

Summary
Expectations of machine learning (ML) are high for discovering new patterns in high-throughput biological data, but most such practices are accustomed to relying on existing knowledge conditions to design experiments. Investigations of the power and limitation of ML in revealing complex patterns from data without the guide of existing knowledge have been lacking. In this study, we conducted systematic experiments on such ab initio knowledge discovery with ML methods on single-cell RNA-sequencing data of early embryonic development. Results showed that a strategy combining unsupervised and supervised ML can reveal major cell lineages with minimum involvement of prior knowledge or manual intervention, and the ab initio mining enabled a new discovery of human early embryonic cell differentiation. The study illustrated the feasibility, significance, and limitation of ab initio ML knowledge discovery on complex biological problems.
Keywords: machine learning, ab initio knowledge discovery, single-cell RNA-seq, embryonic cell lineages
Graphical Abstract
Highlights
-
•
We explore the feasibility of ab initio knowledge discovery from scRNA-seq data
-
•
A combined ML strategy to infer cell lineages with minimum prior knowledge
-
•
It recovers basic developmental knowledge and suggests a new discovery
-
•
We discuss the power and limitation of ab initio knowledge discovery
The Bigger Picture
Machine learning (ML) has been shown to be powerful in many artificial intelligence tasks, so people expect it to be able to reveal patterns that even human experts may have difficulty discovering. Scientists are enthusiastic in using ML to analyze the complex biology underlying various single-cell genomics data, but most existing studies of this type are accustomed to relying on existing knowledge to design experiments. Such practices may miss important discoveries and leave the question open as to how far ML and data may go beyond the sphere of existing knowledge.
This study uses the example of cell lineages in early embryonic development to investigate the feasibility of machine-learning discovery of biological knowledge from data with minimum use of prior knowledge. We call the tasks ab initio knowledge discovery. The strategy and observations can act as a baseline for future efforts of discovering new knowledge from single-cell genomics data.
Machine learning (ML) is highly expected to reveal biological patterns from single-cell omics data, but most existing ML practices involve prior knowledge. We explore the feasibility of ab initio knowledge discovery from scRNA-sequencing data with minimum use of prior knowledge. A strategy combining unsupervised and supervised ML is shown to be powerful in recovering correct embryonic cell lineages and also suggests a new discovery. The observed successes and limitations suggest future directions for ab initio knowledge discovery.
Introduction
Machine learning (ML) has been shown to be powerful in many pattern recognition tasks such as image analysis and computer vision, natural language processing, medical data analysis, and tasks in many other fields.1, 2, 3, 4, 5 The success of ML in those scenarios has led to scientists expecting it to be also powerful in analyzing data in biological research.6 The task may look similar at first glance but in fact there is a significant paradigm shift in the nature of tasks. We are not interested in letting machines learn what scientists already know but hope that ML methods will help us discover unknown patterns underlying the data that challenge human expert analysis. A typical task is to identify unknown structures intrinsic in massive high-dimensional data and to infer underlying principles without the guide of existing knowledge or even without a clearly defined target. Instead of mimicking humans to complete certain tasks as in typical artificial intelligence scenarios, we expect ML methods to help discover new knowledge that human experts cannot find.
Single-cell genomics is playing important roles in current biological studies. High-throughput single-cell RNA sequencing (scRNA-seq) has generated a huge amount of high-dimensional data that are far beyond the capacity of human experts to analyze without the assistance of advanced computational methods. Various ML methods have been playing a major role in analyzing massive single-cell data.7, 8, 9, 10 A typical pipeline for single-cell genomic data analysis is gene selection, dimensionality reduction followed by clustering, visualization, and annotation.10, 11, 12, 13 In most (if not all) published single-cell genomics studies, we are accustomed to relying on existing biological knowledge, human expertise, and interactive tuning in steps such as selecting genes, deciding on reduced dimensionalities, choosing clustering granularity and visualization parameters, selecting trajectory models, and annotation based on known markers.13 Such practices are helpful for confirming the validity of data and ensuring that analyses are compatible with existing knowledge, but raise questions on the capacity of ML methods in discovering new knowledge from the data alone. On the other hand, emerging single-cell omics technologies are providing unprecedented resolution in studying the molecular properties of cells and are pushing the boundary of existing biological knowledge in many directions. The reliance on existing knowledge may bury the value of the new technology in revealing new knowledge that could not be seen with previous technologies. It is unclear in many scenarios whether discoveries from new data have been misled by possible biases in existing knowledge. Efforts are needed to systematically explore the power and limitation of ML methods in discovering biological knowledge from data in an ab initio manner with restricted or controlled involvement of existing knowledge and subjective judgment by human experts.
In this study, we selected a state-of-the-art scRNA-seq dataset of early human embryonic cell development14 and designed an experiment for ab initio knowledge discovery using basic ML methods with controlled involvement of human knowledge. The dataset contains scRNA-seq samples of embryonic day 3 (E3) to day 7 (E7), the important period in embryonic development from the 8-cell stage to pre-implantation embryos. This is a period rich of biological events. The corresponding biological knowledge is also rich, but many existing understandings were obtained from mouse studies.15, 16, 17 It has been reported that there are noticeable differences in many aspects of the early development of human and mouse embryos.14,18,19 We ignored all existing knowledge of embryonic development except the basic assumption that cells of a later day are developed from the earlier day in some unknown lineages. We experimented on the discovery of such lineages from the data using the combination of classic unsupervised and supervised ML methods with minimum involvement of prior knowledge or manual intervention. After a full ML-derived understanding of the developmental process was built, we compared it with existing knowledge and used the knowledge to annotate the ML-derived understanding. Results showed that ML-derived understanding can be well aligned to the latest knowledge, except that the ML-derived understanding included a new discovery on the differentiation of a small fraction of day-4 cells that can augment existing knowledge. We also conducted similar experiments on a mouse dataset20 and a zebrafish dataset21 of embryonic development, and observed various levels of success or failure in discovering more complicated relationships. These experiments highlighted the power and limitation of using current ML methods and scRNA-seq data to discover complicated biological knowledge ab initio, and showed the feasibility and significance of controlling the involvement of existing knowledge and subjective adjustment in mining new biological data.
Results
The Task and Strategy
The task of this ab initio knowledge discovery experiment is to identify the possible lineage relations among cells of each day (or hour) in the early embryonic development data14 without the guide of existing knowledge. We formulated it as a task of clustering and classification: If cells of the same day are of different lineages, there must be distinct clusters in cells of that day; and if the clusters of different days belong to the same lineage, there must be some correspondence between the clusters so that we can classify the lineage on one day using the model trained by the lineage on another day. We decomposed the task into the following subtasks: (1) choosing one day as the candidate reference day for other days; (2) building a candidate developmental process by finding relations among cells of different days based on the reference day; and (3) assessing the plausibility of the candidate developmental process. As no prior knowledge is taken in the experiment, it is difficult to decide beforehand which day is a proper reference for the other days. We took each day as the reference and fulfilled the task for each reference. In this way, we would obtain multiple candidate versions of the development process. We developed a method to infer which one is the most plausible by evaluating the self-consistency of each one.
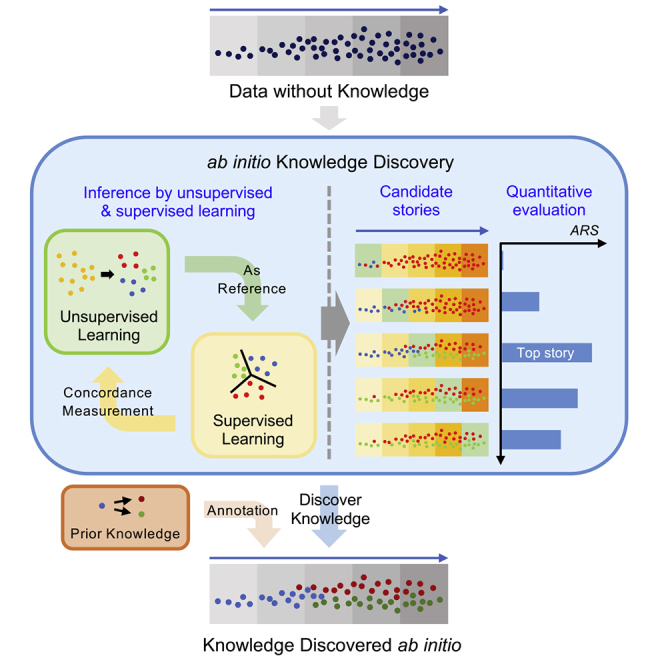
Figure 1 illustrates the overall scheme of the proposed method for ab initio discovery of developmental processes based on a number of samples collected at several time points in a developmental interval. Details of the method are described in Experimental Procedures.
Figure 1.

Overview of the Method
Unsupervised and supervised learning methods were used for building ML-derived understandings of the developmental process with each day as a potential reference. The number of clusters in each reference day can be decided using S-score and scree plots (not shown in figure) or can be exhaustively searched in a range. Multiple versions of developmental processes were constructed. A method was developed for comparing the multiple candidate processes to choose the one with highest self-consistency as the final ML-derived understanding. Existing knowledge was used in the last step to annotate the ML-derived developmental process and detect possible new findings.
We used human early embryo development data14 for the systematic experiment and analyses of this study. Most of the following subsections are based on this dataset. Extra experiments on the mouse and zebrafish data are discussed in the last two subsections.
Number of Clusters for Each Day
We conducted k-means clustering22 on cells of each day by experimenting from k = 1 to k = 9. Figure 2 shows the scree plot of the sum of errors with regard to the choice of k for each day, and Table 1 shows the Silhouette scores23 (S-scores) for each day. We can see that the elbow points on the scree plots are not obvious for most days, which implies that the clustering structures on all days are not very crisp based on the genes we used. Weak elbow points at k = 3 for day 5 and at k = 2 for days 6 and 7 can be perceived, plus an even weaker elbow point at k = 2 for day 4. This agrees with the highest S-scores on those days in Table 1. The S-score at k = 2 is the highest for day 3 but the scree plot of day 3 does not show any elbow point. It should be noted that S-score can only be calculated for k ≥ 2 by definition and therefore cannot be used to rule out the situation that all samples should be taken as one cluster. Based on these observations, we chose the number of clusters for days 3 to 7 as 2, 2, 3, 2, and 2, respectively, but took note that evidence for the existence of two clusters on day 3 and day 4 are weak, especially for day 3. To check the stability of k-means clustering, we did extra experiments with different initial centroids and found that the results were stable (Tables S7–S9).
Figure 2.

Scree Plots of Sum-of-Errors of k-Means Clustering on Each Day
The horizontal axis is the cluster number k. The vertical axis is the sum of errors of samples to cluster centers. Weak elbow points can be identified for day 4, day 5, day 6, and day 7 but not for day 3.
Table 1.
Silhouette Scores of Different Cluster Numbers in Each Day
| k | Day 3 | Day 4 | Day 5 | Day 6 | Day 7 |
|---|---|---|---|---|---|
| 2 | 0.3052a | 0.1275 | 0.1091 | 0.1440 | 0.1536 |
| 3 | 0.1417 | 0.1106 | 0.1139 | 0.0994 | 0.1274 |
| 4 | 0.1255 | 0.1065 | 0.1027 | 0.0810 | 0.0807 |
| 5 | 0.1339 | 0.0842 | 0.0781 | 0.0797 | 0.0808 |
| 6 | 0.1093 | 0.0840 | 0.0772 | 0.0625 | 0.0688 |
The numbers in bold fonts are the highest Silhouette score for each day, respectively.
Candidate Developmental Processes Built on Each Reference Day
Taking the clusters obtained for each day as the seeds for candidate lineages, we trained a support vector machine (SVM)24 classifier with the cells of each reference day and classified cells of all other days to the seed clusters. In this way, each reference day built up one candidate developmental process. We mapped the clustering and classification results on the plane of the first two principal components of each day to visualize the distributions of clusters and classes in the five candidate developmental processes (Figure 3). Table 2 shows the number of cells in each cluster or class of each day in the five candidate developmental processes.
Figure 3.

Distributions of Clusters and Classes in Five Candidate Developmental Processes Derived Using Each Day as the Reference
The plot matrix contains clustering and prediction results of day 3 to day 7 with each day used as reference day. Plots with gray background along the diagonal show the clusters of for each day used as seeds for candidate lineages. The other plots show the classification of cells of the other days to the seed clusters of the reference day in the same row.
Table 2.
Numbers of Cells in the Clusters of Reference Days and in the Classes of the Other Days
| Reference Day (No. of Clusters) | Number of Cells in Clusters/Classes |
||||
|---|---|---|---|---|---|
| Day 3 | Day 4 | Day 5 | Day 6 | Day 7 | |
| Day 3 (2) | (70, 11) | (190, 0) | (377, 0) | (415, 0) | (466, 0) |
| Day 4 (2) | (81, 0) | (110, 80) | (7, 370) | (0, 415) | (0, 466) |
| Day 5 (3) | (81, 0, 0) | (180, 10, 0) | (104, 152, 121) | (0, 261, 154) | (0, 219, 247) |
| Day 6 (2) | (3, 78) | (53, 137) | (212, 165) | (239, 176) | (209, 257) |
| Day 7 (2) | (4, 77) | (145, 45) | (301, 76) | (321, 94) | (236, 230) |
The numbers in bold font at the diagonal are the numbers of cells in each cluster of each reference day. The numbers at other positions of the matrix are the numbers of cells classified to each class using the clusters at the diagonal location of the matrix as the reference.
Five versions of developmental stories can be made up for the five candidate developmental processes. For the one with day 3 as the reference (first row in Figure 3), we see that cells of day 3 are of two clusters in their gene expression patterns, but one cluster disappeared on day 4 and there is only one lineage thereafter. This is biological nonsense considering the order of development, but as we did not involve biological knowledge in this phase we avoided this type of human judgment to evaluate the candidate processes. What makes this candidate process not acceptable from the data themselves is the fact that this story indicates that all cells of other days are of only one cluster. This is in strong conflict with the observation on the number of clusters on other days. For a candidate developmental process to be plausible, we expect it to provide consistent conclusions on the nature of cell heterogeneity on each day from the unsupervised clustering and supervised classification.
The story based on day 4 as reference tells us that all cells are of the same cluster on day 3 and that one new cluster appears on day 4. The cluster on day 3 disappears in later development, leaving most of the cells on day 5 and all cells on day 6 and day 7 being of the other cluster. This candidate process offers a richer storyline, but also has major conflicts with the number of clusters observed for day 5, day 6, and day 7. Similar analyses on the developmental stories based on the other three candidate processes can be done in the same way. Table 3 presents a summary.
Table 3.
Summary of the Five ML-Derived Candidate Developmental Stories
| Story Index | Reference Day | Summary of the ML-Derived Developmental Process |
|---|---|---|
| Story #3 | Day 3 | Two lineages on day 3. One disappears on day 4 and all cells of days 4–7 are of the same lineage of day 3 |
| Story #4 | Day 4 | One lineage on day 3. A new lineage appears on day 4. Most of the cells on day 5 and all cells on days 6 and 7 are of the new lineage from day 4. The lineage from day 3 almost disappears on day 5 and disappears thereafter |
| Story #5 | Day 5 | One lineage on day 3. A minor new lineage appears on day 4. It becomes larger on day 5, and another new lineage appears on day 5. The lineage from day 3 disappears on day 6 and thereafter and the two new lineages continue |
| Story #6 | Day 6 | A major lineage and a minor lineage on day 3. The minor lineage becomes larger from day 4. The two lineages continue thereafter |
| Story #7 | Day 7 | A major lineage and a minor lineage on day 3. The minor lineage becomes much larger from day 4. The two lineages continue thereafter |
By comparing the cell numbers in the unsupervised learning results and supervised learning results, we can come to the conclusion that the candidate developmental process derived with day 5 as the reference is the most plausible. The developmental process can be described in the following way. All cells of day 3 are of the same type (cluster A5). A few cells of a new type (cluster B5) appeared on day 4 while most other cells are still of A5. On day 5, the new B5 cluster becomes larger, a new cell type (cluster C5) appears, and cells of the earlier A5 type become a smaller fraction. On days 6 and 7, cells of the earlier A5 type disappear and only cells of types B5 and C5 remain. Considering the fact that the scree plot indicated the weakest evidence of having two or more clusters on day 3, this story has no major conflict with all other observations.
Quantitative Evaluation of the Candidate Development Processes
The above analyses pointed out the most plausible ML-derived understanding of the developmental process. The reasoning was qualitative and required manual inference and judgment, although no biological knowledge was used. We proposed the following method for automatic judgment on the ML results. We applied quantitative measurement of self-consistency on each candidate process using the reliability scores we defined (Experimental Procedures). Table 4 shows the results. In agreement with the above qualitative analysis, the quantitative evaluation results clearly show that the developmental process with day 5 as reference has the highest adjusted reliability scores (ARSs) and is the most plausible, and we therefore took it as the ML-derived knowledge discovered ab initio from the single-cell gene expression data.
Table 4.
Concordance and Reliability Scores of Each Day and Candidate Development Process
| Reference Day (r) | Concord (i|r) |
Reliab (r) | ARS (r) | ||||
|---|---|---|---|---|---|---|---|
| i = 3 | i = 4 | i = 5 | i = 6 | i = 7 | |||
| Day 3 | – | 0 | 0 | 0 | 0 | 0 | 0 |
| Day 4 | 0 | – | 0.008 | 0 | 0 | 0.002 | 0.003 |
| Day 5 | 0a | 0.04 | – | 0.78 | 0.73 | 0.38 | 0.39 |
| Day 6 | −0.05 | 0.44 | 0.44 | – | 0.75 | 0.39 | 0.25 |
| Day 7 | −0.06 | 0.07 | 0.11 | 0.36 | – | 0.12 | 0.18 |
The bold fonts highlight the row with the reference day that achieves the highest ARS value.
Exhaustive Searching of the Reference Day and Cluster Numbers
The building of the above candidate developmental processes was based on the selection of most proper cluster numbers based on the S-scores and scree plots. To eliminate the influence of the uncertainty in determining the cluster numbers, we conducted an exhaustive search of cluster numbers for each day as a potential reference. For each day, we experimented with cluster numbers k being set from 2 to 10, respectively, and used the obtained clusters as reference to classify cells of other days. For each setting, the predicted classes on target days were compared with clustering results of those days to obtain the concordance scores (“concord”) in the calculation of the ARS for the particular reference day and cluster number. In this way, we enumerated the best possible candidate developmental processes using each day as a reference and each choice of cluster numbers within the given range. Table 5 summarizes the ARSs for all enumeration results. We can see that the developmental process derived using the three clusters of day 5 as reference (“day5_clu3” in Table 5) gives the highest ARS among all enumerations. This confirmed the previous analyses based on manually chosen cluster numbers and provided a strategy for the inference with less manual intervention.
Table 5.
Adjusted Reliability Scores (ARS) of Each Enumerated Candidate Developmental Process
| Reference Day and Cluster Numbera | ARS | Reference Day and Cluster Numbera | ARS | Reference Day and Cluster Numbera | ARS |
|---|---|---|---|---|---|
| day3_clu2 | 0 | day4_clu8 | 0.1367 | day6_clu5 | 0.1426 |
| day3_clu3 | −0.0002 | day4_clu9 | 0.2079 | day6_clu6 | 0.2565 |
| day3_clu4 | −0.0003 | day4_clu10 | 0.2045 | day6_clu7 | 0.1824 |
| day3_clu5 | −0.0003 | day5_clu2 | 0.4220 | day6_clu8 | 0.1848 |
| day3_clu6 | −0.0013 | day5_clu3b | 0.4674b | day6_clu9 | 0.1812 |
| day3_clu7 | −0.0002 | day5_clu4 | 0.1936 | day6_clu10 | 0.1655 |
| day3_clu8 | −0.0004 | day5_clu5 | 0.2130 | day7_clu2 | 0.2434 |
| day3_clu9 | −0.0004 | day5_clu6 | 0.1703 | day7_clu3 | 0.1706 |
| day3_clu10 | −0.0003 | day5_clu7 | 0.2463 | day7_clu4 | 0.2497 |
| day4_clu2 | 0.0011 | day5_clu8 | 0.2408 | day7_clu5 | 0.2199 |
| day4_clu3 | 0.0166 | day5_clu9 | 0.2124 | day7_clu6 | 0.2552 |
| day4_clu4 | 0.0256 | day5_clu10 | 0.2317 | day7_clu7 | 0.1693 |
| day4_clu5 | 0.1123 | day6_clu2 | 0.4099 | day7_clu8 | 0.2074 |
| day4_clu6 | 0.0479 | day6_clu3 | 0.2362 | day7_clu9 | 0.2031 |
| day4_clu7 | 0.0610 | day6_clu4 | 0.1543 | day7_clu10 | 0.1816 |
day3_clu2 means using day 3 cells of 2 clusters as the reference for other days for building the candidate developmental process. The ARS measures the plausibility of each candidate story.
The bold fonts highlight the reference day and cluster number that achieves the highest ARS among all exhaustive search results.
Verification of the Ab Initio Discovery and Alignment to Known Biological Knowledge
Now that we had built up an ML-derived developmental process of embryonic cells from E3 to E7, we compared it with the existing biological knowledge and annotated the ML-derived lineages with biological lineages. According to the current understanding, from E3 to E7 human zygotes differentiate into three major embryonic cell types named pre-lineage, trophectoderm (TE) lineage, and inner cell mass (ICM) lineage.14,25 Cells of the pre-lineage are those that have not started differentiation. TE lineage segregates first, then primitive endoderm (PE) and epiblast (EPI) cells come from the intermediate lineage of ICM.14,26 Cells of different lineages play different roles in the embryogenesis. Cells in E3 and E4 belong to pre-lineage according to the current understanding. TE and ICM cells appear on E5 but there are still pre-lineage cells remaining on E5. ICM further segregates into EPI and PE on E5. By E6 and E7, all pre-lineage cells have differentiated into cells of either TE or ICM (EPI and PE) lineages.
Comparing this existing biological knowledge with the ML-derived developmental story in our discovery, it is straightforward to infer that cluster A5 corresponds to the pre-lineage because it is the sole cell type in E3. A minor disagreement between the ML-derived developmental process with the known biological lineages is that cells in E4 should all be pre-lineage according to the existing knowledge, but the ML-derived knowledge identified 10 “outlier” cells (out of the 190 cells) of E4 that were already differentiated.
The correspondence of clusters B5 and C5 to TE or ICM lineages cannot be inferred from the above reasoning. This is where extra information is needed besides the data themselves. To resolve this question, we took the clustering result of cells on day 5 with k = 4 and compared it with the clusters of k = 3 (Figure 4). Based on the existing knowledge that the ICM lineage is composed of two subtypes PE and EPI, we expected that one of the three clusters in the result of k = 3 would be split into two clusters when k = 4. As we can see in Figure 4, this happened for cluster C5, indicating that cluster C5 corresponds to the ICM lineage and cluster B5 corresponds to the TE lineage. With this extra step of inference guided by existing knowledge and human reasoning, the ML-derived ab initio knowledge discovery in this particular dataset has been fully verified and annotated.
Figure 4.

Comparison of Clustering Results on E5 cells with k = 3 and k = 4
(A) PCA plot of the three clusters.
(B) PCA plot of the four clusters. The cluster C5 when k = 3 is further separated into two subclusters C5 and D5 when k = 4.
New Discovery on Cell Differentiation in E4
The ML-derived understanding of the developmental process indicates that a minor proportion of cells in E4 already differentiated to TE cells (cluster B5). We drew the gene expression heatmap of all E4 cells (Figure 5A), which shows that gene expression patterns for those 10 cells are distinct from the majority of E4 cells. In Figure 5B, we drew the distribution of E5 cells in the plane of the first two principal components of E5 and mapped all E4 cells to this plane. We can see that while most E4 cells map to the region of pre-lineage cells (cluster A5), 10 × 104 cells map to the area of TE cells (cluster B5) on E5. This confirmed the existence of TE cells on E4. We also mapped all E3 cells to this plane, which mapped to the pre-lineage region (cluster A5) (Figure 5B). It is interesting that most E3 cells tend to map to the far end of the pre-lineage cluster while the E4 cells are scattered in an almost linear manner in the cluster with the 10 cells extending to the area of TE cells. Considering the observations from the scree plots that the distinction between clusters in the data are not sharp, we speculated that the gene expression patterns of pre-lineage cells with those of the TE cells are of a continuum rather than a clear switch. A minor proportion of E4 cells grow faster and differentiate to cells with TE properties before E5.
Figure 5.

Visualization of the New Discovery on Cell Differentiation in E4
(A) Expression heatmap of E4 cells. Each row represents one gene and each column represents one cell. The color of scale bar on the right shows the normalized expression level. The 10 “outlier” cells that have been speculated as early TE cells on day 4 by our ab initio discovery are shown at the leftmost side of the heatmap.
(B) PCA plot of day-5 cells with day-3 and day-4 cells mapped onto it. We can see that all E3 cells map to the pre-lineage region of E5 cells, and most E3 cells are in the far end of this cluster. Most E4 cells map to the pre-lineage region along a linear shape, with 10 cells extended into the TE region.
Experiments with Seurat Clustering
k-means is a classic clustering method that has been widely used in many fields, but the clustering method in Seurat10 is more widely used in single-cell data analysis. When we use Seurat clustering to replace k-means by manually adjusting the Seurat parameters for data of each day, the developmental knowledge discovered is almost identical to that described above (Figure S2). However, the tuning of parameters made such a discovery in a non-ab initio manner. We then adopted the exhaustive searching strategy to scan for parameters that lead to results with the highest ARS. The detailed experimental procedure is given in Supplemental Information. The top two choices of the reference day are day 5 with four clusters (ARS = 0.43) and day 5 with three clusters (ARS = 0.32). The corresponding stories visualized in principal component analysis (PCA) plots are shown in Figures S3 and S4, respectively.
It is interesting that the developmental stories with these two solutions are generally compatible with each other and with the story #5 discovered with k-means clustering, except that there is a minor new cluster D discovered among cells of days 3 to 5 in the new top story. Annotating with the prior knowledge, this new cluster tells us that a tiny portion of the pre-lineage cells on day 3 and day 4 belongs to a special subtype. This subtype can be found in a slightly larger proportion among cells on day 5 but disappears from day 6 onward. This subtype has not been reported in the literature but shows noticeable differences in gene expression profiles (Figure S5), perhaps implying some subtle heterogeneity among the pre-lineage cells.
Experiments on Mouse Embryonic Development Data
We applied the same strategy as we did on the human data for ab initio discovery of the candidate developmental processes to the mouse embryonic development data.20 The dataset contains 1,724 cells captured at embryonic days 5.25, 5.5, 6.25, and 6.5 (referred to as E5.25, E5.5, E6.25, and E6.5). The S-scores and scree plots indicated that the best cluster numbers for E5.25, E5.5, E6.25, and E6.5 are 3, 3, 3, and 2, respectively (Table S2 and Figure S6). Using these choices of cluster numbers to infer the candidate developmental process, the highest ARS (2.00) was obtained for the one with E5.25 as the reference, but the ARSs of candidate processes with references of E5.5 and E6.25 are 1.94 and 1.91, respectively, both very close to the highest ARS (Table S3). This suggests that those two time points may also be reasonable choices of reference. Figure 6 shows the plot matrix of the four candidate developmental processes in the same way as for the human data in Figure 3. Table S4 shows the number of cells in each cluster or class of each time point in the four candidate developmental processes. We can see that in fact the ML-derived developmental stories using E5.25, E5.5, or E6.25 as references are almost identical.
Figure 6.

Distributions of Clusters and Classes in Four Candidate Mouse Developmental Processes Derived Using Each Time Point as the Reference
The plot matrix contains clustering and prediction results of E5.25 to E6.5 with each time point used as reference. Plots with gray background along the diagonal show the clusters for each time point used as seeds for candidate lineages. The other plots show the classification of cells of the other time points to the seed clusters of the reference time point in the same row.
We also experimented with the exhaustive searching strategy for all four time points and cluster numbers from 2 to 10. Results also gave the highest ARS (2.2598) for “day5.25_clu3” (Table S5). This confirms that the most plausible developmental process is the one built with the three clusters of E5.25 cells as reference. This ML-derived development process tells us that there are three lineages from E5.25 to E6.5, without a significant differentiation event. Looking into the literature,20 we learned that there are three lineages in the mouse embryonic development from E5.25 to E6.5: epiblast (EPI), extraembryonic ectoderm (ExE), and visceral endoderm (VE). By comparing our data with the lineages reported in the original paper (EPI, ExE, and VE), we found that the ML-derived lineages (clusters A, B, and C) can be annotated with the biological lineages based on their proportion of cells (Table 6). We can see that both the fixed-k strategy and exhaustive-search strategy work well on this dataset in discovering the basic development knowledge ab initio.
Table 6.
Numbers of Cells in Biological Lineages and ML-Derived Clusters
| Total Numbers of Cells in Lineages Reported in Cheng et al.20 | 768 (lineage EPI) | 285 (lineage ExE) | 671 (lineage VE) |
| Total numbers of cells in the ML-derived lineage clusters | 789 (cluster A) | 285 (cluster B) | 650 (cluster C) |
It is interesting that differences between top and following ARS values in the mouse dataset (Table S5) are relatively smaller than those in the human dataset (Table 5). This reflects the different levels of complexity in the two datasets. The human data covered a period when cells develop from one lineage to three lineages, while the mouse data covered a period when cells remain in three lineages.
Experiments on Zebrafish Embryonic Development Data
We conducted the same series of experiments on the zebrafish embryonic development dataset.21 This contains 36,749 cells collected at seven time points during the zebrafish embryonic development, i.e., 4, 6, 8, 10, 14, 18 and 24 h post fertilization (hpf). We observed that S-score and scree plot tend to indicate small cluster numbers (mostly 2–3) for cells of each time point, but exhaustive searching picks up references with larger cluster numbers as the most plausible references. The highest ARSs obtained in the exhaustive searching are also much higher than highest ARSs obtained using the fixed-k strategy. We therefore chose to use the exhaustive searching result as the most plausible candidate developmental process.
With the zebrafish dataset, an exhaustive search identified the time point of 10 hpf of five clusters as the most plausible reference. Figures S7 and S8 show the PCA and t-distributed stochastic neighbor embedding (tSNE) plots of cells at each time point colored with the predicted classes. The ML-derived storyline of the developmental process is as follows. There are two lineages (clusters C and D) at 4 hpf. These two lineages continue all the way to 24 hpf. Two new lineages (clusters B and E) appear at 6 hpf, and another new lineage (cluster A) appear at 8 hpf. All these lineages continue to 24 hpf. In the original paper that published the data,21 the authors identified a total of 198 clusters at all time points (from four clusters in cells of 4 hpf to 72 clusters in cells of 24 hpf), but manually annotated them into 10 cell types using a series of marker genes. The cell types contain many scattered clusters in the tSNE plots, indicating complicated subtype structures and lineage relations. The ML-derived developmental process cannot be annotated to the biological lineages presented in the original paper because the resolutions are different. We used the differentially expressed genes (DEGs) among the cell types reported in the original paper to manually annotate the ML-derived developmental process (Table S6).
The ab initio discovery of the developmental process from this dataset only covers a draft outline of the true biological knowledge lineages with many details missed, and the annotation of the ML-derived process needs the assistance of known DEGs. We compared the nature of the human, mouse, and zebrafish datasets we experimented on in this study to understand why the proposed method works well on the first two datasets but has limited success with the zebrafish data. Looking into the basic knowledge on vertebrate development,27, 28, 29, 30 we realized that the sampling time points in the human data of 3–7 days post coitum (dpc) are approximately from Carnegie stage 2 to 5, long before the development of the first somite. The mouse data of 5.25–6.5 dpc are approximately from Carnegie stage 5–6, still before the first somite occurs. The zebrafish data from 4 to 24 hpf, however, actually span approximately Carnegie stage 7–12. During this period, the zebrafish goes through blastula (2.25–5.25 hpf), gastrula (5.25–10.33 hpf), and segmentation stages (10.33–24 hpf), and enters the pharyngula stage.31 At the end of 24 hpf, the zebrafish embryo already has more than 26 somites. From these facts, we can conceive that the zebrafish development data are beyond the scenario for which the proposed method was designed. The clustering of cells in the zebrafish data are decided not only by the developmental lineages but also by many other developmental factors such as somites and locations. Also, because of the complexity of the late development processes, there is no single time point at which the cells can represent all lineages that have appeared in the long developmental period. Although the ML-derived developmental process from this zebrafish dataset makes basic sense as a coarse outline, it reveals the limitation of the proposed method when the assumptions underlying the method cannot be met.
Discussion
ML has been shown to be powerful in solving many pattern recognition tasks more efficiently than humans and has been afforded great expectations in mining complicated biological data for possible new discoveries. Integrating data with existing knowledge is a convenient strategy for mining the data but may increase the possibility of biased discoveries if existing knowledge is imperfect. It is valuable to have a systematic evaluation of the power and limitation of what can be discovered from the data along with ML methods, without or with controlled involvement of existing knowledge. In this work, we designed an experiment to address this issue by conducting an ab initio knowledge discovery experiment on a set of single-cell gene expression data of early human embryonic development. We developed a method of integrating unsupervised and supervised learning for discovering the possible lineages of embryonic cell differentiation and invented a method to evaluate the reliability of the discovery by checking its self-consistency. The basic ML methods we used were k-means and SVM, but they could also be replaced by other methods (Supplemental Information; Figures S9 and S10). The purpose of these experiments was to investigate to what extent reliable biological knowledge can be derived ab initio from the data with minimum involvement of existing knowledge. Experimental results showed that with a properly designed methodology, ML can reveal the basic biological knowledge from single-cell gene expression data in an automatic manner. However, the discovered patterns need to be annotated with the help of existing knowledge and manual inference. This ab initio mining of single-cell data also revealed a subtle but important new discovery that updates existing knowledge.
We further explored whether the proposed method can be made as a general strategy for ab initio discovery of developmental lineages from time-series data along a developmental course. The basic principle is to combine unsupervised and supervised ML approaches to explore the gene expression heterogeneity of cells within and between time points to infer lineages, and to assess the reliability of the inference based on its self-consistency in the data. The major limitation of the method lies in its basic assumptions: (1) gene expression patterns caused by differentiation of lineages is the major source of heterogeneity in the cells of each sampled time point; and (2) there is a single sampling point at which the cell population can represent all types appearing in the development period. The experiments on the human and mouse developmental data showed that the proposed method works well when the assumptions are generally true. Obviously they are not always true for all experiments, as we have seen with the zebrafish data, which cover a much longer and later period in the development. When multiple sources of heterogeneity exist and no single sampling point can capture cells of all lineages, we will need more sophisticated methods, and more prior knowledge will probably have to be involved in designing the methods.
Trajectory inference (TI) is a category of methods for inferring developmental trajectories for a set of cells believed to be of different developmental time.32 They are usually used for ordering cells according to their inferred developmental time (pseudo-time), visualizing tree structures of developmental lineages and helping to find markers or patterns for certain critical events or processes along the trajectories. Different degrees of prior knowledge and manual adjustments are needed for such tasks. Although some TI methods are regarded as not requiring prior knowledge such as starting points, they often rely on manual choices such as selecting known maker genes, choosing trajectory models (e.g., linear, branching, graph structure), and fine-tuning parameters such as neighbor numbers to make a learned structure better fit human knowledge. We experimented with some TI methods on human early embryonic development data. They could not give the expected lineage tree if the packages were run with default or inappropriate settings, but could produce the expected tree at cell resolution after reasonable settings or adjustments guided by the expectation of the result. Currently most TI applications are not designed for the task of ab initio discovery of developmental processes as we aimed at in this study. Our experiments suggested a promising future solution for finding the outline structure of lineages with the proposed ab initio ML method and then using the structure to guide the inference of detailed trajectories using TI methods.
There are many different scenarios that need the mining of underlying patterns from massive complex data in biology and other fields. Successful applications of ML in many fields may give the illusion that ML has already been proved powerful for knowledge discovery, but in fact most of the successes are the joint products of ML and human knowledge. Involvement of knowledge can come in many forms such as known markers, models, or labeled training data.33 Efforts for using only ML methods to discover knowledge from data are still rare not only in biology but also in many other fields. In a recent work in physics, scientists explored a neural network method for the ab initio discovery of the basic physical understanding that Earth orbits the Sun based on observations on movements of the Sun and Mars appearing from Earth,34 otherwise known as “AI Copernicus.”35 Our experiment shows an example of the ab initio discovery of knowledge on early embryonic development from data with the integration of basic ML methods. The method is still in its infancy if expected to work on more complicated biological processes, but its success sheds light on the future possibilities of developing more advanced ML methods for ab initio scientific discovery from data in fields that lack existing knowledge and challenge manual interpretation. Such advancement will not only empower the discovery of new knowledge in biology and other fields of science but will also move machine intelligence to the higher level of automatic knowledge learning and discovery.
Experimental Procedures
Resource Availability
Lead Contact
Xuegong Zhang (zhangxg@tsinghua.edu.cn).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The original scRNA-seq data of human embryonic cells and corresponding ERCC spike-in reference data can be found at https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-3929/. The original scRNA-seq data of mouse embryonic cells and corresponding ERCC spike-in reference data can be found at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE109071 (GEO: GSE109071). The original scRNA-seq data of zebrafish embryonic cells can be found at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE112294 (GEO: GSE112294).
All third-party software packages used in this study are listed in Table S1. The pseudo-code of proposed self-consistency checking method is provided in Supplemental Information.
Data
The main dataset we worked on was the human early embryo development data published by Petropoulos et al., accession number ArrayExpress: E-MTAB-3929.14 It includes single-cell gene expression data of 26,178 genes in 1,529 cells from 88 human embryos obtained with the Smart-seq2 technology.36 Cells were captured during E3 to E7. Numbers of cells on each day are: E3, 81; E4, 190; E5, 377; E6, 415; and E7, 466. The average number of expressed genes in each cell is 8,500. We adopted a generic strategy to select the top 490 highly variable genes across all cells as the data for our experiment (Figure S1). All gene expression values were measured as log RPKM (reads per kilobase of transcript per million mapped reads). A detailed data pre-processing description is given in Supplemental Information. None of the pre-processing steps is specific to any known biological knowledge or to the question to be studied.
We also used a mouse dataset and a zebrafish dataset for extra experiments to validate the power and limitation of the proposed method. The mouse embryonic development dataset (Cheng et al., accession number GEO: GSE109071) contains 1,724 cells captured at E5.25, E5.5, E6.25, and E6.5.20 The data were also obtained with Smart-seq2. We used the same pre-processing steps on this dataset as we did on the human dataset.
The zebrafish dataset was published by Wanger et al., accession number GEO: GSE112294.21 It contains 36,749 zebrafish embryonic cells collected at seven time points during the development, i.e., 4, 6, 8, 10, 14, 18, and 24 hpf. The data were obtained with inDrops technology.37 We normalized library sizes of cells from all time points and selected the top 500 variable genes using Seurat v3.110 with default parameters. All gene expression values were measured as log UMI (unique molecular identifier) counts. None of the pre-processing steps is specific to any known biological knowledge or to the question to be studied.
Building Candidate Development Processes with Unsupervised and Supervised Learning
Figure 1 illustrates the overall scheme of the proposed method for ab initio discovery of developmental processes based on a number of samples collected at several time points in a developmental interval. It first builds multiple candidate developmental processes with each day as a possible reference, then evaluates the plausibility of each candidate to make the final story. Unsupervised learning is adopted to find clusters in the reference day as seeds for the developmental process. We used the classic k-means clustering22 method for this step. Other clustering methods can also be applied. For the purpose of this study, we chose basic general-purpose methods for the experiments rather than sophisticated methods specifically elaborated for the task. Deciding the number of clusters for each reference day is a key issue. We first adopted Silhouette score23 in combination with the scree plot of sum of errors to help determine the most proper cluster number in each day, then extended this to an exhaustive searching strategy to enumerate through a range of cluster numbers.
Using clusters obtained on the reference day as seeds for candidate lineages, we trained a supervised ML method on the seed data to predict lineages of cells of other days. We used the SVM24 with Gaussian kernel for this task. When there were more than two clusters in the reference day, we adopted the one-versus-all strategy to build a multi-class classifier with SVM. Other classification methods may also be used. Details of the ML packages used are provided in Supplemental Information.
Evaluating Self-Consistency of Candidate Development Processes
When there is no biological knowledge to judge which of the multiple versions of developmental processes is more plausible, the only information we can use is information of the unsupervised and supervised learning. We reasoned that if the differentiation of lineages is the major factor of cell heterogeneity and if an ML-derived developmental process reflects the biological truth, the classification results for each day should tend to be consistent with the clustering results. We designed the following method to check this self-consistency. The pseudo-code is provided in Supplemental Information.
We used the adjusted random index (ARI) to measure the level of agreement between two partitions on the same dataset.38 For example, when using day-r clusters as the reference to predict classes on day i, we define the “concordance of day i based on day r,” or concord score, as the agreement of the day r-based classification of day-i cells with the clustering results of day-i cells themselves. For the convenience of discussion, we denote the clustering results of cells in each day as Si, i = 3,…,7 in the case of the human early embryonic development data, and denote the classification of day i cells using day r clusters as reference as Ci|r, i,r = 3,…,7, i ≠ r. The concord score on day i given day r can then be written as:
| concord (i|r) = ARI(Si,Ci|r). |
The score is 1.0 when clustering scheme Si and classification result Ci|r are identical for all cells of day i. The score is around 0 when classification result is similar to random assignment of the day-i clusters and is <0 when the agreement between two partitions is even less than random chance.
To measure the reliability of the clustering results of day i, we define the reliability score (reliab) of day i as the average of concord scores of all other days using day i as reference:
This measures the compatibility of the clustering results of day i with all other days.
A poor concordance of day i based on day r may be due to the fact that clustering result of day r is not suitable as a reference for day i, and may also be due to a bad clustering result of day i itself. To take both factors into consideration, we further defined an adjusted reliability score (ARS) by weighting the concord score with the reliab score of each target day, i.e.,
We use this ARS to measure the relative level of reliability for choosing the clustering result of a particular day as the reference for building the lineages of all other days. The higher the ARS, the more likely the day is a proper reference.
This reliability evaluation of the reference day has taken into account the classification prediction on all days. We use it as the measure of self-consistency or plausibility of a candidate developmental process built upon the reference day. The one with the highest ARS is selected as the ML-derived developmental process we discovered ab initio from the data.
Besides the above quantitative evaluation, we also visualized the clustering and classification results on the first two principal components or tSNE plots of cells in each day, and manually inspected each candidate ML-derived developmental process to double-check the plausibility of the one selected with high ARS. A storyline can then be made on the candidate developmental process. If there is prior knowledge available, we compare this ab initio discovery with the storyline of the known knowledge to evaluate the power and limitation of the ML method for knowledge discovery.
Acknowledgments
This work is supported by the NSFC grant 61721003 and the National Key R&D Program of China grant 2017YFC0910400. We thank the anonymous reviewers for their suggestions that have helped to improve this work.
Author Contributions
X.Z. conceived the study and initiated the project. N.S., J.L., and F.L. designed the method for inferring the candidate stories. N.S. designed the self-consistency checking method. N.S., J.L., and F.L. conducted experiments. S.C. and K.H. participated in analyzing the results. H.G., S.C., and W.C. conducted the data pre-processing and preparation. X.Z., J.L., N.S., F.L., S.C., and K.H. wrote the manuscript.
Declaration of Interests
The author declares no competing interests.
Published: July 10, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.patter.2020.100071.
Supplemental Information
References
- 1.LeCun Y., Bengio Y., Hinton G. Deep learning. Nature. 2015;521:436–444. doi: 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- 2.Jordan M.I., Mitchell T.M. Machine learning: trends, perspectives, and prospects. Science. 2015;349:255–260. doi: 10.1126/science.aaa8415. [DOI] [PubMed] [Google Scholar]
- 3.Brynjolfsson E., Mitchell T. What can machine learning do? Workforce implications. Science. 2017;358:1530–1534. doi: 10.1126/science.aap8062. [DOI] [PubMed] [Google Scholar]
- 4.Kermany D.S., Goldbaum M., Cai W., Valentim C.C., Liang H., Baxter S.L., McKeown A., Yang G., Wu X., Yan F. Identifying medical diagnoses and treatable diseases by image-based deep learning. Cell. 2018;172:1122–1131. doi: 10.1016/j.cell.2018.02.010. [DOI] [PubMed] [Google Scholar]
- 5.Rampasek L., Goldenberg A. Learning from everyday images enables expert-like diagnosis of retinal diseases. Cell. 2018;172:893–895. doi: 10.1016/j.cell.2018.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Libbrecht M.W., Noble W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015;16:321–332. doi: 10.1038/nrg3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez R., Regier J., Cole M.B., Jordan M.I., Yosef N. Deep generative modeling for single-cell transcriptomics. Nat. Methods. 2018;15:1053–1058. doi: 10.1038/s41592-018-0229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding J., Condon A., Shah S.P. Interpretable dimensionality reduction of single cell transcriptome data with deep generative models. Nat. Commun. 2018;9:1–13. doi: 10.1038/s41467-018-04368-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eraslan G., Simon L.M., Mircea M., Mueller N.S., Theis F.J. Single-cell RNA-seq denoising using a deep count autoencoder. Nat. Commun. 2019;10:1–14. doi: 10.1038/s41467-018-07931-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., III, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luecken M.D., Theis F.J. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 2019;15:e8746. doi: 10.15252/msb.20188746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vieth B., Parekh S., Ziegenhain C., Enard W., Hellmann I. A systematic evaluation of single cell RNA-seq analysis pipelines. Nat. Commun. 2019;10:1–11. doi: 10.1038/s41467-019-12266-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hua K., Zhang X. A case study on the detailed reproducibility of a Human Cell Atlas project. Quant. Biol. 2019;7:162–169. [Google Scholar]
- 14.Petropoulos S., Edsgärd D., Reinius B., Deng Q., Panula S.P., Codeluppi S., Reyes A.P., Linnarsson S., Sandberg R., Lanner F. Single-cell RNA-seq reveals lineage and X chromosome dynamics in human preimplantation embryos. Cell. 2016;165:1. doi: 10.1016/j.cell.2016.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wianny F., Zernicka-Goetz M. Specific interference with gene function by double-stranded RNA in early mouse development. Nat. Cell Biol. 2000;2:70–75. doi: 10.1038/35000016. [DOI] [PubMed] [Google Scholar]
- 16.Hamatani T., Carter M.G., Sharov A.A., Ko M.S. Dynamics of global gene expression changes during mouse preimplantation development. Dev. Cell. 2004;6:117–131. doi: 10.1016/s1534-5807(03)00373-3. [DOI] [PubMed] [Google Scholar]
- 17.Guo F., Li L., Li J., Wu X., Hu B., Zhu P., Wen L., Tang F. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res. 2017;27:967–988. doi: 10.1038/cr.2017.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xue Z., Huang K., Cai C., Cai L., Jiang C.-y., Feng Y., Liu Z., Zeng Q., Cheng L., Sun Y.E. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature. 2013;500:593–597. doi: 10.1038/nature12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith Z.D., Chan M.M., Humm K.C., Karnik R., Mekhoubad S., Regev A., Eggan K., Meissner A. DNA methylation dynamics of the human preimplantation embryo. Nature. 2014;511:611–615. doi: 10.1038/nature13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng S., Pei Y., He L., Peng G., Reinius B., Tam P.P., Jing N., Deng Q. Single-cell RNA-seq reveals cellular heterogeneity of pluripotency transition and X chromosome dynamics during early mouse development. Cell Rep. 2019;26:2593–2607. doi: 10.1016/j.celrep.2019.02.031. [DOI] [PubMed] [Google Scholar]
- 21.Wagner D.E., Weinreb C., Collins Z.M., Briggs J.A., Megason S.G., Klein A.M. Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science. 2018;360:981–987. doi: 10.1126/science.aar4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacQueen, J. (1967). Some methods for classification and analysis of multivariate observations. In Proceedings of the Fifth Berkeley Symposium on Mathematical Statistics and Probability Vol. 1, pp. 281-297.
- 23.Rousseeuw P.J. Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987;20:53–65. [Google Scholar]
- 24.Cortes C., Vapnik V. Support-vector networks. Mach. Learn. 1995;20:273–297. [Google Scholar]
- 25.Cockburn K., Rossant J. Making the blastocyst: lessons from the mouse. J. Clin. Invest. 2010;120:995–1003. doi: 10.1172/JCI41229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niwa H., Toyooka Y., Shimosato D., Strumpf D., Takahashi K., Yagi R., Rossant J. Interaction between Oct3/4 and Cdx2 determines trophectoderm differentiation. Cell. 2005;123:917–929. doi: 10.1016/j.cell.2005.08.040. [DOI] [PubMed] [Google Scholar]
- 27.Otis E.M., Brent R. Equivalent ages in mouse and human embryos. Anat. Rec. 1954;120:33–63. doi: 10.1002/ar.1091200104. [DOI] [PubMed] [Google Scholar]
- 28.O'Rahilly R. Early human development and the chief sources of information on staged human embryos. Eur. J. Obstet. Gynecol. Reprod. Biol. 1979;9:273–280. doi: 10.1016/0028-2243(79)90068-6. [DOI] [PubMed] [Google Scholar]
- 29.Theiler K. Springer-Verlag; 1989. The House Mouse. [Google Scholar]
- 30.O'Rahilly R., Müller F. Developmental stages in human embryos: revised and new measurements. Cells Tissues Organs. 2010;192:73–84. doi: 10.1159/000289817. [DOI] [PubMed] [Google Scholar]
- 31.Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B., Schilling T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 32.Saelens W., Cannoodt R., Todorov H., Saeys Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 2019;37:547–554. doi: 10.1038/s41587-019-0071-9. [DOI] [PubMed] [Google Scholar]
- 33.Abdelaal T., Michielsen L., Cats D., Hoogduin D., Mei H., Reinders M.J., Mahfouz A. A comparison of automatic cell identification methods for single-cell RNA sequencing data. Genome Biol. 2019;20:194. doi: 10.1186/s13059-019-1795-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iten R., Metger T., Wilming H., Del Rio L., Renner R. Discovering physical concepts with neural networks. Phys. Rev. Lett. 2020;124:010508. doi: 10.1103/PhysRevLett.124.010508. [DOI] [PubMed] [Google Scholar]
- 35.Castelvecchi D. AI Copernicus 'discovers' that Earth orbits the Sun. Nature. 2019;575:266–267. doi: 10.1038/d41586-019-03332-7. [DOI] [PubMed] [Google Scholar]
- 36.Picelli S., Faridani O.R., Björklund Å.K., Winberg G., Sagasser S., Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014;9:171. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 37.Klein A.M., Mazutis L., Akartuna I., Tallapragada N., Veres A., Li V., Peshkin L., Weitz D.A., Kirschner M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hubert L., Arabie P. Comparing partitions. J. Class. 1985;2:193–218. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original scRNA-seq data of human embryonic cells and corresponding ERCC spike-in reference data can be found at https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-3929/. The original scRNA-seq data of mouse embryonic cells and corresponding ERCC spike-in reference data can be found at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE109071 (GEO: GSE109071). The original scRNA-seq data of zebrafish embryonic cells can be found at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE112294 (GEO: GSE112294).
All third-party software packages used in this study are listed in Table S1. The pseudo-code of proposed self-consistency checking method is provided in Supplemental Information.