

Abstract
The clearance of apoptotic cells by professional and non-professional phagocytes — a process termed ‘efferocytosis’ — is essential for the maintenance of tissue homeostasis. Accordingly, defective efferocytosis underlies a growing list of chronic inflammatory diseases. Although much has been learnt about the mechanisms of apoptotic cell recognition and uptake, several key areas remain incompletely understood. This Review focuses on new discoveries related to how phagocytes process the metabolic cargo they receive during apoptotic cell uptake; the links between efferocytosis and the resolution of inflammation in health and disease; and the roles of efferocytosis in host defence. Understanding these aspects of efferocytosis sheds light on key physiological and pathophysiological processes and suggests novel therapeutic strategies for diseases driven by defective efferocytosis and impaired inflammation resolution.
Every day, approximately 0.4% of the estimated 37.2 trillion cells in an adult human die1. However, even in tissues where cell turnover is high, apoptotic cells (ACs) are scarce, indicating very high efficiency of and capacity for AC clearance. The clearance of ACs, termed ‘efferocytosis’, is an essential process for the maintenance of tissue homeostasis in normal physiology and for the restoration of homeostasis following disease2,3. In a number of non-resolving, chronic inflammatory diseases, efferocytosis becomes defective, and accumulation of dead cells ensues4. The dead cells can become secondarily necrotic, which can lead to autoimmunity, tissue necrosis and pathological inflammation5,6. Accordingly, a major area of biomedical research is emerging to understand the mechanisms by which efferocytosis is successfully carried out in normal physiology and how it becomes defective in disease.
Genetic, biochemical and imaging approaches have revealed that efferocytosis is morphologically and mechanistically distinct from classic forms of phagocytosis7. Clearance of ACs requires phagocyte expression of receptors that recognize AC-associated ligands, reorganization of the phagocyte cytoskeleton to engulf cell-bound ACs and induction of phagosome–lysosome fusion to degrade AC cargo8 (Box 1). In response to AC ingestion, macrophages restrain the production of proinflammatory cytokines and enhance the production of molecules that dampen inflammation and mediate resolution and repair9,10 (Box 2), processes which fail in settings of defective efferocytosis.
Box 1 | How phagocytes carry out efferocytosis.
The process of efferocytosis is mechanistically distinct from classic forms of phagocytosis and involves several phases: apoptotic cell (AC) finding, AC binding, AC internalization and AC degradation4 (see the figure). in the first phase, ACs induce rapid mobilization of efferocytic immune cells by releasing chemokines (such as CX3C-chemokine ligand 1 (CX3CL1))137, lipids (such as sphingosine 1-phosphate (S1p)138 and lysophosphatidylcholine (LPC)139) and nucleotides (ATP and UTP)140. Second, efferocytes engage ACs through cell-surface receptors that either directly bind molecules on the AC surface or bind bridging molecules that interact with the AC surface7. Efferocyte receptors that directly bind ACs include stabilin 1, stabilin 2, adhesion G protein-coupled receptor B1 (ADGRB1; also known as BAI1), T cell immunoglobulin mucin receptor 1 (TIM1), TIM3, TIM4 and low-density lipoprotein receptor-related protein 1 (LRP1). The receptors that function through bridging molecules include the protein tyrosine kinases TYRO3, AXL and MER proto-oncogene tyrosine kinase (MERTK) (also known as TAMs), the αVβ3 and αVβ5 integrins and the scavenger receptor CD36. The most important bridging molecules are growth arrest-specific protein 6 (GAS6; also known as AXL ligand), protein S and milk fat globule-EGF factor 8 (MFG-E8)7. The most important AC-associated signal is externalized phosphatidylserine, but others include calreticulin, intercellular adhesion molecule 3 (iCAm3) and glycosylation moieties141,142.
Whether direct or indirect, engagement of cell-surface receptors on efferocytes activates the RHO family of small GTPases to mediate AC internalization. For example, the small GTpase RAC1 is activated when the ADGRB1–phosphatidylserine interaction promotes the assembly of a complex of engulfment and cell motility protein 1 (ELMO1) and dedicator of cytokinesis protein 1 (DOCK180)143 or when the stabilin 2–phosphatidylserine interaction drives an association between the PTB domain-containing engulfment adaptor protein 1 (Gulp) and thymosin β4 (REFS144,145). on activation, RAC1 localizes to the site at the efferocyte plasma membrane that has engaged the AC and stimulates verprolin homology domain-containing protein 1 (WAVE1)-dependent activation of actin-related protein 2/3 (ARP2/3) complex, which then polymerizes actin to form the phagocytic cup and internalize the AC146–148.
In the next phase, efferocytes undergo mitochondrial fission to increase the level of cytoplasmic calcium, which is necessary for phagosome sealing, and assemble the class iii phosphoinositide 3-kinase (PI3K) complex to synthesize phosphatidylinositol 3,4,5-trisphosphate (Ptdins(3,4,5)P3) and stabilize the NADPH oxidase 2 (NoX2) complex to produce reactive oxygen species (ROS)22,149. Both Ptdins(3,4,5)P3 and ROS are necessary for conjugating autophagy-related protein LC3-II to lipids at the phagosomal membrane, a process termed ‘LC3-associated phagocytosis’. LC3-associated phagocytosis promotes phagolysosomal assembly and acidification and thereby facilitates AC degradation52. NOX2 deficiency in chronic granulomatous disease has been associated with impaired efferocytosis150.
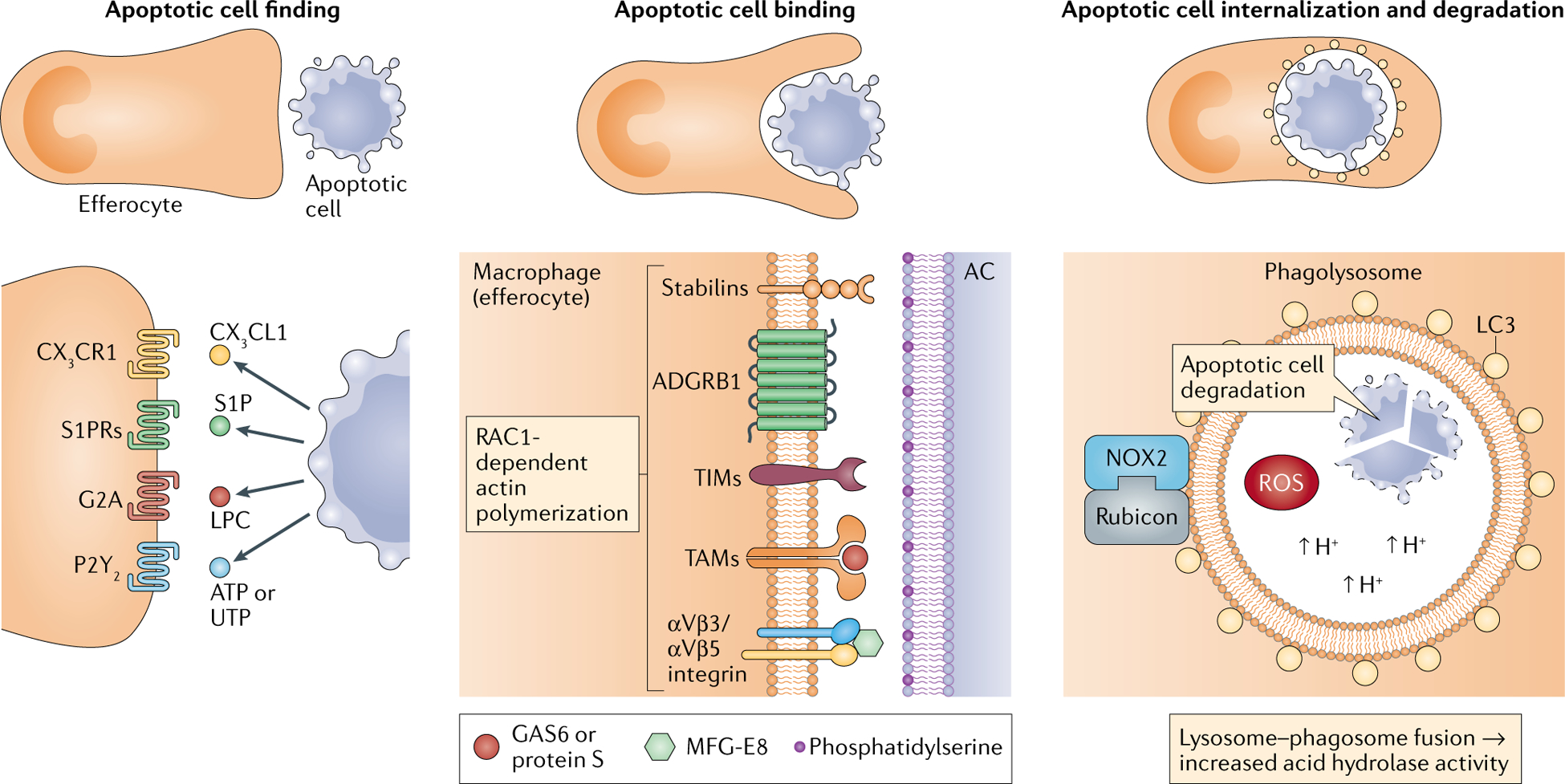
G2A, G protein-coupled receptor G2A; S1PR, sphingosine 1-phosphate receptor.
Box 2 | How efferocytosis prevents inflammation.
During development, organ structure, limb formation and negative selection of T cells demand that cells undergo physiological apoptosis7. However, because apoptotic cells (ACs) can undergo secondary necrosis and trigger immune responses, they must be swiftly removed in a non-inflammatory manner10. Efferocytosis quells inflammation by stimulating the production of anti-inflammatory cytokines while simultaneously repressing proinflammatory cytokines, and it promotes inflammation resolution9. These processes occur through integrated but mechanistically distinct pathways. For example, AC binding to the efferocyte receptor T cell immunoglobulin mucin receptor 1 (TIM1) suppresses the production of tumour necrosis factor (TNF), IL-6 and CC-chemokine ligand 5 (CCL5) by blocking activation of nuclear factor-κB (NF-κB), whereas AC binding to the efferocyte receptor stabilin 2 stimulates the production of transforming growth factor-β (TGFβ)151–153. Binding of ACs to the protein tyrosine kinases mer proto-oncogene tyrosine kinase (MERTK) and AXL can suppress Toll-like receptor (TLR) and type 1 interferon-mediated proinflammatory signalling pathways12. Activation of MERTK by ACs blocks the activity of IκB kinase (IKK), which prevents TLR4-induced, NF-κB-dependent TNF expression154. AXL activation upregulates the transcriptional repressors Twist-related proteins to prevent TLR4-induced TNF promoter activity155. These efferocyte receptors also upregulate the E3 ubiquitin ligases suppressor of cytokine signalling 1 (SOCS1) and SOCS3 and thereby block IFNα-mediated signal transducer and activator of transcription 1 (STAT1) signalling and proinflammatory gene expression156.
Sterols delivered to efferocytes following phagolysosomal AC degradation activate nuclear sterol receptors, such as peroxisome proliferator-activated receptor-γ (PPARγ), PPARδ and liver X receptor-α (LXRα), which stimulate production of the anti-inflammatory cytokines IL-10 and TGFβ and induce differentiation of regulatory T cells and T helper 2 cells to resolve immune responses41,43,57,157–159. Additionally, PPARγ and LXRs bind to nuclear receptor corepressor, which prevents its removal from the promoter sequences of genes encoding certain proinflammatory cytokines160,161, such as TNF and IL-1β. Moreover, interactions between efferocytes and ACs enhance the synthesis of specialized proresolving mediators while suppressing the production of proinflammatory leukotrienes10. For example, engagement of the AC receptor MERTK leads to the translocation of lipoxygenase 5 from the nucleus to the cytoplasm, promoting the synthesis of lipoxin A4 (REF.49). Together, these actions prevent inflammation and promote resolution.
Over the last three decades, much has been learnt about the mechanisms of AC recognition and uptake by professional and non-professional phagocytes, how phagocyte biology is altered by AC uptake to avoid inflammatory responses and the role of impaired efferocytosis in disease. We refer the reader to elegant reviews on these topics2,7,11–15. However, several key areas related to efferocytosis have remained incompletely understood, including how phagocytes respond to the large amounts of metabolic cargo following the degradation of ingested ACs, links between efferocytosis and the resolution of inflammation in health and disease and roles of efferocytosis in successful as well as impaired host defence. The last few years have seen exciting progress in these three areas, which is the focus of this Review.
Continual efferocytosis and metabolism
An interesting and important feature of efferocytosis is the ability of phagocytes to ingest multiple ACs over a relatively short period, referred to as continual efferocytosis. This capability of efferocytes is essential when the AC-to-phagocyte ratio is high (for example, after an acute inflammatory response). There are several challenges faced by phagocytes undergoing continual AC uptake. First, each AC uptake event internalizes a sub-stantial amount of plasma membrane as the phagosome enters the cell, and so subsequent rounds of efferocytosis require rapid restoration of cell surface area16. Second, phagolysosomal degradation of ACs results in large amounts of metabolic cargo, including amino acids, lipids and nucleic acids, that must be processed in a safe and productive manner17.
Membrane recycling.
The phagocytosis of large particles, including ACs, requires adequate cell surface area to engulf and seal the particle16,18,19. For the initial event, inositol phospholipids and phosphoinositide 3-kinases (PI3Ks) promote the fusion of intracellular vesicles, such as recycling endosomes and possibly portions of endoplasmic reticulum (ER)20,21, with the plasma membrane near the site of the developing phagosome. For a subsequent phagocytic event, cell surface area must be restored by membrane recycling from the engulfed phagosome16,18,19. One mechanism involves mitochondrial fission, which occurs after AC engulfment owing to an increase in the level of the mitochondrial fission protein dynamin-related protein 1 (DRP1)22. Mitochondrial fission causes the mitochondria to dissociate from the ER, which facilitates ER calcium release into the cytoplasm rather than into the mitochondria. An increased level of cytoplasmic calcium, probably acting through calcium-dependent vesicular transport proteins such as synaptotagmin VII (REF.23), promotes phagolysosome-to-plasma membrane vesicular transport, which allows phagosome formation around the subsequent AC. In vivo relevance for this process was demonstrated in low-density lipoprotein receptor-deficient mice (LDLR-deficient mice) fed a Western diet and lacking myeloid DRP1 (REF.22). In these mice, atherosclerotic lesional efferocytosis is compromised and plaque necrosis is increased compared with control mice22. An interesting topic for future study is whether atherosclerosis and other diseases driven by defective efferocytosis, such as autoimmune and neurodegenerative disease, acquire defects in mitochondrial fission that contribute to impaired efferocytosis.
Vesicular recycling in cells is also regulated by the RAB family of GTPases. A recent report provided evidence in vitro that RAB17 is recruited to AC-engulfing phagosomes to mediate vesicular transport from AC-containing phagolysosomes to recycling endosomes, which then fuse with the plasma membrane24. This process avoids the intermixing of vesicles containing degraded ACs with the MHC class II-loading compartment, thereby preventing antigen presentation to T cells25. Further studies will be needed to establish the role of RAB17 in continual efferocytosis and avoidance of T cell activation in vivo.
Cholesterol metabolism.
Following the phagolysosomal degradation of ACs, efferocytes are confronted with a large quantity and diversity of potentially cytotoxic macro-molecules that require efflux or metabolism (FIG. 1a). A prime example is related to how efferocytes handle free (unesterified) cholesterol26. Each time an efferocyte degrades an AC, intracellular cholesterol level increases markedly, which is exacerbated if the AC is loaded with cholesterol, as occurs in macrophage foam cells in atherosclerosis27. When macrophages are incubated with apoptotic foam cells in vitro, efficient trafficking of phagolysosomal cholesterol to acyl-CoA cholesterol acyltransferase (ACAT) in the ER catalyses the esterification of free cholesterol to cholesterol fatty acid esters, thereby preventing the membrane-damaging effects of excess free cholesterol28. In addition, macrophages are able to carry out massive cholesterol efflux after ingesting ACs, in part due to induction of the cholesterol efflux transport protein ATP-binding cassette transporter A1 (ABCA1)29. AC-derived sterols, likely through metabolism to oxysterols, can activate the nuclear receptors liver X receptor-α (LXRα) and LXRβ, which induce Abca1 expression. Oxysterols resulting from efferocytosis may have other functions, including blocking inflamma some activation by suppressing mitochondrial oxidative stress and promoting AC internalization by stabilizing the actin GTPase RAC1 (REF.30).
Fig. 1 |. Mechanisms for handling AC-derived metabolic cargo in macrophage efferocytes.
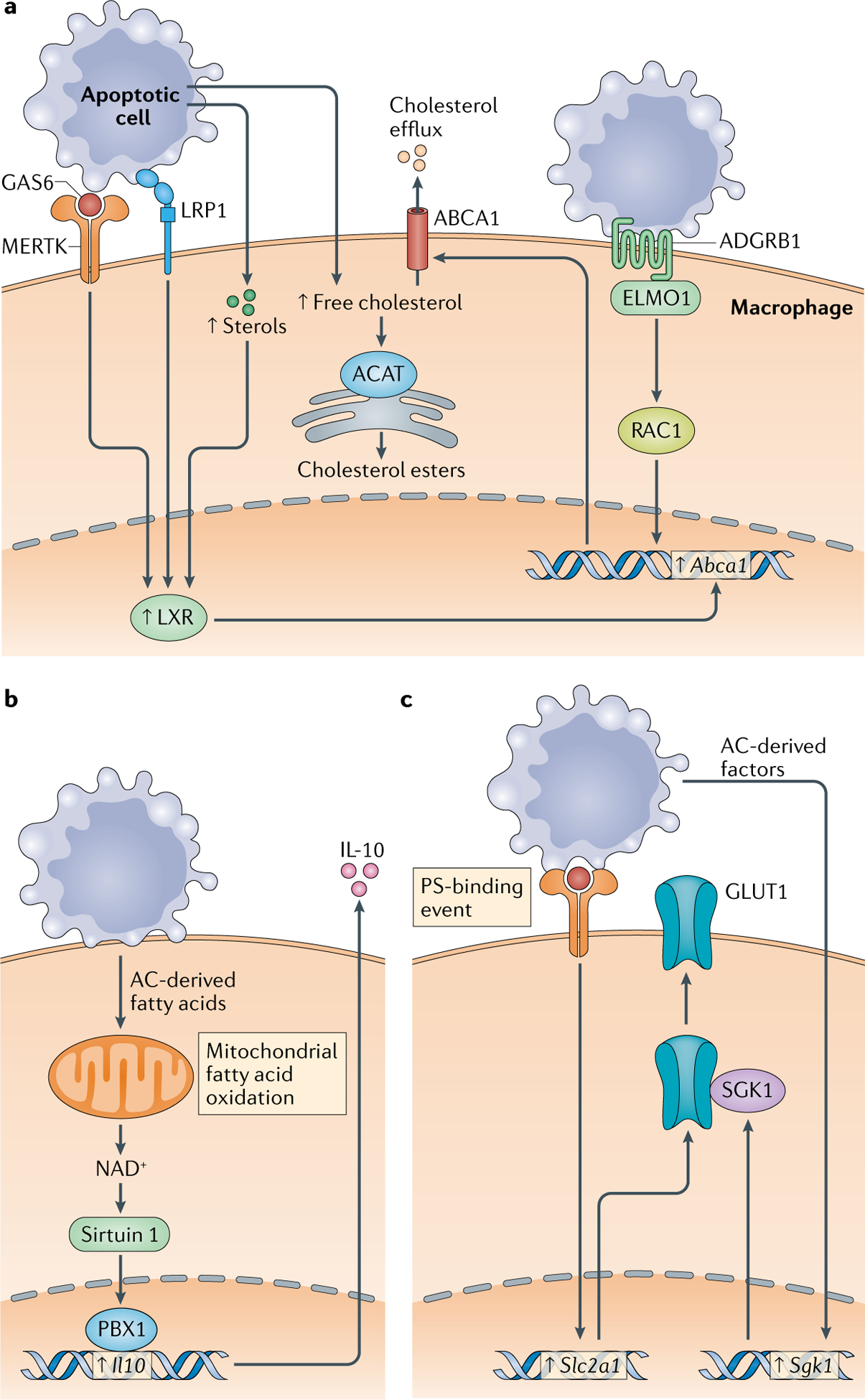
Apoptotic cell (AC)-derived cargo can be cytotoxic, and therefore it needs to be released from the efferocyte or metabolized. Metabolic changes resulting from these processes can be used by the efferocyte to optimize further AC uptake and to trigger resolution pathways. Three examples are illustrated here. a | AC-derived free cholesterol can be esterified by acyl-CoA cholesterol acyltransferase (ACAT) into cholesterol esters and sequestered in neutral lipid droplets or effluxed from the cell to prevent its accumulation. Efflux is facilitated by various efferocytosis receptor signalling pathways and sterol pathways that lead to the induction of cholesterol efflux transporters, such as ATP-binding cassette transporter A1 (ABCA1). b | AC-derived fatty acids undergo oxidation in a mitochondria-mediated pathway that promotes upregulation of expression of proresolving IL-10. c | Binding of ACs to efferocyte receptors that recognize phosphatidylserine (PS) leads to the upregulation of glucose transporter 1 (GLUT1; also known as SLC2A1) and serum/glucocorticoid regulated kinase 1 (SGK1), which is required for GLUT1 transport to the plasma membrane. ADGRB1, adhesion G protein-coupled receptor B1; ELMO1, engulfment cell motility 1; GAS6, growth arrest-specific protein 6; LRP1, LDL-related protein 1; LXR, liver X receptor; MERTK, MER proto-oncogene tyrosine kinase; PBX1, pre-B cell leukaemia homeobox 1.
Another LXR–ABCA1 pathway is mediated by LDLR-related protein 1 (LRP1)-mediated efferocytosis, which activates peroxisome proliferator-activated receptor-γ (PPARγ), an inducer of LXR- and ABCA1-mediated cholesterol efflux31. Finally, engagement of adhesion G protein-coupled receptor B1 (ADGRB1; also known as BAI1) by ACs was also found to induce ABCA1 expression and stimulate cholesterol efflux in cultured cells32. The mechanism was dependent on ADGRB1-mediated AC internalization but, surprisingly, was independent of LXRs, as efflux was not blocked by inhibition or genetic targeting of LXRs32.
Energy metabolism.
Mitochondria are key responders to ingested metabolic loads. One study found that soon after phagocytes degrade ACs, the mitochondrial membrane potential (MMP) increases33. This is related to the delivery of AC cargo, as the uptake of beads does not affect the MMP. Importantly, the increase in MMP is only transient, suggesting that a mechanism eventually becomes activated to guard against excessive MMP. Indeed, this MMP-modulating mechanism was found to involve upregulation of expression of the mitochondrial uncoupling protein UCP2 after the initial increase in MMP33. When UCP2 is silenced using small interfering RNA, MMP remains high, and efferocytosis is impaired. Mice deficient in UCP2 have increased numbers of ACs in two experimental models of apoptosis, which is consistent with defective efferocytosis. Although the mechanism of UCP2 upregulation and the signalling mechanisms linking UCP2-mediated modulation of MMP to specific processes in continual efferocytosis remain to be determined, these findings are important not only for revealing the role of UCP2 but also for showing the importance of continual efferocytosis in vivo.
The mitochondrial response to fatty acids was recently discovered as a novel proresolving mechanism. In efferocytic macrophages isolated from mouse myocardium following infarction as well as in cultured macrophages exposed to ACs in vitro, NAD+ produced by mitochondrial fatty acid oxidation and complex III subunit 5-dependent mitochondrial electron transport leads to sirtuin 1-mediated activation of PBX1, which is an inducer of the gene encoding the resolution mediator IL-10 (REF.34) (FIG. 1b). In mice lacking complex III subunit 5 in myeloid cells, myocardial damage after myocardial infarction is worse and is associated with markedly decreased levels of IL-10. It remains to be determined whether fatty acids originate from degraded ACs, as predicted, and whether the NAD+–sirtuin 1–PBX1 pathway, which was elegantly dissected in vitro, is functional in vivo.
Unlike oxidative phosphorylation, glycolysis is generally considered to be proinflammatory35. A study examining gene changes in efferocytes exposed to ACs versus IgG-coated cells found an increase in glycolysis and a decrease in oxidative phosphorylation in AC-exposed efferocytes, and whereas blocking aerobic glycolysis suppresses efferocytosis, blocking oxidative phosphorylation does not36. Binding of the AC recognition motif, phosphatidylserine, to efferocytes leads to the induction of Slc2a1 mRNA (which encodes the glucose transporter GLUT1), whereas other factors released from ACs induce Sgk1, which encodes a kinase necessary for the localization of GLUT1 at the cell surface (FIG. 1c). Experimental inhibition of Slc2a1 or Sgk1 induction results in the suppression of both initial and continual efferocytosis in vitro and in vivo, including in the setting of atherosclerosis in Western diet-fed Ldlr−/− mice. Inhibition of glycolysis blocked AC-induced actin polymerization, suggesting that blocking GLUT1 inhibits glycolysis and impairs efferocytosis. Furthermore, the later stages of efferocytosis are associated with an increase in the level of lactate, a by-product of glycolysis, and this is associated with an increased level of mRNA encoding the lactate transporter SLC16A1 (also known as MCT1). Silencing of Slc16a1 leads to an increase in cellular lactate level and to impairment of efferocytosis. Furthermore, medium collected from wild-type efferocytes but not from Slc16a1-silenced efferocytes induces Il10 expression in bone marrow-derived macrophages, suggesting a proresolving role of lactate and perhaps other factors released by SLC16A1. Although the identity of the AC-released factors that induce Sgk1, the mechanism linking phosphatidylserine binding to Slc2a1 induction and the molecular pathway downstream of lactate–SLC16A1 remain to be elucidated, this key study is the first to link glycolysis and lactate to efferocytosis and resolution. Although it is somewhat surprising to ascribe a proresolving role to glycolysis, these results are not necessarily at odds with prior findings. Resolution is a natural response to inflammation, and so it may be that inflammation-mediated glycolysis acts as an early trigger to initiate the resolution cascade. Alternatively, glycolysis may be necessary to allow an efferocyte to process the metabolic load resulting from the ingestion of an AC. Additional studies will be needed to more precisely determine the relationship between metabolism and efferocytosis.
Efferocytosis in resolution and repair
In addition to preventing secondary necrosis, efferocytosis has three overall outcomes: termination of inflammatory responses, promotion of self-tolerance and activation of proresolving pathways. When efferocytosis is impaired, these functions are compromised, leading to heightened inflammation, impaired resolution and development of disease. As the effects of efferocytosis on anti-inflammatory and tolerogenic pathways have been reviewed in depth12,37–40, we focus on the proresolving roles of efferocytosis and emerging mechanisms of the failure of this process in disease.
Cell polarization.
Efferocytosis promotes a proresolving phenotype by downregulating proinflammatory cytokine expression, increasing the levels of proresolving mediators, inhibiting inducible nitric oxide synthase (iNOS) and enhancing the production of angiogenic growth factors. These effects occur in part through activation of LXRs, PPARγ and PPARδ41–43. As described in the preceding section, ACs also promote proresolving macrophages through a fatty acid oxidation–IL-10 pathway34. Efferocytosis may also enhance tissue repair by specifying the cell fate of bipotent progenitor cells during chronic liver injury44. In vitro, engulfment of sonicated hepatocyte debris by liver macrophages induces WNT3A expression in these cells44. In vivo, WNT3A promotes canonical WNT signalling in neighbouring progenitor cells and leads to the generation of hepatocytes that repopulate the diseased parenchyma44. Further studies will be needed to link the in vitro and in vivo data, for example, to show that efferocytosis of dead hepatocytes by liver macrophages in vivo is responsible for WNT signalling. In a converse scenario, macrophage polarization to more efficient efferocytes may be triggered by ACs themselves. Apoptotic neutrophils are a rich source of ACs for efferocytosis in inflammation45. In the setting of experimental myocardial infarction, acute depletion of neutrophils resulted in increased fibrosis, greater accumulation of ACs within infarcts and worse cardiac function. Macrophages from these mice express lower levels of the protein tyrosine kinase MERTK, and MERTK expression could be restored in vitro by incubation of macrophages with neutrophil gelatinase-associated lipocalin45.
Production of proresolving mediators.
Efferocytosis also leads to altered production of lipid mediators that play a central role in inflammation resolution. Macrophages incubated with apoptotic neutrophils or neutrophil microparticles increase expression of long-chain fatty acid-derived lipids termed ‘specialized proresolving mediators’ (SPMs), including lipoxin A4, and resolvins D1, D2 and E2, while concomitantly decreasing expression of proinflammatory prostaglandins and leukotriene B4 (REF.46). Lipoxin A4 and resolvin D1 further enhance efferocytosis, setting up a cycle by which they augment their own production46. The increase in SPM production occurs by several mechanisms. First, ACs and their microparticles contain precursors of SPMs, and when ACs are ingested by macrophages, the SPM precursors are converted to mature lipid mediators46. In addition, expression of 12/15-lipoxygenase, a key biosynthetic enzyme for SPMs, is upregulated by efferocytosis47. MERTK–extracellular signal-regulated kinase (ERK) signalling promotes SPM synthesis by increasing the cytoplasmic-to-nuclear ratio of 5-lipoxygenase, another key enzyme in SPM biosynthesis48,49. In vitro and in vivo studies show that deletion of MERTK or cleavage to render MERTK non-functional results in impaired resolution, whereas prevention of MERTK cleavage enhances resolution48. Moreover, activation of LXRα after AC engulfment induces MERTK expression, which implicates MERTK and the SPMs in the proresolving activity of LXR41. MERTK-binding bridging molecules have also been implicated in efferocytosis-induced resolution. For example, protein S is upregulated by macrophages during resolution, and when protein S is deleted from macrophages, they express higher levels of tumour necrosis factor (TNF) and lower levels of IL-10, arginase 1 and resolvin D1 (REF.50).
Two other examples warrant mention. First, after macrophages internalize ACs, MYC is cleaved into MYC-nick, which promotes the recruitment of acetyl-transferases to α-tubulin, facilitating LC3-associated phagocytosis51. LC3-associated phagocytosis promotes phagolysosomal assembly and acidification52, and in this study MYC-nick improved efferocytosis and upregulated Il10, Arg1, Retnla and Mrc1 (REF.51). Second, resolving macrophages in sterile peritonitis increase expression of the secretory integrin-binding protein DEL1 (also known as EDIL3), which promotes efferocytosis by bridging phosphatidylserine on ACs with αVβ3 integrin on macrophages. This process leads to LXR-dependent upregulation of anti-inflammatory and proresolving cytokines and production of lipoxin A4, resolvin D1 and resolvin E1. DEL1 also promotes the proefferocytic function of resolvin D1, suggesting a feedforward mechanism53.
Crosstalk with regulatory T cells.
CD4+CD25+FOXP3+ regulatory T cells (Treg cells) are potent modulators of the immune response. They interact with cells of both the innate immune system and the adaptive immune system to limit T cell proliferation, promote tolerance by deleting autoreactive cells and producing anti-inflammatory cytokines, limit excessive inflammation, foster alternative activation of macrophages towards a proresolving phenotype54,55 and promote resolution56. More recently, Treg cells have been shown to enhance macrophage efferocytosis in vitro and in models of acute lung injury, peritonitis and atherosclerosis57. The mechanism involves Treg cell secretion of IL-13, which upregulates macrophage production of IL-10. IL-10 then promotes macrophage efferocytosis by inducing the GTP-exchange factor VAV1, which enhances RAC1-dependent actin assembly at the phagosome and subsequent AC internalization57. Macrophage efferocytosis also increases the number of Treg cells in vivo, as infusion of ACs leads to expansion of Treg cells in a manner dependent on macrophage-derived transforming growth factor-β (TGFβ)58. A separate in vitro study showed that ingestion of ACs increases phagocyte production of TGFβ and IL-10, which increase the differentiation of Treg cells40. More recently, experimental induction of intestinal epithelial cell apoptosis resulted in uptake of ACs by CD103+ dendritic cells (DCs), leading to the upregulation of genes involved in Treg cell generation and a concomitant increase in the number of T cells in the mesenteric lymph nodes59.When considered together, these studies suggest that Treg cells and macrophages participate in a positive feedback system linked by clearance of ACs.
Prevention of immune activation.
Although efferocytosis of infected ACs by DCs can lead to the presentation of pathogen antigens and activation of effector T cells as part of host defence (see the following section), this process does not usually occur when resolving macrophages ingest non-infected ACs. This concept can be illustrated by a recent study showing that CX3CR1+MERTK+ macrophages in the T cell zone of lymph nodes are inefficient at priming CD4+ T cells, a function instead delegated to phagocytic DCs in the region60. Several mechanisms to explain this observation have been investigated. First, as discussed earlier, internalization of ACs via efferocytosis leads to shunting of cargo towards recycling endosomes and away from the MHC class II-loading compartment, thus avoiding antigen presentation of AC-derived peptides25. Second, phagosomes in proresolving macrophages undergo acidification more efficiently than those in proinflammatory macrophages, leading to proteolysis of AC-derived proteins to a degree that prevents the generation of suitable peptides for MHC class II presentation61.
On the basis of these findings, it is not surprising that autoimmunity can develop when efferocytosis fails (FIG. 2a). There are a few examples of lymphocyte activation and autoimmunity in the setting of defective efferocytosis in mouse models. First, genetic targeting of the AC–efferocyte bridging molecule milk fat globule-EGF factor 8 (MFG-E8) leads to the development of autoantibody-mediated glomerulonephritis that mimics lupus nephritis62. Second, targeting of MERTK results in impaired efferocytosis and increased titres of nuclear autoantibodies63. Third, targeting of complement C1q subcomponent subunit A leads to an accumulation of ACs, high titres of autoantibodies and glomerulonephritis64. Fourth, targeting of PPARδ in macrophages, which impairs efferocytosis owing to defective PPARδ-mediated upregulation of bridging molecules, notably C1q, promotes the production of autoantibodies and lupus-like autoimmune disease43. These data suggest that defective efferocytosis leads to impaired self-tolerance, but it is also possible that uncleared ACs become secondarily necrotic and act as a proinflammatory trigger. Further studies will be necessary to determine the relative contribution of these mechanisms.
Fig. 2 |. Mechanisms of impaired efferocytosis in disease.
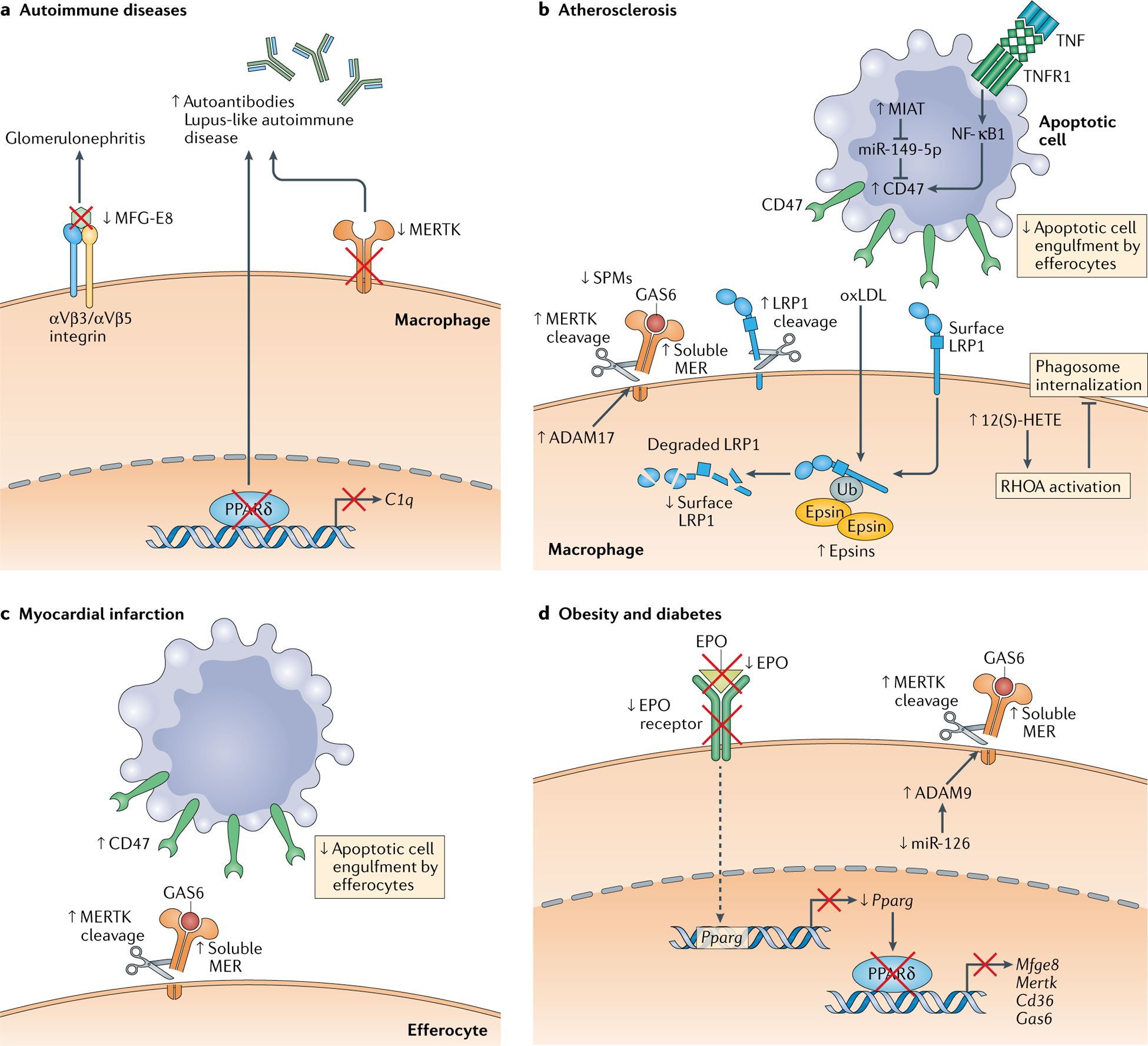
A variety of impairments contribute to defective macrophage efferocytosis and the development and/or progression of disease. a | In the autoimmune disease glomerulonephritis, efferocytosis is impaired due to loss of the efferocytosis bridging molecule milk fat globule-EGF factor 8 (MFG-E8). The development of autoantibodies and lupus-like autoimmune disease in mice is associated with impaired efferocytosis due to reduced levels of the efferocytosis receptor MER proto-oncogene tyrosine kinase (MERTK) or to impaired transcription of the efferocytosis bridging molecule C1q by peroxisome proliferator-activated receptor-δ (PPARδ). b | In atherosclerosis, proteolytic cleavage of the efferocytosis receptors MERTK and low-density lipoprotein receptor-related protein 1 (LRP1) compromises the ability of plaque macrophages to clear dead cells in atherosclerotic lesions, which contributes to plaque necrosis and impaired resolution. The MERTK cleavage product, soluble MER, may be able to compete for the bridging molecule growth arrest-specific protein 6 (GAS6), sequestering it from intact MERTK receptors. Cell-surface LRP1 levels are also decreased by epsin-mediated internalization of the receptor. A particular lipid that accumulates in atherosclerotic lesions called arachidonic acid-derived 12(S)-hydroxyeicosatetraenoic acid (12(S)-HETE) suppresses apoptotic cell internalization by activating RHOA. Finally, apoptotic cells within atherosclerotic plaques upregulate the ‘don’t-eat-me’ signal CD47, impairing their ability to be recognized by phagocytes. Several mechanisms have been proposed, including induction of CD47 expression by a tumour necrosis factor (TNF)−TNF receptor 1 (TNFR1)−nuclear factor-κB1 (NF-κB1) pathway and loss of a CD47-suppressing microRNA, miR-143–5p. c | Similarly to atherosclerosis, both MERTK cleavage in cardiac macrophages and inappropriately high levels of CD47 on apoptotic cardiomyocytes contribute to an impaired resolution response after experimental myocardial infarction. d | In obesity and diabetes, MERTK cleavage also plays an important role in the impaired ability of diabetic macrophages to clear apoptotic cells, although in contrast to the situation in atherosclerosis, cleavage is driven by disintegrin and metalloproteinase domain-containing protein 9 (ADAM9) owing to downregulation of miR-126. Diabetic macrophages also have a downregulation of a proefferocytic pathway involving erythropoietin (EPO)–EPO receptor signalling. Under normal conditions, EPO receptor signalling increases PPARγ expression, leading to enhanced PPARγ-mediated transcription of a number of efferocytosis receptors and bridging molecules. In diabetic mice, however, there is a reduction in levels of both EPO and the EPO receptor that interrupts this pathway and impairs efferocytosis efficiency. MIAT, myocardial infarction-associated transcript; oxLDL, oxidized low-density lipoprotein; SPM, specialized proresolving mediator; Ub, ubiquitin.
Defective efferocytosis in cardiometabolic disease
As illustrated in the previous sections, genetically engineered defects in efferocytosis can impair resolution and predispose an organism to disease. However, there are also a variety of mechanisms by which disease processes themselves cause defective efferocytosis6,65–67. We present here a few examples, focusing on aspects of cardiometabolic disease.
Atherosclerosis.
Atherosclerotic plaques form when modified lipoproteins accumulate within the subendothelial layer of arteries, generating an inflammatory stimulus that drives leukocyte influx into the vessel wall. Many of these leukocytes become apoptotic, and although early in lesion formation they are cleared efficiently, efferocytosis begins to fail in advanced plaques, leading to an accumulation of secondarily necrotic cells in an area of the plaque called the necrotic core68–70. In humans, large necrotic cores are associated with the type of high-risk plaques that are prone to cause myocardial infarction and stroke71. Therefore, elucidating the mechanisms of defective efferocytosis is an important objective of atherosclerosis research4.
One mechanism of defective efferocytosis in atherosclerosis is proteolysis of efferocytosis receptors, such as MERTK and LRP1 (FIG 2b). MERTK is important in lesional efferocytosis, as Western diet-fed Mertk−/−Ldlr−/− mice have increased lesion size, larger necrotic cores and reduced production of SPMs72,73. Lesional macrophages in advanced lesions in humans and mice express high levels of the enzyme calcium/calmodulin-dependent protein kinase IIγ (CaMKIIγ), which suppresses an ATF6–LXRα–MERTK pathway74. Accordingly, the ather osclerotic lesions of Western diet-fed Ldlr−/− mice lacking myeloid CaMKIIγ had higher levels of macrophage MERTK expression, improved efferocytosis and smaller necrotic cores74.
Another potential mechanism of impaired MERTK function in advanced lesions is cell-surface shedding of the receptor on inflammatory macrophages by the protease disintegrin and metalloproteinase domain-containing protein 17 (ADAM17), which leads to defective efferocytosis and the generation of the cleaved extracellular fragment called soluble MER75,76. This soluble fragment can bind to the bridging molecule growth arrest-specific protein 6 (GAS6; also known as AXL ligand), which may prevent its interaction with intact MERTK receptors75, but a role for soluble MER has not yet been demonstrated in an actual disease setting. In progressing atherosclerotic lesions in mice and humans, surface expression of MERTK on macrophages declines, and levels of ADAM17 and soluble MER increase72,77–79, suggesting that MERTK cleavage may be a mechanism for impaired efferocytosis in advanced atherosclerosis. The first evidence suggesting a causative role of MERTK in atheroprogression came from a study using Western diet-fed Ldlr−/− mice in which endogenous MERTK had been replaced by a cleavage-resistant mutant77. The plaques of these mice show enhanced lesional efferocytosis, smaller necrotic cores and higher levels of SPMs compared with control Western diet-fed Ldlr−/− mice77. Genetic targeting of the efferocytosis receptor LRP1 in macrophages or haematopoietic cells in atheroprone mice also leads to increased lesion area and necrotic core size80–82. LRP1-mediated efferocytosis may become impaired during atherosclerosis progression through exposure of lesional macrophages to oxidized LDL, as treatment of cultured macrophages with oxidized LDL promotes epsin-mediated, ubiquitin-dependent internalization and downregulation of LRP1 (REF.83). Consistent with this finding, genetic targeting of myeloid epsin 1 and epsin 2 in apolipoprotein E-deficient mice (Apoe−/− mice) leads to smaller necrotic cores and plaque size83.
Defects in signals derived from ACs have also been identified in atherosclerosis. Inflammatory signalling leads to inappropriate expression of the ‘don’t-eat-me’ signal CD47 in lesional ACs, rendering them resistant to internalization by lesional efferocytes84 (FIG. 2b). Administering an anti-CD47-blocking antibody to atheroprone mice leads to smaller necrotic cores and improves lesional efferocytosis84. Consistent with the links between efferocytosis and inflammation resolution, a more recent study showed that anti-CD47 antibody treatment of Ldlr−/− mice increases the levels of SPMs, particularly resolvin D1, in atherosclerotic lesions85. The study authors also showed that lesional necroptotic cells express very high levels of CD47, which leads to macrophages ‘nibbling’ the cells rather than fully engulfing them. Full engulfment of necroptotic cells was restored by treatment with either anti-CD47 antibody or the resolving mediator resolvin D1, which facilitates the engagement of the ‘eat-me’ signal calreticulin to overcome the CD47 don’t-eat-me signal85.
The mechanism of increased CD47 expression on lesional apoptotic and necroptotic cells is not known, but in vitro studies have suggested contributions of an inflammatory signalling pathway involving the TNF receptor TNFR1 and its downstream mediator nuclear factor-κB1 (REF.84) and of a long non-coding RNA called ‘myocardial infarction-associated transcript’ (MIAT)86. MIAT, the levels of which are increased in patients with symptomatic atherosclerosis, interferes with the post-translational processing of the microRNA miR-149–5p. In vitro, miR-149–5p lowers levels of CD47, and genetic targeting of MIAT in Apoe−/− mice decreases CD47 expression, improves efferocytosis and reduces necrosis in plaques86. Another study showed that CD47 can be therapeutically downregulated by a Toll-like receptor 9 (TLR9) agonist, CpG oligonucleo-tides. TLR9 signalling produces a metabolic shift in macrophages that is associated with enhanced efferocytosis of tumour cells in a mouse model of pancreatic cancer. In this model, CpG oligonucleotide-activated macrophages redirect exogenous fatty acids and glucose towards acetyl-CoA generation and de novo lipid biosynthesis. These macrophages engulf apoptotic tumour cells despite expression of CD47 on the ACs, suggesting that macrophage metabolism can be altered to circumvent the inhibitory function of CD47 (REF.87). Although this has not specifically been investigated in atherosclerosis, TLR9 signalling is known to be athero-protective88, which raises the possibility of a novel therapeutic avenue through which efferocytosis could be enhanced to suppress atherosclerosis progression. In this context, it is interesting to recall the aforementioned study demonstrating a role for fatty acid oxidation in the generation of IL-10 (REF.34), which is consistent with a link between fatty acid metabolism and resolution programmes in macrophages.
More generally, advanced human atherosclerotic lesions have a lower content of SPMs than earlier-stage lesions89, and this defect likely contributes to impaired efferocytosis, as SPMs boost efferocytosis48,90–95. Indeed, treatment of mice with advanced atherosclerosis with a resolving mediator improves lesional efferocytosis96. Furthermore, as mentioned above, resolvin D1 promotes the engulfment of necroptotic cells85. The increase in proinflammatory lipid mediators in advanced atherosclerosis may also compromise efferocytosis. For example, arachidonic acid-derived 12(S)-hydroxyeicosatetraenoic acid, the level of which is increased in progressing atherosclerotic lesions and in serum from individuals with coronary artery disease versus controls, impairs efferocytosis in human monocyte-derived macrophages by activating RHOA, which blocks phagosome internalization97 (FIG. 2b). This defect could be corrected by co-incubation with statins, which inhibit RHOA by blocking a key RHOA-activating modification, isoprenylation.
Myocardial infarction and heart failure.
Efferocytosis plays a key role in the repair of the myocardium after myocardial infarction (FIG. 2c). Mice lacking macrophage MERTK have an exaggerated response to ischaemia–reperfusion injury of the left anterior descending artery, characterized by accumulation of ACs, larger infarct size and more depressed cardiac function. Conversely, mice expressing cleavage-resistant MERTK have a decreased level of ACs in the heart and were more resistant to myocardial injury98,99. A recent study demonstrated that extracellular vesicles secreted by a cardiac progenitor cell population, known as cardiosphere-derived cells, promote the expression of MERTK by macrophages, thereby enhancing efferocytosis and healing after myocardial infarction in rat and mouse models100. As in atherosclerosis, ACs generated during myocardial injury also express inappropriately high levels of CD47. Treatment with anti-CD47 antibody in the period immediately after myocardial infarction leads to improved resolution of cardiac inflammation, reduced infarct size and preserved cardiac function101. In response to ligation of the left anterior descending artery, mice lacking the efferocytosis-bridging molecule MFG-E8 have more cardiac necrosis and inflammation and worse cardiac function compared with control mice, and these effects are attenuated by intracardiac injection of MFG-E8 (REF.102). Finally, a population of cardiac myofibroblasts that appears in the heart after myocardial infarction secretes MFG-E8, which promotes their ability to recognize ACs102. As a consequence, these myofibroblasts downregulate Il1b, Cxcl2 and Il6 expression and upregulate Tgfb expression102. Although TGFβ is generally proresolving when produced by macrophages, production by myofibroblasts may promote excessive fibrosis, which could be deleterious in this setting. Further study will be necessary to determine the myofibroblast-specific role of MFG-E8.
Obesity and diabetes.
Macrophages derived from leptin-deficient ob/ob mice or from mice with diet-induced obesity demonstrate impaired efferocytosis103,104. If this finding were to translate to humans, it might help to explain the predisposition of patients with obesity and diabetes to complications associated with defective resolution, such as impaired wound healing and atherosclerosis. In the case of atherosclerosis, Ldlr−/− ob/ob mice have impaired lesional efferocytosis and larger necrotic cores compared with Ldlr−/− mice. When Ldlr−/− ob/ob mice are fed a diet rich in ω−3 fatty acids, their macrophages show improved efferocytosis104. Although the mechanism is not fully elucidated, it is interesting to hypothesize that SPMs derived from the ω−3 fatty acids are at least partially responsible for the observed improvement. With regard to wound healing, macrophages isolated from wounds of a variety of diabetic mouse models (such as db/db, non-obese diabetic and Akita mice) have impaired efferocytosis, which is associated with higher numbers of ACs and greater expression of proinflammatory cytokines within wounds105.
Another mechanism of defective efferocytosis in obesity may involve loss of MERTK expression. Although ADAM17 is the main protease that cleaves MERTK, a recent study showed that increased expression of ADAM9 in macrophages from db/db mice (which have a genetic dysfunction of the leptin receptor) or macrophages exposed to high levels of exogenous glucose leads to MERTK cleavage and impaired efferocytosis106 (FIG. 2d). The increase in ADAM9 expression is ascribed to decreased expression of miR-126. The translational relevance of these findings is suggested by data comparing hearts from individuals without diabetes and individuals with diabetes and showing that individuals with diabetes had lower expression of both miR-126 and the active form of MERTK (phosphorylated MERTK) and higher expression of ADAM9 (REF.106).
Alternatively, defective efferocytosis in obesity may be related to impaired erythropoietin (EPO) signalling (FIG. 2d). Under normal conditions, the interaction of sphingosine 1-phosphate on ACs with its cog nate recep tor on macrophages promotes an EPO-EPO receptor- PPARγ pathway that enhances efferocytosis. The mechanism involves a JAK2–ERK–phosphorylated CEBP signalling cascade that induces PPARγ, which in turn increases the expression of a variety of efferocytic molecules, including MFG-E8, GAS6, MERTK and CD36 (REF.107). In a model of zymosan-induced peritonitis, macrophages from mice with diet-induced obesity have impaired efferocytosis and reduced levels of both EPO and EPO receptor, which is associated with delayed resolution. Intraperitoneal administration of exogenous EPO to these mice before and during the experimental peritonitis improved efferocytosis and resolution to levels seen in lean mice103. These data raise the possibility that EPO may be a novel therapeutic target for enhancing efferocytosis in obesity.
Efferocytosis in host defence
A common consequence of pathogen invasion is death of host cells, and pathogens often remain alive in these dying cells. If infected dead cells are not rapidly engulfed by phagocytes, live pathogens can be released as a result of pathogen-induced pore formation or overall plasma membrane leakage, such as occurs in secondary cell necrosis or necroptosis, resulting in spread of the infection. If rapid efferocytosis occurs, the fate of the pathogen and the subsequent immune response differ depending on the type of pathogen and the dying cell in which it lives; the type of efferocyte, such as macrophage versus DC; and other host factors, including genetic polymorphisms108,109. In this section, we provide a few examples.
Fate of bacteria after efferocytosis.
When Mycobacterium tuberculosis invades macrophages, bacteria remain alive in phagosomes owing to M. tuberculosis-induced suppression of lysosome–phagosome fusion. Relatively low-virulence strains of M. tuberculosis often induce apoptosis, and when macrophages engulf these ACs, lysosome fusion with AC-containing phagosomes promotes bacterial killing110 (FIG. 3a). Perhaps related to this mechanism, M. tuberculosis infection worsens in mice given a neutralizing antibody against the efferocytosis receptor T cell immunoglobulin mucin receptor 4 (TIM4). Another example of efferocytosis-mediated host defence occurs when certain bacteria, such as Salmonella enterica subsp. enterica serovar Typhimurium and some strains of Klebsiella pneumoniae, trigger pyroptosis in neutrophils111,112. Efferocytosis of the infected neutrophils by macrophages in vitro, which is facilitated by the interaction of pore-induced intracellular traps on pyroptotic cells with scavenger and complement receptors on macrophages112, leads to bacterial neutralization (FIG. 3b). Inhibition of pyroptosis worsens K. pneumoniae infection in mice, but a direct link to impaired efferocytosis has not been shown. This point is important, as molecules released by ACs can affect phagocytes independently of efferocytosis, and this may be particularly important with pore-bearing pyroptotic cells.
Fig. 3 |. The role of efferocytosis in infection and host defence.
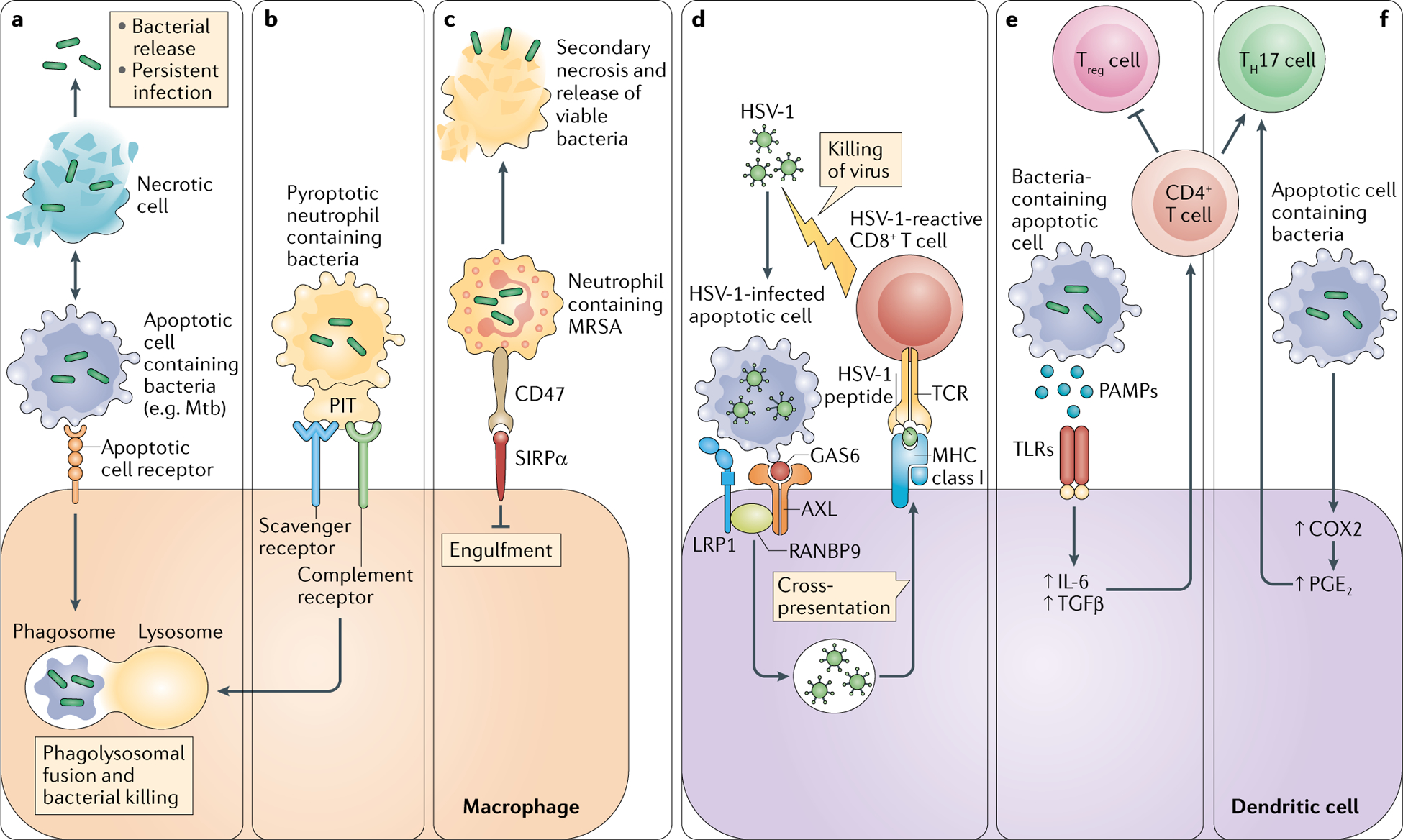
Efferocytosis can mediate host defence against pathogens in some settings but, in other settings, efferocytosis can be usurped by pathogens to promote their spread. Several examples are illustrated. a | Engulfment of infected apoptotic cells by macrophages leads to bacterial killing and prevents secondary necrosis of the infected cells, thereby preventing bacterial release. b | A similar pathway occurs when certain bacteria induce pyroptosis in infected cells, in which pore-induced intracellular traps (PITs) in these cells are recognized by efferocytosis receptors. c | Other types of bacteria, such as methicillin-resistant Staphylococcus aureus (MRSA), can evade efferocytic uptake and killing by inducing expression of the don’t-eat-me signal CD47 on the cells they infect. Efferocytosis of infected cells by dendritic cells can also participate in host defence (not shown). d | Recognition and internalization of herpes simplex virus 1 (HSV-1)-infected apoptotic cells by a protein complex comprising AXL, low-density lipoprotein receptor-related protein 1 (LRP1) and RAN-binding protein 9 (RANBP9) leads to cross-presentation of viral antigen on MHC class I molecules and subsequent generation of antiviral CD8+ T cells. Other types of effector T cells are activated by dendritic cells engulfing bacteria-infected apoptotic cells. e | Pathogen-associated molecular patterns (PAMPs) expressed on infected apoptotic cells activate the production of IL-6 and transforming growth factor-β (TGFβ) via Toll-like receptors (TLRs), which promotes the differentiation of CD4+ T cells to IL-17-producing T helper cells (TH17 cells) and suppresses the generation of regulatory T cells (Treg cells). f | TH17 cells are also activated by prostaglandin E2 (PGE2) resulting from the engulfment of apoptotic cells. COX2, cyclooxygenase 2; GAS6, growth arrest-specific protein 6; Mtb, Mycobacterium tuberculosis; SIRPα, signal-regulatory protein-α; TCR, T cell receptor.
The current literature also contains important examples of how efferocytosis fails to neutralize bacteria. Virulent strains of M. tuberculosis induce mostly cell necrosis, which decreases efferocytosis of infected cells and promotes bacterial spread113. Moreover, an in vitro study suggested that when engulfment of M. tuberculosis-infected necrotic neutrophils by macrophages does occur, it can actually promote M. tuberculosis growth114. In the case of the parasite Leishmania major, macrophage engulfment of infected ACs promotes transformation of promastigotes to amastigotes, which are able to avoid phagolysosomal degradation109. Moreover, promastigotes themselves may impair phagosome maturation. Another example occurs when macrophages engulf apoptotic T cells infected with the parasite that causes Chagas disease — Trypanosoma cruzi. Similarly to uptake of M. tuberculosis-infected necrotic cells, uptake of T. cruzi-infected apoptotic T cells actually promotes parasite replication in macrophages115. T. cruzi-infected ACs engage the vitronectin receptor on macrophages, which stimulates the secretion of prostaglandin E2 (PGE2) and TGFβ, which then increase the infectivity of T. cruzi. Part of the mechanism involves the ability of these molecules to activate ornithine decarboxylase (ODC) in macrophages: ODC converts ornithine into polyamines, and polyamines promote T. cruzi replication. Treatment of T. cruzi-infected mice with inhibitors of PGE2 production markedly suppressed parasitaemia, although a direct link to ODC was not shown115. Finally, the efferocytosis machinery can be usurped by some pathogens to promote direct infection of macrophages. This type of ‘apoptotic mimicry’ is used by vaccinia virus, in which phosphatidylserine expression on the virus membrane facilitates effero cytosis receptor-mediated uptake by macrophages, leading to intracellular viral replication116.
In other cases, pathogens can escape efferocytosis-mediated killing by preventing macrophage engulfment of infected ACs. For example, internalization of methicillin-resistant Staphylococcus aureus by neutrophils induces expression of the don’t-eat-me signal CD47 (REF.117) (FIG. 3c). As a result, dying, infected neutrophils are not engulfed by macrophages, and subsequent necrosis of the neutrophils releases viable bacteria. Although some K. pneumoniae infections are controlled by successful efferocytosis-mediated bacterial killing through the mechanism discussed above, more virulent strains can escape this fate. One such strain of K. pneumoniae blocks pyroptosis in neutrophils and thereby prevents infected neutrophil efferocytosis and subsequent bacterial killing by macrophages118. Another strain of K. pneumoniae activates phospholipid transporter flippases in infected neutrophils, which, when the neutrophils die, prevents externalization of phosphatidylserine and recognition by efferocytic macrophages118. Similarly to methicillin-resistant S. aureus, the bacteria eventually induce necroptosis of the neutrophils, resulting in bacterial spread.
Antigen presentation from infected ACs.
Efferocytosis of infected ACs can help to mount an adaptive immune response against bacteria through the process of antigen cross-presentation. This principle was initially established in vitro by showing that efferocytosis of influenza virus-infected apoptotic monocytes by DCs leads to cross-presentation of influenza virus antigens on MHC class I and activation of CD8+ T cells119. Additional in vitro studies reported similar results using ACs infected with Salmonella Typhimurium, human cytomegalovirus, vaccinia virus, M. tuberculosis, herpes simplex virus (HSV) and HIV-1 (refs120–125). Engulfment of HIV-1-infected or HIV-1-antigen-transduced ACs by DCs in vitro led not only to cross-presentation of antigen on MHC class I, with activation of CD8+ T cells, but also to presentation of antigen on MHC class II, with activation of CD4+ T cells122.
In vivo, proof of this concept has proven to be more challenging. In one study, mice were injected with influenza virus-infected apoptotic 3T3 cells, and this led to T cell activation directed against influenza virus antigen126. However, a direct a link between DC efferocytosis-mediated cross-presentation and protective antipathogen adaptive immunity in an actual infectious setting was not shown. This critical link was finally established following the discovery of a cell-surface protein complex on CD8α+ DCs, comprising AXL, LRP1 and RAN-binding protein 9 (RANBP9), that binds and internalizes HSV-1-infected ACs in a manner that leads to cross-presentation of AC-derived viral antigens to CD8+ T cells127 (FIG. 3d). AXL was necessary for AC binding, LRP1 promoted AC internalization via its interaction with the RAC GTP adaptor GULP1 and RANBP9 facilitated the functional interaction between AXL and LRP1. Genetic targeting of any of these molecules on DCs in mice infected with HSV-1 leads to accumulation of infected ACs, impaired activation of splenic CD8+ T cells directed against HSV-1 antigen, increased viral load and decreased mouse survival127. As these DC-targeted gene knockouts do not affect HSV-1 infectivity itself, the data provide direct molecular-genetic causation evidence linking DC efferocytosis of infected ACs with cross-presentation of antigen and adaptive immunity in pathogen-infected mice.
A subsequent study investigated the role of annexin 1 in efferocytosis-mediated M. tuberculosis cross-presentation in vitro and in vivo128. In cultured DCs exposed to M. tuberculosis-infected ACs, annexin 1 facilitates both AC uptake and antigen cross-presentation. Accordingly, holoannexin 1-deficient mice infected with M. tuberculosis show impaired activation of CD8+ T cells targeting M. tuberculosis antigen, higher bacterial load and lower survival compared with wild-type mice. Further studies are needed to test whether other DC efferocytosis effectors implicated in pathogen antigen cross-presentation in vitro have a role in host defence in vivo.
Efferocytosis of infected ACs can also activate inflammatory IL-17-producing T helper cells (TH17 cells)129 (FIG. 3e). In vitro studies showed that efferocytosis of Escherichia coli-infected ACs by DCs induces the production of several proinflammatory cytokines via TLR activation by AC-associated pathogen-associated molecular patterns (PAMPs). The cytokines, notably IL-6 and TGFβ, promote differentiation of CD4+ T cells into TH17 cells and inhibit production of inflammation-suppressing Treg cells. A subset of these IL-17+CD4+ T cells also produce IL-10, which likely acts as a counter-regulatory mechanism to limit the inflammatory response129. In mice infected with the diarrhoea-causing bacterium Citrobacter rodentium, apoptotic intestinal epithelial cells stimulate the generation of C. rodentium-specific TH17 cells. A recent study comparing DC efferocytosis of sterile ACs versus E. coli-infected ACs corroborated most of these findings, most notably the activation of TH17 cells130. This study also showed that infected ACs increase the expression of CD86 and CCR7 in DCs, which promote antigen presentation and DC migration to lymph nodes, respectively.
The results of these studies seem to imply that the TH17 cell response is a host defence mechanism to limit pathogen damage. However, host defence may be compromised by another consequence of infected AC uptake in this setting; namely, an increase in expression of cyclooxygenase 2 (COX2) and its product PGE2 by efferocytic DCs130 (FIG. 3f). Despite being able to activate TH17 cells in other settings, PGE2 here may actually limit the extent of the antipathogen T 17 cell response131. The mechanism involves a PGE2–EP4 receptor pathway in TH17 cells that downregulates IL-1 receptor and suppresses TH17 cell differentiation. This idea is supported, although not proven, by the finding that treatment of C. rodentium-infected mice with an EP4 receptor antagonist increases colonic TH17 cell numbers and Il17a mRNA levels, suppresses C. rodentium infection and protects mice from colitis. Recall that PGE2 also promotes the replication of T. cruzi in macrophages that have engulfed T. cruzi-infected ACs115, and another study showed that PGE2 resulting from efferocytosis by lung macrophages compromises the ability of these cells to kill phagocytosed S. pneumoniae in vitro132. Thus, whereas postefferocytosis PGE2 production may play a protective role in certain settings133, in other settings it may be maladaptive.
Further complexity is illustrated by the recent finding in C. rodentium-infected mice that efferocytosis of infected ACs by DCs leads to the presentation of AC-derived self-antigens on MHC class II and activation of a subpopulation of self-reactive TH17 cells in the colon134. This subpopulation, which coexists with the C. rodentium-specific TH17 cells described previously129, leads to the production of IgA and IgG1 autoantibodies and autoimmune colitis. As with the C. rodentium-specific TH17 cell response, the autoimmune response is triggered by bacterial ligand-induced TLR activation, which optimizes the ability of DCs to present phagocytosed antigens, and does not occur in mice infected with a strain of C. rodentium that does not cause epithelial cell apoptosis. What determines the proportion of TH17 cells in each subpopulation and how these findings relate to the aforementioned PGE pathway131 remain to be determined. Nonetheless, these findings highlight a crucial issue in our understanding of DC efferocytosis of infected ACs; namely, the importance of the balance among immune tolerance, protective inflammation and maladaptive autoinflammation. Further studies will be necessary to fully understand the mechanisms and possible genetic variations that determine whether DC efferocytosis of infected ACs in infected hosts elicits a proper antipathogen inflammatory response versus one that is either too weak or too strong.
Conclusions
Efferocytosis is crucial for maintaining tissue homeostasis and promoting resolution in response to inflammation and injury. Failed efferocytosis is emerging as a key mechanism driving the development and progression of chronic inflammatory diseases, including atherosclerosis, obesity, diabetes, heart failure, chronic lung disease, neurodegenerative disease and cancer. Accordingly, therapeutic strategies aimed at improving efferocytosis would be predicted to dampen inflammation and improve resolution. As one example, data from genetically engineered mice suggest that inhibiting the cleavage of MERTK and LRP1 would improve efferocytosis, and in the case of MERTK, this strategy would also enhance the production of proresolving mediators. In addition, it may be possible to compensate for MERTK cleavage by increasing its expression. As an example, the endogenous fatty acid amide palmitoylethanolamide increases MERTK expression by activating G protein-coupled receptor 55 (REF.135). When administered to Apoe−/− mice, palmitoylethanolamide promotes features of plaque stability in advanced atherosclerotic lesions, including smaller necrotic cores and increased level of lesional collagen135. As another strategy, antibody-mediated neutralization of CD47 can ameliorate experimental atherosclerosis, and this type of therapy has been used in clinical trials for cancer.
As with all new therapeutic strategies, challenges will emerge. For example, as illustrated in this Review, efferocytosis can promote pathogen virulence in certain settings. Moreover, the fibrotic response following efferocytosis may prove to be damaging in diseases such as pulmonary fibrosis and non-alcoholic steatohepatitis. With anti-CD47 antibody, the major challenge is the development of anaemia due to excessive clearance of erythrocytes. Furthermore, a recent study reported that CD47-deficient mice, which were predicted to be protected from atherosclerosis, actually develop increased lesion formation owing in part to increased lymphocyte activation136. Meeting these challenges will require increased understanding of the molecular and cellular mechanisms and consequences of efferocytosis. One important area that has attracted recent attention and will require further study is how efferocytes process and react to AC-derived metabolic cargo. This area is particularly relevant to chronic inflammatory diseases, in which efferocytes are confronted with large numbers of ACs. Further understanding of efferocyte metabolism may suggest new therapeutic opportunities for diseases driven by excessive apoptosis, defective efferocytosis and impaired resolution and repair.
Acknowledgements
A.C.D. is supported by funding from American Heart Association grant 17FTF33660643. A.Y. is supported by funding from US National Institutes of Health (NIH) grant K99HL145131. The laboratory of I.T. is supported by funding from NIH grants R35HL145228, R01HL127464 and P01HL087123. The authors thank past and present members of the Tabas laboratory who participated in research related to this Review.
Glossary
- Low-density lipoprotein receptor-deficient mice
(LDLR-deficient mice) A mouse model of atherosclerosis in which hypercholesterolaemia is induced by a targeted deletion of the gene encoding LDLr, which functions to clear LDL from the circulation. Ldlr −/− mice fed a high-fat, high-cholesterol diet have a very high level of plasma LDL and develop aortic lesions that are morphologically similar to human atherosclerotic plaques.
- Western diet
A commonly used rodent diet that contains a higher fat, sucrose and cholesterol content than standard chow diet, akin to the fast food diet encountered in the Western hemisphere. ingestion of this diet by Ldlr −/− mice results in weight gain, high glucose levels and elevated levels of circulating cholesterol and triglycerides that drive the development of atherosclerotic plaques.
- Foam cells
Macrophages that localize at sites of early atherosclerotic lesion development and that subsequently ingest apolipoprotein B-containing lipoproteins in the subendothelium. They are called foam cells because lipoprotein uptake and metabolism by these macrophages leads to the accumulation of cholesterol ester droplets in the cytoplasm, which gives the cells a ‘foamy’ appearance.
- Peroxisome proliferator-activated receptor-γ
(PPARγ). A member of a group of nuclear receptor proteins involved in altering lipid and glucose metabolism and inflammation. Their ligands include free fatty acids and eicosanoids.
- Fatty acid oxidation
An important metabolic process used to derive energy through the mobilization and oxidation of fatty acids, mainly in the mitochondrial matrix. fatty acid oxidation is positively and negatively regulated by 5′ AMP-activated protein kinase and mechanistic target of rapamycin, respectively.
- Myocardial infarction
An episode of acute cardiac ischaemia that leads to the death of heart muscle cells. it is usually caused by the rupture or erosion of an atherosclerotic plaque leading to occlusive clot formation.
- Oxidative phosphorylation
The metabolic pathway that occurs at the inner mitochondrial membrane and uses an electrochemical gradient created by the oxidation of electron carriers to generate ATP.
- Glycolysis
A metabolic pathway that generates the cellular high-energy store ATP by oxidizing glucose to pyruvate. in eukaryotic cells, pyruvate is further oxidized to CO2 and H2O in a process known as aerobic respiration, which results in a net yield of 36–38 molecules of ATP per metabolized molecule of glucose.
- Secondary necrosis
A process that occurs in apoptotic cells that are not cleared by phagocytes. The integrity of the plasma membrane is lost, and the constituents of the cell are released.
- WNT signaling
A signalling pathway that regulates cell fate determination, proliferation, adhesion, migration and polarity during development. In addition to the crucial role of this pathway in embryogenesis, WNT ligands and their downstream signalling molecules have been implicated in tumorigenesis and have causative roles in human colon cancers.
- LC3-associated phagocytosis
A non-autophagosomal pathway in which many downstream effector proteins of classic macroautophagy, notably LC3 (the mammalian homologue of yeast Atg8), are used by macrophages to mediate the fusion of lysosomes with phagosomes. it has been shown to facilitate the degradation of internalized apoptotic cells and bacteria by macrophages.
- Apolipoprotein E-deficient mice
(Apoe−/− mice). A widely used mouse model that is prone to develop atherosclerosis because the mice have high levels of remnant lipoproteins (a type of atherogenic lipoprotein). This lipoprotein abnormality is caused by the genetic absence of apolipoprotein e, which normally clears remnant lipoproteins from the bloodstream by interacting with hepatocytes.
- CD47
A plasma membrane molecule that interacts with several receptors on other cells, including signal-regulatory protein-α (SIRPα), thrombospondin and membrane integrins. The interaction of a cell expressing CD47 with SIRPα on a macrophage prevents cell engulfment by the macrophage. This mechanism prevents the internalization of living cells, but can also prevent the uptake of dead cells if CD47 is inappropriately expressed on dead cells.
- Necroptotic cells
Cells undergoing a programmed form of necrotic cell death mediated by receptor-interacting protein kinase 1 (RIPK1), RIPK3 and mixed lineage kinase domain-like protein (MLKL). it can be induced by death receptors and by TIR-domain-containing adaptor protein-inducing interferon-β (TRIF)-dependent Toll-like receptor 3 (TLR3) and TLR4 signalling. inhibition of caspase 8 activation sensitizes cells to necroptosis.
- RHOA
A member of a subfamily of small GTP-binding proteins that have key roles in rearrangement of the cytoskeleton. The nucleotide-bound state of these GTPases is generally regulated by guanine-nucleotide exchange factors, which catalyse GDP–GTP exchange, and GTPase-activating proteins, which facilitate the hydrolysis of the bound GTP. Activation, by extracellular signals through various receptors, results in translocation to the plasma membrane, thereby localizing their activity to discrete sites in the cell.
- Statins
A family of inhibitors targeting 3-hydroxy-3-methylglutaryl-CoA reductase (HMG-CoA reductase), an enzyme that catalyses the conversion of HMg-CoA to l-mevalonate. These molecules are mainly used as cholesterol-lowering drugs, but they also have immunoregulatory and anti-inflammatory properties. l-Mevalonate and its metabolites are implicated in cholesterol synthesis and other intracellular pathways.
- ob/ob mice
A mouse model of metabolic dysregulation and obesity that arises from increased appetite due to a leptin gene mutation that renders these mice functionally leptin deficient.
- Cross-presentation
The ability of certain antigen-presenting cells to load peptides that are derived from exogenous antigens onto MHC class I molecules. This property is atypical, because most cells exclusively present peptides from their endogenous proteins on MHC class I molecules. Cross-presentation is essential for the initiation of immune responses to viruses that do not infect antigen-presenting cells.
- Palmitoylethanolamide
An endogenous fatty acid amide that has potent anti-inflammatory effects through its effects on peroxisome proliferator-activated receptor-α and G protein-coupled receptor 55.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Bianconi E et al. An estimation of the number of cells in the human body. Ann. Hum. Biol 40, 463–471 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Arandjelovic S & Ravichandran KS Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol 16, 907–917 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Back M, Yurdagul A Jr., Tabas I, Oorni K & Kovanen PT Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat. Rev. Cardiol 16, 389–406 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yurdagul A Jr., Doran AC, Cai B, Fredman G & Tabas IA Mechanisms and consequences of defective efferocytosis in atherosclerosis. Front. Cardiovasc. Med 4, 86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawano M & Nagata S Efferocytosis and autoimmune disease. Int. Immunol 30, 551–558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szondy Z, Garabuczi E, Joos G, Tsay GJ & Sarang Z Impaired clearance of apoptotic cells in chronic inflammatory diseases: therapeutic implications. Front. Immunol 5, 354 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elliott MR & Ravichandran KS The dynamics of apoptotic cell clearance. Dev. Cell 38, 147–160 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hochreiter-Hufford A & Ravichandran KS Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb. Perspect. Biol 5, a008748 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kasikara C, Doran AC, Cai B & Tabas I The role of non-resolving inflammation in atherosclerosis. J. Clin. Invest 128, 2713–2723 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fredman G & Tabas I Boosting inflammation resolution in atherosclerosis: the next frontier for therapy. Am. J. Pathol 187, 1211–1221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devitt A & Marshall LJ The innate immune system and the clearance of apoptotic cells. J. Leukoc. Biol 90, 447–457 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Elliott MR, Koster KM & Murphy PS Efferocytosis signaling in the regulation of macrophage inflammatory responses. J. Immunol 198, 1387–1394 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henson PM & Hume DA Apoptotic cell removal in development and tissue homeostasis. Trends Immunol. 27, 244–250 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Maderna P & Godson C Phagocytosis of apoptotic cells and the resolution of inflammation. Biochim. Biophys. Acta 1639, 141–151 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Zent CS & Elliott MR Maxed out macs: physiologic cell clearance as a function of macrophage phagocytic capacity. FEBS J. 284, 1021–1039 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greenberg S & Grinstein S Phagocytosis and innate immunity. Curr. Opin. Immunol 14, 136–145 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Han CZ & Ravichandran KS Metabolic connections during apoptotic cell engulfment. Cell 147, 1442–1445 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aderem A How to eat something bigger than your head. Cell 110, 5–8 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Steinman RM, Mellman IS, Muller WA & Cohn ZA Endocytosis and the recycling of plasma membrane. J. Cell Biol 96, 1–27 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Becker T, Volchuk A & Rothman JE Differential use of endoplasmic reticulum membrane for phagocytosis in J774 macrophages. Proc. Natl Acad. Sci. USA 102, 4022–4026 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campbell-Valois FX et al. Quantitative proteomics reveals that only a subset of the endoplasmic reticulum contributes to the phagosome. Mol. Cell. Proteom 11 M111 016378 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y et al. Mitochondrial fission promotes the continued clearance of apoptotic cells by macrophages. Cell 171, 331–345.e22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that mitochondrial fission enhances continual efferocytosis by stimulating calcium-induced membrane recycling to the cell surface to allow phagosome formation, with demonstration of relevance in vivo.
- 23.Czibener C et al. Ca2+ and synaptotagmin VII-dependent delivery of lysosomal membrane to nascent phagosomes. J. Cell Biol 174, 997–1007 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin C, Argintaru D & Heit B Rab17 mediates intermixing of phagocytosed apoptotic cells with recycling endosomes. Small GTPases 10, 218–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yin C, Kim Y, Argintaru D & Heit B Rab17 mediates differential antigen sorting following efferocytosis and phagocytosis. Cell Death Dis. 7, e2529 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tabas I Cholesterol in health and disease. J. Clin. Invest 110, 583–590 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moore KJ & Tabas I Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cui D et al. Pivotal advance: macrophages become resistant to cholesterol-induced death after phagocytosis of apoptotic cells. J. Leukoc. Biol 82, 1040–1050 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Kiss RS, Elliott MR, Ma Z, Marcel YL & Ravichandran KS Apoptotic cells induce a phosphatidylserine-dependent homeostatic response from phagocytes. Curr. Biol 16, 2252–2258 (2006). [DOI] [PubMed] [Google Scholar]; A key study showing that uptake of ACs stimulates a cholesterol efflux response in efferocytic macrophages.
- 30.Viaud M et al. Lysosomal cholesterol hydrolysis couples efferocytosis to anti-inflammatory oxysterol production. Circ. Res 122, 1369–1384 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xian X et al. LRP1 integrates murine macrophage cholesterol homeostasis and inflammatory responses in atherosclerosis. eLife 6, e29292 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fond AM, Lee CS, Schulman IG, Kiss RS & Ravichandran KS Apoptotic cells trigger a membrane-initiated pathway to increase ABCA1. J. Clin. Invest 125, 2748–2758 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park D et al. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature 477, 220–224 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that continual uptake of ACs by macrophages depends on MMP and is important in vivo.
- 34.Zhang S et al. Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab. 29, 443–456.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; A key study demonstrating how macrophages leverage the metabolites derived from ingested ACs to upregulate anti-inflammatory processes and promote tissue repair.
- 35.Galvan-Pena S & O’Neill LA Metabolic reprograming in macrophage polarization. Front. Immunol 5, 420 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morioka S et al. Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature 563, 714–718 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that AC uptake depends on solute carrier family-mediated aerobic glycolysis and that by-products of this process influence the local microenvironment.
- 37.Voll RE et al. Immunosuppressive effects of apoptotic cells. Nature 390, 350–351 (1997). [DOI] [PubMed] [Google Scholar]
- 38.Savill J & Fadok V Corpse clearance defines the meaning of cell death. Nature 407, 784–788 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Green DR, Ferguson T, Zitvogel L & Kroemer G Immunogenic and tolerogenic cell death. Nat. Rev. Immunol 9, 353–363 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blander JM The many ways tissue phagocytes respond to dying cells. Immunol. Rev 277, 158–173 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.A-Gonzalez N et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 31, 245–258 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; An important study demonstrating a fascinating feedback mechanism in which efferocytosis activates LXR, which in turn induces MERTK.
- 42.Ariel A & Serhan CN New lives given by cell death: macrophage differentiation following their encounter with apoptotic leukocytes during the resolution of inflammation. Front. Immunol 3, 4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mukundan L et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat. Med 15, 1266–1272 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boulter L et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat. Med 18, 572–579 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Horckmans M et al. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J 38, 187–197 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Dalli J & Serhan C Macrophage proresolving mediators-the when and where. Microbiol. Spectr 10.1128/microbiolspec.MCHD-0001-2014 (2016). [DOI] [PMC free article] [PubMed]
- 47.Schif-Zuck S et al. Saturated-efferocytosis generates pro-resolving CD11b low macrophages: modulation by resolvins and glucocorticoids. Eur. J. Immunol 41, 366–379 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cai B et al. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc. Natl Acad. Sci. USA 113, 6526–6531 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai B et al. MerTK signaling in macrophages promotes the synthesis of inflammation resolution mediators by suppressing CaMKII activity. Sci. Signal 11, eaar3721 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lumbroso D et al. Macrophage-derived protein S facilitates apoptotic polymorphonuclear cell clearance by resolution phase macrophages and supports their reprogramming. Front. Immunol 9, 358 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhong X et al. Myc-nick promotes efferocytosis through M2 macrophage polarization during resolution of inflammation. FASEB J. 32, 5312–5325 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Martinez J et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl Acad. Sci. USA 108, 17396–17401 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]; A key study showing that LC3-associated phagocytosis is required for degradation of ingested ACs and suppression of proinflammatory signalling.
- 53.Maekawa T et al. Antagonistic effects of IL-17 and D-resolvins on endothelial Del-1 expression through a GSK-3beta-C/EBPbeta pathway. Nat. Commun 6, 8272 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tiemessen MM et al. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc. Natl Acad. Sci. USA 104, 19446–19451 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weirather J et al. Foxp3+CD4+ T cells improve healing after myocardial infarction by modulating monocyte/ macrophage differentiation. Circ. Res 115, 55–67 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Josefowicz SZ, Lu LF & Rudensky AY Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol 30, 531–564 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Proto JD et al. Regulatory T cells promote macrophage efferocytosis during inflammation resolution. Immunity 49, 666–677 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reveals a molecular cellular crosstalk link between two arms of immune cell-mediated resolution, Treg cells and efferocytic macrophages, with demonstration of in vivo relevance.
- 58.Kleinclauss F et al. Intravenous apoptotic spleen cell infusion induces a TGF-beta-dependent regulatory T-cell expansion. Cell Death Differ. 13, 41–52 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cummings RJ et al. Different tissue phagocytes sample apoptotic cells to direct distinct homeostasis programs. Nature 539, 565–569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; An important study showing that ‘sampling’ of ACs leads to the generation of distinct gene expression signatures between macrophages and DCs, which promotes cell-specific functions related to immune tolerance.
- 60.Baratin M et al. T cell zone resident macrophages silently dispose of apoptotic cells in the lymph node. Immunity 47, 349–362 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Canton J, Khezri R, Glogauer M & Grinstein S Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages. Mol. Biol. Cell 25, 3330–3341 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hanayama R et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science 304, 1147–1150 (2004). [DOI] [PubMed] [Google Scholar]
- 63.Scott RS et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411, 207–211 (2001). [DOI] [PubMed] [Google Scholar]; A landmark study showing that MERTK is an efferocytosis receptor.
- 64.Botto M et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat. Genet 19, 56–59 (1998). [DOI] [PubMed] [Google Scholar]
- 65.Poon IK, Lucas CD, Rossi AG & Ravichandran KS Apoptotic cell clearance: basic biology and therapeutic potential. Nat. Rev. Immunol 14, 166–180 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tajbakhsh A, Gheibi Hayat SM, Butler AE & Sahebkar A Effect of soluble cleavage products of important receptors/ligands on efferocytosis: their role in inflammatory, autoimmune and cardiovascular disease. Ageing Res. Rev 50, 43–57 (2019). [DOI] [PubMed] [Google Scholar]
- 67.Driscoll WS, Vaisar T, Tang J, Wilson CL & Raines EW Macrophage ADAM17 deficiency augments CD36-dependent apoptotic cell uptake and the linked anti-inflammatory phenotype. Circ. Res 113, 52–61 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Geng YJ & Libby P Evidence for apoptosis in advanced human atheroma. Colocalization with interleukin-1 beta-converting enzyme. Am. J. Pathol 147, 251–266 (1995). [PMC free article] [PubMed] [Google Scholar]
- 69.Otsuka F et al. Natural progression of atherosclerosis from pathologic intimal thickening to late fibroatheroma in human coronary arteries: a pathology study. Atherosclerosis 241, 772–782 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schrijvers DM, De Meyer GR, Kockx MM, Herman AG & Martinet W Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler. Thromb. Vasc. Biol 25, 1256–1261 (2005). [DOI] [PubMed] [Google Scholar]; This study uses an innovative method to show that efferocytosis is defective in advanced atherosclerosis in humans.
- 71.Hansson GK, Libby P & Tabas I Inflammation and plaque vulnerability. J. Intern. Med 278, 483–493 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ait-Oufella H et al. Defective mer receptor tyrosine kinase signaling in bone marrow cells promotes apoptotic cell accumulation and accelerates atherosclerosis. Arterioscler. Thromb. Vasc. Biol 28, 1429–1431 (2008). [DOI] [PubMed] [Google Scholar]
- 73.Thorp E, Cui D, Schrijvers DM, Kuriakose G & Tabas I Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe−/− mice. Arterioscler. Thromb. Vasc. Biol 28, 1421–1428 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Doran AC et al. CAMKIIgamma suppresses an efferocytosis pathway in macrophages and promotes atherosclerotic plaque necrosis. J. Clin. Invest 127, 4075–4089 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sather S et al. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood 109, 1026–1033 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thorp E et al. Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cdelta, and p38 mitogen-activated protein kinase (MAPK). J. Biol. Chem 286, 33335–33344 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cai B et al. MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J. Clin. Invest 127, 564–568 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses a cleavage-resistant Mertk-knock-in mouse model to show precisely that MERTK cleavage suppresses inflammation resolution in vivo.
- 78.Garbin U et al. Expansion of necrotic core and shedding of Mertk receptor in human carotid plaques: a role for oxidized polyunsaturated fatty acids? Cardiovasc. Res 97, 125–133 (2013). [DOI] [PubMed] [Google Scholar]
- 79.Zhang Y et al. Angiotensin deteriorates advanced atherosclerosis by promoting MerTK cleavage and impairing efferocytosis through AT1R/ROS/p38MAPK/ ADAM17 pathway. Am. J. Physiol. Cell. Physiol 371, C776–C787 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Yancey PG et al. Macrophage LRP-1 controls plaque cellularity by regulating efferocytosis and Akt activation. Arterioscler. Thromb. Vasc. Biol 30, 787–795 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Overton CD, Yancey PG, Major AS, Linton MF & Fazio S Deletion of macrophage LDL receptor-related protein increases atherogenesis in the mouse. Circ. Res 100, 670–677 (2007). [DOI] [PubMed] [Google Scholar]
- 82.Yancey PG et al. Low-density lipoprotein receptor-related protein 1 prevents early atherosclerosis by limiting lesional apoptosis and inflammatory Ly-6Chigh monocytosis: evidence that the effects are not apolipoprotein E dependent. Circulation 124, 454–464 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brophy ML et al. Myeloid-specific deletion of epsins 1 and 2 reduces atherosclerosis by preventing LRP-1 downregulation. Circ. Res 124, e6–e19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kojima Y et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 536, 86–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This important study shows that CD47 is inappropriately upregulated by ACs in atherosclerosis, rendering them resistant to efferocytosis, and that treatment with an anti-CD47 blocking antibody enhances efferocytosis and ameliorates atherosclerosis.
- 85.Gerlach BD et al. Resolvin D1 promotes the targeting and clearance of necroptotic cells. Cell Death Differ. 10.1038/s41418-019-0370-1 (2019). [DOI] [PMC free article] [PubMed]
- 86.Ye ZM et al. LncRNA MIAT sponges miR-149–5p to inhibit efferocytosis in advanced atherosclerosis through CD47 upregulation. Cell Death Dis. 10, 138 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu M et al. Metabolic rewiring of macrophages by CpG potentiates clearance of cancer cells and overcomes tumor-expressed CD47-mediated ‘don’t-eat-me’ signal. Nat. Immunol 20, 265–275 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Roshan MH, Tambo A & Pace NP The role of TLR2, TLR4, and TLR9 in the pathogenesis of atherosclerosis. Int. J. Inflamm 2016, 1532832 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fredman G et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat. Commun 7, 12859 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dalli J & Serhan CN Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood 120, e60–e72 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chiang N, Dalli J, Colas RA & Serhan CN Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med 212, 1203–1217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dalli J et al. Resolvin D3 and aspirin-triggered resolvin D3 are potent immunoresolvents. Chem. Biol 20, 188–201 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Godson C et al. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol 164, 1663–1667 (2000). [DOI] [PubMed] [Google Scholar]; A seminal study showing that proresolving lipid mediators can stimulate efferocytosis.
- 94.Krishnamoorthy S et al. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl Acad. Sci. USA 107, 1660–1665 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mitchell S et al. Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J. Am. Soc. Nephrol 13, 2497–2507 (2002). [DOI] [PubMed] [Google Scholar]
- 96.Fredman G et al. Targeted nanoparticles containing the proresolving peptide Ac2–26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci. Transl Med 7, 275ra220 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Manega CM et al. 12(S)-Hydroxyeicosatetraenoic acid downregulates monocyte-derived macrophage efferocytosis: new insights in atherosclerosis. Pharmacol. Res 144, 336–342 (2019). [DOI] [PubMed] [Google Scholar]
- 98.DeBerge M et al. MerTK cleavage on resident cardiac macrophages compromises repair after myocardial ischemia reperfusion injury. Circ. Res 121, 930–940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wan E et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelialreproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ. Res 113, 1004–1012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.de Couto G et al. Mechanism of enhanced MerTK-dependent macrophage efferocytosis by extracellular vesicles. Arterioscler. Thromb. Vasc. Biol 39, 2082–2096 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang S et al. Acute CD47 blockade during ischemic myocardial reperfusion enhances phagocytosis-associated cardiac repair. JACC Basic Transl Sci. 2, 386–397 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nakaya M et al. Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J. Clin. Invest 127, 383–401 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Luo B, Wang Z, Zhang Z, Shen Z & Zhang Z The deficiency of macrophage erythropoietin signaling contributes to delayed acute inflammation resolution in diet-induced obese mice. Biochim. Biophys. Acta 1865, 339–349 (2019). [DOI] [PubMed] [Google Scholar]
- 104.Li S et al. Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and reversal by a fish oil diet. Circ. Res 105, 1072–1082 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Khanna S et al. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLOS ONE 5, e9539 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Suresh Babu S et al. MicroRNA-126 overexpression rescues diabetes-induced impairment in efferocytosis of apoptotic cardiomyocytes. Sci. Rep 6, 36207 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Luo B et al. Erythropoeitin signaling in macrophages promotes dying cell clearance and immune tolerance. Immunity 44, 287–302 (2016). [DOI] [PubMed] [Google Scholar]
- 108.Blander JM, Torchinsky MB & Campisi L Revisiting the old link between infection and autoimmune disease with commensals and T helper 17 cells. Immunol. Res 54, 50–68 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Martin CJ, Peters KN & Behar SM Macrophages clean up: efferocytosis and microbial control. Curr. Opin. Microbiol 17, 17–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Martin CJ et al. Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe 12, 289–300 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that efferocytosis and subsequent phagolysosomal degradation of M. tuberculosis-infected ACs can defend against M. tuberculosis infection.
- 111.Codo AC et al. Inhibition of inflammasome activation by a clinical strain of Klebsiella pneumoniae impairs efferocytosis and leads to bacterial dissemination. Cell Death Dis. 9, 1182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jorgensen I, Zhang Y, Krantz BA & Miao EA Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med 213, 2113–2128 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Moraco AH & Kornfeld H Cell death and autophagy in tuberculosis. Semin. Immunol 26, 497–511 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dallenga T et al. M. tuberculosis-induced necrosis of infected neutrophils promotes bacterial growth following phagocytosis by macrophages. Cell Host Microbe 22, 519–530 e513 (2017). [DOI] [PubMed] [Google Scholar]
- 115.Freire-de-Lima CG et al. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature 403, 199–203 (2000). [DOI] [PubMed] [Google Scholar]
- 116.Mercer J & Helenius A Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320, 531–535 (2008). [DOI] [PubMed] [Google Scholar]
- 117.Greenlee-Wacker MC et al. Phagocytosis of Staphylococcus aureus by human neutrophils prevents macrophage efferocytosis and induces programmed necrosis. J. Immunol 192, 4709–4717 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jondle CN, Gupta K, Mishra BB & Sharma J Klebsiella pneumoniae infection of murine neutrophils impairs their efferocytic clearance by modulating cell death machinery. PLOS Pathog. 14, e1007338 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Albert ML, Sauter B & Bhardwaj N Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 392, 86–89 (1998). [DOI] [PubMed] [Google Scholar]
- 120.Arrode G et al. Incoming human cytomegalovirus pp65 (UL83) contained in apoptotic infected fibroblasts is cross-presented to CD8+ T cells by dendritic cells. J. Virol 74, 10018–10024 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bosnjak L et al. Herpes simplex virus infection of human dendritic cells induces apoptosis and allows cross-presentation via uninfected dendritic cells. J. Immunol 174, 2220–2227 (2005). [DOI] [PubMed] [Google Scholar]
- 122.Larsson M et al. Activation of HIV-1 specific CD4 and CD8 T cells by human dendritic cells: roles for cross-presentation and non-infectious HIV-1 virus. AIDS 16, 1319–1329 (2002). [DOI] [PubMed] [Google Scholar]
- 123.Larsson M et al. Efficiency of cross presentation of vaccinia virus-derived antigens by human dendritic cells. Eur. J. Immunol 31, 3432–3442 (2001). [DOI] [PubMed] [Google Scholar]
- 124.Schaible UE et al. Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nat. Med 9, 1039–1046 (2003). [DOI] [PubMed] [Google Scholar]
- 125.Yrlid U & Wick MJ Salmonella-induced apoptosis of infected macrophages results in presentation of a bacteria-encoded antigen after uptake by bystander dendritic cells. J. Exp. Med 191, 613–624 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Blachere NE, Darnell RB & Albert ML Apoptotic cells deliver processed antigen to dendritic cells for cross-presentation. PLOS Biol. 3, e185 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Subramanian M et al. An AXL/LRP-1/RANBP9 complex mediates DC efferocytosis and antigen cross-presentation in vivo. J. Clin. Invest 124, 1296–1308 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reveals a protein complex through which DCs recognize and internalize ACs and presents molecular-genetic causation evidence in vivo that DC efferocytosis of virus-infected ACs triggers an antiviral cross-presentation response.
- 128.Tzelepis F et al. Annexin1 regulates DC efferocytosis and cross-presentation during Mycobacterium tuberculosis infection. J. Clin. Invest 125, 752–768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Torchinsky MB, Garaude J, Martin AP & Blander JM Innate immune recognition of infected apoptotic cells directs T(H)17 cell differentiation. Nature 458, 78–82 (2009). [DOI] [PubMed] [Google Scholar]; An important study showing that phagocytosis of bacteria-infected ACs by DCs leads to proinflammatory cytokine production that drives the generation of TH17 cells.
- 130.Penteado LA et al. Distinctive role of efferocytosis in dendritic cell maturation and migration in sterile or infectious conditions. Immunology 151, 304–313 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dejani NN et al. Intestinal host defense outcome is dictated by PGE2 production during efferocytosis of infected cells. Proc. Natl Acad. Sci. USA 115, E8469–E8478 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Salina AC, Souza TP, Serezani CH & Medeiros AI Efferocytosis-induced prostaglandin E2 production impairs alveolar macrophage effector functions during Streptococcus pneumoniae infection. Innate Immun. 23, 219–227 (2017). [DOI] [PubMed] [Google Scholar]
- 133.Loynes CA et al. PGE2 production at sites of tissue injury promotes an anti-inflammatory neutrophil phenotype and determines the outcome of inflammation resolution in vivo. Sci. Adv 4, eaar8320 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Campisi L et al. Apoptosis in response to microbial infection induces autoreactive TH17 cells. Nat. Immunol 17, 1084–1092 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rinne P et al. Palmitoylethanolamide promotes a proresolving macrophage phenotype and attenuates atherosclerotic plaque formation. Arterioscler. Thromb. Vasc. Biol 38, 2562–2575 (2018). [DOI] [PubMed] [Google Scholar]
- 136.Engelbertsen D et al. Increased lymphocyte activation and atherosclerosis in CD47-deficient mice. Sci. Rep 9, 10608 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Truman LA et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 112, 5026–5036 (2008). [DOI] [PubMed] [Google Scholar]; A key study showing that CX3CL1–CX3CR1 signalling allows macrophages to migrate towards ACs in tissues.
- 138.Gude DR et al. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 22, 2629–2638 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; An important study demonstrating that ACs upregulate sphingosine 1-phosphate as a ‘find-me’ signal to allow their detection by efferocytes.
- 139.Mueller RB, Sheriff A, Gaipl US, Wesselborg S & Lauber K Attraction of phagocytes by apoptotic cells is mediated by lysophosphatidylcholine. Autoimmunity 40, 342–344 (2007). [DOI] [PubMed] [Google Scholar]
- 140.Elliott MR et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; A key study identifying extracellular nucleotides released by ACs as a ‘find-me’ signal to promote efferocyte recruitment.
- 141.Fadok VA et al. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol 148, 2207–2216 (1992). [PubMed] [Google Scholar]
- 142.Fadok VA, de Cathelineau A, Daleke DL, Henson PM & Bratton DL Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J. Biol. Chem 276, 1071–1077 (2001). [DOI] [PubMed] [Google Scholar]; A landmark study showing that phosphatidylserine expression on ACs is required for their recognition and engulfment by phagocytes.
- 143.Park D et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450, 430–434 (2007). [DOI] [PubMed] [Google Scholar]
- 144.Park SY et al. Requirement of adaptor protein GULP during stabilin-2-mediated cell corpse engulfment. J. Biol. Chem 283, 10593–10600 (2008). [DOI] [PubMed] [Google Scholar]
- 145.Lee SJ, So IS, Park SY & Kim IS Thymosin beta4 is involved in stabilin-2-mediated apoptotic cell engulfment. FEBS Lett. 582, 2161–2166 (2008). [DOI] [PubMed] [Google Scholar]
- 146.Miki H, Suetsugu S & Takenawa T WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 17, 6932–6941 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Castellano F, Montcourrier P & Chavrier P Membrane recruitment of Rac1 triggers phagocytosis. J. Cell Sci 113, 2955–2961 (2000). [DOI] [PubMed] [Google Scholar]
- 148.Evans IR, Ghai PA, Urbancic V, Tan KL & Wood W SCAR/WAVE-mediated processing of engulfed apoptotic corpses is essential for effective macrophage migration in Drosophila. Cell Death Differ. 20, 709–720 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Martinez J et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol 17, 893–906 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 150.Fernandez-Boyanapalli RF et al. Impaired apoptotic cell clearance in CGD due to altered macrophage programming is reversed by phosphatidylserine-dependent production of IL-4. Blood 113, 2047–2055 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ichimura T et al. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J. Clin. Invest 118, 1657–1668 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yang L et al. KIM-1-mediated phagocytosis reduces acute injury to the kidney. J. Clin. Invest 125, 1620–1636 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Park SY et al. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 15, 192–201 (2008). [DOI] [PubMed] [Google Scholar]
- 154.Sen P et al. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-kappaB activation in dendritic cells. Blood 109, 653–660 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sharif MN et al. Twist mediates suppression of inflammation by type I IFNs and Axl. J. Exp. Med 203, 1891–1901 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB & Lemke G TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 131, 1124–1136 (2007). [DOI] [PubMed] [Google Scholar]
- 157.A-Gonzalez N & Castrillo A Liver X receptors as regulators of macrophage inflammatory and metabolic pathways. Biochim. Biophys. Acta 1812, 982–994 (2011). [DOI] [PubMed] [Google Scholar]
- 158.Hsu P et al. IL-10 potentiates differentiation of human induced regulatory T cells via STAT3 and Foxo1. J. Immunol 195, 3665–3674 (2015). [DOI] [PubMed] [Google Scholar]
- 159.Oh SA & Li MO TGF-beta: guardian of T cell function. J. Immunol 191, 3973–3979 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jennewein C et al. Sumoylation of peroxisome proliferator-activated receptor gamma by apoptotic cells prevents lipopolysaccharide-induced NCoR removal from kappaB binding sites mediating transrepression of proinflammatory cytokines. J. Immunol 181, 5646–5652 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ghisletti S et al. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol. Cell 25, 57–70 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]