

Abstract
With five histone deacetylase (HDAC) inhibitors approved for cancer treatment, proteolysis-targeting chimeras (PROTACs) for degradation of HDAC are emerging as an alternative strategy for HDAC-targeted therapeutic intervention. Herein, three bestatin-based hydroxamic acids (P1, P2 and P3) were designed, synthesized and biologically evaluated to see if they could work as HDAC degrader by recruiting cellular inhibitor of apoptosis protein 1 (cIAP1) E3 ubiquitin ligase. Among the three compounds, the bestatin-SAHA hybrid P1 exhibited comparable even more potent inhibitory activity against HDAC1, HDAC6 and HDAC8 relative to the approved HDAC inhibitor SAHA. It is worth noting that although P1 could not lead to intracellular HDAC degradation after 6 h of treatment, it could dramatically decrease the intracellular levels of HDAC1, HDAC6 and HDAC8 after 24 h of treatment. Intriguingly, the similar phenomenon was also observed in the HDAC inhibitor SAHA. Cotreatment with proteasome inhibitor bortezomib could not reverse the HDAC decreasing effects of P1 and SAHA, confirming that their HDAC decreasing effects were not due to protein degradation. Moreover, all three bestatin-based hydroxamic acids P1, P2 and P3 exhibited more potent aminopeptidase N (APN, CD13) inhibitory activities than the approved APN inhibitor bestatin, which translated to their superior anti-angiogenic activities. Taken together, a novel bestatin-SAHA hybrid was developed, which worked as a potent APN and HDAC dual inhibitor instead of a PROTAC.
Keywords: hybrid, dual inhibitor, CD13, histone deacetylase, aminopeptidase
1. Introduction
Histone deacetylases (HDACs) can catalyze the removal of acetyl group from lysine residues of histones or some non-histone proteins, therefore playing an essential role in the regulation of gene expression and protein function [1]. To date, five HDAC inhibitors have been approved as anticancer agents [2]. Beyond cancer, HDAC inhibitors are also considered a class of potential therapeutic intervention for neurological disorders, immune diseases, cardiovascular diseases as well as age-related disorders [3,4,5].
An alternative strategy for inhibiting the biological function of proteins is promoting degradation of the target proteins, such as proteolysis-targeting chimeras (PROTACs), a newly developed technique to specifically knockdown the proteins of interest using the cellular ubiquitin-proteasome system [6,7,8,9]. In the aspect of chemical structure, PROTACs are generally bifunctional molecules consisting of three parts: a warhead that binds the protein of interest, an E3-ligase recruiting moiety and a linker that connects the above two parts. In the aspect of biological function, PROTACs can bring the protein of interest and E3-ligase to be adjacent to each other, then E3-ligase facilitates the transfer of ubiquitin to the target protein, consequently leading to the degradation of the ubiquitinated target protein by proteasome [6,7,8,9]. Thus far, the most widely used E3 ligases are cereblon (CRBN) and von Hippel-Lindau (VHL) [8,9]. In the research field of PROTACs for HDAC degradation, several effective degraders recruiting CRBN (compounds 1, 2, 3 and 4) or VHL (compounds 5 and 6) have already been successfully developed, among which PROTACs 1–5 are all HDAC6 degraders, while PROTAC 6 can effectively degrade HDAC1, HDAC2 and HDAC3 (Figure 1) [10,11,12,13,14,15].
Figure 1.

The structures of reported histone deacetylase (HDAC) degraders. The HDAC binding parts are in red, the linker parts are in black and the cereblon (CRBN) and von Hippel-Lindau (VHL) ligands are in blue and green, respectively.
Besides CRBN and VHL, cellular inhibitor of apoptosis protein 1 (cIAP1) is another important E3 ligase used in the design of PROTACs [16,17,18,19,20,21,22]. The PROTACs recruiting the IAPs E3 ligases were also named SNIPERs, which was the abbreviation of specific and nongenetic IAPs-dependent protein eraser [17]. Bestatin ester and bestatin amide were the most extensively investigated cIAP1 ligands in the design of SNIPERs as shown in Figure 2. To be specific, compounds 7 and 8 are both cellular retinoic acid-binding proteins (CRABPs) degraders recruiting cIAP1 [16,17]. Compounds 9, 10 and 11 can lead to the degradation of retinoic acid receptor α (RARα), androgen receptor (AR) and estrogen receptor α (ERα), respectively [18]. Compound 12 is also an ERα degrader [19]. However, to the best of our knowledge, no HDAC degrader recruiting cIAP1 has been reported to date.
Figure 2.

The structures of reported bestatin-based SNIPERs. The target protein binding parts are in red, the linker parts are in black and the bestatin ester and bestatin amide are in green and blue, respectively.
To develop HDAC degrader recruiting cIAP1, we herein designed and synthesized three bestatin-based hydroxamic acids (P1, P2 and P3). As shown in Figure 3, compound P1 was a hybrid molecule of bestatin and the approved pan-HDAC inhibitor SAHA. Compounds P2 and P3 were designed based on the structural feature of two reported HDAC6/8 dual inhibitors with very low molecular weights [23]. All three target compounds contained the bestatin amide scaffold as the cIAP1 recruiting ligand and the hydroxamic acid-based warhead to target HDAC. Biological studies revealed that although the bestatin-SAHA hybrid P1 possessed potent inhibitory activities against HDAC1, HDAC6 and HDAC8, none of the three HDAC isoforms could be degraded by P1 after 6 h of treatment, probably due to the inappropriate length of the linker connecting bestatin and SAHA in P1. Intriguingly, both P1 and SAHA could lead to a proteasome-independent decrease of the intracellular levels of HDAC1, HDAC6 and HDAC8 after 24 h of treatment. Another interesting result was that P1, P2 and P3 all exhibited potent aminopeptidase N (APN) inhibitory activities and anti-angiogenic activities, which were even better than those of the approved APN inhibitor bestatin.
Figure 3.

Design of bestatin-based hydroxamic acids.
2. Results and Discussion
2.1. Chemistry
The synthesis of target compound P1 was described in Scheme 1. Boc-protection of the starting material 13 led to compound 14. Condensation of 15 with N-Boc-p-phenylenediamine using the coupling reagent EDCI/HOBT led to 16. Then, Boc-deprotection in acidic condition gave intermediate 17, which was condensed with suberic acid monomethyl ester to afford compound 18. The deprotection of compound 18 was conducted using ammonium formate as reductive reagent, given to compound 19, which coupled with compound 14 to afford intermediate 20. The methyl ester group of 20 was transformed into hydroxamate moiety with freshly prepared NH2OK to give compound 21. Finally, Boc-deprotection in the presence of CF3COOH gave the target product P1.
Scheme 1.

Reagents and conditions: (a) (Boc)2O, DIPEA, DCM, 83%; (b) N-Boc-p-phenylenediamine, EDCI, HOBT, DCM, 85%; (c) CF3COOH, DCM, 95% for 17, 32% for P1; (d) suberic acid monomethyl ester, EDCI, HOBT, DMF, 80 °C, 62%; (e) 10% Pd/C, ammonium formate, MeOH, THF, 65 °C, 92%; (f) 14, EDCI, HOBT, DMF, 80 °C, 58%; (g) NH2OK/MeOH, 65%.
The synthesis of target compound P2 was illustrated in Scheme 2. Firstly, Boc-protection of compound 22, followed by condensation with 3-(methoxycarbonyl)benzoic acid, led to compound 23. Boc-deprotection of compound 23 using HCl/dioxane led to compound 24. After that, compound 24 coupled with compound 14 to give intermediate 25. Then compound 25 reacted with freshly prepared NH2OK to give compound 26. Finally, Boc-deprotection using HCl/dioxane afforded the target product P2.
Scheme 2.

Reagents and conditions: (a) 1) (Boc)2O, DIPEA, DCM, 66%; 2) 3-(methoxycarbonyl)benzoic acid, HATU, DIPEA, DMF, 88%; (b) HCl/dioxane, 11% for P2; (c) 14, HATU, DIPEA, DMF, 82%; (d) NH2OK/MeOH.
The synthesis of target compound P3 was described in Scheme 3. The starting compound 27 went through Mitsunobu reaction to give compound 28, which then was oxidized to give the carboxylic acid derivatives 29. Then, compound 29 went through hydrazinolysis and Boc-protection to give compound 30. 2-amino benzoic acid 31 coupled with O-benzylhydroxylamine hydrochloride gave compound 32, which went through condensation, Boc-deprotection and condensation to give compound 34. Finally, the Cbz and Boc groups were deprotected to give the target product P3.
Scheme 3.

Reagents and conditions: (a) isoindoline-1,3-dione, PPh3, DIAD, THF, 57%; (b) PhI(AcO)2, TEMPO, CN/H2O, 70 °C; (c) 1) N2H4.H2O, EtOH, 80 °C; 2) (Boc)2O, NaOH, THF; yield: 41% for two steps; (d) O-benzylhydroxylamine hydrochloride, HATU, DIPEA, DMF, 71%; (e) 1) 30, HATU, DIPEA, DMF; 2) HCl/dioxane; (f) 14, HATU, DIPEA, DMF, 50%; (g) 1) 10% Pd/C, H2, MeOH; 2) HCl/dioxane; yield: 8% for two steps.
2.2. In Vitro HDACs Inhibitory Assay
Based on our compound design strategy, we believed that compound P1 could retain the potent pan-HDAC inhibitory activity of SAHA and hoped that P2 and P3 could exhibit HDAC6 and HDAC8 selective profiles. The HDAC inhibitory potency of P1, P2 and P3 was preliminarily evaluated by determining their inhibitory rates against HDAC1/6/8 at a concentration of 10 μM. The results in Table 1 showed that P1 exhibited promising pan-HDAC inhibitory potency with inhibitory rates of over 90% against all three tested HDAC isoforms. However, the HDAC inhibitory rates of compounds P2 and P3 were relatively low, and no obvious isoform selectivity was observed. According to the preliminary results, only P1 was progressed to the determination of IC50. As shown in Table 1, the IC50 values of P1 against HDAC1/6/8 were all lower than those of the positive control SAHA, confirming the potent HDAC inhibitory activity of P1, especially against HDAC1 (IC50 = 30.0 ± 2.8 nM) and HDAC6 (IC50 = 11.5 ± 0.71 nM). The higher potency of P1 compared to SAHA could just come from the capping effect brought by the additional bestatin moiety.
Table 1.
HDAC1/6/8 inhibitory activities of compounds P1, P2, P3 and SAHA.
| Compounds | Inhibitory Rates at 10 μM (IC50) a | ||
|---|---|---|---|
| HDAC1 | HDAC6 | HDAC8 | |
| P1 | 98% (30.0 ± 2.8 nM) | 99% (11.5 ± 0.71 nM) | 90% (694.0 ± 110.3 nM) |
| P2 | 66% | 76% | 64% |
| P3 | 67% | 71% | 51% |
| SAHA | 63.5 ± 3.5 nM | 21.0 ± 1.4 nM | >7000 nM |
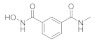
|
NT b | 2.5 ± 1.6 μM c | 14 ± 10 μM c |
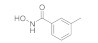
|
NT b | 0.65 ± 0.52 μM c | 3.3 ± 1.7 μM c |
a Assays were performed in replicate (n ≥ 3). The IC50 values were shown as mean ± SD; b NT = not determined; c Reported in [23].
2.3. Western Blot Analysis
Considering its potent HDAC1 and HDAC6 inhibitory activity, compound P1 was further evaluated by western blot analysis to see if it could promote the degradation of HDAC1 and HDAC6 in human multiple myeloma RPMI-8226 cells. As shown in Figure 4A, compound P1 did not lead to HDAC1 or HDAC6 depletion at the concentration up to 20 μM after a 6-hour treatment, though it showed effective intracellular target engagement evidenced by the increased levels of acetyl-histone H3 (the HDAC1 substrate) and acetyl-α-tubulin (the HDAC6 substrate). As expected, the HDAC inhibitor SAHA potently inhibited the intracellular HDAC without influencing the levels of HDAC1 and HDAC6 after a 6-hour treatment (Figure 4A). It was worth noting that 24-hour treatment of P1 at concentrations ranging from 1 μM to 4 μM could significantly decrease the levels of HDAC1, HDAC6 and even HDAC8 in a dose-dependent manner (Figure 4B). More surprisingly, 5 μM SAHA could also effectively lead to the downregulation of HDAC1, HDAC6 and HDAC8, indicating the downregulation was not due to proteasome degradation. This was confirmed by the results that the proteasome inhibitor bortezomib could not block the HDAC downregulation as shown in Figure 4C. Our results showed evidence that some, if not all, HDAC inhibitors possess the HDAC downregulation effects in specific cell lines. Therefore, in the development of HDAC inhibitor-based PROTACs, it is quite important to check if the observed HDAC downregulation is due to the PROTAC-mediated proteasome degradation.
Figure 4.

Western blot analysis of levels of proteins of interest in RPMI-8226 cells. (A) The cells were treated with P1 and SAHA at the indicated concentrations for 6 h. (B) The cells were treated with P1 and SAHA at the indicated concentrations for 24 h. (C) The cells were pre-incubated with 10 nM bortezomib before the treatment of P1 and SAHA at the indicated concentrations for 24 h.
2.4. In Vitro APN Inhibitory Assay
Considering that the structures of P1, P2 and P3 contain the scaffold of bestatin, which is an aminopeptidase N (APN, CD13) inhibitor approved as antitumor immunomodulator [24], the in vitro APN inhibitory activities of P1, P2 and P3 were investigated with bestatin as the positive control. The results in Table 2 showed that all three bestatin-based compounds exhibited potent APN inhibitory activities, among which compound P2 was the most potent one with IC50 value of 1.67 ± 0.03 μM. Notably, the APN inhibitory activities of P1, P2 and P3 were even more potent than that of bestatin, indicating that introduction of large functional groups to the carboxy of bestatin was tolerated for its APN inhibitory activity. These results bring it to our attention that APN inhibition is a potential off-target effect of bestatin-based PROTACs.
Table 2.
APN inhibitory activities of compounds P1, P2, P3 and bestatin.
| Compounds | IC50 (μM) a |
|---|---|
| P1 | 3.43 ± 0.14 |
| P2 | 1.67 ± 0.03 |
| P3 | 2.65 ± 0.16 |
| bestatin | 4.33 ± 0.50 |
a Assays were performed in replicate (n ≥ 3). The data were shown as mean ± SD.
2.5. Ex Vivo Rat Thoracic Aorta Rings (TARs) Assay
APN is a moonlighting metalloenzyme playing crucial roles in various functions such as angiogenesis and metastasis of tumor cells [25]. Encouraged by their remarkable APN inhibitory activities, compounds P1, P2 and P3 were progressed to rat thoracic aorta rings (TARs) assay to evaluate their anti-angiogenesis potencies. As presented in Figure 5, considerable microvessels sprouted from the thoracic aorta ring in the negative control group. In contrast, P1, P2 and P3 effectively inhibited the microvessel outgrowth in a dose-dependent manner. Consistent with the APN inhibitory activities shown in Table 2, compounds P1, P2 and P3 exhibited superior anti-angiogenesis activities relative to bestatin at the same concentration of 5 μM. Note that compound P2, which possessed the most potent APN inhibitory activity among the three analogs, could almost completely inhibit the microvessel outgrowth at the concentration of 5 μM.
Figure 5.

Representative images of the rat thoracic aorta rings treated with compounds at the indicated concentrations.
3. Materials and Methods
3.1. Chemistry
All commercially available starting materials, reagents and solvents were used without further purification unless otherwise stated. All reactions were monitored by thin-layer chromatography on 0.25 mm silica gel plates (60GF-254). UV light, iodine stain and ferric chloride were used to visualize the spots. 1H-NMR and 13C-NMR spectra were recorded on a Bruker DRX spectrometer (Bruker, Billerica, MA, USA) at 400 MHz and 100 MHz, with δ given in parts per million (ppm) and J in hertz (Hz) and using TMS as an internal standard. High-resolution mass spectra were performed by Shandong Analysis and Test Center in Jinan, China. ESI-MS spectra were recorded on an API 4000 spectrometer (SCIEX, Framingham, MA, USA). Silica gel was used for column chromatography purification.
3.1.1. The Synthesis of Target Compound P1
((2S,3R)-3-((Tert-butoxycarbonyl)amino)-2-hydroxy-4-phenylbutanoyl)-l-leucine (14)
To a solution of compound 13 (0.50 g, 1.62 mmol) and DIPEA (0.63 g, 4.86 mmol) in anhydrous DCM (10 mL) was added (Boc)2O (0.43 g, 1.95 mmol) at 20 °C, the reaction mixture was allowed to stir for 12 h. Then, the mixture was concentrated in vacuum to give residue, which was purified by silica gel chromatography to give 0.55 g white solid, yield: 83%. 1H-NMR (400 MHz, DMSO-d6): δ 12.69 (s, 1H), 7.76 (d, J = 8.0 Hz, 1H), 7.29–7.15 (m, 5H), 6.14 (d, J = 12.0 Hz, 1H), 6.04 (s, 1H), 4.33–4.27 (m, 1H), 3.95–3.83 (m, 2H), 2.82–2.77 (m, 1H), 2.67–2.62 (m, 1H), 1.67–1.58 (m, 2H), 1.51–1.46 (m, 1H), 1.29–1.15 (m, 9H), 0.88–0.80 (m, 6H).
Tert-butyl (4-(6-(((benzyloxy)carbonyl)amino)hexanamido)phenyl)carbamate (16)
EDCI (2.11 g, 11.00 mmol) and HOBT (1.49 g, 11.00 mmol) were added into the solution of compound 15 (2.92 g, 11.00 mmol) in anhydrous DCM (50 mL) at 0 °C. The mixture was stirred at 0 °C for 30 min and followed by adding N-Boc-p-phenylenediamine (2.08 g, 10.00 mmol). The mixture was stirred at 0 °C for another 10 min and then stirred at 25 °C for 12 h. Filtration and the residue was washed with DCM to give 3.87 g of compound 16 as a white solid, yield: 85%. 1H-NMR (400 MHz, DMSO-d6): δ 9.65 (s, 1H), 9.14 (s, 1H), 7.37–7.35 (d, J = 8.0 Hz, 2H), 7.29–7.15 (m, 7H), 4.91 (s, 2H), 2.92–2.88 (m, 2H), 2.18–2.14 (m, 2H), 1.50–1.44 (m, 2H), 1.38–1.30 (m, 11H), 1.21–1.17 (m, 2H).
Benzyl (6-((4-aminophenyl)amino)-6-oxohexyl)carbamate (17)
Compound 16 (2.28 g, 5.00 mmol) was dissolved in DCM (5 mL) and trifluoroacetic acid (10 mL) at 0 °C. The mixture was stirred at 25 °C for 2 h. The solvent was evaporated in vacuum and the residue was re-dissolved in water (50 mL). The pH was adjusted to 6 with saturated NaHCO3. The precipitate was filtered and dried in vacuum to give 1.69 g white solid, yield: 95%. 1H-NMR (400 MHz, DMSO-d6): δ 9.42 (s, 1H), 7.38–7.25 (m, 5H), 7.20 (d, J = 8.0 Hz, 2H), 6.47 (d, J = 8.0 Hz, 2H), 5.00 (s, 2H), 4.82 (s, 2H), 3.00–2.96 (m, 2H), 2.21–2.17 (m, 2H), 1.56–1.50 (m, 2H), 1.43–1.40 (m, 2H), 1.28–1.24 (m, 2H).
Methyl 8-((4-(6-(((benzyloxy)carbonyl)amino)hexanamido)phenyl)amino)-8-oxooctanoate (18)
EDCI (2.11 g, 11.00 mmol) and HOBT (1.49 g, 11.00 mmol) were added into the solution of suberic acid monomethyl ester (2.07 g, 11.00 mmol) in DMF (20 mL) at 0 °C. The mixture was stirred at 0 °C for 30 min and followed by adding compound 17 (3.55 g, 10.00 mmol). The mixture was stirred at 0 °C for 10 min and then stirred at 80 °C for 7 h. The mixture was poured into water (100 mL). The formed precipitate was filtered off and washed with DCM (20 mL), then dried in vacuum to give 3.26 g white solid, yield: 62%. 1H-NMR (400 MHz, DMSO-d6): δ 9.78 (s, 2H), 7.73–7.67 (m, 1H), 7.48 (s, 4H), 7.38–7.24 (m, 5H), 5.00 (s, 2H), 3.58 (s, 3H), 3.01–2.96 (m, 2H), 2.31–2.24 (m, 6H), 1.58–1.49 (m, 6H), 1.46–1.39 (m, 2H), 1.30–1.28 (m, 4H), 0.89–0.84 (m, 2H).
Methyl 8-((4-(6-aminohexanamido)phenyl)amino)-8-oxooctanoate (19)
Compound 18 (1.05 g, 2.00 mmol) was dissolved in THF (10 mL) and MeOH (10 mL) and followed by the addition of ammonium formate (0.50 g, 8.00 mmol) and 10% Pd/C (0.10 g). The mixture was stirred at 65 °C for 3 h. Then Pd/C was filtered out and the solvent was evaporated in vacuum to give 0.72 g white solid, yield: 92%. 1H-NMR (400 MHz, DMSO-d6): δ 9.78 (d, J = 3.6 Hz, 2H), 7.48 (s, 4H), 3.58 (s, 3H), 2.54–2.51 (m, 2H), 2.31–2.24 (m, 6H), 1.60–1.50 (m, 6H), 1.39–1.24 (m, 8H).
Methyl 8-((4-((6R,7S,10S)-6-benzyl-7-hydroxy-10-isobutyl-2,2-dimethyl-4,8,11-trioxo-3-oxa-5,9,12-triazaoctadecan-18-amido)phenyl)amino)-8-oxooctanoate (20)
EDCI (0.42 g, 2.20 mmol) and HOBT (0.30 g, 2.20 mmol) were added into the solution of compound 14 (0.86 g, 2.10 mmol) in DMF (20 mL) at 0 °C. The mixture was stirred at 0 °C for 30 min and followed by the addition of compound 19 (0.78 g, 2.00 mmol). The mixture was stirred at 0 °C for another 10 min and then stirred at 80 °C for 7 h. The mixture was poured into water (100 mL). The formed precipitate was filtered off to afford the crude product, which was purified by column chromatography (DCM/MeOH = 20:1). The obtained product was further recrystallized from ethyl acetate/petroleum ether (1:1) to give 0.91 g white solid, yield: 58%. 1H-NMR (400 MHz, DMSO-d6): δ 9.78 (s, 2H), 8.01–7.98 (m, 1H), 7.59 (d, J = 8.0 Hz, 1H), 7.48 (s, 4H), 7.27–7.18 (m, 5H), 6.20 (d, J = 8.0 Hz, 1H), 5.96 (d, J = 8.0 Hz, 1H), 4.34–4.28 (m, 1H),3.98–3.92 (m, 1H), 3.82–3.80 (m, 1H), 3.58 (s, 3H), 3.04–3.01 (m, 2H), 2.81–2.76 (m, 1H), 2.69–2.64 (m, 1H), 2.31–2.24 (m, 6H), 1.58–1.51 (m, 6H), 1.44–1.39 (m, 2H), 1.30–1.15 (m, 18H), 0.86–0.81 (m, 6H).
Tert-butyl ((2R,3S)-3-hydroxy-4-(((S)-1-((6-((4-(8-(hydroxyamino)-8-oxo-octanamido)phenyl) amino)-6-oxohexyl)amino)-4-methyl-1-oxopentan-2-yl)amino) -4-oxo-1-phenylbutan-2-yl)carbamate (21)
Compound 20 (0.78 g, 1.00 mmol) was added into the freshly prepared potassium hydroxylamine methanol solution (5 mL), and the mixture was stirred at 25 °C for 0.5 h. The mixture was poured into water (30 mL) and was acidified with 5% citric acid to pH = 6. The formed precipitate was filtered and dried to give 0.51 g white solid, yield: 65%. 1H-NMR (400 MHz, DMSO-d6): δ 10.34 (s, 1H), 9.78 (s, 2H), 8.67 (s, 1H), 7.99 (t, J = 4.0 Hz, 1H), 7.59 (d, J = 8.0 Hz, 1H), 7.48 (s, 4H), 7.27–7.18 (m, 5H), 6.20 (d, J = 8.0 Hz, 1H), 5.98 (d, J = 8.0 Hz, 1H), 4.34–4.28 (m, 1H), 3.98–3.92 (m, 1H), 3.80 (s, 1H), 3.07–2.99 (m, 2H), 2.81–2.76 (m, 1H), 2.69–2.63 (m, 1H), 2.27–2.23 (m, 4H), 1.93 (t, J = 8.0 Hz, 2H), 1.59–1.36 (m, 8H), 1.28–1.15 (m, 18H), 0.86–0.79 (m, 6H).
N1-(4-(6-((S)-2-((2S,3R)-3-Amino-2-hydroxy-4-phenylbutanamido)-4-methylpentanamido) hexanamido)phenyl)-N8-hydroxyoctanediamide trifluoroacetate (P1)
Compound 21 (0.39 g, 0.50 mmol) was dissolved in DCM (2 mL) and CF3COOH (2 mL). The mixture was stirred at 25 °C for 1 h. The solvent was evaporated and the residue was added EtOAc. The formed precipitate was filtered and dried to give the desired product P1 (0.13 g, yield: 32%) as white solid (CF3COOH salt, purity: 95.2%). 1H-NMR (400 MHz, DMSO-d6): δ 10.34 (s, 1H), 9.79 (s, 2H), 8.67 (s, 1H), 8.09 (t, J = 8.0 Hz, 1H), 8.01 (d, J = 8.0 Hz, 1H), 7.89 (s, 3H), 7.48 (s, 4H), 7.37–7.26 (m, 5H), 6.60 (d, J = 4.0 Hz, 1H), 4.28–4.23 (m, 1H), 4.00–3.97 (m, 1H), 3.54 (s, 1H), 3.09–2.93 (m, 2H), 2.82–2.63 (m, 1H), 2.27–2.23 (m, 4H), 1.93 (t, J = 8.0 Hz, 2H), 1.57–1.40 (m, 10H), 1.29–1.23 (m, 7H), 0.89–0.86 (m, 6H). 13C-NMR (100 MHz, DMSO-d6): δ 171.85 (C=O), 171.35 (C=O), 171.26 (C=O), 170.98 (C=O), 169.57 (C=O), 136.64 (ArC), 135.06 (ArC), 129.88 (ArC), 129.10 (ArC), 127.41 (ArC), 119.83 (ArC), 68.66, 54.74, 51.73, 41.56, 38.95, 36.73, 36.67, 35.22, 32.70, 29.23, 28.90, 28.87, 26.51, 25.54, 25.51, 25.30, 24.72, 23.25, 22.44.
3.1.2. The Synthesis of Target Compound P2
Methyl 3-((2,2-dimethyl-4-oxo-3,8,11-trioxa-5-azatridecan-13-yl)carbamoyl)-benzoate (23)
To a solution of compound 22 (1.00 g, 6.76 mmol) and DIPEA (2.61 g, 20.27 mmol) in anhydrous DCM (20 mL) was added (Boc)2O (1.47 g, 6.76 mmol) in portions at 20 °C over 15 min. The reaction mixture was stirred at 20 °C for 12 h. After that, the mixture was concentrated in vacuum to give residue, which was purified by silica gel chromatography to give the key intermediate (1.10 g, yield: 66%). To a solution of 3-(methoxycarbonyl)benzoic acid (0.80 g, 4.43 mmol) in anhydrous DMF (10 mL) was added HATU (1.68 g, 4.43 mmol) and DIPEA (1.71 g, 13.29 mmol) in portions at 20 °C. The reaction mixture was stirred at 20 °C for 10 min, then the key intermediate (1.10 g, 4.43 mmol) was added and stirred at 20 °C for another 2 h. The reaction mixture was poured into water (25 mL). The aqueous layer was extrated with EtOAc (50 mL × 3), dried over sodium sulfate and concentrated in vacuum to give residue. The residue was purified by silica gel chromatography to give desired compound 23 (1.60 g, yield: 88%). ESI-MS, m/z = 411.4 [M + H]+.
Methyl 3-((2-(2-(2-aminoethoxy)ethoxy)ethyl)carbamoyl)benzoate hydrochloride (24)
To a solution of compound 23 (1.60 g, 3.90 mmol) in anhydrous dioxane (10 mL) was added HCl/dioxane (20 mL, 4 M) in portions at 20 °C. The reaction mixture was stirred at 20 °C for 2 h, followed by the concentrationin vacuum to give 1.35 g crude product as HCl salt, which was used for next step without further purification. ESI-MS, m/z = 311.0 [M + H]+.
Methyl 3-(((6R,7S,10S)-6-benzyl-7-hydroxy-10-isobutyl-2,2-dimethyl-4,8,11-trioxo-3,15,18-trioxa-5,9,12-triazaicosan-20-yl)carbamoyl)benzoate (25)
To a solution of compound 14 (0.50 g, 1.23 mmol) in anhydrous DMF (7 mL), HATU (0.47 g, 1.23 mmol) and DIPEA (0.48 g, 3.69 mmol) were added in portions at 20 °C. The reaction mixture was stirred at 20 °C for 10 min, then compound 24 (0.43 g, 1.23 mmol) was added and stirred at 20 °C for 2 h. The reaction mixture was poured into water (25 mL). The aqueous layer was extrated with EtOAc (50 mL × 3), dried over sodium sulfate, and concentrated in vacuum to give residue, which was purified by silica gel chromatography to afford the desired product 25 (0.71 g, 82% yield). ESI-MS, m/z = 701.4 [M + H]+.
Tert-butyl ((13S,16S,17R)-16-hydroxy-1-(3-(hydroxycarbamoyl)phenyl)-13-isobutyl-1,12,15-trioxo-18-phenyl-5,8-dioxa-2,11,14-triazaoctadecan-17-yl)carbamate (26)
Using the similar synthetic method of compound 21, compound 25 gave compound 26 (0.51 g, crude). The residue was used for the next step reaction without purification. ESI-MS, m/z = 602.3 [M-Boc + H]+.
N1-((11S,14S,15R)-15-Amino-14-hydroxy-11-isobutyl-10,13-dioxo-16-phenyl-3,6-dioxa-9,12-diazahexadecyl)-N3-hydroxyisophthalamide trifluoroacetate (P2)
To a solution of compound 26 (0.51 g, crude) in anhydrous dioxane (10 mL), HCl/dioxane was added (20 mL, 4M) in portions at 20 °C. After being stirred at 20 °C for 2 h, the reaction mixture was concentrated in vacuum to give residue, which was purified by Pre-HPLC (gradient elution with acetonitrile/water containing 0.225% CF3COOH) to afford the desired product P2 (51 mg, yield: 11%) as white solid (CF3COOH salt, purity: 97.4%). 1H-NMR (400 MHz, DMSO-d6): δ 11.32 (s, 1H), 9.14 (s, 1H), 8.66 (t, J = 5.6 Hz, 1H), 8.24 (d, J = 1.8 Hz, 1H), 8.19 (t, J = 5.6 Hz, 1H), 8.03 (d, J = 8.2 Hz, 1H), 7.96 (d, J = 7.8 Hz, 1H), 7.88 (dd, J = 12.4 Hz, J = 6.6 Hz, 4H), 7.54 (t, J = 7.7 Hz, 1H), 7.36 (dd, J = 8.8 Hz, J = 6.0 Hz, 2H), 7.32–7.24 (m, 3H), 6.64 (s, 1H), 4.32–4.26 (m, 1H), 3.98 (d, J = 3.3 Hz, 1H), 3.60–3.34 (m, 11H), 3.28–3.07 (m, 2H), 2.93 (dd, J = 13.8 Hz, J = 7.8 Hz, 1H), 2.80 (dd, J = 13.8 Hz, J = 6.5 Hz, 1H), 1.64–1.39 (m, 3H), 0.87 (dd, J = 8.0 Hz, J = 6.4 Hz, 6H). 13C-NMR (100 MHz, DMSO-d6): δ 172.20 (C=O), 171.01 (C=O), 166.26 (C=O), 164.19 (C=O), 158.95 (CF3COOH), 136.66 (ArC), 135.10 (ArC), 133.42 (ArC), 130.16 (ArC), 129.87 (ArC), 129.75 (ArC), 129.10 (ArC), 128.88 (ArC), 127.40 (ArC), 126.52 (ArC), 115.77 (CF3COOH), 70.01, 69.98, 69.30, 68.67, 54.71, 51.69, 41.53, 39.08, 35.22, 24.67, 23.27, 22.44. HRMS (AP-ESI) m/z, calcd. for C30H44N5O8 [M + H]+ 602.3190, found 602.3186.
3.1.3. The Synthesis of Target Compound P3
2-(2-(2-(2-Hydroxyethoxy)ethoxy)ethyl)isoindoline-1,3-dione (28)
To a solution of compound 27 (1.50 g, 10.00 mmol) and isoindoline-1,3-dione (1.47 g, 10.00 mmol) and PPh3 (2.62 g, 10.00 mmol) in THF (40 mL), DIAD was added (2.02 g, 10.00 mmol). After being stirred at 20 °C for 5 h, the mixture was concentrated to give a residue, which was purified by silica gel chromatography to give desired product (1.60 g, yield: 57%). ESI-MS, m/z = 280.2 [M+H]+.
2-(2-(2-(1,3-Dioxoisoindolin-2-yl)ethoxy)ethoxy)acetic acid (29)
To a solution of compound 28 (1.40 g, 5.00 mmol) and PhI(AcO)2 (6.44 g, 20.00 mmol) in 50 mL CH3CN/H2O (V:V = 4:1) was added TEMPO (0.16 g, 1.00 mmol). The mixture was stirred at 70 °C for 5 h, then the saturated Na2SO3 aqueous (50 mL) was added. The aqueous layer was extracted with EtOAc, then the organic layer was discarded. The aqueous was adjusted pH = 3 with 2 M HCl, then extracted with EtOAc. The organic layer was dried and concentrated to give the crude (1.00 g, 3.41 mmol) used for the next step without purification. ESI-MS, m/z = 294.1 [M + H]+.
2,2-Dimethyl-4-oxo-3,8,11-trioxa-5-azatridecan-13-oic acid (30)
To a solution of compound 29 (1.00 g, 3.41 mmol) in EtOH (20 mL), N2H4.H2O was added (0.50 g, 7.00 mmol). The reaction mixture was stirred at 80 °C for 5 h. The formed white solid was filtered out and the filtrate was concentrated to give a pale yellow oil. To a solution of the pale yellow oil in THF (15 mL) and 1N NaOH (7 mL) was added (Boc)2O (0.89 g, 4.09 mmol). The mixture was stirred at room temperature for 17 h, then water (20 mL) was added and extracted with EtOAc. The aqueous layer was adjusted pH = 4 with citric acid, then the aqueous layer was extracted with EtOAc. The mixture was dried and concentrated to give a residue, which was purified by silica gel chromatography EtOAc/PE/HCOOH (2/1/0.5% to 10/1/0.5%) to give desired compound 30 (0.37 g, yield: 41% two steps). ESI-MS, m/z = 164.4 [M-Boc + H]+.
3-Amino-N-(benzyloxy)benzamide (32)
To a solution of compound 31 (0.37 g, 2.80 mmol) in anhydrous DMF (5 mL), HATU (1.14 g, 3.00 mmol) and DIPEA (0.77 g, 6.00 mmol) was added at 20 °C. The reaction mixture was stirred at 20 °C for 10 min, then O-benzylhydroxylamine hydrochloride (0.48 g, 3.00 mmol) was added. After being stirred at 20 °C for another 2 h, the reaction mixture was poured into water (25 mL). The aqueous layer was extracted with EtOAc, dried over sodium sulfate and concentrated in vacuum to give residue. The residue was purified by silica gel chromatography to give desired compound 32 (0.48 g, yield: 71%). ESI-MS, m/z = 243.3 [M + H]+.
3-(2-(2-(2-Aminoethoxy)ethoxy)acetamido)-N-(benzyloxy)benzamide hydrochloride (33)
Using the similar synthetic method for compound 32, compound 30 (0.37 g, 1.40 mmol) reacted with compound 32 to give a key intermediate. The solution of intermediate (0.48 g, 0.99 mmol) in HCl/dioxane (20 mL, 4 M) was allowed to stir at 20 °C for 6 h. The reaction mixture was concentrated in vacuum to give 0.40 g crude product 33 as HCl salt. ESI-MS, m/z = 388.0 [M + H]+.
Tert-butyl ((11S,14S,15R)-1-((3-((benzyloxy)carbamoyl)phenyl)amino)-14-hydroxy-11-isobutyl- 1,10,13-trioxo-16-phenyl-3,6-dioxa-9,12-diazahexadecan-15-yl)carbamate (34)
Using the similar synthetic method for compound 32, compound 14 (0.40 g, 0.98 mmol) reacted with compound 33 to give desired product 34 (0.38 g, yield: 50%). ESI-MS, m/z = 678.5 [M-Boc + H]+.
3-((11S,14S,15R)-15-Amino-14-hydroxy-11-isobutyl-10,13-dioxo-16-phenyl-3,6-dioxa-9,12- diazahexadecanamido)-N-hydroxybenzamide (P3)
To a solution of compound 34 (0.38 g, 0.56 mmol) in MeOH (10 mL) wet 10% Pd/C was added (0.10 g). The reaction mixture was stirred at 20 °C under H2 atmosphere for 2 h, then the Pd/C was filtered out and the mixture was concentrated to give the residue used in the next step. HCl/dioxane (5 mL, 4M) was added into the residue and stirred at 20 °C for 6 h. The reaction mixture was concentrated in vacuum and purified by Pre-HPLC (gradient elution with acetonitrile/water containing 0.225% NH3·H2O) to afford the desired product P3 (26 mg, yield: 8%) as white solid (free base, purity: 96.1%). 1H-NMR (400 MHz, DMSO-d6): δ 9.83 (s, 1H), 8.30 (t, J = 5.6 Hz, 1H), 8.09–8.02 (m, 1H), 7.82–7.72 (m, 2H), 7.46–7.34 (m, 2H), 7.33–7.13 (m, 5H), 4.34–4.16 (m, 1H), 4.08 (s, 2H), 3.75 (dd, J = 12.5 Hz, J = 2.4 Hz, 1H), 3.67–3.51 (m, 4H), 3.41 (dt, J = 12.1 Hz, J = 6.0 Hz, 2H), 3.22–3.12 (m, 2H), 2.83–2.73 (m, 1H), 1.64–1.40 (m, 3H), 0.91–0.75 (m, 6H). HRMS (AP-ESI) m/z, calcd. for C29H42N5O8 [M + H]+ 588.3033, found 588.3032.
3.2. In Vitro HDACs Inhibitory Assay
All the HDAC enzymes were brought from BPS Bioscience. Ten μL of enzyme solution (HDAC1, HDAC6 or HDAC8) was mixed with different concentrations of tested compound (50 μL). The mixture was incubated at 37 °C for 5 min, followed by adding 40 μL fluorogenic substrate (Boc-Lys(acetyl)-AMC for HDAC1 and HDAC6, Boc-Lys(triflouroacetyl)-AMC for HDAC8). After incubation at 37 °C for 30 min, the mixture was quenched by addition of 100 μL of developer containing trypsin and Trichostatin A. Over another incubation at 37 °C for 20 min, fluorescence intensity was measured using a microplate reader at excitation and emission wavelengths of 390 nm and 460 nm, respectively. The inhibition rates were calculated from the fluorescence intensity readout of tested wells relative to those of control wells, and the IC50 values were calculated using Prism non-linear curve fitting method.
3.3. Western Blot Analysis
The RPMI-8226 cells were plated in flat-bottom six-well plates and allowed to grow for 12 h, and then treated with compounds or DMSO for a specific time period. Then, the cells were washed twice with cold PBS and lysed in ice-cold RIPA buffer, which comprised 50 mM Tris Base, 150 mM NaCl, 5 mM EDTA, 0.1% (v/v) SDS, 0.5% (v/v) sodium deoxycholate and 1% (v/v) Triton-x-100. Lysates were cleared by centrifugation. Protein concentrations were determined using the BCA assay. Equal amounts of cell extracts were then resolved by SDS-PAGE, transferred to nitrocellulose membrane and probed with HDAC1 antibody, HDAC6 antibody, HDAC8 antibody, acetyl-histone H3 antibody, acetyl-α-tubulin antibody and β-actin antibody, respectively. The membrane was washed twice with TBST buffer before being incubated with secondary antibodies. Images were acquired using a GE ImageQuant LAS 4000.
3.4. In Vitro APN Inhibitory Assay
The inhibitory activities of target compounds against APN were determined using l-leu-p-nitroanilide as substrate and microsomal APN from porcine kidney microsomes (Biocol) in 50 mM PBS, pH 7.2 as enzyme. Briefly, inhibitors (40 μL), PBS (145 μL), substrate (5 μL) and APN solution (10 μL) were added into 96-well plates. The mixture was incubated at 37 °C for 30 min. The hydrolysis product p-nitroanilide was measured at 405 nm. The inhibition rates were calculated based on the ultraviolet absorption readout of tested wells relative to those of control wells, and the IC50 values were calculated using Prism non-linear curve fitting method.
3.5. Ex Vivo Rat Aortic Rings Assay
Matrigel (100 μL; BD biosciences, NJ) was added into test well of 96-well plates and then allowed to polymerize for 0.5 h at 37 °C. Sprague–Dawley rats that were 4 to 6 weeks old were sacrificed and aortas were harvested. Each aorta was cut into 1 mm slices and embedded in additional 100 μL of Matrigel in 96-well plates. After that, the rings were incubated for 30 min at 37 °C and 5% CO2. Aorta rings were treated with the vehicle (0.5% DMSO) or compounds each day for 5 days and photographed on the sixth day at ×200 magnification. All experiments involving laboratory animals were performed with the approval by the institutional guidelines of Animal Care and Use Committee at Shandong University.
4. Conclusions
In summary, three hydroxamic acids containing the bestatin amide scaffold were designed and synthesized. Biological evaluation revealed that the bestatin-SAHA hybrid P1 was a potent pan-HDAC inhibitor, while the HDAC inhibitory potencies and isoform-selective profiles of P2 and P3 with m-substituted N-hydroxybenzamide warhead were far from satisfactory. Although P1 exhibited potent HDAC1/6/8 inhibitory activities, it could not induce proteasome-mediated degradation of the above HDAC isoforms. It is worth noting that prolong period of treatment of P1 or SAHA could dramatically decrease the intracellular levels of many HDAC isoforms including HDAC1/6/8 in RPMI-8226 cells through an unconfirmed mechanism. Note that this effect might be cell-type-dependent. Additionally, P1, P2 and P3 all exhibited potent APN inhibitory activities, indicating that attaching large functional group to the carboxy group of bestatin would not compromise its APN inhibitory potency. It is worth noting that a previous study reported that the combination of HDAC and aminopeptidase inhibitors is highly synergistic in multiple myeloma via the disruption of the NFκB signalling pathway [26], which indicates that our dual inhibitor P1 might be useful as anti-multiple myeloma agent.
Though no HDAC degrader was obtained, our research drew much needed attentions for the development of PROTACs. First, pan-HDAC inhibitor SAHA is capable of downregulating the cellular levels of multi-HDAC isoforms after long period of treatment. Therefore, it is indispensable to do mechanism study to confirm that the HDAC downregulation effect of HDAC inhibitor-based PROTACs is derived from the PROTAC-mediated degradation rather than from their intrinsic HDAC inhibitory activities. Secondly, explaining the cellular effects of bestatin-based PROTACs with caution is necessary because the bestatin-based PROTACs can also inhibit APN and even other aminopeptidases.
Abbreviations
| HDACs | histone deacetylases |
| PROTACs | proteolysis-targeting chimeras |
| VHL | von Hippel-Lindau |
| cIAP1 | cellular inhibitor of apoptosis protein 1 |
| SNIPERs | specific and nongenetic IAPs-dependent protein eraser |
| CRABPs | cellular retinoic acid-binding proteins |
| RARα | retinoic acid receptor α |
| AR | androgen receptor |
| ERα | estrogen receptor α |
| APN | aminopeptidase N |
| TARs | rat thoracic aorta rings |
| ppm | parts per million |
Author Contributions
Conceptualization, Y.Z. and C.J.C.; investigation, J.C. and W.Z.; writing—original draft preparation, J.C. and W.Z.; writing—review and editing, C.Z., Q.L., S.L. and G.Z.; supervision, Y.Z. and C.J.C.; funding acquisition, Y.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Natural Science Foundation of Shandong Province (Grant No. ZR2018QH007), Key Research and Development Program of Shandong Province (2017CXGC1401) and the Young Scholars Program of Shandong University (YSPSDU, 2016WLJH33).
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability: Samples of the compound P1 are available from the authors.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Sangwan R., Rajan R., Mandal P.K. HDAC as onco target: Reviewing the synthetic approaches with SAR study of their inhibitors. Eur. J. Med. Chem. 2018;158:620–706. doi: 10.1016/j.ejmech.2018.08.073. [DOI] [PubMed] [Google Scholar]
- 2.Zagni C., Floresta G., Monciino G., Rescifina A. The search for potent, small-molecule HDACIs in cancer treatment: A decade after vorinostat. Med. Res. Rev. 2017;37:1373–1428. doi: 10.1002/med.21437. [DOI] [PubMed] [Google Scholar]
- 3.Falkenberg K.J., Johnstone R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug. Discov. 2014;13:673–691. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- 4.Li P., Ge J.B., Li H. Lysine acetyltransferases and lysine deacetylases as targets for cardiovascular disease. Nat. Rev. Cardiol. 2020;17:96–115. doi: 10.1038/s41569-019-0235-9. [DOI] [PubMed] [Google Scholar]
- 5.McIntyre R.L., Daniels E.G., Molenaars M., Houtkooper R.H., Janssens G.E. From molecular promise to preclinical results: HDAC inhibitors in the race for healthy aging drugs. Embo. Mol. Med. 2019;11:e9854. doi: 10.15252/emmm.201809854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burslem G.M., Crews C.M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell. 2020;181:102–114. doi: 10.1016/j.cell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chamberlain P.P., Hamann L.G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019;15:937–944. doi: 10.1038/s41589-019-0362-y. [DOI] [PubMed] [Google Scholar]
- 8.Schapira M., Calabrese M.F., Bullock A.N., Crews C.M. Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug Discov. 2019;18:949–963. doi: 10.1038/s41573-019-0047-y. [DOI] [PubMed] [Google Scholar]
- 9.Toure M., Crews C.M. Small-molecule PROTACS: New approaches to protein degradation. Angew. Chem. Int. Ed. Engl. 2016;55:1966–1973. doi: 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- 10.Yang K., Song Y., Xie H., Wu H., Wu Y.T., Leisten E.D., Tang W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg. Med. Chem. Lett. 2018;28:2493–2497. doi: 10.1016/j.bmcl.2018.05.057. [DOI] [PubMed] [Google Scholar]
- 11.Wu H., Yang K., Zhang Z., Leisten E.D., Li Z., Xie H., Liu J., Smith K.A., Novakova Z., Barinka C., et al. Development of multifunctional histone deacetylase 6 degraders with potent antimyeloma activity. J. Med. Chem. 2019;62:7042–7057. doi: 10.1021/acs.jmedchem.9b00516. [DOI] [PubMed] [Google Scholar]
- 12.Yang K., Wu H., Zhang Z., Leisten E.D., Nie X., Liu B., Wen Z., Zhang J., Cunningham M.D., Tang W. Development of selective histone deacetylase 6 (HDAC6) degraders recruiting von hippel-lindau (VHL) E3 ubiquitin ligase. ACS Med. Chem. Lett. 2020;11:575–581. doi: 10.1021/acsmedchemlett.0c00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.An Z., Lv W., Su S., Wu W., Rao Y. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell. 2019;10:606–609. doi: 10.1007/s13238-018-0602-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang H., Lv W., He M., Deng H., Li H., Wu W., Rao Y. Plasticity in designing PROTACs for delective and potent degradation of HDAC6. Chem. Commun. 2019;55:14848–14851. doi: 10.1039/C9CC08509B. [DOI] [PubMed] [Google Scholar]
- 15.Smalley J.P., Adams G.E., Millard C.J., Song Y., Norris J.K.S., Schwabe J.W.R., Cowley S.M., Hodgkinson J.T. PROTAC-mediated degradation of class I histone deacetylase enzymes in corepressor complexes. Chem. Commun. 2020;56:4476–4479. doi: 10.1039/D0CC01485K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Itoh Y., Ishikawa M., Naito M., Hashimoto Y. Protein knockdown using methyl bestatin-ligand hybrid molecules: Design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J. Am. Chem. Soc. 2010;132:5820–5826. doi: 10.1021/ja100691p. [DOI] [PubMed] [Google Scholar]
- 17.Itoh Y., Ishikawa M., Kitaguchi R., Sato S., Naito M., Hashimoto Y. Development of target protein-selective degradation inducer for protein knockdown. Bioorg. Med. Chem. 2011;19:3229–3241. doi: 10.1016/j.bmc.2011.03.057. [DOI] [PubMed] [Google Scholar]
- 18.Itoh Y., Kitaguchi R., Ishikawa M., Naito M., Hashimoto Y. Design, synthesis and biological evaluation of nuclear receptor-degradation inducers. Bioorg. Med. Chem. 2011;19:6768–6778. doi: 10.1016/j.bmc.2011.09.041. [DOI] [PubMed] [Google Scholar]
- 19.Demizu Y., Okuhira K., Motoi H., Ohno A., Shoda T., Fukuhara K., Okuda H., Naito M., Kurihara M. Design and synthesis of estrogen receptor degradation inducer based on a protein knockdown strategy. Bioorg. Med. Chem. Lett. 2012;22:1793–1796. doi: 10.1016/j.bmcl.2011.11.086. [DOI] [PubMed] [Google Scholar]
- 20.Itoh Y., Ishikawa M., Kitaguchi R., Okuhira K., Naito M., Hashimoto Y. Double protein knockdown of cIAP1 and CRABP-II using a hybrid molecule consisting of ATRA and IAPs antagonist. Bioorg. Med. Chem. Lett. 2012;22:4453–4457. doi: 10.1016/j.bmcl.2012.04.134. [DOI] [PubMed] [Google Scholar]
- 21.Tomoshige S., Naito M., Hashimoto Y., Ishikawa M. Degradation of halotag-fused nuclear proteins using bestatin-halotag ligand hybrid molecules. Org. Biomol. Chem. 2015;13:9746–9750. doi: 10.1039/c5ob01395j. [DOI] [PubMed] [Google Scholar]
- 22.Tinworth C.P., Lithgow H., Dittus L., Bassi Z.I., Hughes S.E., Muelbaier M., Dai H., Smith I.E.D., Kerr W.J., Burley G.A., et al. PROTAC-mediated degradation of bruton’s tyrosine kinase is inhibited by covalent binding. ACS Chem. Biol. 2019;14:342–347. doi: 10.1021/acschembio.8b01094. [DOI] [PubMed] [Google Scholar]
- 23.Olson D.E., Wagner F.F., Kaya T., Gale J.P., Aidoud N., Davoine E.L., Lazzaro F., Weïwer M., Zhang Y.-L., Holson E.B. Discovery of the first histone deacetylase 6/8 dual inhibitors. J. Med. Chem. 2013;56:4816–4820. doi: 10.1021/jm400390r. [DOI] [PubMed] [Google Scholar]
- 24.Hitzerd S.M., Verbrugge S.E., Ossenkoppele G., Jansen G., Peters G.J. Positioning of aminopeptidase inhibitors in next generation cancer therapy. Amino Acids. 2014;46:793–808. doi: 10.1007/s00726-013-1648-0. [DOI] [PubMed] [Google Scholar]
- 25.Amin S.A., Adhikari N., Jha T.J. Design of aminopeptidase n inhibitors as anti-cancer agents. J. Med. Chem. 2018;61:6468–6490. doi: 10.1021/acs.jmedchem.7b00782. [DOI] [PubMed] [Google Scholar]
- 26.Smith E.M., Zhang L., Warker B.A., Davenport E.L., Aronson L.I., Krige D., Hooftman L., Drummond A.H., Morgan G.J., Davies F.E. The combination of HDAC and aminopeptidase inhibitors is highly synergistic in myeloma and leads to disruption of the NFκB signalling pathway. Oncotarget. 2015;6:17314–17327. doi: 10.18632/oncotarget.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]