They also explain how this tumor-resident CTL subset can locally participate in anti-tumor CD8 T cell immunity and contribute to the response to anti-PD-1 therapy.
Figure 7.

Tumor CD8+ TRM Cells Display an Oligoclonal TCR Repertoire
(A) Frequency of productive TCR sequences of the top 10 clonotypes (gray) and all other clonotypes (white) in paired NSCLC TRM and non-TRM cells from each patient sample (patients 22–30). TCR-β CDR3 region in tumor TRM and non-TRM cells were analyzed by TCR-seq (n = 9).
(B) Frequency of the top 10 TCR sequences in tumor TRM and non-TRM cells from both TCR-seq and RNA-seq (n = 14).
(C) Pie charts illustrating distribution of TCRV-β families in TRM and non-TRM cells from tumors (n = 9) and autologous TRM cells from healthy lungs (n = 4).
(D) Correlation between productive frequency of the top 10 TCR clonotypes and the percentage of PD-1+ cells in tumor TRMs (n = 11).
Horizontal lines correspond to means ± SEMs.
∗p < 0.05 paired t test; ns, not significant. The r value indicates the Pearson correlation coefficient.
See also Figure S6.
Discussion
In this report, we show that NSCLC tumors are strongly infiltrated by CD8+ TRM cells, most of which express a homogeneous CD103+CD49a+CD69+ phenotype. This TRM subset was also found in adjacent healthy lung tissues, but at lower densities than in autologous tumor samples, with no correlation between the frequency of either, suggesting that they are two distinct TRM subsets. Tumor CD103+CD8+ TILs displayed transcriptomic and phenotypic signatures characteristic of TRM cells, including the downregulation of (1) Klf2 (regulates genes involved in T cell trafficking such as SELL, CCR7, and S1PR123), (2) S1pr1 (mediates the egress of T cells from lymphoid organs24), and (3) CD62L and CCR7 (key regulators of T cell migration to lymphoid organs) and the upregulation of (1) CD103 (favors T cell recruitment and retention in epithelial tumor islets through binding to its ligand, epithelial cell marker E-cadherin25), (2) CD49a (responsible for retaining CD8 TRM cells in peripheral tissues via attachment to the extracellular matrix11), and (3) CD69 C-type lectin (modulates the TRM cell capacity to exit non-lymphoid tissues through interaction and degradation of S1pr126). NSCLC CD8+ TRM cells also overexpress several T cell inhibitory receptors and exhaustion surface markers, with co-expression of PD-1 and CD39, supporting the conclusion that they are enriched in activated tumor antigen-reactive T cells. In this regard, it has been demonstrated that PD-1 accurately identifies the repertoire of clonally expanded tumor-specific CD8+ T cells.22 The expression of CD39, together with CD103, has been reported to identify truly tumor-reactive CD8 T cells in human solid tumors20 and to distinguish tumor antigen-specific CD8+ T cells from viral antigen-specific bystander T cells present in the TME.27 The TOX gene, recently identified in tumor-reactive murine T cells and in human PD1hiCD39hi TILs,28 was also upregulated in human NSCLC TRM cells, further indicating that they are enriched in tumor-specific T cells. NSCLC TRM cells also frequently expressed 4-1BB (CD137) co-stimulatory receptor characteristics of antigen-experienced TILs.21 These molecular features likely cooperate in retaining CD8+ TRM cells in tumor tissues to detect and mount robust local immune responses to transformed cells.
T-bet, together with Eomes T-box transcription factors, were demonstrated to control the development and survival of skin and lung CD103+ TRM cells by controlling TGF-β and IL-15 expression.29 However, while extinguishing Eomes is necessary for CD103+CD8+ TRM cell formation, residual T-bet expression is required for IL-15-mediated CD103+CD8+ TRM survival. Consistently, we show that lung tumor CD103+CD8+ TRM cells express T-bet at levels similar to healthy lung CD103+CD8+ TRM, but they only weakly express Eomes. ZEB1 was strongly expressed in human NSCLC CD103+CD8+ TRM cells, and this expression is likely induced by TGF-β at the tumor site. In this regard, Zeb1 has emerged as a critical transcription factor for memory T cell survival and function.30 At least a subset of NSCLC CD8+ TRM cells displays a transcription factor profile characteristic of Th17/Tc17 lymphocytes, with expression of Aiolos, pSTAT3, and AhR. The upregulation of Aiolos by STAT3 and AhR was reported to promote Th17 differentiation by silencing IL-2 expression.31 In contrast, TCF7/TCF-1 was less abundant in NSCLC TRMs than in non-TRM cells. The downregulation of TCF7/TCF-1 in CD103+CD8+ TILs is consistent with previous single-cell analyses of TRM cells from breast cancer and NSCLC.7,8 Exhausted PD1+CD8+ TILs from human melanoma are also TCF7neg and enriched with CD39- and CD103-expressing cells.20,32 Notably, the activation of TCR leads to the downregulation of TCF-1 in human CD8+ T cells,33 suggesting recent TCR engagement of TCF-1low NSCLC TRM cells. These TCF-1low/neg CD8+ cells, as opposed to bystander TCF-1+ TILs, are tumor-reactive CTLs,8,20,32,34 highlighting their anti-tumor potential and suggesting that TCF-1-expressing (stem-like or memory precursor-like) cells are not the only source of T cell expansion following ICB,35, 36, 37, 38, 39, 40 and that TCF-1low (TRM) cells are also reactivated to participate in the response to anti-PD-1.
TCF7 emerges as a central transcription factor in CD8+ TRM differentiation in IL-17-producing lymphocytes, as its downregulation results in enhanced Tc17-cell development.41 Consistently, our functional experiments showed that a small subset of NSCLC CD8+ TRM cells produces IL-17 after PMA/ionomycin stimulation at frequencies similar to heathy donor Th17/Tc17-polarized T cells. Human CD8+CD49a− TRM cells were also found to produce IL-17 in skin epithelia and psoriasis lesions, promoting local inflammation.42 Differentiation of a subpopulation of CD8+ TRM cells into the Tc17 lineage is likely associated with the secretion of IL-6 and TGF-β by lung tumor cells themselves and the inhibition of IFNγ, conditions known to promote the formation of murine CD4+ Th17 lineage.43 Murine Tc17/IFNγ CD8+ T cells mediate improved the anti-tumor immunity that is associated with enhanced survival.44 A small subset of NSCLC CD103+CD8+ TRM cells also produces IL-17 and IFN-γ, displays increased proliferative potential, and mediates cytotoxic function toward autologous cancer cells correlated with the production of granzyme B and the formation of stable conjugates with the cognate target.
The notion that CD103 is a marker of antigen-specific T cells is supported by studies of antiviral and anti-tumor CTL responses, demonstrating that the integrin is induced on CD8+ T cells specific to influenza virus in the lung,45 Epstein-Barr virus (EBV) in the tonsil,46 cancer testis antigen NY-ESO-1 in ovarian cancer,47 and the α-actinin 4 (actn-4) neoepitope in NSCLC.48 The tumor antigen specificity of CD103+CD8+ TRM cells was further emphasized by TCR-seq analyses revealing the expansion of particular TCR clonotypes in CD103+CD8+ TILs, but to a lesser extent in KLRG1+CD8+ TILs. TCR clonality in tumor CD8+ TRM cells correlated with high PD-1 expression, which reflects their strong activation state and suggests high TCR avidity for the tumor antigen, resulting in antigen-driving T cell proliferation. Remarkably, CD103+CD8+ TRM cells were shown to expand during anti-PD-1 immunotherapy of melanoma49 and lung carcinoma patients.8 Here, we provide evidence that a high density of CD103+CD8+ TRM cells in tumors was associated with greatly improved PFS in NSCLC patients treated with anti-PD-(L)1 as a single agent. We also provide evidence that the density of CD103+CD8+ TIL was increased in tumors after anti-PD-1 treatment, suggesting that PD-1 blockade reactivates this tumor-specific TRM subset, which proliferates and exerts cytotoxic activity so as to participate in anti-tumor immunity and response to ICB. The reason for intertumor TRM density variation is not understood, but it may reflect interpatient variations in the number of antigen-specific T cell precursors, the tumor mutational burden, or the amount of active TGF-β required for CD103 expression in TCR-engaged T lymphocytes.16,48,50 In regard to epithelial tumor regions, enhanced infiltration by CD103+CD8+ TRM cells did not correlate with E-cadherin expression levels by cancer cells. Consistent results were obtained in ovarian cancer,3 but not in bladder cancer, where intratumoral infiltration of CD103+ TIL was associated with the expression of E-cadherin on tumor cells.51 Although the enhanced expression of ICAM-1 on cancer cells was not associated with the increased density of CD103+CD8+ TIL in epithelial tumor islets, the combination with high TRM infiltration improved patient PFS. This may be associated with strong adhesion between tumor cells and specific TRM cells provided by the ICAM-1-LFA-1 interaction and required for effective target cell lysis by activated CTLs.17,52 In contrast, the infiltration of epithelial tumor regions by CD103−CD8+ TIL did not result in the improved survival of anti-PD-1-treated patients. This is surprising, since previous studies reported that CD8+ T cell infiltration is associated with better clinical outcomes and response to ICB.1 Although the status of CD8+ TILs for CD103 expression was not investigated, these T cells likely expressed the integrin, since it is required for their recruitment and functionality within epithelial tumor regions.53
Overall, our study demonstrates a major role for CD103+CD8+ TRM cells in anti-tumor immunity and the response to anti-PD-1 in lung cancer. It also delineates CD103 as a biomarker that is able to accurately identify the repertoire of clonally expanded tumor-reactive CD8+ T cells. These CD103+ CD8+ T cells are activated, proliferate, a subset of which can produce IL-17 and IFNγ, and effectively kill autologous malignant cells. Thus, investigating CD103+CD8+ TRM cells in cancer patients not only contributes to our understanding of the local anti-tumor CTL response but it also provides insight into the role of this T cell population in response to ICB. Improved responses to anti-PD-1 in patients with high densities of tumor CD103+CD8+ TRM cells, even after having undergone resection of their tumor, may be associated with a pool of cancer-specific CD8+ T cells present in bone marrow, as has been described in infectious diseases.54 This non-circulating memory CD8+ T cell subset develops in response to a wide variety of antigens, including tumor antigens, and expands upon antigen rechallenge. Thus, CD103+CD8+ TRM cells appear to be crucial components in immune protection against cancer and in the response of NSCLC to ICB, opening up new avenues in cancer immunotherapy.
Limitations of Study
The total CD8+high population in the stroma was as correlative as the CD103+CD8+high population (both with 30.62 months of PFS), most likely due to the frequent expression of CD103 on CD8+ TILs even in the stroma. The study lacks functional features on IL-17-producing TRM cells. Isolation from the same tumor of IL-17+ and IL-17− CD8+CD103+ TILs and sufficient numbers of autologous tumor cells will help to further characterize this small TRM cell subset. Single-cell RNA-seq of the tumor TRM population may help define the molecular features of Tc17 cells. TCR-γδ lymphocytes may contribute to IL-17 production in the TME, even though their total number is very weak.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposit Data | ||
| Raw RNAseq data | This study | EGAS00001004707 |
| Antibodies | ||
| anti-human CD8 Pacific blue (RPA-T8) | BioLegend | Cat# 301023; RRID:AB_493110 |
| anti-human CD103-FITC (Ber-ACT8) | BioLegend | Cat# 350204; RRID:AB_10639865 |
| anti-human KLRG1-PE (13F12F2) | ebioscience | Cat# 12-9488-42; RRID:AB_2572716 |
| anti-human CD3-Alexa700 (UCHT1) | BioLegend | Cat# 300424; RRID:AB_493741 |
| anti-human CD69-APC-Cy7 (FN50) | BioLegend | Cat# 310914; RRID:AB_314849 |
| anti-human granzyme-B-FITC (GB11) | BioLegend | Cat#515403; RRID:AB_2114575 |
| anti-human TNFα-PE/Dazzle594 (Mab11) | BioLegend | Cat# 502945; RRID:AB_2564172 |
| anti-human Aiolos-Alexa488 (16D9C97) | BioLegend | Cat# 371107; RRID:AB_2616975 |
| anti-human Vα7.2-APC-Cy7 (3C10) | BioLegend | Cat# 351713; RRID:AB_2561995 |
| anti-human CD161-Alexa488 (HP-3G10) | BioLegend | Cat# 339923; RRID:AB_2563938 |
| anti-human TCF-1-PE (7F11A10) | BioLegend | Cat#655207; RRID:AB_2728491 |
| anti-human 4-1BB-PE-Dazzle594 (clone 4B4-1) | BioLegend | Cat#309825; RRID:AB_2566259 |
| anti-human TCRγδ-PerCPCy5.5 (B1) | BioLegend | Cat# 331223; RRID:AB_2563012 |
| anti-human CD103-BV711 (Ber-ACT8) | BD Biosciences | Cat# 563162; RRID:AB_2738039 |
| anti-human CD49-PerCPefluor710 (TS2/7) | Thermo Fisher Scientific | Cat# 46-9490-41; RRID:AB_2573890 |
| anti-human S1PR1-efluor660 (SW4GYPP) | Thermo Fisher Scientific | Cat# 50-3639-41; RRID:AB_2574207 |
| anti-human PD-1-PeCyanine7 (eBioJ105) | Thermo Fisher Scientific | Cat# 25-2799-42; RRID:AB_10853804 |
| anti-human AHR-PerCPefluor710 (FF3399) | Thermo Fisher Scientific | Cat# 46-9854-41; RRID:AB_2573905 |
| anti-human Ki67-PerCPefluor710 (20Raj1) | Thermo Fisher Scientific | Cat# 46-5699-41; RRID:AB_10804404 |
| anti-human IFNγ-APC (B27) | BD Biosciences | Cat# 554702; RRID:AB_398580 |
| anti-human Hobit-Alexa647 (Sanquin-Hobit/1) | BD Biosciences | Cat# 566250; RRID:AB_2739629 |
| anti-human CD45RA-APC (T6D11) | Miltenyi Biotec | Cat#130-098-187; RRID:AB_2660978 |
| anti-human pSTAT3pS727-APC (REA324) | Miltenyi Biotec | Cat# 130-105-000; RRID:AB_2653576 |
| anti-human CD39-APC (MZ18-23C8) | Miltenyi Biotec | Cat#130-100-459; RRID:AB_2660867 |
| anti-human IL17A-PeVio770 | Miltenyi Biotec | Cat#130-100-327; RRID:AB_2659809 |
| anti-human/mouse Tbet-APC (REA102) | Miltenyi Biotec | Cat# 130-119-821; RRID:AB_2784465 |
| mouse anti-human phospho-tyrosine (PY20) | BD Biosciences | Cat# 610000; RRID:AB_397423 |
| rabbit anti-human CD8 | Thermo Fisher Scientific | Cat#PA1-37298; RRID:AB_2075396 |
| Goat anti-mouse IgG AlexaFluor-488 | Thermo Fisher Scientific | Cat# A-11001; RRID:AB_2534069 |
| Goat anti-rabbit AlexaFluor-647 | Thermo Fisher Scientific | Cat# A-21244; RRID:AB_2535812 |
| anti-human Pan-keratin (clones AE1/AE3) | Agilent | Cat# M3515; RRID:AB_2132885 |
| IHC anti-human CD8 (clone SP16) | Spring Bioscience | Cat#M3164; RRID:AB_1660846 |
| IHC anti-human CD103 (clone EPR4166-2) | Abcam | Cat#ab129202; RRID:AB_11142856 |
| anti-human ICAM-1 (clone N1C2) | GeneTex | Cat#GTX100450; RRID:AB_1950536 |
| anti-human E-cadherin (clone EP6) | BioSB | Cat#BSB5466 |
| LEAF Purified anti-human CD3 antibody (OKT3) | BioLegend | Cat#317304; RRID:AB_571925 |
| Purified anti-human CD28 antibody (CD28.2) | BioLegend | Cat#302901; RRID:AB_314303 |
| Biological Samples | ||
| Resected lung tumors and adjacent healthy lung tissue samples | Institut mutualiste Montsouris and the Hôpital Marie-Lannelongue | See Tables S1 and S4 for details |
| Human healthy blood | Établissement français du sang, Paris, France | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Phorbol 12-myristate 13-acetate (PMA) | Sigma-Aldrich | Cat#P1585 |
| ionomycine | Sigma-Aldrich | Cat#10634 |
| recombinant IL-2 | Sanofi recherche | Cat#1A1 |
| recombinant human TGF-β1 | R&D Systems | Cat#240-B-002 |
| recombinant human IL-6 | Miltenyi Biotec | Cat# 130-095-365 |
| BrefeldinA | Thermo Fisher Scientific | Cat#00-4506-51 |
| Chromium-51 Radionuclide | Perkin Elmer | Cat#NEZ030005MC |
| Critical Commercial Assays | ||
| CD8 Microbeads, human | Miltenyi Biotec | Cat#130-045-201 |
| Tumor cells Isolation Kit, human | Miltenyi Biotec | Cat#130-108-339 |
| Human Tumor dissociation kit | Miltenyi Biotec | Cat#130-095-929 |
| Foxp3/Transcription Factor Staining Buffer Set | Thermo Fisher Scientific | Cat#00-5523-00 |
| Single cell RNA purification kit | Norgen | Cat#51800 |
| SureSelect Automated Strand-Specific RNA Library Preparation Kit | Agilent | Cat#G9691A |
| QIAamp DNA micro kit | QIAGEN | Cat#56304 |
| Maxima first strand cDNA synthesis kit | Thermo-Fischer Scientific | Cat#K1641 |
| Maxima SYBR Green Master Mix | ThermoFischer Scientific | Cat#4472908 |
| LIVE/DEAD Fixable UV dead cell stain kit | ThermoFischer | Cat#L34962 |
| Human IL-17A ELISA Kit | Biotechne | Cat#DY317-05 |
| Opal 7-color Automation IHC Kit | Akoya/Perkin Elmer | NEL801001KT |
| Oligonucleotides | ||
| Primer sequences for IKZF3, RORC,AHR, IL17A, TCF7, S1PR1 and 18S | This paper; See Table S10 | N/A |
| Software and Algorithms | ||
| GraphPad Prism software V8 | GraphPad Software Inc. | RRID:SCR_002798 |
| FlowJo V10 | Tree Star Inc | RRID: SCR_008520 |
| RStudio software | The R Foundation | RRID:SCR_000432 |
| InForm software | Akoya/PerkinElmer | N/A |
| R (v 3.2.3) | The R Foundation | RRID: SCR_001905 |
| FastQC (v 0.11.3) | Babraham bioinformatics | RRID:SCR_014583 |
| Salmon (v 0.8.2) | 55 | N/A |
| sleuth (v 0.28.1) | 56 | RRID:SCR_002555 |
| clusterProfiler (v 3.8.0) | 57 | RRID:SCR_016884 |
| MiXCR (v 2.1.10) | 58 | RRID:SCR_018725 |
| KEGG pathway | https://www.genome.jp/kegg-bin/show_pathway?hsa04660 | RRID:SCR_012773 |
| ImmunoSEQ Analyzer Platform Adaptive Biotechnologie | Adaptive Biotechnologie | RRID:SCR_014709 |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directly to and will be fulfilled by the Lead contact, Fathia Mami-Chouaib (fathia.mami-chouaib@gustaveroussy.fr)
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The RNA sequencing dataset comparing the transcriptome of CD8+CD103+ cells with CD8+CD103- cells, as shown in Figures 3, 4, 5, S3, and S4, Tables generated during this study are available at European Genome-phenome Archive (EGA) under EGAS00001004707 access number study.
Experimental Model and Subject Details
Patient cohort characteristics and tumor samples
We conducted a monocentric retrospective study of a discovery cohort of 111 and a validation cohort of 41 advanced NSCLC patients receiving treatment with PD-1/PD-L1 inhibitors in a variety of settings covering routine clinical care, expanded access, compassionate-use programs and clinical trials (nivolumab, pembrolizumab, atezolizumab or durvalumab) at Gustave Roussy between November 2012 and February 2020. Demographic, clinical, pathological and molecular data were also collected (Tables S1 and S4). Radiological assessments were performed every 8 weeks per RECIST v1.1 and per the investigator’s discretion. Fast-progressors were defined as patients who died during the first 12 weeks since the start of immunotherapy. This study was approved by the Institutional Review Board of Gustave Roussy (Commission scientifique des Essais thérapeutiques [CSET]) and informed consent was obtained. Freshly resected lung tumors and adjacent healthy lung tissue samples were obtained from the Institut mutualiste Montsouris and the Hôpital Marie-Lannelongue. For these resected tissues, the sex and the gender of the subjects are not known.
Healthy donor blood samples were collected from the French blood bank (Etablissement Français du Sang (EFS); agreement number N°12/EFS/079). The sex and the gender of the subjects are not known.
Method Details
Immunohistochemical staining
IHC was performed on archived FFPE tumor tissues using Ventana Benchmark and Discovery automated platforms. Briefly, after deparaffinisation and epitope retrieval in CC1 buffer (pH = 8, 36 min at 95°C), tissue sections were incubated with primary mAb for ICAM-1 (clone N1C2, GeneTex, 1:200 dilution) or E-cadherin (clone EP6, Bio SB, prediluted) for 1 h at room temperature. Amplification and detection steps used the ultraview kit with amplification, and 3,3′-diaminobenzidine was used as a chromogen. ICAM-1 staining was evaluated as H-score (0 to 300) based on the percentage of tumor cells stained at each intensity (scale from 0 to 3). Since the intensity of E-cadherin staining was homogeneous within each individual tumor sample, we scored E-cadherin staining as the prominent intensity for each tumor on a semiquantitative scale from 0 to 3.
Multiplexed fluorescent IHC for CD8 TRM was performed by sequential staining of a single tissue section with anti-CD8 (clone SP16, Spring Bioscience, 1:200), anti-CD103 (clone EPR4166-2, Abcam, 1:200) and anti-cytokeratin (clones AE1/AE3, Agilent, 1:100). For each staining, the HRP-conjugated amplification system was associated with a tyramide-coupled fluorophore: Opal 690, Opal 250 and Opal 570, respectively. Multispectral fluorescent images were captured using the Vectra 3 microscope (PerkinElmer) and regions of interest were selected. Image analysis using InForm software (PerkinElmer) included spectral unmixing, tissue segmentation (stromal versus epithelial areas) using a trainable classifier, nuclei detection based on dapi staining and cell segmentation followed by cell phenotyping for identification of cell populations defined by the combination of individual markers. The density (number of cells per square mm) of CD8+, CD103+ CD8+ and CD103-CD8+ cells was determined for each tumor sample in the total tumor area, as well as in the stromal and intraepithelial compartments, based on tissue segmentation. Results from image analysis were validated for all cases.
Tumor cells and tissue-infiltrating lymphocytes isolation
Freshly resected lung tumors and adjacent healthy lung tissue samples were immediately cut into small fragments and digested for 40 min at 37°C using the tumor dissociation kit (Miltenyi Biotech). The dissociated samples were smashed on 100 μm cell strainers, washed, and red blood cell lysis was performed. CD8 T lymphocytes were positively selected using CD8 microbeads according to the manufacturer’s instructions (Miltenyi Biotec). Recovered cells were either used for phenotypic analyses or further sorted by BD FACSAriaIII or BDFusion cell sorter (BD Biosciences) using anti-CD8-Pacific blue (RPA-T8, Biolegend), anti-CD103-FITC (Ber-ACT8, Biolegend) and anti-KLRG1-PE (clone 13F12F2, ebioscience) mAb. Dead cells were excluded using DAPI. CD103+CD8+ and KLRG1+CD8+ T cell populations were isolated and then either stored at −80°C for further DNA or RNA isolation or cultured for 2-5 days in the presence of low doses of IL-2 (20 U/ml) for functional studies. Tumor cells were recovered from the negative fraction of the CD8 T cell isolation described above, and then purified using a human tumor cell isolation kit (Miltenyi Biotec).
RNA and TCR sequencing and analyses
Total RNA was extracted from each sorted CD103+CD8+ and KLRG1+CD8+ TIL population pair and autologous healthy lung CD103+CD8+ lymphocytes when available; 150,000 lymphocytes per sample were processed using a single cell RNA purification kit (Norgen) according to the manufacturer’s instructions. RNA integrity (RNA Integrity Score ≥ 00007.0) was checked on the Agilent 2100 Bioanalyzer and quantity was determined using Qubit (Invitrogen). The SureSelect Automated Strand-Specific RNA Library Preparation Kit was used according to the manufacturer’s instructions with the Bravo Platform. Briefly, 50 ng of total RNA sample were used for poly-A mRNA selection using oligo(dT) beads and subjected to thermal mRNA fragmentation. The fragmented mRNA samples were subjected to cDNA synthesis and were further converted into double-stranded DNA using the reagents supplied in the kit, and the resulting dsDNA was used for library preparation. The final libraries were bar-coded, purified, pooled in equal concentrations and subjected to paired-end sequencing on a HiSeq-2000 sequencer (Illumina). Fast quality was assessed using FastQC (v 0.11.3) and did not require further trimming or adaptor removal. Counting of reads over the transcriptome was performed over Gencode (v 19, GRCh37.p13) with Salmon55 (v 0.8.2), using non-oriented library, 100 bootstrap and sequence bias correction. All other parameters were left to default. The pseudo-mapping rates were between 79% and 88% of overall reads. Differential analysis was performed within the R (v 3.2.3) environment with sleuth56 (v 0.28.1), wasabi (v 0.1) and in-house scripts. PCA highlights inter-individual variations that are far above any variation of interest, which was confirmed by a likelihood-test ratio and taken into account within the Wald test.
Primary functional analyses were performed using Ingenuity Pathway Analysis and GSEA was performed with clusterProfiler57 (v 3.8.0). RNaseq are available at the European Molecular Biology Laboratory European Bioinformatics Institute database (https://www.ebi.ac.uk/arrayexpress). TCR repertoire analysis was performed with a MiXCR package58 (v 2.1.10). KEGG pathway: https://www.genome.jp/kegg-bin/show_pathway?hsa04660.
For TCRseq, total DNA from CD103+CD8+ and KLRG1+CD8+ TIL subset pairs and autologous healthy lung CD103+CD8+ cells when available, sorted from nine NSCLC patient tissues, was purified using a QIAamp DNA micro kit (QIAGEN). DNA was quantified with fluorescence-based measurement Qubit (Life Technologies). TCRβ-CDR3 sequencing was performed by ImmunoSEQ, Adaptive Biotechnologies (Seattle). Raw data of TCR reads and sequences were uploaded on the ImmunoSEQ Analyzer Platform (Adaptive Biotechnologies).
For quantitative (q) RT-PCR, total RNA was extracted from sorted cell populations using the single cell RNA purification kit (Norgen Biotek). cDNA were synthesized using the Maxima first strand cDNA synthesis kit (Thermo-Fischer Scientific). qRT-PCR was performed on a step-one plus (Applied Biosystems) using Maxima SYBR Green Master Mix (ThermoFischer Scientific). Expression levels of transcripts were normalized to 18S housekeeping gene. PCR primers for human, TCF7, IKZF3, AHR, RORC, IL17 and 18S genes were designed by Sigma-Aldrich and used according to the manufacturer’s recommendations (Table S10).
Flow cytometry and ELISA
Phenotypic analyses were performed by direct immunofluorescence with a panel of fluorochrome-conjugated antibodies. Anti-CD3-Alexa700 (UCHT1), anti-CD8-PacificBlue (RPA-T8), anti-CD69-APC-Cy7 (FN50), anti-granzyme-B-FITC (GB11), anti-TNFα-PE/Dazzle594 (Mab11), anti-Aiolos-Alexa488 (16D9C97), anti-Vα7.2-APC-Cy7 (3C10), anti-CD161-Alexa488 (HP-3G10), anti-TCF-1-PE (7F11A10), anti-4-1BB-PE-Dazzle594 (clone 4B4-1) and anti-TCRγδ-PerCPCy5.5 (B1) were supplied by BioLegend. Anti-CD103-BV711 (Ber-ACT8), anti-IFNγ-APC (B27) and anti-Hobit-Alexa647 (Sanquin-Hobit/1) was purchased from BD Biosciences, anti-KLRG1-PE (13F12F2), anti-CD49-PerCPefluor710 (TS2/7), anti-S1PR1-APC (SW4GYPP), anti-PD-1-PeCy7 (eBioJ105), anti-AHR-PerCPefluor710 (FF3399), anti-Ki67-PerCPefluor710 (20Raj1) were supplied by Thermo Fisher Scientific. Anti-CD45RA-APC, anti-pSTAT3pS727-APC, anti-CD39-APC, anti-IL17A-PeVio770 and anti-Tbet-APC (REA102) were purchased from Miltenyi. For intracellular expression of IFNγ, TNFα and IL-17A, cells were stimulated for 4 h with PMA (50 ng/ml) plus ionomycine (1 μg/ml) in the presence of BrefeldinA (1 μg/ml, ebioscience). Cells were fixed, permeabilized (FoxP3 buffers Kit, ebioscience) and then stained with fluorochrome-conjugated mAb. Dead cells were excluded using a LIVE/DEAD Fixable UV dead cell stain kit (Thermo Fisher Scientific). Stained cells were analyzed by flow cytometry using a BD FACS Fortessa flow cytometer (BD Biosciences). Data were processed using FlowJo V10 software (Tree Star Inc.).
For ELISA, sorted CD8+ T lymphocytes were stimulated overnight with PMA plus ionomycine, and then supernatants were stored until IL-17 dosage (eBiosciences).
Th17-positive control cells were generated from healthy donor’s CD4+ peripheral blood lymphocytes (PBL) stimulated with anti-CD3 and anti-CD28 in the presence of TGF-β and IL-6.
Conjugate formation and cytotoxic experiments
Formation of stable conjugates between T cells and autologous tumor cells was analyzed by confocal microscopy. Effector and target cells were co-cultured for 30 min at 1:1 E:T ratio, and then plated on poly-(L-lysine)-coated coverslips (Sigma-Aldrich, Saint-Louis, MO). Cells were then fixed, permeabilized as described16 and stained with mouse anti-phospho-tyrosine (PY20, BD Biosciences) and rabbit anti-CD8 (Thermo Fisher Scientific), followed by anti-mouse AlexaFluor-488 and anti-rabbit AlexaFluor-647 (Thermo Fisher Scientific). Coverslips were mounted and analyzed using a fluorescence microscope (Leica, HR Sp8) with x63 lenses, and polarization of phospho-Tyr to the immune synapse between T cells and target cells was calculated. Stable conjugates were defined by polarization of p-Tyr at the contact zone between effector cells and tumor cells. Cytotoxic activity was evaluated using the conventional 4 h 51Cr-release assay as described.16
Quantification and Statistical Analysis
ORR was defined as complete (CR) plus partial response (PR). OS was calculated from the date of first immunotherapy administration until death due to any cause. PFS was calculated from the date of first immunotherapy until disease progression or death due to any cause. Comparisons between patient characteristics were performed using Chi-square or Fisher’s exact test for discrete variables and the unpaired t test, Wilcoxon sign-rank test or analysis of variance for continuous variables. The best cut-point for total CD8+, CD103+ and CD103- TIL was assessed using the log-rank maximization method.59 Survival analyses were performed using the Kaplan-Meier method and the log-rank test. All P values inferior to 0.05 were considered statistically significant.
A Cox proportional hazards regression model was used to evaluate independent prognostic factors for OS and PFS. Variables included in the final multivariate model were selected according to their clinical relevance and statistical significance in univariate analysis (p value cut-off = 0.10). The proportional hazard hypothesis was verified with the Schoenfeld residual method. Predictive factors of disease control were tested with logistic regression in univariate and multivariate analyses. The alpha level was 5%. Statistical analyses were performed with R (free software environment for statistical computing and graphics).
Statistical significance was determined with the paired or unpaired Student t test, or with the one-way analysis of variance (ANOVA) test with Bonferroni correction. Data are presented as mean ± SEM. All statistical details of experiments can be found in Figures and Figure legends; n = number of patient or tumor/lung samples. Statistical analyses were performed with GraphPad Prism software V8 (GraphPad Software Inc., San Diego, CA). ∗ p < 0.05; ∗∗ p < 0.01; ∗∗∗ p < 0.001 and RStudio software.
Acknowledgments
This work was supported by grants from the Institut National du Cancer (INCa; PLBIO016-080 grant no. 10557); the “Association pour la Recherche sur le Cancer (ARC; grant nos. PJA20161204720, SIGN’IT20181007792, and PJA20181208049), Ligue Contre le Cancer (Comité des Yvelines, grant no. 9FI12414QLCZ and Comité du Val de Marne, 2019) and Bristol-Myers Squibb (BMS, France; grant no. CA209-942). S. Corgnac was supported by a grant from the Groupement des Entreprises Françaises dans la Lutte Contre le Cancer (GEFLUC; grant no. 2016-R16180LL) and Cancéropôle Ile de France, and was a recipient of a fellowship from Fondation Recherche Medicale (FRM), Gustave Roussy (SIRIC-SOCRATE), and INCa (PLBIO016-080 grant no. 10557). I.M. was the recipient of a fellowship from the Ligue National Contre le Cancer. M.L. was a recipient of a MENRT fellowship from the French Ministry of Research, a fellowship from the Ligue Contre le Cancer and Gustave Roussy (SIRIC-SOCRATE), and a Taxe d’apprentissage TA-2016 from École des Sciences du Cancer de Gustave Roussy/Université Paris-Sud. We thank Georges Bismuth from Cochin Hospital and Eric Tartour from Hôpital Européen Georges Pompidou for critically reading this manuscript. We also thank Yann Lecluse, Cyril Catelain and Philippe Rameau from the cytometry facility (Plateforme d’Imagerie-Cytométrie), Virginie Marty from the platform of histocytopathology (Centre de Ressources Biologiques [CRB]), and Yahia Adnani, Aziza Caidi, Guillaume Meurice and Marc Deloger from the platform of bioinformatics (INSERM US23, CNRS UMS3655) of Gustave Roussy for their help with flow cytometry, lung tumor specimens, and RNA-seq analyses and data file deposit, respectively. We are grateful to José-Carlos Benitez-Montanez, Jihène Lahmar-Bach-Hamba, Jordi Remon-Masip, Maud Ngocamus, and Claudio Nicotra from the Department of Cancer Medicine of Gustave Roussy for their help in establishing patient tumor cohorts. We also thank Vassili Soumelis from Institut Curie for help with the RNA extraction protocol and Caroline Communaux from the CRB of Marie Lannelongue Hospital for help with fresh human NSCLC tumors. The graphical abstract was created with BioRender.
Author Contributions
Conception and design, S. Corgnac. and F.M.-C. Development of Methodology, S. Corgnac, I.M., N.D., J.A., and F.M.-C. Data Acquisition (e.g., providing animals, acquiring and managing patients, providing facilities), S. Corgnac, I.M., L.M., E.A., H.H., T.D., M.L., E.V., J.K., L.G., N.S., P.V., V.d.M., O.M., J.-Y.S., C.M., S. Chouaib, D.P., J.A., B.B., and F.M.-C. Data Analysis & Interpretation (e.g., statistical analysis, biostatistics, computational analysis), S. Corgnac, E.A., L.M., J.A., B.B., and F.M.-C. Writing, Reviewing, and/or Revision of Manuscript, S. Corgnac and F.M.-C. Administrative, Technical, and Material Support (e.g., reporting and organizing data, constructing databases), S. Corgnac, I.M., L.M., E.A., P.V., V.d.M., O.M., J.-Y.S., C.M., B.B., and F.M.-C. Study Supervision, F.M.-C.
Declaration of Interests
B.B.’s sponsored research at Gustave Roussy Cancer Center consists of the following: Abbvie, Amgen, AstraZeneca, BeiGene, Blueprint Medicines, BMS, Boehringer Ingelheim, Celgene, Cristal Therapeutics, Daiichi-Sankyo, Eli Lilly, GSK, Ignyta, IPSEN, Inivata, Janssen, Merck KGaA, MSD, Nektar, Onxeo, OSE Immunotherapeutics, Pfizer, Pharma Mar, Roche-Genentech, Sanofi, Servier, Spectrum Pharmaceuticals, Takeda, Tiziana Pharma, and Tolero Pharmaceuticals.
Published: October 20, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrm.2020.100127.
Supplemental Information
References
- 1.Tumeh P.C., Harview C.L., Yearley J.H., Shintaku I.P., Taylor E.J., Robert L., Chmielowski B., Spasic M., Henry G., Ciobanu V. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGranahan N., Furness A.J., Rosenthal R., Ramskov S., Lyngaa R., Saini S.K., Jamal-Hanjani M., Wilson G.A., Birkbak N.J., Hiley C.T. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Webb J.R., Milne K., Watson P., Deleeuw R.J., Nelson B.H. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin. Cancer Res. 2014;20:434–444. doi: 10.1158/1078-0432.CCR-13-1877. [DOI] [PubMed] [Google Scholar]
- 4.Djenidi F., Adam J., Goubar A., Durgeau A., Meurice G., de Montpréville V., Validire P., Besse B., Mami-Chouaib F. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J. Immunol. 2015;194:3475–3486. doi: 10.4049/jimmunol.1402711. [DOI] [PubMed] [Google Scholar]
- 5.Ganesan A.P., Clarke J., Wood O., Garrido-Martin E.M., Chee S.J., Mellows T., Samaniego-Castruita D., Singh D., Seumois G., Alzetani A. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat. Immunol. 2017;18:940–950. doi: 10.1038/ni.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nizard M., Roussel H., Diniz M.O., Karaki S., Tran T., Voron T., Dransart E., Sandoval F., Riquet M., Rance B. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun. 2017;8:15221. doi: 10.1038/ncomms15221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Savas P., Virassamy B., Ye C., Salim A., Mintoff C.P., Caramia F., Salgado R., Byrne D.J., Teo Z.L., Dushyanthen S., Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab) Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018;24:986–993. doi: 10.1038/s41591-018-0078-7. [DOI] [PubMed] [Google Scholar]
- 8.Clarke J., Panwar B., Madrigal A., Singh D., Gujar R., Wood O., Chee S.J., Eschweiler S., King E.V., Awad A.S. Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J. Exp. Med. 2019;216:2128–2149. doi: 10.1084/jem.20190249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mackay L.K., Stock A.T., Ma J.Z., Jones C.M., Kent S.J., Mueller S.N., Heath W.R., Carbone F.R., Gebhardt T. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc. Natl. Acad. Sci. USA. 2012;109:7037–7042. doi: 10.1073/pnas.1202288109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hofmann M., Pircher H. E-cadherin promotes accumulation of a unique memory CD8 T-cell population in murine salivary glands. Proc. Natl. Acad. Sci. USA. 2011;108:16741–16746. doi: 10.1073/pnas.1107200108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ray S.J., Franki S.N., Pierce R.H., Dimitrova S., Koteliansky V., Sprague A.G., Doherty P.C., de Fougerolles A.R., Topham D.J. The collagen binding alpha1beta1 integrin VLA-1 regulates CD8 T cell-mediated immune protection against heterologous influenza infection. Immunity. 2004;20:167–179. doi: 10.1016/s1074-7613(04)00021-4. [DOI] [PubMed] [Google Scholar]
- 12.McNamara H.A., Cai Y., Wagle M.V., Sontani Y., Roots C.M., Miosge L.A., O’Connor J.H., Sutton H.J., Ganusov V.V., Heath W.R. Up-regulation of LFA-1 allows liver-resident memory T cells to patrol and remain in the hepatic sinusoids. Sci. Immunol. 2017;2:eaaj1996. doi: 10.1126/sciimmunol.aaj1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mackay L.K., Minnich M., Kragten N.A., Liao Y., Nota B., Seillet C., Zaid A., Man K., Preston S., Freestone D. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science. 2016;352:459–463. doi: 10.1126/science.aad2035. [DOI] [PubMed] [Google Scholar]
- 14.Hombrink P., Helbig C., Backer R.A., Piet B., Oja A.E., Stark R., Brasser G., Jongejan A., Jonkers R.E., Nota B. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat. Immunol. 2016;17:1467–1478. doi: 10.1038/ni.3589. [DOI] [PubMed] [Google Scholar]
- 15.Milner J.J., Toma C., Yu B., Zhang K., Omilusik K., Phan A.T., Wang D., Getzler A.J., Nguyen T., Crotty S. Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature. 2017;552:253–257. doi: 10.1038/nature24993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le Floc’h A., Jalil A., Vergnon I., Le Maux Chansac B., Lazar V., Bismuth G., Chouaib S., Mami-Chouaib F. Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J. Exp. Med. 2007;204:559–570. doi: 10.1084/jem.20061524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anikeeva N., Somersalo K., Sims T.N., Thomas V.K., Dustin M.L., Sykulev Y. Distinct role of lymphocyte function-associated antigen-1 in mediating effective cytolytic activity by cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA. 2005;102:6437–6442. doi: 10.1073/pnas.0502467102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnston R.J., Comps-Agrar L., Hackney J., Yu X., Huseni M., Yang Y., Park S., Javinal V., Chiu H., Irving B. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. 2014;26:923–937. doi: 10.1016/j.ccell.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 19.Guo X., Zhang Y., Zheng L., Zheng C., Song J., Zhang Q., Kang B., Liu Z., Jin L., Xing R. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018;24:978–985. doi: 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 20.Duhen T., Duhen R., Montler R., Moses J., Moudgil T., de Miranda N.F., Goodall C.P., Blair T.C., Fox B.A., McDermott J.E. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018;9:2724. doi: 10.1038/s41467-018-05072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parkhurst M., Gros A., Pasetto A., Prickett T., Crystal J.S., Robbins P., Rosenberg S.A. Isolation of T-Cell Receptors Specifically Reactive with Mutated Tumor-Associated Antigens from Tumor-Infiltrating Lymphocytes Based on CD137 Expression. Clin. Cancer Res. 2017;23:2491–2505. doi: 10.1158/1078-0432.CCR-16-2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gros A., Robbins P.F., Yao X., Li Y.F., Turcotte S., Tran E., Wunderlich J.R., Mixon A., Farid S., Dudley M.E. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Invest. 2014;124:2246–2259. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takada K., Wang X., Hart G.T., Odumade O.A., Weinreich M.A., Hogquist K.A., Jameson S.C. Kruppel-like factor 2 is required for trafficking but not quiescence in postactivated T cells. J. Immunol. 2011;186:775–783. doi: 10.4049/jimmunol.1000094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Skon C.N., Lee J.Y., Anderson K.G., Masopust D., Hogquist K.A., Jameson S.C. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013;14:1285–1293. doi: 10.1038/ni.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cepek K.L., Shaw S.K., Parker C.M., Russell G.J., Morrow J.S., Rimm D.L., Brenner M.B. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E beta 7 integrin. Nature. 1994;372:190–193. doi: 10.1038/372190a0. [DOI] [PubMed] [Google Scholar]
- 26.Shiow L.R., Rosen D.B., Brdicková N., Xu Y., An J., Lanier L.L., Cyster J.G., Matloubian M. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440:540–544. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- 27.Simoni Y., Becht E., Fehlings M., Loh C.Y., Koo S.L., Teng K.W.W., Yeong J.P.S., Nahar R., Zhang T., Kared H. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557:575–579. doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 28.Scott A.C., Dundar F., Zumbo P., Chandran S.S., Klebanoff C.A., Shakiba M., Trivedi P., Menocal L., Appleby H., Camara S.J. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019;571:270–274. doi: 10.1038/s41586-019-1324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mackay L.K., Braun A., Macleod B.L., Collins N., Tebartz C., Bedoui S., Carbone F.R., Gebhardt T. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J. Immunol. 2015;194:2059–2063. doi: 10.4049/jimmunol.1402256. [DOI] [PubMed] [Google Scholar]
- 30.Guan T., Dominguez C.X., Amezquita R.A., Laidlaw B.J., Cheng J., Henao-Mejia J., Williams A., Flavell R.A., Lu J., Kaech S.M. ZEB1, ZEB2, and the miR-200 family form a counterregulatory network to regulate CD8+ T cell fates. J. Exp. Med. 2018;215:1153–1168. doi: 10.1084/jem.20171352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quintana F.J., Jin H., Burns E.J., Nadeau M., Yeste A., Kumar D., Rangachari M., Zhu C., Xiao S., Seavitt J. Aiolos promotes TH17 differentiation by directly silencing Il2 expression. Nat. Immunol. 2012;13:770–777. doi: 10.1038/ni.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H., van der Leun A.M., Yofe I., Lubling Y., Gelbard-Solodkin D., van Akkooi A.C.J., van den Braber M., Rozeman E.A., Haanen J., Blank C.U. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell. 2019;176:775–789.e18. doi: 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willinger T., Freeman T., Herbert M., Hasegawa H., McMichael A.J., Callan M.F. Human naive CD8 T cells down-regulate expression of the WNT pathway transcription factors lymphoid enhancer binding factor 1 and transcription factor 7 (T cell factor-1) following antigen encounter in vitro and in vivo. J. Immunol. 2006;176:1439–1446. doi: 10.4049/jimmunol.176.3.1439. [DOI] [PubMed] [Google Scholar]
- 34.Brummelman J., Mazza E.M.C., Alvisi G., Colombo F.S., Grilli A., Mikulak J., Mavilio D., Alloisio M., Ferrari F., Lopci E. High-dimensional single cell analysis identifies stem-like cytotoxic CD8+ T cells infiltrating human tumors. J. Exp. Med. 2018;215:2520–2535. doi: 10.1084/jem.20180684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Im S.J., Hashimoto M., Gerner M.Y., Lee J., Kissick H.T., Burger M.C., Shan Q., Hale J.S., Lee J., Nasti T.H. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537:417–421. doi: 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sade-Feldman M., Yizhak K., Bjorgaard S.L., Ray J.P., de Boer C.G., Jenkins R.W., Lieb D.J., Chen J.H., Frederick D.T., Barzily-Rokni M. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell. 2018;175:998–1013.e20. doi: 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kurtulus S., Madi A., Escobar G., Klapholz M., Nyman J., Christian E., Pawlak M., Dionne D., Xia J., Rozenblatt-Rosen O. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1(-)CD8(+) Tumor-Infiltrating T Cells. Immunity. 2019;50:181–194.e6. doi: 10.1016/j.immuni.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller B.C., Sen D.R., Al Abosy R., Bi K., Virkud Y.V., LaFleur M.W., Yates K.B., Lako A., Felt K., Naik G.S. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019;20:326–336. doi: 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siddiqui I., Schaeuble K., Chennupati V., Fuertes Marraco S.A., Calderon-Copete S., Pais Ferreira D., Carmona S.J., Scarpellino L., Gfeller D., Pradervand S. Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity. 2019;50:195–211.e10. doi: 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 40.Yost K.E., Satpathy A.T., Wells D.K., Qi Y., Wang C., Kageyama R., McNamara K.L., Granja J.M., Sarin K.Y., Brown R.A. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019;25:1251–1259. doi: 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mielke L.A., Liao Y., Clemens E.B., Firth M.A., Duckworth B., Huang Q., Almeida F.F., Chopin M., Koay H.F., Bell C.A. TCF-1 limits the formation of Tc17 cells via repression of the MAF-RORγt axis. J. Exp. Med. 2019;216:1682–1699. doi: 10.1084/jem.20181778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheuk S., Schlums H., Gallais Sérézal I., Martini E., Chiang S.C., Marquardt N., Gibbs A., Detlofsson E., Introini A., Forkel M. CD49a Expression Defines Tissue-Resident CD8+ T Cells Poised for Cytotoxic Function in Human Skin. Immunity. 2017;46:287–300. doi: 10.1016/j.immuni.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ichiyama K., Sekiya T., Inoue N., Tamiya T., Kashiwagi I., Kimura A., Morita R., Muto G., Shichita T., Takahashi R., Yoshimura A. Transcription factor Smad-independent T helper 17 cell induction by transforming-growth factor-β is mediated by suppression of eomesodermin. Immunity. 2011;34:741–754. doi: 10.1016/j.immuni.2011.02.021. [DOI] [PubMed] [Google Scholar]
- 44.Tajima M., Wakita D., Satoh T., Kitamura H., Nishimura T. IL-17/IFN-γ double producing CD8+ T (Tc17/IFN-γ) cells: a novel cytotoxic T-cell subset converted from Tc17 cells by IL-12. Int. Immunol. 2011;23:751–759. doi: 10.1093/intimm/dxr086. [DOI] [PubMed] [Google Scholar]
- 45.Piet B., de Bree G.J., Smids-Dierdorp B.S., van der Loos C.M., Remmerswaal E.B., von der Thüsen J.H., van Haarst J.M., Eerenberg J.P., ten Brinke A., van der Bij W. CD8+ T cells with an intraepithelial phenotype upregulate cytotoxic function upon influenza infection in human lung. J. Clin. Invest. 2011;121:2254–2263. doi: 10.1172/JCI44675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woodberry T., Suscovich T.J., Henry L.M., August M., Waring M.T., Kaur A., Hess C., Kutok J.L., Aster J.C., Wang F. Alpha E beta 7 (CD103) expression identifies a highly active, tonsil-resident effector-memory CTL population. J. Immunol. 2005;175:4355–4362. doi: 10.4049/jimmunol.175.7.4355. [DOI] [PubMed] [Google Scholar]
- 47.Webb J.R., Wick D.A., Nielsen J.S., Tran E., Milne K., McMurtrie E., Nelson B.H. Profound elevation of CD8+ T cells expressing the intraepithelial lymphocyte marker CD103 (alphaE/beta7 Integrin) in high-grade serous ovarian cancer. Gynecol. Oncol. 2010;118:228–236. doi: 10.1016/j.ygyno.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 48.Franciszkiewicz K., Le Floc’h A., Jalil A., Vigant F., Robert T., Vergnon I., Mackiewicz A., Benihoud K., Validire P., Chouaib S. Intratumoral induction of CD103 triggers tumor-specific CTL function and CCR5-dependent T-cell retention. Cancer Res. 2009;69:6249–6255. doi: 10.1158/0008-5472.CAN-08-3571. [DOI] [PubMed] [Google Scholar]
- 49.Edwards J., Wilmott J.S., Madore J., Gide T.N., Quek C., Tasker A., Ferguson A., Chen J., Hewavisenti R., Hersey P. CD103+ Tumor-Resident CD8+ T Cells Are Associated with Improved Survival in Immunotherapy-Naïve Melanoma Patients and Expand Significantly During Anti-PD-1 Treatment. Clin. Cancer Res. 2018;24:3036–3045. doi: 10.1158/1078-0432.CCR-17-2257. [DOI] [PubMed] [Google Scholar]
- 50.El-Asady R., Yuan R., Liu K., Wang D., Gress R.E., Lucas P.J., Drachenberg C.B., Hadley G.A. TGF-beta-dependent CD103 expression by CD8(+) T cells promotes selective destruction of the host intestinal epithelium during graft-versus-host disease. J. Exp. Med. 2005;201:1647–1657. doi: 10.1084/jem.20041044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang B., Wu S., Zeng H., Liu Z., Dong W., He W., Chen X., Dong X., Zheng L., Lin T., Huang J. CD103+ Tumor Infiltrating Lymphocytes Predict a Favorable Prognosis in Urothelial Cell Carcinoma of the Bladder. J. Urol. 2015;194:556–562. doi: 10.1016/j.juro.2015.02.2941. [DOI] [PubMed] [Google Scholar]
- 52.Franciszkiewicz K., Le Floc’h A., Boutet M., Vergnon I., Schmitt A., Mami-Chouaib F. CD103 or LFA-1 engagement at the immune synapse between cytotoxic T cells and tumor cells promotes maturation and regulates T-cell effector functions. Cancer Res. 2013;73:617–628. doi: 10.1158/0008-5472.CAN-12-2569. [DOI] [PubMed] [Google Scholar]
- 53.Boutet M., Gauthier L., Leclerc M., Gros G., de Montpreville V., Théret N., Donnadieu E., Mami-Chouaib F. TGFβ Signaling Intersects with CD103 Integrin Signaling to Promote T-Lymphocyte Accumulation and Antitumor Activity in the Lung Tumor Microenvironment. Cancer Res. 2016;76:1757–1769. doi: 10.1158/0008-5472.CAN-15-1545. [DOI] [PubMed] [Google Scholar]
- 54.Pascutti M.F., Geerman S., Collins N., Brasser G., Nota B., Stark R., Behr F., Oja A., Slot E., Panagioti E. Peripheral and systemic antigens elicit an expandable pool of resident memory CD8(+) T cells in the bone marrow. Eur. J. Immunol. 2019;49:853–872. doi: 10.1002/eji.201848003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pimentel H., Bray N.L., Puente S., Melsted P., Pachter L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods. 2017;14:687–690. doi: 10.1038/nmeth.4324. [DOI] [PubMed] [Google Scholar]
- 57.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bolotin D.A., Poslavsky S., Mitrophanov I., Shugay M., Mamedov I.Z., Putintseva E.V., Chudakov D.M. MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods. 2015;12:380–381. doi: 10.1038/nmeth.3364. [DOI] [PubMed] [Google Scholar]
- 59.Hothorn T., Lausen B. On the Exact Distribution of Maximally Selected Rank Statistics. Comput. Stat. Data Anal. 2003;43:121–137. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing dataset comparing the transcriptome of CD8+CD103+ cells with CD8+CD103- cells, as shown in Figures 3, 4, 5, S3, and S4, Tables generated during this study are available at European Genome-phenome Archive (EGA) under EGAS00001004707 access number study.