

Abstract
A BF3·OEt2 catalyzed intramolecular Povarov reaction was used to synthesize 15 chromenopyridine fused thiazolino-2-pyridone peptidomimetics. The reaction works with several O-alkylated salicylaldehydes and amino functionalized thiazolino-2-pyridones, to generate polyheterocycles with diverse substitution. The synthesized compounds were screened for their ability to bind α-synuclein and amyloid β fibrils in vitro. Analogues substituted with a nitro group bind to mature amyloid fibrils, and the activity moreover depends on the positioning of this functional group.
The thiazolino fused 2-pyridone represents a privileged scaffold which can be modified to display various biological activities.1 It was initially designed as a peptidomimetic to combat the virulence of uropathogenic E. coli by inhibiting the formation of pili.1a With alternative substitution patterns, compounds based on this scaffold have also demonstrated activity against Chlamydia trachomatis,1bListeria monocytogenes,1c and Mycobacterium tuberculosis.1d Rigidifying the scaffold by equipping it with sterically demanding aryl groups provides compounds with the ability to modulate formation of bacterial and human amyloid fibrils.2 Extension of the bicyclic thiazolino fused 2-pyridone with nitrogen containing aromatic heterocycles offers another way of rigidifying the peptidomimetic scaffold,3 as exemplified by compounds 1–4 (Figure 1A). These analogues are able to modulate α-synuclein amyloid fibril formation, which is associated with Parkinson’s disease, a human neurodegenerative disorder,2a,3a,3b or bind to mature α-synuclein fibrils.3c
Figure 1.

(A) Bicyclic 2-pyridones capable of modulating (compounds 1–3) and binding (compound 4) to α-synuclein and amyloid β fibrils. (B) Chromenopyridine containing bioactive polyheterocycles 5–8 and retrosynthetic strategy devised for construction of chromenopyridine fused 2-pyridone polyheterocycle 11.
Chromenopyridine is a versatile structural motif present in a variety of polyheterocycles with applications in biology as estrogenic, antibacterial, and anticancer agents and biosensors (compounds 5–8, respectively, Figure 1B).4 Recently, the merging of two different active fragments to develop scaffolds with improved biological properties has received considerable attention.5 As mentioned above, annulation of bicyclic thiazolino fused 2-pyridone with different heterocycles has resulted in scaffolds capable of modulating amyloid fibrils.3 We envisaged that fusing thiazolino 2-pyridone and chromenopyridine, by combining units 9 and 10, could afford scaffolds with the ability to target amyloid structures (Figure 1B) and, in addition, constitute a new central fragment with great potential for drug discovery in general.
Since its discovery, the Povarov reaction6 has been used widely for the synthesis of nitrogen containing, six-membered heterocycles with biological relevance.7 Recently, we utilized the Lewis acid catalyzed Povarov reaction to construct a tricyclic pyridine fused 2-pyridone peptidomimetic scaffold,3c whose analogues have been shown to bind α-synuclein and Aβ fibrils by ThT displacement8in vitro (e.g., compound 4, Figure 1A). We envisioned that performing the Povarov reaction in an intramolecular fashion9 could result in the desired chromenopyridine annulated 2-pyridones 11 in a single operation (Figure 1B). The new scaffold 11 being equipped with the tetrahedral carbon, and the oxygen, could decrease planarity of the structures and enable increased hydrogen bonding, respectively.10 In addition, these features could potentially confer selectivity between different amyloid structures, a very desired property in diagnostic and therapeutic applications of amyloid binding small molecules.11
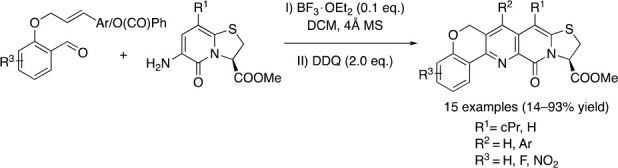
We perceived that chromenopyridine ring fused 2-pyridone scaffold 11 would be accessible from amino 2-pyridone 9 and O-alkylated salicylaldehyde 10. Hence, we began our work by investigating the feasibility of the reaction between 9a and O-cinnamyl salicylaldehyde 10a in an intramolecular Povarov setup using BF3·OEt2 as catalyst, followed by oxidation with DDQ (Scheme 1).3c As hypothesized, the intramolecular reaction worked smoothly, and the desired product 11a was isolated in excellent yield. To explore the substrate scope of the reaction, substituted O-cinnamyl salicylaldehydes 10b–i (Schemes S1 and S2) were allowed to react with 6-amino-2-pyridones 9a,b to construct 11b–k in good to excellent yields (Scheme 1).
Scheme 1. Synthesis of 2-Pyridone Based Polyheterocycles 11a–k.

Aware of the importance of the 4-nitrophenyl substituent for amyloid binding activity of the tricyclic scaffold 4,3c we prepared 11b–f and 11k with nitro groups in various positions. Notably, salicylaldehydes with electron withdrawing R3 substituents underwent faster Povarov reaction due to lowering of LUMO in the electrophilic imine intermediate, providing 11b–d, 11g, and 11k.12 Electron donating R4 substituents rendered the alkene more nucleophilic and likewise decreased the reaction times, while electron withdrawing R4 substituents increased them. Heating was required to achieve synthetically useful reaction times for synthesis of 11e and 11h, where, in the former case, the moderate yield reflects a less clean conversion. In the preparation of 11f, the effect of the electron withdrawing R3 substituent compensated for poor nucleophilicity of the alkene moiety, and heating was not required to complete the reaction in 1 day. To test the scalability, 11b was also prepared from 1.6 mmol of 9a. The yield (88%) is comparable to that at 0.4 mmol scale (86%).
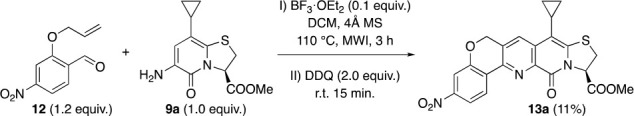
Next, we turned our attention toward the synthesis of C-13 unsubstituted target molecules 13 (Scheme 2). SAR from our previous study suggested that the best amyloid binding properties are achieved when the corresponding position is unsubstituted.3c It was approached by Povarov reaction between 9a and O-allyl salicylaldehyde 12. To our dismay, we were only able to isolate small amounts (7%) of the desired product 13a, from the complex reaction mixture after 4 days at 70 °C. Microwave irradiation, 120 °C for 3 h, shared the same lack of success, and 13a was isolated in only 11% yield. Though fruitful results are reported,9e attempts with terminal allyl moieties often suffer from low yields.7a,9a,12a,12b
Scheme 2. Unsuccessful Attempt to Synthesize C-13 Unsubstituted Compound 13a.
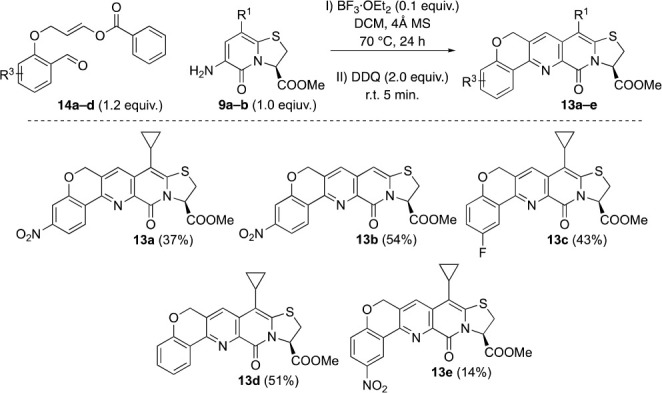
Familiar with the mechanistic features of the Lewis acid catalyzed Povarov reaction,7a,7d,7e,12c,13 we realized that use of the terminal allyl group as alkene components would require the reaction to go via high energy carbocations or operate via an alternative mechanism.9a,9b,9d,14 With our previous strives in mind where we had made use of ethyl vinyl ether as alkene component, for synthesis of unsubstituted tricyclic analogues 4,3c we naturally thought of employing a vinyl ester moiety as electron donating auxiliary. With 3-bromopropenyl benzoate,15 we were able to alkylate salicylic aldehydes to synthesize the required intermediates 14a–d (Schemes 3 and S3). With the alkene component now armed with an ester group, capable of mesomeric contributions, we attempted Povarov reactions between 14a–d and 9a,b. Still, heating the reaction mixtures to 70 °C was required to achieve reasonable reaction times.
Scheme 3. Synthesis of C-13 Unsubstituted Compounds 13a–e.
The benzoate functionality was eliminated during oxidation to provide the desired products.
The desired compounds 13a–e were isolated in low to moderate yields after 24 h of heating, followed by oxidation with DDQ at room temperature. The unusually low yield of 13e results from the formation of side products, which complicated the purification. The low to moderate yields motivated us to try a slightly different approach, using CuBr2 catalysis and O-propargyl salicylaldehyde (Scheme S4).7b,7c,7f,9a Unfortunately, the method suffered from undesired side reactions and 13a was only isolated in 31% yield.
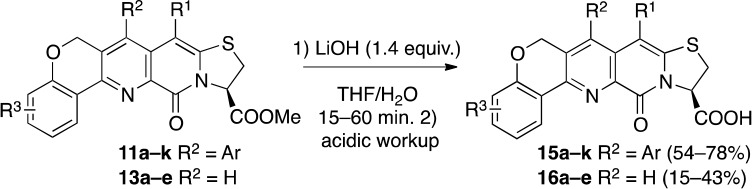
Having both sets of compounds in hand, we hydrolyzed the methyl ester to deprotect the carboxylic acid and reveal the peptidomimetic (Scheme 4). The limited solubility of 16a–e in organic solvents complicated handling, and these compounds were thus obtained in lower yields.
Scheme 4. Deprotection of Methyl Esters through Saponification of 11a–k and 13a–e.
The carboxylic acids 15a–k and 16a–e were initially screened in an α-synuclein fibrilization assay with Thioflavin T (ThT) to probe for fibril binding and modulation of amyloid fibril formation (Figure 2A and Figure S1).8,16 Compounds 15b,c, 15e,f, 15k, 16a,b, and 16e bind to the α-synuclein fibrils and had a significant effect on the fluorescence intensity, by competing with ThT for binding. In addition, compound 15e has a mild inhibitory effect upon the fibril formation, as the lag phase was slightly extended. In order to verify that amyloid fibrils were indeed present, samples were taken upon the end point of the assay and visualized with transmission electron microscopy (TEM) (Figure S2). No visible difference was observed between the control experiment (black trace) and the mixture with 16a (green trace) (Figures 2B and S2).
Figure 2.

(A) Representative selection of ThT fluorescence traces. Each experiment was performed in triplicate (Figure S1) and normalized to the average. Compounds 15b and 16a appear to bind fibrils strongly, whereas 15a does not seem to bind to any significant extent. 15e and 16b are borderline. (B) TEM picture of fibrils formed in the presence of compound 16a (green trace).
In a similar assay, compounds (15a–c, 15e,f, 15k, 16a–c, 16e) were added after 70 h, when the fluorescence traces had reached the plateau phase (Figures 3A and S3). The compounds were observed to bind to the mature α-synuclein fibrils and displace bound ThT, indicated by reduction of the ThT fluorescence, to varying degrees (Figures 3B and S4).
Figure 3.

(A) ThT trace for compound 16a added after 70 h (green), α-Syn control (black), and ThT background fluorescence (gray). (B) Retained ThT fluorescence 5 h after addition of compounds 15a–c, 15e,f, 15k, 16a–c, and 16e to mature α-synuclein fibrils, compared to the fluorescence intensity 1 h before addition. For comparison, 15a and 16c were also included.
Interestingly, all compounds equipped with a nitro group except 15d showed binding activity. The strongest binding was observed for compounds with the nitro group situated in position C-9 (15b, 15c, 16a, and 16b). Moving the nitro group to position C-8 (16e) or to the 4′ position of the C-13 aryl group (15e,f) is accompanied by a decrease in fibril binding ability. Notably, the presence of the C-13 phenyl group abolishes all binding activity rendered by the nitro group in position C-8 (15d vs 16e) but not C-9 (15b vs 16a, Figure S3). The cyclopropyl group appears to be the favored C-14 substituent, over methoxy and proton (15b vs 15c and 15k, and 16a vs 16b). This observation is in agreement with the SAR on scaffold 4, where cyclopropyl as R1 substituent was found somewhat superior to phenyl, hydrogen, and methoxy substituents.3c Taken together, these observations fit with the hypothesis that binding sites on amyloid fibrils are made up of shallow hydrophobic clefts flanked by polar groups, situated between the rows of side chains and running parallel to the fiber axis.11a,11c,11e
Compounds were also evaluated against Aβ40 in a similar manner (Figure 4 and Figures S5–S7). Compounds 15b,c, 15f, and 16a, which bind strongly to α-synuclein fibrils, were found to bind mature Aβ fibrils as well, indicated by reduced ThT fluorescence, compared to the control experiments.
Figure 4.

Retained ThT fluorescence after addition of selected compounds to mature Aβ40 fibrils in vitro. (A) ThT fluorescence trace for addition of compound 15b (magenta), α-Syn control (black), and ThT background fluorescence (gray). (B) Bar chart representation of the retained fluorescence (60 h), compared to the intensity before compound addition (40 h).
O
In summary, we have developed methods to fuse two privileged scaffolds, namely, chromenopyridines and thiazolino 2-pyridones. These new peptidomimetic polyheterocycles constitute a new scaffold with potential for diverse substitution patterns. The intramolecular Povarov reaction between -alkylated salicylaldehydes and amino functionalized thiazolino-2-pyridones afforded chromenopyridine fused 2-pyridone polyheterocycles in moderate to excellent yields. Rewardingly, biological evaluation of chromenopyridine fused thiazolino 2-pyridones revealed compounds capable of binding to α-synuclein and Aβ40 amyloid fibrils in vitro. An interesting SAR was observed with respect to groups at position C-9. The polyheterocycles equipped with a nitro group at position C-9 showed the strongest binding to α-synuclein and Aβ40 amyloid fibrils. However, changing the position of the nitro group resulted in decreased binding ability versus both amyloid fibrils. As binding to mature amyloid fibrils is a property of pharmacological relevance,11 we intend to investigate these promising compounds further in future biological studies.
Experimental Section
General
Unless otherwise stated, purchased reactants and reagents were used as received from commercial suppliers. Molecular sieves and LiCl were dried at 300 °C under high vacuum for 4 h prior to use. Acetonitrile was dried over activated 3 Å molecular sieves (5% w/v) for 48 h, then transferred via syringe to new 3 Å molecular sieves (5% w/v) for storage until use. DMF, THF, and diethyl ether were dried using an SG Water solvent drying tower according to the manufacturer’s instructions and stored over activated 3 Å (DMF) or 4 Å (THF and diethyl ether) MS for 48 h or more before use. Amberlyst was rinsed prior to use with THF/MeOH 1:1 in a cylindrical sintered funnel until the filtrate was transparent, then dried briefly by passing air through. Microwave reactions were performed in sealed vessels using a Biotage Initiator microwave synthesizer, temperatures were monitored by an internal IR probe, and stirring was mediated magnetically. TLC was performed on purchased aluminum backed silica gel plates (median pore size 60 Å, fluorescent indicator 254 nm) and detected with UV light at 254 and 366 nm. Flash column chromatography was performed using silica gel (0.063–0.200 mesh). Automated flash column chromatography was performed using a Biotage Isolera One system and purchased prepacked silica gel cartridges (Biotage SNAP Cartridge, KP-Sil). Preparative HPLC was performed on a Gilson instrument with a Phenomenex column (250 × 21.2 mm; Gemini 5 μm NX-C18, 110 Å). MeCN/water, with 0.75% HCOOH in the mobile phase. 30–100% MeCN in water over 30 min with a flow rate of 20 mL/min. The elution was monitored with UV-abs. at 254 nm. Freeze-drying was accomplished by freezing the diluted MeCN/water solutions in liquid nitrogen and then employing a Scanvac CoolSafe freeze-dryer connected to an Edwards 28 rotary vane oil pump. Optical rotation was measured with a Rudolph Autopol IV polarimeter 343 at 22 °C and 589 nm. [α] is reported in deg·mL·g–1·dm–1; concentrations (c) are given in g/100 mL. IR spectra were recorded on a Bruker Alpha-t spectrometer. The samples were prepared as KBr pellets or between NaCl plates; absorbances are given in reciprocal cm. 1H, 13C, and 19F NMR spectra were recorded on a Bruker Avance III 400 MHz spectrometer with a BBO-F/H Smartprobe or a Bruker Avance III HD 600 MHz spectrometer with a CP BBO-H/F, 5 mm cryoprobe, at 298 K, unless another temperature is given. All spectrometers were operated by Topspin 3.5. Resonances are given in ppm relative to TMS, and calibrated to solvent residual signals [CDCl3: δH = 7.26 ppm; δC = 77.16 ppm. (CD3)2SO: δH = 2.50 ppm; δC = 39.51 ppm. (CD3)CO δH = 2.05 ppm; δC = 29.84 ppm]. The following abbreviations are used to indicate splitting patterns: s = singlet; d = doublet; dd = double doublet; t = triplet; m = multiplet; bs = broad singlet. LC-MS was conducted on a Micromass ZQ mass spectrometer with ES+ and ES– ionization. HRMS was performed on a mass spectrometer with ESI-TOF (ES+/ES–). Human wild-type α-synuclein was expressed and purified as described previously,3c expressed and purified Aβ40 was supplied by Alexotech AB.
General Procedure for Synthesis of O-Alkylated Salicylaldehydes 10a–i
Salicylaldehydes were O-alkylated according to established procedures with modifications.17 Under an atmosphere of nitrogen, cinnamyl bromide II (2.34 mmol, 1.30 equiv) was dissolved in DMF (1.0 mL) and transferred with a syringe to a mixture of salicylaldehyde I (1.80 mmol, 1.00 equiv) and K2CO3 (496 mg, 2.00 equiv) under nitrogen. The reaction mixture was stirred at r.t. until completion was indicated by TLC analysis. The mixture was then transferred to a beaker of ice-cold water (60 mL) while stirring; the resulting precipitate was filtered off and washed with water (5 mL). The precipitate was then dissolved in EtOAc (50 mL) and washed with brine (5 × 25 mL), dried with sodium sulfate, filtered, and evaporated. Unless otherwise stated, the compounds were used in the following synthetic step without further purification.
2-(Cinnamyloxy)benzaldehyde (10a)
The compound was prepared by following the general procedure. The mixture was stirred for 6.5 h. The crude product was purified with automated flash column chromatography (25 g cartridge, 10–20% EtOAc in heptane). White, low melting solid (373 mg, 87.2%). IR (KBR): ν 3028, 2862, 1684, 1598, 1580, 1481, 1455, 1375, 1286, 1236 cm–1. 1H NMR (400 MHz, CDCl3): δ 10.58 (s, 1H), 7.86 (dd, J = 7.9, 1.8 Hz, 1H), 7.55 (td, J = 8.0, 1.8 Hz, 1H), 7.43 (d, J = 7.0 Hz, 2H), 7.35 (t, J = 7.6 Hz, 2H), 7.31–7.27 (m, 1H), 7.05 (dt, J = 7.5, 3.2 Hz, 2H), 6.77 (d, J = 16.0 Hz, 1H), 6.43 (dt, J = 16.0, 5.7 Hz, 1H), 4.84 (dd, J = 5.7, 1.4 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 189.9, 161.1, 136.2, 136.0, 133.7, 128.8, 128.7, 128.3, 126.8, 125.3, 123.6, 121.1, 113.1, 69.3. HRMS (ESI-TOF) m/z: [M – H]− Calcd for C16H13O2– 237.0921; Found 237.0917.
2-(Cinnamyloxy)-4-nitrobenzaldehyde (10b)
The compound was prepared by following the general procedure. The reaction mixture was stirred for 5 h. Light yellow solid (425 mg, 83.6%). IR (KBr): ν 3116, 3042, 2903, 2864, 1693, 1612, 1592, 1527, 1480, 1428, 1408, 1386, 1347, 1301, 1208, 1182, 971 cm–1. 1H NMR (400 MHz, CDCl3): δ 10.60 (s, 1H), 8.01 (d, J = 8.4 Hz, 1H), 7.92 (s, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.44 (d, J = 7.7 Hz, 2H), 7.36 (t, J = 7.5 Hz, 2H), 7.31 (t, J = 7.3 Hz, 1H), 6.83 (d, J = 15.9 Hz, 1H), 6.43 (dt, J = 15.9, 6.0 Hz, 1H), 4.95 (d, J = 5.9 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 188.4, 161.0, 152.3, 135.8, 135.3, 129.8, 129.0, 128.9, 128.7, 126.9, 122.0, 115.9, 108.5, 70.2. HRMS (ESI-TOF) m/z: [M – H]− Calcd for C16H12NO4– 282.0772; Found 282.0774.
2-(Cinnamyloxy)-5-nitrobenzaldehyde (10c)
The compound was prepared by following the general procedure. The reaction mixture was stirred for 2.75 h. Light yellow solid (446 mg; 87.7%). IR (KBr): ν 3075, 2908, 1688, 1610, 1588, 1516, 1487, 1450, 1429, 1391, 1340, 1274, 1177, 1157, 1141, 1078, 1063 cm–1. 1H NMR (400 MHz, CDCl3): δ 10.53 (s, 1H), 8.73 (d, J = 2.9 Hz, 1H), 8.43 (dd, J = 9.2, 2.9 Hz, 1H), 7.48–7.41 (m, 2H), 7.41–7.28 (m, 3H), 7.18 (d, J = 9.2 Hz, 1H), 6.85–6.76 (m, 1H), 6.42 (dt, J = 15.9, 6.0 Hz, 1H), 4.97 (dd, J = 6.0, 1.5 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 187.6, 164.8, 141.8, 135.7, 135.3, 130.7, 129.0, 128.9, 126.9, 125.0, 124.9, 121.8, 113.5, 70.4. HRMS (ESI-TOF) m/z: [M – H]− Calcd for C16H12NO4– 282.0772; Found 282.0767.
2-(Cinnamyloxy)-5-fluorobenzaldehyde (10d)
The compound was prepared by following the general procedure starting from 5-fluoro-2-hydroxybenzaldehyde (251 mg, 1.79 mmol). The reaction was stirred for 4.5 h, and the product was purified with automated flash column chromatography (50 g cartridge, 3–20% EtOAc in n-heptane) as white solid in 84% yield (388 mg, 1.51 mmol). IR (KBr): ν 3415, 3339, 3065, 3030, 2940, 2868, 2054, 1976, 1883, 1780, 1679, 1610, 1580, 1485, 1461 cm–1. 1H NMR (400 MHz, CDCl3) δ 10.54 (d, J = 3.1 Hz, 1H), 7.56 (dd, J = 8.3, 3.3 Hz, 1H), 7.47–7.41 (m, 2H), 7.41–7.34 (m, 2H), 7.34–7.24 (m, 3H), 7.04 (dd, J = 9.1, 3.9 Hz, 1H), 6.78 (dd, J = 16.0, 1.6 Hz, 1H), 6.43 (dt, J = 16.0, 5.8 Hz, 1H), 4.84 (dd, J = 5.8, 1.5 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3) δ 188.7, 188.6, 135.9, 133.8, 128.7, 128.2, 126.6, 126.0, 125.9, 123.1, 122.5, 122.3, 114.7, 114.6, 114.2, 114.0, 77.3, 77.0, 76.7, 69.9. 19F{1H} NMR (376 MHz, CDCl3) δ −122.14. HRMS (ESI-TOF) m/z: [M – H]− Calcd for C16H12FO2– 255.0827; Found 255.0819.
(E)-4-Nitro-2-((3-(4-nitrophenyl)allyl)oxy)benzaldehyde (10e)
The compound was prepared by following the general procedure but with 1.8 equiv of alkyl bromide. The reaction mixture was stirred overnight. During workup, the precipitate was dissolved in DCM instead of EtOAc. The crude product was purified with flash column chromatography (30 × 70 mm silica gel, DCM). Light yellow solid (431 mg, 73.1%). IR (KBr): ν 3113, 1692, 1596, 1526, 1511, 1391, 1345, 1309, 1248, 1183, 1109 cm–1. 1H NMR (400 MHz, CDCl3): δ 10.61 (d, J = 0.7 Hz, 1H), 8.23 (d, J = 8.8 Hz, 2H), 8.03 (d, J = 8.3 Hz, 1H), 7.92 (dt, J = 11.4, 1.5 Hz, 2H), 7.58 (d, J = 8.8 Hz, 2H), 6.91 (d, J = 16.1 Hz, 1H), 6.61 (dt, J = 16.0, 5.5 Hz, 1H), 5.00 (dd, J = 5.5, 1.6 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 188.1, 160.6, 152.3, 147.7, 142.1, 132.2, 130.1, 129.0, 127.5, 126.9, 124.3, 116.3, 108.3, 69.4. HRMS (ESI-TOF) m/z: [M – H]− Calcd for C16H11N2O6– 327.0622; Found 327.0622.
(E)-2-((3-(4-Nitrophenyl)allyl)oxy)benzaldehyde (10f)
The compound was prepared by following the general procedure but with 1.8 equiv of crude alkyl bromide. The reaction mixture was stirred overnight. The crude product was purified with automated flash column chromatography (25 g cartridge, 8–25% EtOAc in heptane. Off white solid (311 mg, 61.1%). IR (KBr): ν 3106, 3078, 2869, 1688, 1597, 1513, 1484, 1458, 1450, 1340, 1284, 1251, 1235, 1189, 1165, 1105, 1072, 1041, 1002 cm–1. 1H NMR (400 MHz, CDCl3): δ 10.58 (d, J = 0.7 Hz, 1H), 8.21 (d, J = 8.8 Hz, 2H), 7.88 (dd, J = 7.7, 1.8 Hz, 1H), 7.62–7.51 (m, 3H), 7.12–6.99 (m, 2H), 6.86 (d, J = 16.1 Hz, 1H), 6.61 (dt, J = 16.1, 5.2 Hz, 1H), 4.89 (dd, J = 5.2, 1.7 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 189.6, 160.7, 147.4, 142.7, 136.1, 130.8, 129.0, 128.6, 127.3, 125.3, 124.3, 121.5, 112.9, 68.5. HRMS (ESI-TOF) m/z: [M – H]− Calcd for C16H12NO4– 282.0772; Found 282.0767.
(E)-2-((3-(3-(Trifluoromethyl)phenyl)allyl)oxy)benzaldehyde (10g)
The compound was prepared by following the general procedure but with 1.8 equiv of crude alkyl bromide. The reaction mixture was stirred overnight. The crude product was purified with automated flash column chromatography (50 g cartridge, 3–10% EtOAc in heptane). White, low melting solid (434 mg, 78.8%). IR (KBr): ν 3071, 2936, 2878, 2770, 1682, 1598, 1484, 1459, 1441, 1404, 1378, 1322, 1306, 1287, 1243, 1201, 1164, 1122, 1075, 1041, 1007 cm–1. 1H NMR (400 MHz, CDCl3): δ 10.58 (d, J = 0.8 Hz, 1H), 7.87 (dd, J = 7.7, 1.9 Hz, 1H), 7.66 (s, 1H), 7.62–7.50 (m, 3H), 7.47 (t, J = 7.7 Hz, 1H), 7.10–7.00 (m, 2H), 6.81 (d, J = 16.1 Hz, 1H), 6.51 (dt, J = 15.9, 5.4 Hz, 1H), 4.86 (dd, J = 5.5, 1.6 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 189.8, 160.9, 137.1, 136.1, 131.9, 131.3 (q, J = 32.2 Hz), 129.9 (d, J = 1.5 Hz), 129.3, 128.8, 125.7, 125.3, 124.8 (q, J = 3.8 Hz), 124.2 (q. J = 272.4 Hz), 123.4 (q, J = 3.8 Hz), 121.3, 113.0, 68.8. 19F NMR (376 MHz, CDCl3): δ −62.79. HRMS (ESI-TOF) m/z: [M – H]− Calcd for C17H12F3O2– 305.0795; Found 305.0782.
(E)-2-((3-(3-Methoxyphenyl)allyl)oxy)benzaldehyde (10h)
The compound was prepared by following the general procedure but with 1.8 equiv of crude alkyl bromide. The reaction mixture was stirred overnight. The crude product was purified with automated flash column chromatography (50 g cartridge, 5–20% diethyl ether in heptane). The fractions containing the pure desired product were combined and evaporated. The residue was then co-evaporated with chloroform twice. White, low melting solid (446 mg, 93%) calculated from 494 mg containing chloroform. IR (KBr): ν 3003, 2967, 2876, 2839, 1683, 1600, 1575, 1484, 1464, 1435, 1375, 1308, 1290, 1244, 1155, 1048, 1002, 972 cm–1. 1H NMR (400 MHz, CDCl3): δ 10.57 (d, J = 0.7 Hz, 1H), 7.86 (dd, J = 8.0, 1.9 Hz, 1H), 7.55 (ddd, J = 8.3, 7.4, 1.9 Hz, 1H), 7.30–7.21 (m, 1H), 7.09–6.98 (m, 3H), 6.95 (t, J = 2.1 Hz, 1H), 6.84 (ddd, J = 8.2, 2.6, 0.9 Hz, 1H), 6.74 (d, J = 15.9 Hz, 1H), 6.42 (dt, J = 16.0, 5.7 Hz, 1H), 4.83 (dd, J = 5.7, 1.6 Hz, 2H), 3.83 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 189.9, 161.1, 160.0, 137.7, 136.0, 133.5, 129.8, 128.7, 125.3, 123.9, 121.1, 119.4, 114.0, 113.1, 112.1, 69.2, 55.4. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C16H16NaO3+ 291.0997; Found 291.1003.
(E)-2-((3-(4-Ethoxyphenyl)allyl)oxy)benzaldehyde (10i)
The compound was prepared by following the general procedure but with 1.8 equiv of crude alkyl bromide. The reaction mixture was stirred overnight. Only about 50% conversion of salicylaldehyde was indicated by TLC, but the alkyl bromide was consumed. The crude product was purified with automated flash column chromatography (25 g cartridge, 5–20% diethyl ether in heptane). Off white solid (124 mg, 24.5%). IR (KBr): ν 2980, 2934, 2879, 1681, 1599, 1512, 1485, 1455, 1388, 1287, 1239, 1179, 1047, 978, cm–1. 1H NMR (400 MHz, CDCl3): δ 10.59 (s, 1H), 7.88 (dd, J = 8.0, 1.8 Hz, 1H), 7.60–7.52 (m, 1H), 7.37 (d, J = 8.7 Hz, 2H), 7.07 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 8.7 Hz, 2H), 6.73 (d, J = 16.0 Hz, 1H), 6.31 (dt, J = 15.9, 5.9 Hz, 1H), 4.83 (dd, J = 6.0, 1.5 Hz, 2H), 4.07 (q, J = 7.0 Hz, 2H), 1.44 (t, J = 7.0 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 190.0, 161.3, 159.2, 136.0, 133.6, 128.8, 128.6, 128.0, 125.3, 121.1, 121.0, 114.8, 113.1, 69.6, 63.7, 15.0. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H18NaO3+ 305.1154; Found 305.1160.
General Procedure for Synthesis of C-13 Aryl Substituted Povarov Reaction Products 11a–j
The 6-amino thizolo-2-pyridone 9 (0.40 mmol, 1.00 equiv) and 2-(cinnamyloxy)-benzaldehyde 10 (0.48 mmol, 1.20 equiv) were weighed up in a Biotage Microwave reaction tube and dissolved in DCM (4 mL). 4 Å MS (8–10 pellets) was added, followed by BF3·OEt2 (10.8 μL; 0.100 equiv), whereupon the tube was sealed and left stirring at r.t. The reaction was monitored with TLC until complete consumption of 1, and the corresponding imine intermediate, was indicated. The tube was then opened and DDQ (182 mg; 2.00 equiv) was added. The reaction mixture was stirred until complete oxidation was visible by TLC, then transferred to a separation funnel and diluted with DCM (25 mL). The mixture was washed with saturated aqueous bicarbonate solution (2 × 10 mL), followed by brine (10 mL). The aq. phases were re-extracted once each with DCM (3 mL). The organic phase was dried over sodium sulfate, filtered, and evaporated to dryness. The crude residue was redissolved in DCM and purified with automated flash column chromatography. The fractions containing pure desired product were combined, evaporated, and co-evaporated with distilled chloroform twice. The residue was put under high vacuum for several hours before storage at −20 °C. The yield was calculated from the 1H NMR spectrum recorded in d6-DMSO.
Methyl (R)-8-Cyclopropyl-13-oxo-7-phenyl-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylate (11a)
The compound was prepared by following the general procedure. The reaction was finished by TLC analysis after 9.5 h. DDQ was added, and the mixture was stirred for 25 min. The crude product was purified with automated flash column chromatography (25 g cartridge, 25–75% EtOAc in heptane). Yellow-orange solid (185 mg, 93%). The compound contained chloroform; yield calculated from 1H NMR sample in (CD3)2SO. [α]D −226° (c 0.35, CHCl3). IR (KBr): ν 3451, 1001, 2950, 1753, 1666, 1579, 1461, 1436, 1382, 1300, 1258, 1220, 1183, cm–1. 1H NMR (400 MHz, CDCl3): δ 8.51 (dd, J = 7.8, 1.7 Hz, 1H), 7.43 (dq, J = 4.7, 3.2, 2.5 Hz, 3H), 7.37–7.28 (m, 2H), 7.12 (td, J = 7.5, 1.1 Hz, 1H), 6.91 (dd, J = 8.1, 1.1 Hz, 1H), 5.73 (dd, J = 8.2, 2.6 Hz, 1H), 5.12–4.98 (m, 2H), 3.81 (s, 3H), 3.69 (dd, J = 11.6, 8.2 Hz, 1H), 3.50 (dd, J = 11.6, 2.6 Hz, 1H), 1.01–0.89 (m, 1H), 0.34–0.11 (m, 4H). 13C{1H} NMR (100 MHz, CDCl3): δ 168.8, 159.6, 156.4, 146.9, 143.5, 142.3, 140.8, 137.5, 133.6, 131.8, 130.0, 129.9, 129.5, 128.4, 128.0, 126.5, 123.0, 122.7, 116.6, 109.5, 66.6, 63.5, 53.5, 31.6, 15.7, 11.6, 11.6. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C28H23N2O4S+ 483.1373; Found 483.1379.
Methyl 8-Cyclopropyl-3-nitro-13-oxo-7-phenyl-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylate (11b)
The compound was prepared by following the general procedure. Upon addition of boron trifluoride, the stirred solution turned dark purple and a precipitate emerged within a few minutes. The resulting suspension thickened so that the stirring was compromised. The tube was shaken manually with regular intervals for 20 min until the suspension was lighter and the magnetic stirring could be continued. The precipitate eventually dissolved completely, and the reaction was then followed with TLC until complete consumption of 9a was indicated, after 2 h reaction time. DDQ (197 mg, 2.17 equiv) was added and stirred for 50 min. The residue was purified with automated flash column chromatography (25 g SNAP Cartridge, 20–65% EtOAc in heptane). Yellow solid (181 mg, 85.8%) The compound contained chloroform; yield calculated from 1H NMR sample in (CD3)2SO. 11b (746 mg, 88.4%) was also prepared at 1.6 mmol scale, according to the general procedure. The reaction was complete after 3 h. [α]D −228° (c 0.289, CHCl3); IR (KBr): ν 3452, 1752, 1668, 1573, 1526, 1433, 1342, 1227 cm–1; 1H NMR (400 MHz, CDCl3): δ 8.65 (d, J = 8.6 Hz, 1H), 7.95 (dd, J = 8.6, 2.2 Hz, 1H), 7.76 (d, J = 2.2 Hz, 1H), 7.49–7.42 (m, 3H), 7.36–7.30 (m, 1H), 7.24–7.29 (m, 1H), 5.74 (dd, J = 8.3, 2.7 Hz, 1H), 5.22–5.07 (m, 2H), 3.83 (s, 3H), 3.71 (dd, J = 11.7, 8.3 Hz, 1H), 3.52 (dd, J = 11.6, 2.6 Hz, 1H), 1.03–0.90 (m, 1H), 0.36–0.23 (m, 2H), 0.23–0.14 (m, 1H). 13C{1H} NMR (100 MHz, CDCl3): δ 168.6, 159.4, 156.3, 149.7, 145.3, 144.5, 142.7, 141.2, 137.0, 134.6, 129.9, 129.8, 129.6, 128.74, 128.65, 128.2, 127.1, 117.5, 112.5, 109.4, 67.1, 63.6, 53.5, 31.6, 15.7, 11.68, 11.60. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C28H22N3O6S+ 528.1224; Found 528.1229.
Methyl (R)-3-Nitro-13-oxo-7-phenyl-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylate (11c)
The compound was prepared by following the general procedure. A precipitate emerged after 10 min stirring upon addition of boron trifluoride. The magnetic stirring was compromised as the mixture got viscous, and the tube was shaken manually for 10 min, until the viscosity had decreased to a point where magnetic stirring was possible. The reaction was finished after 5.8 h. DDQ was added, and the mixture was stirred for 10 min. The crude product was dissolved in a larger volume of DCM and loaded onto a column (30 × 80 mm silica gel, equilibrated with DCM) and purified with flash column chromatography (0–70% EtOAc in heptane). Yellow solid (179 mg, 92%). The compound contained chloroform; yield was calculated from 1H NMR spectrum recorded in (CD3)2SO. [α]D −256° (c 0.55, CHCl3). IR (KBr): ν 3443, 1754, 1680, 1595, 1535, 1514, 1451, 1432, 1342, 1322, 1222, 1179, 1041 cm–1. 1H NMR [400 MHz, (CD3)2SO]: δ 8.48 (d, J = 8.6 Hz, 1H), 8.06 (dd, J = 8.6, 2.3 Hz, 1H), 7.79 (d, J = 2.2 Hz, 1H), 7.66–7.53 (m, 3H), 7.37 (ddt, J = 13.3, 7.8, 1.7 Hz, 2H), 6.02 (s, 1H), 5.75 (dd, J = 8.7, 2.0 Hz, 1H), 5.20 (d, J = 3.4 Hz, 2H), 3.94 (dd, J = 11.9, 8.7 Hz, 1H), 3.75 (s, 3H), 3.66 (dd, J = 11.9, 2.0 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3): δ 168.4, 159.8, 156.4, 149.8, 145.2, 142.6, 142.3, 139.6, 133.9, 133.5, 129.5, 129.5, 129.4, 129.0, 128.8, 128.5, 128.5, 127.1, 117.5, 112.7, 96.1, 66.9, 63.0, 53.6, 32.4. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C25H18N3O6S+ 488.0911; Found 488.0901.
Methyl (R)-8-Cyclopropyl-2-nitro-13-oxo-7-phenyl-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylate (11d)
The compound was prepared by following the general procedure. The reaction was finished after 6.6 h. DDQ was added, and the mixture was stirred for 20 min. The crude product was purified with automated flash column chromatography (25 g cartridge, 15–55% EtOAc in heptane). Yellow solid (148 mg, 70%). The compound contained chloroform; yield was calculated from 1H NMR spectrum recorded in (CD3)2SO. [α]D −197° (c 0.29, CHCl3). IR (KBr): ν 3444, 3001, 2953, 1753, 1669, 1618, 1580, 1549, 1518, 1494, 1482, 1459, 1443, 1384, 1340, 1299, 1250, 1229, 1088, 1022 cm–1; 1H NMR [400 MHz, (CD3)2SO]: δ 8.99 (d, J = 2.9 Hz, 1H), 8.26 (dd, J = 9.0, 2.9 Hz, 1H), 7.55–7.44 (m, 3H), 7.36 (dt, J = 5.4, 2.9 Hz, 1H), 7.22 (d, J = 9.0 Hz, 1H), 5.72 (dd, J = 8.8, 2.6 Hz, 1H), 5.39–5.14 (m, 2H), 3.86 (dd, J = 11.8, 8.8 Hz, 1H), 3.76 (s, 3H), 3.58 (dd, J = 11.8, 2.6 Hz, 1H), 0.87 (m, 1H), 0.32–-0.02 (m, 4H). 13C{1H} NMR (100 MHz, CDCl3): δ 168.6, 160.8, 159.3, 145.0, 144.4, 143.4, 142.6, 141.2, 137.0, 134.4, 129.8, 129.7, 128.7, 128.4, 128.2, 126.8, 123.3, 122.4, 117.6, 109.2, 67.3, 63.5, 53.5, 31.6, 15.7, 11.7, 11.6. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C28H22N3O6S+ 528.1224; Found 528.1216.
Methyl (R)-8-Cyclopropyl-7-(4-nitrophenyl)-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylate (11e)
The compound was prepared by following the general procedure, but the mixture was heated in an oil bath at 70 °C for 23 h. DDQ was added, and the mixture was stirred at room temperature for 15 min. The crude product was purified with automated flash column chromatography (25 g cartridge, 15–60% EtOAc in heptane). Yellow solid (114 mg, 54%). The compound contained chloroform; yield was calculated from 1H NMR spectrum recorded in (CD3)2SO. [α]D −163° (c 0.21, CHCl3). IR (KBr): ν 2999, 2953, 2848, 1752, 1667, 1579, 1552, 1519, 1491, 1462, 1438, 1381, 1347, 1299, 1258, 1220, 1179, 1149, 1107, 1070, 1035, 1005 cm–1. 1H NMR [400 MHz, (CD3)2SO]: δ 8.33 (dt, J = 6.4, 2.3 Hz, 2H), 8.21 (dd, J = 7.7, 1.7 Hz, 1H), 7.79 (dd, J = 8.7, 2.0 Hz, 1H), 7.69 (dd, J = 8.7, 2.0 Hz, 1H), 7.41 (td, J = 7.7, 1.7 Hz, 1H), 7.20 (td, J = 7.5, 1.1 Hz, 1H), 7.00 (dd, J = 8.2, 1.1 Hz, 1H), 5.73 (dd, J = 8.7, 2.5 Hz, 1H), 5.20–4.97 (m, 2H), 3.86 (dd, J = 11.9, 8.8 Hz, 1H), 3.76 (s, 3H), 3.58 (dd, J = 11.9, 2.6 Hz, 1H), 0.92–0.80 (m, 1H), 0.34–0.17 (m, 3H), 0.07 (q, J = 7.2, 6.1 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3): δ 168.6, 159.3, 156.3, 147.8, 147.1, 145.0, 144.4, 141.0, 139.3, 133.2, 132.1, 131.1, 131.1, 128.9, 126.5, 123.3, 122.9, 122.7, 116.7, 108.3, 66.2, 63.6, 53.5, 31.6, 16.0, 12.3, 12.2. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C28H22N3O6S+ 528.1224; Found 528.1228.
Methyl (R)-3-Nitro-7-(4-nitrophenyl)-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylate (11f)
The compound was prepared by following the general procedure. A red precipitate emerged upon addition of boron trifluoride. The mixture got thick, so the tube was sonicated in an ultrasonic water bath at room temperature for 5 min, then shaken manually with regular intervals during several hours, until the viscosity had decreased to a point where magnetic stirring was no longer compromised. The reaction was finished after 24.5 h. DDQ was added, and the mixture was stirred for 10 min. The crude product was purified with flash column chromatography (80 × 30 mm SiO2, 0–17% EtOAc in DCM. Orange solid (167 mg, 78.5%). The compound contained chloroform; yield was calculated from 1H NMR spectrum recorded in (CD3)2SO. [α]D −239° (c 0.28, CHCl3). IR (KBr): ν 3447, 1756, 1670, 1595, 1532, 1433, 1348, 1227, 1042 cm–1. 1H NMR [400 MHz, (CD3)2SO]: δ 8.51–8.40 (m, 3H), 8.07 (dd, J = 8.7, 2.3 Hz, 1H), 7.80 (d, J = 2.3 Hz, 1H), 7.75–7.66 (m, 2H), 6.06 (s, 1H), 5.79–5.72 (m, 1H), 5.22 (d, J = 1.6 Hz, 2H), 3.94 (dd, J = 11.9, 8.8 Hz, 1H), 3.76 (s, 3H), 3.66 (dd, J = 11.8, 2.0 Hz, 1H). 13C{1H} NMR [151 MHz, (CD3)2SO]: δ 168.6, 158.3, 155.9, 149.1, 147.9, 144.7, 143.5, 139.9, 139.6, 138.6, 133.1, 130.8, 130.6, 128.3, 128.1, 125.9, 124.3 (2C), 117.5, 112.3, 94.2, 79.2, 66.3, 62.5, 53.1, 31.5. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C25H17N4O8S+ 533.0762; Found 533.0748.
Methyl (R)-8-Cyclopropyl-2-fluoro-13-oxo-7-phenyl-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylate (11g)
The compound was prepared by following the general procedure starting from 9a (106 mg, 0.40 mmol). The reaction was stirred for 4.5 h, and the product was purified with automated flash column chromatography (50 g cartridge, 10–70% EtOAc in n-heptane) in 92% yield as a light-yellow powder (170 mg, 0.36 mmol). [α]D −160° [c 0.33, (CD3)2SO]. IR (KBr): ν 3548, 3473, 3414, 3081, 3000, 2953, 2844, 2041, 1752, 1665, 1617, 1578, 1550, 1520, 1494, 1483, 1461, 1429 cm–1. 1H NMR [400 MHz, (CH3)2SO] δ 7.66 (dd, J = 9.0, 3.2 Hz, 1H), 7.27 (ttd, J = 7.7, 5.6, 5.1, 3.0 Hz, 4H), 7.15 (dt, J = 5.6, 3.0 Hz, 1H), 7.05 (td, J = 8.6, 3.2 Hz, 1H), 6.84 (dd, J = 8.9, 4.5 Hz, 1H), 5.50 (dd, J = 8.8, 2.5 Hz, 1H), 4.99–4.75 (m, 2H), 3.65 (dd, J = 11.8, 8.8 Hz, 1H), 3.55 (s, 3H), 3.36 (dd, J = 11.9, 2.6 Hz, 1H), 0.72–0.61 (m, 1H), 0.10 to −0.08 (m, 3H), −0.12 to −0.24 (m, 1H). 13C{1H} NMR [100 MHz, (CD3)2SO] δ 169.2, 158.5, 152.5, 145.9, 144.6, 142.4, 140.4, 137.0, 134.2, 130.3, 130.2, 129.4, 128.8, 128.3, 128.2, 119.1, 119.0, 108.0, 79.6, 66.63, 63.64, 53.4, 31.1, 15.7, 11.5, 11.4. 19F{1H} NMR [376 MHz, (CD3)2SO] δ −120.83. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C28H22FN2O4S+ 501.1279; Found 501.1287.
Methyl (R)-8-Cyclopropyl-13-oxo-7-(3-(trifluoromethyl)phenyl)-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylate (11h)
The compound was prepared by following the general procedure, but the mixture was heated in an oil bath at 70 °C for 8 h. DDQ was added, and the mixture was stirred at room temperature for 10 min. The crude product was purified with automated flash column chromatography (25 g cartridge, 20–65% EtOAc in heptane). Yellow solid (163 mg, 74%). The compound contained chloroform; yield was calculated from 1H NMR spectrum recorded in (CD3)2SO. [α]D −165° (c 0.37, CHCl3). IR (KBr): ν 3552, 3475, 3414, 3236, 3002, 2955, 1753, 1668, 1611, 1580, 1553, 1518, 1489, 1464, 1437, 1382, 1329, 1296, 1257, 1221, 1178, 1168, 1126, 1108, 1073, 1046 cm–1. 1H NMR [400 MHz, (CD3)2SO]: δ 8.21 (dd, J = 7.8, 1.7 Hz, 1H), 7.96–7.64 (m, 4H), 7.40 (td, J = 7.7, 1.7 Hz, 1H), 7.19 (td, J = 7.6, 1.1 Hz, 1H), 6.99 (d, J = 8.1 Hz, 1H), 5.71 (d, J = 8.7 Hz, 1H), 5.20–4.89 (m, 2H), 3.86 (ddd, J = 11.9, 8.7, 6.4 Hz, 1H), 3.75 (s, 3H), 3.57 (ddd, J = 11.8, 4.0, 2.6 Hz, 1H), 0.77 (m, 1H), 0.39 to −0.16 (m, 4H). 13C{1H} NMR [100 MHz, (CD3)2SO, 343 K]: δ 168.4 (d, J = 3.3 Hz), 157.8, 155.5, 145.1, 144.8, 139.9, 139.7, 137.6, 133.6 (d, J = 10.1 Hz), 132.9 (d, J = 6.0 Hz), 131.4, 128.7 (d, J = 7.5 Hz), 128.6 (d, J = 2.7 Hz), 126.26–125.97 (m), 124.6, 124.6 (d, J = 4.0 Hz), 122.1 (d, J = 7.4 Hz), 116.4, 106.7 (d, J = 7.2 Hz), 78.8, 65.6, 63.0 (d, J = 4.7 Hz), 52.5, 30.3 (d, J = 4.2 Hz), 14.9 (d, J = 5.7 Hz), 11.0, 10.9. The multiplets in the13C NMR spectrum are caused by atropoisomers.19F NMR (376 MHz, CDCl3) δ −62.68. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C29H22F3N2O4S+ 551.1247; Found 551.1249.
Methyl (R)-8-Cyclopropyl-7-(3-methoxyphenyl)-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylate (11i)
The compound was prepared by following the general procedure. The reaction was finished after 5.5 h. DDQ was added, and the mixture was stirred for 10 min. The crude product was purified with automated flash column chromatography (25 g cartridge, 20–70% EtOAc in heptane). Yellow solid (172 mg, 83.7%). The compound contained chloroform; yield was calculated from 1H NMR spectrum recorded in (CD3)2SO. [α]D −160° (c 0.31, CHCl3). IR (KBr): ν 3442, 3000, 2953, 2837, 1753, 1667, 1606, 1579, 1552, 1517, 1489, 1463, 1436, 1381, 1371, 1298, 1284, 1260, 1217, 1180, 1149, 1107, 1071, 1035 cm–1. 1H NMR [400 MHz, (CD3)2SO, 343 K]: δ 8.23 (dd, J = 7.8, 1.7 Hz, 1H), 7.45–7.33 (m, 2H), 7.17 (td, J = 7.5, 1.1 Hz, 1H), 7.07–6.94 (m, 3H), 6.94–6.87 (m, 1H), 5.68 (dt, J = 8.7, 3.1 Hz, 1H), 5.19–4.95 (m, 2H), 3.89–3.79 (m, 4H), 3.76 (s, 3H), 3.54 (dd, J = 11.8, 2.8 Hz, 1H), 1.02–0.93 (m, 1H), 0.30 (m, 3H), 0.21–0.09 (m, 1H). 13C{1H} NMR [100 MHz, (CD3)2SO, 343 K]: 168.4, 158.4 (d, J = 7.5 Hz), 157.9, 155.6, 144.8, 144.3, 141.4, 139.9, 137.8, 133.0, 131.3, 128.7 (d, J = 7.9 Hz), 128.5, 124.6, 122.3, 122.0, 121.9, 116.4, 115.4 (d, J = 13.4 Hz), 113.6, 107.3, 78.8, 65.8, 62.9, 55.1, 52.5, 30.3, 14.7, 10.83–10.45 (m). The multiplets in the13C NMR spectrum are caused by atropoisomers. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C29H25N2O5S+ 513.1479; Found 513.1476.
Methyl (R)-8-Cyclopropyl-7-(4-ethoxyphenyl)-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylate (11j)
The compound was prepared by following the general procedure. The reaction was finished after 70 min. DDQ was added, and the mixture was stirred for 10 min. The crude product was purified with automated flash column chromatography (25 g cartridge, 20–60% EtOAc in heptane). The column was colored orange as the desired product eluted; we are hence suspecting that the compound binds to or breaks down on silica gel. Yellow solid (112 mg, 53.0%). The compound contained chloroform; yield was calculated from 1H NMR spectrum recorded in (CD3)2SO. [α]D −188° (c 0.32, CHCl3). IR (KBr): ν 3443, 2979, 1753, 1666, 1607, 1581, 1507, 1437, 1381, 1245, 1176, 1108, 1044 cm–1. 1H NMR [400 MHz, (CD3)2SO]: δ 8.19 (dd, J = 7.8, 1.7 Hz, 1H), 7.43–7.30 (m, 2H), 7.24 (dd, J = 8.7, 2.3 Hz, 1H), 7.18 (td, J = 7.5, 1.1 Hz, 1H), 7.05–6.95 (m, 3H), 5.69 (dd, J = 8.7, 2.5 Hz, 1H), 5.22–5.00 (m, 2H), 4.09 (d, J = 7.0 Hz, 2H), 3.85 (dd, J = 11.8, 8.7 Hz, 1H), 3.75 (s, 3H), 3.56 (dd, J = 11.8, 2.6 Hz, 1H), 1.37 (t, J = 6.9 Hz, 3H), 0.98–0.84 (m, 1H), 0.40–0.26 (m,1H), 0.26–0.03 (m, 3H). 13C{1H} NMR [100 MHz, (CD3)2SO]: δ 168.8, 158.4, 158.3, 155.8, 145.1, 144.6, 141.8, 140.0, 133.6, 131.7, 131.1, 129.1, 128.7, 125.0, 122.8, 122.5, 116.8, 113.7, 113.5, 107.8, 79.2, 66.0, 63.1, 52.9, 30.6, 15.1, 14.6, 10.9, 10.9. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C30H27N2O5S+ 527.1635; Found 527.1632.
(R)-Methyl 8-Methoxy-3-nitro-13-oxo-7-phenyl-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylate (11k)
The compound was prepared by following the general procedure. The reaction was finished after 2 h. DDQ was added, and the mixture was stirred for 10 min. The crude product was purified with automated flash column chromatography (25 g cartridge, 0–100% EtOAc in heptane). Yellow solid (155 mg, 73%). [α]D25 −241 (c 0.4, CHCl3). IR (KBr): ν 3468, 3000, 1742, 1667, 1592, 1552, 1531, 1494, 1456, 1410, 1375, 1226, 922, 799, 740, 705 cm–1. 1H NMR (600 MHz, CDCl3): δ 8.68 (d, J = 8.3 Hz, 1H), 7.94 (d, J = 7.3 Hz, 1H), 7.75 (d, J = 2.0 Hz, 1H), 7.52–7.43 (m, 3H), 7.28 (d, J = 6.5 Hz, 2H), 5.78 (d, J = 6.2 Hz, 1H), 5.03 (q, J = 15.0 Hz, 2H), 3.83 (s, 3H), 3.81–3.76 (m, 1H), 3.61 (d, J = 11.1 Hz, 1H), 2.98 (s, 3H). 13C NMR (151 MHz, CDCl3): δ 168.2, 158.4, 156.4, 149.8, 145.0, 140.9, 140.4, 135.8, 135.7, 132.5, 130.0, 129.9, 128.5, 128.2, 128.2, 128.2, 128.1, 127.1, 117.4, 112.6, 66.8, 63.4, 60.1, 53.6, 32.0. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C26H20N3O7S+ 518.1016; Found 518.1016.
2-(Allyloxy)-4-nitrobenzaldehyde (12)
Under an atmosphere of nitrogen, 2-hydroxy-4-nitrobenzaldehyde Ib (300 mg, 1.00 equiv) and K2CO3 (498 mg, 2.01 equiv) were suspended in DMF (1.5 mL). While stirring at r.t., allyl bromide (198 μL, 1.30 equiv) was added. The reaction mixture was stirred for 3:40 h at r.t. until completion was indicated by TLC analysis. The mixture was transferred to a beaker of ice-cold water (60 mL) while stirring; the resulting precipitate was filtered off and washed with water (5 mL). The precipitate was then dissolved in EtOAc (50 mL) and washed with brine (5 × 25 mL), dried with sodium sulfate, filtered, and evaporated. Yellow solid (326 mg, 87.7%). IR (KBr): ν 3102, 3086, 2886, 1689, 1614, 1590, 1532, 1482, 1433, 1420, 1395, 1369, 1350, 1308, 1270, 1246, 1184, 1111, 1087, 995, 937, 88, 815, 747 cm–1. 1H NMR (600 MHz, CDCl3): δ 10.57 (s, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.88 (d, J = 8.5 Hz, 1H), 7.85 (s, 1H), 6.09 (ddt, J = 16.6, 10.6, 5.3 Hz, 1H), 5.51 (d, J = 17.3 Hz, 1H), 5.42 (d, J = 10.5 Hz, 1H), 4.78 (d, J = 4.7 Hz, 2H). 13C{1H} NMR (151 MHz, CDCl3): δ 188.4, 160.9, 152.2, 131.3, 129.7, 129.0, 119.6, 115.8, 108.5, 70.2. HRMS (ESI-TOF) m/z: [M – H]− Calcd for C10H8NO4– 206.0459; Found 206.0465.
General Procedure for Synthesis of Povarov Reaction Products 13a–e
The 6-amino thizolo-2-pyridone 9 (0.40 mmol, 1.00 equiv) and O-(propenyl benzoate)-salicylaldehyde 14 (0.48 mmol, 2.00 equiv) were weighed up in a Biotage Microwave reaction tube and dissolved in DCM (4 mL). 4 Å MS (8–10 pellets) was added, followed by BF3·OEt2 (10.8 μL; 0.10 equiv), whereupon the tube was sealed and left stirring in an oil bath at 70 °C. The reaction was monitored with TLC until complete consumption of 9 was indicated. The tube was then cooled down to r.t., opened, and DDQ (182 mg; 2.00 equiv) was added. The reaction mixture was stirred for 5 min; complete oxidation was indicated by TLC. Then it was transferred to a separation funnel and diluted with DCM (25 mL). The mixture was washed with saturated aqueous bicarbonate solution (2 × 10 mL), followed by brine (10 mL). The aq. phases were re-extracted once each with DCM (3 mL). The organic phase was dried over sodium sulfate, filtered, and evaporated to dryness. The crude residue was redissolved in DCM and purified with automated flash column chromatography. The fractions containing pure desired product were combined, evaporated, and co-evaporated with distilled chloroform twice. The residue was put under high vacuum for several hours before storage at −20 °C. The yield was calculated from 1H NMR spectrum recorded in d6-DMSO.
Methyl 8-Cyclopropyl-3-nitro-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylate (13a)
The compound was prepared by following the general procedure starting from 9a (106 mg, 0.40 mmol). The reaction mixture was stirred for 24 h, and the product was purified on automated flash column chromatography (50 g cartridge, 10–75% EtOAc in n-heptane) in 37% yield (66 mg, 0.146 mmol), isolated as a light yellow solid. [α]D −235° (c 0.264, CHCl3); IR (KBr): ν 3086, 6005, 2955, 2924, 2852, 1754, 1670, 1577, 1531, 1460, 1343, 1237, 1032, 918, 805, 738 cm–1; 1H NMR (600 MHz, CDCl3): δ 8.64 (d, J = 8.6 Hz, 1H), 8.09 (s, 1H), 7.96–7.91 (m, 1H), 7.84–7.80 (m, 1H), 5.77 (dd, J = 8.2, 1.8 Hz, 1H), 5.45 (s, 2H), 3.80 (s, 3H), 3.76 (dd, J = 11.5, 8.4 Hz, 1H), 3.58 (d, J = 11.6 Hz, 1H), 1.79–1.73 (m, 1H), 1.15–1.04 (m, 2H), 0.76–0.68 (m, 2H). 13C{1H} NMR (151 MHz, CDCl3): δ 168.6, 159.5, 156.6, 149.7, 144.9, 143.3, 140.2, 135.9, 130.4, 128.2, 127.3, 126.8, 117.5, 112.9, 108.1, 68.6, 63.1, 53.5, 31.8, 10.1, 7.7, 7.6. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C22H18N3O6S+ 452.0911; Found 452.0911.
Methyl (R)-3-Nitro-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylate (13b)
The compound was prepared by following the general procedure starting from 9a (106 mg, 0.40 mmol). The reaction mixture was stirred for 24 h, and the product was purified on automated flash column chromatography (50 g cartridge, 10–50% EtOAc in n-heptane) in 54% yield (89 mg, 0.216 mmol) as a yellow powder. [α]D −216° [c 0.16, (CH3)2SO]. IR (KBr): ν 3548, 3415, 3083, 3006, 2955, 2851, 1749, 1672, 1605, 1589, 1533, 1514, 1457, 1433 cm–1. 1H NMR [400 MHz, (CD3)2SO] δ 8.38 (d, J = 8.6 Hz, 1H), 8.01 (dd, J = 8.6, 2.3 Hz, 1H), 7.87 (d, J = 1.1 Hz, 1H), 7.79 (d, J = 2.3 Hz, 1H), 6.71 (s, 1H), 5.73 (dd, J = 8.7, 2.0 Hz, 1H), 5.54 (s, 2H), 3.98 (dd, J = 11.8, 8.7 Hz, 1H), 3.75 (s, 3H), 3.69 (dd, J = 11.9, 2.0 Hz, 2H). 13C{1H} NMR [100 MHz, (CD3)2SO] δ 169.1, 156.6, 149.4, 144.0, 138.9, 135.2, 131.5, 130.1, 117.8, 112.8, 97.2, 68.0, 62.9, 53.5, 32.0. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C19H13N3NaO6S+ 434.0423; Found 434.0428.
Methyl (R)-8-Cyclopropyl-2-fluoro-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylate (13c)
The compound was prepared by following the general procedure starting from 9a (106 mg, 0.40 mmol). The reaction mixture was stirred for 24 h, and the product was purified on automated flash column chromatography (50 g cartridge, 10–50% EtOAc in n-heptane) isolated in 43% yield (73 mg, 0.172 mmol) as a light-yellow powder. [α]D −200° [c 0.33, (CH3)2SO]. IR (KBr): ν 3549, 3473, 3415, 3004, 2247, 1753, 1663, 1606, 1582, 1523, 1490, 1470 cm–1. 1H NMR [600 MHz, (CD3)2SO] δ 8.29 (s, 1H), 7.90–7.79 (m, 1H), 7.26 (d, J = 3.3 Hz, 1H), 7.09 (dd, J = 8.9, 4.5 Hz, 1H), 5.71 (dd, J = 8.9, 2.1 Hz, 1H), 5.52–5.42 (m, 2H), 3.88 (dd, J = 11.8, 8.9 Hz, 1H), 3.74 (s, 3H), 3.62 (dd, J = 11.8, 2.2 Hz, 1H), 1.83–1.74 (m, 1H), 1.13–0.99 (m, 2H), 0.70–0.55 (m, 2H). 13C{1H} NMR [151 MHz, (CD3)2SO] δ 169.3, 158.7, 158.7, 157.1, 153.1, 145.2, 145.2, 143.2, 139.2, 135.5, 130.8, 128.1, 124.0, 124.0, 119.4, 119.4, 118.9, 118.7, 110.5, 110.3, 107.2, 67.9, 63.0, 53.4, 31.2, 10.2, 7.9, 7.7. 19F NMR [565 MHz, (CD3)2SO] δ −120.86 (td, J = 8.8, 8.3, 4.4 Hz). HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C22H18FN2O4S+ 425.0966; Found 425.0972.
Methyl (R)-8-Cyclopropyl-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylate (13d)
The compound was prepared by following the general procedure starting from 9a (106 mg, 0.40 mmol). The reaction mixture was stirred for 24 h, and the product was purified with automated flash column chromatography (50 g cartridge, 20–80% EtOAc in n-heptane) isolated in 51% yield (85 mg, 0.20 mmol) as a yellow powder. [α]D −135° [c 0.33, (CH3)2SO]. IR (KBr): ν 3415, 3082, 3003, 2953, 2927, 2844, 1747, 1663, 1603, 1588, 1578, 1519, 1493, 1461 cm–1. 1H NMR [600 MHz, (CD3)2SO] δ 8.27 (s, 1H), 8.21 (dd, J = 7.8, 1.7 Hz, 1H), 7.41 (td, J = 7.7, 1.7 Hz, 1H), 7.18 (t, J = 7.5 Hz, 1H), 7.04 (d, J = 8.1 Hz, 1H), 5.71 (dd, J = 8.8, 2.2 Hz, 1H), 5.50–5.45 (m, 2H), 3.89 (dd, J = 11.8, 8.8 Hz, 1H), 3.74 (s, 3H), 3.62 (dd, J = 11.8, 2.2 Hz, 1H), 1.85–1.75 (m, 1H), 1.10–1.05 (m, 2H), 0.71–0.56 (m, 2H). 13C{1H} NMR [151 MHz, (CD3)2SO] δ 169.3, 158.8, 156.9, 146.2, 142.6, 139.3, 135.0, 132.2, 130.8, 127.9, 125.0, 122.8, 122.8, 117.6, 107.2, 67.8, 63.0, 53.4, 31.2, 10.2, 7.9, 7.7. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C22H19N2O4S+ 407.1060; Found 407.1063.
Methyl (R)-8-Cyclopropyl-2-nitro-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylate (13e)
The compound was prepared by following the general procedure starting from 9a (106 mg, 0.40 mmol). The reaction mixture was stirred for 24 h, and the product was purified with automated flash column chromatography (50 g cartridge, 10–50% EtOAc in n-heptane) in 14% yield (25 mg, 0.05 mmol) as a yellow powder. [α]D −83° [c 0.12, (CH3)2SO]. IR (KBr): ν 3417, 3082, 30009, 2954, 2852, 2247, 1753, 1665, 1617, 1606, 1592, 1578, 1520, 1488, 1467 cm–1. 1H NMR [600 MHz, (CD3)2SO] δ 8.98 (d, J = 2.9 Hz, 1H), 8.34 (s, 1H), 8.27 (dd, J = 9.0, 2.9 Hz, 1H), 7.27 (d, J = 9.0 Hz, 1H), 5.78–5.67 (m, 3H), 3.91 (dd, J = 11.8, 8.9 Hz, 1H), 3.75 (s, 3H), 3.64 (dd, J = 11.8, 2.2 Hz, 1H), 1.85–1.78 (m, 1H), 1.15–1.05 (m, 2H), 0.73–0.60 (m, 2H). 13C{1H} NMR [151 MHz, (CD3)2SO] δ 169.2, 161.6, 158.6, 143.8, 143.8, 142.7, 139.4, 135.8, 130.1, 128.4, 127.3, 122.8, 120.4, 119.0, 107.2, 68.6, 63.0, 53.4, 31.2, 10.2, 7.9, 7.7. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C22H17N3NaO6S+ 474.0736; Found 474.0714.
General Procedure for Synthesis of O-Alkylated Salicylaldehydes 14a–d
Under an atmosphere of nitrogen, 3-bromopropenyl benzoate (868 mg, 3.60 mmol, 1.80 equiv) was dissolved in DMF (2.0 mL) and transferred with a syringe to a mixture of salicylaldehyde I (2.00 mmol, 1.00 equiv) and K2CO3 (553 mg, 2.00 equiv) under nitrogen. The reaction mixture was stirred at r.t. until completion was indicated by TLC analysis. The mixture was diluted with CH2Cl2 (50 mL) and washed with brine (5 × 50 mL), filtered through sodium sulfate, and evaporated. The crude product was purified with automated flash column chromatography.
(E)-3-(2-Formyl-4-nitrophenoxy)prop-1-en-1-yl Benzoate (14a)
The compound was prepared by following the general procedure starting from 5-nitro-2-hydroxybenzaldehyde (334 mg, 2.00 mmol). The reaction mixture was stirred for 1.0 h, and the product was purified with automated flash column chromatography (50 g cartridge, 5–85% EtOAc in n-heptane) in 61% yield (400 mg, 1.22 mmol) as a white powder. (E)-3-Bromoprop-1-en-1-yl benzoate was prepared according to literature reports and crystallized from heptane.15a IR (KBr): ν 3412, 3111, 3069, 2947, 2898, 2631, 1718, 1687, 1609, 1588, 1526, 1481, 1451, 1428, 1406 cm–1. 1H NMR (600 MHz, CDCl3) δ 10.51 (s, 1H), 8.75 (d, J = 2.9 Hz, 1H), 8.47 (dd, J = 9.1, 2.9 Hz, 1H), 8.18–8.10 (m, 2H), 7.83 (d, J = 12.4 Hz, 1H), 7.67 (t, J = 7.5 Hz, 1H), 7.53 (t, J = 7.8 Hz, 2H), 7.19 (d, J = 9.1 Hz, 1H), 5.95 (dt, J = 12.5, 7.1 Hz, 1H), 4.89 (d, J = 7.1 Hz, 2H). 13C{1H} NMR (151 MHz, CDCl3) δ 187.3, 164.3, 163.2, 141.8, 141.1, 130.5, 130.1, 130.1, 128.7, 128.5, 128.3, 128.2, 127.0, 124.9, 124.8, 113.0, 107.7, 66.6. HRMS (ESI-TOF) m/z: [M – H]− Calcd for C17H12NO6– [M – H]− 326.0670; Found 326.0657.
(E)-3-(2-Formyl-5-nitrophenoxy)prop-1-en-1-yl Benzoate (14b)
The compound was prepared by following the general procedure starting from 4-nitro-2-hydroxybenzaldehyde (334 mg, 2.00 mmol). The reaction mixture was stirred for 2.0 h, and the product was purified with automated flash column chromatography (50 g cartridge, 10–90% EtOAc in n-heptane) in 68% yield (450 mg, 1.37 mmol) as a white powder. (E)-3-Bromoprop-1-en-1-yl benzoate was prepared according to literature reports and crystallized from heptane.15a IR (KBr): ν 3420, 3098, 3086, 3036, 2953, 2868, 2756, 1730, 1694, 1613, 1601, 1586, 1523, 1493, 1477, 1452, 1428 cm–1. 1H NMR (600 MHz, CDCl3) δ 10.58 (s, 1H), 8.15 (dt, J = 8.2, 0.9 Hz, 2H), 8.03 (d, J = 8.9 Hz, 1H), 7.93 (dq, J = 5.0, 2.0 Hz, 2H), 7.83 (dd, J = 12.5, 1.2 Hz, 1H), 7.70–7.59 (m, 1H), 7.52 (t, J = 7.7 Hz, 2H), 5.96 (dt, J = 12.6, 7.0 Hz, 1H), 4.87 (d, J = 7.0 Hz, 2H). 13C{1H} NMR (151 MHz, CDCl3) δ 188.1, 163.2, 160.5, 140.9, 134.0, 130.1, 129.7, 128.8, 128.6, 128.2, 115.9, 108.1, 107.9, 66.4. HRMS (ESI-TOF) m/z: [M – H]− Calcd for C17H12NO6– 326.0670; Found 326.0668.
(E)-3-(4-Fluoro-2-formylphenoxy)prop-1-en-1-yl Benzoate (14c)
The compound was prepared by following the general procedure starting from 5-fluoro-2-hydroxybenzaldehyde (251 mg, 1.79 mmol). The reaction mixture was stirred for 2.5 h, and the product was purified with automated flash column chromatography (50 g cartridge, 2–50% EtOAc in n-heptane) as a white powder in 84% yield (388 mg, 1.51 mmol). (E)-3-Bromoprop-1-en-1-yl benzoate was prepared according to literature reports and crystallized from heptane.15a IR (KBr): ν 3548, 3415, 3091, 3059, 2946, 2892, 2870, 2766, 1727, 1685, 1614, 1599, 1584, 1495, 1452 cm–1. 1H NMR (600 MHz, CDCl3) δ 10.48 (s, 1H), 8.18–8.09 (m, 2H), 7.76 (dd, J = 12.5, 1.3 Hz, 1H), 7.70–7.62 (m, 1H), 7.56 (dd, J = 8.2, 3.3 Hz, 1H), 7.52 (t, J = 7.8 Hz, 2H), 7.31–7.27 (m, 3H), 7.04 (dd, J = 9.1, 3.9 Hz, 1H), 5.92 (dt, J = 12.5, 7.0 Hz, 1H), 4.74 (dd, J = 7.0, 1.2 Hz, 2H). 13C{1H} NMR (151 MHz, CDCl3) δ 188.58, 188.57, 163.3, 157.9, 156.98, 156.97, 156.3, 140.2, 133.9, 130.10, 128.6, 128.4, 122.5, 122.3, 114.68, 114.63, 114.3, 114.1, 108.9. 19F{1H} NMR (376 MHz, CDCl3) δ −121.79. HRMS (ESI-TOF) m/z: [M – H]− Calcd for C17H12FO4– 299.0725; Found 299.0725.
3-(2-Formylphenoxy)prop-1-en-1-yl Benzoate (14d)
The compound was prepared by following the general procedure starting from 2-hydroxybenzaldehyde (244 mg, 2.00 mmol). The reaction mixture was stirred for 1.5 h, and the product was purified with automated flash column chromatography (50 g cartridge, 2–25% EtOAc in n-heptane) in 79% yield as a white powder (450 mg, 1.59 mmol). 3-Bromoprop-1-en-1-yl benzoate was prepared by following literature reports.15a Instead of recrystallization, it was purified on automated flash column chromatography (50 g cartridge, 0–10% EtOAc in n-heptane) and isolated as a mixture of E and Z isomers. Thus, the alkylated salicylaldehyde was also isolated as a mixture of E and Z isomers. IR (KBr): ν 3415, 3093, 3067, 2941, 2859, 2761, 1734, 1691, 1599, 1582, 1482, 1453, 1400 cm–1. 1H NMR (400 MHz, CDCl3) δ 10.55 (s, 1H), 10.25 (s, 1H), 8.30–8.23 (m, 1H), 8.19–8.09 (m, 2H), 8.00–7.98 (m, 1H), 7.90–7.87 (m, 1H), 7.80–7.42 (m, 9H), 7.37–7.35 (m, 1H), 7.12–7.02 (m, 2H), 5.98–5.91 (m, 1H), 5.42–5.37 (m, 1H), 5.03–5.01 (m, 1H), 4.77–4.75 (m, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ 189.7, 189.6, 188.4, 164.9, 163.3, 162.8, 160.7, 152.3, 140.0, 137.1, 135.8, 135.8, 135.3, 134.0, 134.0, 133.9, 130.3, 130.2, 130.1, 130.0, 128.7, 128.7, 128.6, 128.6, 128.6, 128.5, 128.4, 128.3, 126.5, 125.2, 125.2, 123.5, 121.1, 121.0, 112.8, 112.7, 109.2, 108.7, 65.4, 62.2. HRMS (ESI-TOF) m/z: [M – H]− Calcd for C17H13O4– 281.0819; Found 281.0819.
General Procedure for Hydrolysis of Methyl Esters. Synthesis of 15a–j and 16a–e
The methyl ester was dissolved in THF (4 mL), and LiOH (0.10 M, 1.40 equiv) was added to the stirred solution. The hydrolysis was monitored with TLC. Upon completion, HCl (1.00 M, 1.50 equiv) was added. The resulting mixture was stirred for 5 min, then concentrated partially, until most of the THF was removed. The residue was partitioned between water and CHCl3/MeOH 9:1 (10 mL). The phases were separated, and the aqueous phase was extracted once more (5 mL). The combined organic extracts were dried with sodium sulfate, filtered, and evaporated. The residue was redissolved in DMSO (1–3 mL) and purified with preparative HPLC. The fractions containing the pure desired product were combined, diluted with water, and freeze-dried.
(R)-8-Cyclopropyl-13-oxo-7-phenyl-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylic Acid (15a)
The compound was prepared by following the general procedure. 50 mg of 11a was hydrolyzed. The reaction was finished after 42 min. Orange solid (32.6 mg, 67.2%). [α]D −90.40 [c 0.20, (CD3)2SO]; IR (KBr): ν 3441, 3003, 1743, 1648, 1610, 1576, 1519, 1493, 1463, 1437, 1382, 1299, 1253, 1221, 1182, 1036 cm–1. 1H NMR [600 MHz, (CD3)2SO]: δ 13.52 (s, 1H), 8.21 (dd, J = 7.7, 1.6 Hz, 1H), 7.55–7.31 (m, 6H), 7.19 (t, J = 7.5 Hz, 1H), 6.99 (d, J = 8.0 Hz, 1H), 5.60 (dd, J = 8.7, 1.9 Hz, 1H), 5.13 (d, J = 14.7 Hz, 1H), 5.00 (d, J = 14.6 Hz, 1H), 3.83 (dd, J = 11.7, 8.7 Hz, 1H), 3.54 (dd, J = 11.9, 1.9 Hz, 1H), 0.85–0.75 (m, 1H), 0.30 to −0.01 (m, 4H). 13C{1H} NMR [151 MHz, (CD3)2SO] δ 169.6, 158.2, 155.8, 145.1, 145.0, 141.7, 140.1, 136.8, 133.3, 131.7, 129.9, 129.8, 128.7, 128.3, 127.8, 127.7, 125.0, 122.7, 122.5, 116.8, 107.3, 66.0, 63.2, 30.9, 15.2, 11.2, 11.0. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C27H21N2O4S+ 469.1217; Found 469.1228.
8-Cyclopropyl-3-nitro-13-oxo-7-phenyl-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylic Acid (15b)
The compound was prepared by following the general procedure. 62 mg of methyl ester 11b was hydrolyzed. The reaction was found complete after 50 min. Yellow solid (30.9 mg, 51.2%). [α]D −122.3° [c 0.308, (CD3)2SO]; IR (KBr): ν 3448, 1638, 1573, 1530, 1438, 1343, 1226, 1042, 741 cm–1. 1H NMR [600 MHz, (CD3)2SO]: δ 13.63 (bs, 1H), 8.42 (dd, J = 8.6, 1.8 Hz, 1H), 8.05 (dd, J = 8.6, 2.2 Hz, 1H), 7.76 (d, J = 2.2 Hz, 1H), 7.54–7.41 (m, 4H), 7.40–7.33 (m, 1H), 5.61 (d, J = 8.7 Hz, 1H), 5.31–5.09 (m, 2H), 3.84 (dd, J = 11.7, 8.7 Hz, 1H), 3.56 (dd, J = 11.7, 1.9 Hz, 1H), 0.90–0.82 (m, 1H), 0.30–0.15 (m, 3H), 0.12–0.00 (m, 1H). 13C{1H} NMR [151 MHz, (CD3)2SO]: δ 169.5, 157.9, 155.7, 149.1, 146.7, 142.7, 142.1, 140.4, 136.4, 134.3, 129.8, 129.7, 129.2, 128.5, 128.4, 127.9, 127.8, 126.0, 117.5, 112.0, 107.3, 66.7, 63.3, 31.1, 15.2, 11.2, 11.1. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C27H20N3O6S+ 514.1068; Found 514.1062.
(R)-3-Nitro-13-oxo-7-phenyl-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylic Acid (15c)
The compound was prepared by following the general procedure but with 1.60 equiv of LiOH. 50 mg of 11c was hydrolyzed. The reaction was finished after 2 h, whereupon 1.72 equiv of HCl was added. The mixture was partitioned between DCM/n-BuOH (2:1) and brine/water (1:1). Yellow solid (38 mg, 78%). [α]D −6.79 [c 0.18, (CD3)2SO]. IR (KBr): ν 3440, 3082, 2935, 1754, 1666, 1623, 1592, 1577, 1536, 1517, 1226, 1043 cm–1. 1H NMR [400 MHz, (CD3)2SO, 343 K]: δ 8.50 (d, J = 8.6 Hz, 1H), 8.02 (dd, J = 8.6, 2.3 Hz, 1H), 7.74 (d, J = 2.3 Hz, 1H), 7.65–7.55 (m, 3H), 7.40–7.32 (m, 2H), 5.95 (s, 1H), 5.62 (dd, J = 8.5, 1.8 Hz, 1H), 5.19 (s, 2H), 3.93 (dd, J = 11.8, 8.5 Hz, 1H), 3.62 (dd, J = 11.7, 1.8 Hz, 1H). 13C{1H} NMR [100 MHz, (CD3)2SO, 343 K]: δ 168.9, 158.1, 155.6, 148.9, 144.0, 143.1, 141.3, 138.6, 133.2, 132.8, 128.82, 128.79, 128.75, 128.5, 128.4, 128.1, 127.7, 125.6, 116.9, 111.8, 93.6, 66.3, 62.6, 31.6. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H16N3O6S+ 474.0755; Found 474.0757.
(R)-8-Cyclopropyl-2-nitro-13-oxo-7-phenyl-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylic Acid (15d)
The compound was prepared by following the general procedure. 50 mg of 11d was hydrolyzed. The reaction was finished after 40 min. Yellow solid (36.9 mg, 75.8%). [α]D −92.47 [c 0.23, (CH3)2SO]. IR (KBr): ν 3442, 3082, 3002, 1743, 1631, 1589, 1574, 1550, 1518, 1494, 1481, 1460, 1443, 1385, 1341, 1297, 1251, 1201, 1091, 1023 cm–1. 1H NMR [600 MHz, (CD3)2SO]: δ 13.62 (s, 1H), 9.00 (d, J = 2.9 Hz, 1H), 8.25 (dd, J = 8.9, 2.5 Hz, 1H), 7.47 (dd, J = 17.9, 4.9 Hz, 4H), 7.36 (d, J = 5.2 Hz, 1H), 7.21 (d, J = 8.8 Hz, 1H), 5.62 (d, J = 8.5 Hz, 1H), 5.27 (dd, J = 87.1, 14.9 Hz, 2H), 3.84 (dd, J = 11.7, 8.7 Hz, 1H), 3.56 (d, J = 11.6 Hz, 1H), 0.86 (s, 1H), 0.30–0.15 (m, 3H), 0.10–0.00 (m, 1H). 13C{1H} NMR [151 MHz, (CD3)2SO]: δ 169.5, 160.6, 158.0, 146.3, 142.5, 142.4, 142.2, 140.2, 136.4, 134.1, 129.7, 129.7, 128.5, 128.0, 127.9, 127.8, 126.9, 122.6, 120.2, 118.2, 107.2, 66.9, 63.4, 31.0, 15.2, 11.2, 11.1. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C27H20N3O6S+ 514.1068; Found 514.1088.
(R)-8-Cyclopropyl-7-(4-nitrophenyl)-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylic Acid (15e)
The compound was prepared by following the general procedure. 50 mg of 11e was hydrolyzed. The reaction was finished after 30 min. Yellow solid (34.5 mg, 70.9%). [α]D −94.72° [c 0.26, (CD3)2SO]. IR (KBr): ν 3442, 3075, 3002, 2850, 2578, 1758, 1665, 1605, 1561, 1519, 1491, 1464, 1442, 1385, 1348, 1298, 1258, 1225, 1183, 1109, 1045 cm–1. 1H NMR [600 MHz, (CD3)2SO]: δ 13.58 (s, 1H), 8.37–8.27 (m, 2H), 8.21 (d, J = 7.7 Hz, 1H), 7.78 (d, J = 7.7 Hz, 1H), 7.68 (d, J = 7.9 Hz, 1H), 7.40 (t, J = 7.7 Hz, 1H), 7.20 (t, J = 7.5 Hz, 1H), 7.00 (d, J = 8.1 Hz, 1H), 5.62 (d, J = 8.6 Hz, 1H), 5.17–4.97 (m, 2H), 3.89–3.78 (m, 1H), 3.55 (d, J = 11.6 Hz, 1H), 0.89–0.82 (m, 1H), 0.32–0.17 (m, 3H), 0.10–0.03 (m, 1H). 13C{1H} NMR [151 MHz, (CD3)2SO]: δ 169.5, 158.0, 155.8, 147.2, 146.1, 144.9, 143.7, 140.1, 139.4, 133.0, 131.8, 131.4, 131.3, 128.5, 125.0, 123.0, 122.9, 122.6, 122.5, 116.8, 106.7, 65.8, 63.3, 31.0, 15.5, 11.8, 11.7. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C27H20N3O6S+ 514.1068; Found 514.1072.
(R)-3-Nitro-7-(4-nitrophenyl)-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylic Acid (15f)
The compound was prepared by following the general procedure but with 1.60 equiv of LiOH. 50 mg of 11f was hydrolyzed. The reaction was finished after 2 h, whereupon 1.72 equiv of HCl was added. The mixture was partitioned between DCM/n-BuOH (1:1) and brine/water (1:1). Yellow solid (26.1 mg, 53.6%) [α]D −3.55 [c 0.51, (CD3)2SO]. IR (KBr): ν 3435, 3107, 1748, 1666, 1592, 1566, 1533, 1453, 1431, 1384, 1346, 1227, 1180, 1043 cm–1. 1H NMR [400 MHz, (CD3)2SO]: δ 13.74 (s, 1H), 8.54–8.35 (m, 3H), 8.04 (dd, J = 8.7, 2.3 Hz, 1H), 7.77 (d, J = 2.3 Hz, 1H), 7.73–7.67 (m, 1H), 5.98 (s, 1H), 5.62 (dd, J = 8.6, 1.5 Hz, 1H), 5.20 (s, 2H), 3.90 (dd, J = 11.8, 8.6 Hz, 1H), 3.62 (dd, J = 11.8, 1.6 Hz, 1H). 13C{1H} NMR [100 MHz, (CD3)2SO]: δ 169.4, 158.3, 155.8, 149.0, 147.9, 144.9, 143.2, 140.0, 139.5, 138.6, 133.1, 130.8, 130.6, 128.1, 128.0, 125.8, 124.3, 117.5, 112.2, 93.9, 66.3, 62.8, 31.9. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H15N4O8S+ 519.0605; Found 519.0612.
(R)-8-Cyclopropyl-2-fluoro-13-oxo-7-phenyl-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylic Acid (15g)
The compound was prepared by following the general procedure starting from 11g (50 mg, 0.09 mmol). The reaction mixture was stirred for 4 h, and the product was isolated as a yellow powder in 38% yield (38 mg, 0.07 mmol). [α]D −113° [c 0.12, (CH3)2SO]. IR (KBr): ν 3415, 3081, 3001, 2921, 2852, 1713, 1656, 1619, 1575, 1550, 1520, 1494, 1482, 1462, 1429 cm–1. 1H NMR [600 MHz, (CD3)2SO] δ 7.67 (dd, J = 9.0, 3.2 Hz, 1H), 7.31–7.19 (m, 5H), 7.14 (dt, J = 6.8, 2.4 Hz, 1H), 7.03 (td, J = 8.6, 3.2 Hz, 1H), 6.83 (dd, J = 8.9, 4.5 Hz, 1H), 5.25 (t, J = 7.8 Hz, 1H), 4.94–4.76 (m, 2H), 3.54 (dd, J = 11.3, 8.4 Hz, 1H), 3.33 (dt, J = 11.5, 2.9 Hz, 1H), 0.70–0.55 (m, 1H), 0.15 to −0.10 (m, 3H), −0.13 to −0.25 (m, 1H). 13C{1H} NMR [151 MHz, (CD3)2SO] δ 169.5, 158.8, 158.46, 158.44, 157.2, 152.4, 146.8, 144.0, 142.0, 140.7, 140.6, 137.2, 134.06, 134.04, 130.3, 130.25, 130.22, 128.9, 128.7, 128.2, 128.16, 128.12, 124.6, 124.5, 119.0, 118.9, 118.6, 118.5, 110.8, 110.7, 107.1, 66.64, 66.62, 64.8, 32.1, 15.67, 15.65, 11.6, 11.5. 19F NMR [565 MHz, (CD3)2SO] δ −120.83 HRMS (ESI) m/z: [M + H]+ Calcd for C27H20FN2O4S+ 487.1123; Found 487.1103.
(R)-8-Cyclopropyl-13-oxo-7-(3-(trifluoromethyl)phenyl)-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylic Acid (15h)
The compound was prepared by following the general procedure, but upon complete saponification, the mixture was neutralized with Amberlyst 15 until around pH = 6 by pH paper, then filtered through a pad of THF-wet Celite. The amberlyst and Celite were rinsed with MeOH until the filtrate was transparent. The filtrate was evaporated and extracted according to the general procedure. The residue was triturated with Et2O (0.5 mL) and filtered. The solid was washed with more Et2O (0.5 mL) and dried under vacuum overnight. 25 mg of 11h was hydrolyzed to provide 15h (15.45 mg, 63.4%) as a yellow solid. [α]D −51.71° [c 0.29, (CD3)2SO]. IR (KBr): ν 3442, 3082, 3003, 1734, 1629, 1577, 1518, 1490, 1446, 1382, 1330, 1169, 1127 cm–1. 1H NMR [600 MHz, (CD3)2SO, 343 K]: δ 8.25 (dd, J = 7.7, 1.8 Hz, 1H), 7.85–7.65 (m, 4H), 7.37 (td, J = 7.8, 1.7 Hz, 1H), 7.17 (t, J = 7.5 Hz, 1H), 6.96 (d, J = 8.1 Hz, 1H), 5.38 (d, J = 8.2 Hz, 1H), 5.14–4.92 (m, 2H), 3.68 (dt, J = 10.0, 7.3 Hz, 1H), 3.55 (d, J = 10.9 Hz, 1H), 0.80–0.70 (m, 1H), 0.34 to −0.07 (m, 4H). 13C{1H} NMR [151 MHz, (CD3)2SO, 343 K]: δ 168.4, 157.7, 155.4, 146.6, 144.0, 140.3, 137.9, 133.6, 132.6 (d, J = 13.3 Hz), 131.0, 128.6 (d, J = 9.9 Hz), 127.8, 126.1, 124.7, 124.6, 124.3, 122.9, 122.5, 122.0, 116.3, 105.3, 65.6, 65.1, 31.8 (d, J = 8.3 Hz), 14.8 (d, J = 9.5 Hz), 11.2–0.9 (m). The multiplets in the13C NMR spectrum are caused by atropoisomerism.19F NMR [565 MHz, (CD3)2SO, 343 K]: δ −61.20. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C28H20F3N2O4S+ 537.1091; Found 537.1094.
(R)-8-Cyclopropyl-7-(3-methoxyphenyl)-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylic Acid (15i)
The compound was prepared by following the general procedure, but upon complete saponification, the mixture was neutralized with Amberlyst 15 until around pH = 6 by pH paper, then filtered through a pad of THF-wet Celite. The amberlyst and Celite were rinsed with MeOH until the filtrate was transparent. The filtrate was evaporated and extracted according to the general procedure. The residue was triturated in Et2O (0.5 mL) and filtered. The solid was washed with more Et2O (0.5 mL) and dried under vacuum overnight. 25 mg of 11i was converted to 15i (17.5 mg, 72%) isolated as a yellow solid. [α]D −91.56° [c 0.21, (CD3)2SO]. IR (KBr): ν 3442, 3390, 2941, 2836, 1757, 1646, 1608, 1577, 1519, 1463, 1439, 1439, 1218, 1038 cm–1. 1H NMR [600 MHz, (CD3)2SO, 343 K]: δ 8.24 (dd, J = 7.8, 1.7 Hz, 1H), 7.44–7.33 (m, 2H), 7.16 (td, J = 7.5, 1.1 Hz, 1H), 7.03 (dd, J = 8.2, 2.5 Hz, 1H), 7.00–6.87 (m, 3H), 5.51–5.45 (m, 1H), 5.17–4.96 (m, 2H), 3.82 (d, J = 8.6 Hz, 3H), 3.78–3.71 (m, 1H), 3.53 (dd, J = 11.3, 2.0 Hz, 1H), 0.99–0.92 (m, 1H), 0.34–0.21 (m, 3H), 0.20–0.09 (m, 1H). 13C{1H} NMR [151 MHz, (CD3)2SO, 343 K]: δ 168.7, 158.4 (d, J = 9.2 Hz), 157.8, 155.5, 145.2, 144.3, 141.0, 140.1, 138.0, 132.8, 131.1, 128.6 (d, J = 10.8 Hz), 128.0, 124.6, 122.5, 121.9, 116.3, 115.4 (d, J = 12.2 Hz), 113.5, 106.4, 65.8, 64.1, 55.1, 31.2, 14.6, 11.14–10.10 (m). The multiplets in the13C NMR spectrum are caused by atropoisomerism. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C28H22N2O5S+ 499.1322; Found 499.1326.
(R)-8-Cyclopropyl-7-(4-ethoxyphenyl)-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylic Acid (15j)
The compound was prepared by following the general procedure. 50 mg of 11j was hydrolyzed. The reaction was finished after 65 min. Yellow powder (35.5 mg, 72.9%). [α]D −78.30° [c 0.19, (CD3)2SO]. IR (KBr): ν 3413, 2980, 1745, 1640, 1608, 1577, 1508, 1463, 1441, 1383, 1299, 1283, 1245, 1223, 1178, 1111, 1045 cm–1. 1H NMR [600 MHz, (CD3)2SO] δ 13.52 (s, 1H), 8.19 (d, J = 7.8 Hz, 1H), 7.39 (t, J = 7.7 Hz, 1H), 7.33 (d, J = 7.9 Hz, 1H), 7.24 (d, J = 8.2 Hz, 1H), 7.18 (t, J = 7.5 Hz, 1H), 7.07–6.95 (m, 3H), 5.59 (d, J = 8.7 Hz, 1H), 5.21–5.01 (m, 2H), 4.09 (q, J = 6.8 Hz, 2H), 3.86–3.78 (m, 1H), 3.53 (d, J = 11.6 Hz, 1H), 1.37 (t, J = 6.9 Hz, 3H), 1.00–0.90 (m, 1H), 0.36–0.08 (m, 4H). 13C{1H} NMR [151 MHz, (CD3)2SO] δ 169.6, 158.4, 158.2, 155.8, 144.9, 144.9, 141.7, 140.1, 133.5, 131.6, 131.1, 129.0, 128.8, 125.0, 122.8, 122.4, 116.8, 113.6, 113.5, 107.5, 66.0, 63.2, 63.1, 30.9, 15.0, 14.6, 11.0, 10.9. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C29H25N2O5S+ 513.1479; Found 513.1496.
(R)-8-Methoxy-3-nitro-13-oxo-7-phenyl-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylic Acid (15k)
The compound was prepared by following the general procedure. Compound precipitated during the workup and was filtered through a sintered funnel and dried. The compound was redissolved in DMSO (3 mL) and purified with preparative HPLC. 105 mg of 11k was hydrolyzed. The reaction was finished after 16 h. Yellow powder (66 mg, 64.6%). [α]D −52° [c 0.4, (CD3)2SO]. IR (KBr): ν 3449, 3057, 1740, 1665, 1635, 1583, 1556, 1533, 1515, 1458, 1430, 1382, 1229, 1181, 925, 820, 740, 707 cm–1. 1H NMR [600 MHz, (CD3)2SO] δ 8.51–8.37 (m, 1H), 8.04 (dd, J = 8.6, 2.2 Hz, 1H), 7.75 (d, J = 2.2 Hz, 1H), 7.49–7.48 (m, 3H), 7.39–7.36 (m, 2H), 5.63 (d, J = 8.5 Hz, 1H), 5.10–5.01 (m, 2H), 3.90 (dd, J = 11.6, 8.6 Hz, 1H), 3.64 (d, J = 11.3 Hz, 1H), 2.88 (s, 3H). 13C{1H} NMR [151 MHz, (CD3)2SO] δ 169.2, 157.1, 155.7, 149.0, 143.0, 140.1, 139.6, 137.2, 135.5, 130.6, 129.6, 129.4, 128.3, 128.1, 128.0, 127.7, 127.6, 125.9, 117.4, 112.1, 66.3, 63.4, 59.4, 31.7. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C25H18N3O7S+ 504.0860; Found 504.0842.
8-Cyclopropyl-3-nitro-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylic Acid (16a)
The compound was prepared by following the general procedure. 33 mg of 13a was hydrolyzed. The reaction was finished after 40 min. Orange solid (9.1 mg, 28.5%). [α]D −162.3° [c 0.186, (CD3)2SO]; IR (KBr): ν 3438, 1731, 1652, 1578, 1531, 1432, 1345, 1238, 1034, 919, 739 cm–1. 1H NMR [600 MHz, (CD3)2SO]: δ 8.38 (dd, J = 8.6, 2.1 Hz, 1H), 8.29 (s, 1H), 8.01 (dd, J = 8.6, 2.3 Hz, 1H), 7.80 (d, J = 2.2 Hz, 1H), 5.63–5.60 (m, 3H), 3.86 (dd, J = 11.7, 8.7 Hz, 1H), 3.61 (dd, J = 11.7, 1.6 Hz, 1H), 1.80–1.74 (m, 1H), 1.12–1.02 (m, 2H), 0.68–0.63 (m, 1H), 0.56–0.51 (m, 1H). 13C{1H} NMR [151 MHz, (CD3)2SO]: δ 169.6, 158.2, 156.3, 148.9, 144.1, 143.3, 139.6, 135.5, 130.6, 128.2, 127.7, 125.6, 117.3, 112.3, 106.5, 67.9, 62.8, 31.3, 9.7, 7.5, 7.3. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C21H16N3O6S+ 438.0755; Found 438.0769.
(R)-3-Nitro-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylic Acid (16b)
The compound was prepared by following the general procedure starting from 13b (28 mg, 0.07 mmol). The reaction mixture was stirred for 0.5 h, and the product was isolated as a yellow powder in 15% yield (4.200 mg, 0.01 mmol). [α]D −100° [c 0.10, (CD3)2SO]. IR (KBr): ν 3418, 3085, 2927, 2597, 1742, 1642, 1614, 1584, 1535, 1516, 1456 cm–1. 1H NMR [600 MHz, (CD3)2SO] δ 8.41 (d, J = 8.6 Hz, 1H), 8.03 (dd, J = 8.6, 2.3 Hz, 1H), 7.87 (s, 1H), 7.81 (d, J = 2.2 Hz, 1H), 6.68 (s, 1H), 5.55 (d, J = 17.8 Hz, 3H), 3.93 (dd, J = 11.5, 8.5 Hz, 1H), 3.66 (d, J = 11.4 Hz, 1H). 13C{1H} NMR [151 MHz, (CD3)2SO] δ 169.3, 158.5, 156.2, 148.9, 144.0, 143.5, 138.7, 134.7, 130.8, 129.5, 128.2, 125.6, 117.3, 112.3, 96.3, 67.6, 63.0, 32.1. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C18H12N3O6S+ 398.0442; Found 398.0439.
(R)-8-Cyclopropyl-2-fluoro-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylic Acid (16c)
The compound was prepared by following the general procedure starting from 13c (47 mg, 0.11 mmol). The reaction mixture was stirred for 1 h, and the product was isolated as a yellow powder in 43% yield (20 mg, 0.04 mmol). [α]D −145° [c 0.22, (CD3)2SO]. IR (KBr): ν 3415, 3070, 3002, 2846, 1744, 1626, 1601, 1569, 1525, 1473 cm–1. 1H NMR [400 MHz, (CD3)2SO] δ 13.49 (s, 1H), 8.27 (s, 1H), 7.85 (dd, J = 9.0, 3.2 Hz, 1H), 7.26 (td, J = 8.6, 3.2 Hz, 1H), 7.09 (dd, J = 8.9, 4.6 Hz, 1H), 5.61 (dd, J = 8.8, 1.6 Hz, 1H), 5.47 (s, 2H), 3.86 (dd, J = 11.8, 8.7 Hz, 1H), 3.60 (dd, J = 11.8, 1.7 Hz, 1H), 1.85–1.70 (m, 1H), 1.13–1.02 (m, 2H), 0.70–0.54 (m, 2H). 13C{1H} NMR [100 MHz, (CD3)2SO] δ 170.1, 159.1, 158.7, 156.7, 153.0, 145.0, 143.5, 139.4, 135.4, 130.6, 127.9, 124.1, 124.0, 119.4, 119.3, 118.8, 118.6, 110.5, 110.3, 106.9, 67.9, 63.2, 31.6, 10.2, 7.9, 7.7. 19F{1H} NMR [376 MHz, (CD3)2SO] δ −120.90. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C21H16FN2O4S+ 411.0810; Found 411.0801.
(R)-8-Cyclopropyl-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylic Acid (16d)
The compound was prepared by following the general procedure starting from 13d (47 mg, 0.11 mmol). The reaction mixture was stirred for 2 h, and the product was isolated as a white powder in 28% yield (13 mg, 0.03 mmol). [α]D −60° [c 0.16, (CD3)2SO]. IR (KBr): ν 3432, 3001, 2548, 1742, 1625, 1599, 1582, 1565, 1524, 1493, 1467 cm–1. 1H NMR [600 MHz, (CD3)2SO] δ 8.26 (s, 1H), 8.21 (dd, J = 7.8, 1.7 Hz, 1H), 7.40 (td, J = 7.7, 1.7 Hz, 1H), 7.18 (t, J = 7.5 Hz, 1H), 7.04 (d, J = 8.1 Hz, 1H), 5.61 (dd, J = 8.8, 1.6 Hz, 1H), 5.47 (d, J = 1.5 Hz, 2H), 3.86 (dd, J = 11.7, 8.7 Hz, 1H), 3.59 (dd, J = 11.7, 1.6 Hz, 1H), 1.83–1.74 (m, 1H), 1.13–1.02 (m, 2H), 0.70–0.54 (m, 2H). 13C{1H} NMR [151 MHz, (CD3)2SO] δ 168.0, 156.7, 154.7, 143.9, 140.7, 137.3, 132.9, 130.0, 128.5, 125.6, 122.9, 120.75, 120.73, 115.5, 104.8, 65.7, 61.0, 29.4, 8.1, 5.7, 5.6. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C21H16N2NaO4S+ 415.0729; Found 415.0712.
(R)-8-Cyclopropyl-2-nitro-13-oxo-6,10,11,13-tetrahydrochromeno[4,3-b]thiazolo[2,3-g][1,7]naphthyridine-11-carboxylic Acid (16e)
The compound was prepared by following the general procedure starting from 13e (20.76 mg, 0.046 mmol). The reaction was stirred for 1.5 h, and the product was isolated as yellow powder in 20% yield (4.200 mg, 0.009 mmol). [α]D −198° [c 0.10, (CD3)2SO]. IR (KBr): ν 3550, 3475, 3414, 3236, 3079, 2844, 2497, 1754, 1634, 1617, 1587 cm–1. 1H NMR [600 MHz, (CD3)2SO] δ 8.95 (d, J = 2.8 Hz, 1H), 8.24 (dd, J = 9.0, 2.9 Hz, 1H), 7.25 (d, J = 9.0 Hz, 1H), 5.69–5.59 (m, 4H), 3.87–3.84 (m, 1H), 3.61 (dd, J = 11.6, 1.7 Hz, 1H), 1.79–1.75 (m, 1H), 1.11–1.03 (m, 2H), 0.68–0.56 (m, 2H). 13C{1H} NMR [151 MHz, (CD3)2SO] δ 169.5, 161.1, 158.2, 143.7, 143.0, 142.2, 139.0, 135.2, 129.3, 127.7, 126.7, 122.3, 119.8, 118.5, 106.3, 68.1, 62.9, 31.2, 9.7, 7.4, 7.2. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C21H16N3O6S+ 438.0755; Found 438.0766.
2-(Hydroxymethyl)-5-nitrophenol
The compound was prepared from 2-hydroxy-4-nitro-benzoic acid by following a published procedure18 but with only 3 equiv of BH3·SMe2. 10.00 g was converted to 8.39 g (91%) after purification. NMR data were in agreement with published data.
2-Hydroxy-4-nitrobenzaldehyde (Ib)
2-(Hydroxymethyl)-5-nitrophenol (8.20 g, 1.00 equiv) was dissolved in THF (100 mL). (Diacetoxyiodo)benzene (17.27 g, 1.11 equiv) and TEMPO (158 mg, 0.02 equiv) were weighed up and added. The resulting suspension was stirred at r.t. After 30 min, a clear solution remained, but TLC indicated some remaining acid. No further progress was indicated after 65 min, whereupon (diacetoxyiodo)benzene (3.14 g, 0.20 equiv) was added. After 50 min of stirring, the reaction was found complete by TLC. Saturated aqueous sodium thiosulfate solution (100 mL) was added, and the mixture was stirred for 5 min, then transferred to a separation funnel and extracted with EtOAc (150 + 50 + 50 mL). The organic phases were combined, washed with brine (200 mL), and evaporated. The residue was redissolved in chloroform (300 mL) and extracted with aqueous potassium hydroxide solution (5% w/v, 3 × 200 mL). The combined extracts were cooled in an ice-water bath and neutralized with HCl (12 M, 44 mL). The precipitate was filtered off and dissolved in EtOAc, dried with sodium sulfate, filtered, and evaporated. The residue was redissolved in DCM/MeOH (99:1) and purified with flash column chromatography (70 × 120 mm silica gel, DCM/MeOH 99:1). 5.23 g (64.5%) isolated as a light-yellow powder. NMR data recorded in (CD3)2CO are in close agreement with published data,18 and the phenolic proton is visible as well. 1H NMR (600 MHz, (CD3)2CO): δ 11.06 (s, 1H), 10.27 (s, 1H), 8.10 (d, J = 8.4 Hz, 1H), 7.93–7.81 (m, 1H), 7.76 (s, 1H). IR (KBr): ν 3452, 3224, 3110, 3046, 2885, 1676, 1537, 1476, 1353, 1299, 1219, 1172, 1116, 958, 882, 827, 793, 740 cm–1. 1H NMR (600 MHz, CDCl3): δ 11.16 (s, 1H), 10.08 (s, 1H), 7.79–7.89 (m, 3H). 13C{1H} NMR (151 MHz, CDCl3): δ 196.1, 162.0, 152.6, 134.9, 123.8, 114.5, 113.6. HRMS (ESI-TOF) m/z: [M – H]− Calcd for C7H4NO4– 166.0146; Found 166.0145.
General Procedure for Synthesis of Cinnamyl Esters IVa–c
The cinnamyl esters were synthesized according to a literature procedure with slight modifications.19 LiCl (509 mg; 12 mmol, 1.2 equiv) was weighed up in an oven-dried 100 mL round-bottom flask and put under vacuum. The flask was heated with a hot air gun for several minutes and allowed to cool down to room temperature, then back-filled with nitrogen. The flask was put under vacuum and back-filled with nitrogen twice more. Dried MeCN (30 mL) was added with a syringe, and the resulting suspension was cooled to 0 °C in an ice-water bath. Triethylphosphonoacetate (2.4 mL, 12 mmol, 1.2 equiv) was added with a syringe while stirring. After 20 min was added benzaldehyde III (10 mmol, 1.0 equiv), followed by DBU (1.8 mL, 12 mmol, 1.2 equiv) dropwise. The resulting reaction mixture was allowed to warm up to room temperature and stirred until completion was indicated by TLC analysis. Upon complete consumption of the aldehyde, the mixture was concentrated under reduced pressure and partitioned between diethyl ether (30 mL) and NH4Cl solution (sat. aq.) (40 mL). The aq. phase was extracted with diethyl ether (2 × 30), and the combined organic phase was dried over sodium sulfate, filtered, and concentrated by rotary evaporation. The residue was redissolved in DCM and purified with automated flash column chromatography.
Ethyl (E)-3-(3-Methoxyphenyl)acrylate (IVa)
The compound was prepared by following the general procedure. The reaction was finished after 3.5 h. The crude product was purified with automated flash column chromatography (50 g cartridge, 5–15% EtOAc in heptane). Transparent oil (1.94 g, 94%). IR (KBr): ν 2981, 2940, 2906, 2837, 1712, 1638, 1581, 1490, 1466, 1434, 1367, 1311, 1293, 1261, 1179, 1041, 982 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 16.0 Hz, 1H), 7.30 (t, J = 7.9 Hz, 1H), 7.12 (dt, J = 7.7, 1.2 Hz, 1H), 7.04 (s, 1H), 6.93 (ddd, J = 8.3, 2.5, 0.9 Hz, 1H), 6.42 (d, J = 16.0 Hz, 1H), 4.27 (q, J = 7.1 Hz, 2H), 3.83 (s, 3H), 1.34 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 167.1, 160.0, 144.6, 136.0, 130.0, 120.9, 118.7, 116.3, 113.0, 60.7, 55.4, 14.5. The NMR data are in agreement with published data for this compound.20a
Ethyl (E)-3-(3-(Trifluoromethyl)phenyl)acrylate (IVb)
The compound was prepared by following the general procedure. The reaction was finished after 1.3 h. The crude product was purified with automated flash column chromatography (50 g cartridge, 5% EtOAc in heptane). Colorless oil that crystallizes at room temperature (1.20 g, 89%). IR (KBr): ν 3048, 2986, 2912, 1715, 1675, 1642, 1478, 1441, 1367, 1335, 1308, 1281, 1202, 1157, 1125, 1098, 1079, 1035, 996 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.77 (s, 1H), 7.74–7.66 (m, 2H), 7.63 (d, J = 7.8 Hz, 0H), 7.52 (t, J = 7.8 Hz, 1H), 6.50 (d, J = 16.0 Hz, 1H), 4.28 (q, J = 7.1 Hz, 2H), 1.35 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 166.6, 142.9, 135.4, 131.6 (q, J = 32.5 Hz), 131.2 (d, J = 1.4 Hz), 129.6, 126.7 (q, J = 3.7 Hz), 124.7 (q, J = 3.8 Hz), 123.9 (q, J = 272.5 Hz), 120.4, 60.9, 14.4. 19F NMR (376 MHz, CDCl3) δ −62.9. The NMR data are in agreement with published data for this compound.20b
Ethyl (E)-3-(4-Ethoxyphenyl)acrylate (IVc)
The compound was prepared by following the general procedure but at 15 mmol scale. The reaction was finished after 2.2 h. The crude product was purified with automated flash column chromatography (50 g cartridge, 5% EtOAc in heptane). White solid (3.03 g, 91%). IR (KBr): ν 3395, 2988, 2932, 1710, 1631, 1604, 1573, 1482, 1425, 1394, 1364, 1289, 1253, 1168, 1043 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.64 (d, J = 15.9 Hz, 1H), 7.46 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 8.8 Hz 2H), 6.30 (d, J = 15.9 Hz, 1H), 4.25 (q, J = 7.1 Hz, 2H), 4.06 (q, J = 7.0 Hz, 2H), 1.43 (t, J = 7.0 Hz, 3H), 1.33 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 167.5, 160.9, 144.5, 129.8, 127.2, 115.8, 114.9, 63.8, 60.5, 14.9, 14.52.
General Procedure for Synthesis of Cinnamyl Alcohols Va–d
The cinnamyl alcohols were synthesized according to a literature procedure.19c The cinnamyl ester IV (10 mmol, 1.0 equiv) was weighed up in an oven-dried 100 mL round-bottom flask, put under nitrogen, and dissolved in dried THF (30 mL). The reaction mixture was cooled to −78 °C in a dry ice/acetone bath, and DiBAl-H (1 M in toluene, 24 mL, 24 mmol. 2.4 equiv) was added slowly with a syringe. The mixture was stirred at −78 °C while the reaction was monitored with TLC. Upon complete consumption of the cinnamyl ester, saturated Rochelle salt solution (20 mL) was added to the mixture at −78 °C. The mixture was allowed to warm up to room temperature, transferred to a separation funnel, and extracted with EtOAc (5 × 40 mL). The organic phases were combined, dried over sodium sulfate, filtered through a pad of EtOAc-wet Celite, and concentrated by rotary evaporation.
(E)-3-(3-Methoxyphenyl)prop-2-en-1-ol (Va)
The compound was prepared at 9.30 mmol scale by following the general procedure. The reaction was finished after 1 h. Transparent liquid (1.54 g, quant.). IR (KBr): ν 3361, 2939, 2836, 1599, 1580, 1490, 1465, 1454, 1433, 1289, 1257, 1156, 1046, 1011, 970 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.27–7.20 (m, 1H), 7.02–6.96 (m, 1H), 6.93 (t, J = 2.1 Hz, 1H), 6.81 (ddd, J = 8.2, 2.6, 0.9 Hz, 1H), 6.59 (d, J = 15.9 Hz, 1H), 6.36 (dt, J = 15.9, 5.7 Hz, 1H), 4.33 (dd, J = 5.7, 1.5 Hz, 2H), 3.82 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 159.9, 138.3, 131.1, 129.7, 129.0, 119.3, 113.5, 111.9, 63.8, 55.4. The NMR data are in agreement with published data for this compound.20c
(E)-3-(3-(Trifluoromethyl)phenyl)prop-2-en-1-ol (Vb)
The compound was prepared at 8.89 mmol scale by following the general procedure. The reaction was complete after 1 h. Transparent liquid (1.76 g, quant.). IR (KBr): ν 3345, 2869, 1487, 1441, 1334, 1270, 1199, 1165, 1125, 1099, 1072, 1011, 967 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.63 (s, 1H), 7.55 (d, J = 7.4 Hz, 1H), 7.52–7.39 (m, 2H), 6.71–6.61 (m, 1H), 6.44 (dt, J = 15.9, 5.4 Hz, 1H), 4.36 (dd, J = 5.4, 1.6 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 137.6, 131.2, (q, J = 32.3 Hz), 130.7, 129.7 (d, J = 1.4 Hz), 129.5, 129.2, 124.3 (q, J = 3.8 Hz), 124.2 (q, J = 136.2 Hz), 123.2 (q, J = 3.8 Hz), 63.5. 19F NMR (376 MHz, CDCl3): δ −62.81. The NMR data are in agreement with published data for this compound.20d
(E)-3-(4-Ethoxyphenyl)prop-2-en-1-ol (Vc)
The compound was prepared at 8.57 mmol scale by following the general procedure. The reaction was complete after 2 h. White solid (1.52 g, quant.). IR (KBr): ν 3363, 3274, 2979, 2931, 1605, 1512, 1476, 1447, 1397, 1305, 1269, 1246, 1175, 1114, 1087, 1048, 1007, 970 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.38–7.29 (m, 2H), 6.92–6.80 (m, 2H), 6.62–6.48 (m, 1H), 6.23 (dt, J = 15.9, 6.0 Hz, 1H), 4.35–4.27 (m, 2H), 4.04 (q, J = 7.0 Hz, 2H), 1.41 (t, J = 7.0 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 158.9, 131.2, 129.4, 127.8, 126.2, 114.7, 64.1, 63.6, 15.0. The NMR data are in agreement with published data for this compound.20e
(E)-3-(4-Nitrophenyl)prop-2-en-1-ol (Vd)
The compound was prepared at 10.4 mmol scale by following the general procedure. The reaction was finished after 1 h. Light yellow solid (1.89 g, quant.). IR (KBr): ν 3508, 3108, 3080, 1652, 1596, 1504, 1455, 1411, 1338, 1101, 972, 958 cm–1. 1H NMR (400 MHz, CDCl3): δ 8.23–8.10 (m, 2H), 7.59–7.44 (m, 2H), 6.72 (dt, J = 16.0, 1.8 Hz, 1H), 6.54 (dt, J = 16.0, 5.1 Hz, 1H), 4.40 (dd, J = 5.1, 1.7 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 147.1, 143.4, 133.7, 128.4, 127.1, 124.2, 63.3. The NMR data are in agreement with published data for this compound.20f
General Procedure for Synthesis of Cinnamyl Bromides IIa–d
The cinnamyl bromides were synthesized according to a literature procedure with slight modifications.19c The cinnamyl alcohol V (2.0 mmol, 1.0 equiv) was weighed up in an oven-dried Biotage microwave reaction tube and put under nitrogen. Dried diethyl ether (4 mL) was added, and the resulting suspension was cooled to 0 °C in an ice/water bath. PBr3 (0.20 mL, 2.0 mmol, 1.0 equiv) was added dropwise with a syringe. The resulting mixture was stirred at 0 °C until complete consumption of the starting material was indicated by TLC. The reaction was quenched at 0 °C by slow and careful addition of a concentrated phosphate buffer at pH 7.4 (4 mL). The reaction mixture was transferred to a separation funnel, and the phases were separated. The aq. phase was extracted with diethyl ether (2 × 5 mL). The combined organic phases were dried over sodium sulfate, filtered, and concentrated by rotary evaporation. The crude cinnamyl bromide was used for the next step without further purification or storage.
(E)-1-(3-Bromoprop-1-en-1-yl)-3-methoxybenzene (IIa)
The compound was prepared at 4.45 mmol scale by following the general procedure but with 0.915 equiv of PBr3 used. The reaction was complete after 5 min. Transparent liquid (0.938 g, 92.8% crude yield). 1H NMR (400 MHz, CDCl3): δ 7.25 (t, J = 7.9 Hz, 1H), 6.98 (dt, J = 7.6, 1.2 Hz, 1H), 6.92 (t, J = 2.1 Hz, 1H), 6.86–6.80 (m, 1H), 6.62 (d, J = 15.5 Hz, 1H), 6.39 (dt, J = 15.5, 7.7 Hz, 1H), 4.16 (dd, J = 7.8, 1.0 Hz, 2H), 3.82 (s, 3H). The NMR data are in agreement with published data for this compound.20g
(E)-1-(3-Bromoprop-1-en-1-yl)-3-(trifluoromethyl)benzene (IIb)
The compound was prepared at 3.98 mmol scale by following the general procedure. The reaction was complete after 25 min. Off white solid (0.977 g, 92.6% crude yield). 1H NMR (600 MHz, CDCl3): δ 7.63 (s, 1H), 7.54 (ddd, J = 19.3, 7.7, 1.7 Hz, 2H), 7.45 (t, J = 7.7 Hz, 1H), 6.67 (d, J = 15.6 Hz, 1H), 6.47 (dt, J = 15.5, 7.7 Hz, 1H), 4.15 (dd, J = 7.7, 1.1 Hz, 2H). The NMR data are in agreement with published data for this compound.20h
(E)-1-(3-Bromoprop-1-en-1-yl)-4-ethoxybenzene (IIc)
The compound was prepared at 5.03 mmol scale by following the general procedure. The reaction was finished after 5 min. White solid (1.10 g, 91% crude yield). 1H NMR (400 MHz, CDCl3): δ 7.31 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H), 6.59 (d, J = 15.6 Hz, 1H), 6.26 (dt, J = 15.6, 7.9 Hz, 1H), 4.17 (dd, J = 7.9, 1.0 Hz, 2H), 4.04 (q, J = 7.0 Hz, 2H), 1.41 (t, J = 7.0 Hz, 3H). This compound is unstable and has to be used in the next step immediately.
(E)-1-(3-Bromoprop-1-en-1-yl)-4-nitrobenzene (IId)
The compound was prepared at 6.0 mmol scale by following the general procedure but with DCM as solvent and 1.22 equiv of PBr3 used. The reaction was complete after 40 min. Light yellow solid (1.39 g, 96% crude yield). IR (KBr): ν 1595, 1515, 1341, 1197, 1107, 971 cm–1. 1H NMR (400 MHz, CDCl3): δ 8.20 (d, J = 8.8 Hz, 2H), 7.53 (d, J = 8.8 Hz, 2H), 6.71 (d, J = 15.7 Hz, 1H), 6.61–6.51 (m, 1H), 4.16 (dd, J = 7.5, 0.9 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3) δ 147.5, 142.3, 132.2, 130.0, 127.4, 124.2, 32.0. The NMR data are in agreement with published data for this compoud.20i
2-(Prop-2-yn-1-yloxy)benzaldehyde (VI)
Under an atmosphere of nitrogen, salicylaldehyde Ia (1.07 mL, 1.00 equiv) and K2CO3 (2.76 g, 2.00 equiv) were suspended in DMF (5.0 mL). While stirring at r.t., propargyl bromide (80% w/w in toluene, 1.45 mL, 1.30 equiv) was added. The reaction mixture was stirred for 2.5 h at r.t. until completion was indicated by TLC analysis, then diluted with DCM (100 mL) and washed with water (50 mL), followed by brine (3 × 50 mL). The organic phase was dried, filtered, and concentrated. The crude product was purified with automated flash column chromatography (50 g cartridge, 0–4% EtOAc in heptane). Off white solid (1.50 g, 94%). NMR spectra were in agreement with published data.21
(R)-8-Cyclopropyl-6-nitro-5-oxo-2,3-dihydro-5H-thiazolo[3,2-a]pyridine-3-carboxylic Acid (VII)
The ester (methyl (R)-8-cyclopropyl-6-nitro-5-oxo-2,3-dihydro-5H-thiazolo[3,2-a]pyridine-3-carboxylate, 50 mg, 0.169 mmol, 1.0 equiv) was dissolved in THF (4 mL) and put under stirring at r.t. LiOH solution (2.36 mL, 0.1M, 1.4 equiv) was added. TLC after 70 min indicated complete conversion. HCl (1 M aq., 253 μL, 1.5 equiv) was added. After 5 min stirring, the mixture was evaporated to water, diluted with brine (3 mL), and extracted with CHCl3/IPA 9:1 (10 + 5 mL). The organic phases were combined, dried, filtered, and evaporated (47.4 mg). The residue was trituated with diethyl ether (2 × 2,5 mL) and dried under vacuum. Yellow solid (35 mg, 74%). [α]D −1.3° [c 0.15, (CD3)2SO]. IR (KBr): ν 3393, 3010, 1738, 1679, 1496, 1391, 1313, 1266, 1240, 1008 cm–1. 1H NMR [600 MHz, (CD3)2SO] δ 13.84 (s, 1H), 8.13 (s, 1H), 5.67 (dd, J = 9.6, 1.9 Hz, 1H), 4.03 (dd, J = 12.1, 9.6 Hz, 1H), 3.75 (dd, J = 12.1, 1.9 Hz, 1H), 1.63 (tt, J = 8.3, 5.1 Hz, 1H), 0.92–0.84 (m, 2H), 0.73–0.66 (m, 1H), 0.66–0.60 (m, 1H). 13C{1H} NMR [151 MHz, (CD3)2SO] δ 168.8, 161.1, 152.2, 138.7, 132.1, 112.2, 64.2, 32.3, 11.9, 6.4, 6.2. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C11H10N2NaO5S+ 305.0208; Found 305.0198.
Acknowledgments
F.A. is grateful to the Swedish Research Council (2017-02339, 2017-00695, and 2018-04589), the Knut and Alice Wallenberg foundation (KAW 2013.0031), the Göran Gustafsson foundation, the Kempe Foundation (SMK-1755), the Swedish Foundation for Strategic Research (SB12-0070), the National Institutes of Health (R01AI134847-01A1), the Erling Perssons Stiftelse and the Michael J Fox foundation for financial support. The authors acknowledge the facilities and technical assistance of the Umeå Core Facility Electron Microscopy (UCEM) at the Chemical Biology Centre (KBC), Umeå University, a part of the National Microscopy Infrastructure (NMI).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c01699.
Schemes S1–S4; 1H NMR, 13C NMR, and 19F NMR spectra for all new compounds; and fibrils binding results for carboxylic acids 15a–k and 16a–e (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Uropathogenic E. coli:; a Chorell E.; Pinkner J. S.; Phan G.; Edvinsson S.; Buelens F.; Remaut H.; Waksman G.; Hultgren S. J.; Almqvist F. Design and Synthesis of C-2 Substituted Thiazolo and Dihydrothiazolo Ring-Fused 2-Pyridones: Pilicides with Increased Antivirulence Activity. J. Med. Chem. 2010, 53, 5690–5695. and references cited within 10.1021/jm100470t. [DOI] [PMC free article] [PubMed] [Google Scholar]; Chlamydia:; b Good J. A. D.; Kulén M.; Silver J.; Krishnan K. S.; Bahnan W.; Núñez-Otero C.; Nilsson I.; Wede E.; de Groot E.; Gylfe Å.; Bergström S.; Almqvist F. Thiazolino 2-Pyridone Amide Isosteres As Inhibitors of Chlamydia trachomatis Infectivity. J. Med. Chem. 2017, 60, 9393–9399. and references cited within 10.1021/acs.jmedchem.7b00716. [DOI] [PubMed] [Google Scholar]; Listeria:; c Kulén M.; Lindgren M.; Hansen S.; Cairns A. G.; Grundström C.; Begum A.; van der Lingen I.; Brännström K.; Hall M.; Sauer U. H.; Johansson J.; Sauer-Eriksson A. E.; Almqvist F. Structure-Based Design of Inhibitors Targeting PrfA, the Master Virulence Regulator of Listeria monocytogenes. J. Med. Chem. 2018, 61 (9), 4165–4175. and references cited within 10.1021/acs.jmedchem.8b00289. [DOI] [PubMed] [Google Scholar]; Tuberculosis:; d Flentie K.; Harrison G. A.; Tükenmez H.; Livny J.; Good J. A. D.; Sarkar S.; Zhu D. X.; Kinsella R. L.; Weiss L. A.; Solomon S. D.; Schene M. E.; Hansen M. R.; Cairns A. G.; Kulén M.; Wixe T.; Lindgren A. E. G.; Chorell E.; Bengtsson C.; Krishnan K. S.; Hultgren S. J.; Larsson C.; Almqvist F.; Stallings C. L. Chemical disarming of isoniazid resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (21), 10510–10517. 10.1073/pnas.1818009116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Andersson E. K.; Bengtsson C.; Evans M. L.; Chorell E.; Sellstedt M.; Lindgren A. E. G.; Hufnagel D. A.; Bhattacharya M.; Tessier P. M.; Wittung-Stafshede P.; Almqvist F.; Chapman M. R. Modulation of Curli Assembly and Pellicle Biofilm Formation by Chemical and Protein Chaperones. Chem. Biol. 2013, 20 (10), 1245–1254. and references cited within 10.1016/j.chembiol.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Horvath I.; Sellstedt M.; Weise C.; Nordvall L.-M.; Prasad G. K.; Olofsson A.; Larsson G.; Almqvist F.; Wittung-Stafshede P. Modulation of α-Synuclein Fibrillization by Ring-Fused 2-Pyridones: Templation and Inhibition Involve Oligomers with Different Structure. Arch. Biochem. Biophys. 2013, 532 (2), 84–90. and references cited within 10.1016/j.abb.2013.01.012. [DOI] [PubMed] [Google Scholar]
- a Singh P.; Chorell E.; Krishnan K. S.; Kindahl T.; Åden J.; Wittung-Stafshede P.; Almqvist F. Synthesis of Multiring Fused 2-Pyridones via a Nitrene Insertion Reaction: Fluorescent Modulators of α-Synuclein Amyloid Formation. Org. Lett. 2015, 17 (24), 6194–6197. 10.1021/acs.orglett.5b03190. [DOI] [PubMed] [Google Scholar]; b Sellstedt M.; Almqvist F. Synthesis of a Novel Tricyclic Peptidomimetic Scaffold. Org. Lett. 2008, 10 (18), 4005–4007. 10.1021/ol801506y. [DOI] [PubMed] [Google Scholar]; c Singh P.; Adolfsson D. E.; Ådén J.; Cairns A. G.; Bartens C.; Brännström K.; Olofsson A.; Almqvist F. Pyridine-Fused 2-Pyridones via Povarov and A3 Reactions: Rapid Generation of Highly Functionalized Tricyclic Heterocycles Capable of Amyloid Fibril Binding. J. Org. Chem. 2019, 84 (7), 3887–3903. 10.1021/acs.joc.8b03015. [DOI] [PubMed] [Google Scholar]
- a Thapa U.; Thapa P.; Karki R.; Yun M.; Choi J. H.; Jahng Y.; Lee E.; Jeon K.-H.; Na Y.; Ha E.-M.; Cho W.-J.; Kwon Y.; Lee E.-S. Synthesis of 2, 4-diaryl chromenopyridines and evaluation of their topoisomerase I and II inhibitory activity, cytotoxicity, and structure–activity relationship. Eur. J. Med. Chem. 2011, 46, 3201–3209. 10.1016/j.ejmech.2011.04.029. [DOI] [PubMed] [Google Scholar]; b Vu A. T.; Campbell A. N.; Harris H. A.; Unwalla R. J.; Manas E. S.; Mewshaw R. E. ERβ ligands. Part 6:6H-Chromeno [4, 3-b] quinolines as a new series of estrogen receptor β-selective ligands. Bioorg. Med. Chem. Lett. 2007, 17, 4053–4056. 10.1016/j.bmcl.2007.04.068. [DOI] [PubMed] [Google Scholar]; c Huang W.; Lin W.; Guan X. Development of ratiometric fluorescent pH sensors based on chromenoquinoline derivatives with tunable pKa values for bioimaging. Tetrahedron Lett. 2014, 55, 116–119. 10.1016/j.tetlet.2013.10.130. [DOI] [Google Scholar]; d Liu X.; Li Y.; Ren X.; Yang Q.; Su Y.; He L.; Song X. Methylated chromenoquinoline dyes: synthesis, optical properties, and application for mitochondrial labeling. Chem. Commun. 2018, 54, 1509–1512. 10.1039/C7CC08154E. [DOI] [PubMed] [Google Scholar]; e El-Essawy F. A.; El-Etrawy A.-A. S. Synthesis of New Chromeno [4, 3-b] pyrazolo [4, 3-e] pyridines Derivatives with Antimicrobial Evaluation. J. Heterocycl. Chem. 2014, 51, 191–195. 10.1002/jhet.1687. [DOI] [Google Scholar]; f Hu H.; Chen X.; Sun K.; Wang J.; Liu Y.; Liu H.; Fan L.; Yu B.; Sun Y.; Qu L.; Zhao Y. Silver-Catalyzed Radical Cascade Cyclization toward 1, 5-/1, 3-Dicarbonyl Heterocycles: An Atom-/Step-Economical Strategy Leading to Chromenopyridines and Isoxazole-/Pyrazole-Containing Chroman-4-Ones. Org. Lett. 2018, 20, 6157–6160. 10.1021/acs.orglett.8b02627. [DOI] [PubMed] [Google Scholar]; g Nunez-Vergara L. J.; Squella J. A.; Navarrete-Encina P. A.; Vicente-García E.; Preciado S.; Lavilla R. Chromenopyridines: promising scaffolds for medicinal and biological chemistry. Curr. Med. Chem. 2011, 18, 4761–4785. 10.2174/092986711797535272. [DOI] [PubMed] [Google Scholar]
- Shaveta; Mishra S.; Singh P. Hybrid molecules: The privileged scaffolds for various pharmaceuticals. Eur. J. Med. Chem. 2016, 124, 500–536. 10.1016/j.ejmech.2016.08.039. [DOI] [PubMed] [Google Scholar]
- a Povarov L. S.; Grigos V. I.; Mikhailov B. M. Reaction of Benzylideneaniline with some Unsaturated Compounds. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1963, 12, 1878–1880. 10.1007/BF00843814. [DOI] [Google Scholar]; b Povarov L. S. αβ-Unsaturated ethers and their analogues in reactions of diene synthesis. Russ. Chem. Rev. 1967, 36, 656–670. and references cited within 10.1070/RC1967v036n09ABEH001680. [DOI] [Google Scholar]
- a Powell D. A.; Batey R. A. Total Synthesis of the Alkaloids Martinelline and Martinellic Acid via a Hetero Diels–Alder Multicomponent Coupling Reaction. Org. Lett. 2002, 4 (17), 2913–2916. 10.1021/ol026293d. [DOI] [PubMed] [Google Scholar]; b Chen M.; Sun N.; Liu Y. Environmentally Benign Synthesis of Indeno[1,2-b]quinolines via an Intramolecular Povarov Reaction. Org. Lett. 2013, 15 (21), 5574–5577. 10.1021/ol402775k. [DOI] [PubMed] [Google Scholar]; c Twin H.; Batey R. A. Intramolecular Hetero Diels-Alder (Povarov) Approach to the Synthesis of the Alkaloids Luotonin A and Camptothecin. Org. Lett. 2004, 6 (26), 4913–4916. 10.1021/ol0479848. [DOI] [PubMed] [Google Scholar]; d Bello D.; Ramon R.; Lavilla R. Mechanistic Variations of the Povarov Multicomponent Reaction and Related Processes. Curr. Org. Chem. 2010, 14 (4), 332–356. and references cited within 10.2174/138527210790231883. [DOI] [Google Scholar]; e Glushkov V. A.; Tolstikov A. G. Synthesis of Substituted 1,2,3,4-Tetrahydroquinones by the Povarov Reaction. New Potentials of the Classical Reaction. Russ. Chem. Rev. 2008, 77 (2), 137–159. and references cited within 10.1070/RC2008v077n02ABEH003749. [DOI] [Google Scholar]; f Kouznetsov V. V. Recent Synthetic Developments in a Powerful Imino Diels–Alder Reaction (Povarov reaction): Application to the Synthesis of N-Polyheterocycles and Related Alkaloids. Tetrahedron 2009, 65, 2721–2750. and references cited within 10.1016/j.tet.2008.12.059. [DOI] [Google Scholar]
- Meng F.; Marek P.; Potter K. J.; Verchere C. B.; Raleigh D. P. Rifampicin Does Not Prevent Amyloid Fibril Formation by Human Islet Amyloid Polypeptide but Does Inhibit Fibril Thioflavin-T Interactions: Implications for Mechanistic Studies of β-Cell Death. Biochemistry 2008, 47, 6016–6024. 10.1021/bi702518m. [DOI] [PubMed] [Google Scholar]
- a Kudale A. A.; Miller D. O.; Dawe L. N.; Bodwell G. J. Intramolecular Povarov Reactions Involving 3-Aminocoumarins. Org. Biomol. Chem. 2011, 9, 7196–7206. 10.1039/c1ob05867c. [DOI] [PubMed] [Google Scholar]; b Belal M. D.; Das D. K.; Khan A. T. Synthesis of Pyrido[2,3-c]coumarin Derivatives by an Intramolecular Povarov Reaction. Synthesis 2015, 47 (08), 1109–1116. 10.1055/s-0034-1380131. [DOI] [Google Scholar]; c Anniyappan M.; Muralidharan D.; Perumal P. T. Triphenylphosphonium Perchlorate as an Efficient Catalyst for Mono- and Bis-Intramolecular Imino Diels–Alder Reactions: Synthesis of Tetrahydrochromanoquinolines. Tetrahedron Lett. 2003, 44, 3653–3657. 10.1016/S0040-4039(03)00707-X. [DOI] [Google Scholar]; d Laschat S.; Lauterwein J. Intramolecular Hetero-Diels-Alder Reaction of N-Arylimines. Applications to the Synthesis of Octahydroacridine Derivatives. J. Org. Chem. 1993, 58, 2856–2861. 10.1021/jo00062a033. [DOI] [Google Scholar]; e Wölfling J.; Frank E.; Schneider G.; Tietze L. F. Synthesis of Novel Steroid Alkaloids by Cyclization of Arylimines from Estrone. Eur. J. Org. Chem. 1999, 1999, 3013–3020. 10.1002/(SICI)1099-0690(199911)1999:11<3013::AID-EJOC3013>3.0.CO;2-H. [DOI] [Google Scholar]; f Magomedov N. A. Efficient Construction of Cyclopenta[b]quinoline Core of Isoschizozygane Alkaloids via Intramolecular Formal Hetero-Diels–Alder Reaction. Org. Lett. 2003, 5, 2509–2512. 10.1021/ol034776r. [DOI] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- a Jiang L.; Liu C.; Leibly D.; Landau M.; Zhao M.; Hughes M. P.; Eisenberg D. S. Structure-Based Discovery of Fiber-Binding Compounds that Reduce the Cytotoxicity of Amyloid Beta. eLife 2013, 2, e00857. 10.7554/eLife.00857. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chu W.; Zhou D.; Gaba V.; Liu J.; Li S.; Peng X.; Xu J.; Dhavale D.; Bagchi D. P.; d’Avignon A.; Shakerdge N. B.; Bacskai B. J.; Tu Z.; Kotzbauer P. T.; Mach R. H. Design, Synthesis, and Characterization of 3-(Benzylidene)indolin-2-one Derivatives as Ligands for α-Synuclein Fibrils. J. Med. Chem. 2015, 58, 6002–6017. 10.1021/acs.jmedchem.5b00571. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jensen J. R.; Cisek K.; Funk K. E.; Naphade S.; Schafer K. N.; Kuret J. Research Towards Tau Imaging. J. Alzheimer's Dis. 2011, 26, 147–157. 10.3233/JAD-2011-0003. [DOI] [PubMed] [Google Scholar]; d Wu C.; Wang Z.; Lei H.; Duan Y.; Bowers M. T.; Shea J.-E. The Binding of Thioflavin T and Its Neutral Analog BTA-1 to Protofibrils of the Alzheimer’s Disease Aβ16–22 Peptide Probed by Molecular Dynamics Simulations. J. Mol. Biol. 2008, 384, 718–729. 10.1016/j.jmb.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Krebs M. R. H.; Bromley E. H. C.; Donald A. M. The Binding of Thioflavin-T to Amyloid Fibrils: Localisation and Implications. J. Struct. Biol. 2005, 149, 30–37. 10.1016/j.jsb.2004.08.002. [DOI] [PubMed] [Google Scholar]; f Honson N. S.; Johnson R. L.; Huang W.; Inglese J.; Austin C. P.; Kuret J. Kuret, Differentiating Alzheimer Disease-Associated Aggregates with Small Molecules. Neurobiol. Dis. 2007, 28, 251–260. 10.1016/j.nbd.2007.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Neal K. L.; Shakerdge N. B.; Hou S. S.; Klunk W. E.; Mathis C. A.; Nesterov E. E.; Swager T. M.; McLean P. J.; Bacskai B. J. Development and Screening of Contrast Agents for In Vivo Imaging of Parkinson’s Disease. Mol. Imaging Biol. 2013, 15, 585–595. 10.1007/s11307-013-0634-y. [DOI] [PubMed] [Google Scholar]; h Zhang X.; Jin H.; Padakanti P. K.; Li J.; Yang H.; Fan J.; Mach R. H.; Kotzbauer P.; Tu Z. Radiosynthesis and in Vivo Evaluation of Two PET Radioligands for Imaging α-Synuclein. Appl. Sci. 2014, 4 (1), 66–78. 10.3390/app4010066. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Bagchi D. P.; Yu L.; Perlmutter J. S.; Xu J.; Mach R. H.; Tu Z.; Kotzbauer P. T. Binding of the Radioligand SIL23 to α-Synuclein Fibrils in Parkinson Disease Brain Tissue Establishes Feasibility and Screening Approaches for Developing a Parkinson Disease Imaging Agent. PLoS One 2013, 8, e55031. 10.1371/journal.pone.0055031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tomashevskaya M. M.; Tomashenko O. A.; Tomashevskii A. A.; Sokolov V. V.; Potekhin A. A. New One-Step Procedure for the Synthesis of 6H-Chromeno[4,3-b]quinolines and 8a,9,14a-Tetrahydro-8H-[benzo5,6]chromeno[4,3-b]quinolines. Russ. J. Org. Chem. 2007, 43 (1), 77–82. 10.1134/S1070428007010095. [DOI] [Google Scholar]; b Maiti S.; Panja S. K.; Sadhukhan K.; Ghosh J.; Bandyopadhyay C. Effects of Substituent and Catalyst on the Intramolecular Povarov Reaction—Synthesis of Chromenonaphthyridines. Tetrahedron Lett. 2012, 53, 694–696. 10.1016/j.tetlet.2011.11.130. [DOI] [Google Scholar]; c Kobayashi S.; Ishitani H.; Nagayama S. Lanthanide Triflate Catalyzed Imino Diels-Alder Reactions; Convenient Syntheses of Pyridine and Quinoline Derivatives. Synthesis 1995, 1995, 1195–1202. 10.1055/s-1995-4066. [DOI] [Google Scholar]; d Makioka Y.; Shindo T.; Taniguchi Y.; Takaki K.; Fujiwara Y. Ytterbium(III) Triflate Catalyzed Synthesis of Quinoline Derivatives from N-Arylaldimines and Vinyl Ethers. Synthesis 1995, 1995, 801–804. 10.1055/s-1995-4002. [DOI] [Google Scholar]
- a Sartori G.; Bigi F.; Maggi R.; Mazzacani A.; Oppici G. Clay/Water Mixtures – A Heterogeneous and Ecologically Efficient Catalyst for the Three-Component Stereoselective Synthesis of Tetrahydroquinolines. Eur. J. Org. Chem. 2001, 2001, 2513–2518. 10.1002/1099-0690(200107)2001:13<2513::AID-EJOC2513>3.0.CO;2-A. [DOI] [Google Scholar]; b Hermitage S.; Jay D. A.; Whiting A. Evidence for the Non-Concerted [4 + 2]-Cycloaddition of N-Aryl Imines When Acting as both Dienophiles and Dienes under Lewis Acid-Catalysed Conditions. Tetrahedron Lett. 2002, 43, 9633–9636. 10.1016/S0040-4039(02)02392-4. [DOI] [Google Scholar]; c Hermitage S.; Howard J. A. K.; Jay D.; Pritchard R. G.; Probert M. R.; Whiting A. Mechanistic studies on the formal aza-Diels-Alder reactions of N-aryl imines: evidence for the non-concertedness under Lewis-acid catalysed conditions. Org. Biomol. Chem. 2004, 2, 2451–2460. 10.1039/B407293F. [DOI] [PubMed] [Google Scholar]; d Alves M. J.; Azoia N. G.; Fortes A. G. Regio- and Stereo-Selective Aza-Diels–Alder Reaction of Ethyl Glyoxylate 4-Methoxyphenylimine with 1,3-Dienes in the Presence of BF3·Et2O. Evidence for a Non-Concerted Mechanism. Tetrahedron 2007, 63, 727–734. 10.1016/j.tet.2006.10.085. [DOI] [Google Scholar]; e Lucchini V.; Prato M.; Scorrano G.; Stivanello M.; Valle G. Acid-Catalysed Addition of N-Aryl Imines to Dihydrofuran. Postulated Dependence of the Reaction Mechanism on the Relative Face of Approach of Reactants. J. Chem. Soc., Perkin Trans. 2 1992, 2, 259–266. 10.1039/p29920000259. [DOI] [Google Scholar]; f Jiménez O.; de la Rosa G.; Lavilla R. Straightforward access to a structurally diverse set of oxacyclic scaffolds through a four-component reaction. Angew. Chem., Int. Ed. 2005, 44, 6521–6525. 10.1002/anie.200501548. [DOI] [PubMed] [Google Scholar]
- a Beifuss U.; Ledderhose S.; Ondrus V. Generation of Cationic 2-Azabutadienes from N,S-Acetals and their use for the Regio- and Diastereoselective Synthesis of 1,2,3,4-Tetrahydroquinolines by Intermolecular [4π+ + 2π] Cycloadditions. ARKIVOC 2005, 2005, 147–173. 10.3998/ark.5550190.0006.514. [DOI] [Google Scholar]; b Stevenson P. J.; Nieuwenhuyzen M.; Osborne D. Multi Component Coupling Reactions of N-Acetyl-2-Azetine. ARKIVOC 2000, 11, 129–144. 10.3998/ark.5550190.0008.b12. [DOI] [PubMed] [Google Scholar]
- a Lombardo M.; Morganti S.; Trombini C. 3-Bromopropenyl Esters in Organic Synthesis: Indium- and Zinc-Mediated Entries to Alk-1-ene-3,4-diols. J. Org. Chem. 2003, 68, 997–1006. 10.1021/jo0262457. [DOI] [PubMed] [Google Scholar]; b Ulich L. H.; Adams R. The reaction between acid halides and aldehydes. III. J. Am. Chem. Soc. 1921, 43 (3), 660–667. 10.1021/ja01436a036. [DOI] [Google Scholar]; c Neuenschwander M.; Bigler P.; Christen K.; Iseli R.; Kyburz R.; Mühle H. Chloracylierung und Bromacylierung von Carbonylverbindungen: Eine in Vergessenheit geratene Carbonylreaktion. I. Präparative Anwendungsbreite. Helv. Chim. Acta 1978, 61, 2047–2058. 10.1002/hlca.19780610612. [DOI] [Google Scholar]
- a Vassar P. S.; Culling C. F. Fluorescent stains, with special reference to amyloid and connective tissues. Arch. Pathol. 1959, 68, 487–498. [PubMed] [Google Scholar]; b Biancalana M.; Koide S. Molecular Mechanism of Thioflavin-T Binding to Amyloid Fibrils. Biochim. Biophys. Acta, Proteins Proteomics 2010, 1804, 1405–1412. 10.1016/j.bbapap.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; c LeVine H. Thioflavine T Interaction with Amyloid β-Sheet Structures. Amyloid 1995, 2, 1–6. 10.3109/13506129509031881. [DOI] [Google Scholar]; d Chorell E.; Andersson E.; Evans M.; Jain N.; Götheson A.; Åden J.; Chapman M. R.; Almqvist F.; Wittung-Stafshede P. Bacterial Chaperones CsgE and CsgC Differentially Modulate Human α-Synuclein Amyloid Formation via Transient Contacts. PLoS One 2015, 10 (10), e0140194 10.1371/journal.pone.0140194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Claisen L.; Eisleb O. Über die Umlagerung von Phenolallyläthern in die isomeren Allylphenole. Liebigs Ann. 1913, 401, 21–119. 10.1002/jlac.19134010103. [DOI] [Google Scholar]; b Black M.; Cadogan J. I. G.; McNab H.; MacPherson A. D.; Roddam V. P.; Smith C.; Swenson H. R. Synthesis of fused furans by gas-phase pyrolysis of 2-allyloxyarylpropenoic esters 1. J. Chem. Soc., Perkin Trans. 1 1997, 2483–2494. 10.1039/a702451g. [DOI] [Google Scholar]; c Hirano K.; Biju A. T.; Piel I.; Glorius F. N-Heterocyclic Carbene-Catalyzed Hydroacylation of Unactivated Double Bonds. J. Am. Chem. Soc. 2009, 131, 14190–14191. 10.1021/ja906361g. [DOI] [PubMed] [Google Scholar]
- MacMillan K. S.; Nguyen T.; Hwang I.; Boger D. L. Total Synthesis and Evaluation of iso-Duocarmycin SA and iso-Yatakemycin. J. Am. Chem. Soc. 2009, 131 (3), 1187–1194. 10.1021/ja808108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Blanchette M. A.; Choy W.; Davis J. T.; Essenfeld A. P.; Masamune S.; Roush W. R.; Sakai T. Horner-Wadsworth-Emmons reaction: use of lithium chloride and an amine for base sensitive compounds. Tetrahedron Lett. 1984, 25, 2183–2186. 10.1016/S0040-4039(01)80205-7. [DOI] [Google Scholar]; b Li J.-Q.; Peters B.; Andersson P. G. Highly enantioselective asymetric isomerization of primary allylic alcohol with an iridium-N, P complex. Chem. - Eur. J. 2011, 17, 11143–11145. 10.1002/chem.201101524. [DOI] [PubMed] [Google Scholar]; c Chan Y.-C.; Yeung Y.-Y. Halogen bond catalyzed bromocarbocyclization. Angew. Chem., Int. Ed. 2018, 57, 3483–3487. 10.1002/anie.201800261. [DOI] [PubMed] [Google Scholar]
- a Ganapathy D.; Sekar G. Palladium nanoparticles stabilized by metal–carbon covalent bond: An efficient and reusable nanocatalyst in cross-coupling reactions. Catal. Commun. 2013, 39, 50–54. 10.1016/j.catcom.2013.04.028. [DOI] [Google Scholar]; b Kumar N. S. C. R.; Raj I. V. P.; Sudalai A. Sulfonamide- and hydrazine-based palladium catalysts: Stable and efficient catalysts for C–C coupling reactions in aqueous medium. J. Mol. Catal. A: Chem. 2007, 269 (1–2), 218–224. 10.1016/j.molcata.2006.12.023. [DOI] [Google Scholar]; c Kim E.; Koh M.; Lim B. J.; Park S. B. Emission wavelength prediction of a full-color-tunable fluorescent core skeleton, 9-aryl-1,2-dihydropyrrolo[3,4-b]indolizin-3-one. J. Am. Chem. Soc. 2011, 133 (17), 6642–6649. 10.1021/ja110766a. [DOI] [PubMed] [Google Scholar]; d Li H.; Chen H.; Zhou Y.; Huang J.; Yi J.; Zhao H.; Wang W.; Jing L. Selective Synthesis of Z-Cinnamyl Ethers and Cinnamyl Alcohols through Visible Light-Promoted Photocatalytic E to Z Isomerization. Chem. - Asian J. 2020, 15 (5), 555–559. 10.1002/asia.201901778. [DOI] [PubMed] [Google Scholar]; e Hjelmgaard T.; Faure S.; Lemoine P.; Viossat B.; Aitken D. J. Rapid Assembly of the Polyhydroxylated β-Amino Acid Constituents of Microsclerodermins C, D, and E. Org. Lett. 2008, 10 (5), 841–844. 10.1021/ol702962z. [DOI] [PubMed] [Google Scholar]; f Zhang M. J.; Tan D. W.; Li H. X.; Young D. J.; Wang H. F.; Li H. Y.; Lang J. P. Switchable Chemoselective Transfer Hydrogenations of Unsaturated Carbonyls Using Copper (I) N-Donor Thiolate Clusters. J. Org. Chem. 2018, 83 (3), 1204–1215. 10.1021/acs.joc.7b02676. [DOI] [PubMed] [Google Scholar]; g Cho H.; Jeon H.; Shin J. E.; Lee S.; Park S.; Kim S. Asymmetric Synthesis of Cα-Substituted Prolines through Curtin–Hammett-Controlled Diastereoselective N-Alkylation. Chem. - Eur. J. 2019, 25 (10), 2447–2451. 10.1002/chem.201804965. [DOI] [PubMed] [Google Scholar]; h Zhang Y.; Xiong W.; Cen J.; Yan W.; Wu Y.; Qi C.; Wu W.; Jiang H. Direct bromocarboxylation of arynes using allyl bromides and carbon dioxide. Chem. Commun. 2019, 55 (82), 12304–12307. 10.1039/C9CC05495B. [DOI] [PubMed] [Google Scholar]; i van Zijl A. W.; Arnold L. A.; Minnaard A. J.; Feringa B. L. Highly Enantioselective Copper-Catalyzed Allylic Alkylation with Phosphoramidite Ligands. Adv. Synth. Catal. 2004, 346 (4), 413–420. 10.1002/adsc.200303207. [DOI] [Google Scholar]
- Martin-Acosta P.; Feresin G.; Tapia A.; Estevez-Braun A. Microwave-Assisted Organocatalytic Intramolecular Knoevenagel/Hetero Diels-Alder Reaction with O-(Arylpropynyloxy)-Salicylaldehydes: Synthesis of Polycyclic Embelin Derivatives. J. Org. Chem. 2016, 81, 9738–9756. 10.1021/acs.joc.6b01818. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.