

ABSTRACT
Nonalcoholic steatohepatitis (NASH) is one of the most common chronic liver diseases in the world, yet no pharmacotherapies are available. The lack of translational animal models is a major barrier impeding elucidation of disease mechanisms and drug development. Multiple preclinical models of NASH have been proposed and can broadly be characterized as diet-induced, deficiency-induced, toxin-induced, genetically induced, or a combination of these. However, very few models develop advanced fibrosis while still reflecting human disease etiology or pathology, which is problematic since fibrosis stage is considered the best prognostic marker in patients and an important endpoint in clinical trials of NASH. While mice and rats predominate the NASH research, several other species have emerged as promising models. This review critically evaluates animal models of NASH, focusing on their ability to develop advanced fibrosis while maintaining their relevance to the human condition.
Keywords: nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, fibrosis, animal models, translational research
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a nutritional disease driven by overnutrition and/or consumption of an unhealthy diet, which affects ∼25% of the world's population (1–4). NAFLD covers a spectrum of disease states, including bland steatosis, nonalcoholic steatohepatitis (NASH), and fibrosis (5). Notably, the extent of liver fibrosis constitutes the most important prognostic hallmark and is a principal endpoint in several clinical trials of NASH (6, 7). However, although several compounds are in clinical trials, no efficient pharmacological therapy is yet available.
One of the major obstacles limiting our understanding of disease mechanisms, identification of relevant therapeutic targets, and improvement in treatment options is the poor translational validity of many of the available animal models. Preclinical models of NASH show considerable variability in the development and severity of hepatic fibrosis and only a few exhibit the defining histological features of human NASH. Progression toward advanced hepatic fibrosis is often challenging, commonly requiring micronutrient-deficient diets or hepatotoxins to induce severe fibrosis, which poorly reflects disease etiology and lacks important construct validity (8). While mice and rats are frequently used as NASH research models, and do offer many advantages, other species have shown promise in reflecting different or broader aspects of the disease and its progression. In this review, we critically evaluate animal models of NASH with an emphasis on their ability to develop hepatic fibrosis and their relevance to human disease.
Current Status of Knowledge
Hallmarks of NASH-associated fibrosis in humans
The diagnosis of NASH is based on histology and requires the simultaneous presence of steatosis, ballooning hepatocytes, and lobular inflammation (9). Although fibrosis is not strictly part of the histological definition of NASH, it remains the best predictor of both overall and liver-related mortality in patients (7, 10). Importantly, risk of death or liver transplantation increases with fibrosis stage (7), scored according to the recommendations from the NASH Clinical Research Network (Table 1) (11). NAFLD patients exhibit a distinct distribution pattern of hepatic fibrosis. In contrast to other liver diseases, in which central veins may only be involved at late stages, NAFLD-related fibrosis originates pericentrally in zone 3 with the formation of delicate perisinusoidal fibrosis (12–14). Another hallmark is the presence of lobular perisinusoidal fibrosis and thin collagen fibers surrounding hepatocytes in a characteristic chicken-wire pattern (12–14) (Figure 1). As the disease progresses, central veins and portal areas are linked by fibrous bridges denoting the stage of advanced fibrosis (F3), which culminates in hepatic cirrhosis (F4) where fibrotic septa link portal and central areas to form nodules that isolate islands of hepatic parenchyma (13, 18). Portal areas may be spared during progression, but periportal fibrosis is usually present (13). Notably, early fibrosis distribution may differ in adult and pediatric NAFLD. Portal fibrosis predominates in children (68.4%, 26 of 38 patients), although the characteristic pattern of “adult” fibrosis has also been reported (28.9%, 11 of 38 patients) (19).
TABLE 1.
Histopathological scoring of NASH-related fibrosis1
| Score | Definition |
|---|---|
| 0 | None |
| 1 | Perisinusoidal or periportal |
| 1A | Mild, zone 3, perisinusoidal fibrosis |
| 1B | Moderate, zone 3, perisinusoidal fibrosis |
| 1C | Portal/periportal fibrosis |
| 2 | Portal/periportal and perisinusoidal fibrosis |
| 3 | Bridging fibrosis |
| 4 | Cirrhosis |
1Fibrosis scores defined according to the NASH Clinical Research Network (11). NASH, nonalcoholic steatohepatitis.
FIGURE 1.

Histopathology of human NASH and models with advanced fibrosis following a Western diet. (A) Perisinusoidal fibrosis (F1) located around a central vein in a patient with NASH (Sirius Red Hemalun stain, ×10 magnification) Modified from reference 14 with permission. (B) Bridging fibrosis (F3) and (C) cirrhosis (F4) with thick and extensive bridging in patients with NASH. Arrows denote fibrous bridges and arrowheads indicate perisinusoidal fibrosis, P: portal tract, V: hepatic vein (Elastica van Gieson stain; scale bar, 250 μm). Modified from reference 15 with permission from Wiley. (D) Bridging fibrosis in the DIAMOND mouse (Sirius Red stain, ×10 magnification). Modified from reference 16 with permission. (E) Bridging fibrosis in the guinea pig NASH model also showing the prototypical perisinusoidal (chicken-wire) pattern (Picro Sirius Red stain and Weigert's hematoxylin; scale bar, 200 μm). (F) Bridging fibrosis (arrows) with perisinusoidal collagen deposition in hamsters, inset is part of the original figure, but is not used in this reproduction (Sirius Red stain, ×10 magnification). Modified from reference 17 with permission. Permissions to reuse have been obtained for all figures and the documentation is provided in the Supplemental Material. DIAMOND, diet-induced animal model of nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis.
Activation of the hepatic stellate cells (HSCs) enhances their expression of α-smooth muscle actin (αSMA) and is the fundamental event in hepatic fibrogenesis (Figure 2). Several core fibrogenic pathways are conserved across various liver diseases, including transforming growth factor β (TGFβ), connective tissue growth factor (CTGF), oxidative stress, platelet-derived growth factor (PDGF), vascular endothelial growth factor, and integrins (20). Pathways more specific to NASH and NASH fibrosis have also emerged and include accumulation of free cholesterol, advanced glycation end-products, hedgehog signaling, Toll-like receptors (TLRs), and inflammasome activation (20, 21). In addition, endoplasmic reticulum (ER) stress and apoptosis are also important to NASH progression (22, 23). Microarray analysis of patients with mild (F0–1, n = 23) and severe (n = 32, F3–4) NASH identified several pathways (gene ontology analysis) associated with advanced fibrosis (29). Patients with advanced fibrosis displayed enhanced biological function pathways such as “cell adhesion and migration” and “development and extracellular matrix (ECM) organization,” whereas functions related to “metabolism” were decreased (29). Furthermore, 64 genes could consistently differentiate severe from mild NASH and were associated with pathways related to “tissue remodeling/regeneration,” “progenitor cells,” “cancer,” and “cardiovascular disease.” A detailed description of the specific genes and pathways regulated in human NASH and NASH-associated fibrosis is not within the scope of the current review, but additional information can be found in selected references (20, 21, 29–31). Importantly, these genes and pathways may serve as a reference when validating experimental models of NASH with advanced fibrosis.
FIGURE 2.
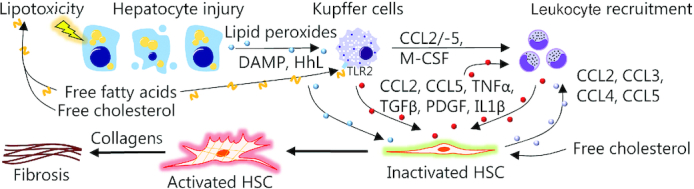
Initiation of NASH-related fibrosis. Nutrient overload results in lipotoxicity and hepatocyte injury leading to the release of a plethora of danger signals (e.g., DAMPs, HhL, and reactive oxygen species) that interact with HSCs and Kupffer cells (resident liver macrophages) in conjunction with free fatty acids and free cholesterol to promote inflammation (24–26). Monocytes are recruited from the circulation, in part, via CCL2, CCL5, and M-CSF released by Kupffer cells. Following differentiation into monocyte-derived macrophages, these infiltrating cells promote hepatic inflammation and recruitment of additional circulating leucocytes by contributing to the release of CCL2, CCL3, and CCL5 (27). Hepatocyte injury, Kupffer cells, and infiltrating macrophages facilitate activation of HSCs, which normally reside in a quiescent state in the healthy liver, but differentiate into ECM-producing myofibroblasts upon activation. HSC collagen production is promoted by TGFβ while PDGF promotes HSC proliferation and migration (27, 28). Macrophages also enhance HSC survival via TNFα and IL1β. In addition to this, HSCs themselves exert immunoregulatory effects by releasing CCL2, CCL3, CCL4, and CCL5, which recruit monocytes from the circulation, thereby sustaining their own activation (27). Thus, chronic inflammation and continual activation of HSCs ultimately result in liver fibrosis or even cirrhosis. CCL, chemokine (c-c motif) ligand; DAMP, damage-associated molecular pattern; ECM, extracellular matrix; HhL, hedgehog ligand; HSC, hepatic stellate cells; M-CSF, macrophage colony-stimulating factor; NASH, nonalcoholic steatohepatitis; PDGF, platelet-derived growth factor; TGFβ, transforming growth factor β; TLR2, Toll-like receptor 2.
During fibrogenesis, the composition of the hepatic ECM changes considerably and ECM components directly contribute to the remodeling process by communicating with surrounding cells (32). Collagens constitute the major part of the fibrotic ECM, with type I collagen as the most abundant component (33). In general, mild fibrosis is characterized by an initial increase in collagen types I and III, which increase further during fibrosis progression. In advanced fibrosis and cirrhosis, other ECM components are increased such as collagen types V, VI, X, and XVIII, elastin, fibronectin, and laminin, while collagen type XII may be decreased (32).
Preclinical models of NASH-related fibrosis
NASH research is challenged by the lack of animal models that adequately mimic etiology, pathogenesis, and, that due to the importance of histology in diagnosis and trial outcomes, reproduce the histological features of human NASH (8). Furthermore, there is substantial interstrain and interspecies variation in fibrosis development and severity: for example, some mouse strains readily develop hepatic fibrosis, while others are practically resistant (34). In general, animal models of NASH-associated fibrosis can be characterized as diet-induced, deficiency-induced, toxin-induced, genetically induced, or as a mixture of these modalities. The most commonly applied models are discussed below.
Diet-induced models
The diet-induced models apply a broad range of dietary regimens ranging from alternating contents and sources of fat to different amounts of cholesterol and additional supplements promoting a NASH-like phenotype. The following sections describe selected dietary approaches to exemplify principles of disease induction and associated mechanisms including potential limitations to consider when translating findings to human patients.
High-fat diets
The straightforward high-fat diets (HFDs) contain up to 70% of calories from fat. The HFD often leads to obesity and insulin resistance; however, development of NAFLD in laboratory animals requires prolonged feeding periods and the condition is typically less severe than in humans (35, 36). Accordingly, mice fed an HFD (60% of calories from fat) developed steatosis within 10 wk, mild inflammation after 34 wk, and slight perivenular fibrosis after 50 wk (37). Similar to patients (38), gene expression suggested enhanced de novo lipogenesis in HFD-fed animals while genes related to inflammation and prototypical fibrosis monocyte chemoattractant protein 1 [(Mcp1/Ccl2), Tnfa, Acta2 (actin alpha 2, encoding αSMA), Tgfb1, and collagen 1a1 (Col1a1)] were also increased (37). In rats, steatosis, mild inflammation, and increased apoptosis were present after 4 wk of being fed an HFD (58% of calories from fat) and mild pericellular and sinusoidal fibrosis (F1) was detected after 24 wk (39). Although capturing some of the central fibrogenic mechanisms, these models are limited by the mild phenotype and only reflect the earliest stages of NASH, indicating that other nutritional triggers are required to induce disease progression, especially fibrosis (Table 2).
TABLE 2.
Diet-induced NASH-associated fibrosis1
| Diet and species (ref) | Fibrosis (time to development) | Comments/applicable to advanced NASH fibrosis? |
|---|---|---|
| HFD | ||
| Mice (37) | Slight perivenular fibrosis (50 wk) | No progression beyond early/mild fibrosis; not applicable |
| Rats (39) | Mild perisinusoidal fibrosis (24 wk) | — |
| High-fat, high-cholesterol ± cholate | ||
| Hamsters (43) | Bridging fibrosis (12 wk) | Periportal microvesicular steatosis unlike adult NASH; not applicable if using toxic levels of dietary cholesterol |
| Mice (42) | Mild perisinusoidal fibrosis (week 30); bridging fibrosis (week 24, with dietary cholate) | Increased glucose and insulin tolerance; very high cholesterol content; cholate may be liver toxic; other models seem more applicable |
| Rabbits (48) | Central-central and portal-central bridging fibrosis (week 36) | Liver triglycerides not increased; nominal reduction of ALT and AST; improved insulin sensitivity and blood glucose; not applicable |
| Rats (45, 46) | Mild perisinusoidal fibrosis in one-third of animals (week 16); mild/moderate central and portal fibrosis (week 24–48); mild/moderate fibrosis in most and cirrhosis in some animals (week 9 with dietary cholate) | No progression beyond early fibrosis unless using very high levels of cholesterol and cholate; cholate may be liver toxic; not applicable |
| Western | ||
| DIAMOND mice (16) | Central perisinusoidal fibrosis (week 16–24); bridging fibrosis (week 52) | Histology and transcriptome mirror human NASH; long feeding time to achieve advanced fibrosis; applicable |
| Guinea pig (55) | Central perisinusoidal fibrosis (week 16); central-central and central-portal bridging fibrosis (week 25) | Histology and gene expression mirror human NASH; not obese; glucose tolerance not compromised; applicable |
| Hamster (17) | Bridging fibrosis (week 12–16) | No ballooning and predominantly microvesicular; could be applicable |
| Mice (52–54) | Central and portal perisinusoidal fibrosis (week 16–50); minority of animals (2/9) develop advanced fibrosis (week 50) | Advanced fibrosis requires lengthy feeding regimens and only develops in few animals; no advantages over other models |
| Rat (51) | Absent (week 16) | No fibrosis; periportal steatosis (unlike adult NASH); not applicable |
| Ossabaw pigs and Ossabaw miniature pig(56–58) | Mild fibrosis in half (3/6) of animals (week 16); periportal sinusoidal/moderate fibrosis (week 24); possibly bridging fibrosis (week 34)2 | Metabolic profile mimics human; predominantly microvesicular steatosis unlike humans; normal liver anatomy differs substantially from humans; portal-portal bridges present in normal liver; fibrosis pattern does not mimic adult NASH; not applicable |
| MCD | ||
| Mice (59) | Periportal fibrosis (week 10); perisinusoidal and periportal fibrosis (week 14); bridging fibrosis (week 16) | No ballooning; extensive weight loss; enhanced glucose and insulin tolerance; dissimilar molecular signaling pathways; fibrosis originating periportally (mice) unlike adult NASH; disease etiology does not mimic humans; not applicable |
| Rat (60, 61) | Central pericellular fibrosis (week 12); bridging fibrosis (week 17); cirrhosis (week 20) | — |
| CD-HFD | ||
| Mice (62) | Mild perisinusoidal (52 wk) | No advanced fibrosis; disease etiology does not mimic humans; not applicable |
| CDAA | ||
| Mice (63) | Mild/moderate (week 20) | No advanced fibrosis; disease etiology does not mimic humans; glucose/insulin tolerance not affected; not applicable |
| Rats (54) | Bridging fibrosis with nodular formation (week 12) | Rapid induction of advanced fibrosis and histology and mirror human NASH; disease etiology does not mimic humans; weight gain is decreased; other models may reflect dietary NASH etiology more closely |
| CDAA-HFD | ||
| Mice (64–66) | Perisinusoidal and periportal fibrosis (week 24); portal-portal bridging fibrosis (week 48) | Disease induction is faster compared with CDAA in mice; weight gain decreased; could be applicable, but other models seem more suitable |
| Göttingen minipigs (67) | Chicken-wire fibrosis (week 8) | Normal liver anatomy differs substantially from humans; weight gain decreased; no hepatocyte ballooning; no advanced fibrosis; not applicable |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; CD-HFD, choline-deficient, high-fat diet; CDAA, choline-deficient, l-amino acid–defined; CDAA-HFD, choline-deficient, l-amino acid–defined high-fat diet; DIAMOND, diet-induced animal model of nonalcoholic fatty liver disease; HFD, high-fat diet; MCD, methionine and choline deficient; NASH, nonalcoholic steatohepatitis; Ref, reference.
Scoring system not defined, complicating the determination of fibrosis stage.
High-fat and -cholesterol diets
Cholesterol appears to drive the progression from steatosis to NASH (24) and may act synergistically with lipids to induce NASH, possibly by directly damaging organelle membranes and increasing production of reactive oxygen species. In turn, this leads to hepatocellular damage, which promotes inflammation, hepatocellular decay, and eventually activates repair mechanisms including fibrogenesis (40, 41). Indeed, cholesterol and free cholesterol concentrations correlate with hepatic fibrosis and a sharp increase in intracellular free cholesterol accompany the onset of fibrosis (34).
In line with the dietary characteristics associated with human NAFLD/NASH, cholesterol has been included in experimental diets to enhance disease severity. Although applied amounts (>0.5%) of cholesterol are significantly higher than those found in a human diet, studies show a clear effect on hepatic endpoints including fibrosis (Table 2). Severe steatosis, inflammation, and mild perisinusoidal fibrosis (F1) were induced in mice following high-fat (33% of calories from fat), high-cholesterol (1%) feeding for 30 wk (42). The addition of cholate (0.5%), which promotes intestinal fat and cholesterol absorption, to a high-cholesterol (1.25%) diet induced hepatic inflammation, HSC activation, and bridging fibrosis (F3) after 24 wk in mice (42). Pathway analysis suggested increased TGFβ signaling, but not any other pathways directly related to fibrosis. However, hepatic triglyceride concentrations and cholesterol synthesis decreased while insulin and glucose tolerance actually improved compared with controls, thereby diverging from human NASH (42).
Hamsters rapidly developed bridging fibrosis alongside inflammation and microvesicular steatosis located in portal areas when fed a diet containing fat (∼23%), cholesterol (1%), and deoxycholate (0.25%) over 12 wk (43). However, high amounts of dietary cholesterol (≥1%) are potentially hepatotoxic in hamsters and impair normal lipoprotein metabolism, which may affect the translatability of findings (44). Following high-fat (30% calories from fat)/high-cholesterol (2%) feeding, 33% (4/12) of rats developed mild perisinusoidal fibrosis after 16 wk. From week 24 to 48, fibrosis progressed, with the majority (80%, 8/10) of animals displaying mild-to-moderate central and portal perisinusoidal fibrosis and a few (20%, 2/10) developing bridging fibrosis (45). Cirrhosis was induced in 40% of rats (2/5) after only 9 wk by increasing the cholesterol content further (2.5%) in combination with high fat and cholate (2%), although the reported findings were from a relatively small group size, potentially affecting the reproducibility of results (46). Hepatic Tgfb1, Col1a1, and Col4a1 gene expression was increased, but in contrast to humans, de novo lipogenesis was unaltered and/or downregulated and insulin concentrations tended to decrease (38, 46). Furthermore, it should be considered whether high cholesterol (>0.5%) amounts that by far exceed human consumption and the addition of cholate truly reflect human patho-etiology (8). Importantly, compared with an HFD alone (60% of calories from fat), the addition of cholate (0.5%) reduced hepatic triglyceride concentrations in a fibroblast growth factor 21–dependent manner (47). Moreover, body fat accumulation, insulin resistance, and glucose tolerance were also improved by cholate (47). This suggests that cholate may reverse the metabolic changes associated with NAFLD, further questioning the usefulness of cholate in these studies. In rabbits, a high-fat and high-cholesterol (0.75%) diet induced formation of central-central and central-portal bridges after 36 wk (48). Similar to patients, Tlr2, Tlr4, Tnfa, Tgfb1, Col1a1, Col3a1, and tissue inhibitor of metalloproteinases (Timp) 1 (Timp1) and Timp2 gene levels were increased in the liver (48). In contrast to what is reported in humans with NASH, plasma concentrations of hepatic injury markers alanine transaminase and aspartate transaminase decreased nominally, hepatic triglyceride concentrations were not increased, and insulin sensitivity was enhanced (48).
Overall, high-cholesterol diets can be used to induce advanced fibrosis. However, a common challenge is the simultaneous suppression of endogenous cholesterol synthesis (42, 46, 48) contrary to humans, where cholesterol synthesis is increased and associated with disease severity (49).
Western/cafeteria diets
Other diets attempt to mimic the unhealthy eating habits of humans more closely by containing a mixture of fat, sugar, and cholesterol. However, the compositions of the individual regimens of these so-called Western or cafeteria diets vary substantially between studies, making interstudy comparisons difficult. Sugar-containing drinking water may also be added as increased consumption of soft drinks (i.e., high in sugars) has been associated with NAFLD (50). Feeding rats a Western diet (40% fat, 40% sugar, and 2% cholesterol) for 16 wk induced periportal steatosis (i.e., not the same origin as is most commonly recorded in adult NASH patients) and inflammation, but not hepatocyte ballooning or fibrosis (51). While liver MCP1 protein concentrations were increased, TNFA concentrations were reduced compared with healthy controls (51). Portal and bridging fibrosis was observed in hamsters alongside panlobular microvesicular steatosis, obesity, and insulin resistance when fed a Western diet (40.8% fat, 0.5% cholesterol, and sugar water) for 12–16 wk (Figure 1) (17). However, the underlying fibrogenic mechanisms were not assessed in the study (17). Mice fed a Western diet (40% of calories from fat, added sucrose/fructose, and 0.15–2% cholesterol) with and without sugar water for 24–50 wk developed obesity, insulin resistance, and progressive hepatic steatosis and inflammation (52, 53). Similar to humans, perisinusoidal and -cellular (chicken-wire) fibrosis was noted around central veins and portal areas (F2) after 16 wk. Advanced fibrosis (≥F3) was absent (52, 53) or only seen in a few mice (22%, 2/9) after 50 wk (54). In these models, expression of genes related to inflammation (Tnfa and Mcp1) and fibrosis [Acta2 (αSMA), Col1a1, Col3a1, and Timp1] was increased in the liver (52, 53, 68), indicating activation of core fibrogenic pathways. Hepatocyte ballooning was present in some models (68), while not in others (52), or more closely resembling microvesicular steatosis than hepatocellular ballooning (53). This reflects another central point of criticism in many of the current preclinical models: is human NASH sufficiently well modeled in the absence of ballooning hepatocytes?
Recently, the diet-induced animal model of nonalcoholic fatty liver disease (DIAMOND) mouse was reported to reflect human NASH after ingestion of a more physiologically relevant diet [42% of calories from fat, 34% sucrose, 0.1% cholesterol, and sugar water (4.2% fructose/glucose solution equivalent of commercial soft drinks)] (16). The DIAMOND mouse is an isogenic cross between C57BL/6J and 129S1/SvImJ mice and this isogenic strain (B6/129) develops obesity, insulin resistance, and hepatic steatosis, which sequentially progresses to NASH with histopathology and molecular pathways similar to humans. Pericellular fibrosis located around the central vein (F1) was present after 16–24 wk, with some animals displaying both portal and central fibrosis (F2). By week 52, the majority of the animals had developed bridging fibrosis (Figure 1). Furthermore, the DIAMOND mice displayed enhanced de novo lipogenesis, ER stress, as well as apoptosis and transcriptomic analysis supported that the mice mimic the transcriptome of human NASH (16). Accordingly, the model is promising for the study of disease mechanisms and intervention strategies, although the feeding period required to reach advanced fibrosis is long and weight loss appeared after 44 wk in animals fed both the Western and control diet, possibly indicating the onset of senescence (16).
Guinea pigs fed a Western diet (45% of calories from fat, 15% sucrose, 0.35% cholesterol) also develop the central histological hallmarks (steatosis, ballooning, and inflammation) of human NASH, constituting a relatively novel and perhaps underappreciated model of NASH (55). Molecular pathways reflect human pathophysiology and suggest enhanced de novo lipogenesis [sterol-regulatory element-binding transcription factor (Srebf1; encoding sterol regulatory element-binding protein 1) and fatty acid synthase (Fasn)], inflammation (Mcp1, Il8, and Tnfa) and upregulation of core fibrogenic pathways [Tgfb1, Pdgfb, Acta2 (αSMA), Col1a1, and Col3a1] (69). The model is further supported by a response to a pharmacological intervention similar to humans (70). While guinea pigs were not obese relative to feed pellet–fed controls and glucose homeostasis did not appear compromised (55, 71), impaired glucose tolerance and insulin resistance have previously been reported in high-fat–fed guinea pigs (72). These animals were fed exclusively with carbohydrates originating from sucrose and fructose (72), which may explain differences between studies. Central perisinusoidal (chicken-wire) fibrosis (F1) was present after 16 wk, progressing to bridging fibrosis (F3) between central and portal areas after 25 wk (55) (Figure 1). Furthermore, in contrast to the HDL-dominated plasma profile of rats, mice, hamsters, and rabbits, both guinea pigs and humans have an LDL-dominant profile, adding to their translational relevance (73).
Considering larger animal models, Ossabaw pigs fed a Western diet (43% of calories from fat, ≤20% fructose/high-fructose corn syrup, 2% cholesterol) or Ossabaw miniature pigs fed the same diet with decreased amounts of choline and 0.5% cholate for 16–34 wk developed obesity, insulin resistance, and high concentrations of circulating cholesterol (56–58), thereby, mimicking the metabolic disturbances often seen in human NASH. However, in contrast to patients, the hepatic lipid accumulation predominantly presented as microvesicular steatosis. While ballooning hepatocytes were reported (56–58), they may not have completely mimicked typical human-like ballooning [as also mentioned by others (74)], which is also a challenge in many of the small-animal models, as noted previously. Mild pericellular fibrosis was evident in 50% (3/6) of animals at week 16 and progressed to moderate fibrosis in 83.3% (5/6) of animals at week 24 (57). In contrast, Iberian pigs fed a Western diet (9.1% fat, 12% fructose, 0.7% cholesterol) for 10 wk did not develop fibrosis (75), but this may have been due to the relatively short study duration. At week 34, mRNA levels of TLR4, TLR9, TNFA, and TGFB were increased and F3 fibrosis was reported, indicating that bridging fibrosis may be present in Ossabaw pigs (56). However, the scoring system was not described, making it difficult to ascertain the reported fibrosis stages. Bridging fibrosis was reported in a few (2/14) Göttingen minipigs fed a Western diet (43% fat, 17.8% fructose, 1–2% cholesterol) for 13 mo, with the majority of the animals (9/14) developing mild-to-moderate (F1–2) fibrosis (74). However, steatosis was absent, thereby deviating considerably from the human disease. Thus, induction of advanced fibrosis in pigs is possible, but requires prolonged feeding periods and does not develop in all animals. This may be a challenge considering the smaller group sizes often resulting from the cost of maintaining large-animal models. Importantly, the anatomy of the pig liver differs significantly from that of human livers and almost all other small-animal models. In pigs, the hepatic lobuli are clearly defined and outlined by fibrotic tissue that connects the portal areas [illustrated in (75, 76)]. Thus, portal–portal bridges are already present under physiological conditions and the amount of connective tissue in the normal porcine liver is comparable with amounts in fibrotic livers from humans (76). In pigs with NASH, fibrosis was predominantly located periportally and originated from the septa connecting the portal areas (58), which contrasts with findings from humans (12–14, 18). Furthermore, the quantity and distribution of hepatic connective tissue change from birth until 6 mo of age (77) and may complicate study comparison. It should also be noted that the pigs used in these studies were usually quite young (ranging from 13 d to 10 mo) (56–58, 74, 75). Combined with their periportal fibrosis patterns, which are similar to human pediatric NASH (19), these porcine models may reflect pediatric/juvenile NASH more than adult NASH.
While HFD and Westernized diets attempt to mimic human disease etiology, differences in metabolic profile and hepatic histopathological phenotype compromise reproducibility and limit the translational potential of findings. Similar to high-cholesterol diets, Western diets utilizing even low amounts of cholesterol (0.1%) suppress endogenous cholesterol synthesis (16, 68, 69).
Interestingly, compared with toxin- and deficiency-induced models, mice fed a Western diet displayed a much greater overlap of molecular pathways compared with patients with NASH (78), suggesting that they might better reflect the mechanisms underlying human NASH. However, other analyses did not confirm such a close relation and, as stated by the authors, the overall overlap between the different mouse models and human livers is small (78). Consequently, there is still a need for improvement and critical consideration of biochemical read-outs and pathological endpoints associated with model characteristics and concurrent disease progression.
Role of macronutrients in diet-induced NASH-fibrosis
The amount of sugar used in so-called Western diets varies widely and is usually substantially higher than that of actual human diets in the Western world that they supposedly model, where added sugars are estimated to account for ∼15% of the overall energy intake (79). In rats fed a diet with free fructose (30%) and glucose (30%) or sucrose (60%) for 4 mo, hepatic collagen-III did not differ between groups (80). Similarly, mice fed an HFD (60% fat) for 10 wk supplemented with either 30% fructose or glucose in drinking water developed NASH and mild hepatic fibrosis independent of the sugar type, although insulin sensitivity, glucose tolerance, and obesity were all worsened in fructose-consuming mice (81). While daily fructose consumption has been shown to correlate with increased fibrosis in individuals with NAFLD (82), a meta-analysis from 2014 concluded that there was insufficient evidence to draw conclusions on the effect of sucrose, fructose, or high-fructose corn syrup on NAFLD, including liver fibrosis markers (83). Consequently, it remains unclear if certain types of sugar are more fibrogenic than others.
The type of fatty acids in the diet also affects the development of NASH fibrosis and is an important consideration when formulating experimental diets. Only a Western diet (40% of energy from fat, 22% fructose, 2% cholesterol) with trans-fatty acids but not lard (containing primarily a mixture of saturated and unsaturated fatty acids) induced hepatic fibrosis in ob/ob mice (84). In contrast, mice fed a Western diet (40% of energy from fat, 20% of energy from fructose, and 2% cholesterol) developed more severe NASH with fibrosis using a non–trans- compared with a trans-fat lipid source (85). Together with the much lower consumption of trans fats by humans and since regulatory bodies are limiting their use (discussed below), this advocates the use of other fat sources to model human disease. Interestingly, the amount of SFAs (especially palmitic acid (C16H32O2, hexadecanoic acid)) was much higher in the non–trans-fat diet described previously (44.5%) compared with the trans-fat diet (26.6%) (85). Long-term injections with the SFA palmitate induced liver fibrosis in mice fed a normal diet or HFD, suggesting a role of palmitate in NASH-related fibrosis (86). Moreover, SFAs may be more fibrogenic than PUFA as they induced higher expression of fibrogenic genes [Acta2 (αSMA), Tgfb, and Col1a1] in rats fed an HFD for 8 wk (87). Accordingly, increased accumulation of SFAs (palmitic acid and stearic acid) (C18H36O2, octadecanoic acid) relative to MUFAs was found in patients with NAFLD and this accumulation correlated with disease severity (88). However, it is important to note that the effect of dietary fat and other macronutrients is confounded by other factors, such as micronutrient content and palatability, that differ between the diets being compared, thus further complicating interpretation (89). Clearly, the impact of the various dietary fat sources in relation to NASH fibrosis is complex, but collectively, the data suggest that SFAs are useful in inducing NASH and liver fibrosis in animal models.
Micronutrient-deficiency models
Methionine- and choline-deficient diets
The methionine- and choline-deficient (MCD) diet induces steatohepatitis and fibrosis by impairing β-oxidation and VLDL particle production, both pivotal for hepatic lipid disposal, consequently “trapping” lipids in the liver (36, 38). Compared with mice fed a Western diet (45% of calories from fat, 0.2% cholesterol, and sugar water), liver inflammation, fibrosis, and HSC activation were more severe in MCD mice (90). Steatosis and hepatic inflammation were observed in MCD mice after only 2 wk with periportal (F1) fibrosis depositions present after 10 wk; thus, the initial fibrotic pattern resembled pediatric rather than adult NASH (59). Perisinusoidal and periportal fibrosis (F2, week 14) progressed to bridging fibrosis (F3) at week 16 (59). Similar to mice, MCD rats exhibited progressive development of steatohepatitis with steatosis (week 2), inflammation (week 5), central pericellular fibrosis (week 12), bridging fibrosis (week 17), and cirrhosis, with nodule formation in 89.5% (17/19) of rats by week 20 (60, 61). This was accompanied by an increase in αSMA-positive cells, TGFβ1 protein concentrations, and fibrogenic gene expression [Ccn2 (encoding CTGF), Timp1, Timp2, Col1a1, and Col1a2] in the liver (60, 61). Thus, compared with HFD and Western diets, steatohepatitis and advanced fibrosis (≥F3) are more rapidly induced by the MCD diet, which is an appealing feature of this model. However, enhanced insulin sensitivity and glucose tolerance in addition to weight loss (≤40% within 8 wk and ≤70% after 20 wk) are common (8, 59–61, 91), raising serious concerns regarding animal welfare and questioning the translational relevance of the model. Furthermore, hepatic gene expression patterns suggest that the pathogenesis is different, as MCD models exhibit suppressed de novo lipogenesis (92, 93) in contrast to the enhanced de novo lipogenesis reported in patients (38, 94, 95). Using gene-set enrichment analysis, none or only few KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways overlapped between mice fed an MCD diet for 4 or 8 wk, respectively, and patients with NASH, even when only comparing with patients with fibrosis (78). Consequently, while the MCD diet may rapidly induce a NASH-like liver phenotype with advanced fibrosis and cirrhosis, the disease mechanisms and metabolic context are not similar to those in humans. For example, the substantial weight loss is not clinically relevant and a substantial drawback of the model. Given the availability of other models, particularly Western diet–based models, that more closely mimic human NASH etiology and develop advanced hepatic fibrosis, there seem to be better alternatives and results from the MCD model should be interpreted with caution (8, 35).
To overcome the weight loss associated with the MCD diet, choline deficiency has been combined with an HFD (45% of calories from fat, 17% of calories from sucrose) (62). The choline-deficient–HFD (CD-HFD) induced obesity, glucose intolerance (measured at week 26), hepatic steatosis, and NASH with hepatocellular ballooning and inflammation. In addition, 25% of animals had liver tumors after 52 wk (62). At this time point, CD-HFD animals developed mild pericellular fibrosis and increased hepatic expressions of fibrogenic genes [e.g., Acta2 (αSMA)], macrophage inflammatory protein 1α, Col1a2, Mmp2, Timp1, and Timp2, thereby reflecting the mechanistic pathways of human liver fibrosis (62).
Choline-deficient, l-amino acid–defined diets
Like the MCD diet, the choline-deficient, l-amino acid–defined (CDAA) diet is deficient in choline. In addition, proteins are substituted with an equivalent mixture of l-amino acids (36). In CDAA-fed mice, weight gain is stagnated or similar to that of controls (63, 96), an advantage compared with the MCD diet. After 8 wk, hepatic steatosis was more severe in CDAA mice and inflammation was similar compared with MCD mice. While absent at week 8 (96), fibrosis was reported in CDAA-fed mice after 20 wk with a concomitant increase in fibrogenic and inflammatory gene expression [e.g., Acta2 (αSMA), Tgfb1, Col1a1, Timp1, Tnfa, and Mcp1] (63). However, glucose tolerance, insulin sensitivity, and Srebf1cmRNA levels (regulating de novo lipogenesis) were not affected by the CDAA diet (63). Compared with mice, rats appear to be more susceptible to the fibrogenic stimuli imposed by the CDAA diet and developed NASH with severe bridging fibrosis including the formation of hepatic nodules after only 12 wk (54). The addition of 1–2% cholesterol to the CDAA diet nominally enhanced collagen deposition but did not otherwise affect liver histology (54). Thus, the CDAA diet captures several aspects of NASH, including progression of advanced fibrosis, and the absence of weight loss is a considerable improvement over the MCD diet. The CDAA diet can also be combined with an HFD. After 24 wk fed this choline-deficient, l-amino acid–defined high-fat diet (CDAA-HFD), mice developed NASH with moderate (F2, perisinusoidal and periportal) fibrosis, which progressed to bridging fibrosis (F3) and hepatocellular carcinoma after 48 wk of being fed the diet (64). Although not necessarily specific to NASH, expression of fibrogenic and inflammatory genes [Tnfa, Tgfb1, Acta2 (αSMA), Col1a1, and Col3a1] mirrored those commonly observed in human liver fibrosis (64). However, weight gain was significantly decreased compared with standard feed pellets, which is a common feature in this model (64, 65, 97). Opposite to what is generally observed in patients with NASH, glucose and insulin concentrations were decreased and circulating cholesterol and triglyceride concentrations were lower in CDAA-HFD mice compared with feed-pellet–fed controls (65, 97). In contrast to CDAA alone, alanine aminotransferase and aspartate aminotransferase were elevated in mice fed a CDAA-HFD for 6 wk (66). Furthermore, hepatic steatosis, collagen content, and αSMA-positive cells were higher in CDAA-HFD mice, which also displayed hepatocellular ballooning and mild (F1) fibrosis at week 6 or 8 (66, 98). Thus, the CDAA-HFD may have more rapidly induced NASH compared with the CDAA diet alone. However, the decreased weight gain compared with feed pellet–fed animals seemed more pronounced in the CDAA-HFD compared with the CDAA diet alone (66, 98). Similar to rodents, a CDAA-HFD (30% fat, 20% sucrose or fructose) supplemented with 1% cholesterol rapidly induced steatohepatitis with macrovesicular steatosis and lobular inflammation in young Göttingen minipigs (67). However, hepatocyte ballooning was absent, insulin concentrations nominally decreased, and the CDAA-HFD animals weighed less than normal controls (67). Gene expression suggested decreased endogenous cholesterol synthesis, similar to other models utilizing high amounts of dietary cholesterol (42, 48, 46, 16, 68, 69). αSMA-positive HSC and relative fibrosis area were increased alongside enhanced expression of genes [COL1A1, COL3A1, TIMP1, and ACTA2 (αSMA)] related to core fibrogenic pathways. The fibrotic tissue formed the classical chicken-wire pattern observed in patients with NASH, but fibrotic bridges were not noted (67).
Even though, the CDAA, CDAA-HFD, and CD-HFD circumvent the weight loss associated with the MCD diet it is questionable if hepatic steatosis caused by defective VLDL-triglyceride export secondary to choline deficiency reflects the mechanisms underlying human NASH. Thus, nutritional deficiency of this magnitude is far from human NASH etiology and results should be interpreted with this in mind.
Toxin-induced models
Classic models of liver fibrosis include carbon tetrachloride (CCl4) or thioacetamide (TAA) administration that rapidly and robustly induce advanced fibrosis and cirrhosis (Table 3). However, in particular when used by itself, acute toxicity-induced liver fibrosis is not a relevant model of NASH and the translational value of CCl4 or TAA in relation to NASH-associated fibrosis is highly questionable. Nevertheless, they are still used and therefore briefly discussed below.
TABLE 3.
Chemically and genetically (hyperphagic) induced fibrosis1
| Chemical/genetic | Species (ref) | Fibrosis (time to development) | Comments/applicable to advanced NASH fibrosis? |
|---|---|---|---|
| CCL4 | Mice and rats (101, 102) | Centrilobular fibrosis (week 2), central-central bridging fibrosis (week 4–6); cirrhosis (week 9–16). | Disease etiology differs significantly from humans; weight loss; not applicable |
| TAA | Mice and rats (101) | Periportal fibrosis (week 6); portal-portal and portal-central bridging fibrosis (week 12); cirrhosis (week 16) | Disease etiology differs significantly from humans; fibrosis can have portal origin; weight loss; not applicable |
| CCl4 + Western diet | Mice (108) | Bridging fibrosis (week 12); cirrhosis in some animals (week 24) | Transciptomic pathways similar to humans; CCl4 decreases weight gain, insulin, and dyslipidemia; no role of CCl4 in human NASH; potential applicable for specific research questions, but other models may be more advantageous |
| DMN and DEN | Rats (110, 111) | Central fibrosis (week 3) and portal-central bridging fibrosis and cirrhosis (week 8–12) | Bridging necrosis, weight loss, and high mortality; weight loss; not applicable |
| Streptozotocin | Rats (112–114) | Central and portal fibrosis (week 20); bridging fibrosis in some animals if administered postnatally (week 20) | Disease etiology differs from humans; hypoinsulinemic; not applicable |
| Ob/ob | Mice (115, 116) | Absent on feed pellet diet; mild perisinusoidal fibrosis in portal areas on HFD (week 12); bridging fibrosis on AMLN diet (week 12) | Fibrosis has portal origin; leptin important during fibrogenesis; could be applicable on an ALMN diet |
| Db/db | Mice (115, 116) | Absent on feed pellet diet; mild perisinusoidal fibrosis in portal areas on HFD (week 12); central and portal fibrosis on MCD diet (week 4) | Fibrosis has portal origin and is only mild/moderate; leptin important during fibrogenesis; not applicable |
| Fa/fa | Rats (117) | Mild periportal fibrosis on HFD (week 8); resistant to MCD-induced fibrosis | Fibrosis has portal origin and is only mild; leptin important during fibrogenesis; not applicable |
| Foz/foz (Alms1) | Mice (118, 119) | Portal and central perisinusoidal fibrosis (week 24) | Fibrosis is only mild/moderate; not applicable |
| KK-ay | Mice (97, 120) | Fibrosis absent on feed pellet diet; mild/moderate fibrosis on MCD diet (week 8); mild/moderate fibrosis on CDAA diet (week 30) | Weight loss and decreased glucose concentrations on MCD diet; CDAA decreased insulin/glucose concentrations and fibrosis not increased in KK-ay compared with wild-type; not applicable |
| Mc4r−/− | Mice (121) | Periportal and -central fibrosis (week 20) | Could be applicable if advance fibrosis develops with longer study duration |
1 Alms1, Alström gene; AMLN, Amylin liver NASH; CDAAD, choline-deficient, l-amino acid–defined; CCl4, carbon tetrachloride; DEN, diethylnitrosamine; DMN, dimethylnitrosamine; HFD, high-fat diet; MCD, methionine and choline deficient; Mc4r, melanocortin 4 receptor; NASH, nonalcoholic steatohepatitis; Ref, reference; TTA, thioacetamide.
CCl4 and TAA
CCl4 is activated via cytochrome P450 2E1, expressed mainly by centrilobular hepatocytes, generating the trichloromethyl free radical (99, 100). Consequently, the toxicity induces central fibrosis, which is similar to NASH, although the underlying mechanisms differ considerably (99). This is followed by central-central bridges and the development of pseudo-lobuli with initial sparing of portal areas (99). Notably, the zonal pattern should be characterized with care as pseudo-lobulization may cause the zonation to be misinterpreted as portal (99). In mice, centrilobular fibrosis can be observed already 2 wk post–CCl4 administration and central-central bridges are formed after 4 wk. These are accentuated by week 12 alongside formation of central-portal bridges that progressed to cirrhosis by week 16 (101). In rats, bridging fibrosis (week 6) and cirrhosis (week 9) have also been reported (102). Unlike the fibrosis observed in NASH patients, the advanced fibrosis formed after CCl4 treatment is not persistent and prone to spontaneous regression upon ceasing administration (103, 104). In TAA-treated rats and mice, periportal fibrosis was present after 6 wk and progressed with portal-portal and portal-central bridging after week 12 and cirrhosis at week 16 (101). In this model, the periportal origin of the fibrosis is not comparable to the central origin associated with adult NASH. Fibrosis induced by TAA may be more persistent compared with CCL4 (105, 106), and no regression of cirrhosis was reported 8 wk post–TAA treatment (107). Despite the appeal of rapidly induced advanced fibrosis, these models do not clearly reflect human NASH or NASH-associated fibrosis as the nutritional and metabolic components are completely absent. To attempt to overcome these shortcomings, weekly CCL4 administration has been combined with a Western diet (42% of calories from fat, 41% sucrose, 1.25% cholesterol, and sugar water), which induced histopathological and transcriptomic changes similar to those of human NASH (108). Bridging fibrosis was observed after 12 wk and “some animals” were recorded to progress to cirrhosis after 24 wk, allowing for the study of NASH-related advanced fibrosis. Pathway analysis at week 24 showed that the Western diet/CCl4 model resembled human NASH (e.g., increased “focal adhesion,” “ECM receptor interaction,” and “Toll-like receptor pathway”) and did so better than 16 other models including toxin-induced (streptozotocin), genetic (e.g., Pten-/- and ob/ob), and dietary models (MCD, high-fat/-cholesterol and high-fat/-sugar/-cholesterol) (108).
Similar to Western diets alone, cholesterol synthesis pathways were decreased, which is not observed in human NASH (49, 108). However, CCl4 treatment decreased food intake, weight gain, and circulating insulin and somewhat improved insulin resistance compared with animals fed a Western diet alone (108). Thus, although the model mimics the hepatic phenotype and the underlying molecular mechanisms of patients, it is important to consider if the loss of metabolic context and difference in disease etiology are worth the rapid induction of advanced NASH fibrosis, especially since dietary models can produce similar hepatic phenotypes while maintaining metabolic context, albeit more time is required. Furthermore, CCl4 has no known role in human NASH and may even induce drug-metabolizing enzymes, which could complicate the interpretation of pharmacological studies in this model (109).
Dimethylnitrosamine and diethylnitrosamine
Dimethylnitrosamine (DMN) and diethylnitrosamine (DEN) are mutagenic compounds that cause fibrosis, cirrhosis, and hepatocellular carcinoma (122, 123). Unlike CCl4, the injury is auto-progressive and fibrosis and tumors can form after drug cessation (123). In rats, DMN causes hemorrhage, bridging necrosis, and collagen deposition around central veins with occasional portal-central bridging (110). Similarly, DEN administration results in inflammation followed by perilobular fibrosis, which progresses to cirrhosis, hepatocellular carcinoma, and upregulation of genes [Acta2 (αSMA), Tgfb1, Col1, and Col3] related to general fibrosis (111). In addition to the bridging necrosis, which is not a feature of NASH (12), the administration of DMN/DEN has been associated with high mortality, weight loss (≤40%), and lethargy (110, 111). While these compounds are most often used to study liver cancer, the models are rarely applied in isolated studies of fibrosis (105) and alone they do not reflect NAFLD/NASH. Additionally, it is still unclear if these models reflect NAFLD-related hepatocellular carcinoma as the underlying carcinogenic stimulus differs (36). Analogous to CCL4 and TAA, isolated administration of DMN and DEN fails to reproduce the metabolic context of NASH-induced fibrosis and, due to the substantial pathophysiological differences (e.g., extensive necrosis), they are unlikely to model NASH fibrosis.
Streptozotocin
As NAFLD is prevalent in patients with diabetes (4), some models attempt to mimic the hyperglycemia associated with the disease by destroying the pancreatic β-cells through administration of the toxic glucose analog streptozotocin (124). Compared with an HFD (45% of calories from fat) alone, streptozotocin-treated mice fed an HFD further increased fibrosis around central veins, portal tracts, and in zone 2 after 20 wk (112). In addition, hepatic mRNA levels of Ccn2 (CTGF), Timp1, Col1, and Col3 were increased (112). Postnatal streptozotocin treatment of mice followed by an HFD (57% of calories from fat) induces slight fibrosis after 5 wk, without progression at week 10 (113), and, although infrequent, some mice developed bridging fibrosis after 20 wk (114). In contrast to patients, these models are associated with hypoinsulinemia, weight loss, or reduced weight gain (112–114) and seem to lack hepatocellular ballooning (8). Furthermore, HFD-streptozotocin–treated mice (59% of calories from fat + sugar water) did not reflect the pathways dysregulated in human NASH (78). Moreover, streptozotocin may be directly liver toxic, questioning whether the observed effect on liver fibrosis is mediated by the induced hyperglycemia or rather by direct hepatic toxicity.
Thus, while hepatotoxic compounds may be used to rapidly induce advanced fibrosis, these models differ considerably from NASH etiology and pathogenesis and are generally more likely to reflect an effect of local hepatic toxicity rather than the more systemic metabolic origin of NASH-related fibrosis (8).
Genetic models
Animals overexpressing genes related to disease development, such as TGFβ1, can be useful in validating specific disease mechanisms, but may override or bypass normal counter-regulatory mechanisms, induce ectopic gene expression, and generally represent an oversimplification of NASH pathophysiology (125). Given the nutritional origin of the disease, the following discussion only focuses on genetic models of hyperphagia-driven obesity attempting to mirror the metabolic context of NASH (Table 3).
Leptin- and leptin receptor–deficient models
A mutation in the gene coding for leptin (ob/ob mice) or the leptin receptor (db/db mice and fa/fa rats) results in obesity, hepatic steatosis, and mild inflammation, but not fibrosis (115 –117). Ingesting an HFD (37% of calories as fat, 11% saccharose), most ob/ob and db/db mice developed mild pericellular fibrosis in portal areas after 3 mo (115). Similarly, mild periportal fibrosis and increased hepatic protein expression of COL1, αSMA and TIMP1 can be induced in fa/fa rats by feeding them an HFD (60% of calories from fat) for 8 wk (117). Notably, this fibrosis pattern does not mimic adult human NASH (19). The induction of fibrosis can be accelerated by an MCD diet, and after 4 wk ob/ob mice exhibited more steatosis but less severe inflammation than db/db mice. However, while central and portal pericellular fibrosis were observed in db/db mice, advanced fibrosis was absent and ob/ob mice were resistant to fibrosis development (116). Accordingly, mRNA levels of Tgfb were significantly increased following the MCD diet in db/db but not in ob/ob mice (116). It is possible to induce advanced fibrosis in ob/ob mice by feeding them a Western-type diet (40% of calories from fat, 18% of fat calories from trans fat, 20% fructose, and 2% cholesterol), as developed by Amylin Pharmaceuticals and subsequently termed the Amylin liver NASH (AMLN) diet (126). This diet rapidly induces NASH and ≥F3 fibrosis in the majority of animals in only 12 wk, a benefit of this model considering the otherwise mild/moderate fibrosis usually observed in ob/ob mice (127). At week 26, genes related to human NASH [e.g., Mcp1, C-C chemokine receptor type 2 (Ccr2), and various Tlr] and general fibrosis (e.g., Col1a1, Col3a1, and Col4a1) were increased, but, unlike in humans, cholesterol synthesis was suppressed (127). Notably, the amounts of trans fat in these diets are disproportionate compared with human intake, where trans fat has been estimated to account for 0.3–4.2% of total energy intake (128). The increased attention on the negative consequences of trans fatty acids in human health, and subsequent restricted use as stated by the US FDA and in the European Union (effective from 2021), have forced the dietary composition to be changed, which may affect disease outcome. It should also be noted that leptin is an important mediator of fibrosis and leptin deficiency or resistance due to gene mutations is extremely rare in humans (129–131). Thus, these models likely differ fundamentally in disease-related etiology and the molecular mechanisms underlying development of NASH-associated fibrosis.
Alms1 (foz/foz) mice
A spontaneous mutation in the Alström gene (Alms1) renders foz/foz mice hyperphagic, obese, and hyperglycemic (118, 119). Feeding the animals a high-fat (43% of calories from fat)/high-cholesterol (0.19%) diet exacerbates the obese phenotype and results in hepatic steatosis and inflammation after 12 wk (118). After 24 wk, foz/foz mice developed central and portal perisinusoidal fibrosis (F2) and livers were characterized by increased hepatocyte apoptosis alongside higher mRNA levels of Mcp1, Tgfb, Pdgfa, Pdgfb, and Col1a1, similar to human liver fibrosis (118, 119). Unexpectedly, protein concentrations of TIMP1 and TIMP2 were decreased (119). Liver injury was greater on a C57BL/6 compared with Balb/c background in which HSC activation and fibrosis were absent (132).
KK-ay mice
KK-ay mice are hyperphagic due to a heterozygous mutation in the Agouti gene and develop obesity, hyperglycemia, and insulin resistance as well as hepatic steatosis and mild lobular inflammation (120, 133). The steatohepatitis can be aggravated by an MCD diet, which promotes mild to moderate fibrosis after 8 wk, but also the weight loss and decreased glucose concentrations characteristic of the MCD diet (120). To mitigate the weight loss, KK-ay mice were fed a CDAA-HFD (61% of calories from fat) or Western diet (41% of calories from fat, 30% of calories from fructose, 2% cholesterol) for ≤30 wk (97). Hepatic inflammation and fibrosis were more pronounced with the CDAA-HFD, but not compared with CDAA-HFD–fed wild-type mice (97). In this regard, the genetic model offered little advantage with respect to liver histology and disease development. Fibrosis did not progress to advanced stages and the CDAA-HFD decreased insulin and glucose concentrations, as opposed to the phenotype usually encountered in patients (97).
Melanocortin 4 receptor–deficient mice
Melanocortin 4 receptor regulates food intake and deficient mice (due to receptor knock-out; Mc4r-/-) fed an HFD (60% fat) for 20 wk develop NASH with steatosis, ballooning, inflammation, and fibrosis (121). Furthermore, hepatic gene expression seemed to mirror patients with increased de novo lipogenesis [Fasn and acetyl-CoA carboxylase 1 (Acc1)], fibrogenesis (Tgfb1, Timp1, and Col1a1), and inflammation (Tnfa) (121). However, fibrosis did not progress beyond moderate (F2), suggesting that longer feeding periods might be needed to induce advanced fibrosis or limiting this model to the study of early fibrogenesis.
Although these genetic models reproduce many of the metabolic and pathological changes associated with human NASH, the apparent lack of advanced fibrosis limits their usefulness in the pursuit of targets for this key endpoint. In addition, the genetic alterations exploited in these models rarely reflect the patient population, which could affect translational validity.
Conclusions
NASH and the closely related metabolic disturbances (e.g., obesity and type 2 diabetes) are nutritional diseases. Consequently, the induction of NASH in preclinical models should ideally rely on human-like diets that produce similar metabolic disturbances, both systemically and in the liver, rather than supraphysiological levels of nutrients (e.g., extremely high concentrations of cholesterol), toxins, or micronutrient deficiencies that are not relevant for human NASH. An unhealthy diet rich in fat and sugar is an important risk factor for NAFLD (134). Accordingly, models utilizing “Western diets” seem promising as they can induce NASH (with steatosis, ballooning, and inflammation) and advanced fibrosis. A closer association with the molecular pathways of human NASH compared with other models further supports the use of such dietary regimens (78). With this in mind, the DIAMOND mouse, guinea pigs, and perhaps hamsters fed Western diets are promising models of NASH and NASH-associated fibrosis. Ob/ob mice fed the AMLN diet may also be applicable, although it is necessary to carefully consider the relevance of leptin deficiency and high amounts of trans fat in relation to human disease.
Nevertheless, the mechanisms of human and mouse NASH differ significantly (78), and while models may often reflect core fibrogenic pathways, relatively little is known about their ability to mirror the fibrogenic mechanisms more specifically associated with NASH. Thus, it is essential to keep improving and validating these models, particularly with regard to the underlying molecular mechanisms, to ensure that they properly reflect human NASH-related advanced fibrosis and to increase their predictive validity.
Supplementary Material
ACKNOWLEDGEMENTS
The authors’ responsibilities were as follows—DHI, JL, and PT-N: conceptualized this review and interpreted the data; DHI: wrote the draft manuscript, which was subsequently edited by all authors; PT-N: had responsibility for the final content; and all authors: read and approved the final manuscript.
Notes
JL is supported in part by the LifePharm Centre for In Vivo Pharmacology under the University of Copenhagen.
Author disclosures: The authors report no conflicts of interest. LifePharm Centre had no role in the design, implementation, analysis, or interpretation of the data.
Supplemental Material is available from “Supplementary data” link in the online posting of the article and from the same link in the online table of contents at https://academic.oup.com/advances.
Abbreviations used: ACTA2, actin alpha 2; AMLN, Amylin liver NASH; CCl4, carbon tetrachloride; CD-HFD, choline-deficient, high-fat diet; CDAA, choline-deficient, l-amino acid–defined; CDAA-HFD, choline-deficient, l-amino acid–defined high-fat diet; Col, collagen; CTGF, connective tissue growth factor; DEN, diethylnitrosamine; DIAMOND, diet-induced animal model of nonalcoholic fatty liver disease; DMN, dimethylnitrosamine; ECM, extracellular matrix; ER, endoplasmic reticulum; Fasn, fatty acid synthase; HFD, high-fat diet; HSC, hepatic stellate cell; MCD, methionine- and choline-deficient; Mcp1/Ccl2, monocyte chemoattractant protein 1; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; PDGF, platelet-derived growth factor; TAA, thioacetamide; TGF, transforming growth factor; TIMP, tissue inhibitor of metalloproteinases; TLR, Toll-like receptor; αSMA, α-smooth muscle actin.
Contributor Information
David H Ipsen, Department of Veterinary and Animal Sciences, Faculty of Health and Medical Sciences, University of Copenhagen, Frederiksberg C, Denmark.
Jens Lykkesfeldt, Department of Veterinary and Animal Sciences, Faculty of Health and Medical Sciences, University of Copenhagen, Frederiksberg C, Denmark.
Pernille Tveden-Nyborg, Department of Veterinary and Animal Sciences, Faculty of Health and Medical Sciences, University of Copenhagen, Frederiksberg C, Denmark.
References
- 1. Zivkovic AM, German JB, Sanyal AJ. Comparative review of diets for the metabolic syndrome: implications for nonalcoholic fatty liver disease. Am J Clin Nutr. 2007;86(2):285–300. [DOI] [PubMed] [Google Scholar]
- 2. Schuppan D, Surabattula R, Wang XY. Determinants of fibrosis progression and regression in NASH. J Hepatol. 2018;68(2):238–50. [DOI] [PubMed] [Google Scholar]
- 3. Asrih M, Jornayvaz FR. Diets and nonalcoholic fatty liver disease: the good and the bad. Clin Nutr. 2014;33(2):186–90. [DOI] [PubMed] [Google Scholar]
- 4. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84. [DOI] [PubMed] [Google Scholar]
- 5. Hardy T, Oakley F, Anstee QM, Day CP. Nonalcoholic fatty liver disease: pathogenesis and disease spectrum. Annu Rev Pathol. 2016;11:451–96. [DOI] [PubMed] [Google Scholar]
- 6. Ratziu V. A critical review of endpoints for non-cirrhotic NASH therapeutic trials. J Hepatol. 2018;68(2):353–61. [DOI] [PubMed] [Google Scholar]
- 7. Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Keach JC, Lafferty HD, Stahler A et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. 2015;149(2):389–97..e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Farrell G, Schattenberg JM, Leclercq I, Yeh MM, Goldin R, Teoh N, Schuppan D. Mouse models of nonalcoholic steatohepatitis: toward optimization of their relevance to human nonalcoholic steatohepatitis. Hepatology. 2019;69(5):2241–57. [DOI] [PubMed] [Google Scholar]
- 9. Kleiner DE, Bedossa P. Liver histology and clinical trials for nonalcoholic steatohepatitis—perspectives from 2 pathologists. Gastroenterology. 2015;149(6):1305–8. [DOI] [PubMed] [Google Scholar]
- 10. Loomba R, Chalasani N. The hierarchical model of NAFLD: prognostic significance of histologic features in NASH. Gastroenterology. 2015;149(2):278–81. [DOI] [PubMed] [Google Scholar]
- 11. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313–21. [DOI] [PubMed] [Google Scholar]
- 12. Kleiner DE, Makhlouf HR. Histology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in adults and children. Clin Liver Dis. 2016;20(2):293–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bedossa P. Pathology of non-alcoholic fatty liver disease. Liver Int. 2017;37(Suppl 1):85–9. [DOI] [PubMed] [Google Scholar]
- 14. Bedossa P. Histological assessment of NAFLD. Dig Dis Sci. 2016;61(5):1348–55. [DOI] [PubMed] [Google Scholar]
- 15. Masugi Y, Abe T, Tsujikawa H, Effendi K, Hashiguchi A, Abe M, Imai Y, Hino K, Hige S, Kawanaka M et al. Quantitative assessment of liver fibrosis reveals a nonlinear association with fibrosis stage in nonalcoholic fatty liver disease. Hepatol Commun. 2018;2(1):58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, Kumar DP, Daita K, Min HK, Mirshahi F et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J Hepatol. 2016;65(3):579–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Briand F, Brousseau E, Quinsat M, Burcelin R, Sulpice T. Obeticholic acid raises LDL-cholesterol and reduces HDL-cholesterol in the Diet-Induced NASH (DIN) hamster model. Eur J Pharmacol. 2018;818:449–56. [DOI] [PubMed] [Google Scholar]
- 18. Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371(9615):838–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Skoien R, Richardson MM, Jonsson JR, Powell EE, Brunt EM, Neuschwander-Tetri BA, Bhathal PS, Dixon JB, O'Brien PE, Tilg H et al. Heterogeneity of fibrosis patterns in non-alcoholic fatty liver disease supports the presence of multiple fibrogenic pathways. Liver Int. 2013;33(4):624–32. [DOI] [PubMed] [Google Scholar]
- 20. Wallace MC, Friedman SL, Mann DA. Emerging and disease-specific mechanisms of hepatic stellate cell activation. Semin Liver Dis. 2015;35(2):107–18. [DOI] [PubMed] [Google Scholar]
- 21. Friedman SL. Liver fibrosis in 2012: Convergent pathways that cause hepatic fibrosis in NASH. Nat Rev Gastroenterol Hepatol. 2013;10(2):71–2. [DOI] [PubMed] [Google Scholar]
- 22. Wieckowska A, Zein NN, Yerian LM, Lopez AR, McCullough AJ, Feldstein AE. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology. 2006;44(1):27–33. [DOI] [PubMed] [Google Scholar]
- 23. Kim JY, Garcia-Carbonell R, Yamachika S, Zhao P, Dhar D, Loomba R, Kaufman RJ, Saltiel AR, Karin M. ER stress drives lipogenesis and steatohepatitis via caspase-2 activation of S1P. Cell. 2018;175(1):133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC, Garcia-Ruiz C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4(3):185–98. [DOI] [PubMed] [Google Scholar]
- 25. Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut. 2015;64(5):830–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tomita K, Teratani T, Suzuki T, Shimizu M, Sato H, Narimatsu K, Okada Y, Kurihara C, Irie R, Yokoyama H et al. Free cholesterol accumulation in hepatic stellate cells: mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology. 2014;59(1):154–69. [DOI] [PubMed] [Google Scholar]
- 27. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. 2014;14(3):181–94. [DOI] [PubMed] [Google Scholar]
- 28. Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, Brenner DA. The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J Hepatol. 1999;30(1):77–87. [DOI] [PubMed] [Google Scholar]
- 29. Moylan CA, Pang H, Dellinger A, Suzuki A, Garrett ME, Guy CD, Murphy SK, Ashley-Koch AE, Choi SS, Michelotti GA et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology. 2014;59(2):471–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gerhard GS, Legendre C, Still CD, Chu X, Petrick A, DiStefano JK. Transcriptomic profiling of obesity-related nonalcoholic steatohepatitis reveals a core set of fibrosis-specific genes. J Endocr Soc. 2018;2(7):710–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ryaboshapkina M, Hammar M. Human hepatic gene expression signature of non-alcoholic fatty liver disease progression, a meta-analysis. Sci Rep. 2017;7(1):12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Karsdal MA, Nielsen SH, Leeming DJ, Langholm LL, Nielsen MJ, Manon-Jensen T, Siebuhr A, Gudmann NS, Ronnow S, Sand JM et al. The good and the bad collagens of fibrosis—their role in signaling and organ function. Adv Drug Deliv Rev. 2017;121:43–56. [DOI] [PubMed] [Google Scholar]
- 33. Schuppan D, Ashfaq-Khan M, Yang AT, Kim YO. Liver fibrosis: direct antifibrotic agents and targeted therapies. Matrix Biol. 2018;68-69:435–51. [DOI] [PubMed] [Google Scholar]
- 34. Hui ST, Kurt Z, Tuominen I, Norheim F, R CD, Pan C, Dirks DL, Magyar CE, French SW, Chella Krishnan K et al. The genetic architecture of diet-induced hepatic fibrosis in mice. Hepatology. 2018;68(6):2182–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Santhekadur PK, Kumar DP, Sanyal AJ. Preclinical models of non-alcoholic fatty liver disease. J Hepatol. 2018;68(2):230–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Van Herck MA, Vonghia L, Francque SM. Animal models of nonalcoholic fatty liver disease—a starter's guide. Nutrients. 2017;9(10):1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ito M, Suzuki J, Tsujioka S, Sasaki M, Gomori A, Shirakura T, Hirose H, Ito M, Ishihara A, Iwaasa H et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol Res. 2007;37(1):50–7. [DOI] [PubMed] [Google Scholar]
- 38. Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol Life Sci. 2018;75(18):3313–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Svegliati-Baroni G, Candelaresi C, Saccomanno S, Ferretti G, Bachetti T, Marzioni M, De Minicis S, Nobili L, Salzano R, Omenetti A et al. A model of insulin resistance and nonalcoholic steatohepatitis in rats: role of peroxisome proliferator-activated receptor-alpha and n-3 polyunsaturated fatty acid treatment on liver injury. Am J Pathol. 2006;169(3):846–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Savard C, Tartaglione EV, Kuver R, Haigh WG, Farrell GC, Subramanian S, Chait A, Yeh MM, Quinn LS, Ioannou GN. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology. 2013;57(1):81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tirosh O. Hypoxic signaling and cholesterol lipotoxicity in fatty liver disease progression. Oxid Med Cell Longev. 2018;2018:2548154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Matsuzawa N, Takamura T, Kurita S, Misu H, Ota T, Ando H, Yokoyama M, Honda M, Zen Y, Nakanuma Y et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology. 2007;46(5):1392–403. [DOI] [PubMed] [Google Scholar]
- 43. Lai YS, Yang TC, Chang PY, Chang SF, Ho SL, Chen HL, Lu SC. Electronegative LDL is linked to high-fat, high-cholesterol diet-induced nonalcoholic steatohepatitis in hamsters. J Nutr Biochem. 2016;30:44–52. [DOI] [PubMed] [Google Scholar]
- 44. Dillard A, Matthan NR, Lichtenstein AH. Use of hamster as a model to study diet-induced atherosclerosis. Nutr Metab (Lond). 2010;7:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xu ZJ, Fan JG, Ding XD, Qiao L, Wang GL. Characterization of high-fat, diet-induced, non-alcoholic steatohepatitis with fibrosis in rats. Dig Dis Sci. 2010;55(4):931–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ichimura M, Kawase M, Masuzumi M, Sakaki M, Nagata Y, Tanaka K, Suruga K, Tamaru S, Kato S, Tsuneyama K et al. High-fat and high-cholesterol diet rapidly induces non-alcoholic steatohepatitis with advanced fibrosis in Sprague-Dawley rats. Hepatol Res. 2015;45(4):458–69. [DOI] [PubMed] [Google Scholar]
- 47. Ippagunta SM, Kharitonenkov A, Adams AC, Hillgartner FB. Cholic acid supplementation of a high-fat obesogenic diet suppresses hepatic triacylglycerol accumulation in mice via a fibroblast growth factor 21-dependent mechanism. J Nutr. 2018;148(4):510–7. [DOI] [PubMed] [Google Scholar]
- 48. Ogawa T, Fujii H, Yoshizato K, Kawada N. A human-type nonalcoholic steatohepatitis model with advanced fibrosis in rabbits. Am J Pathol. 2010;177(1):153–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Min HK, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, Kellum J, Warnick R, Contos MJ, Sanyal AJ. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012;15(5):665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Abid A, Taha O, Nseir W, Farah R, Grosovski M, Assy N. Soft drink consumption is associated with fatty liver disease independent of metabolic syndrome. J Hepatol. 2009;51(5):918–24. [DOI] [PubMed] [Google Scholar]
- 51. Jensen VS, Hvid H, Damgaard J, Nygaard H, Ingvorsen C, Wulff EM, Lykkesfeldt J, Fledelius C. Dietary fat stimulates development of NAFLD more potently than dietary fructose in Sprague-Dawley rats. Diabetol Metab Syndr. 2018;10:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Krishnan A, Abdullah TS, Mounajjed T, Hartono S, McConico A, White T, LeBrasseur N, Lanza I, Nair S, Gores G et al. A longitudinal study of whole body, tissue, and cellular physiology in a mouse model of fibrosing NASH with high fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol. 2017;312(6):G666–G80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Charlton M, Krishnan A, Viker K, Sanderson S, Cazanave S, McConico A, Masuoko H, Gores G. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol. 2011;301(5):G825–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tølbøl KS, Stierstorfer B, Rippmann JF, Veidal SS, Rigbolt KTG, Schonberger T, Gillum MP, Hansen HH, Vrang N, Jelsing J et al. Disease progression and pharmacological intervention in a nutrient-deficient rat model of nonalcoholic steatohepatitis. Dig Dis Sci. 2019;64(5):1238–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ipsen DH, Tveden-Nyborg P, Rolin B, Rakipovski G, Beck M, Mortensen LW, Faerk L, Heegaard PM, Moller P, Lykkesfeldt J. High-fat but not sucrose intake is essential for induction of dyslipidemia and non-alcoholic steatohepatitis in guinea pigs. Nutr Metab (Lond). 2016;13(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Panasevich MR, Meers GM, Linden MA, Booth FW, Perfield JW, 2nd Fritsche KL, Wankhade UD, Chintapalli SV, Shankar K, Ibdah JA et al. High-fat, high-fructose, high-cholesterol feeding causes severe NASH and cecal microbiota dysbiosis in juvenile Ossabaw swine. Am J Physiol Endocrinol Metab. 2018;314(1):E78–E92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liang T, Alloosh M, Bell LN, Fullenkamp A, Saxena R, Van Alstine W, Bybee P, Werling K, Sturek M, Chalasani N et al. Liver injury and fibrosis induced by dietary challenge in the Ossabaw miniature Swine. PLoS One. 2015;10(5):e0124173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee L, Alloosh M, Saxena R, Van Alstine W, Watkins BA, Klaunig JE, Sturek M, Chalasani N. Nutritional model of steatohepatitis and metabolic syndrome in the Ossabaw miniature swine. Hepatology. 2009;50(1):56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Itagaki H, Shimizu K, Morikawa S, Ogawa K, Ezaki T. Morphological and functional characterization of non-alcoholic fatty liver disease induced by a methionine-choline-deficient diet in C57BL/6 mice. Int J Clin Exp Pathol. 2013;6(12):2683–96. [PMC free article] [PubMed] [Google Scholar]
- 60. George J, Pera N, Phung N, Leclercq I, Yun Hou J, Farrell G. Lipid peroxidation, stellate cell activation and hepatic fibrogenesis in a rat model of chronic steatohepatitis. J Hepatol. 2003;39(5):756–64. [DOI] [PubMed] [Google Scholar]
- 61. Kajikawa S, Imada K, Takeuchi T, Shimizu Y, Kawashima A, Harada T, Mizuguchi K. Eicosapentaenoic acid attenuates progression of hepatic fibrosis with inhibition of reactive oxygen species production in rats fed methionine- and choline-deficient diet. Dig Dis Sci. 2011;56(4):1065–74. [DOI] [PubMed] [Google Scholar]
- 62. Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, Ringelhan M, Simonavicius N, Egger M, Wohlleber D et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. 2014;26(4):549–64. [DOI] [PubMed] [Google Scholar]
- 63. Kodama Y, Kisseleva T, Iwaisako K, Miura K, Taura K, De Minicis S, Osterreicher CH, Schnabl B, Seki E, Brenner DA. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology. 2009;137(4):1467–77 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ikawa-Yoshida A, Matsuo S, Kato A, Ohmori Y, Higashida A, Kaneko E, Matsumoto M. Hepatocellular carcinoma in a mouse model fed a choline-deficient, L-amino acid-defined, high-fat diet. Int J Exp Pathol. 2017;98(4):221–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Min-DeBartolo J, Schlerman F, Akare S, Wang J, McMahon J, Zhan Y, Syed J, He W, Zhang B, Martinez RV. Thrombospondin-I is a critical modulator in non-alcoholic steatohepatitis (NASH). PLoS One. 2019;14(12):e0226854–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Matsumoto M, Hada N, Sakamaki Y, Uno A, Shiga T, Tanaka C, Ito T, Katsume A, Sudoh M. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int J Exp Path. 2013;94(2):93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pedersen HD, Galsgaard ED, BØ Christoffersen, Cirera S, Holst D, Fredholm M, Latta M. NASH-inducing diets in Göttingen minipigs. J Clin Exp Hepatol. 2020;10(3):211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tølbøl KS, Kristiansen MN, Hansen HH, Veidal SS, Rigbolt KT, Gillum MP, Jelsing J, Vrang N, Feigh M. Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet-induced obese mouse models of biopsy-confirmed nonalcoholic steatohepatitis. World J Gastroenterol. 2018;24(2):179–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ipsen DH, Skat-Rordam J, Tsamouri MM, Latta M, Lykkesfeldt J, Tveden-Nyborg P. Molecular drivers of non-alcoholic steatohepatitis are sustained in mild-to-late fibrosis progression in a guinea pig model. Mol Genet Genomics. 2019;294(3):649–61. [DOI] [PubMed] [Google Scholar]
- 70. Ipsen DH, Rolin B, Rakipovski G, Skovsted GF, Madsen A, Kolstrup S, Schou-Pedersen AM, Skat-Rordam J, Lykkesfeldt J, Tveden-Nyborg P. Liraglutide decreases hepatic inflammation and injury in advanced lean non-alcoholic steatohepatitis. Basic Clin Pharmacol Toxicol. 2018;123(6):704–13. [DOI] [PubMed] [Google Scholar]
- 71. Tveden-Nyborg P, Birck MM, Ipsen DH, Thiessen T, Feldmann LB, Lindblad MM, Jensen HE, Lykkesfeldt J. Diet-induced dyslipidemia leads to nonalcoholic fatty liver disease and oxidative stress in guinea pigs. Transl Res. 2016;168:146–60. [DOI] [PubMed] [Google Scholar]
- 72. Podell BK, Ackart DF, Richardson MA, DiLisio JE, Pulford B, Basaraba RJ. A model of type 2 diabetes in the guinea pig using sequential diet-induced glucose intolerance and streptozotocin treatment. Dis Model Mech. 2017;10(2):151–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Fernandez ML, Volek JS. Guinea pigs: a suitable animal model to study lipoprotein metabolism, atherosclerosis and inflammation. Nutr Metab (Lond). 2006;3:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schumacher-Petersen C, Christoffersen BO, Kirk RK, Ludvigsen TP, Zois NE, Pedersen HD, Vyberg M, Olsen LH. Experimental non-alcoholic steatohepatitis in Gottingen minipigs: consequences of high fat-fructose-cholesterol diet and diabetes. J Transl Med. 2019;17(1):110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hernandez GV, Smith VA, Melnyk M, Burd MA, Sprayberry KA, Edwards MS, Peterson DG, Bennet DC, Fanter RK, Columbus DA et al. Dysregulated FXR-FGF19 signaling and choline metabolism are associated with gut dysbiosis and hyperplasia in a novel pig model of pediatric NASH. Am J Physiol Gastrointest Liver Physiol. 2020;318(3):G582–G609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mik P, Tonar Z, Maleckova A, Eberlova L, Liska V, Palek R, Rosendorf J, Jirik M, Mirka H, Kralickova M et al. Distribution of connective tissue in the male and female porcine liver: histological mapping and recommendations for sampling. J Comp Pathol. 2018;162:1–13. [DOI] [PubMed] [Google Scholar]
- 77. Bal HS, Getty R. Changes in the histomorphology of the uterus of the domestic pig (Sus scrofa domesticus) with advancing age. J Gerontol. 1973;28(2):160–72. [DOI] [PubMed] [Google Scholar]
- 78. Teufel A, Itzel T, Erhart W, Brosch M, Wang XY, Kim YO, von Schonfels W, Herrmann A, Bruckner S, Stickel F et al. Comparison of gene expression patterns between mouse models of nonalcoholic fatty liver disease and liver tissues from patients. Gastroenterology. 2016;151(3):513. [DOI] [PubMed] [Google Scholar]
- 79. Jensen T, Abdelmalek MF, Sullivan S, Nadeau KJ, Green M, Roncal C, Nakagawa T, Kuwabara M, Sato Y, Kang DH et al. Fructose and sugar: a major mediator of non-alcoholic fatty liver disease. J Hepatol. 2018;68(5):1063–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sanchez-Lozada LG, Mu W, Roncal C, Sautin YY, Abdelmalek M, Reungjui S, Le M, Nakagawa T, Lan HY, Yu X et al. Comparison of free fructose and glucose to sucrose in the ability to cause fatty liver. Eur J Nutr. 2010;49(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Softic S, Gupta MK, Wang GX, Fujisaka S, O'Neill BT, Rao TN, Willoughby J, Harbison C, Fitzgerald K, Ilkayeva O et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest. 2017;127(11):4059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Abdelmalek MF, Suzuki A, Guy C, Unalp-Arida A, Colvin R, Johnson RJ, Diehl AM. Nonalcoholic Steatohepatitis Clinical Research Network. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51(6):1961–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chung M, Ma J, Patel K, Berger S, Lau J, Lichtenstein AH. Fructose, high-fructose corn syrup, sucrose, and nonalcoholic fatty liver disease or indexes of liver health: a systematic review and meta-analysis. Am J Clin Nutr. 2014;100(3):833–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Trevaskis JL, Griffin PS, Wittmer C, Neuschwander-Tetri BA, Brunt EM, Dolman CS, Erickson MR, Napora J, Parkes DG, Roth JD. Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am J Physiol Gastrointest Liver Physiol. 2012;302(8):G762–72. [DOI] [PubMed] [Google Scholar]
- 85. Drescher HK, Weiskirchen R, Fulop A, Hopf C, de San Roman EG, Huesgen PF, de Bruin A, Bongiovanni L, Christ A, Tolba R et al. The influence of different fat sources on steatohepatitis and fibrosis development in the Western diet mouse model of non-alcoholic steatohepatitis (NASH). Front Physiol. 2019;10:770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ogawa Y, Imajo K, Honda Y, Kessoku T, Tomeno W, Kato S, Fujita K, Yoneda M, Saito S, Saigusa Y et al. Palmitate-induced lipotoxicity is crucial for the pathogenesis of nonalcoholic fatty liver disease in cooperation with gut-derived endotoxin. Sci Rep. 2018;8(1):11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gentile CL, Weir TL, Cox-York KA, Wei Y, Wang D, Reese L, Moran G, Estrada A, Mulligan C, Pagliassotti MJ et al. The role of visceral and subcutaneous adipose tissue fatty acid composition in liver pathophysiology associated with NAFLD. Adipocyte. 2015;4(2):101–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Musso G, Cassader M, Paschetta E, Gambino R. Bioactive lipid species and metabolic pathways in progression and resolution of nonalcoholic steatohepatitis. Gastroenterology. 2018;155(2):282. [DOI] [PubMed] [Google Scholar]
- 89. Warden CH, Fisler JS. Comparisons of diets used in animal models of high-fat feeding. Cell Metab. 2008;7(4):277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Machado MV, Michelotti GA, Xie G, Almeida Pereira T, Boursier J, Bohnic B, Guy CD, Diehl AM. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS One. 2015;10(5):e0127991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Rinella ME, Green RM. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol. 2004;40(1):47–51. [DOI] [PubMed] [Google Scholar]
- 92. Pierce AA, Pickens MK, Siao K, Grenert JP, Maher JJ. Differential hepatotoxicity of dietary and DNL-derived palmitate in the methionine-choline-deficient model of steatohepatitis. BMC Gastroenterol. 2015;15:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, Green RM. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J Lipid Res. 2008;49(5):1068–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Diraison F, Moulin P, Beylot M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab. 2003;29(5):478–85. [DOI] [PubMed] [Google Scholar]
- 95. Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146(3):726–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ishioka M, Miura K, Minami S, Shimura Y, Ohnishi H. Altered gut microbiota composition and immune response in experimental steatohepatitis mouse models. Dig Dis Sci. 2017;62(2):396–406. [DOI] [PubMed] [Google Scholar]
- 97. Abe N, , Kato S, Tsuchida T, Sugimoto K, Saito R, Verschuren L, Kleemann R, Oka K. Longitudinal characterization of diet-induced genetic murine models of non-alcoholic steatohepatitis with metabolic, histological, and transcriptomic hallmarks of human patients. Biol Open. 2019;8(5):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wei G, An P, Vaid KA, Nasser I, Huang P, Tan L, Zhao S, Schuppan D, Popov YV. Comparison of murine steatohepatitis models identifies a dietary intervention with robust fibrosis, ductular reaction, and rapid progression to cirrhosis and cancer. Am J Physiol Gastrointest Liver Physiol. 2020;318(1):G174–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hammad S, Braeuning A, Meyer C, Mohamed F, Hengstler JG, Dooley S. A frequent misinterpretation in current research on liver fibrosis: the vessel in the center of CCl4-induced pseudolobules is a portal vein. Arch Toxicol. 2017;91(11):3689–92. [DOI] [PubMed] [Google Scholar]
- 100. Weber LW, Boll M, Stampfl A. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol. 2003;33(2):105–36. [DOI] [PubMed] [Google Scholar]
- 101. Liu F, Chen L, Rao HY, Teng X, Ren YY, Lu YQ, Zhang W, Wu N, Liu FF, Wei L. Automated evaluation of liver fibrosis in thioacetamide, carbon tetrachloride, and bile duct ligation rodent models using second-harmonic generation/two-photon excited fluorescence microscopy. Lab Invest. 2017;97(1):84–92. [DOI] [PubMed] [Google Scholar]
- 102. Oh Y, Park O, Swierczewska M, Hamilton JP, Park JS, Kim TH, Lim SM, Eom H, Jo DG, Lee CE et al. Systemic PEGylated TRAIL treatment ameliorates liver cirrhosis in rats by eliminating activated hepatic stellate cells. Hepatology. 2016;64(1):209–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, Iwaisako K, Moore-Morris T, Scott B, Tsukamoto H et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci. 2012;109(24):9448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S, Hovell C, Arthur MJ. Mechanisms of spontaneous resolution of rat liver fibrosis: hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. 1998;102(3):538–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Starkel P, Leclercq IA. Animal models for the study of hepatic fibrosis. Best Pract Res Clin Gastroenterol. 2011;25(2):319–33. [DOI] [PubMed] [Google Scholar]
- 106. Salguero Palacios R, Roderfeld M, Hemmann S, Rath T, Atanasova S, Tschuschner A, Gressner OA, Weiskirchen R, Graf J, Roeb E. Activation of hepatic stellate cells is associated with cytokine expression in thioacetamide-induced hepatic fibrosis in mice. Lab Invest. 2008;88(11):1192–203. [DOI] [PubMed] [Google Scholar]
- 107. Reif S, Aeed H, Shilo Y, Reich R, Kloog Y, Kweon YO, Bruck R. Treatment of thioacetamide-induced liver cirrhosis by the Ras antagonist, farnesylthiosalicylic acid. J Hepatol. 2004;41(2):235–41. [DOI] [PubMed] [Google Scholar]
- 108. Tsuchida T, Lee YA, Fujiwara N, Ybanez M, Allen B, Martins S, Fiel MI, Goossens N, Chou HI, Hoshida Y et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J Hepatol. 2018;69(2):385–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Castro RE, Diehl AM. Towards a definite mouse model of NAFLD. J Hepatol. 2018;69(2):272–4. [DOI] [PubMed] [Google Scholar]
- 110. George J, Rao KR, Stern R, Chandrakasan G. Dimethylnitrosamine-induced liver injury in rats: the early deposition of collagen. Toxicology. 2001;156(2-3):129–38. [DOI] [PubMed] [Google Scholar]
- 111. Ding YF, Wu ZH, Wei YJ, Shu L, Peng YR. Hepatic inflammation-fibrosis-cancer axis in the rat hepatocellular carcinoma induced by diethylnitrosamine. J Cancer Res Clin Oncol. 2017;143(5):821–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Lo L, McLennan SV, Williams PF, Bonner J, Chowdhury S, McCaughan GW, Gorrell MD, Yue DK, Twigg SM. Diabetes is a progression factor for hepatic fibrosis in a high fat fed mouse obesity model of non-alcoholic steatohepatitis. J Hepatol. 2011;55(2):435–44. [DOI] [PubMed] [Google Scholar]
- 113. Saito T, Muramatsu M, Ishii Y, Saigo Y, Konuma T, Toriniwa Y, Miyajima K, Ohta T. Pathophysiological analysis of the progression of hepatic lesions in STAM mice. Physiol Res. 2017;66(5):791–9. [DOI] [PubMed] [Google Scholar]
- 114. Fujii M, Shibazaki Y, Wakamatsu K, Honda Y, Kawauchi Y, Suzuki K, Arumugam S, Watanabe K, Ichida T, Asakura H et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med Mol Morphol. 2013;46(3):141–52. [DOI] [PubMed] [Google Scholar]
- 115. Trak-Smayra V, Paradis V, Massart J, Nasser S, Jebara V, Fromenty B. Pathology of the liver in obese and diabetic ob/ob and db/db mice fed a standard or high-calorie diet. Int J Exp Pathol. 2011;92(6):413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sahai A, Malladi P, Pan X, Paul R, Melin-Aldana H, Green RM, Whitington PF. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: role of short-form leptin receptors and osteopontin. Am J Physiol Gastrointest Liver Physiol. 2004;287(5):G1035–43. [DOI] [PubMed] [Google Scholar]
- 117. Carmiel-Haggai M, Cederbaum AI, Nieto N. A high-fat diet leads to the progression of non-alcoholic fatty liver disease in obese rats. FASEB J. 2005;19(1):136–8. [DOI] [PubMed] [Google Scholar]
- 118. Larter CZ, Yeh MM, Van Rooyen DM, Teoh NC, Brooling J, Hou JY, Williams J, Clyne M, Nolan CJ, Farrell GC. Roles of adipose restriction and metabolic factors in progression of steatosis to steatohepatitis in obese, diabetic mice. J Gastroenterol Hepatol. 2009;24(10):1658–68. [DOI] [PubMed] [Google Scholar]
- 119. Larter CZ, Yeh MM, Haigh WG, Van Rooyen DM, Brooling J, Heydet D, Nolan CJ, Teoh NC, Farrell GC. Dietary modification dampens liver inflammation and fibrosis in obesity-related fatty liver disease. Obesity. 2013;21(6):1189–99. [DOI] [PubMed] [Google Scholar]
- 120. Okumura K, Ikejima K, Kon K, Abe W, Yamashina S, Enomoto N, Takei Y, Sato N. Exacerbation of dietary steatohepatitis and fibrosis in obese, diabetic KK-A(y) mice. Hepatol Res. 2006;36(3):217–28. [DOI] [PubMed] [Google Scholar]
- 121. Itoh M, Suganami T, Nakagawa N, Tanaka M, Yamamoto Y, Kamei Y, Terai S, Sakaida I, Ogawa Y. Melanocortin 4 receptor-deficient mice as a novel mouse model of nonalcoholic steatohepatitis. Am J Pathol. 2011;179(5):2454–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. George J, Tsuchishima M, Tsutsumi M. Molecular mechanisms in the pathogenesis of N-nitrosodimethylamine induced hepatic fibrosis. Cell Death Dis. 2019;10(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Delire B, Starkel P, Leclercq I. Animal models for fibrotic liver diseases: what we have, what we need, and what is under development. J Clin Transl Hepatol. 2015;3(1):53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Lenzen S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia. 2008;51(2):216–26. [DOI] [PubMed] [Google Scholar]
- 125. Trautwein C, Friedman SL, Schuppan D, Pinzani M. Hepatic fibrosis: concept to treatment. J Hepatol. 2015;62(1 Suppl):S15–24. [DOI] [PubMed] [Google Scholar]
- 126. Hansen HH, Feigh M, Veidal SS, Rigbolt KT, Vrang N, Fosgerau K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov Today. 2017;22(11):1707–18. [DOI] [PubMed] [Google Scholar]
- 127. Kristiansen MN, Veidal SS, Rigbolt KT, Tolbol KS, Roth JD, Jelsing J, Vrang N, Feigh M. Obese diet-induced mouse models of nonalcoholic steatohepatitis-tracking disease by liver biopsy. World J Hepatol. 2016;8(16):673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Wanders AJ, Zock PL, Brouwer IA. trans Fat intake and its dietary sources in general populations worldwide: a systematic review. Nutrients. 2017;9(8):840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Leclercq IA, Farrell GC, Schriemer R, Robertson GR. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J HepatolJ. Hepatol. 2002;37(2):206–13. [DOI] [PubMed] [Google Scholar]
- 130. Paz-Filho G, Mastronardi C, Delibasi T, Wong ML, Licinio J. Congenital leptin deficiency: diagnosis and effects of leptin replacement therapy. Arq Bras Endocrinol Metabol. 2010;54(8):690–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Lee UE, Friedman SL. Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol. 2011;25(2):195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Farrell GC, Mridha AR, Yeh MM, Arsov T, Van Rooyen DM, Brooling J, Nguyen T, Heydet D, Delghingaro-Augusto V, Nolan CJ et al. Strain dependence of diet-induced NASH and liver fibrosis in obese mice is linked to diabetes and inflammatory phenotype. Liver Int. 2014;34(7):1084–93. [DOI] [PubMed] [Google Scholar]
- 133. Denk H, Abuja PM, Zatloukal K. Animal models of NAFLD from the pathologist's point of view. Biochim Biophys Acta Mol Basis Dis. 2019;1865(5):929–42. [DOI] [PubMed] [Google Scholar]
- 134. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J, Bugianesi E. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15(1):11–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.