

Abstract
We showed that intrarenal suppression of TNF production under low salt (LS) conditions increases renal cortical AGT mRNA and protein expression. Intrarenal injection of murine recombinant TNF attenuated increases of AGT in mice ingesting LS. Moreover, AGT mRNA and protein expression increased approximately 6-fold and 2-fold, respectively, in mice ingesting LS that also received an intrarenal injection of a lentivirus construct that specifically silenced TNF in the kidney (U6-TNF-ex4). Silencing of TNF under normal salt (NS) and high salt (HS) conditions also resulted in increased AGT expression. Since renal TNF production decreases in response to LS and increases in response to HS, the data suggest that alterations in TNF production under these conditions modulates the degree of AGT expression. We also tested the hypothesis that TNF inhibits intrarenal AGT expression by a mechanism involving miR-133a. Expression of miR-133a decreased in mice given LS and increased in response to HS for 7 days. Intrarenal silencing of TNF reversed the effects of HS on miR-133a-dependent AGT expression. In contrast, intrarenal TNF administration increased miR-133a expression in the kidney. Collectively, the data suggest that miR-133a is a salt-sensitive miRNA that inhibits AGT in the kidney and is increased by TNF. The HS-induced increase in blood pressure observed following silencing of TNF was markedly reduced upon intrarenal administration of miR-133a suggesting that intrinsic effects of TNF in the kidney to limit the blood pressure response to HS include an increase in miR-133a, which suppresses AGT expression.
Keywords: TNF, angiotensinogen, miR-133a, dietary salt
Graphical Abstract
INTRODUCTION
We recently showed that tumor necrosis factor-alpha (TNF) produced by the kidney under non-inflammatory, normotensive conditions modulates salt-induced changes in Na+-K+-2Cl− cotransporter (NKCC2) isoform A and B expression and function (1;2). Intrarenal TNF contributes to the regulation of Na+ excretion as part of an adaptive mechanism involving NKCC2A and acts as a negative regulator of blood pressure in response to increases in NaCl intake (1). Additionally, TNF inhibits NKCC2B expression and activity, which regulates the extent of Na+ reabsorption in the context of salt restriction (2). Overall, these findings indicate that the actions of TNF along both the medullary (m) and cortical (c) thick ascending limb (TAL) limits Na+ reabsorption in response to changes in dietary salt intake by regulating NKCC2A and B isoforms (2).
The intrarenal RAS (renin-angiotensin-system) is important for regulating sodium transport and blood pressure, and the proximal tubule (PT) is a nephron segment that contains the essential molecules needed for full expression of this system (3–7). Intrarenal angiotensinogen (AGT), the precursor of angiotensin (Ang) peptides, is produced mainly in the cortical PT and is important for activation of the intrarenal RAS (8;9). For instance, overexpression of AGT in the PT causes hypertension, and AGT synthesis by this segment of the nephron is regulated by high Na+ intake, Ang II, and inflammatory cytokines (7;10). In vitro studies showed that TNF inhibits AGT mRNA accumulation and protein expression in human kidney-2 (HK-2) cells, an immortalized proximal tubule cell line (11). Cardiac specific overexpression of TNF also suppresses AGT mRNA accumulation in the heart (12). However, the in vivo effects of TNF on the regulation of intrarenal AGT and its contribution to blood pressure homeostasis are not known.
Multifactorial, tissue-specific mechanisms regulate the expression and function of AGT, and additional regulatory pathways remain to be uncovered. Emerging evidence suggests that microRNAs (miRNAs), noncoding RNAs of 21–25 nucleotides that contribute to the regulation of gene expression by repressing target gene translation and/or facilitating target gene mRNA degradation, are important modulators of renal function (13–16). Indeed, several miRNAs have been associated with either the development or protection against hypertension, although how they are regulated and which components of the RAS they interact with is not well understood (17–19). miR-133a is abundantly expressed in cardiac and skeletal muscle, however, the expression and function of this miRNA in the kidney is not well defined (20). Recently, miR-133a was reported to attenuate AGT expression in the brain and inhibit (pro) renin receptor (PRR) expression in human umbilical vein endothelial cells (21;22). In the present study, we determined the effects of dietary salt intake on miR133a expression and its contribution to the mechanism by which TNF regulates AGT expression and function in the kidney.
METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request. A detailed description of all methods is provided in the online Data Supplement.
Chemicals-
All chemicals including collagenase (type 1A), tissue culture media, and polyclonal antibodies for AGT were purchased from Sigma (St Louis, MO). pLKO.1, psPAX2, and pMD2.G plasmids were from Addgene (MIT, Cambridge, MA). The polyvinylidene difluoride membranes and the lentivirus purification kit (cat. no. 631234) were purchased from Amersham (Arlington Heights, IL) and Clontech (Mountain View, CA), respectively. Lentivirus targeting vector and constructs were generated using standard cloning procedures as previously described (23).
Animals-
C57BL/6 mice (male, B.W. 20-22g ) were purchased from Jackson Laboratory. Mice were fed either low-salt diet (LS, 0.02% NaCl; Envigo) normal-salt diet (NS; 0.4% NaCl; Envigo), or 1% NaCl in the drinking water (HS) with NS and tap water ad libitum. All protocols were approved by the New York Medical College IACUC committee and conducted in accordance with National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Quantitative RT-PCR analysis-
RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA). The amplification of cDNA fragments, TaqMan MicroRNA Assays and quantitative RT-PCR analysis were performed using PCR or CYBR Green Chemistry using an Applied Biosystems real-time PCR instrument as in previous studies (23).
Western blot analysis-
Proteins were extracted from kidney cortical samples after homogenization with protease inhibitors and quantified as described (2); see Supplement.
shTNF and miR-133a constructs-
The lentivirus encoding shTNF was designed and constructed using a short hairpin (sh)RNA-expressing construct targeting exon 4 of murine TNF (U6-TNF-ex4) as previously described (1). Constructs for miR-133a were designed, generated, and verified by DNA sequencing using procedures similar to those previously described (23–25).
Administration of lentivirus, miR-133a, or TNF products in vivo-
Intrarenal administration of lentivirus, filter-purified miR-133a (~5 × 107 transducing units), or recombinant TNF was performed as previously described (2) and detailed in the Supplement.
Radiotelemetry measurements-
Mean arterial pressure (MAP) was measured using radiotelemetry as previously described (1); see Supplement.
Statistical Analysis-
Data were analyzed using Student’s t test for comparisons between 2 groups or One-way ANOVA (followed by Tukey or Dunnett post hoc test) was used for comparisons among groups by GraphPad Prism 6 software. Radiotelemetry data obtained on consecutive days were analyzed by repeated-measures ANOVA. P values <0.05 were considered statistically significant. All data are expressed as mean ± SE.
RESULTS
Downregulation of renal TNF production increases renal cortical AGT-
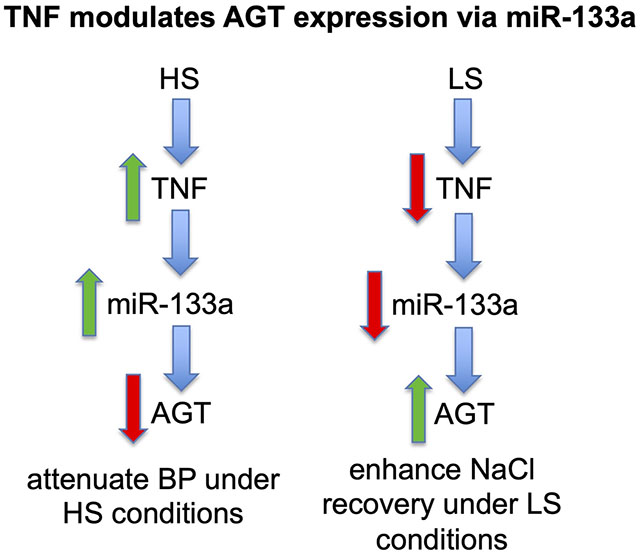
Our previous studies showed that LS intake downregulates TNF production in renal cortex, an effect that is reflected by reduced urinary levels of TNF (2). In the present study, we measured AGT mRNA and protein expression in renal cortex by RT-PCR and Western blotting, respectively, after mice ingested either LS or NS diet for 7 days. AGT mRNA expression in the renal cortex was significantly increased (Fig. 1A), although AGT protein expression did not change in response to LS compared with mice given NS (Fig. 1B). We have previously achieved specific silencing of TNF in the kidney using a shRNA-TNF lentivirus silencing construct (U6-TNF-ex4) (1). This approach was used to further determine the link between intrarenal levels of TNF and AGT (Figs. S1A and B). Silencing TNF with U6-TNF-ex4 increased both AGT mRNA accumulation (Fig. 1C) and protein expression (Fig. 1D) in cortex from mice ingesting LS for 7 days. In contrast, the relative abundance of cortical AGT mRNA and protein expression decreased after mice received a single intrarenal injection of murine recombinant TNF-α (5 ng/g body wt) into both kidneys followed by LS intake for 7 days (Fig. 1E). This effect of TNF also was observed in primary cultures of mouse PT cells incubated in LS isotonic medium (Figs. S2A and B). Collectively, these data indicate that TNF acts as a negative regulator of AGT in the renal cortex in vivo and PT cells in vitro.
Figure 1: Effects of TNF on renal cortical AGT mRNA and protein expression during LS intake.
Mice received LS [0.02% NaCl (wt/wt)] or NS [0.4% NaCl (wt/wt)] for 7 days. A): AGT mRNA expression was measured by RT-PCR after mice ingested LS or NS for 7 days. B): Western blotting of AGT using anti-AGT antibody (top); anti-β-actin antibody was used as a loading control (bottom). C and D): AGT mRNA and protein expression in renal cortex was measured by RT-PCR or Western blotting, respectively, after intrarenal injection of either ~5 × 107 transducing units (TU) lentivirus U6-TNF-ex4 or U6 control constructs into both kidneys of mice that ingested LS for 7 days. E and F): AGT mRNA and protein expression was measured after a single intrarenal injection of murine recombinant TNF-α (5 ng/g body wt) into both kidneys followed by LS for 7 days. Data are shown as mean ± SE; (n=10).
Renal TNF attenuates AGT levels under normal and high salt conditions:
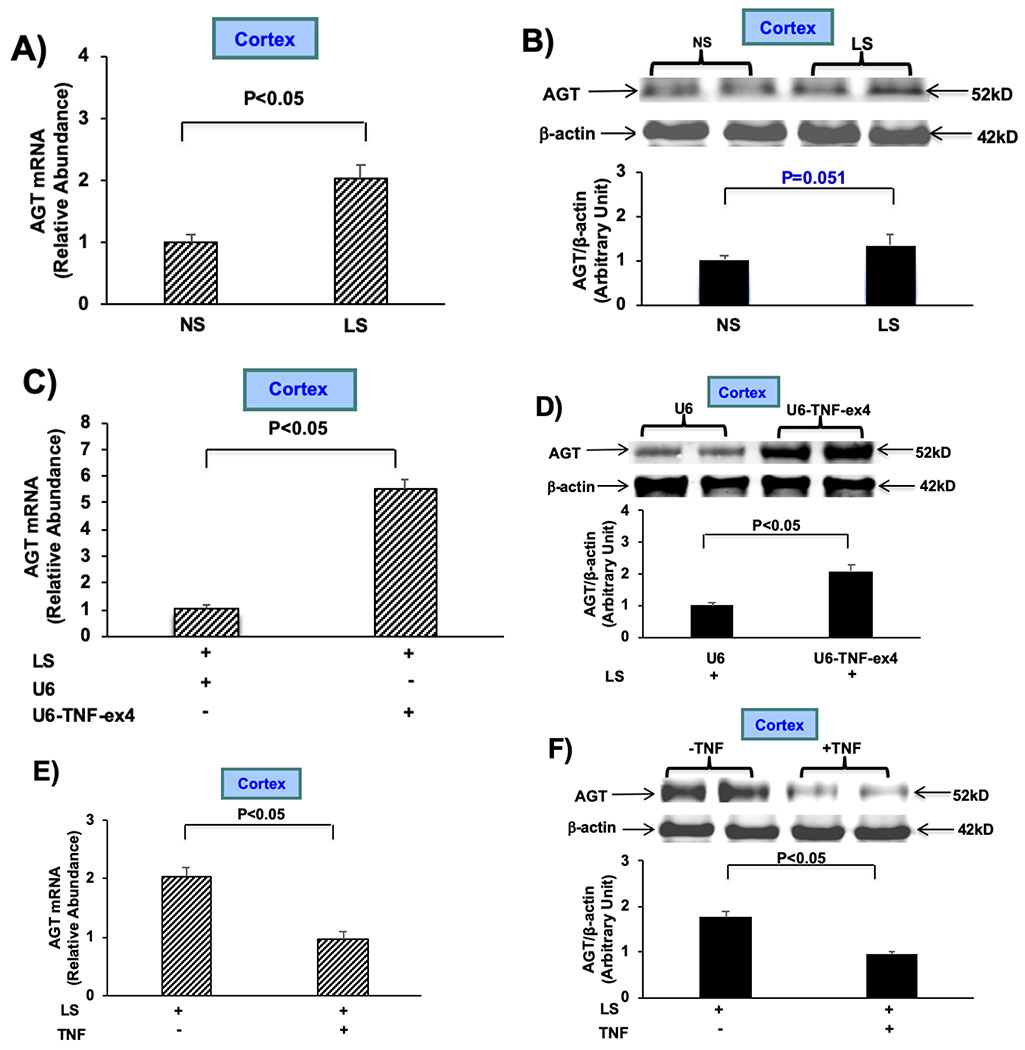
To determine if the ability of TNF to modulate AGT levels is limited to LS conditions, we determined the effects of TNF on AGT mRNA and protein expression in renal cortex obtained from mice given NS or HS for 7 days. The levels of AGT mRNA and protein did not differ in mice given HS compared to those given NS (Fig. 2A and B). In contrast, the relative abundance of AGT mRNA increased more than 2-fold after intrarenal injection of lentivirus U6-TNF-ex4 into both kidneys followed by NS for 7 days, although AGT protein expression did not change significantly (Fig. 2C and D). Moreover, AGT mRNA accumulation increased after intrarenal injection of the U6-TNF-ex4 lentivirus construct followed by HS for 7 days (Fig. 2E). AGT protein expression also increased after intrarenal injection of lentivirus U6-TNF-ex4 to mice given HS for 7 days (Fig. 2F). The data indicate that TNF attenuates AGT expression under NS and HS conditions and suggest that renal TNF production, which increases in response to HS conditions (26), is part of the mechanism that suppresses activation of the intrarenal RAS during HS conditions.
Figure 2: Effects of TNF on renal cortical AGT expression during normal and high salt intake:
A): AGT mRNA expression was measured by RT-PCR after mice were given NS or HS for 7 days. B): Western blotting of AGT (top) and anti-β-actin used as a loading control (bottom) was performed in mice given NS or HS for 7 days. C and D): AGT mRNA and protein expression was determined after intrarenal injection of either lentivirus U6-TNF-ex4 (~5 × 107 TU) or U6 constructs into both kidneys of mice followed given NS for 7 days. E and F): AGT mRNA and protein expression was measured after intrarenal injection of either lentivirus U6-TNF-ex4 or U6 constructs into both kidneys in mice given HS for 7 days. Data are shown as mean ± SE; (n=10).
miR-133a downregulates target gene AGT expression in the renal cortex-
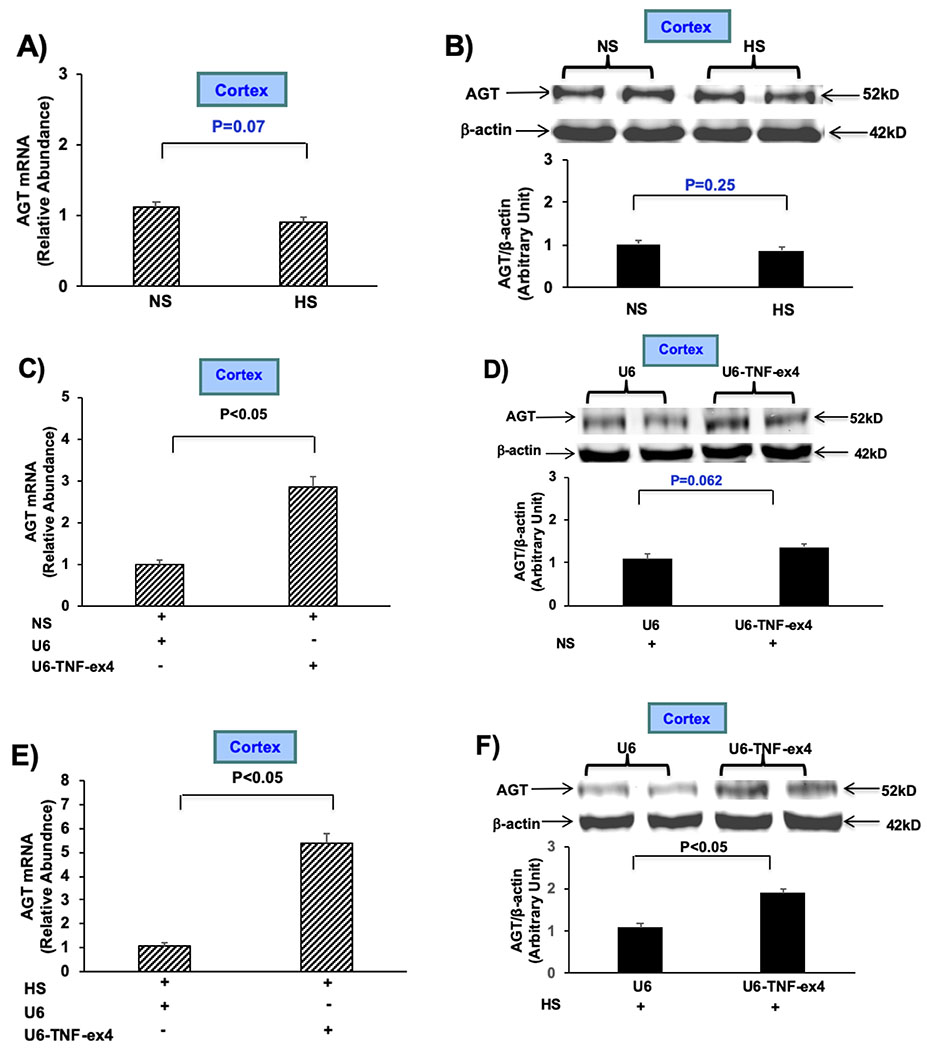
We identified a potential miR-133a-binding site within the 3’-untraslated region (3’UTR) of AGT using the prediction tool TargetScan v5.1 (Fig. 3A), determined the effects of miR-133a on AGT expression, and addressed whether its expression can be modulated by changes in salt intake. AGT mRNA in renal cortex decreased after intrarenal injection of miR-133a into both kidneys followed by LS intake for 7 days (Fig. 3B). Next miR-133a expression was measured after mice were given NS or LS diet for 7 days. Expression of miR-133a decreased in mice given LS for 7 days compared to those given NS, supporting the concept that downregulation of miR-133a could contribute to an increase in AGT mRNA under LS conditions (Fig. 3C). To determine if miR-133a was reciprocally regulated by high and low salt intake, miR-133a expression was determined in mice that ingested HS for 7 days (Fig. 3D). Indeed, a two-fold increase in miR-133a expression was observed in mice ingesting HS (Fig. 3D). Additionally, AGT mRNA decreased after injection of miR-133a followed by ingestion of HS for 7 days (Fig. 3E). Collectively, these data indicate that miR-133a levels are inhibited by LS, increased by HS intake, and reveal that the ability of miR-133a to downregulate AGT expression is not affected by salt intake.
Figure 3: miR-133a downregulates target gene AGT expression in renal cortex.
A) Schematic diagram showing the putative miR-133a binding sites within the 3’-untraslated region (3’UTR) of AGT; coding sequence (CD), and microRNA response element (MRE). B) AGT mRNA expression was measured by RT-PCR after intrarenal injection of either miR-133a (~2 × 108 TU) or scramble control into both kidneys of mice given LS for 7 days. C) miR-133a expression was measured after mice were given NS (−) or LS for 7 days. D) miR-133a expression was measured after mice were given NS (−) or HS for 7 days. E) AGT mRNA expression was determined after intrarenal injection of either miR-133a or scramble control into both kidneys of mice given HS for 7 days. Data are shown as mean ± SE; (n=10).
TNF induces miR-133a expression-
The relationship between TNF and miR-133a, both of which inhibited the expression of AGT, was determined in mice given either LS or HS for 7 days. Mice were given a single intrarenal injection of murine recombinant TNF (5ng/g body weight) or saline control into each kidney for 24h, which increased miR-133a expression in cortex obtained from mice given LS (Fig. 4A). Moreover, silencing TNF with U6-TNF-ex4 inhibited miR-133a expression in mice given LS, as well as in mice given HS (Fig. 4B and C, respectively). AGT mRNA and protein expression was reduced in primary cultures of mPT cells, when miR-133a was added to cells in which TNF was silenced (Figs. S3A and B, respectively). Suppression of TNF mRNA in U6-TNF-ex4 pretreated mPT cells challenged with HS is shown as a positive control (Fig. S3C). Collectively, these data indicate that TNF is a positive regulator of miR-133a expression and are consistent with the hypothesis that TNF regulates AGT in a miR-133a-dependent manner.
Figure 4: TNF upregulates miR-133a expression in the renal cortex.
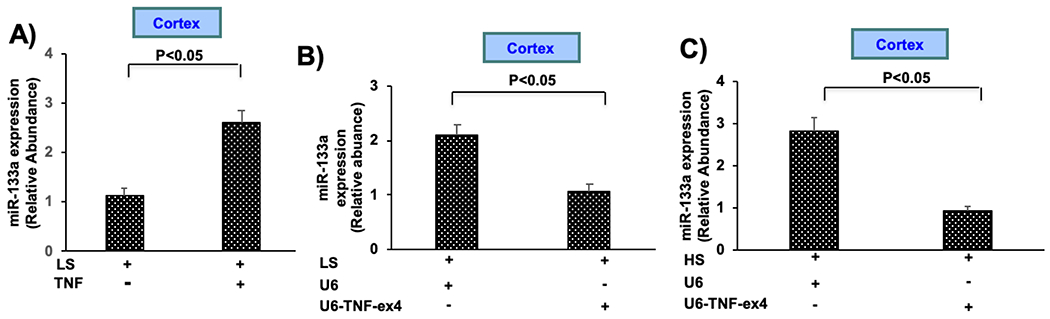
A) miR-133a expression was measured after a single intrarenal injection of murine recombinant TNF into both kidneys followed by ingestion of LS for 7 days. B) miR-133a expression was determined after intrarenal injection of either lentivirus U6-TNF-ex4 (~5 × 107 TU) or U6 constructs into both kidneys followed by ingestion of LS for 7 days. C) miR-133a expression was measured after intrarenal injection of either lentivirus U6-TNF-ex4 or U6 into both kidneys of mice given HS for 7 days. Data are shown as mean ± SE; (n=10).
Administration of miR-133a reverses the increase in blood pressure induced by silencing renal TNF-
We previously showed that silencing TNF in the kidney results in elevated blood pressure responses to high salt that are rapid, reversible, and sustained (1). In the present study, we determined the relationship between miR-133a-dependent suppression of AGT by TNF on the increase in blood pressure induced by renal silencing of TNF in mice receiving HS. Mice received intrarenal injections of either a mixture of scramble miRNA control and U6-TNF-ex4, or miR-133a and U6-TNF-ex4 for 3 days then were given HS for an additional 7 days. Baseline MAP was similar in both groups of mice while ingesting tap water for 3 days (Fig. 5A). However, the HS-induced increase in MAP observed in mice injected with U6-TNF-ex4 and miR-133a was markedly reduced compared to responses seen in mice that received a scrambled control miRNA along with the TNF silencing vector (Fig. 5A). Similarly, MAP averaged over 7 days of HS intake was lower in mice that received both miR-133a and U6-TNF-ex4 compared to mice in which only TNF was silenced (Fig. 5B). Accumulation of AGT mRNA was inhibited in mice that received the combination of U6-TNF-ex4 and miR-133a compared to the scramble control group (Fig. 5C). Interestingly, HS intake did not increase miR-133a in the liver, nor did AGT mRNA accumulation in the liver change in mice given the U6-TNF-ex4 and miR-133a combination (Figs. 5D and E, respectively). The silencing effect of U6-TNF-ex4 on TNF mRNA also was restricted to the kidney (Figs. S4A and B, respectively). Taken together, these data support the concept that induction of miR-133a by TNF regulates intrarenal AGT expression as part of a mechanism that attenuates increases in blood pressure in response to HS intake.
Figure 5: Intrarenal administration of miR-133a inhibits HS-induced increases in blood pressure subsequent to knockdown of TNF in the kidney.
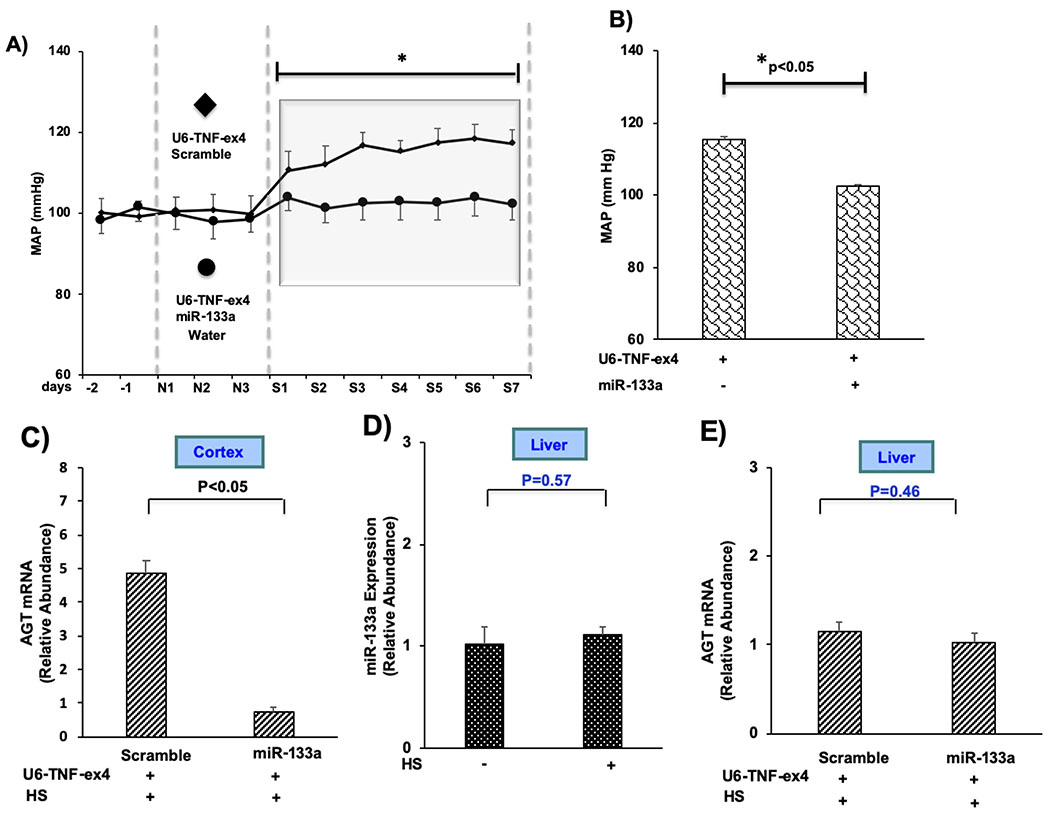
A) Daily blood pressure measurements for mice implanted with radiotelemetry probes, injected with either U6-TNF-ex4 (~5 × 107 TU) or a combination of U6-TNF-ex4 and miR-133a (~2 × 108 TU) into both kidneys, and given HS for 7 days. B) Summation of MAP for mice given HS for 7 days. C) AGT mRNA expression was determined after intrarenal injection of either U6-TNF-ex4 or a combination of U6-TNF-ex4 and miR-133a in mice given HS for 7 days. D) miR-133a expression in liver was measured in mice given NS (−) or HS for 7 days. E): AGT mRNA expression in liver was measured following intrarenal injection of either U6-TNF-ex4 or a combination of U6-TNF-ex4 and miR-133a in mice given HS for 7 days. Data are shown as mean ± SE; (n=10, ANOVA and t test).
DISCUSSION
We demonstrated that TNF production within the kidney attenuates renal AGT mRNA and protein expression. Suppression of renal TNF by LS conditions (2) contributed to an increase in AGT mRNA accumulation in the renal cortex, an effect that was enhanced by additional suppression of TNF production by intrarenal administration of a TNF lentivirus silencing vector, and was reversed by an intrarenal injection of TNF. The lentiviral-mediated suppression of TNF also increased AGT protein expression. Similarly, silencing of HS-dependent increases in TNF production augmented AGT mRNA and protein expression. The inhibitory effects of TNF on AGT expression were dependent on its ability to increase miR-133a, which suppresses AGT mRNA in the kidney. Moreover, the increase in blood pressure induced by HS intake in mice with low levels of TNF in the kidney, was reversed by intrarenal administration of miR-133a, which inhibited AGT in the kidney but not liver. Collectively, the data indicate that limiting endogenous renal production of TNF and its ability to increase miR-133a sensitizes mice to HS-mediated increases in blood pressure.
TNF exhibits regulatory effects in diverse epithelial cells including those in kidney, lung, and colon where active Na+ transport is important for fluid clearance and other homeostatic mechanisms (27–29). We previously showed that HS increases TNF production in the kidney and renal-specific silencing of TNF unmasks salt-dependent increases in blood pressure via a mechanism involving NKCC2A (1;26). We also demonstrated that downregulation of renal TNF production in response to LS intake contributes to the regulation of NaCl reabsorption via a selective increase in NKCC2B expression, an effect that was enhanced when TNF production was further suppressed with a TNF-specific silencing lentivirus (2). Collectively, the studies showed that TNF prevents NaCl reabsorption via mechanisms functioning in the mTAL or cTAL/MD regions of the kidney, respectively, and are consistent with previous studies showing a TNR receptor 1-dependent natriuretic effect of TNF (30;31). The present study reveals that intrarenal effects of TNF are neither limited to transporter molecules nor distal segments of the nephron given the ability of this cytokine to suppress AGT expression in the renal cortex and primary mPT cells. These data support those showing that inhibitory effects of TNF on AGT are observed in human proximal tubule cells and human kidney-2 (HK-2) cells in vitro (11). However, the in vivo implications of TNF acting as an endogenous negative regulator of AGT on blood pressure homeostasis, for example, have not previously been described. Since TNF modulates the response of salt intake on NKCC2 isoform expression, and AGT also is sensitive to changes in salt intake (32;33), we evaluated the effects of TNF on AGT expression in response to low- and high-salt intake. The present study is the first to assign an effect of TNF involving AGT that regulates the blood pressure response to HS intake.
The inhibitory effects of TNF on AGT expression may be one of multiple mechanisms that regulate Na+ reabsorption during dietary Na+ restriction. We recently showed that downregulation of TNF production as a result of LS intake resulted in increased NaCl reabsorption via NKCC2B expressed in the MD/cTAL region (2). Consequently, the TNF-dependent downregulation of AGT, together with the effects of TNF on NKCC2B expression, are consistent with the finding that changes in NKCC2B isoform abundance in response to LS intake was partly mediated by Ang II and AT1 receptor activation (34). The coordinated effects of TNF on NKCC2B and AGT also are intriguing as they are consistent with previous studies showing that TNF inhibits renin expression (35). AGT synthesis occurs in the proximal straight tubule (36), and while some studies have shown that LS increases AGT expression (32;33) others have not (37–39). The present study suggests that a progressive decrease in TNF levels allow for an initial increase in the level of AGT mRNA (LS intake) followed by an increase in AGT protein expression as TNF levels decrease further (LS + shTNF silencing). Moreover, intrarenal administration of TNF attenuates the increase in AGT expression induced in response to LS intake and intrarenal silencing of TNF under HS conditions increases AGT expression.
Recent findings indicate that miR-133a is expressed in brain, heart, renal cells, and also is found in urine (22;40–43). miRNA133a protects against myocardial fibrosis, heart failure, controls vascular smooth muscle cell phenotypic switching, and may prevent diabetes-induced increases in extracellular matrix protein expression (20;41;44–46). Based on data illustrating the 3’-UTR of AGT, AGT 3’ UTR miR-133a-binding prediction among species, schematic diagram of in silico analysis of putative miR-133a binding sites within the 3’-UTR of AGT, luciferase assays to demonstrate the binding of miR-133a to the 3’-UTR of AGT in NG108 cells transfected with the appropriate overexpression plasmid and scramble controls over several different doses, and the ability of miR-133a to attenuate AGT expression in the brain and other tissues (22;41), we tested the hypothesis that TNF inhibits changes in renal AGT expression induced by alterations in dietary salt intake via a mechanism involving miR-133a. Expression of several miRNAs are sensitive to increases in Na+ and K+ intake (47–49), and we show reciprocal regulation of miR-133a by high and low salt in the kidney. Thus, LS-mediated suppression of TNF production was associated with a concommitant reduction of miR-133a expression, and was accompanied by an increase in AGT that was reversed by intrarenal administration of TNF. In contrast, HS intake increased TNF-dependent miR-133a and suppressed AGT expression. The present findings suggest that TNF orchestrates an epigenetic mechanism that regulates AGT expression in the kidney via a mechanism involving miRNA-133a. TNF has been shown to alter the expression of additional miRNAs in diverse tissues. For instance, TNF increases the expression of miR-155 in osteosarcoma cells, regulates miRNAs in endothelial cells as part of a regulatory network, induces miR-98 in fibroblast like synoviocytes, and inhibits bone marrow stem cell proliferation/differentiation by reducing expression of miR-34a (50–53). The molecular details of how TNF increases the expression of miRNAs requires further investigation. However, it is intriguing to speculate that there may be a link between the miR-133a-dependent inhibition of AGT by TNF and the previously reported p50/p50 homodimer-dependent inhibition of AGT by TNF (11).
Formation of AGT, renin, and Ang II in the renal cortex is critical for the maintenance of the intrarenal RAS, which plays an important role in kidney function, but promotes hypertension and renal disease when stimulated inappropriately (4;5;54;55). Regulation of renal function and blood pressure also is dependent on the ability of miRNAs to influence the expression of diverse molecules (19;43;56). Since ingestion of HS suppresses the RAS and silencing TNF inhibits miR-133a, we determined if we could reverse the increase in blood pressure induced by HS in the absence of TNF by injecting miR-133a directly into the kidney. This approach was pursued rather than attempting to raise blood pressure by inhibiting miR-133a in mice given HS since there are multiple mechanisms that regulate suppression of the RAS and the blood pressure lowering effects of TNF via NKCC2A would remain intact (1). Indeed, blood pressure was lower in mice given an intrarenal injection of miR-133a compared to mice in which TNF was silenced in the kidney. Under these conditions administration of miR-133a reduced AGT mRNA levels in the kidney, but not liver, suggesting that the interaction of TNF with the intrarenal RAS regulates blood pressure. These findings are consistent with those showing that downregulation of miR-133a in the heart was associated with Ang II-dependent increases in blood pressure (45). The present study suggests TNF-mediated regulation of AGT via miR-133a functions as a mechanism that mitigates increases in blood pressure following elevated NaCl intake.
Perspectives
Renal-derived TNF, which attenuates Na+ reabsorption and the blood pressure response to high salt intake via the NKCC2A isoform, also inhibits AGT expression in the kidney. These findings suggest that TNF regulates function in the PT as well as the mTAL and cTAL/MD regions of the kidney. The newly emerging intratubular TNF system intersects with the intratubular RAS via induction of miRNA-133a, which is expressed in the PT, inhibits intrarenal AGT expression, and inhibits salt-induced increases in blood pressure. The present findings suggest TNF acts as an endogenous inhibitor of AGT expression in response to changes in dietary salt intake and supports the concept that dysregulation of an intratubular TNF system contributes to salt-sensitive hypertension associated with activation of the RAS.
Supplementary Material
Novelty and Significance.
What is New?
-TNF inhibits intrarenal AGT in vivo and regulates the blood pressure response to high salt intake.
-TNF acts as an endogenous regulator in the kidney that increases miR-133a expression as part of a novel mechanism that inhibits AGT expression.
-miR-133a in the kidney is a salt-sensitive miRNA whose expression is increased by high salt intake and decreased by low salt intake.
-miR133a attenuates high salt-mediated increases in blood pressure by inhibiting AGT expression in the kidney.
What is Relevant?
TNF activation of miR-133a functions as a protective mechanism to limit expression of AGT and attenuate salt-induced increases in blood pressure. This finding is consistent with the protective role of miR-133a, which previously has been shown to attenuate cardiac hypertrophy and fibrosis. The study shows that TNF modulates the response of AGT to changes in dietary salt intake in the proximal tubule, thus, the intrinsic effects of TNF in the kidney are not limited to effects on ion transporters or distal segments of the nephron.
Summary
This study illustrates an effect on blood pressure regulation via intersection between the intrarenal RAS and TNF systems. The intrinsic effects of TNF within the kidney include an increased expression of miR-133a, which regulates changes in AGT expression in response to dietary salt intake.
Acknowledgments:
Sources of Funding: This work was supported by NIH grant R01HL133077.
Footnotes
Disclosures: Nothing to disclose for all authors.
Reference List
- (1).Hao S, Hao M, Ferreri NR. Renal-Specific Silencing of TNF (Tumor Necrosis Factor) Unmasks Salt-Dependent Increases in Blood Pressure via an NKCC2A (Na(+)-K(+)-2Cl(-) Cotransporter Isoform A)-Dependent Mechanism. Hypertension 2018; 71(6):1117–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hao S, Salzo J, Hao M, Ferreri NR. Regulation of NKCC2B by TNF-alpha in response to salt restriction. Am J Physiol Renal Physiol 2020; 318(1):F273–F282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 2007; 59(3):251–287. [DOI] [PubMed] [Google Scholar]
- (4).Lavoie JL, Lake-Bruse KD, Sigmund CD. Increased blood pressure in transgenic mice expressing both human renin and angiotensinogen in the renal proximal tubule. Am J Physiol Renal Physiol 2004; 286(5):F965–F971. [DOI] [PubMed] [Google Scholar]
- (5).Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension 2011; 57(3):355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rohrwasser A, Morgan T, Dillon HF, Zhao L, Callaway CW, Hillas E, Zhang S, Cheng T, Inagami T, Ward K, Terreros DA, Lalouel JM. Elements of a paracrine tubular renin-angiotensin system along the entire nephron. Hypertension 1999; 34(6):1265–1274. [DOI] [PubMed] [Google Scholar]
- (7).Ying J, Stuart D, Hillas E, Gociman BR, Ramkumar N, Lalouel JM, Kohan DE. Overexpression of mouse angiotensinogen in renal proximal tubule causes salt-sensitive hypertension in mice. Am J Hypertens 2012; 25(6):684–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Majid DS, Prieto MC, Navar LG. Salt-Sensitive Hypertension: Perspectives on Intrarenal Mechanisms. Curr Hypertens Rev 2015; 11(1):38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Reinhold SW, Kruger B, Barner C, Zoicas F, Kammerl MC, Hoffmann U, Bergler T, Banas B, Kramer BK. Nephron-specific expression of components of the renin-angiotensin-aldosterone system in the mouse kidney. J Renin Angiotensin Aldosterone Syst 2012; 13(1):46–55. [DOI] [PubMed] [Google Scholar]
- (10).Ramkumar N, Kohan DE. Proximal tubule angiotensinogen modulation of arterial pressure. Curr Opin Nephrol Hypertens 2013; 22(1):32–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Satou R, Miyata K, Katsurada A, Navar LG, Kobori H. Tumor necrosis factor-{alpha} suppresses angiotensinogen expression through formation of a p50/p50 homodimer in human renal proximal tubular cells. Am J Physiol Cell Physiol 2010; 299(4):C750–C759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Flesch M, Hoper A, Dell'Italia L, Evans K, Bond R, Peshock R, Diwan A, Brinsa TA, Wei CC, Sivasubramanian N, Spinale FG, Mann DL. Activation and functional significance of the renin-angiotensin system in mice with cardiac restricted overexpression of tumor necrosis factor. Circulation 2003; 108(5):598–604. [DOI] [PubMed] [Google Scholar]
- (13).Liang M, Liu Y, Mladinov D, Cowley AW Jr, Trivedi H, Fang Y, Xu X, Ding X, Tian Z. MicroRNA: a new frontier in kidney and blood pressure research. Am J Physiol Renal Physiol 2009; 297(3):F553–F558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Liu Y, Usa K, Wang F, Liu P, Geurts AM, Li J, Williams AM, Regner KR, Kong Y, Liu H, Nie J, Liang M. MicroRNA-214–3p in the Kidney Contributes to the Development of Hypertension. J Am Soc Nephrol 2018; 29(10):2518–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Mladinov D, Liu Y, Mattson DL, Liang M. MicroRNAs contribute to the maintenance of cell-type-specific physiological characteristics: miR-192 targets Na+/K+-ATPase beta1. Nucleic Acids Res 2013; 41(2):1273–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Trionfini P, Benigni A, Remuzzi G. MicroRNAs in kidney physiology and disease. Nat Rev Nephrol 2015; 11(1):23–33. [DOI] [PubMed] [Google Scholar]
- (17).Baker MA, Wang F, Liu Y, Kriegel AJ, Geurts AM, Usa K, Xue H, Wang D, Kong Y, Liang M. MiR-192–5p in the Kidney Protects Against the Development of Hypertension. Hypertension 2019; 73(2):399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kriegel AJ, Baker MA, Liu Y, Liu P, Cowley AW Jr, Liang M. Endogenous microRNAs in human microvascular endothelial cells regulate mRNAs encoded by hypertension-related genes. Hypertension 2015; 66(4):793–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wu G, Jose PA, Zeng C. Noncoding RNAs in the Regulatory Network of Hypertension. Hypertension 2018; 72(5):1047–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM, Dorn GW. MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ Res 2010; 106(1):166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Liu B, Lan M, Wei H, Zhang D, Liu J, Teng J. Downregulated microRNA133a induces HUVECs injury: Potential role of the (pro) renin receptor in angiotensin IIdependent hypertension. Mol Med Rep 2019; 20(3):2796–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Sharma NM, Nandi SS, Zheng H, Mishra PK, Patel KP. A novel role for miR-133a in centrally mediated activation of the renin-angiotensin system in congestive heart failure. Am J Physiol Heart Circ Physiol 2017; 312(5):H968–H979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hao S, Zhao H, Darzynkiewicz Z, Battula S, Ferreri NR. Expression and function of NFAT5 in medullary thick ascending limb (mTAL) cells. Am J Physiol Renal Physiol 2009; 296(6):F1494–F1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Liao XB, Zhang ZY, Yuan K, Liu Y, Feng X, Cui RR, Hu YR, Yuan ZS, Gu L, Li SJ, Mao DA, Lu Q, Zhou XM, de Jesus Perez VA, Yuan LQ. MiR-133a modulates osteogenic differentiation of vascular smooth muscle cells. Endocrinology 2013; 154(9):3344–3352. [DOI] [PubMed] [Google Scholar]
- (25).Mughal W, Martens M, Field J, Chapman D, Huang J, Rattan S, Hai Y, Cheung KG, Kereliuk S, West AR, Cole LK, Hatch GM, Diehl-Jones W, Keijzer R, Dolinsky VW, Dixon IM, Parmacek MS, Gordon JW. Correction to: Myocardin regulates mitochondrial calcium homeostasis and prevents permeability transition. Cell Death Differ 2020; 27(1):404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hao S, Bellner L, Ferreri NR. NKCC2A and NFAT5 regulate renal TNF production induced by hypertonic NaCl intake. Am J Physiol Renal Physiol 2013; 304(5):F533–F542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Amasheh S, Barmeyer C, Koch CS, Tavalali S, Mankertz J, Epple HJ, Gehring MM, Florian P, Kroesen AJ, Zeitz M, Fromm M, Schulzke JD. Cytokine-dependent transcriptional down-regulation of epithelial sodium channel in ulcerative colitis. Gastroenterology 2004; 126(7):1711–1720. [DOI] [PubMed] [Google Scholar]
- (28).Escalante BA, Ferreri NR, Dunn CE, McGiff JC. Cytokines affect ion transport in primary cultured thick ascending limb of Henle's loop cells. Am J Physiol 1994; 266(6 Pt 1):C1568–C1576. [DOI] [PubMed] [Google Scholar]
- (29).Rezaiguia S, Garat C, Delclaux C, Meignan M, Fleury J, Legrand P, Matthay MA, Jayr C. Acute bacterial pneumonia in rats increases alveolar epithelial fluid clearance by a tumor necrosis factor-alpha-dependent mechanism. J Clin Invest 1997; 99(2):325–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Castillo A, Islam MT, Prieto MC, Majid DS. Tumor necrosis factor-alpha receptor type 1, not type 2, mediates its acute responses in the kidney. Am J Physiol Renal Physiol 2012; 302(12):F1650–F1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Chen CC, Pedraza PL, Hao S, Stier CT, Ferreri NR. TNFR1-deficient mice display altered blood pressure and renal responses to ANG II infusion. Am J Physiol Renal Physiol 2010; 299(5):F1141–F1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ingelfinger JR, Pratt RE, Ellison K, Dzau VJ. Sodium regulation of angiotensinogen mRNA expression in rat kidney cortex and medulla. J Clin Invest 1986; 78(5):1311–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Singh I, Grams M, Wang WH, Yang T, Killen P, Smart A, Schnermann J, Briggs JP. Coordinate regulation of renal expression of nitric oxide synthase, renin, and angiotensinogen mRNA by dietary salt. Am J Physiol 1996; 270(6 Pt 2):F1027–F1037. [DOI] [PubMed] [Google Scholar]
- (34).Schiessl IM, Rosenauer A, Kattler V, Minuth WW, Oppermann M, Castrop H. Dietary salt intake modulates differential splicing of the Na-K-2Cl cotransporter NKCC2. Am J Physiol Renal Physiol 2013; 305(8):F1139–F1148. [DOI] [PubMed] [Google Scholar]
- (35).Todorov V, Muller M, Schweda F, Kurtz A. Tumor necrosis factor-alpha inhibits renin gene expression. Am J Physiol Regul Integr Comp Physiol 2002; 283(5):R1046–R1051. [DOI] [PubMed] [Google Scholar]
- (36).Pohl M, Kaminski H, Castrop H, Bader M, Himmerkus N, Bleich M, Bachmann S, Theilig F. Intrarenal renin angiotensin system revisited: role of megalin-dependent endocytosis along the proximal nephron. J Biol Chem 2010; 285(53):41935–41946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Lantelme P, Rohrwasser A, Gociman B, Hillas E, Cheng T, Petty G, Thomas J, Xiao S, Ishigami T, Herrmann T, Terreros DA, Ward K, Lalouel JM. Effects of dietary sodium and genetic background on angiotensinogen and Renin in mouse. Hypertension 2002; 39(5):1007–1014. [DOI] [PubMed] [Google Scholar]
- (38).Pratt RE, Zou WM, Naftilan AJ, Ingelfinger JR, Dzau VJ. Altered sodium regulation of renal angiotensinogen mRNA in the spontaneously hypertensive rat. Am J Physiol 1989; 256(3 Pt 2):F469–F474. [DOI] [PubMed] [Google Scholar]
- (39).Shao W, Seth DM, Prieto MC, Kobori H, Navar LG. Activation of the renin-angiotensin system by a low-salt diet does not augment intratubular angiotensinogen and angiotensin II in rats. Am J Physiol Renal Physiol 2013; 304(5):F505–F514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ben-Dov IZ, Tan YC, Morozov P, Wilson PD, Rennert H, Blumenfeld JD, Tuschl T. Urine microRNA as potential biomarkers of autosomal dominant polycystic kidney disease progression: description of miRNA profiles at baseline. PLoS One 2014; 9(1):e86856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Chen S, Puthanveetil P, Feng B, Matkovich SJ, Dorn GW, Chakrabarti S. Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J Cell Mol Med 2014; 18(3):415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol 2002; 12(9):735–739. [DOI] [PubMed] [Google Scholar]
- (43).Wei Q, Mi QS, Dong Z. The regulation and function of microRNAs in kidney diseases. IUBMB Life 2013; 65(7):602–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Castaldi A, Zaglia T, Di M,V, Carullo P, Viggiani G, Borile G, Di SB, Schiattarella GG, Gualazzi MG, Elia L, Stirparo GG, Colorito ML, Pironti G, Kunderfranco P, Esposito G, Bang ML, Mongillo M, Condorelli G, Catalucci D. MicroRNA-133 modulates the beta1-adrenergic receptor transduction cascade. Circ Res 2014; 115(2):273–283. [DOI] [PubMed] [Google Scholar]
- (45).Castoldi G, Di Gioia CR, Bombardi C, Catalucci D, Corradi B, Gualazzi MG, Leopizzi M, Mancini M, Zerbini G, Condorelli G, Stella A. MiR-133a regulates collagen 1A1: potential role of miR-133a in myocardial fibrosis in angiotensin II-dependent hypertension. J Cell Physiol 2012; 227(2):850–856. [DOI] [PubMed] [Google Scholar]
- (46).Torella D, Iaconetti C, Catalucci D, Ellison GM, Leone A, Waring CD, Bochicchio A, Vicinanza C, Aquila I, Curcio A, Condorelli G, Indolfi C. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ Res 2011; 109(8):880–893. [DOI] [PubMed] [Google Scholar]
- (47).Huang W, Liu H, Wang T, Zhang T, Kuang J, Luo Y, Chung SS, Yuan L, Yang JY. Tonicity-responsive microRNAs contribute to the maximal induction of osmoregulatory transcription factor OREBP in response to high-NaCl hypertonicity. Nucleic Acids Res 2011; 39(2):475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Lin DH, Yue P, Pan C, Sun P, Wang WH. MicroRNA 802 stimulates ROMK channels by suppressing caveolin-1. J Am Soc Nephrol 2011; 22(6):1087–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Liu Y, Taylor NE, Lu L, Usa K, Cowley AW Jr., Ferreri NR, Yeo NC, Liang M. Renal medullary microRNAs in Dahl salt-sensitive rats: miR-29b regulates several collagens and related genes. Hypertension 2010; 55(4):974–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Li Z, Chen H, Wang F, Wang Z, Zhao Q, Liu S, Huang B, Lou S, Zuo J. Down-regulation of microRNA-98 promoted apoptosis of TNF-alpha stimulated human fibroblast-like synoviocytes via up-regulating IL-10. Gene 2019; 706:124–130. [DOI] [PubMed] [Google Scholar]
- (51).Xin W, Wang X, Zhang W, Zhu H, Dong R, Zhang J. Tumor Necrosis Factor-alpha Inhibits Bone Marrow Stem Cell Differentiation into Osteoblasts by Downregulating microRNA-34a Expression. Ann Clin Lab Sci 2019; 49(3):324–329. [PubMed] [Google Scholar]
- (52).Yao J, Lin J, He L, Huang J, Liu Q. TNF-alpha/miR-155 axis induces the transformation of osteosarcoma cancer stem cells independent of TP53INP1. Gene 2020; 726:144224. [DOI] [PubMed] [Google Scholar]
- (53).Zhu H, Li Y, Wang MX, Wang JH, Du WX, Zhou F. Analysis of cardiovascular disease-related NF-kappaB-regulated genes and microRNAs in TNFalpha-treated primary mouse vascular endothelial cells. J Zhejiang Univ Sci B 2019; 20(10):803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Majid DS. Tumor necrosis factor-alpha and kidney function: experimental findings in mice. Adv Exp Med Biol 2011; 691:471–480. [DOI] [PubMed] [Google Scholar]
- (55).Reverte V, Gogulamudi VR, Rosales CB, Musial DC, Gonsalez SR, Parra-Vitela AJ, Galeas-Pena M, Sure VN, Visniauskas B, Lindsey SH, Katakam PVG, Prieto MC. Urinary angiotensinogen increases in the absence of overt renal injury in high fat diet-induced type 2 diabetic mice. J Diabetes Complications 2020; 34(2):107448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Liang M Epigenetic Mechanisms and Hypertension. Hypertension 2018; 72(6):1244–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.