

Abstract
Background
Patients at increased risk for coronary artery disease and adverse prognosis during heart failure exhibit increased levels of circulating trimethylamine N‐oxide (TMAO), a metabolite formed in the metabolism of dietary phosphatidylcholine. We investigated the efficacy of dietary withdrawal of TMAO as well as use of a gut microbe‐targeted inhibitor of TMAO production, on cardiac function and structure during heart failure.
Methods and Results
Male C57BLK/6J mice were fed either control diet, a diet containing TMAO (0.12% wt/wt), a diet containing choline (1% wt/wt), or a diet containing choline (1% wt/wt) plus a microbial choline trimethylamine lyase inhibitor, iodomethylcholine (0.06% wt/wt), starting 3 weeks before transverse aortic constriction. At 6 weeks after transverse aortic constriction, a subset of animals in the TMAO group were switched to a control diet for the remainder of the study. Left ventricular structure and function were monitored at 3‐week intervals. Withdrawal of TMAO from the diet attenuated adverse ventricular remodeling and improved cardiac function compared with the TMAO group. Similarly, inhibiting gut microbial conversion of choline to TMAO with a choline trimethylamine lyase inhibitor, iodomethylcholine, improved remodeling and cardiac function compared with the choline‐fed group.
Conclusions
These experimental findings are clinically relevant, and they demonstrate that TMAO levels are modifiable following long‐term exposure periods with either dietary withdrawal of TMAO or gut microbial blockade of TMAO generation. Furthermore, these therapeutic strategies to reduce circulating TMAO levels mitigate the negative effects of dietary choline and TMAO in heart failure.
Keywords: cardiac fibrosis, dietary choline, gut microbiota, metabolomics
Subject Categories: Heart Failure, Hypertrophy, Remodeling
Nonstandard Abbreviations and Acronyms
- CVD
cardiovascular disease
- HF
heart failure
- LV
left ventricular
- PSR
Picrosirius red
- TAC
transverse aortic constriction
- TMA
trimethylamine
- TMAO
trimethylamine N‐oxide
- WGA
wheat germ agglutinin
Clinical Perspective
What Is New?
Following development of heart failure in a pressure overload heart failure mouse model, dietary modification from a high to low content of choline or trimethylamine N‐oxide leads to lower circulating trimethylamine N‐oxide levels and attenuation of adverse cardiac remodeling.
Similar anti–adverse ventricular remodeling effects are observed with direct nonlethal inhibition of gut microbiota‐dependent trimethylamine N‐oxide generation, providing proof of concept that dietary nutrient‐mediated gut microbial metabolism is mechanistically linked to adverse cardiac remodeling.
What Are the Clinical Implications?
Despite significant morbidity and mortality, dietary and lifestyle modification in the management of heart failure have primarily focused on salt restriction and exercise.
The present studies support a role of gut microbe‐generated metabolites as contributors to cardio‐renal disease progression.
They also support further studies to target specific dietary nutrients or microbial enzymes to prevent disease progression in patients with heart failure.
Introduction
Heart failure (HF) remains a leading cause of death, afflicting >6.5 million people in the United States1. It is predicted that by 2030 the prevalence of HF will increase ≈50% and will have an economic burden that exceeds ≈$70 billion in the United States alone.1 With a limited number of effective drugs to treat HF, there is a necessity to focus on nutrition and lifestyle alterations in patients with HF. Currently, there is a limited set of dietary guidelines that have concentrated on limiting sodium and fluid consumption for HF patients.2 While this has been the primary focus, there is a paucity in outcomes data to support these alterations. The increasing prevalence of HF and mounting economic burden provide an impetus to discover novel therapeutic targets and lifestyle modifications to treat HF and the associated underlying cardiovascular diseases (CVDs)/comorbidities.
A therapeutic target of great interest is the human microbiome. It is estimated that the number of bacteria in the body is approximately equal to the number of human cells3 and that bacteria outnumber eukaryotes and archaea in the human microbiome by 2 to 3 orders of magnitude.4, 5 The microbiome has evolved concurrently over time with humans to perform vital functions in their host environments. The most significant portion of human commensals reside in the large intestine and serve numerous functions that human enzymes are incapable of carrying out, such as deriving nutrients from indigestible plant materials. In addition to this essential function, the gut microbiome also interacts with and educates the host immune system, helping to protect against invasive pathogens.6, 7
The interplay among the gut microbiome, diet, and cardiovascular health has recently emerged as an important interaction in many CVDs. Phosphatidylcholine is a dietary component found at high concentrations in food sources such as cheese, egg yolk, meat, and shellfish. It is converted in the gut to choline, and subsequently to trimethylamine (TMA) via intestinal microbiota. TMA then enters the portal circulation and is converted to TMAO via flavin‐containing monooxygenases in the liver.8, 9 Choline itself is an essential nutrient that plays several roles in human physiology. It is the precursor to the neurotransmitter acetylcholine, and plays a critical role in neurological function and fetal development.10, 11, 12 However, it has been shown in humans that the metabolite choline, found in high concentrations in the Western diet and meat, is associated with HF and increased cardiovascular risk.13, 14 Furthermore, studies in both mice and humans have elucidated the metabolic pathway of transforming choline into TMAO, revealing TMAO as a terminal, deleterious end product13, 14, 15, 16 Preclinical and human intervention studies have shown that dietary choline increases circulating levels of TMAO and consequently increases atherosclerotic plaque formation, thrombosis, and inflammatory signaling, which all play a critical role in the pathogenesis of CVDs.16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27 Clinical studies have also positively correlated circulating TMAO levels with the development and severity of HF.13, 28, 29, 30, 31 Multiple retrospective analyses of clinical trials corroborate the prognostic value and association of increased TMAO levels and CVD burden32, 33 Our group has previously shown that dietary choline and TMAO exacerbate the progression of cardiac dysfunction in the setting of HF following transverse aortic constriction (TAC).34 At present, there are no US Food and Drug Administration–approved pharmacotherapies that alter the gut microbiome to prevent or attenuate CVD progression and severity.
In the current study, we investigated the impact of lowering TMAO levels via withdrawing TMAO after chronic exposure to dietary TMAO following TAC to evaluate the reversibility of the pathological effects of TMAO in HF. Furthermore, we sought to investigate the effects of inhibiting microbial production of TMAO using a novel TMA lyase inhibitor in the setting of HF in mice fed dietary choline. To test these dietary and pharmacologic modifications, we used a well‐established murine model of pressure overload–induced chronic HF.
Methods
Experimental Animals
C57BL6/J male mice aged 10 to 12 weeks (Jackson Labs, Bar Harbor, ME) were used for these studies. All animals were housed and maintained in an on‐site temperature‐controlled animal facility adhering to a 12‐hour light/dark cycle and provided with water and rodent chow ad libitum. All animals were humanely cared for in accordance with the Principles of Laboratory Animal Care dictated by the National Society of Medical Research and the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health (Publication No. 85‐23, Revised 1996). All animal procedures were approved by the Institutional Animal Care and Use Committee of both the Louisiana State University Health Sciences Center and the Cleveland Clinic. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Experimental Diets
Mice were maintained on either a chemically defined control “chow” diet (TD.130104) (n=26,TMAO study; n=19, iodomethylcholine study), chow supplemented with 0.12% (wt/wt) TMAO (TD.07865) (n=35, TMAO study), chow supplemented with 1% (wt/wt) choline (TD.09041) (n=22), or chow containing 1% (wt/wt) choline+0.06% (wt/wt) iodomethylcholine (choline TMA lyase inhibitor) (TD.150812) (n=19) from Envigo (Madison, WI). TD.07865, TD.09041, and TD.150812 are the same background diet as TD.130104. In each protocol, the diet was initiated 3 weeks before TAC and maintained per study parameters. Those animals subject to diet withdrawal (n=16, TMAO study) were randomized and allocation concealment was performed. All investigators were blinded to randomization until all data were fully analyzed.
TAC Protocol
Cardiac pressure overload and HF were induced using TAC surgery, as described previously.34, 35, 36, 37, 38 The experimental protocol for these studies is shown in Figure 1A.
Figure 1. (A), Experimental protocol for transverse aortic constriction (TAC) heart failure studies in control, trimethylamine N‐oxide (TMAO) fed, and TMAO withdrawal study groups.
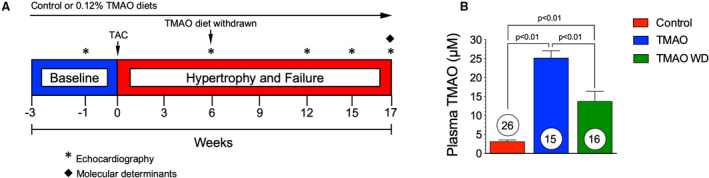
Mice were fed experimental diets starting at 3 weeks before TAC surgery. In a subset of the TMAO group, dietary TMAO was withdrawn at 6 weeks following TAC. Left ventricular function and structure were evaluated via echocardiography at 3‐week intervals following baseline echocardiography studies at 1 week before TAC surgery. (B), Circulating levels of TMAO in control, TMAO, and TMAO withdrawal groups. Data from plasma samples were collected at the 17‐week time point. Results are shown as mean± SEM. Numbers in circles represent the total number analyzed per group.
Echocardiography Assessment
One week before the TAC procedure, a baseline transthoracic echocardiogram was performed; following the TAC procedure, echocardiography was performed for 15 to 17 weeks at 2‐ to 6‐week intervals using a 30‐MHz probe on a Vevo 2100 (Visualsonics), as described previously34, 39 Briefly, while mice were under light anesthesia with isoflurane (0.25–0.50%) supplemented with 100% O2, and a heart rate >400 bpm, a series of 5 M‐mode long‐axis images were taken along the left ventricle from base to apex to measure the thicknesses of interventricular septal wall, left ventricular (LV) posterior wall, and LV chamber diameters at systole or diastole (LVESD and LVEDD). The average of the five M‐mode measurements were used to calculate LV chamber volume at systole or diastole, LV fractional shortening, LV ejection fraction, and LV mass with the following equations:
Measurements of Plasma B‐type Natriuretic Peptide
B‐type natriuretic peptide (BNP; BNP EIA kit, Phoenix Pharmaceuticals, Inc.) plasma levels were determined by ELISA at 17 weeks following TAC, as described previously40
Quantification of Plasma TMAO Levels
Stable isotope dilution liquid chromatography–mass spectrometry on a Shimadzu LCMS 8060 triple quadrupole mass spectrometer interfaced with a Nexera ultra‐high‐performance liquid chromatography system was used for quantification of TMAO in plasma taken at the study end point, as previously described.16, 41, 42, 43
Iodomethylcholine
Iodomethylcholine is a structural analog of choline that acts as a TMA lyase inhibitor, which was first developed and characterized in our previous work by Roberts et al.44 Iodomethylcholine was designed as a suicide substrate inhibitor with several key features: (1) Iodomethylcholine is initially nonreactive and can be transported into the intact gut microbe; (2) iodomethylcholine is nonlethal to the microbe; and (3) iodomethylcholine possesses a structure that, when cleaved, generates a reactive species that can irreversibly inhibit choline TMA lyase. Iodomethylcholine is an irreversible TMA lyase inhibitor and does not affect commensal viability. It was developed as a tool drug and has previously been shown to be safe when screened in a battery of pharmacological safety tests (ie, human Ether‐à‐go‐go–related gene channel function, mitochondria safety test, Ames testing, and human kidney‐2 cell viability assay). While iodomethylcholine is a tool drug and is not appropriate for use in humans at the present time, ongoing work is moving forward for first‐in‐human studies with a member of this new class of gut microbiota‐targeting inhibitor.
Histologic Staining and Quantitative Analysis
Heart tissue was fixed in 10% buffered formalin and processed into paraffin wax blocks. Sections (5 μm) were mounted onto glass slides and stained for Picrosirius red (PSR) to quantify total fibrosis or wheat germ agglutinin (WGA) for identification of extracellular matrix for myocyte cross‐sectional area as previously described35 in all groups. A mean of >10 images was taken from each slide and quantified in a blinded manner per animal per group. Image J was used to quantify PSR positive tissue and myocyte cross‐sectional area. Kidney tissue was fixed in 10% buffered formalin and processed into paraffin wax blocks. Sections were mounted onto glass slides and stain for Masson's Trichrome to quantify kidney fibrosis, as previously described.35
Quantitative Real‐Time Polymerase Chain Reaction
Heart tissue was frozen in liquid N2 and stored at −80°C until RNA isolation was performed, as previously described.35 Gene expression for collagen 1a1, collagen 3a1, fibronectin‐1, transforming growth factor‐β, interleukin‐6, connective tissue growth factor, matrix metallopeptidase 2, metallopeptidase inhibitor 1 and 2 were analyzed.
Blinding and Randomization
An alphanumeric code was assigned to each animal several times over the course of the study to ensure proper blinding and allocation concealment of treatment groups. In the TMAO withdrawal study, animals were randomized after the 6‐week echocardiographic data were collected. All experimental analyses were performed in a blinded manner, and all investigators were blinded as to treatment or diet until all data were analyzed and the study was completed.
Statistical Analysis
All data are presented as mean±SEM. Repeated‐measures ANOVA analyses with Tukey correction for multiple comparisons were performed using Prism 6 (GraphPad Software). A P<0.05 was considered statistically significant.
Results
Withdrawal of Dietary of TMAO Reduces Circulating TMAO Levels
We evaluated whether the withdrawal of dietary TMAO (0.12%) could modulate plasma TMAO levels following initial exposure in mice (Figure 1A). Administration of dietary TMAO (0.12% wt/wt) for 17 weeks resulted in a significant (P<0.01) 8‐fold increase in plasma TMAO to ≈25 μmol/L compared with the control diet (Figure 1B) with plasma TMAO levels of ≈3 μmol/L. Circulating TMAO levels measured at 17 weeks were significantly (P<0.01) reduced in the TMAO withdrawal group; however, TMAO levels remain elevated compared with control diet (P<0.01) following removal of dietary TMAO at 6 weeks.
Withdrawal of Dietary TMAO Attenuates Adverse Remodeling and Cardiac Dysfunction in HF
To evaluate the effect of TMAO dietary withdrawal on cardiac function, we fed either control chow or 0.12% (wt/wt) supplemented TMAO chow and induced chronic HF using a TAC procedure (Figure 1A). At 6 weeks following TAC, in a subset of the TMAO group, the 0.12% (wt/wt) TMAO diet was withdrawn and mice were maintained on control chow for the remainder of the study. Dietary TMAO significantly worsened cardiac function in comparison with control diet (Figure 2) (Table 1). In Table 1, at the 17‐week time point, all measured end points are significantly different (P<0.01) compared with baseline, within group. At the 17‐week time point, adverse cardiac remodeling was worsened in the TMAO group compared with control as seen by increased LV dilation (Figure 2A and 2B) (P<0.01). However, when TMAO was withdrawn from the diet, these negative effects were attenuated. Cardiac remodeling was inhibited when TMAO was withdrawn from the diet following TAC, as evidenced by reduced cardiac dilation (P<0.01) in the TMAO withdrawal group compared with the TMAO group at 17 weeks (Figure 2A). Cardiac dysfunction was attenuated as early as 9 weeks following TMAO withdrawal (P<0.05) and LV fractional shortening was significantly improved compared with the TMAO group at 17 weeks (P<0.01) (Figure 2C). Furthermore, in the TMAO withdrawal group, the interventricular septum did not exhibit the same extent of pathological remodeling as observed in the control and TMAO groups (Figure 2D) (P<0.01 at 17 weeks). At 17 weeks, there was no significant difference in the LV posterior wall thickness in any of the treatment groups (Figure 2E). B‐type natriuretic peptide (BNP) was measured in the plasma at 17 weeks following TAC to further evaluate HF severity. The TMAO group had significantly (P<0.05) increased circulating BNP levels compared with control (Figure 2F). Interestingly, the TMAO withdrawal group had significantly reduced (P<0.05) circulating BNP levels compared with TMAO at 17 weeks following TAC. This finding substantiates our echocardiographic findings of LV function, which demonstrate that TMAO withdrawal attenuates LV dilatation in HF as compared with chronic TMAO feeding. Representative photographs (Figure 3A) demonstrate the extent of cardiac enlargement in the chronic TMAO and TMAO withdrawal study groups compared with the control groups at 17 weeks following TAC‐induced HF. However, TMAO withdrawal did not significantly reduce heart weight:body weight (Figure 3B) or heart weight:tibia length compared with the TMAO group (Figure 3C). Furthermore, we investigated the potential of pulmonary edema in this HF model and observed no change in lung weight:tibia length (data not shown). At 17 weeks, there was a significant (P<0.05) increase in the left atrial weight:tibia length in the TMAO group, and while not significant, there was a trend in reduction of left atrial weight:tibia length in the TMAO withdrawal group (Figure 3D). LV tissue sections obtained from animals at 17 weeks after TAC were stained with WGA and myocyte cross‐sectional area was quantified. There was no significant difference in myocyte cross‐sectional area between all groups, as detailed in Figure 3E and 3F.
Figure 2. Withdrawing trimethylamine N‐oxide (TMAO) improves cardiac function and remodeling.
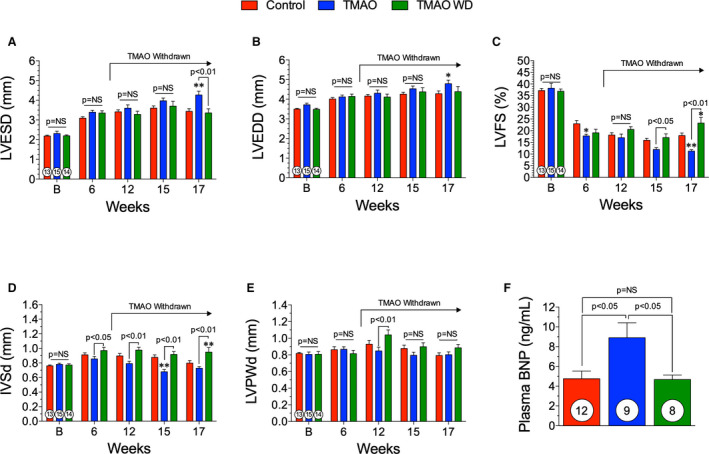
(A), left ventricular (LV) end‐systolic diameter (LVESD), (B) LV end‐diastolic diameter (LVEDD), (C) LV fractional shortening (LVFS), and (D) interventricular septal wall thickness (IVSd) and (E) LV posterior wall thickness at diastole (LVPWd) following transverse aortic constriction (TAC) in control, TMAO, and TMAO withdrawal groups. (F), Plasma B‐type natriuretic peptide (BNP) levels at 17 weeks following TAC. All results are shown as mean± SEM. P=NS (not significant), *P<0.05 vs control, **P<0.01 vs control. Numbers in circles represent the total number analyzed per group.
Table 1.
TMAO Withdrawal Study Echocardiographic Measurements
| Control | TMAO | TMAO Withdrawal | |
|---|---|---|---|
| Baseline | |||
| LV end‐systolic volume, μL | 15.8±1.53 | 22.0±1.63 | 16.9±2.49 |
| LV end‐diastolic volume, μL | 37.0±2.72 | 44.5±2.36 | 34.7±3.52 |
| LV ejection fraction, % | 67.2±1.29 | 66.4±2.45 | 67.8±1.06 |
| LV mass, mg | 90.6±1.90 | 106±7.52 | 87.46±3.16 |
| 17 weeks after TAC | |||
| LV end‐systolic volume, μL | 40.0±4.37 | 57.0±5.04a | 43.9±6.88b |
| LV end‐diastolic volume, μL | 60.2±5.23 | 74.3±5.23 | 69.0±7.98 |
| LV ejection fraction, % | 36.3±2.05 | 24.4±1.36a | 45.8±4.43c , d |
| LV mass, mg | 128±7.83 | 151±9.70 | 160±9.65c |
Data shown as the mean±SEM. LV indicates left ventricular; TAC, transverse aortic constriction; and TMAO, trimethylamine N‐oxide.
P<0.01 compared with control group at that time point.
P<0.05 compared with TMAO group at that time point.
P<0.05 compared with control group at that time point.
P<0.01 compared with TMAO group at that time point.
Figure 3. Hypertrophic Remodeling following TMAO withdrawal.(A), Representative images of hearts taken at 17 weeks.
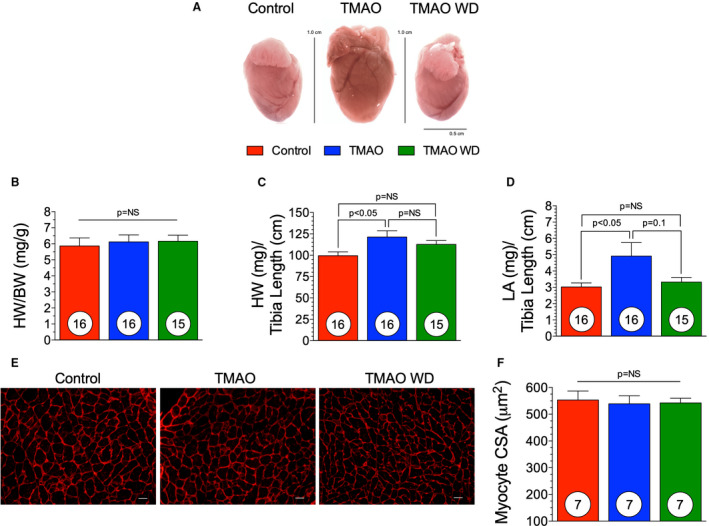
(B), Heart weight (HW) to body weight (BW) of control, trimethylamine N‐oxide (TMAO) and TMAO withdrawal groups. (C), Heart weight to tibia length of control, TMAO, and TMAO withdrawal groups. (D), left atrial (LA) weight to tibia length. (E), Representative photomicrographs (×40) of WGA staining for myocyte cross sectional area (CSA) (scale bar=20 μm) and (F) quantification in all 3 groups. Results are shown as mean± SEM. P=NS (not significant).
Withdrawal of Dietary TMAO Did Not Influence Cardiac or Renal Fibrosis but Significantly Reduced the Fibrotic Response
At 17 weeks after TAC, hearts were collected and processed for histologic analysis. PSR staining was performed on cross sections of hearts from control, TMAO, and TMAO withdrawal groups. Representative photomicrographs (Figure 4A) demonstrate the level of LV fibrosis. PSR‐positive areas were quantified as a percentage of total tissue area. TMAO diet did not increase the total cardiac fibrotic area compared with control, and withdrawal at 6 weeks after TAC did not significantly reduce cardiac fibrosis compared with control or TMAO (Figure 4B). Furthermore, we performed quantitative real‐time polymerase chain reaction to elucidate the levels of expression of profibrotic genes in the heart. There was a significant increase in transforming growth factor‐β in the TMAO group (P<0.01) that was attenuated in the TMAO withdrawal group (Figure 4C) (P<0.01). Furthermore, collagen 1a1 and metallopeptidase inhibitor 2 were significantly reduced in the TMAO withdrawal group compared with TMAO group (Figure 4C, P<0.05 and P<0.01, respectively). We investigated the effect that TMAO diet withdrawal on kidney fibrosis. Representative photomicrographs (Figure 4D) demonstrate kidney fibrosis in control, TMAO, and TMAO withdrawal groups. At 17 weeks after TAC, there was a significant increase in total kidney fibrosis in the TMAO group compared with control (P<0.05) (Figure 4E). After withdrawal of the TMAO diet, there was no significant difference from the TMAO group (Figure 4E).
Figure 4. Fibrotic response following TMAO withdrawal. (A), Representative photomicrographs (×40) of Picrosirius red staining in control, trimethylamine N‐oxide (TMAO), and TMAO withdrawal groups.
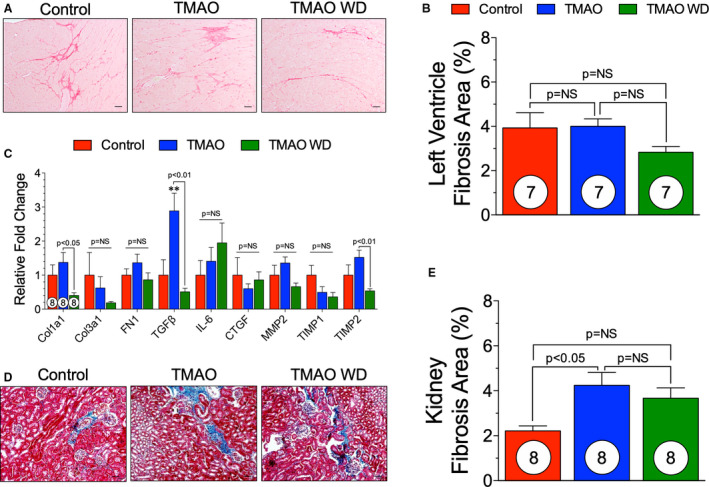
(B), Quantification of fibrosis as a percentage of total tissue area. (C), Gene expression levels of fibrotic markers in control, TMAO, and TMAO withdrawal groups. Interleukin‐6 (IL6): control (n=5), TMAO (n=6) and TMAO withdrawal (n=7). Metallopeptidase inhibitor 1 (TIMP1): TMAO withdrawal (n=7). Metallopeptidase inhibitor 2 (TIMP2): control (n=7). (D), Representative photomicrographs (×40) of renal Masson's trichrome staining in control, TMAO and TMAO withdrawal groups. (E), Quantification of kidney fibrosis as a percentage of total tissue area. Scale bar=20 μm. Results are shown as mean±SEM. P=NS (not significant). Col1A1 indicates collagen 1A1; Col3A1, collagen 3A1; CTGF, connective tissue growth factor; FN1, fibronectin 1; MMP2, matrix metalloprotease 2; TGFβ, transforming growth factor β; and TIMP, metallopeptidase inhibitor.
TMAO Production Is Attenuated by a Novel TMA Lyase Inhibitor in the Setting of a Choline Diet and TAC
In additional experimental studies, we investigated a novel gut microbial‐targeting TMA lyase inhibitor (iodomethylcholine)44 in mice fed a high‐choline diet and subjected to TAC‐mediated HF (Figure 5A). Microbial choline TMA lyase inhibition provides a novel mechanism to inhibit gut microbe conversion of choline to TMA, the immediate precursor to TMAO.44 Mass spectrometry analyses revealed that iodomethylcholine levels were detectable only at negligible levels in plasma (submicromolar; data not shown), but in high levels within the cecal contents in the choline+iodomethylcholine group (Figure 5B). To evaluate the effectiveness of the inhibitor, we measured circulating TMAO levels at 15 weeks following TAC. In animals fed a 1% (wt/wt) choline diet, circulating plasma levels of TMAO were significantly (P<0.01) increased (>15‐fold) compared with mice fed a control diet, while TMAO levels in the choline+iodomethylcholine group were at control levels (Figure 5C). These data display that choline is effectively converted into TMAO and that the microbe‐targeting choline TMA lyase inhibitor, iodomethylcholine, given orally, effectively blocks the conversion of choline into TMA and ultimately TMAO.
Figure 5. Experimental Protocol and Cecum IMC and Circulating TMAO Level.(A), Experimental protocol for transverse aortic constriction (TAC) heart failure studies in control, choline, and choline+iodomethylcholine study groups.
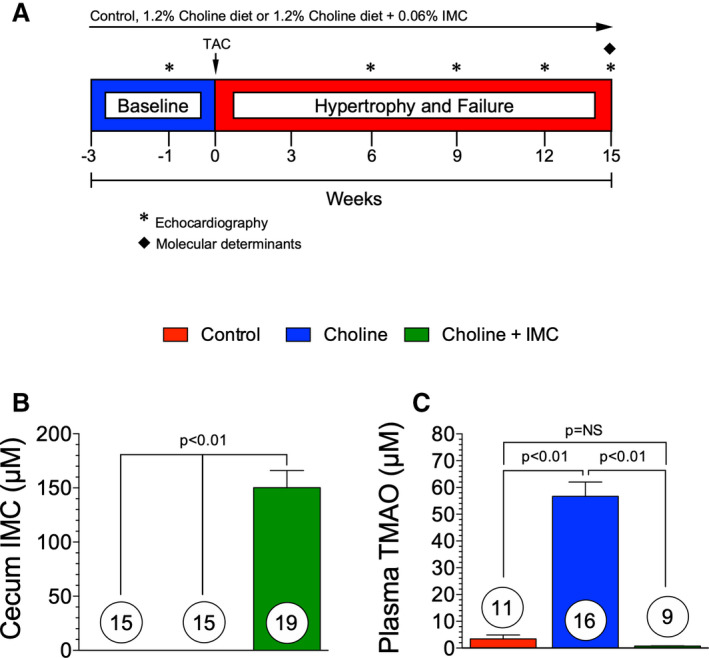
Mice were fed experimental diets starting at 3 weeks prior to TAC surgery. Left ventricular (LV) function and structure were evaluated via echocardiography at 3‐week intervals following baseline echocardiography studies at 1 week before TAC surgery. (B), Cecum levels of iodomethylcholine were measured in all 3 groups at 15 weeks. (C), Circulating levels of trimethylamine N‐oxide (TMAO) in control, choline, and choline+iodomethylcholine groups at 15 weeks. Results are shown as mean± SEM. P=NS (not significant).
Microbial Choline TMA Lyase Inhibition Attenuates Choline Diet‐Induced Cardiac Dysfunction in HF
We performed TAC surgery to induce LV hypertrophy and HF and followed the animals for a period of 15 weeks, while they were maintained on their respective diets. We found that the choline‐alone group had worse cardiac remodeling and dysfunction compared with the control groups at 15 weeks after TAC (Figure 6, Table 2). In Table 2, at the 15‐week time point, all measured end points are significantly different (P<0.05 or P<0.01, respectively) compared with baseline, within group. The left ventricular end‐systolic dimension was significantly (P<0.05) increased in the choline‐fed group compared with choline+iodomethylcholine (Figure 6A). However, no changes were observed in the LV end‐diastolic diameter (Figure 6B). There was also a significant (P<0.01) reduction in the LV fractional shortening in the choline diet group at 15‐weeks after TAC compared with control (Figure 6C). Importantly, we found that there were no significant differences in cardiac structure or function following TAC HF between the control group and the choline+iodomethylcholine group at any time point in the study. Further, LV fractional shortening significantly (P<0.05) improved with iodomethylcholine at 15 weeks after TAC compared with the choline diet group (Figure 6C). These data suggest that the inhibition of gut microbial TMA lyase attenuates the pathological actions of dietary choline on cardiac structure and function in the setting of pressure overload–induced HF. The pathological remodeling and cardiac myocyte loss were largely abrogated in the choline+iodomethylcholine group compared with the choline‐fed group in the intraventricular septum (Figure 6D). We observed no significant changes in the LV posterior wall thickness (Figure 6E). At 15 weeks, we observed an ≈3‐fold increase in circulating BNP levels in the choline group compared with control and a significant (P<0.01) reduction in BNP levels in the choline+iodomethylcholine group compared with choline (Figure 6F). Representative photographs of hearts from control, choline diet, and choline diet+iodomethylcholine groups are shown in Figure 7A. There were no significant changes in heart weight:body weight or tibia length at 15 weeks following TAC among groups (Figure 7B and 7C). At 15 weeks, there was no significant difference in the left atrial weight:tibia length in groups (Figure 7D). At 15 weeks, LV sections were stained for WGA and myocyte cross‐sectional area was quantified. There was no significant difference in myocyte cross‐sectional area among groups (Figure 7E and 7F). Taken together, these data suggest that dietary choline via TMAO generation has detrimental effects on cardiac function and remodeling during HF. Moreover, blocking the gut microbial metabolic pathway of choline transformation into TMAO can, in large part, blunt these effects.
Figure 6. The trimethylamine lyase inhibitor iodomethylcholine inhibited choline‐induced exacerbation of cardiac dysfunction and adverse remodeling following transverse aortic constriction.
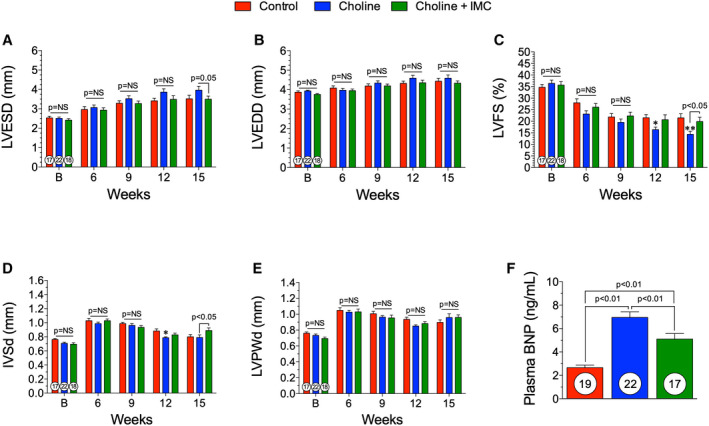
(A), left ventricular (LV) end‐systolic diameter (LVESD), (B) LV end‐diastolic diameter (LVEDD), (C) LV fractional shortening (LVFS), and (D) interventricular septal wall thickness (IVSd) and (E) LV posterior wall thickness at diastole (LVPWd) following transverse aortic constriction (TAC) in control, trimethylamine N‐oxide (TMAO), and TMAO withdrawal groups. (F), plasma B‐type natriuretic peptide (BNP) levels at 15 weeks following TAC. All results are shown as mean± SEM. P=NS (not significant), *P<0.05 vs control, **P<0.01 vs control.
Table 2.
Choline+Iodomethylcholine Study Echocardiographic Measurements
| Control | Choline | Choline+Iodomethylcholine | |
|---|---|---|---|
| Baseline | |||
| LV end‐systolic volume, μL | 23.3±1.70 | 23.4±1.50 | 20.8±1.58 |
| LV end‐diastolic volume, μL | 65.0±2.33 | 67.7±1.64 | 60.3±1.71 |
| LV ejection fraction, % | 64.4±1.63 | 65.9±1.72 | 66.0±1.96 |
| LV mass, mg | 104±3.25 | 99.8±2.85 | 86.5±2.80 |
| 15 wks after TAC | |||
| LV end‐systolic volume, μL | 53.4±5.00 | 73.8±9.24a | 51.9±5.42b |
| LV end‐diastolic volume, μL | 90.8±6.24 | 100±8.79 | 84.4±5.25 |
| LV ejection fraction, % | 43.6±2.69 | 30.3±2.45c | 41.4±3.23b |
| LV mass, mg | 155±9.29 | 169±12.5 | 161±10.0 |
Data shown as the mean±SEM. LV indicates left ventricular; and TAC, transverse aortic constriction.
P<0.05 compared with control at that time point.
P<0.05 compared with the choline group at that time point.
P<0.01 compared with control at that time point.
Figure 7. Hypertrophic Remodeling following IMC Administration.(A), Representative images of hearts taken at 15 weeks.
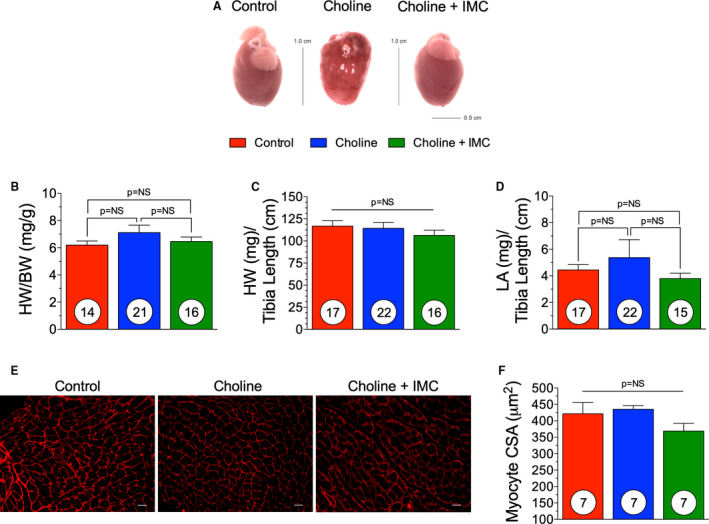
(B), Heart weight (HW) to body weight (BW) of control, trimethylamine N‐oxide (TMAO) and TMAO withdrawal groups. (C), Heart weight to tibia length of control, TMAO, and TMAO withdrawal groups. (D), left atrial (LA) weight to tibia length. (E), Representative photomicrographs (×40) of wheat germ agglutinin staining for myocyte cross sectional area (CSA) (scale bar=20 μm) and (F) quantification in all three groups. Results are shown as mean± SEM. P=NS (not significant).
Inhibition of TMA Lyase Reduced the Fibrotic Response to TAC and Choline‐Enriched Diet
PSR staining was performed on cross sections of hearts from control, choline, and choline+iodomethylcholine groups. Representative photomicrographs (Figure 8A) of whole heart section demonstrate the level of fibrosis. The PSR‐positive area was quantified as a percentage of total tissue area. Choline‐fed animals had a significant increase in the cardiac fibrotic response at 15 weeks (P<0.05) compared with control (Figure 8B). Choline+iodomethylcholine significantly inhibited the fibrosis generated by TAC and choline (P<0.05) and was not significantly different than control. Furthermore, we looked at profibrotic gene expression in the left ventricles of animals in control, choline, and choline+iodomethylcholine groups (Figure 8C). We observed an overall increase in gene expression within the choline‐fed group. transforming growth factor‐β and matrix metallopeptidase 2 were significantly increased in the choline‐fed group compared with control (P<0.05). Additionally, choline+iodomethylcholine significantly reduced collagen 1a1, collagen 3a1, fibronectin‐1, transforming growth factor‐β, connective tissue growth factor‐1, matrix metallopeptidase 2, and metallopeptidase inhibitor 2 compared with the choline‐fed group (Figure 8C). These data suggest that the gut microbial choline TMA lyase inhibitor iodomethylcholine has the capacity to inhibit the exacerbated fibrotic response to a choline‐rich diet in the presence of TAC‐induced HF.
Figure 8. Iodomethylcholine attenuates the fibrotic response induced via choline enriched diet.
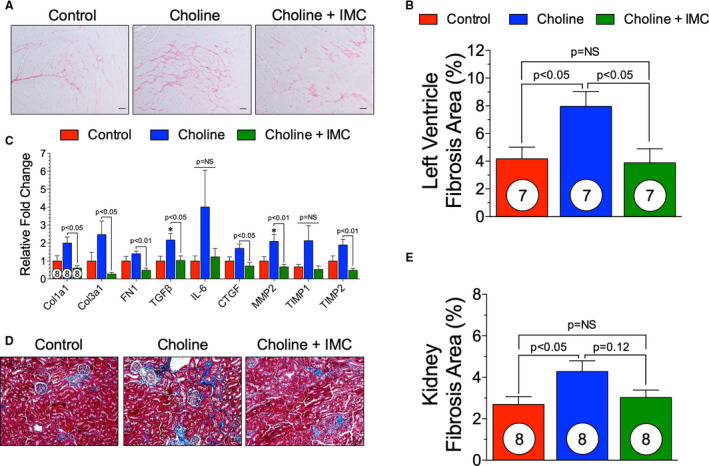
(A), Representative photomicrographs (×40) of Picrosirius red staining in control, choline, and choline+iodomethylcholine groups. (B), Quantification of fibrosis as a percentage of total tissue area. (C), Gene expression levels of fibrotic markers in control, trimethylamine N‐oxide (TMAO), and TMAO withdrawal groups. (D), Representative photomicrographs (×40) of renal Masson's trichrome staining in control, choline and choline+iodomethylcholine groups. (E), Quantification of kidney fibrosis as a percentage of total tissue area. Scale bar=20 μm. Results are shown as mean± SEM. P=NS (not significant). Col1A1 indicates collagen 1A1; Col3A1, collagen 3A1; CTGF, connective tissue growth factor; FN1, fibronectin 1; MMP2, matrix metalloprotease 2; TGFβ, transforming growth factor β; and TIMP, metallopeptidase inhibitor.
Masson's trichrome staining of the kidney was performed to investigate if TMA lyase inhibition would mitigate the adverse kidney fibrosis observed in a TAC‐induced HF model ± choline diet. Figure 8D shows representative photomicrographs of kidney fibrosis. Quantification of fibrotic area in control, choline‐fed, and choline+iodomethylcholine groups demonstrated that a choline‐rich diet significantly (P<0.05) increased kidney fibrosis compared with control. The total fibrotic area was reduced within the choline+iodomethylcholine group compared with a choline diet; however, this did not reach statistical significance (Figure 8E).
Discussion
Lifestyle decisions, specifically dietary choices, have an impact on the development of cardiovascular comorbidities, such as diabetes mellitus and obesity. These 2 comorbidities alone have a profound influence on the development and progression of HF.45 Examining the effect of withdrawing dietary TMAO following chronic exposure has allowed us to evaluate the pathological effects of TMAO in the setting of HF. In this study, we extend our earlier findings of the detrimental role of TMAO in HF independent from the progression of atherosclerosis. Our studies herein reveal that removing dietary TMAO during HF has beneficial effects on the myocardium. This is evidenced by improvements in LV structure and function and reductions in circulating levels of both BNP and TMAO following withdrawal of dietary TMAO. The ability to modify circulating TMAO levels after an initial long‐term exposure period is an important finding for the treatment of HF. This finding supports the idea that further dietary modifications and new guidelines are necessary, notwithstanding changes to sodium and fluid restriction.2 Our study also shows that the effects of TMAO on the myocardium are modifiable and that myocardium can recover in terms of structure and function following withdrawal of TMAO in the setting of HF. These data lend credence to the notion that more developed and well‐established guidelines are necessary with respect to nutrition and dietary alterations for patients suffering from HF.
The gut microbiome is an active area of research for regulating pathological dietary metabolites of cardiovascular disease, specifically targeting the production of TMAO. Studies in apolipoprotein E −/− mice have shown that treatment with broad‐spectrum bactericidal antibiotics effectively reduces circulating TMAO levels and reduces the number of atherosclerotic lesions.16 However, when treated with antibiotics, the reduction in TMAO levels is only temporary. The transient suppression of circulating TMAO is attributable to repopulation of the intestines by antibiotic‐resistant bacteria. In addition, antibiotics also can cause adverse gastrointestinal complications with long‐term use, such as nausea, diarrhea, and colitis. Despite the limitations of antibiotics as an effective therapeutic for modulating the gut microbiome in HF, these experiments confirmed that blocking TMAO production with the novel nonlethal TMA lyase inhibitor iodomethylcholine improves cardiac function in the setting of HF.
Because of the many beneficial components of the gut microbiome, complete eradication is not a desirable therapeutic approach. However, targeting 1 noncritical functional component safeguards essential functions. Deciding where to intercept the choline metabolic pathway is critical, since choline is a vital nutrient. Thus, blocking choline adsorption would lead to its deficiency and adverse neurological effects. Moreover, blocking the pathway too far downstream, for instance, at the liver, by inhibiting flavin‐containing monooxygenase 3 (blocking the conversion of TMA to TMAO) results in trimethylaminuria or “fish odor syndrome.” Flavin‐containing monooxygenase 3 is also involved in the metabolism and inactivation of several compounds, for example, multiple drug classes, rendering flavin‐containing monooxygenase 3 an even less appealing target. A more desirable step to target in the choline metabolic pathway is the gut microbial conversion of choline to TMA. The enzyme responsible for the conversion of choline to the volatile metabolite TMA is choline TMA lyase, and this conversion is gut microbe dependent16, 46, 47 Choline TMA lyase is a widely distributed microbial enzyme found in the human gut48 In vitro experiments have shown that choline TMA lyase has specificity for choline and suggest its main role in physiology is the conversion of choline to TMA within a subset of gut microbial community members.
The present studies suggest that beyond reduction in atherosclerosis and thrombosis potential24, 44 targeting the gut microbial TMA/TMAO pathway can prove beneficial in helping to prevent the development or progression of HF. Iodomethylcholine is a recently developed mechanism‐based suicide substrate inhibitor of microbial choline TMA‐lyase activity44 In our model, iodomethylcholine successfully attenuated the negative effects of dietary choline on cardiac function. We found that mice receiving both dietary choline and the inhibitor (choline+iodomethylcholine) had plasma TMAO levels nearly identical to controls. This suggests that iodomethylcholine is effectively inhibiting the gut microbial enzymatic conversion of choline to TMA, the rate‐limiting step in TMAO production. Dietary choline worsened cardiac dysfunction in the setting of HF compared with control, evidenced by significant reductions in LV fractional shortening compared with control diet, while iodomethylcholine attenuated this effect. The choline+iodomethylcholine group had a significantly higher LV ejection fraction than choline alone, which was preserved to the same degree as the control diet group.
The present study corroborates and extends our previous findings that dietary choline and its gut microbe‐dependent metabolite, TMAO, can exacerbate the development of adverse ventricular remodeling and the fibrotic response during pressure overload34 In using the TMA lyase inhibitor, iodomethylcholine, we demonstrate the effective inhibition of circulating TMAO levels, which led to an attenuation of adverse cardiac remodeling and fibrosis in the setting of HF. We did not see any change in the cardiac fibrotic response to a TMAO‐enriched diet or its withdrawal, despite significant alterations in cardiac structure and function. This does not preclude such effects from being detectable when followed over longer periods of time, but confirm that preventative strategies to attenuate the onset of fibrosis are far more effective than reversal after development. This functional change in response to TMAO withdrawal, despite no significant change in fibrosis, may also be due to submaximal levels of circulating TMAO when mice were fed a 0.12% (wt/wt) TMAO diet. Previous in vitro work by Savi et al49 demonstrated that a similar level of TMAO (20 μmol/L TMAO) can affect myocyte energetics, contractility, and calcium dynamics. Of importance, the TMAO levels achieved in the present study with TMAO and choline diets (reaching ≈25–60 μmol/L) are similar to the elevated levels observed in clinical studies13, 14, 43 This previous work and our current study would suggest that clinically observed levels of circulating TMAO have the capacity to alter myocyte function before eliciting major changes to the fibrotic response.
Our previous work15 and others50 have demonstrated that increased TMAO levels have been linked to renal fibrosis and dysfunction, and is elevated in subjects with advanced chronic kidney disease. The data herein corroborate these findings; we demonstrate that with a TMAO‐ or choline‐rich diet, there is a significant increase in circulating TMAO levels, which exacerbates kidney fibrosis when compared with the control groups. Withdrawal of TMAO had no significant effect on kidney fibrosis when compared with the TMAO group. This may be attributable to the time of TMAO diet withdrawal. When the choline TMA lyase inhibitor iodomethylcholine was administered, there was a reduction in kidney fibrosis toward levels observed in control group. However, these data did not reach statistical significance when compared with the choline‐rich diet group.
Limitations of the present study are worth noting. The murine model is not fully comparable to the pathology of HF in patients and does not reproduce the pathophysiology of HF induced by myocardial infarction and myocardial cell death. In the present study, we used the TAC protocol that induces a pressure overload model of HF, which is a well‐established preclinical model35, 36, 37, 38, 51 but does not represent the full spectrum of human disease. Additionally, there are modest differences among the LV function data, HF biomarkers, and cardiac fibrosis data following TAC heart failure presented herein and our previous work34 Of importance is that we did observe significant reductions in LV function, cardiac fibrosis, and severe HF, as well as powerful beneficial effects of TMAO withdrawal and gut microbial choline TMA lyase inhibition. Another limitation of the present study is that beyond induction of multiple profibrotic gene expression changes, we have not fully elucidated the molecular mechanisms by which TMAO exerts its adverse effects on cardiac remodeling and function. Notably, a recent study by Chen and colleagues52 reported the discovery of the endoplasmic reticulum stress kinase PERK (EIF2AK3) as a TMAO receptor for glucose‐related metabolic effects associated with the metabolite. It remains unknown if PERK, an alternative as of yet unidentified TMAO receptors, or TMAO itself (acting solely via other proposed actions on protein conformation and stability) participate in the physiological effects observed herein21, 53
As our understanding of the gut microbiome and its interaction with host physiology expands, it is important that we understand how dietary components are metabolized. Our data suggest that TMAO is not merely a by‐product that is increased in subjects with HF but that it can directly play a causative role in the pathogenesis of HF. This study is of significance because it shows that circulating TMAO levels are modifiable after long‐term exposure periods, and TMAO is available as an in vitro clinical diagnostic test. We also have shown that the heart is able to recover from long‐term exposure to high circulating levels of TMAO and choline via improvements in cardiac function. Moreover, blocking the conversion of choline to TMAO at the level of the gut microbiota via the microbial enzyme choline TMA lyase is an effective mechanism to attenuate the negative effects of dietary choline on the cardiovascular system. The choline TMA lyase inhibitor iodomethylcholine, when given in conjunction with choline, successfully blocked choline's conversion to TMAO and significantly lowered circulating TMAO levels and attenuated cardiac dysfunction. Further studies aimed at manipulating this pathway and monitoring HF risks seem warranted to better understand the role of TMAO in human physiology. It is remarkable to note that in a recent dietary intervention study involving chronic exposure to 3 different isocaloric diets, selection of protein source (red meat versus white meat versus nonmeat) had a profound impact on circulating TMAO levels among healthy volunteers—with TMAO levels observed on the chronic red meat diet well within the range of values observed in the present study54 Together, we believe that choline/TMAO‐enriched diets lead to exacerbation of adverse remodeling and cardiac dysfunction, and that pharmacological targeting of TMA lyase and dietary modification both represent viable strategies for mitigating HF manifestation and progression.
Sources of Funding
This work was supported by grants from the National Heart, Lung, and Blood Institute (National Institutes of Health; R01 HL092141, R01 HL093579, R01 HL11657, and U24 HL 094373 to Lefer). S.L.H. reports support by grants P01 HL147823, R01HL103866, and R01HL126827 from the National Heart, Lung, and Blood Institute of the National Institutes of Health and the Office of Dietary Supplements, as well as from the Fondation Leducq. Mass spectrometry instrumentation used was housed within the Cleveland Clinic Mass Spectrometry Facility with partial support through a Center of Innovation by Shimadzu Corporation.
Disclosures
Dr Hazen reports being named as coinventor on pending and issued patents held by the Cleveland Clinic relating to cardiovascular diagnostics and therapeutics. Dr Hazen also reports being a paid consultant for P&G, having received research funds from P&G, and Roche Diagnostics, and being eligible to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics or therapeutics from Cleveland HeartLab, Quest Diagnostics, and P&G. The remaining authors have no disclosures to report.
(J Am Heart Assoc. 2020;9:e016223 DOI: 10.1161/JAHA.119.016223.)
For Sources of Funding and Disclosers, see page 13.
References
- 1. Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, et al. Heart disease and stroke statistics—2020 update: a report from the American Heart Association. Circulation. 2020;141:e139–e596. [DOI] [PubMed] [Google Scholar]
- 2. Vest AR, Chan M, Deswal A, Givertz MM, Lekavich C, Lennie T, Litwin SE, Parsly L, Rodgers JE, Rich MW, et al. Nutrition, obesity, and cachexia in patients with heart failure: a consensus statement from the heart failure society of America Scientific Statements Committee. J Card Fail. 2019;25:380–400. [DOI] [PubMed] [Google Scholar]
- 3. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14:e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez‐Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. 1977;31:107–133. [DOI] [PubMed] [Google Scholar]
- 7. Luckey TD. Introduction to intestinal microecology. Am J Clin Nutr. 1972;25:1292–1294. [DOI] [PubMed] [Google Scholar]
- 8. Shih DM, Wang Z, Lee R, Meng Y, Che N, Charugundla S, Qi H, Wu J, Pan C, Brown JM, et al. Flavin containing monooxygenase 3 exerts broad effects on glucose and lipid metabolism and atherosclerosis. J Lipid Res. 2015;56:22–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Warrier M, Shih DM, Burrows AC, Ferguson D, Gromovsky AD, Brown AL, Marshall S, McDaniel A, Schugar RC, Wang Z, et al. The TMAO‐generating enzyme flavin monooxygenase 3 is a central regulator of cholesterol balance. Cell Rep. 2015;10:326–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sheard NF, Zeisel SH. Choline: an essential dietary nutrient? Nutrition. 1989;5:1–5. [PubMed] [Google Scholar]
- 11. Zeisel SH, da Costa KA. Choline: an essential nutrient for public health. Nutr Rev. 2009;67:615–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zeisel SH. Choline: essential for brain development and function. Adv Pediatr. 1997;44:263–295. [PubMed] [Google Scholar]
- 13. Tang WH, Wang Z, Fan Y, Levison B, Hazen JE, Donahue LM, Wu Y, Hazen SL. Prognostic value of elevated levels of intestinal microbe‐generated metabolite trimethylamine‐N‐oxide in patients with heart failure: refining the gut hypothesis. J Am Coll Cardiol. 2014;64:1908–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tang WH, Wang Z, Kennedy DJ, Wu Y, Buffa JA, Agatisa‐Boyle B, Li XS, Levison BS, Hazen SL. Gut microbiota‐dependent trimethylamine N‐oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res. 2015;116:448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, et al. Intestinal microbiota metabolism of L‐carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Caesar R, Fak F, Backhed F. Effects of gut microbiota on obesity and atherosclerosis via modulation of inflammation and lipid metabolism. J Intern Med. 2010;268:320–328. [DOI] [PubMed] [Google Scholar]
- 19. Koren O, Spor A, Felin J, Fak F, Stombaugh J, Tremaroli V, Behre CJ, Knight R, Fagerberg B, Ley RE, et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci USA. 2011;108(suppl 1):4592–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gregory JC, Buffa JA, Org E, Wang Z, Levison BS, Zhu W, Wagner MA, Bennett BJ, Li L, DiDonato JA, et al. Transmission of atherosclerosis susceptibility with gut microbial transplantation. J Biol Chem. 2015;290:5647–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seldin MM, Meng Y, Qi H, Zhu W, Wang Z, Hazen SL, Lusis AJ, Shih DM. Trimethylamine N‐oxide promotes vascular inflammation through signaling of mitogen‐activated protein kinase and nuclear factor‐Kappab. J Am Heart Assoc. 2016;5:002767 DOI: 10.1161/JAHA.115.002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, Li L, Fu X, Wu Y, Mehrabian M, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell. 2016;165:111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhu W, Wang Z, Tang WHW, Hazen SL. Gut microbe‐generated trimethylamine N‐oxide from dietary choline is prothrombotic in subjects. Circulation. 2017;135:1671–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, Gu X, Huang Y, Zamanian‐Daryoush M, Culley MK, et al. Non‐lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell. 2015;163:1585–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shih DM, Zhu W, Schugar RC, Meng Y, Jia X, Miikeda A, Wang Z, Zieger M, Lee R, Graham M, et al. Genetic deficiency of flavin‐containing monooxygenase 3 (FMO3) protects against thrombosis but has only a minor effect on plasma lipid levels‐brief report. Arterioscler Thromb Vasc Biol. 2019;39:1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Skye SM, Zhu W, Romano KA, Guo CJ, Wang Z, Jia X, Kirsop J, Haag B, Lang JM, DiDonato JA, et al. Microbial transplantation with human gut commensals containing CutC is sufficient to transmit enhanced platelet reactivity and thrombosis potential. Circ Res. 2018;123:1164–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu W, Buffa JA, Wang Z, Warrier M, Schugar R, Shih DM, Gupta N, Gregory JC, Org E, Fu X, et al. Flavin monooxygenase 3, the host hepatic enzyme in the metaorganismal trimethylamine N‐oxide‐generating pathway, modulates platelet responsiveness and thrombosis risk. J Thromb Haemost. 2018;16:1857–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Suzuki T, Heaney LM, Bhandari SS, Jones DJ, Ng LL. Trimethylamine N‐oxide and prognosis in acute heart failure. Heart. 2016;102:841–848. [DOI] [PubMed] [Google Scholar]
- 29. Troseid M, Ueland T, Hov JR, Svardal A, Gregersen I, Dahl CP, Aakhus S, Gude E, Bjorndal B, Halvorsen B, et al. Microbiota‐dependent metabolite trimethylamine‐N‐oxide is associated with disease severity and survival of patients with chronic heart failure. J Intern Med. 2015;277:717–726. [DOI] [PubMed] [Google Scholar]
- 30. Tang WH, Wang Z, Shrestha K, Borowski AG, Wu Y, Troughton RW, Klein AL, Hazen SL. Intestinal microbiota‐dependent phosphatidylcholine metabolites, diastolic dysfunction, and adverse clinical outcomes in chronic systolic heart failure. J Card Fail. 2015;21:91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou X, Jin M, Liu L, Yu Z, Lu X, Zhang H. Trimethylamine N‐oxide and cardiovascular outcomes in patients with chronic heart failure after myocardial infarction. ESC Heart Fail. 2020;7:188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qi J, You T, Li J, Pan T, Xiang L, Han Y, Zhu L. Circulating trimethylamine N‐oxide and the risk of cardiovascular diseases: a systematic review and meta‐analysis of 11 prospective cohort studies. J Cell Mol Med. 2018;22:185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schiattarella GG, Sannino A, Toscano E, Giugliano G, Gargiulo G, Franzone A, Trimarco B, Esposito G, Perrino C. Gut microbe‐generated metabolite trimethylamine‐N‐oxide as cardiovascular risk biomarker: a systematic review and dose‐response meta‐analysis. Eur Heart J. 2017;38:2948–2956. [DOI] [PubMed] [Google Scholar]
- 34. Organ CL, Otsuka H, Bhushan S, Wang Z, Bradley J, Trivedi R, Polhemus DJ, Tang WH, Wu Y, Hazen SL, et al. Choline diet and its gut microbe‐derived metabolite, trimethylamine N‐oxide, exacerbate pressure overload‐induced heart failure. Circ Heart Fail. 2016;9:e002314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li Z, Organ CL, Kang J, Polhemus DJ, Trivedi RK, Sharp TE III, Jenkins JS, Tao YX, Xian M, Lefer DJ. Hydrogen sulfide attenuates renin angiotensin and aldosterone pathological signaling to preserve kidney function and improve exercise tolerance in heart failure. JACC Basic Transl Sci. 2018;3:796–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Polhemus D, Kondo K, Bhushan S, Bir SC, Kevil CG, Murohara T, Lefer DJ, Calvert JW. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ Heart Fail. 2013;6:1077–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bradley JM, Li Z, Organ CL, Polhemus DJ, Otsuka H, Islam KN, Bhushan S, Gorodnya OM, Ruchko MV, Gillespie MN, et al. A novel MTDNA repair fusion protein attenuates maladaptive remodeling and preserves cardiac function in heart failure. Am J Physiol Heart Circ Physiol. 2018;314:H311–H321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bradley JM, Spaletra P, Li Z, Sharp TE III, Goodchild TT, Corral LG, Fung L, Chan KW, Sullivan RW, Swindlehurst CA, et al. A novel fibroblast activation inhibitor attenuates left ventricular remodeling and preserves cardiac function in heart failure. Am J Physiol Heart Circ Physiol. 2018;315:H563–H570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Polhemus DJ, Gao J, Scarborough AL, Trivedi R, McDonough KH, Goodchild TT, Smart F, Kapusta DR, Lefer DJ. Radiofrequency renal denervation protects the ischemic heart via inhibition of GRK2 and increased nitric oxide signaling. Circ Res. 2016;119:470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Polhemus DJ, Trivedi RK, Gao J, Li Z, Scarborough AL, Goodchild TT, Varner KJ, Xia H, Smart FW, Kapusta DR, et al. Renal sympathetic denervation protects the failing heart via inhibition of neprilysin activity in the kidney. J Am Coll Cardiol. 2017;70:2139–2153. [DOI] [PubMed] [Google Scholar]
- 41. Koeth RA, Levison BS, Culley MK, Buffa JA, Wang Z, Gregory JC, Org E, Wu Y, Li L, Smith JD, et al. Gamma‐butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of L‐carnitine to TMAO. Cell Metab. 2014;20:799–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang Z, Levison BS, Hazen JE, Donahue L, Li XM, Hazen SL. Measurement of trimethylamine‐N‐oxide by stable isotope dilution liquid chromatography tandem mass spectrometry. Anal Biochem. 2014;455:35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Z, Bergeron N, Levison BS, Li XS, Chiu S, Jia X, Koeth RA, Li L, Wu Y, Tang WHW, et al. Impact of chronic dietary red meat, white meat, or non‐meat protein on trimethylamine N‐oxide metabolism and renal excretion in healthy men and women. Eur Heart J. 2018;40:583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roberts AB, Gu X, Buffa JA, Hurd AG, Wang Z, Zhu W, Gupta N, Skye SM, Cody DB, Levison BS, et al. Development of a gut microbe‐targeted nonlethal therapeutic to inhibit thrombosis potential. Nat Med. 2018;24:1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bozkurt B, Aguilar D, Deswal A, Dunbar SB, Francis GS, Horwich T, Jessup M, Kosiborod M, Pritchett AM, Ramasubbu K, et al. Contributory risk and management of comorbidities of hypertension, obesity, diabetes mellitus, hyperlipidemia, and metabolic syndrome in chronic heart failure: a scientific statement from the American Heart Association. Circulation. 2016;134:e535–e578. [DOI] [PubMed] [Google Scholar]
- 46. Cashman JR. Human flavin‐containing monooxygenase: substrate specificity and role in drug metabolism. Curr Drug Metab. 2000;1:181–191. [DOI] [PubMed] [Google Scholar]
- 47. Krueger SK, Williams DE. Mammalian flavin‐containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol Ther. 2005;106:357–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Craciun S, Marks JA, Balskus EP. Characterization of choline trimethylamine‐lyase expands the chemistry of glycyl radical enzymes. ACS Chem Biol. 2014;9:1408–1413. [DOI] [PubMed] [Google Scholar]
- 49. Savi M, Bocchi L, Bresciani L, Falco A, Quaini F, Mena P, Brighenti F, Crozier A, Stilli D, Del Rio D. Trimethylamine‐N‐oxide (TMAO)‐induced impairment of cardiomyocyte function and the protective role of urolithin B‐glucuronide. Molecules. 2018;23:549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Missailidis C, Hällqvist J, Qureshi AR, Barany P, Heimbürger O, Lindholm B, Stenvinkel P, Bergman P. Serum trimethylamine‐N‐oxide is strongly related to renal function and predicts outcome in chronic kidney disease. PLoS One. 2016;11:e0141738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G Sr, Gojon G Jr, et al. H(2)s protects against pressure overload‐induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. 2013;127:1116–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen S, Henderson A, Petriello MC, Romano KA, Gearing M, Miao J, Schell M, Sandoval‐Espinola WJ, Tao J, Sha B, et al. Trimethylamine N‐oxide binds and activates PERK to promote metabolic dysfunction. Cell Metab. 2019;30:1141–1151. e1145. [DOI] [PubMed] [Google Scholar]
- 53. Moore JO, Hendrickson WA. Structural analysis of sensor domains from the TMAO‐responsive histidine kinase receptor TorS. Structure. 2009;17:1195–1204. [DOI] [PubMed] [Google Scholar]
- 54. Wang Z, Bergeron N, Levison BS, Li XS, Chiu S, Jia X, Koeth RA, Li L, Wu Y, Tang WHW, et al. Impact of chronic dietary red meat, white meat, or non‐meat protein on trimethylamine N‐oxide metabolism and renal excretion in healthy men and women. Eur Heart J. 2019;40:583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]