

Abstract
Rationale: Cystic fibrosis (CF) is a life-shortening, multisystem hereditary disease caused by abnormal chloride transport. CF lung disease is driven by innate immune dysfunction and exaggerated inflammatory responses that contribute to tissue injury. To define the transcriptional profile of this airway immune dysfunction, we performed the first single-cell transcriptome characterization of CF sputum.
Objectives: To define the transcriptional profile of sputum cells and its implication in the pathogenesis of immune function and the development of CF lung disease.
Methods: We performed single-cell RNA sequencing of sputum cells from nine subjects with CF and five healthy control subjects. We applied novel computational approaches to define expression-based cell function and maturity profiles, herein called transcriptional archetypes.
Measurements and Main Results: The airway immune cell repertoire shifted from alveolar macrophages in healthy control subjects to a predominance of recruited monocytes and neutrophils in CF. Recruited lung mononuclear phagocytes were abundant in CF and were separated into the following three archetypes: activated monocytes, monocyte-derived macrophages, and heat shock–activated monocytes. Neutrophils were the most prevalent in CF, with a dominant immature proinflammatory archetype. Although CF monocytes exhibited proinflammatory features, both monocytes and neutrophils showed transcriptional evidence of abnormal phagocytic and cell-survival programs.
Conclusions: Our findings offer an opportunity to understand subject-specific immune dysfunction and its contribution to divergent clinical courses in CF. As we progress toward personalized applications of therapeutic and genomic developments, we hope this inflammation-profiling approach will enable further discoveries that change the natural history of CF lung disease.
Keywords: neutrophils, RNA-seq, macrophages, monocytes, cystic fibrosis
At a Glance Commentary
Scientific Knowledge on the Subject
Functionally different subsets of neutrophils and mononuclear phagocytes with defective bacterial killing, impaired phagocytic function, and enhanced cytokine production have been described in cystic fibrosis (CF). However, the broad spectrum of transcriptional alterations underlying immune dysfunction in individual CF airway cells has not been characterized.
What This Study Adds to the Field
This is the first single-cell RNA sequencing characterization of airway immune cells from subjects with CF and healthy control subjects. We observed a shift in the airway immune cell repertoire of subjects with CF from alveolar macrophages to a predominance of recruited monocytes and neutrophils. We identified a novel population of recruited lung mononuclear phagocytes in CF, with three distinct transcriptional archetypes (activated monocytes, monocyte-derived macrophages, and heat shock–activated monocytes) and characterized neutrophil subpopulations, highlighting a dominant immature proinflammatory archetype. Our findings offer an opportunity to understand subject-specific immune dysfunction and its potential contribution to CF pathogenesis.
Cystic fibrosis (CF) is a life-shortening, multiorgan hereditary disease affecting over 33,000 individuals in the United States (1, 2). Clinical manifestations of CF are caused by mutations in the CFTR (CF transmembrane conductance regulator) gene that cause abnormal chloride and bicarbonate transport on epithelial surfaces (3, 4). The disruption of epithelial and innate immune functions is a key contributor to CF lung disease, the primary cause of morbidity and mortality in CF (5, 6). Non-CFTR disease-modifying genes also contribute to immune dysfunction, clinical phenotype, and disease progression in CF (7, 8).
Airway inflammation is crucial in the development of CF lung disease, in which recruited cells cause tissue damage (9–11). Inflammatory cell populations are heterogeneous, with increasingly recognized CF-specific polymorphonuclear neutrophil (PMN) and macrophage (MΦ) subclasses (10). CF immune cells from blood and lung biopsies have been profiled using bulk RNA sequencing to characterize the transcriptional profiles associated with disease progression and clinical outcomes (12–15). Flow-cytometry studies, including our group’s mass cytometry characterization of CF immune cells, also shed light on the functional defects of CF immune cell subsets and distinct patterns of immune activation across subpopulations (10, 16–18). These studies have been constrained by the limited number of protein or genetic markers available per assay to define population clusters and assess immune responses. A study providing individualized cellular data on sputum cell types with the granularity afforded by single-cell RNA sequencing (scRNAseq) has not been reported in CF or any other lung disease.
Airway PMNs in CF have been characterized in the past (19–24). However, progress in high-throughput single-cell immune profiling has been slow relative to that in other immune cells such as peripheral blood mononuclear cells. This may be in part due to the overall limited viability and increased fragility of airway PMNs ex vivo. CF PMNs generally have a proinflammatory profile, yet some studies reveal functionally different subsets, including populations with abnormal immune function and defective bacterial killing (10). Airway MΦs and other mononuclear phagocytes are also present in CF airway secretions (25–27). Specifically, CF airway monocytes have impaired phagocytic function and enhanced cytokine production (28, 29), playing an important role in driving exaggerated airway inflammation in CF (9, 25).
Single-cell transcriptome profiling is a powerful tool for studying innate immune defects and defining cell subpopulations that contribute to pathogenesis (30). The use of immune cells from sputum instead of circulating cells or cells differentiated in vitro allows us to investigate gene expression profiles that reflect airway transmigration, response to the airway microenvironment, and cell–cell and cell–pathogen interactions that are key to CF pathogenesis.
Previously identified CF inflammatory cell subpopulations from other studies suggested to us that these cells exist as a continuum of immune maturation and function rather than as isolated, clearly defined subpopulations. To define this spectrum, we applied scRNAseq followed by pseudotime analysis and novel approaches to visualize high-dimensional data. In the continuum of sputum inflammatory cells, those with most extreme gene expression features defined functional and maturity trajectories, herein called transcriptional archetypes (31). These archetypes constitute a dynamic, more inclusive way of understanding transcriptional differences within immune cells. Our approach also allowed us to investigate the relationship between transcription factors and genes involved in immune activation and cell maturation, which was not previously possible because of an inability to sequence the full cellular transcriptome.
This work is the first to characterize the spectrum of maturation and immune activation states of inflammatory cell populations in CF airways at an unprecedented resolution, enabled by scRNAseq. Transcriptional profiling of inflammatory cell archetypes could open the door for highly targeted therapeutic interventions in subjects with similar CF-causing mutations who experience divergent clinical courses.
Some of the results of these studies have been previously reported in the form of a preprint (https://doi.org/10.1101/2020.03.06.20032292).
Methods
Detailed methods are provided in the online supplement.
Results
Disease-Specific Cell Distributions of CF Airway Inflammatory and Epithelial Cells
The primary objective of this study was to characterize sputum cell subpopulations in CF using unbiased transcriptome analysis of single cells obtained from subjects with CF and healthy control (HC) subjects. Our recruitment period extended from December 2018 to December 2019. Nine subjects with a confirmed CF diagnosis from the Yale Adult CF Program provided sputum samples. We also recruited five HC subjects to undergo sputum induction according to previous protocols (16).
Study subjects were closely age matched, with a higher inclusion of female subjects in the CF group (67% CF, n = 6; 40% HC, n = 2). The CF cohort was comprised primarily of F508del homozygous subjects (78%, n = 7), with only two F508del heterozygotes harboring either one deletion or one frameshift mutation in one CFTR allele and an F508del in the other. The CF cohort’s degree of lung function impairment, as determined by FEV1, ranged from mild to severe (FEV1 19–84% of predicted), with a mean FEV1 of 57%. All subjects with CF had pancreatic exocrine insufficiency, and 44% (n = 4) carried a diagnosis of CF-related diabetes. Pseudomonas aeruginosa was isolated in the sputum of 56% of subjects with CF (n = 5). The majority of subjects with CF were receiving CFTR-modulator therapy (89%, n = 8) with a combination of either ivacaftor/tezacaftor (67%, n = 6) or ivacaftor/lumacaftor (22%, n = 2). For further demographic and clinical details, see Table 1.
Table 1.
Demographic Characteristics of Study Subjects from the Yale Adult Cystic Fibrosis Program and Healthy Control Subjects
| Characteristics | HC Subjects (n = 5) | Subjects with CF (n = 9) |
|---|---|---|
| Age, yr | ||
| Mean ± SD | 35.4 ± 5.9 | 30.6 ± 6.5 |
| Range | 26–42 | 24–43 |
| Sex, n (%) | ||
| F | 2 (40) | 6 (67) |
| M | 3 (60) | 3 (33) |
| Mutation background, n (%) | ||
| F508del/F508del | NA | 7 (77.8) |
| F508del/other | NA | 2 (22.2) |
| No F508del mutations | NA | 0 (0) |
| FEV1, L | ||
| Mean ± SD | NA | 1.9 ± 0.7 |
| Range | NA | 0.68–2.85 |
| FEV1, % | ||
| Mean ± SD | NA | 57 ± 21.5 |
| Range | NA | 19–84 |
| BMI, kg/m2 | ||
| Mean ± SD | NA | 22.2 ± 2.1 |
| Range | NA | 19.11–25.73 |
| CF comorbidities, n (%) | ||
| Pancreatic exocrine insufficiency | NA | 9 (100) |
| CF-related diabetes | NA | 4 (44.4) |
| Liver disease | NA | 1 (11.1) |
| Microbiology, n (%) | ||
| P. aeruginosa colonization | NA | 5 (55.6) |
| CFTR modulators, n (%) | ||
| Ivacaftor/tezacaftor | NA | 6 (66.7) |
| Ivacaftor/lumacaftor | NA | 2 (22.2) |
| No modulator | NA | 1 (11.1) |
Definition of abbreviations: BMI = body mass index; CF = cystic fibrosis; CFTR= cystic fibrosis transmembrane conductance regulator; HC = healthy control; NA = not applicable; P. aeruginosa = Pseudomonas aeruginosa.
We developed a standardized scRNAseq workflow for sputum sample analysis (Figure 1A) and profiled a total of 20,095 sputum cells (12,494 CF and 7,601 HC). We identified the following nine distinct sputum cell populations based on known transcriptomic markers (Figure 1C and Data file E1 in the online supplement): mononuclear phagocytes (recruited lung monocytes, monocyte-derived MΦs [MoMΦs], and alveolar MΦs [alvMΦs]); classical and plasmacytoid dendritic cells; PMNs; lymphocytes (B, T, and NK cells); and airway epithelial cells from buccal and tracheobronchial mucosa (Figures 1B–1D). The expression of CFTR in sputum cells was overall very low, and CFTR was detected in most cell types in frequencies ranging from 0 to 6.84% (Figure E1).
Figure 1.
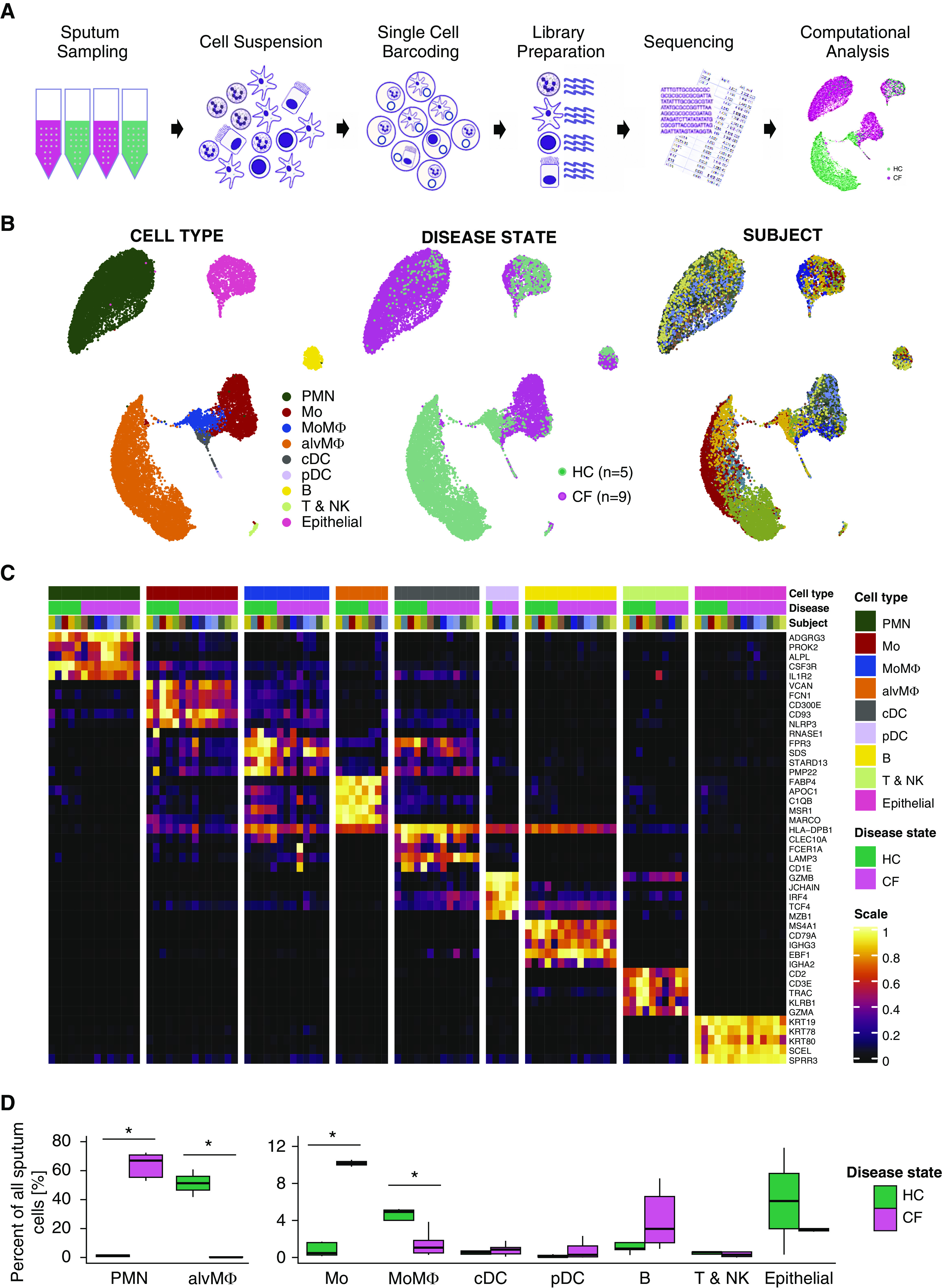
Single-cell RNA sequencing reveals an immune cell repertoire shift from alveolar macrophages to recruited monocytes and polymorphonuclear neutrophils in cystic fibrosis (CF). (A) Schematic of the experimental design. Spontaneously expectorated sputum from patients with CF and induced sputum from healthy control subjects was collected. Sputum was processed into a single-cell suspension. Droplet-based single-cell RNA sequencing barcoding, library preparation, sequencing, and computational analysis were performed. (B) Uniform manifold approximation and projection visualization of 20,095 sputum cells from nine patients with CF and five control subjects. Each dot represents a single cell, and cells are labeled by 1) cell type, 2) disease state, and 3) subject. (C) Heatmap of marker genes for all cell types identified. Each column represents the average expression value of one subject grouped by disease status and cell type. Gene expression values are unity-normalized from 0 to 1. (D) Boxplots showing percentages of all identified cell types to all cells profiled per subject, separated by disease state. Whiskers represent 1.5× interquartile range. *P < 0.05 determined by a Wilcoxon rank sum test comparing cell percentages of patients with CF and control subjects. alvMΦ = alveolar macrophage; B = B-lymphocyte; cDC = classical dendritic cell; HC = healthy control; Mo = monocyte; MoMΦ = monocyte-derived macrophage; pDC = plasmacytoid dendritic cell; PMN = polymorphonuclear neutrophil; T and NK = T-lymphocytes and NK cells.
The Inflammatory Cell Repertoire of CF Sputum Displays a Shift from alvMΦs to Airway Monocytes and PMNs
The dominant cell populations in the CF and HC samples were strikingly different. PMNs contributed 64% of all CF cells, with minimal numbers of alvMΦs (0.4%). In contrast, the HC samples were composed of 80.2% alvMΦs with almost no detectable PMNs (<2%; P < 0.002 for both). Furthermore, subjects with CF also exhibited increased numbers of airway monocytes (19% CF and 1% HC; P = 0.001) and B cells (4% CF and 1% HC; not significant) and lower numbers of MoMΦs (1% CF and 6% HC; P = 0.007) (Figures 1B–1D). Disease-associated PMNs, MΦs, and monocyte cellular distributions were confirmed on mass cytometry data from a previously published study by our group that compared surface markers of inflammatory sputum cells in CF and HC cells (Figure E2) (16). Furthermore, correlation of cell-type gene classifiers in this study and analogous cell types in the largest scRNAseq dataset of the distal lung available to date (n = 28) revealed a greater correlation between HC cell types from each dataset than that within other cell types from the same dataset, confirming our cell annotations (Figure E3) (31). Our findings indicate that immune cell populations in CF sputum are distinguishable from HC sputum through scRNAseq and that our cell annotations and shifts in major cell distributions in CF are consistent with other mass cytometry and scRNAseq studies.
Recruited CF Lung Mononuclear Phagocytes Display Distinct Maturation and Immune Activation Archetypes
AlvMΦs were rare in CF sputum; however, we identified a distinct subpopulation of recruited lung mononuclear phagocytes (RLPs) (Figure 1B) that included recruited lung monocytes and MoMΦs. These RLPs were defined by a high expression of mononuclear phagocyte–associated genes (LYZ, CTSB, CTSH, CTSL, CTSS, CTSZ, HLA-DRA, HLA-DRB1, LGALS1, FTL, and CD74). RLPs were relatively abundant in CF (20% of CF cells) and were rarely identified in HC sputum (7% of HC cells; P = 0.06). RLPs were a heterogeneous group, with pronounced and notably different plasticity in CF. This suggested that RLPs would differ not only in abundance but also in transcriptional profiles between HC and CF cells.
To characterize the spectrum of immune activation and maturation of monocytes and MoMΦs contained within CF and HC RLPs, we performed a pseudotime analysis using potential of heat diffusion for affinity-based transition embedding. Pseudotime analysis is a computational technique that allows the distribution of single-cell expression profiles along the continuum of a biologic process marked by gene expression changes (in this case, cell maturation, immune activation, and heat-shock response gene expression). The pseudotime analysis demonstrated three distinct gene expression trajectories, and in turn, the most extreme phenotypes of these trajectories defined three RLP transcriptional archetypes in the sputum (Figure 2A) (31, 32). Two of these archetypes were CF-predominant archetypes (activated proinflammatory monocytes and heat shock–activated monocytes). The third RLP archetype, mature resting MoMΦs, was more prevalent in HC cells.
Figure 2.
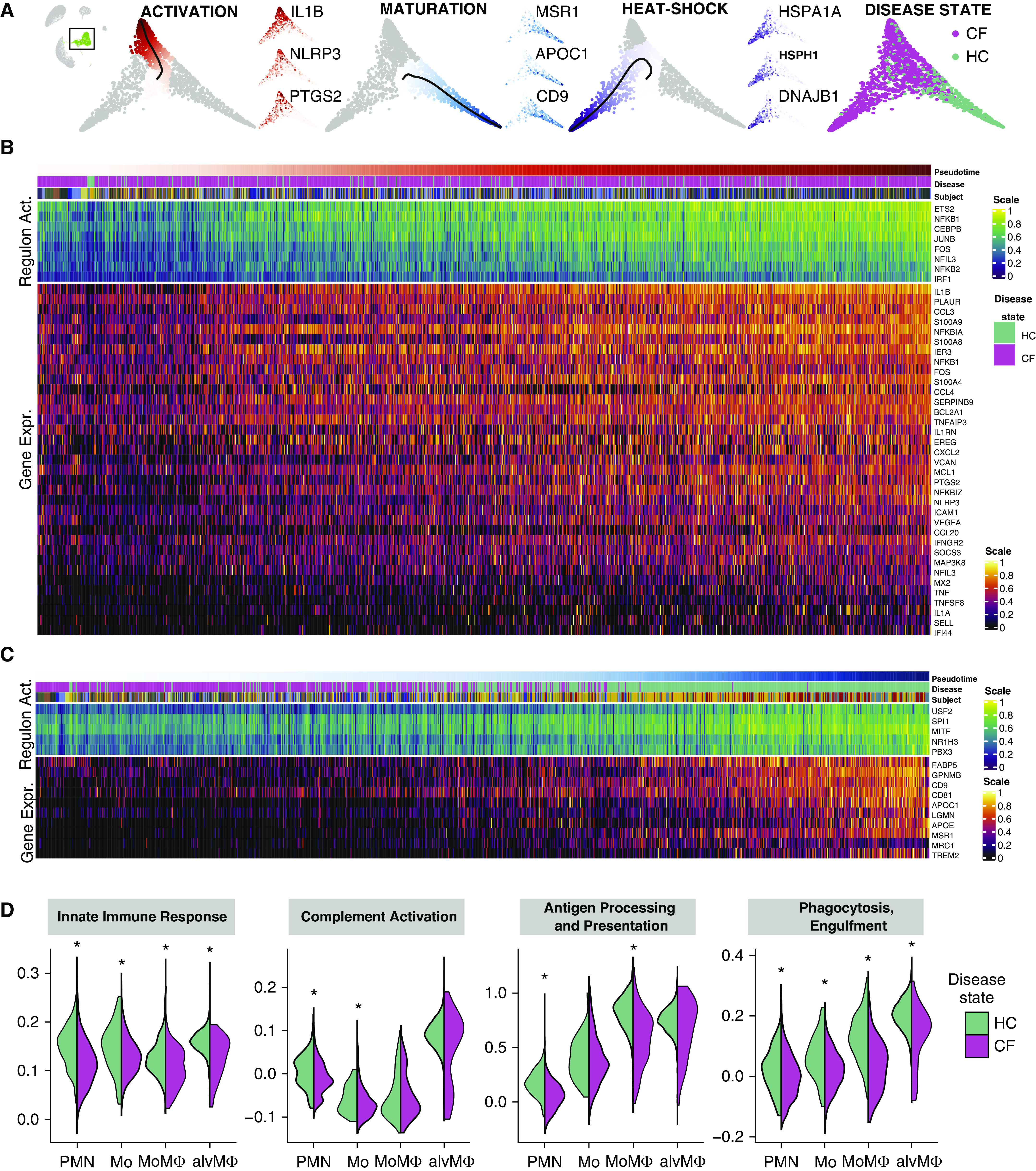
Recruited lung mononuclear phagocytes are a distinct cell population with a broad spectrum of maturity and immune activation in cystic fibrosis airways. (A) Potential of heat diffusion for affinity-based transition embedding (PHATE) of monocytes and monocyte-derived macrophages colored by pseudotime, all starting from quiescent monocytes toward 1) activated monocytes, 2) mature monocyte-derived macrophages, 3) monocytes expressing a heat-shock response, and 4) monocytes and monocyte-derived macrophages, colored by disease state. All three archetypes are accompanied by three PHATE plots colored by the gene expression of typical genes, ramping up along a specific pseudotime. For the corresponding uniform manifold approximation and projection embedding colored by the gene expressions of the same genes, see Figure E4. For the corresponding PHATE colored by cell type and subjects, see Figure E5. (B) Heatmap of gene expression and regulon activity in monocytes undergoing activation (ordered by pseudotime distances along PHATE manifolds that transition from quiescent monocytes toward an activated monocyte archetype). (C) Heatmap of gene expression and regulon activity in monocytes undergoing maturation (ordered by pseudotime distances along PHATE manifolds that transition from quiescent monocytes toward a control-enriched mature monocyte-derived macrophage archetype). In both heatmaps, annotation bars represent the pseudotime distance, disease status, and subject for each cell; expression values are centered and scaled. (D) Violin plots of pathway activity scores grouped by cell type and separated by disease state. Depicted pathway scores from left to right are as follows: GO:0045087 innate immune response, GO:0006958 complement activation and classical pathway, GO:0019882 antigen processing and presentation, and GO:0006911 phagocytosis and engulfment. *False discovery rate–adjusted P values < 0.05 calculated using the Wilcoxon signed-rank test. alvMΦ = alveolar macrophage; CF = cystic fibrosis; Gene Expr. = gene expression; HC = healthy control; Mo = monocyte; MoMΦ = monocyte-derived macrophage; PMN = polymorphonuclear neutrophil; Regulon Act. = regulon activity.
Next, we examined the sequence of gene expression changes leading to the mature resting MoMΦ and activated proinflammatory monocyte archetypes, correlating gene expression changes with pseudotime distance values. The trajectory toward activated proinflammatory monocytes was characterized by a gradual and steady increase of proinflammatory chemokine and cytokine gene expression. This trajectory was characterized by increasing expression of IL1B, CXCL2, CCL3, CCL4, CCL20, VEGFA, EREG, calprotectin (S100A8 and S100A9) (33), the antiapoptotic proteins MCL1 and BCL2L1, the inflammasome subunit NLRP3 (34), inducible cyclooxygenase 2 (PTGS2), and the transcription factor NFKB1 (Figures 2B and E4 and E5 and Data file E2). In the activated monocyte archetype, imputed regulating factors of common activator/repressor genes (i.e., regulons) suggested increased activity of NFKB1 and the proinflammatory transcription factors NFKB2, ETS, and IRF1. Proinflammatory cytokines TNF and IL1A were expressed only toward the extreme end of the trajectory in the most activated monocytes. In contrast to those found in CF RLPs, we did not observe similar immune activation archetypes in MoMΦs or in alvMΦs from HC RLPs. Remarkably, although proinflammatory CF monocytes exhibited increased overall cytokine expression, they also showed impaired expression of key phagocytic and cytolytic components of the immune response (complement C1Q), markers of maturation toward a MΦ phenotype (APOC1 and APOE), and phagocytic function (MARCO) compared with other RLP archetypes (Figures 2B and 2D).
The mature resting MoMΦ archetype was enriched in HC cells, and none of the CF MΦs reached the distal end of this archetype (Figure 2C). Key regulons involved in monocyte to MΦ maturation were active and increasingly expressed toward the distal end of the archetype trajectory, including canonical SPI1 (PU.1) as well as MITF and USF2. The maturation of MoMΦs was accompanied by a gradual transcriptional increase of scavenger and pattern-recognition receptors MSR1 and MRC1, surface markers CD9 and CD81, apolipoproteins APOC1 and APOE, and FABP5.
MoMΦs were rare in sputum overall but were more evenly distributed between subjects with CF and HC subjects; these were distinguished by expression of PLA2G7, an enzyme that inactivates platelet-activating factor, monocyte chemokine CCL2, LGMN (a cysteine protease involved in MHC-II presentation and differentiation toward dendritic cells), and activated-leukocyte cell adhesion molecule ALCAM. The majority of sputum cells in HC were alvMΦs. These highly abundant HC alvMΦs expressed the expected levels of phagocytosis-associated genes, underscoring the transcriptional readiness of healthy immune cells to participate in phagocytic functions and coordinate inflammatory cell recruitment, without the basal proinflammatory activity noted in the CF-predominant monocytes. Taken together, these findings show that CF RLPs have high proinflammatory gene expression but limited phagocytosis-associated transcriptional responses, which is consistent with excessive inflammation and impaired host defense responses known to occur on CF airways.
An Immature Proinflammatory Archetype Prevails among CF Airway PMNs
CF sputum contained 64% PMN in contrast with HC sputum, in which PMNs constituted 2% of sputum cells (Figure 1D). The potential of heat diffusion for affinity-based transition embedding of the PMN spectrum of gene expression (PMN manifold) enabled us to identify three PMN archetypes based on canonical markers of PMN immaturity (CXCR4 and IGF2R) and maturity (FCGR3B, ALPL, and CXCR2) as well as a heat-shock response archetype (Figures 3A and 3B and E6). To analyze gradual changes within the PMN manifold, we applied trajectory inference and correlated the resulting pseudotime distances with gene expression and regulon activity. When tracing PMN maturation, we observed that expression of calprotectin (S100A8 and S100A9), S100A11, CSF3R, and antiapoptotic factor BL2A1 are gained relatively early, in contrast to the classical maturation markers FCGR3B, ALPL, CXCR2, and CD14, which ramp up in expression relatively late (Figure 3B and Data file E2) (35, 36). In immature PMNs, we observed a gradual increase of the transcription factors TFEC, MITF, and STAT3 and the maturation-associated transcription factors CEBPB and NFIL3. The CF-predominant immature PMN archetype was further defined by increased expression of the PMN-activating chemokine MIP (CCL3 and CCL4) and the downstream transcription factor and adapter molecules IRAK3 and TRAF3. These findings suggest that CF airway PMNs have an overall proinflammatory phenotype, with a large subpopulation of PMNs exhibiting a functional and maturational transcriptional shift, which is consistent with an immature PMN gene expression profile.
Figure 3.
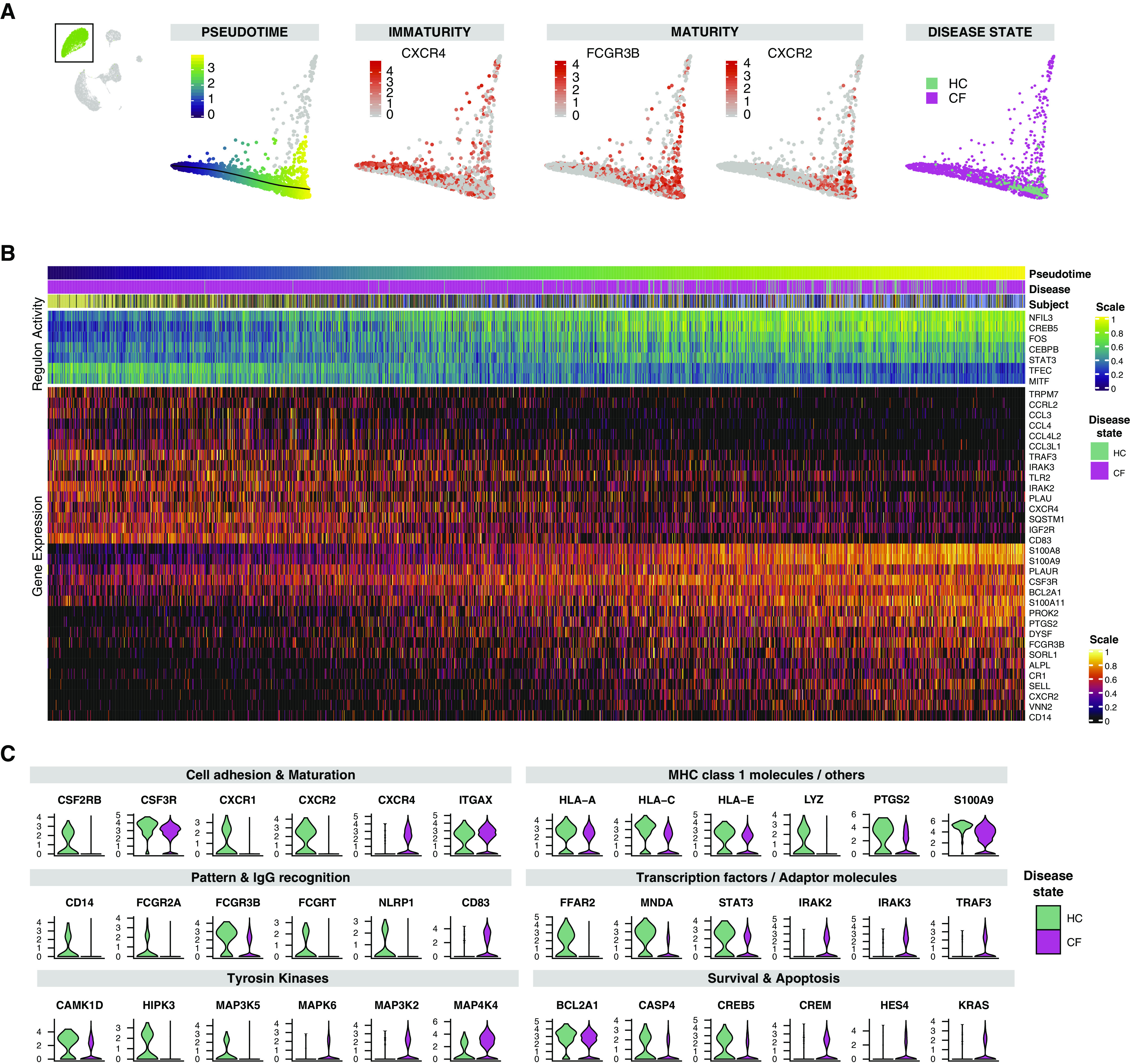
An immature proinflammatory archetype prevails among cystic fibrosis (CF) airway polymorphonuclear neutrophils (PMNs). (A) Potential of heat diffusion for affinity-based transition embedding (PHATE) of PMNs colored by 1) pseudotime from immature to mature PMNs, 2) examples of canonical marker features of immaturity (CXCR4) and maturity (FCGR3B and CXCR2) in peripheral PMNs, and 3) disease state. The cells deviating upward are PMNs expressing heat-shock response genes. For PHATE colored by gene expression of HSPA1A, HSPH1, and DNAJB1, see Figure E6A. For corresponding PHATE colored by disease state and subjects, see Figure E6B. (B) Heatmap of gene expression and regulon activity in PMNs ordered by pseudotime distances along PHATE manifolds that transition from CF-enriched regions of the immature and activated PMN archetype toward control-enriched mature PMN archetype. Annotation bars represent the pseudotime distance, disease status, and subject for each cell; expression values are centered and scaled. (C) Violin plots of differentially expressed genes comparing CF and control PMN populations (for P values, see Data file E3) grouped by disease state and sorted thematically. HC = healthy control; MHC = major histocompatibility complex.
CF PMN Archetypes Have Decreased Phagocytic Marker and Tyrosine Kinase Expression
We compared the gene expression profiles of CF and HC PMNs to understand transcriptomic differences associated with their immune function (Data file E3). We categorized the top gene expression differences between CF and HC PMNs accordingly into the following: 1) cell adhesion and maturation markers, 2) MHC class I molecules, 3) pattern and IgG recognition, 4) transcription factors and adaptor molecules, 5) tyrosine kinase expression, and 6) survival and apoptosis genes (Figure 3C). In CF PMNs, cell adhesion and maturation markers were lower overall than in HC PMNs (CSF2RB, CSF3R, CXCR2, ICAM3, and PECAM1), except for ITGAX. The decreased expression of these markers in CF reflects a higher prevalence of the immature PMN archetype described above. In addition to decreased CXCR- and CSF-receptor expression, CF PMNs also expressed lower CXCR1, IL1RN, and IL1B that could condition further defects in phagocytosis and inflammatory cell recruitment. We identified striking differences in antigen presentation, pathogen recognition, and phagocytosis-associated genes between CF and HC PMNs. CF PMNs showed decreased expression of numerous members of the MHC-I molecules (HLA-A/B/C/E); immunoglobin receptors (FCGR3B, FCGR2A, and FCGRT); pathogen recognition receptors CD14, TLR2, and NLRP1; and lysozyme (LYZ). Interestingly, two genes involved in the assembly of lipid rafts and primary neutrophil granule release were increased (SYK and CD63), suggesting that although PMNs may suffer from defective phagocytic activity, the transcriptional infrastructure needed to express tissue proteases and inflammatory mediators in the airways is preserved. CF PMNs demonstrated increased transcriptomic activation characterized by the expression of transcription factors and proinflammatory adapter molecules (increased PI3, IRAK2/3, TRAF3, and TANK), yet this activation did not translate into increased expression of inflammatory cytokines. Interestingly, the downstream response to cytokine activation appeared to be blunted, as shown by decreased overall tyrosine kinase gene expression (ITPK1, MAP3K5, MAP2K4, CAMK1D, PIK3CD, and HIPK3). Finally, we observed the induction of genes involved in the hypoxic response (HIF1A, VEGFA, FGF13, and PTGS2) and diverging proapoptotic signals with lower expression of CASP4, RPS6KA5, CREB5, and BCL2A and increased expression of HES4, KRAS, and CREM in CF. These observations underscore the presence of a hypoxic airway environment in CF and a dysfunctional cell-death program that enhances the survival of functionally ineffective PMNs. Taken together, these findings indicate that CF PMNs do not carry out an effective transcriptional response to inflammatory stimuli and lack essential components for pathogen recognition and removal.
Discussion
This is the first single-cell transcriptome characterization of immune cells in CF sputum. We identified CF-specific differences in cell subpopulations, including alvMΦs, RLPs, and PMNs. Furthermore, these cells had markedly different transcriptional profiles when compared with their HC counterparts. Previous CF studies have used transcriptomic analysis to determine the likelihood of adverse outcomes in CF lung disease; however, they have not focused on establishing differences between healthy and CF airway inflammatory cells or characterizing their immune activation profiles (12–15). The most remarkable finding from this study is the discovery of novel archetypes of RLPs, enabled by an unprecedented depth of gene expression profiling. These inflammatory cell subpopulations exhibit a wide spectrum of maturity and immune activation in CF. Airway MΦs and other mononuclear cells have been described in human CF airway secretions (25, 26), and their role in driving exaggerated airway inflammation in CF has been well characterized in animal models (9, 25). However, a broader genomics approach to define sputum RLPs, their potential functional impairments, and pathogenic role has not been reported.
We identified three novel archetypes of CF RLPs, including activated monocytes, mature MoMΦs, and heat shock–activated monocytes. Airway monocytes in CF have impaired ion transport and phagocytic function; however, their role in CF lung disease remains undefined (28, 37). Others have described dramatic changes in monocyte cell adhesion and chemotaxis that perpetuate inflammation in CF lungs together with enhanced chemokine production that sustains PMN recruitment and injury (38). In agreement with these studies, we observe that monocytes are rather abundant in CF sputum but are deficient in monocyte maturation gene expression markers (MITF and SPI1). Furthermore, CF monocytes were not only abundant but also highly active from the immune perspective, expressing high levels of inflammation-related genes (CXCL8, IL1B, CCL3, and calprotectin). These observations underscore a defect in CF monocyte maturation that preserves a highly proinflammatory phenotype and contributes to airway damage and aberrant inflammatory cell recruitment (39).
MΦs recovered from CF lungs are relatively small in size and express minimal levels of the mannose receptor MRC1 or MARCO typically detected on alvMΦs (Figure E7) (26, 27). This has been interpreted as an indication that CF airway MΦs are recruited from the circulation, as opposed to tissue-resident alvMΦs, which are of embryonic origin. Here, we show that most CF airway MΦs originate from recruited monocytes, whereas the majority of HC airway cells were bona fide tissue-resident alvMΦs.
In contrast to CF airway monocytes, more mature CF phagocytes (MoMΦs and alvMΦs) showed low levels of immune activation markers observed in CF monocytes and of key phagocytic and cytolytic components of the immune response (complement C1Qs and MARCO). This underscores that in CF, RLPs that reach maturity exhibit transcriptomic evidence of impaired or limited phagocytic function, accounting for the known impaired phagocytic abilities of these cells in CF.
We did not detect a distinct acute exacerbation signature in CF samples. This may reflect our stringent gene expression analysis strategy, a lack of paired sputum samples, and sample size limitations to perform this subgroup analysis. This is an important question to pursue in the future because paired samples in stable and exacerbation states from the same individual may reveal critical genetic modifiers of a patient's clinical course.
PMNs were the most abundant immune cells in the sputum of patients with CF, which is consistent with reports in the CF literature and similar to the predominance of alvMΦs in HC sputum (19–24). Here, we report the discovery of new archetypes of CF PMNs based on inflammatory and maturity gene expression markers; one is characterized by high maturity and a limited proinflammatory transcriptional state, and another is characterized with higher proinflammatory activity and delayed expression of maturity markers. Overall, the increased expression of proinflammatory genes in immature PMNs highlights a highly activated and proinflammatory state, which is clearly distinguishable from the transcriptional profile of HC PMNs. The immature airway PMN archetype shares features of a previously described subpopulation of transmigrated PMNs with increased granule release, immunoregulatory and metabolic activity, and defective bacterial killing in in vitro studies, referred to as “granule-releasing, immunomodulatory, and metabolically active (GRIM)” neutrophils (10, 40). We identified cells with similar characteristics but as part of a spectrum of granulocyte maturation that encompasses vigorously activated PMNs on one extreme and PMNs with decreased expression of maturity markers and evidence of recent airway migration on the other extreme. Adding to the complexity of these PMN subpopulations, counterproductive proapoptotic and antiapoptotic signals were present across the CF PMNs when compared with the HC PMNs (increased UVRAG, PLPP3, and ATG7; decreased CASP4, RPS6KA5, CREB5, and BCL2A). Taken together, these findings underscore an aberrant proinflammatory state in CF PMNs, which is exacerbated by the disruption of immunomodulatory and antiinflammatory mechanisms such as apoptosis and transcription factor suppression.
The presence of B cells in the CF sputum was an intriguing finding. SNPs in the class II major histocompatibility complex (MHCII) of the F508del population are associated with delayed P. aeruginosa colonization and slower lung function decline (41–44). Although we observed no differences in MHCII gene expression in the B cells of subjects with CF (Figure E8), a focused study on MHCII SNPs could identify B-cell subpopulations with a protective role against P. aeruginosa and its associated impact on pulmonary health.
This work includes two technical advances. First, this is the only reported scRNAseq study of CF sputum, a notoriously complex biological sample with high variability in cell viability and cellularity between subjects. Second, our sputum-processing protocol avoids the use of reducing agents to solubilize sputum and instead minimizes immune cell activation and injury by using mechanical disruption and filtering. Importantly, ours is the first report of a sputum cryopreservation protocol allowing the retrieval of live cells for scRNAseq analysis and avoiding sputum-solubilizing agents typically used in sputum sample processing (online supplement, Figure E9) (45–49). The ability to use cryopreserved cells overcomes a major limitation of previous single-cell studies that required fresh samples (13, 16); this is particularly important for the recovery of PMNs, which known for their short lifespan ex vivo and their susceptibility to immune activation. Our study has several limitations, including the following: 1) Large differences in predominant cell types between subjects with CF and HC subjects make it difficult to generalize gene expression changes between disease and control groups. Although we present these comparisons, our focus is on understanding CF-specific cell distributions and their spectrum of maturity and activation markers. 2) Because HC subjects express minimal sputum if any at all, we used a standardized approach for sputum induction in these subjects, whereas CF cells were obtained from spontaneously expectorated sputum. Because single-cell suspensions are standardized for number of cells before any analysis, these sampling differences likely have a minor impact on our observations. 3) There was an uneven sex distribution across the study groups. This may be of particular importance in CF because female sex in CF is associated with disparities in life expectancy, frequency of exacerbations, and early acquisition of respiratory pathogens (50). However, of the differentially expressed genes between subjects with CF and control subjects, we did not observe divergent differential gene expression changes in women or men. Finally, 4) our study has a small sample size; however, we sought to match subjects according to age and sex, and HC subjects were compared with a relatively homogeneous CF cohort in terms of CFTR mutation background, CF comorbidities, and ongoing therapy. Although a small number of patients were recruited for this study, we believe they are representative of patients with CF based on the F508del allele frequency in our cohort and the identification of nine distinct cell types representative of airway cells in CF. Despite these limitations, our findings are robust and representative of the CF airway compartment.
CF research is progressing rapidly toward clinical, molecular, and functional characterization based on individualized high-throughput diagnosis and functional profiling. Our application of scRNAseq enabled the discovery of transcriptional archetypes in CF-specific cell subpopulations that may underlie subject-specific differences in disease progression and response to therapy. As we advance toward early applications of therapeutic and genomic technologies, we hope this approach to individualized airway inflammation profiling will serve as a foundation for further discoveries that transform the natural history of CF lung disease.
Supplementary Material
Acknowledgments
Acknowledgment
The authors thank their patients, the medical staff at the Yale Adult Cystic Fibrosis Program, Dr. Farida Ahangari, and Dr. Jonathan Koff, the director of the Yale Adult Cystic Fibrosis Program, for their support and contributions to this project. Sequencing was conducted by Mei Zhong at the Yale Stem Cell Center Genomics Core facility, which was supported by the Connecticut Regenerative Medicine Research Fund and the Li Ka Shing Foundation.
Footnotes
Supported by the NIH and NHLBI through grants NIH T32-HL007778 and K01-HL125514-01 (C.J.B.); the Cystic Fibrosis Foundation through its Fifth Year Clinical Fellowship Award (C.J.B.); the American Thoracic Society Foundation's Unrestricted Research Award (C.J.B.); NIH U01 HL145567 and R01 HL127349 (N.K.); and DoD W81XWH-19-1-0131 (J.C.S.).
Author Contributions: N.K. and C.J.B. conceptualized, acquired funding, and supervised the study. S.K., M.E.E., and C.J.B. performed sample collection, phenotyping, and sputum processing. G.L.C. facilitated sputum collection infrastructure and processing protocols. J.C.S. and T.S.A. performed single-cell barcoding library construction. Data was processed, curated, and visualized by J.C.S. under the supervision of X.Y., N.K., and C.J.B., and was analyzed by J.C.S., M.S., C.S.D.C., E.M.B., and C.J.B. Mass cytometry data were reanalyzed by J.L.G., R.R.M., and E.M.B. J.C.S., T.S.A., S.P., I.O.R., and N.K. created and provided single-cell RNA sequencing data of control distal lungs. T.S.A. calculated the correlation matrix. Y.Z. and R.R.M. performed the sample processing comparison experiments. The manuscript was drafted by J.C.S. and C.J.B. and was reviewed and edited by all other authors.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202004-0991OC on June 30, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Cystic Fibrosis Foundation. Cystic Fibrosis Foundation patient registry 2017 annual data report. 2018 [2020 Jan 15] Available from: https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2017-Patient-Registry-Annual-Data-Report.pdf. [Google Scholar]
- 2.Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet. 2015;16:45–56. doi: 10.1038/nrg3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 4.Welsh MJ, Ramsey BW, Accurso F, Cutting GR. Cystic fibrosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Vogelstein B, editors. The metabolic and molecular basis of inherited disease. New York: McGraw-Hill; 2001. pp. 5121–5189. [Google Scholar]
- 5.McCague AF, Raraigh KS, Pellicore MJ, Davis-Marcisak EF, Evans TA, Han ST, et al. Correlating cystic fibrosis transmembrane conductance regulator function with clinical features to inform precision treatment of cystic fibrosis. Am J Respir Crit Care Med. 2019;199:1116–1126. doi: 10.1164/rccm.201901-0145OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med. 2015;372:351–362. doi: 10.1056/NEJMra1300109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corvol H, Blackman SM, Boëlle PY, Gallins PJ, Pace RG, Stonebraker JR, et al. Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat Commun. 2015;6:8382. doi: 10.1038/ncomms9382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Polineni D, Dang H, Gallins PJ, Jones LC, Pace RG, Stonebraker JR, et al. Airway mucosal host defense is key to genomic regulation of cystic fibrosis lung disease severity. Am J Respir Crit Care Med. 2018;197:79–93. doi: 10.1164/rccm.201701-0134OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruscia EM, Zhang PX, Ferreira E, Caputo C, Emerson JW, Tuck D, et al. Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator-/- mice. Am J Respir Cell Mol Biol. 2009;40:295–304. doi: 10.1165/rcmb.2008-0170OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forrest OA, Ingersoll SA, Preininger MK, Laval J, Limoli DH, Brown MR, et al. Frontline Science: pathological conditioning of human neutrophils recruited to the airway milieu in cystic fibrosis. J Leukoc Biol. 2018;104:665–675. doi: 10.1002/JLB.5HI1117-454RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Margaroli C, Garratt LW, Horati H, Dittrich AS, Rosenow T, Montgomery ST, et al. Elastase exocytosis by airway neutrophils is associated with early lung damage in children with cystic fibrosis. Am J Respir Crit Care Med. 2019;199:873–881. doi: 10.1164/rccm.201803-0442OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chesné J, Danger R, Botturi K, Reynaud-Gaubert M, Mussot S, Stern M, et al. COLT Consortium. Systematic analysis of blood cell transcriptome in end-stage chronic respiratory diseases. PLoS One. 2014;9:e109291. doi: 10.1371/journal.pone.0109291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang K, Poppenberg KE, Wong L, Chen Y, Borowitz D, Goetz D, et al. RNA sequencing data from neutrophils of patients with cystic fibrosis reveals potential for developing biomarkers for pulmonary exacerbations. J Cystic Fibros. 2019;18:194–202. doi: 10.1016/j.jcf.2018.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kormann MSD, Dewerth A, Eichner F, Baskaran P, Hector A, Regamey N, et al. Transcriptomic profile of cystic fibrosis patients identifies type I interferon response and ribosomal stalk proteins as potential modifiers of disease severity. PLoS One. 2017;12:e0183526. doi: 10.1371/journal.pone.0183526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levy H, Jia S, Pan A, Zhang X, Kaldunski M, Nugent ML, et al. Identification of molecular signatures of cystic fibrosis disease status with plasma-based functional genomics. Physiol Genomics. 2019;51:27–41. doi: 10.1152/physiolgenomics.00109.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yao Y, Welp T, Liu Q, Niu N, Wang X, Britto CJ, et al. Multiparameter single cell profiling of airway inflammatory cells. Cytometry B Clin Cytom. 2017;92:12–20. doi: 10.1002/cyto.b.21491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang S, Shrestha CL, Kopp BT. Cystic fibrosis transmembrane conductance regulator (CFTR) modulators have differential effects on cystic fibrosis macrophage function. Sci Rep. 2018;8:17066. doi: 10.1038/s41598-018-35151-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sorio C, Montresor A, Bolomini-Vittori M, Caldrer S, Rossi B, Dusi S, et al. Mutations of cystic fibrosis transmembrane conductance regulator gene cause a monocyte-selective adhesion deficiency. Am J Respir Crit Care Med. 2016;193:1123–1133. doi: 10.1164/rccm.201510-1922OC. [DOI] [PubMed] [Google Scholar]
- 19.Ramsey BW, Downey GP, Goss CH. Update in cystic fibrosis 2018. Am J Respir Crit Care Med. 2019;199:1188–1194. doi: 10.1164/rccm.201902-0310UP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexis NE, Muhlebach MS, Peden DB, Noah TL. Attenuation of host defense function of lung phagocytes in young cystic fibrosis patients. J Cystic Fibros. 2006;5:17–25. doi: 10.1016/j.jcf.2005.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Konstan MW, Hilliard KA, Norvell TM, Berger M. Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinically mild lung disease suggest ongoing infection and inflammation. Am J Respir Crit Care Med. 1994;150:448–454. doi: 10.1164/ajrccm.150.2.8049828. [DOI] [PubMed] [Google Scholar]
- 22.Pohl K, Hayes E, Keenan J, Henry M, Meleady P, Molloy K, et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood. 2014;124:999–1009. doi: 10.1182/blood-2014-02-555268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenfeld M, Gibson RL, McNamara S, Emerson J, Burns JL, Castile R, et al. Early pulmonary infection, inflammation, and clinical outcomes in infants with cystic fibrosis. Pediatr Pulmonol. 2001;32:356–366. doi: 10.1002/ppul.1144. [DOI] [PubMed] [Google Scholar]
- 24.Witko-Sarsat V, Allen RC, Paulais M, Nguyen AT, Bessou G, Lenoir G, et al. Disturbed myeloperoxidase-dependent activity of neutrophils in cystic fibrosis homozygotes and heterozygotes, and its correction by amiloride. J Immunol. 1996;157:2728–2735. [PubMed] [Google Scholar]
- 25.Bruscia EM, Bonfield TL. Cystic fibrosis lung immunity: the role of the macrophage. J Innate Immun. 2016;8:550–563. doi: 10.1159/000446825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garratt LW, Wright AK, Ranganathan SC, Grigg J, Sly PD on behalf of AREST CF. Small macrophages are present in early childhood respiratory disease. J Cystic Fibros. 2012;11:201–208. doi: 10.1016/j.jcf.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 27.Wright AK, Rao S, Range S, Eder C, Hofer TP, Frankenberger M, et al. Pivotal advance: expansion of small sputum macrophages in CF: failure to express MARCO and mannose receptors. J Leukoc Biol. 2009;86:479–489. doi: 10.1189/jlb.1108699. [DOI] [PubMed] [Google Scholar]
- 28.Riquelme SA, Lozano C, Moustafa AM, Liimatta K, Tomlinson KL, Britto C, et al. CFTR-PTEN-dependent mitochondrial metabolic dysfunction promotes Pseudomonas aeruginosa airway infection. Sci Transl Med. 2019;11:eaav4634. doi: 10.1126/scitranslmed.aav4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Riquelme SA, Liimatta K, Wong Fok Lung T, Fields B, Ahn D, Chen D, et al. Pseudomonas aeruginosa utilizes host-derived itaconate to redirect its metabolism to promote biofilm formation. Cell Metab. 2020;31:1091–1106, e6. doi: 10.1016/j.cmet.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med. 2019;199:1517–1536. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohammadi S, Ravindra V, Gleich DF, Grama A. A geometric approach to characterize the functional identity of single cells. Nat Commun. 2018;9:1516. doi: 10.1038/s41467-018-03933-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohammadi S, Davila-Velderrain J, Kellis M. Multi-resolution single-cell state characterization via joint archetypal/network analysis [preprint] doi: 10.1101/746339. bioRxiv 2020 [accessed 2020 Feb 12]. Available from: [DOI] [Google Scholar]
- 33.Reid PA, McAllister DA, Boyd AC, Innes JA, Porteous D, Greening AP, et al. Measurement of serum calprotectin in stable patients predicts exacerbation and lung function decline in cystic fibrosis. Am J Respir Crit Care Med. 2015;191:233–236. doi: 10.1164/rccm.201407-1365LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McElvaney OJ, Zaslona Z, Becker-Flegler K, Palsson-McDermott EM, Boland F, Gunaratnam C, et al. Specific inhibition of the NLRP3 inflammasome as an antiinflammatory strategy in cystic fibrosis. Am J Respir Crit Care Med. 2019;200:1381–1391. doi: 10.1164/rccm.201905-1013OC. [DOI] [PubMed] [Google Scholar]
- 35.Evrard M, Kwok IWH, Chong SZ, Teng KWW, Becht E, Chen J, et al. Developmental analysis of bone marrow neutrophils reveals populations specialized in expansion, trafficking, and effector functions. Immunity. 2018;48:364–379, e8. doi: 10.1016/j.immuni.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 36.Grassi L, Pourfarzad F, Ullrich S, Merkel A, Were F, Carrillo-de-Santa-Pau E, et al. Dynamics of transcription regulation in human bone marrow myeloid differentiation to mature blood neutrophils. Cell Rep. 2018;24:2784–2794. doi: 10.1016/j.celrep.2018.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van de Weert-van Leeuwen PB, Van Meegen MA, Speirs JJ, Pals DJ, Rooijakkers SHM, Van der Ent CK, et al. Optimal complement-mediated phagocytosis of Pseudomonas aeruginosa by monocytes is cystic fibrosis transmembrane conductance regulator-dependent. Am J Respir Cell Mol Biol. 2013;49:463–470. doi: 10.1165/rcmb.2012-0502OC. [DOI] [PubMed] [Google Scholar]
- 38.Kreisel D, Nava RG, Li W, Zinselmeyer BH, Wang B, Lai J, et al. In vivo two-photon imaging reveals monocyte-dependent neutrophil extravasation during pulmonary inflammation. Proc Natl Acad Sci USA. 2010;107:18073–18078. doi: 10.1073/pnas.1008737107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bruscia EM, Zhang PX, Satoh A, Caputo C, Medzhitov R, Shenoy A, et al. Abnormal trafficking and degradation of TLR4 underlie the elevated inflammatory response in cystic fibrosis. J Immunol. 2011;186:6990–6998. doi: 10.4049/jimmunol.1100396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ingersoll SA, Laval J, Forrest OA, Preininger M, Brown MR, Arafat D, et al. Mature cystic fibrosis airway neutrophils suppress T cell function: evidence for a role of arginase 1 but not programmed death-ligand 1. J Immunol. 2015;194:5520–5528. doi: 10.4049/jimmunol.1500312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aron Y, Polla BS, Bienvenu T, Dall’ava J, Dusser D, Hubert D. HLA class II polymorphism in cystic fibrosis: a possible modifier of pulmonary phenotype. Am J Respir Crit Care Med. 1999;159:1464–1468. doi: 10.1164/ajrccm.159.5.9807046. [DOI] [PubMed] [Google Scholar]
- 42.Laki J, Laki I, Németh K, Ujhelyi R, Bede O, Endreffy E, et al. The 8.1 ancestral MHC haplotype is associated with delayed onset of colonization in cystic fibrosis. Int Immunol. 2006;18:1585–1590. doi: 10.1093/intimm/dxl091. [DOI] [PubMed] [Google Scholar]
- 43.O’Neal WK, Gallins P, Pace RG, Dang H, Wolf WE, Jones LC, et al. Gene expression in transformed lymphocytes reveals variation in endomembrane and HLA pathways modifying cystic fibrosis pulmonary phenotypes. Am J Hum Genet. 2015;96:318–328. doi: 10.1016/j.ajhg.2014.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu S, Song K, Bomberger J, Kolls JK.Functional studies to understand immune modifiers in cystic fibrosis J Immunol 2019;202(1 Suppl):182.81 [Google Scholar]
- 45.Hector A, Jonas F, Kappler M, Feilcke M, Hartl D, Griese M. Novel method to process cystic fibrosis sputum for determination of oxidative state. Respiration. 2010;80:393–400. doi: 10.1159/000271607. [DOI] [PubMed] [Google Scholar]
- 46.Sagel SD, Kapsner R, Osberg I, Sontag MK, Accurso FJ. Airway inflammation in children with cystic fibrosis and healthy children assessed by sputum induction. Am J Respir Crit Care Med. 2001;164:1425–1431. doi: 10.1164/ajrccm.164.8.2104075. [DOI] [PubMed] [Google Scholar]
- 47.Hisert KB, Liles WC, Manicone AM. A flow cytometric method for isolating cystic fibrosis airway macrophages from expectorated sputum. Am J Respir Cell Mol Biol. 2019;61:42–50. doi: 10.1165/rcmb.2018-0236MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mayer-Hamblett N, Aitken ML, Accurso FJ, Kronmal RA, Konstan MW, Burns JL, et al. Association between pulmonary function and sputum biomarkers in cystic fibrosis. Am J Respir Crit Care Med. 2007;175:822–828. doi: 10.1164/rccm.200609-1354OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ordoñez CL, Stulbarg M, Grundland H, Liu JT, Boushey HA. Effect of clarithromycin on airway obstruction and inflammatory markers in induced sputum in cystic fibrosis: a pilot study. Pediatr Pulmonol. 2001;32:29–37. doi: 10.1002/ppul.1085. [DOI] [PubMed] [Google Scholar]
- 50.Han MK, Arteaga-Solis E, Blenis J, Bourjeily G, Clegg DJ, DeMeo D, et al. Female sex and gender in lung/sleep health and disease: increased understanding of basic biological, pathophysiological, and behavioral mechanisms leading to better health for female patients with lung disease. Am J Respir Crit Care Med. 2018;198:850–858. doi: 10.1164/rccm.201801-0168WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.