

Abstract
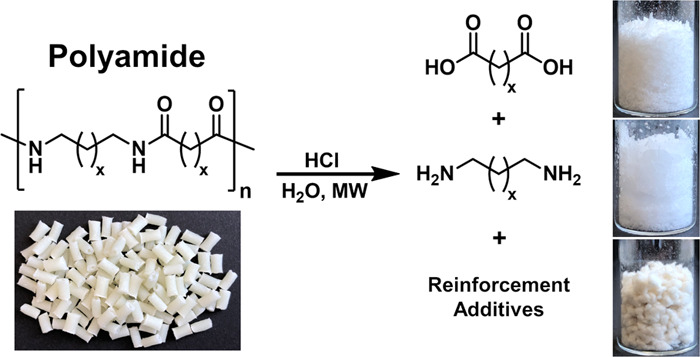
We report on a simple and efficient chemical recycling process for aliphatic polyamides (PA 66, PA 1010, PA 11, and PA 12), whereby PAs are converted exclusively into their constituent monomers even in the presence of reinforcement additives, such as carbon- and glass-fibers. In this process, the rate of PA hydrolysis reaction, performed under microwave irradiation in the presence of HCl as an acid catalyst, depends on the PA type, the HCl/amide mole ratio, and the type and amount of reinforcement additives. PA 66 is completely converted into the constituent monomers at 200 °C and a 1.25 HCl/amide mole ratio in 10 min. Long-chain PAs (PA 11, PA 12, and PA 1010) and PAs containing glass- or carbon-fiber reinforcement additives need at the same experimental conditions longer reaction times. Alternatively, they can be completely hydrolyzed at 200 °C within a comparable reaction time at a higher HCl/amide mole ratio of 2.5. Complete and straightforward conversion of PAs into the constituent monomers in the absence of side reactions simplifies the isolation and purification of monomers and reinforcement additives, which have been recovered in high yields and quality comparable to those of commercially available chemicals.
Keywords: polymer waste, chemical recycling, microwave chemistry, hydrolysis, aliphatic polyamides, composites
Short abstract
Aliphatic polyamides were completely degraded by microwave-assisted hydrolysis to recover the monomers in high yields and purity.
Introduction
Increasing production of plastic waste presents an ever-growing environmental issue because most of it ends up in landfills or is incinerated to recover at least a part of its value in the form of energy. To alleviate the impact of plastic waste on the environment, new recycling methods are being continuously developed.1,2 Mechanical recycling is already well established; however, after a certain number of reprocessing cycles, a decrease in the quality of recycled products has been observed, mainly due to the thermomechanical degradation of polymer chains.3 Alternatively, polymer waste is converted to feedstock for monomers, fuel production, or other value-added products and intermediates by chemical recycling,4−6 where the main challenge is to make the depolymerization processes economically viable.7
Polyamides (PAs) are a class of polymers suitable for chemical recycling, with PA 6 and PA 66 being the most commonly studied and recycled on an industrial scale already. The amide group in the PA backbone is stable enough to make PAs useful in various application fields ranging from fibers in carpets and textiles, engineering plastic in automotive, electrical, electronic, and construction industry to packaging and coating sectors. On the other hand, PAs can undergo depolymerization by different degradation agents, such as water, ammonia, or alcohols/glycols in combination with various catalysts, facilitating the depolymerization processes.8−10 Most of the studies address the depolymerization of PA 6 with the aim to recover the cyclic ε-caprolactam (CPL) monomer, which is then isolated by distillation. For PAs prepared from dicarboxylic acids and diamines (PA 66 and PA 1010) or long-chain amino acid monomers (PA 11 and PA 12), complete depolymerization is preferred because oligomers are difficult to separate from monomers. Partially depolymerized PA mixtures are more suitable for less demanding applications, for example, as curing agents for phenolic resins.11
For complete depolymerization of PAs into the constituent monomers, extreme reaction conditions and long reaction times are usually required, which inadvertently lead to undesired side products, again complicating the purification steps. Such processes often prove to be economically unfeasible due to the low yields of recovered monomers. Chen et al. were able to recover CPL from PA 6 in 78% yield by hydrolysis at 300 °C in 85 min reaction time using phosphotungstic heteropolyacid as a catalyst, with 6-aminocaproic acid and water-soluble oligomers being the main side products.12 Similar results were reported by Huang et al. for hydrolysis of PA 6 at 354 °C for 75 min in the absence of a catalyst, whereby the final reaction mixture consisted of a large amount of residual oligomers.13 Further increase in the reaction temperature to 400 °C led to a decreased CPL yield due to its decomposition.14 PA 66 degradation in supercritical water at 380 °C and 28 MPa for 30 min resulted in complete decomposition of hexamethylene diamine (HMDA) into cycloheptylamine, while adipic acid (AA) was partially converted to cyclopentanone.15 Kamimura et al. developed a method for direct conversion of PA 6 into the CPL monomer using supercritical iPrOH. Complete degradation of amide bonds was achieved in 1 h at 350 °C or in 4 h at 330 °C; however, the resulting reaction mixture consisted of several constituents, the share of which depended on the reaction conditions used.16 Hommez et al. examined glycolysis of PA 6 in ethylene glycol (EG) as a decomposition agent and H3PO4 as an acid catalyst. In this way, PA 6 was converted into CPL and linear oligomers terminated with free and EG esterified carboxyl groups.17 Glycolysis of PA 66 with EG for 1.5 h at normal pressure and temperature of 190 °C in the presence of diammonium hydrogen phosphate as a catalyst resulted in incomplete degradation. Thus, the obtained mixture of glycosylates was used as a substitute for commercial polyols in the synthesis of polyurethanes.18 PA-based waste was also transformed into hydroxyalkanoates or a mixture of diols and diesters in supercritical MeOH at 330 °C.19−21 Ammonolysis, on the other hand, enables conversion of both PA 6 and PA 66 simultaneously to HMDA by transformation of carboxylic groups via the amides to nitrile groups, which are then hydrogenated to the final amine groups.22,23
Because depolymerization processes of PAs demand high reaction temperature, microwave heating, allowing for reduced energy consumption and shorter reaction times compared to the processes based on conventional heating, is becoming increasingly attractive.24,25 The advantage of microwave heating is reported in a study dealing with PA 6 hydrolysis, where after 15 min of microwave irradiation at 200 W (temperature logarithmically increased to a maximum value of 300 °C) in the presence of H3PO4 as a catalyst, more than 90% of PA 6 was converted into the water-soluble oligomers and 6-aminocaproic acid.25 Microwave-assisted depolymerization of PA 6 was also performed in acetic acid with a catalytic amount of 4-dimethylaminopyridine at 250 °C for 15 min. The resulting N-acetylcaprolactam was further transformed into CPL by a transfer of the acetyl functionality to 2-aminoethanol.26 PA 6 depolymerization in hydrophilic ionic liquids using N,N-dimethylaminopyridine as a catalyst resulted after 1 h of microwave heating at 300 °C in 55% CPL yield.27
Acid-catalyzed hydrolysis of PA 6 below 200 °C results almost exclusively in water-soluble acyclic 6-aminocaproic acid and, depending on reaction conditions, also in oligomers.28,29 Since PA 6 is synthesized by ring-opening polymerization of CPL, additional steps are required to cyclize 6-aminocaproic acid, making the depolymerization pathways resulting in direct CPL formation economically more viable. On the other hand, exclusive formation of linear monomers in depolymerization of PA 66, PA 1010, and PA 11 is preferential, considering that these PAs are synthesized by polycondensation. An exception is PA 12, prepared from dodecanolactam via a cyclotrimerization of butadiene. However, due to the low ring strain of dodecanolactam, a prepolymer is prepared first at high temperature by addition of water and acid catalyst,8 indicating that a similar polycondensation synthetic route as the one used for PA 11 synthesis is also viable for the linear 12-dodecanoic acid (12-ADDA) monomer.
Here, we report on a simple and efficient recycling process for converting different types of aliphatic PAs (PA 66, PA 1010, PA 11, and PA 12) into their constituent monomers. PAs were hydrolyzed under microwave irradiation using hydrochloric acid (HCl) as an acid catalyst. Complete and straightforward hydrolysis of PAs into the constituent monomers was used by our group to develop a method for the quantitative determination of PA 6 and/or PA 66 in postconsumer wastes.28 Complete conversion of PAs into the constituent monomers facilitates their isolation and purification, which consequently results in high yields and purity of recovered monomers. Furthermore, the influence of reinforcement additives, such as carbon- and glass-fibers, on the PA depolymerization process has been studied, as well as a possibility to recycle the reinforcement additives.
Experimental Section
Materials
A list of PA samples, which were received in the form of pellets (4.2 × 3.4 × 2.7 mm3 average size), is collected in Table 1. The 37% HCl used for preparation of HCl solutions of lower concentrations for PA degradation experiments was purchased from Sigma-Aldrich. Sodium hydroxide (NaOH, Honeywell Fluka) was used for neutralization of reaction mixtures composed of diacid and diamine dihydrochloride. Ethanol (EtOH, 99.9%, Carlo Erbo, Italy) was used for recrystallization of diacid and amino acid hydrochloride monomers recovered from PA composite materials. AA (≥99.5%, Sigma-Aldrich) and sebacic acid (SA, 99%, Sigma-Aldrich) were used for comparison of their properties with those of recycled monomers. 11-Aminoundecanoic acid (11-AUDA) and 12-ADDA (11-AUDA ≥ 99%, 12-ADDA ≥ 98%, both Sigma-Aldrich) were used to prepare 11-aminoundecanoic acid hydrochloride (11-AUDAxHCl) and 12-dodecanoic acid hydrochloride (12-ADDAxHCl), respectively, the properties of which were compared to those of recycled monomers. HMDA and 1,10-decanediamine (1,10-DDA) monomers (HMDA and 1,10-DDA ≥ 98%, both Sigma-Aldrich) were freshly distilled on a Kugelrohr short-path distillation apparatus (Glass oven B-585 Kugelrohr, Büchi, Switzerland) and then used for comparison of their properties with those of recycled monomers. Acetonitrile (ACN) from Honeywell, Riedel de Haen, Chromasolv, HPLC gradient grade, ≥99.9%; trifluoroacetic acid (TFA) from ACROS Organics, ≥99.5%; or formic acid (FA) from Honeywell, ≥99%, were used as received for preparation of mobile phases. Water was purified using a Milli-Q reverse osmosis system (Millipore, Watford, UK) to obtain MQ water (MQ) with typically 18.2 MΩ cm resistivity and < 4 ppb carbon.
Table 1. PAs Used for Depolymerization Experiments.
| sample | composition | trade name | supplier |
|---|---|---|---|
| PA 66 | PA 66 | Sigma-Aldrich | |
| PA 66-GF35 | PA 66 containing 35 wt % glass-fibers | Kordsa, Turkey | |
| PA 11 | PA 11 | Rilsan KNO | Arkema, France |
| PA 11-CF30 | PA 11 containing 30 wt % carbon-fibers | Rilsan BSR 30 | Arkema, France |
| PA 11-GF30 | PA 11 containing 30 wt % glass-fibers | Rilsan BZM 30 O TLD | Arkema, France |
| PA 12 | PA 12 | Rilsamid AECHVO | Arkema, France |
| PA 12-GF50 | PA 12 containing 50 wt % glass-fibers and black pigment | Rilsamid AZM 50 black | Arkema, France |
| PA 1010 | PA 1010 | Rilsan TMFO F | Arkema, France |
Microwave-Assisted Depolymerization Process
PA depolymerizations were carried out in 30 mL glass vials sealed with PTFE-coated silicone septa, which were mounted in a laboratory microwave reactor Monowave 400 (Anton Paar GmbH, Austria) equipped with temperature and pressure sensors as well as a video camera. In a typical depolymerization procedure, PA pellets and a magnetic stirring bar were put into the glass vial, which was then filled with 8 mL of HCl solution, prepared by diluting 37% HCl with MQ water. The mole ratio between HCl and amide groups in the PA backbone was varied by HCl concentration or PA weight. To prevent overheating, the reaction mixtures were heated to a predetermined temperature gradually in 5 min. When the reaction temperature had been reached, it was kept constant for the whole duration of the experiment by a minimal microwave power input required. After the depolymerization experiment had been completed, the reaction vessel was rapidly cooled down by a stream of compressed air (Figure S1). Because of the microwave reactor design, it was not possible to take aliquots from the reaction mixture during the reaction. Therefore, the course of reaction was followed by performing identical experiments with different depolymerization times. Reaction mixtures used to determine the PA degradation degree at different depolymerization times were transferred into 100 mL round-bottom flasks to remove solvent on a rotary evaporator. Thus-obtained solid residues were further dried at 50 °C in a vacuum oven for 12 h. Dried PA hydrolysates were further homogenized in a mortar. Afterward, the dry reaction products were analyzed by 1H NMR and HPLC on a reversed-phase C18 column.
Isolation and Purification of Recycled Monomers
Reaction mixtures consisting of water-insoluble reinforcement additives (carbon- or glass-fibers) were reheated to 80–95 °C to dissolve the monomer(s). Then, they were filtered through a glass fritted funnel to remove insoluble sample constituents.
PA 66 and PA 1010
The reaction mixtures were cooled to 0 °C to precipitate diacid monomers, which were then isolated by filtration using a glass fritted funnel and washed with cold water. The diacid monomers were recrystallized twice from water (yields: AA, 90%; SA, 89%). Diamine monomers were isolated from the filtrates, which were first concentrated on a rotary evaporator. Solid NaOH was then added to the concentrated filtrates to reach the pH of 12–13. Afterward, the residual water was removed by vacuum distillation at 110 °C and 290 mbar on a Kugelrohr distillation apparatus, followed by distillation of HMDA or 1,10-DDA by slowly increasing the temperature to 140 or 160 °C and reducing the pressure to 70 or 50 mbar (HMDA yield, 86%; 1,10-DDA yield, 78%).
PA 11 and PA 12
The reaction mixtures were cooled to 0 °C to precipitate the amino acids, which were then isolated by filtration using a glass fritted funnel and washed with cold water. The recycled 11-AUDAxHCl and 12-ADDAxHCl monomers were further purified by recrystallization from water (11-AUDAxHCl yield, 93%; 12-ADDAxHCl yield, 97%).
Diacids and amino acid hydrochlorides recovered from the PA-based composites were additionally recrystallized from EtOH between the two recrystallizations from water to remove the trace amounts of impurities originating from the modified fibers and/or additives present in PA-based composites.
Determination of the PA Degradation Degree
The degree of PA degradation was determined from 1H NMR spectra of dried reaction mixtures, which were recorded at room temperature in DMSO-d6 with added TFA on a DD2 600 MHz instrument (Agilent Technologies). If reaction mixtures were not completely soluble in DMSO-d6 due to the presence of oligomers, their 1H NMR spectra were recorded in hexafluoroisopropanol (HFIP) using deuterated benzene (C6D6) as an inset. The degree of PA degradation was assessed from the signal integrals of the methylene group next to the carbonyl of the amide bond (−CH2CONH−) of the oligomers and the methylene group next to the carboxyl (−CH2COOH) functional group of the monomer according to eq 1:
| 1 |
The presence of oligomers in PA hydrolysates was also assessed by a reversed-phase HPLC on an Agilent Zorbax Eclipse XDB C18 column (4.6 × 150 mm2; 5 μm, Agilent Technologies). The chromatographic system consisted of an Agilent 1100 Series binary pump equipped with the model 7725i Rheodyne (Bensheim, Germany) manual injector and an evaporative light-scattering (ELS) detector (Agilent Technologies). The mobile phase used was a gradient of ACN as solvent A and MQ containing 1 vol % ACN and 0.1 vol % FA as solvent B. Linear gradient of the mobile phase composition ran from A/B 1/99 (v/v) to A/B 30/70 (v/v) in the first 20 min for PA 66 and from A/B 1/99 (v/v) to A/B 40/60 (v/v) in the first 15 min for PA 11 and PA 1010. Then, an isocratic elution was held for 5 min, after which the gradient was restored back to the initial A/B ratio. For HPLC measurements on the RP-C18 column, 10 mg of dry and homogenized hydrolysate was dissolved in 10 mL of the mobile phase of the starting composition (A/B = 1/99; v/v) during constant stirring for at least 2 h. Afterward, the solution was filtered if necessary. The injection volume of thus-prepared solutions onto the column was 15 μL. The mobile phase was delivered at a flow rate of 0.5 mL/min. The column temperature was 30 °C, and the temperatures of the evaporator and nebulizer of the ELS detector was set to 50 °C.
Characterization of Recovered Monomers and Reinforcement Additives
Isolated monomers were characterized by 1H and 13C NMR spectra, FTIR, HPLC, and determination of melting point (Tm). 1H and 13C NMR spectra of monomers were recorded at room temperature in DMSO-d6 with added TFA and DMSO-d6, respectively, on a DD2 600 MHz instrument. FTIR transmittance spectra were recorded on an FTIR spectrometer Spectrum One (Perkin-Elmer, Waltham, MA) in an ATR mode in the spectral range from 400 to 4000 cm–1 with a 4 cm–1 spectral resolution. Purity of AA and SA compared to the commercially available diacids was determined by a reversed-phase HPLC on an Agilent Zorbax Eclipse XDB C18 column using an Agilent Technologies chromatographic system equipped with a binary pump and an Agilent 1260 Infinity UV detector operating at a wavelength of 210 nm. The mobile phase used was a gradient of ACN as solvent A and MQ containing 1 vol % ACN and 0.1 vol % TFA as solvent B. In the first 10 min, a linear gradient of the mobile phase compositions ran from A/B 1/99 (v/v) to A/B 30/70 (v/v) for AA and from A/B 20/80 (v/v) to A/B 40/60 (v/v) for SA. Then, isocratic elusion was held for 2 min, after which the gradient was restored back to the initial value (v/v). The mobile phase was delivered at a flow rate of 0.5 mL/min. Solution concentrations of AA and SA monomers were typically 0.5 and 0.1 mg/mL, respectively, while the injection volume was 100 μL. Purity of HMDA and 1,10-DDA compared to the freshly distilled commercially available diamines was determined on a Primesep 100 analytical column (4.6 × 150 mm2; 5 μm, SIELC Technologies). The mobile phase consisted of MQ/ACN/TFA in a volume ratio of 60/40/0.27 and was supplied at a flow rate of 1 mL/min. Purity of recovered 11-AUDAxHCl and 12-ADDAxHCl was determined compared to the amino acid hydrochlorides prepared from the commercially available 11-AUDA and 12-ADDA. For this purpose, HPLC measurements were performed on a Primesep 200 analytical column (4.6 × 150 mm2; 5 μm, SIELC Technologies). The optimal mobile phase composition was MQ/ACN/TFA in a volume ratio of 70/30/0.2 and was delivered at a flow rate of 1 mL/min. Isocratic HPLC measurements on the Primesep columns were performed using an Agilent Technologies chromatographic system equipped with an isocratic pump, a column heater, and an Optilab rEX RI detector (Wyatt Technology Corporation). The column temperature was 30 °C, and the temperature of the RI detector was set to 35 °C. Monomers were dissolved in the appropriate solvent at a concentration of 1 mg/mL, while the injection volume of the sample solutions onto the columns was 50 μL. For each HPLC method, a good linear relationship was observed between the area under the monomer peak in the chromatogram and the concentration of commercially available diacids, diamines, or amino acid hydrochlorides as indicated by the correlation coefficients approaching unity and low values of the line intercepts at the y-axis. HPLC methods were used to determine the purity of the recovered monomers using an external standard calibration method. For this purpose, commercial diamines were freshly distilled, amino acids were transformed into hydrochloric salts, and diacids were dried in a vacuum oven until constant weight. Tm values of recovered monomers were determined by a Leica Galen III melting point apparatus (Wetzlar, Germany) and are reported uncorrected.
The morphology of recovered reinforcement additives was investigated by SEM on an HR-SEM Zeiss Ultra plus instrument (Carl Zeiss, Germany). TGA measurements were performed on a TGA/DSC 1 thermogravimetric analyzer (Mettled Toledo, Switzerland) at a heating rate of 10 °C min–1 from 50 to 600 °C in a nitrogen atmosphere. At 600 °C, the atmosphere was switched to oxygen (20 mL/min) and the samples were further heated to 1000 °C at the same heating rate.
Results and Discussion
Complete Degradation of Aliphatic PAs into the Constituent Monomers
In this work, aliphatic PAs are completely depolymerized by microwave-assisted hydrolysis using HCl as an acid catalyst to obtain the dicarboxylic acid and diamine monomers from PA 66 and PA 1010 or the amino acid hydrochlorides from PA 11 and PA 12 after isolation (Scheme 1). Heating of reaction mixtures was performed by microwave irradiation because direct heating was shown to be more efficient than indirect one using conventional heating.
Scheme 1. Degradation of PAs to Dicarboxylic Acid (1: AA or SA) and Diamine Dihydrochloride (2: HMDAx2HCl or 1,10-DDAx2HCl) or to Amino Acid Hydrochloride (3: 11-AUDAxHCl or 12-ADDAxHCl) by Hydrolysis Using HCl as a Catalyst.

In a first set of experiments, the effect of the mole ratio of HCl to the amide bonds in the PA backbone on the degradation rate of PA 66 at the temperature of 200 °C was studied. 1H NMR spectra of completely depolymerized PA 66 recorded in DMSO-d6 with added TFA are typical for mixtures of AA and protonated HMDA. Incompletely depolymerized reaction mixtures show additional signals at 3.03 and 2.06 ppm, corresponding to the protons of the methylene groups near the amide bond of oligomers, i.e., −CH2NHCO– and −CH2CONH–, respectively (Figure 1). The degree of degradation was evaluated from the peak integrals of the methylene group beside the carbonyl group (−CH2CONH−) of PA oligomers and the methylene group beside the acid functional group of the AA monomer (−CH2COOH) according to eq 1. At the equimolar ratio of HCl to the amide groups, the depolymerization of PA 66 was fast, as indicated by 95.6% degree of degradation after a reaction time of 5 min (Table 2; entry 4, Figure 1). Although the reaction time was extended to 40 min, there was still ∼1% oligomers present in the reaction mixture (Table 2; entry 6, Figure 1). When 0.5 equiv of HCl to the amide bonds had been used, 1H NMR spectra recorded in HFIP reveal 50.8% degree of degradation after 5 min, which did not improve significantly even after 40 min reaction time (Table 2; entries 1 and 3, respectively). These results show a significant effect of the HCl/amide mole ratio on the rate of PA depolymerization. Namely, HCl not only serves as a catalyst for the PA hydrolysis reaction, where it protonates the amide groups to facilitate the attack of water on the carbonyl carbon, but also is consumed during depolymerization for the protonation of the formed amine groups. Therefore, ensuring a sufficient amount of acid is crucial for complete PA degradation, and in this respect, HCl is a suitable acid due to its accessibility and low cost. For complete PA 66 degradation, 1.25 excess of HCl per amide group was used. After 5 min reaction time at 200 °C, only a trace amount of oligomers was still visible in the 1H NMR spectrum of this reaction mixture, and a complete PA 66 degradation was observed after 10 min (Table 2; entries 12 and 13). Two times diluted acid solution at a reduced sample amount, and thus an unchanged HCl/amide mole ratio (1.25), resulted in a comparable degree of PA 66 degradation after reaction times of 5 and 10 min (Table 2; entries 14 and 15, Figure 1), indicating efficient PA depolymerization even in more diluted acid.
Figure 1.
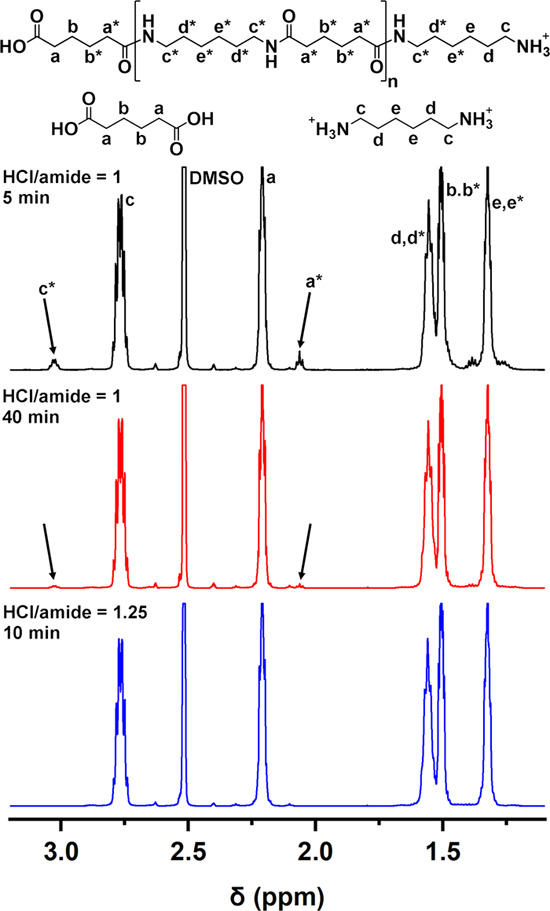
1H NMR spectra of dried reaction mixtures after depolymerization of PA 66 at 200 °C and different mole ratios of HCl to the amide bonds after different reaction times. Methylene groups of monomers and oligomers are assigned in the spectra (denoted a-e and a*-e*). Arrows indicate nonoverlapping methylene groups (a* and c*) specific for oligomers.
Table 2. Reaction Conditions for Microwave-Assisted, Acid-Catalyzed Hydrolysis of Aliphatic PAs in the Presence and Absence of Reinforcement Additivesa.
| irradiation
conditions |
|||||||
|---|---|---|---|---|---|---|---|
| entry | sample | m(PA) (g) | 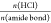 |
c(HCl) (g/mol) | T (°C) | t (min) | degree of conversion (%) |
| 1 | PA 66 | 5.03 | 0.5 | 2.27 | 200 | 5 | 50.8 |
| 2 | 20 | 54.1 | |||||
| 3 | 40 | 54.7 | |||||
| 4 | 5.03 | 1 | 5.56 | 200 | 5 | 95.6 | |
| 5 | 20 | 98.3 | |||||
| 6 | 40 | 99.1 | |||||
| 7 | 4.00 | 1.25 | 5.56 | 170 | 3.5b | 80.9 | |
| 8 | 5 | 87.9 | |||||
| 9 | 10 | 95.2 | |||||
| 10 | 20 | 99.4 | |||||
| 11 | 30 | 100 | |||||
| 12 | 200 | 5 | 99.9 | ||||
| 13 | 10 | 100 | |||||
| 14 | 2.00 | 1.25 | 2.27 | 200 | 5 | 99.7 | |
| 15 | 10 | 100 | |||||
| 16 | PA 66-GF35 | 6.18 | 1.25 | 5.56 | 170 | 60 | 95.2 |
| 17 | 3.09 | 2.27 | 200 | 10 | 99.8 | ||
| 18 | 15 | 100 | |||||
| 19 | 4.00 | 1.92 | 5.56 | 200 | 10 | 100 | |
| 20 | PA 11 | 3.27 | 1.25 | 2.27 | 200 | 5 | 64.9 |
| 21 | 20 | 94.9 | |||||
| 22 | 40 | 100 | |||||
| 23 | 3.27 | 2.5 | 5.56 | 200 | 5 | 92.7 | |
| 24 | 6b | 98.2 | |||||
| 25 | 7.5 | 99.3 | |||||
| 26 | 10 | 100 | |||||
| 27 | PA 11-GF30 | 2.33 | 2.5 | 2.27 | 200 | 10 | 99.6 |
| 28 | 15 | 100 | |||||
| 29 | PA 11-CF30 | 2.33 | 2.5 | 2.27 | 200 | 10 | 98.6 |
| 30 | 15 | 99.5 | |||||
| 31 | 20 | 100 | |||||
| 32 | PA 12 | 3.51 | 2.5 | 5.56 | 200 | 5 | 86.9 |
| 33 | 6.5b | 98.7 | |||||
| 34 | 10 | 100 | |||||
| 35 | PA 12-GF50 | 3.51 | 5 | 5.56 | 200 | 10 | 99.4 |
| 36 | 15 | 100 | |||||
| 37 | PA 1010 | 3.00 | 2.5 | 5.56 | 200 | 7.5b | 94.7 |
| 38 | 10 | 96.2 | |||||
| 39 | 15 | 99.7 | |||||
| 40 | 17 | 100 | |||||
Volume of the HCl solution was 8 mL in all experiments.
Reaction time when all pellets were dissolved at the selected reaction temperature.
Next, the effect of temperature on PA 66 depolymerization was studied at 1.25 mol equiv of HCl per amide bond. In contrast to PA 66 degradation at 200 °C, where the pellets completely dissolved when the reaction temperature had been reached (reaction time 0 min), it took 3.5 min for complete dissolution of PA 66 pellets at 170 °C, where 80.9% degree of degradation was determined (Table 2; entry 7). Complete hydrolysis of PA 66 at 170 °C was achieved in 30 min (Table 2; entry 11). 1H NMR results are supported by HPLC results, where the chromatograms of incompletely hydrolyzed reaction mixtures show, beside the characteristic peaks of AA and HMDA, also the additional ones corresponding to the PA 66 oligomers, the intensity of which decreases with prolonged depolymerization time (Figure 2). The presence of oligomers in the final reaction products complicates the isolation of the AA monomer because oligomers coprecipitate with AA upon cooling. Due to this reason, a complete degradation of PA 66 to the constituent monomers is essential. Furthermore, even at a prolonged reaction time of 60 min at 170 or 200 °C, no additional signals were observed in 1H NMR spectra of reaction products, indicating straightforward PA depolymerization into the constituent monomers without formation of any side products (e.g., cycloheptylamine, cyclopentanone, alkene), which are commonly formed during depolymerization of PA 66 or PA 12 in supercritical fluids.15,19−21
Figure 2.
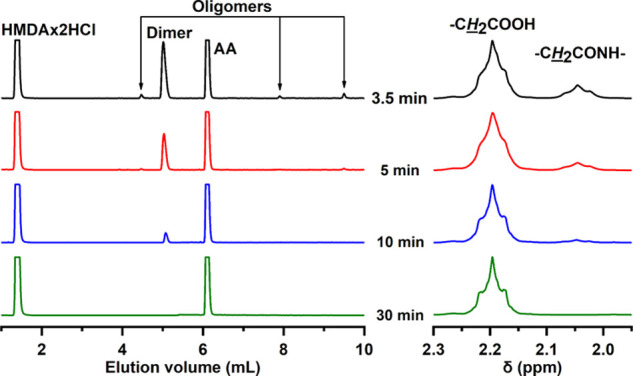
(Left) HPLC chromatograms and (right) enlarged 1H NMR spectra of dried reaction mixtures obtained by acid-catalyzed hydrolysis of PA 66 under microwave irradiation at 170 °C and a 1.25 HCl/amide mole ratio after different reaction times.
Microwave-assisted, acid-catalyzed hydrolysis was also applied for the degradation of long-chain aliphatic PAs (PA 11, PA 12, and PA 1010). PA 11 and PA 12 are synthesized from 11-AUDA and 12-ADDA amino acids, respectively, while PA 1010 is synthesized from long-chain diacid (SA) and diamine (1,10-DDA). PA 11 was first depolymerized under the same experimental conditions as they were determined to be optimal for PA 66 (Table 2; entry 15). Compared to PA 66, the depolymerization of PA 11 was significantly slower as indicated by only 64.9% degree of degradation after 5 min reaction time at 200 °C (Table 2; entry 20), whereby solid pellets were still present in a turbid reaction mixture. Full degradation of PA 11 was achieved after 40 min (Table 2; entry 22), which is a four times longer reaction time than that necessary to completely degrade PA 66. For complete hydrolysis of long-chain PAs into the constituent monomers within a comparable reaction time as it was needed for PA 66, larger equivalents of acid to the amide bonds were used. At the HCl/amide mole ratio of 2.5 instead of 1.25, only 10 min reaction time at 200 °C was necessary to completely depolymerize PA 11 (Table 2, entry 26, Figure 3) and PA 12, while PA 1010 required 17 min (Table 2; entries 34 and 40, respectively). The ability of a particular PA to depolymerize into the constituent monomer(s) depends mainly on the length of the alkyl segments between the adjacent amide bonds since it defines the PA polarity and thus the effectiveness of PA hydration by water.8,30 Generally, longer alkyl segments between the amide bonds result in less polar PAs, which are more resistant to chemicals and have a lower tendency toward hydrolysis. Other important parameters are the Tm, degree of crystallinity, and density of PA, which are defined by the ability of PA to form intermolecular hydrogen bonds between amide groups.8 Because of the low density of long-chain PAs, the pellets float on the top of reaction mixtures, while on the other hand, PA 66 pellets of higher density sink to the bottom of the microwave vial. For this reason, effective stirring of the reaction mixtures during PA hydrolysis is essential. Longer reaction times needed for complete conversion of long-chain PAs are also connected with the longer times required for the pellets of PA 11, PA 12, and PA 1010 (Table 2, entries 24, 33, and 37) to completely dissolve. Slower degradation of long-chain PAs is most likely also associated with a poor solubility of oligomers that remain on the surface of the pellets until more soluble monomer is formed. The depolymerization of long-chain PAs therefore proceeds mainly on the interface between the pellets and water, while in the case of PA 66 soluble oligomers depolymerize mainly in solution.
Figure 3.
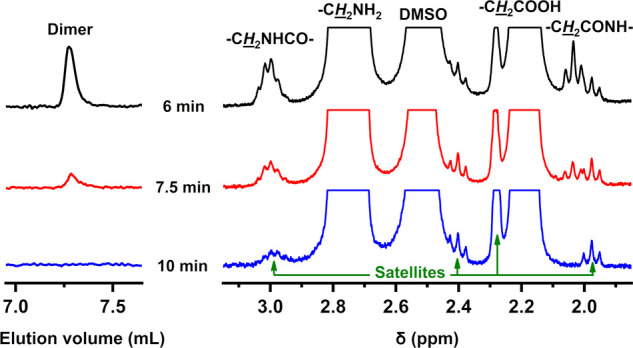
(Left) Enlarged HPLC chromatograms and (right) enlarged 1H NMR spectra of dried reaction mixtures obtained by acid-catalyzed hydrolysis of PA 11 under microwave irradiation at 200 °C and a 2.5 HCl/amide mole ratio after different reaction times.
Because one of the goals of chemical recycling is to recycle the composite materials,31 we investigated the impact of reinforcement additives (glass- and carbon-fibers) on the PA depolymerization process. A series of experiments on PA 66-GF35 was performed at 200 °C. At optimal experimental conditions for neat PA 66 (Table 2; entry 15), some oligomers were still present in the reaction mixture of PA 66-GF35 after 10 min reaction time (Table 2; entry 17), indicating slower depolymerization of PA 66 in the presence of glass-fibers, most probably due to poorer PA solvation. Complete hydrolysis of PA 66-GF35 at 200 °C was achieved after 15 min (Table 2; entry 18). Similar results were observed for PA 11-based composites (PA 11-GF30 and PA 11-CF30), where the reaction time for full conversion of PA 11 to 11-AUDAxHCl at 200 °C and a 2.5 HCl/amide mole ratio had to be prolonged from 10 min, as optimal for neat PA 11 (Table 2; entry 26), to 15 and 20 min, respectively (Table 2; entries 28 and 31). Faster depolymerization of PA 66-GF35 was observed at a higher HCl/amide mole ratio (Table 2; entry 19). Higher contents of reinforcement additives in PA-based composites further slowed down the PA depolymerization. For example, it took 15 min to completely hydrolyze PA 12-GF50 at 200 °C although the HCl/amide mole ratio had been increased to 5 (Table 2; entry 36). These results reveal great influence of glass-fiber and especially carbon-fiber reinforcement additives on the PA depolymerization process because extended reaction time in combination with a higher mole excess of acid to the amide bonds is necessary for complete hydrolysis of PA composites compared to the optimal reaction conditions, allowing for complete conversion of neat PA analogues to the constituent monomers.
Isolation and Purification of Recovered Monomers
The monomers were isolated in high yields from the fully degraded PA reaction mixtures obtained by microwave-assisted hydrolysis under the optimal reaction conditions (Table 3). PA 66 and PA 1010 depolymerize by acid-catalyzed hydrolysis into dicarboxylic acid and diamine dihydrochloride. Therefore, the isolation and purification procedures of both monomers were similar. AA and HMDAx2HCl recovered from PA 66 as well as SA and 1,10-DDAx2HCl recovered from PA 1010 were isolated sequentially from the completely depolymerized reaction mixtures. AA and SA precipitated from the acidic solutions of hydrolysates during cooling to room temperature. Further precipitation of AA and SA was enhanced by cooling down the reaction mixtures to 0 °C, whereby HMDA and 1,10-DDA in the form of salts were still completely soluble. The precipitated AA and SA were isolated by filtration. Complete removal of HMDA from AA and 1,10-DDA from SA was achieved by two sequential recrystallizations of diacids from water (Table 3). HMDA and 1,10-DDA were isolated from the solutions obtained after AA and SA filtration, respectively. The filtrates had been concentrated before NaOH was added to the solutions to deprotonate the amine groups. HMDA and 1,10-DDA were isolated by vacuum distillation. Complete hydrolysis of PA 11 and PA 12 results in hydrochloric salts of the amino acid monomers, which were simply isolated by filtration. The isolated monomers (AA, SA, 12-ADDAxHCl, 11-AUDAxHCl) were further purified by recrystallization from water to reach the quality comparable to that of commercially available monomers.
Table 3. Optimized Reaction Conditions for Complete Microwave-Assisted, Acid-Catalyzed Hydrolysis of Aliphatic PAs Together with Yields, Tm, and Purities of Recovered Monomers and Reinforcement Additivesa.
| MW conditions | recovered
monomer/reinforcement additive |
|||||||
|---|---|---|---|---|---|---|---|---|
| entry | sample | 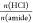 |
T (°C) | t (min) | yield (%) | Tm (°C)b | purity (%)c | |
| 1 | PA 66 | 1.25 | 200 | 10 | AA | 90 | 152.6–153.3 | 100.6 ± 0.2 |
| HMDA | 86 | 100.1 ± 0.3 | ||||||
| 2 | PA 66-GF35 | 1.25 | 200 | 15 | AA | 83 | 152.3–152.9 | 100.7 ± 0.2 |
| HMDA | 81 | 100.1 ± 0.4 | ||||||
| GF | 97 | |||||||
| 3 | PA 11 | 2.5 | 200 | 10 | 11-AUDAxHCl | 93 | 147.3–148.7 | 100.1 ± 0.2 |
| 4 | PA 11-GF30 | 2.5 | 200 | 15 | 11-AUDAxHCl | 71 | 147.0–148.6 | 99.8 ± 0.1 |
| GF | 97 | |||||||
| 5 | PA 11-CF30 | 2.5 | 200 | 20 | 11-AUDAxHCl | 72 | 146.7–147.0 | 99.3 ± 0.2 |
| CF | 99 | |||||||
| 6 | PA 12 | 2.5 | 200 | 10 | 12-ADDAxHCl | 97 | 160.9–163.5 | 99.9 ± 0.5 |
| 7 | PA 12-GF50 | 5 | 200 | 15 | 12-ADDAxHCl | 77 | 159.6–162.5 | 97.1 ± 0.3 |
| GF | 97 | |||||||
| 8 | PA 1010 | 2.5 | 200 | 17 | SA | 89 | 133.1–134.0 | 100.4 ± 0.8 |
| 1,10-DDA | 78 | 60.4–61.0 | 100.0 ± 0.2 | |||||
Volume of HCl solution was 8 mL in all experiments.
Tm of commercially available monomers: AA: 152.4–153.3 °C; SA: 133.1–134.0 °C; 1,10-DDA: 61.2–62.1 °C; 11-AUDAxHCl: 146.3–149.7 °C; 12-ADDAxHCl: 160.7–163.5 °C.
HPLC purity was determined comparatively to commercial monomers.
High purity of all recovered monomers was confirmed by 1H and 13C NMR spectra, where no additional signals indicating the presence of side products were detected, as well as by FTIR spectra and determination of Tm, which are in good agreement with those of commercial monomers (Figures S2–S19). High purity of the monomers was further confirmed by HPLC using an external calibration method based on commercial monomers (Table 3).
The isolation of recovered monomer(s) from the hydrolysates of PA composites was similar, except for an additional step, in which the reinforcement additives (carbon- or glass-fibers) were removed immediately after reaction completion by filtration of hot reaction mixtures containing completely soluble monomer(s). Amino acid and dicarboxylic acid monomers recovered from PA composites were after recrystallization from water, additionally recrystallized from EtOH to remove specific additives present in the composite samples. In this way, the purity of recycled monomers was improved by about 1–2% at the expense of lower yields of recovered monomers (by ∼15%, Table 3). The absence of side products and additives in the monomers recovered from the PA-based composites was assessed from FTIR and NMR spectra (Figures S20–S34), while their high purity was confirmed by HPLC and determination of Tm, which match those of commercially available chemicals (Table 3). Furthermore, the reinforcement additives were successfully recovered as indicated by SEM (Figure 4) in high yields (above 97%). High inorganic content (above 97%) of the recovered glass-fibers as well as carbon-fibers from the PA composites was confirmed by TGA and FTIR (Figures S35 and S36), indicating a possibility to reuse them in further applications.
Figure 4.
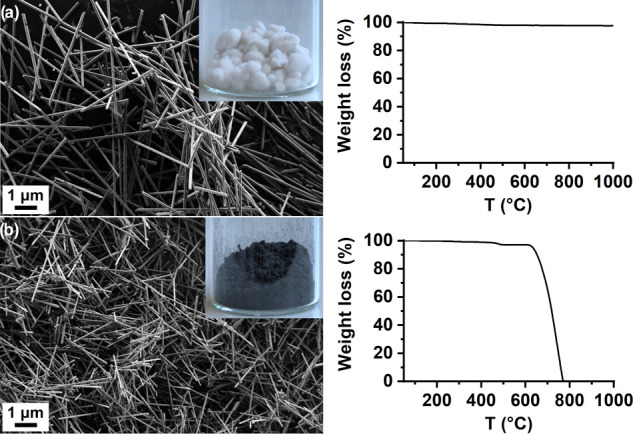
SEM images and TGA curves of (a) glass-fibers recovered from PA 11-GF30, and (b) carbon-fibers recovered from PA 11-CF30.
Conclusions
The efficient and simple depolymerization procedure for green chemical recycling of aliphatic PAs is disclosed. Hydrolysis in the presence of HCl as a catalyst under microwave irradiation converts PAs into the constituent monomers in the absence of any side-product formation. In this process, PAs were completely hydrolyzed at 200 °C in a relatively short time (less than 20 min), even in the presence of reinforcement additives (glass- or carbon-fibers). The rate of hydrolysis reaction depends on the PA type (the length of the alkyl segments between the adjacent amide bonds), the HCl/amide mole ratio, the presence of reinforcement additives, as well as their type and content. At the same reaction conditions, less polar long-chain PAs depolymerize slower than short-chain PAs; however, the rate of hydrolysis reaction can be accelerated using a higher mole ratio of HCl to the amide bonds in the PA backbone. Uncomplicated and efficient procedures for isolation and purification of recovered monomers resulted in high yields, especially for PAs synthesized from amino acid monomers. The recovered monomers are of high quality, comparable to that of the commercial-grade raw materials. The disclosed process also has a great potential for recycling the reinforcement additives and thus meets the requirements of circular economy.
Acknowledgments
This project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement no. 820665 (PolynSPIRE). The authors acknowledge the financial support from the Slovenian Research Agency (Research Core Funding no. P2-0145) and thank Arkema and Kordsa for providing PA samples.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssuschemeng.0c05706.
1H NMR, 13C NMR, and FTIR spectra of recovered monomers and FTIR spectra of recovered reinforcement additives (glass- and carbon-fibers) (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Garcia J. M.; Robertson M. L. The Future of Plastics Recycling. Science 2017, 358, 870–872. 10.1126/science.aaq0324. [DOI] [PubMed] [Google Scholar]
- Okan M.; Aydin H. M.; Barsbay M. Current Approaches to Waste Polymer Utilization and Minimization: A Review: Current Approaches to Waste Polymer Utilization and Minimization. J. Chem. Technol. Biotechnol. 2019, 94, 8–21. 10.1002/jctb.5778. [DOI] [Google Scholar]
- Ragaert K.; Delva L.; Van Geem K. Mechanical and Chemical Recycling of Solid Plastic Waste. Waste Manage. 2017, 69, 24–58. 10.1016/j.wasman.2017.07.044. [DOI] [PubMed] [Google Scholar]
- Coates G. W.; Getzler Y. D. Y. L. Chemical Recycling to Monomer for an Ideal, Circular Polymer Economy. Nat. Rev. Mater. 2020, 10.1038/s41578-020-0190-4. [DOI] [Google Scholar]
- Datta J.; Kopczyńska P. From Polymer Waste to Potential Main Industrial Products: Actual State of Recycling and Recovering. Crit. Rev. Environ. Sci. Technol. 2016, 46, 905–946. 10.1080/10643389.2016.1180227. [DOI] [Google Scholar]
- Garcia-Manyes S.; Beedle A. E. M. Steering Chemical Reactions with Force. Nat. Rev. Chem. 2017, 1, 0083 10.1038/s41570-017-0083. [DOI] [Google Scholar]
- Hong M.; Chen E. Y.-X. Chemically Recyclable Polymers: A Circular Economy Approach to Sustainability. Green Chem. 2017, 19, 3692–3706. 10.1039/C7GC01496A. [DOI] [Google Scholar]
- Herzog B.; Kohan M. I.; Mestemacher S. A.; Pagilagan R. U.; Redmond K.. Polyamides. Ullmann’s Encyclopedia of Industrial Chemistry; Barbara E.Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp 697–732. [Google Scholar]
- Achilias D. S.; Andriotis L.; Koutsidis I. A.; Louka D. A.; Nianias N. P.; Siafaka P.; Tsagkalias I.; Tsintzou G.. Recent Advances in the Chemical Recycling of Polymers (PP, PS, LDPE, HDPE, PVC, PC, Nylon, PMMA). In Material Recycling – Trends and Perspectives; Achilias D., Ed.; InTech, 2012; pp 3–64. [Google Scholar]
- Mihut C.; Captain D. K.; Gadala-Maria F.; Amiridis M. D. Review: Recycling of Nylon from Carpet Waste. Polym. Eng. Sci. 2001, 41, 1457–1470. 10.1002/pen.10845. [DOI] [Google Scholar]
- Emik S.; Yılmaz B. Y.; İyim T. B. Investigation of the Usage of Depolymerized Nylon 66 Intermediate in Phenolic Resin Modification. Polym-Plast. Technol. Mater. 2019, 58, 454–463. 10.1080/03602559.2018.1482920. [DOI] [Google Scholar]
- Chen J.; Liu G.; Jin L.; Ni P.; Li Z.; He H.; Xu Y.; Zhang J.; Dong J. Catalytic Hydrolysis of Waste Nylon 6 to Produce ε-Caprolactam in Sub-Critical Water. J. Anal. Appl. Pyrolysis 2010, 87, 50–55. 10.1016/j.jaap.2009.10.004. [DOI] [Google Scholar]
- Wang W.; Meng L.; Huang Y. Hydrolytic Degradation of Monomer Casting Nylon in Subcritical Water. Polym. Degrad. Stab. 2014, 110, 312–317. 10.1016/j.polymdegradstab.2014.09.014. [DOI] [Google Scholar]
- Goto M.; Umeda M.; Kodama A.; Hirose T.; Nagaoka S. Monomerization of Nylon 6 in Sub- and Supercritical Water. Kobunski Ronbunsku 2001, 58, 548–551. 10.1295/koron.58.548. [DOI] [Google Scholar]
- Meng L.; Zhang Y.; Huang Y.; Shibata M.; Yosomiya R. Studies on the Decomposition Behavior of Nylon-66 in Supercritical Water. Polym. Degrad. Stab. 2004, 83, 389–393. 10.1016/j.polymdegradstab.2003.08.001. [DOI] [Google Scholar]
- Kamimura A.; Oishi Y.; Kaiso K.; Sugimoto T.; Kashiwagi K. Supercritical Secondary Alcohols as Useful Media To Convert Polyamide into Monomeric Lactams. ChemSusChem 2008, 1, 82–84. 10.1002/cssc.200700024. [DOI] [PubMed] [Google Scholar]
- Hommez B.; Goethals E. J. Degradation of Nylon-6 By Glycolysis. Part 1: Identification of Degradation Products. J. Macromol. Sci., Part A: Pure Appl.Chem.: Pure Appl.Chem. 1998, 35, 1489–1505. 10.1080/10601329808007312. [DOI] [Google Scholar]
- Datta J.; Błażek K.; Włoch M.; Bukowski R. A New Approach to Chemical Recycling of Polyamide 6.6 and Synthesis of Polyurethanes with Recovered Intermediates. J. Polym. Environ. 2018, 26, 4415–4429. 10.1007/s10924-018-1314-4. [DOI] [Google Scholar]
- Kamimura A.; Kaiso K.; Suzuki S.; Oishi Y.; Ohara Y.; Sugimoto T.; Kashiwagi K.; Yoshimoto M. Direct Conversion of Polyamides to ω-Hydroxyalkanoic Acid Derivatives by Using Supercritical MeOH. Green Chem. 2011, 13, 2055–2061. 10.1039/c1gc15172j. [DOI] [Google Scholar]
- Kamimura A.; Ikeda K.; Suzuki S.; Kato K.; Akinari Y.; Sugimoto T.; Kashiwagi K.; Kaiso K.; Matsumoto H.; Yoshimoto M. Efficient Conversion of Polyamides to ω-Hydroxyalkanoic Acids: A New Method for Chemical Recycling of Waste Plastics. ChemSusChem 2014, 7, 2473–2477. 10.1002/cssc.201402125. [DOI] [PubMed] [Google Scholar]
- Matsumoto H.; Akinari Y.; Kaiso K.; Kamimura A. Efficient Depolymerization and Chemical Conversion of Polyamide 66 to 1,6-Hexanediol. J. Mater. Cycles Waste Manage. 2017, 19, 326–331. 10.1007/s10163-015-0425-4. [DOI] [Google Scholar]
- Kalfas G. A. Mathematical Modeling of the Depolymerization of Polyamide Mixtures - Part I: Kinetic Mechanism and Parametric Studies in Batch Reactors. Polym. React. Eng. 1998, 6, 41–67. 10.1080/10543414.1998.10744482. [DOI] [Google Scholar]
- Duch M. W.; Allgeier A. M. Deactivation of Nitrile Hydrogenation Catalysts: New Mechanistic Insight from a Nylon Recycle Process. Appl. Catal., A 2007, 318, 190–198. 10.1016/j.apcata.2006.11.003. [DOI] [Google Scholar]
- Dallinger D.; Kappe C. O. Automated Generation of a Dihydropyrimidine Compound Library Using Microwave-Assisted Processing. Nat. Protoc. 2007, 2, 1713–1721. 10.1038/nprot.2007.224. [DOI] [PubMed] [Google Scholar]
- Klun U.; Kržan A. Rapid Microwave Induced Depolymerization of Polyamide-6. Polymer 2000, 41, 4361–4365. 10.1016/S0032-3861(99)00658-8. [DOI] [Google Scholar]
- Alberti C.; Figueira R.; Hofmann M.; Koschke S.; Enthaler S. Chemical Recycling of End-of-Life Polyamide 6 via Ring Closing Depolymerization. ChemistrySelect 2019, 4, 12638–12642. 10.1002/slct.201903970. [DOI] [Google Scholar]
- Kamimura A.; Shiramatsu Y.; Kawamoto T. Depolymerization of Polyamide 6 in Hydrophilic Ionic Liquids. Green Energy Environ. 2019, 4, 166–170. 10.1016/j.gee.2019.01.002. [DOI] [Google Scholar]
- Žagar E.; Češarek U.; Drinčić A.; Sitar S.; Shlyapnikov I. M.; Pahovnik D. Quantitative Determination of PA6 and/or PA66 Content in Polyamide-Containing Wastes. ACS Sustainable Chem. Eng. 2020, 10.1021/acssuschemeng.0c04190. [DOI] [Google Scholar]
- Shukla S. R.; Harad A. M.; Mahato D. Depolymerization of Nylon 6 Waste Fibers. J. Appl. Polym. Sci. 2006, 100, 186–190. 10.1002/app.22775. [DOI] [Google Scholar]
- Biron M.Biobricks: The Breakthrough of Drop-In Solutions. Renewable Polyamides. In Industrial Applications of Renewable Plastics: Environmental, Technological, and Economic Advances; Elsevier Science: Oxford, 2016; pp 214–235. [Google Scholar]
- Oliveux G.; Dandy L. O.; Leeke G. A. Current Status of Recycling of Fibre Reinforced Polymers: Review of Technologies, Reuse and Resulting Properties. Prog. Mater. Sci. 2015, 72, 61–99. 10.1016/j.pmatsci.2015.01.004. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.