

Abstract
The NEIL3 DNA glycosylase maintains genome integrity during replication by excising oxidized bases from single-stranded DNA (ssDNA) and unhooking interstrand cross-links (ICLs) at fork structures. In addition to its N-terminal catalytic glycosylase domain, NEIL3 contains two tandem C-terminal GRF-type zinc fingers that are absent in the other NEIL paralogs. ssDNA binding by the GRF–ZF motifs helps recruit NEIL3 to replication forks converged at an ICL, but the nature of DNA binding and the effect of the GRF–ZF domain on catalysis of base excision and ICL unhooking is unknown. Here, we show that the tandem GRF–ZFs of NEIL3 provide affinity and specificity for DNA that is greater than each individual motif alone. The crystal structure of the GRF domain shows that the tandem ZF motifs adopt a flexible head-to-tail configuration well-suited for binding to multiple ssDNA conformations. Functionally, we establish that the NEIL3 GRF domain inhibits glycosylase activity against monoadducts and ICLs. This autoinhibitory activity contrasts GRF–ZF domains of other DNA-processing enzymes, which typically use ssDNA binding to enhance catalytic activity, and suggests that the C-terminal region of NEIL3 is involved in both DNA damage recruitment and enzymatic regulation.
Keywords: base excision repair (BER), DNA glycosylase, interstrand DNA cross-link, GRF, zinc finger, DNA repair, DNA-binding protein, DNA damage, crystallography, nucleic acid enzymology, DNA–protein interaction, X-ray crystallography
The NEIL (endonuclease VIII like) family of DNA glycosylases, which include NEIL1, NEIL2, and NEIL3, are important for repair of oxidative DNA damage in vertebrates (1). They are bifunctional enzymes that catalyze hydrolysis of the N-glycosidic bond (base excision activity) and β- and δ-elimination of the resulting apurinic/apyrimidinic (AP, or abasic) site (AP lyase activity), which leads to cleavage of the DNA backbone (1–5). Like the prokaryotic Nei/Fpg (formamidopyrimidine DNA glycosylase) orthologs, the NEIL glycosylases are specific for oxidized bases, including spiroiminodihydantoin and guanidinohydantoin, formamidopyrimidines, 5,6-dihydrothymine (DHT), 5,6-dihydrouracil, 5-hydroxycytosine, 5-hydroxyuracil, and thymine glycol, many of which are mutagenic (6–12).
NEIL3 plays a critical role in protecting cells during DNA replication. NEIL3 is expressed in early S phase, and the protein is found primarily in proliferating cells and co-localizes with Replication Protein A (RPA) (13–16). NEIL3 is the only DNA glycosylase of the Nei/Fpg superfamily to have a strong preference for lesions in ssDNA and at branched structures expected to be found at replication forks (4, 7, 8, 11, 17). In addition, NEIL3 repairs damaged telomeric DNA during S phase and excises thymine glycol, spiroiminodihydantoin, and guanidinohydantoin lesions from G-quadruplexes (18–20). NEIL3 is highly expressed in various human cancer cells and in primary malignant melanomas associated with metastasis (21, 22), exhibits a high frequency of loss of heterozygosity in hepatocellular carcinomas (23), and is frequently mutated in colorectal and breast cancer (24, 25). Loss of NEIL3 was shown to increase deleterious strand breaks during DNA replication and to enhance sensitivity to Ataxia telangiectasia and Rad3-related protein (ATR) and poly(ADP-ribose) polymerase inhibitors (26).
Consistent with its role in DNA replication, NEIL3 participates in the repair of interstrand DNA cross-links (ICLs) (27–29). ICLs are highly toxic lesions that covalently link opposing DNA strands and consequently inhibit replication (30). ICL repair is initiated by unhooking the tethered strands, either by dual incisions on one strand by endonucleases associated with Fanconi anemia and nucleotide excision repair, or alternatively by cleavage of the cross-linked nucleotide by a specialized DNA glycosylase (5, 31). NEIL3 unhooks ICLs derived from the natural product psoralen in both Xenopus cell extracts and in human cells in a replication-dependent manner (27–29). Moreover, NEIL3 unhooks ICLs derived from highly abundant endogenous AP lesions (17, 27). AP-ICLs are formed between a ring-opened AP aldehyde on one strand and the exocyclic N6 amine of adenine on the opposite strand (32–35). NEIL3 cleaves the nonnative N-glycosidic bond from the AP site (27) and preferably acts on AP-ICLs residing on the leading-strand template at a forked DNA structure (17). This substrate specificity is unique to vertebrate NEIL3, because an unrelated bacterial ICL glycosylase does not cleave AP-ICLs but recognizes ICLs independent of DNA structure (36–38).
NEIL3 is a distinct member of the Nei DNA glycosylases, which contain a helix–two-turn–helix glycosylase domain (GD) (1). The structure of the NEIL3–GD explains the enzyme's preference for ssDNA, because it lacks two of the three residues important for intercalating into the dsDNA during excision of the damaged nucleobase (9). Unlike NEIL1, NEIL2, and the other Nei members, NEIL3 further contains a 323-residue C-terminal extension with three zinc finger (ZF) motifs—an internal RanBP/NPL4-type (NZF) and two glycine–arginine–phenylalanine (GRF) ZFs (10, 12). Both the NZF and GRF–ZF domains are required for recruitment of NEIL3 to replication forks that have converged at an ICL in Xenopus extracts (28). The NZF does this through direct interaction with ubiquitylated CMG helicase found at convergent forks, whereas the GRFs do so presumably through their interaction with ssDNA (28).
GRF–ZFs are ssDNA-binding elements present at the C-terminus of several DNA-processing enzymes (39, 40). In several cases these motifs have been shown to enhance enzymatic activity (39, 41–43). For example, disrupting the GRF–ssDNA interaction in AP endonuclease 2 (APE2) inhibits nuclease activity in vitro (39) and is required for full functional complementation of APE2 synthetic lethality with BRCA-deficient cell lines (44). Despite our understanding of the role of the GRF domain on recruitment of NEIL3 to converged forks at an ICL (28), how the GRF domain cooperates to bind DNA and regulate NEIL3 DNA glycosylase activity is unknown. Here, we report the first crystal structure of the NEIL3 GRF domain and probe the role for GRF DNA binding on NEIL3 ICL unhooking and base excision. We find that ssDNA binding by NEIL3's GRF motifs inhibits NEIL3 enzymatic activity. In addition, the structural flexibility between tandem GRF motifs suggests that this domain can adapt to a variety of ssDNA conformations.
Results
GRF zinc fingers bind ssDNA and fork-like structures
The individual NEIL3 GRF motifs have been shown to bind ssDNA (28), but the DNA binding specificity of the tandem GRF domain (GRF12), has not been investigated. We purified GST fusions (Fig. S1) of the Mus musculus (m)NEIL3 individual GRF1 and GRF2 and tandem GRF12 proteins and used electrophoretic mobility shift assays (EMSAs) to probe DNA binding to 40-mer ssDNA, dsDNA, and fork (splayed arm) DNA structures, the latter of which contained a 20-nt dsDNA region with 20-nt ssDNA arms. Each GRF construct bound ssDNA, but not dsDNA (Fig. 1, A–C), consistent with previous results from Xenopus laevis (x)NEIL3 (28). We also observed that the individual and tandem GRF motifs bound fork DNA with an affinity similar to that of ssDNA. Notably, the tandem GRF12 protein displayed 5–10-fold higher affinity for ssDNA and fork DNA (apparent Kd = 10 nm) compared with the isolated GRF1 or GRF2 constructs (Kd,app = 50–100 nm). In addition, the GRF12 domain showed only one major protein–DNA complex band in EMSAs (Fig. 1A), whereas the individual GRF1 and GRF2 motifs exhibited multiple protein–DNA bands indicative of nonspecific binding at higher protein concentrations (Fig. 1, B and C). Despite the appearance of the plotted data shown in Fig. 1 (B and C), there is no statistically significant difference between the affinities of GRF1 and GRF2 for either ssDNA or forked DNA. In control experiments, neither purified recombinant GST (Fig. 1D) nor a NEIL3 GST–NZF domain fusion (Fig. 1E) bound DNA with appreciable affinity. These results indicate that the NEIL3 GRF12 domain binds ssDNA with no apparent specificity for a fork structure and that the tandem GRF motifs bind with higher affinity and specificity than either GRF motif alone.
Figure 1.
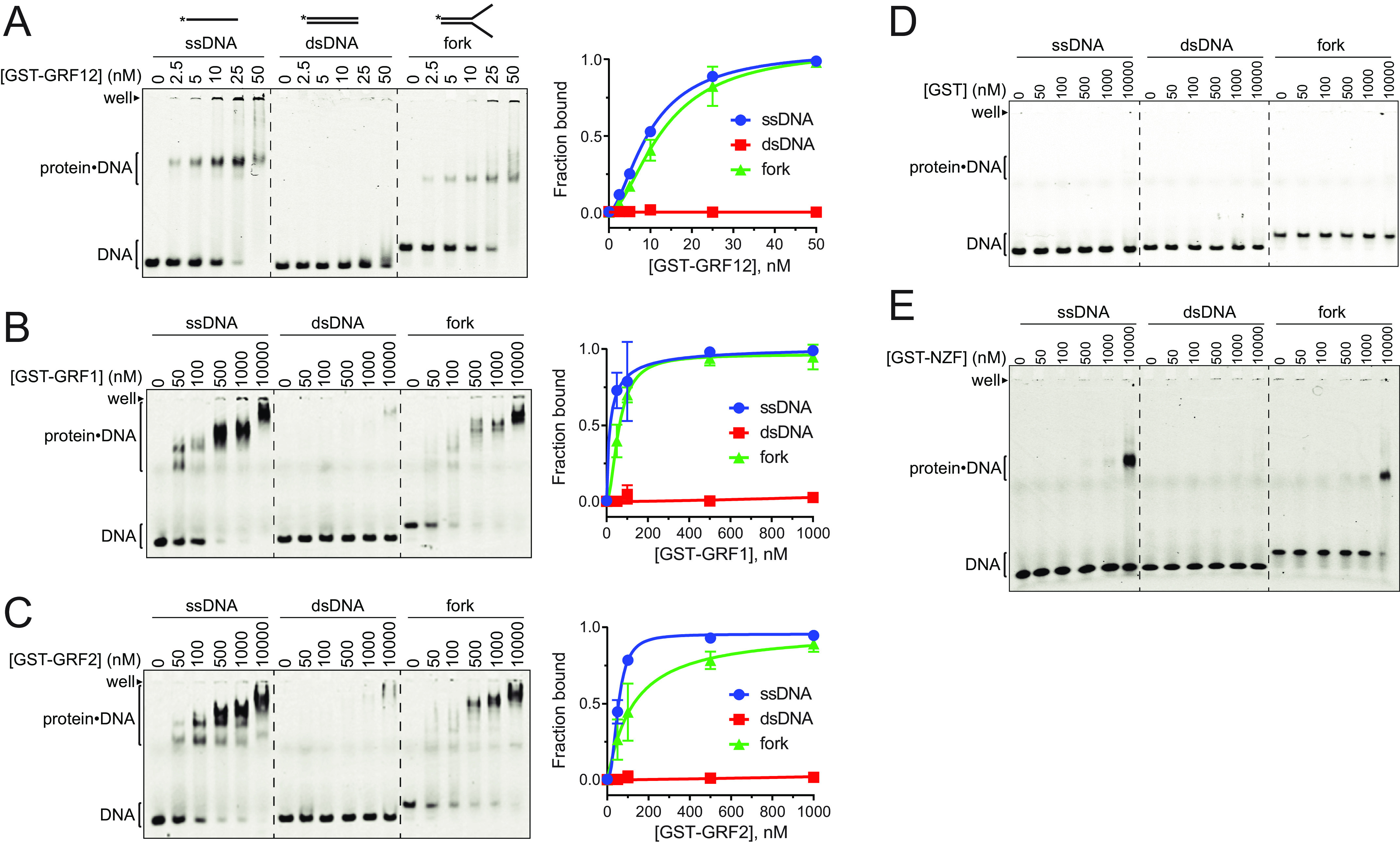
NEIL3 GRF motifs bind ssDNA. A–E, EMSAs for mNEIL3 ZF motifs binding to ssDNA, dsDNA, and splayed arm (fork). Gels in A–C are quantified in the plots to the right. The data are means ± S.D. for n = 3 replicates. A, GST–GRF12. B, GST–GRF1. C, GST–GRF2. D, GST control. E, GST–NZF.
Structural basis for ssDNA binding by the NEIL3 GRF domain
To shed light on the basis for cooperative DNA binding by the GRF12 domains, we crystallized and determined a 2.6 Å crystal structure of human (h)NEIL3 GRF12 domain using single-wavelength anomalous dispersion at the zinc edge (Fig. 2A and Fig. S2). The final model, which contains two GRF12 protomers per asymmetric unit, was refined to a crystallographic residual of 22.7% (Rwork) and 26.6% (Rfree) with reasonable statistics (Table 1). The structure resembles a butterfly, in which two side-by-side GRF motifs are joined by a three-residue flexible linker and oriented in a head-to-tail fashion, with the zinc-binding loops on opposite corners of the molecule (Fig. 2A). Each GRF forms a crescent-shaped structure composed of a three-stranded β-sheet with a CHCC-type zinc finger on one end and a 7–10-residue loop on the other. The concave surfaces of both GRF1 and GRF2 contain a preponderance of basic residues (Fig. 2B), several of which have been shown to bind DNA in xNEIL3 and xAPE2 (28, 39). In NEIL3 GRF12, the concave surfaces face outward and form a continuous surface that likely binds DNA (Fig. 2B). The relative orientation between the GRF1 and GRF2 motifs is different in the two protomers in the asymmetric unit, indicating a large degree of flexibility between them that could accommodate different DNA conformations (Fig. 2C).
Figure 2.
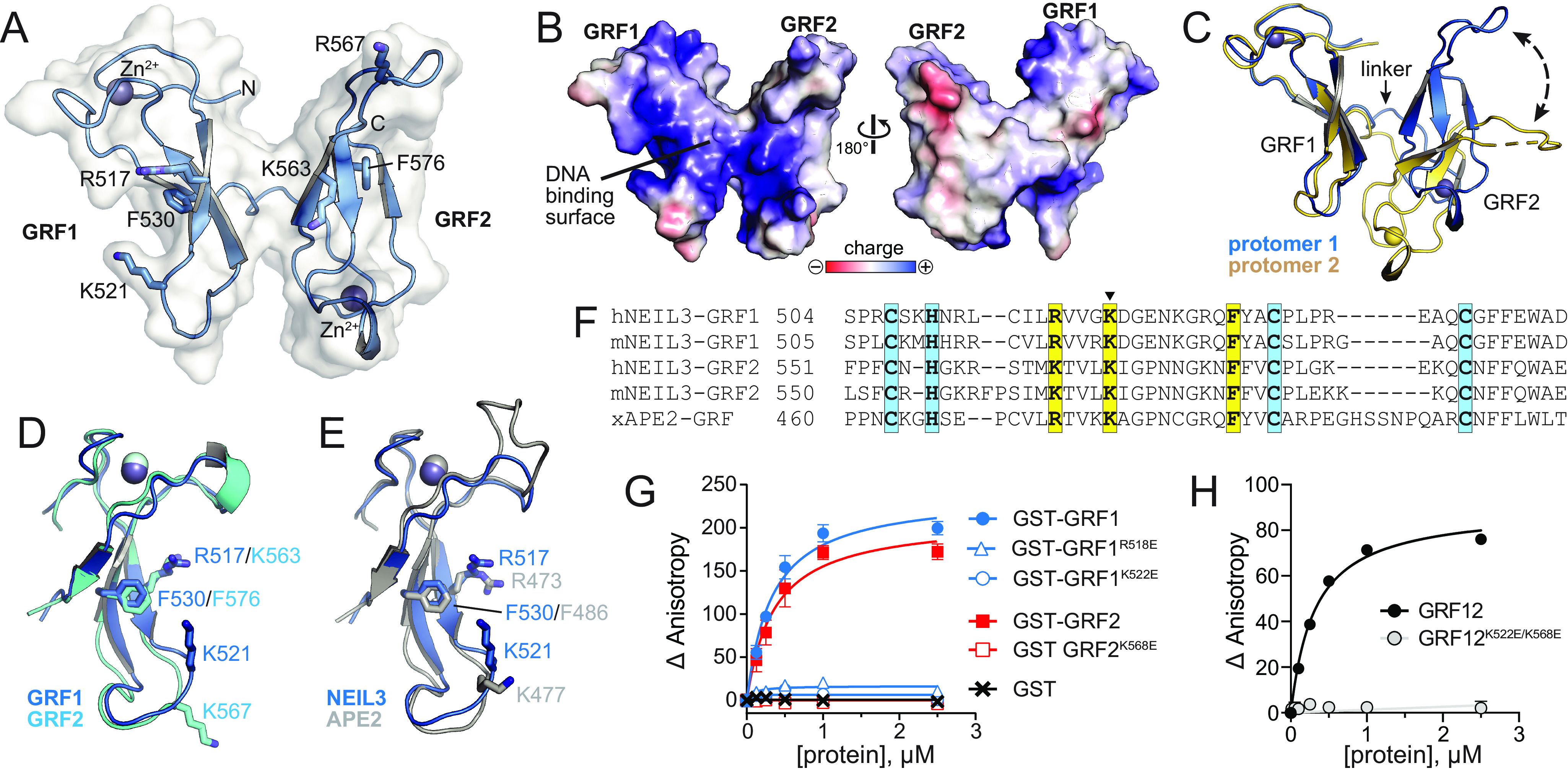
Crystal structure of hNEIL3 GRF domain. A, structure of one of the two protomers in the asymmetric unit. The molecular surface is shown in white, and the positions of several conserved DNA binding residues are indicated. B, The two faces of the GRF12 structure, colored by electrostatic potential. The image on the left is in the same orientation as in A. C, the two GRF12 molecules in the asymmetric unit, superimposed by GRF1. The difference in relative position of GRF2 is highlighted by a dashed arrow. D, superposition of GRF1 and GRF2. E, superposition of GRF motifs from hNEIL3 (blue) and xAPE2 (silver, PDB ID 5U6Z). F, structure-based sequence alignment of hNEIL3 and xAPE2 GRF structures, together with aligned sequences from mNEIL3. Zn2+-coordinating and DNA-binding residues are highlighted blue and yellow, respectively. The lysine mutated in the GRFmut constructs is marked with a triangle. G and H, DNA binding of individual mNEIL3 GRF motifs as GST fusion proteins (G) and the tandem, untagged GRF12 domain (H). The data are means ± S.D. (n = 3).
Table 1.
Data collection and refinement statistics. The data were generated from a single crystal
| Zn-SAD | Native | |
|---|---|---|
| Data collection | ||
| Space group | I4 | I4 |
| Cell dimensions | ||
| a, b, c (Å) | 93.608, 93.608, 63.718 | 93.494, 93.494, 63.646 |
| α, β, γ (°) | 90.00, 90.00, 90.00 | 90.00, 90.00, 90.00 |
| Wavelength | 1.27059 | 1.0000 |
| Resolution (Å)a | 2.80 (2.85–2.80) | 2.60 (2.64–2.60) |
| Rsyma | 0.056 (0.727) | 0.038 (0.656) |
| Rmeasa | 0.060 (0.799) | 0.043 (0.742) |
| Rpima | 0.022 (0.326) | 0.020 (0.344) |
| CC1/2 | 0.810 | 0.840 |
| I/σIa | 30.8 (2.2) | 31.4 (2.0) |
| Completeness (%)a | 99.9 (99.7) | 99.8 (100.0) |
| Redundancya | 7.4 (5.9) | 4.5 (4.6) |
| Refinement | ||
| Resolution (Å)a | 34.95–2.60 (2.70–2.60) | |
| No. reflectionsa | 7550 (469) | |
| Rwork/Rfreea | 0.227/0.266 (0.271/0.306) | |
| No. atoms | ||
| Protein | 1332 | |
| Solvent | 0 | |
| B-factors | ||
| Protein | 51.9 | |
| Solvent | ||
| RMSDs | ||
| Bond lengths (Å) | 0.003 | |
| Bond angles (°) | 0.675 | |
| Ramachandran plot (%) | ||
| Favored | 89.9 | |
| Allowed | 10.1 | |
| Outliers | 0 | |
aNumbers in parentheses refer to data in the highest-resolution shell.
The structures of GRF1 and GRF2 are virtually identical, with an RMSD of 0.89 Å for all atoms (Fig. 2D). In addition, both NEIL3-GRFs are similar to that of xAPE2, with RMSD values of 0.86 Å (GRF1) and 0.60 Å (GRF2) for all atoms (Fig. 2E). The positions of GRF1/GRF2 residues Arg517/Lys563, Lys521/Lys567, and Phe530/Phe576 correspond to the DNA binding residues in xAPE2 (Arg473, Lys477, and Phe486) (39) (Fig. 2, D–F). We validated the importance of these residues for DNA binding by mutating each of the corresponding residues in mNEIL3–GRF proteins to glutamate and quantifying DNA affinity in solution by fluorescence anisotropy. Individual glutamate substitutions were made in GST–GRF1 or GST–GRF2 fusion proteins. WT GST–GRF1 and GST–GRF2 bound to 40-mer ssDNA with Kd values of 0.3 ± 0.04 and 0.4 ± 0.06 μm, respectively, and the GST tag alone showed no binding (Fig. 2G). Glutamate substitution of either mNEIL3 Arg518 or Lys522, which correspond to hNEIL3 Arg517 and Lys521, or mNEIL3 Lys568, which corresponds to hNEIL3 Lys567, completely abrogated DNA binding in the individual GST–GRF1 and GST–GRF2 proteins (Fig. 2G). The lack of binding by the GRF1 K522E mutant is consistent with that shown previously by the corresponding mutant (K500E) in an xNEIL3 MBP-GRF1 construct (28). We next tested binding of a K522E/K568E double mutant in GRF12 that had the GST tag removed (Fig. 2H). WT GRF12 bound the 40-mer ssDNA with a Kd value of 0.2 ± 0.01, whereas the double mutant showed at least 100-fold reduced DNA binding affinity. Thus, the continuous concave surface of the GRF12 domain is likely the main DNA-binding site.
A Dali search for structural homologs identified the GRF-like zinc ribbon (ZR) motifs in Escherichia coli topoisomerase I (TopI) in addition to that of APE2 (45, 46). NEIL3, APE2, and TopI all contain similar GRF–ZF or GRF-like ZR motifs at their extreme C terminus (Fig. 3A). TopI contains five ZR motifs, four of which bind ssDNA in linear fashion (45) (Fig. 3B). Like NEIL3–GRF12, the TopI–ZR1–5 motifs are highly similar to one another, are joined by flexible linkers, and present a contiguous DNA-binding surface. This similarity suggests that NEIL3 will bind to ssDNA across the two GRF motifs in a similar way. The NEIL3–GRFs are most similar to TopI–ZR1 (RMSD = 1.86 Å for backbone atoms), although all five TopI–ZRs have the same general GRF-like architecture (Fig. 3C). TopI–ZR1 interacts with ssDNA by an arginine that corresponds to Arg517/Lys563 in hNEIL3, as well as several aromatic residues from the β strands, including Phe616, which corresponds to Phe530/Phe576 in hNEIL3 (Fig. 3D). TopI–ZR3, –ZR4, and –ZR5 show similar interactions, whereby the arginine side chains contact the deoxyribose-phosphate backbone and a phenylalanine or tyrosine at the center of the concave cleft stacks against the nucleobases (Fig. 3, C and D). Despite the wide variety of relative orientations between consecutive ZRs, the ssDNA binds TopI in a linear fashion with two or three nucleotides per ZR. To gain insight into the footprint of ssDNA across NEIL3–GRF12, we used fluorescence anisotropy to monitor binding to ssDNA of various lengths from 10 to 40 nucleotides (Fig. 3E). We observe a correlation in binding affinity and DNA length and a clear distinction in binding affinities between 20 and 30 nucleotides. The calculated Kd values (in µm) are 1.8 ± 0.3 (10-mer), 1.3 ± 0.2 (20-mer), 0.4 ± 0.1 (30-mer), and 0.2 ± 0.01 (40-mer). A stoichiometric binding experiment with 40-mer ssDNA revealed saturation of binding at a GRF12:DNA ratio of 8:1, which corresponds to five nucleotides per GRF12, consistent with the two or three nucleotides bound to each TopI ZR motif (Fig. 3F).
Figure 3.
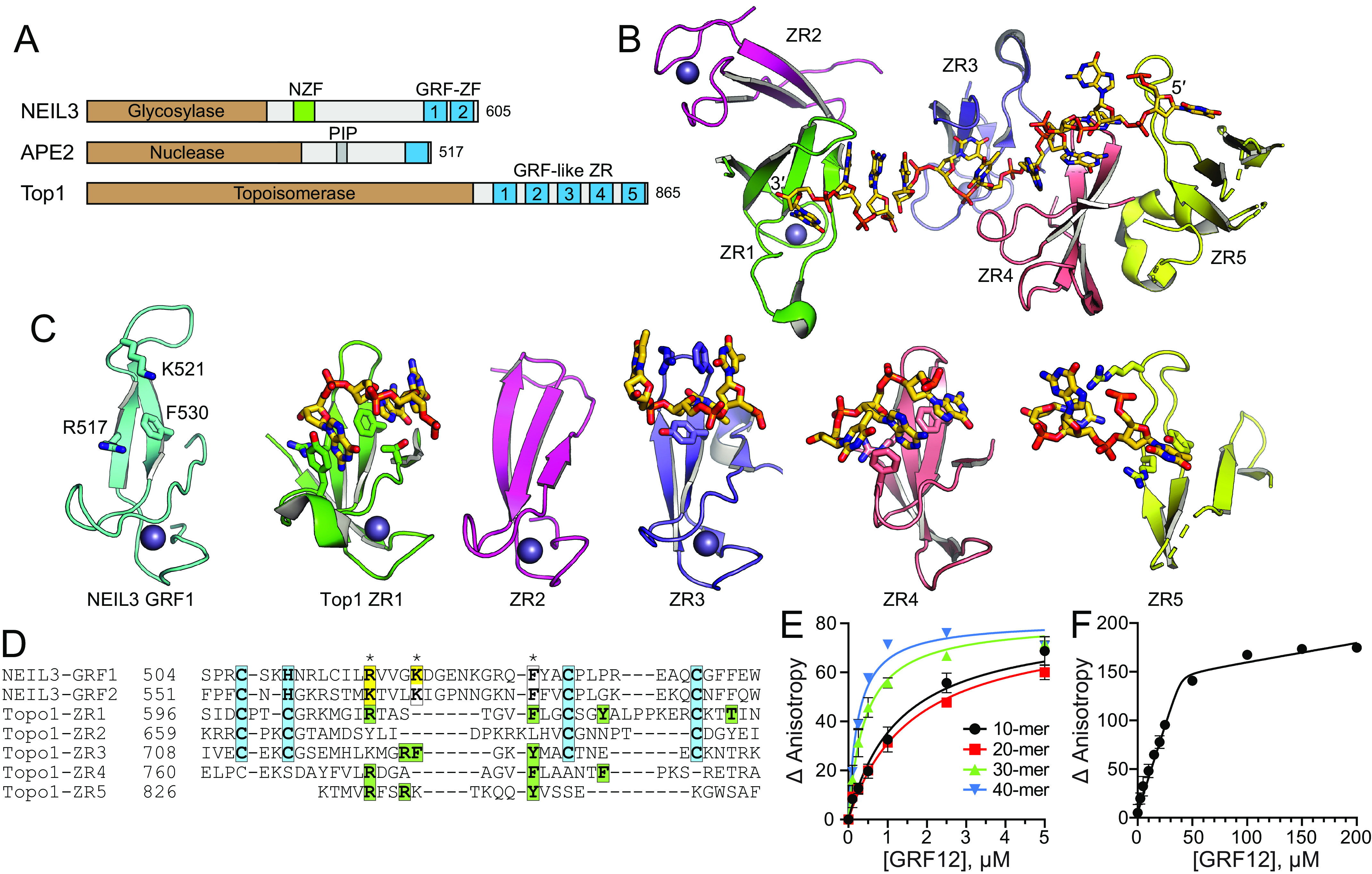
DNA-binding modes by GRF motifs. A, NEIL3, APE2, and E. coli TopI contain GRF–ZF and GRF-like ZR motifs at their C terminus. B, the structure of the TopI ZR domain, with DNA shown as sticks. C, comparison of hNEIL3 GRF1 (cyan) with individual TopI ZR motifs (colored as in B). DNA-binding residues and DNA are shown as sticks. D, structure-based sequence alignment. Zn2+-coordinating residues are highlighted cyan. NEIL3 residues that affect DNA binding upon mutation are highlighted yellow, and DNA interacting residues in the TopI structure are green. Asterisks mark the highest conservation among DNA-binding positions. E, DNA binding of mNEIL3 GRF12 to ssDNA of varying lengths. Total [DNA] used was 25 nm. The data are means ± S.D. (n = 3). F, stoichiometry of binding of GRF12 to 40-mer ssDNA. Total [DNA] = 5.05 μm (≫ Kd). The inflection point in the titration curve fits to [GRF12] = 40 μm, which equals an 8:1 GRF12:DNA molar ratio.
The GRF domain inhibits NEIL3 glycosylase activity
The GRF–ZF of APE2 and GRF-like ZR domains of TopI and human topoisomerase III have been shown to enhance catalytic activity (39, 41–43). We therefore investigated whether the tandem GRF motifs have an impact on NEIL3 DNA glycosylase activity. We first tested the effect of the GRF DNA binding mutations on ICL unhooking activity by full-length mNEIL3 under single-turnover conditions. We compared WT protein against a K522E/K568E double mutant (GRFmut) and a GRF12 deletion mutant (ΔGRF) (Fig. 4A). We previously found that the mNEIL3–GD is able to unhook AP-ICLs at forks only when the AP site is on the strand corresponding to the leading-strand template (17). Here, we found that the full-length protein has the same specificity. We saw no unhooking of the lagging-strand AP-ICL substrate (Fig. 4B) but observed both unhooking and AP lyase activity when NEIL3 was incubated with the leading-strand AP-ICL substrate (Fig. 4C). Interestingly, both the GRFmut and ΔGRF mutants showed significantly faster ICL unhooking kinetics relative to the WT protein (Fig. 4D), suggesting an autoinhibition of ICL unhooking activity by the GRF domain.
Figure 4.

DNA binding by the GRF domain inhibits NEIL3 ICL unhooking activity. A, schematic of proteins used in this experiment. B, representative gel for ICL unhooking by WT mNEIL3 against a lagging-strand AP-dA ICL fork substrate. C, ICL unhooking activity of WT, GRFmut, and ΔGRF mNEIL3 proteins against the leading-strand AP-dA ICL fork. D, quantification of data shown in B and C (means ± S.D. for three independent experiments).
Because the mNEIL3–GD has base excision activity on its own (8, 17), we tested the mechanism by which the GRF–ZF inhibits catalytic activity by adding the purified GRF12 domain in trans to mNEIL3–GD enzymatic reactions containing ssDNA and fork substrates harboring a single DHT residue (Fig. 5A). Compared with reactions that contained no GRF12 protein, the presence of an equimolar amount of GRF12 caused a severe reduction in DHT excision kinetics (Fig. 5B). To test whether this reduction of enzymatic activity was caused by competition of GD and GRF binding to the DNA substrate, we performed the same reaction with the GRF12 K522E/K568E double mutant (GRF12mut). The addition of GRF12mut did not inhibit the reaction (Fig. 5B), indicating that the presumed DNA-binding surface of GRF12 inhibits base excision by the glycosylase domain.
Figure 5.
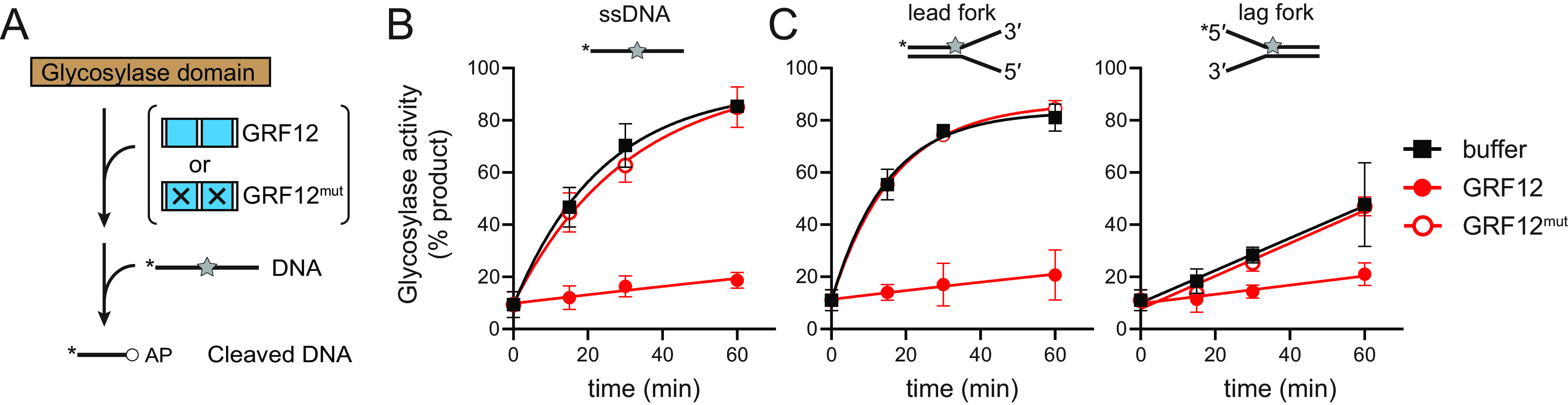
DNA binding by GRF inhibits NEIL3 glycosylase activity in trans. A, experimental design. Glycosylase domain was mixed with either GRF12, GRF12K522E/K568E (GRF12mut), or buffer prior to incubating with either ssDNA or splayed arms containing a DHT lesion. B and C, quantification of DNA glycosylase activity for ssDNA (B) and fork (C) substrates. The data are means ± S.D. (n = 3). Raw data are shown in Fig. S3.
The same preference that exists for AP-ICLs on the leading template strand of forks also exists for DHT monoadducts (17). Unlike with ICL forks, however, mNEIL3–GD retains a low level of activity for monoadducts on the lagging-strand template (17). We therefore tested whether GRF DNA binding would affect this specificity by altering the relative activities for forks containing a monoadduct on the leading versus the lagging strand (Fig. 5C). Similar to the result with ssDNA, the addition of GRF12, but not GRF12mut, caused the same inhibition of glycosylase activity against both DHT fork substrates, indicating that the GRF domain has no effect on the preference for leading-strand damage at forks (Fig. 5C).
Discussion
GRF–ZFs are ssDNA-binding elements present at the C-terminus of several DNA repair enzymes (39, 45). We show here that the GRF domain of NEIL3 is similar in structure and ssDNA-binding properties to the GRF-like motifs of metazoan APE2 and bacterial TopI. However, unlike APE2 and TopI, in which GRF ssDNA binding enhances activity of the catalytic domains (39, 41–43), we find that the NEIL3 GRF–ssDNA interaction inhibits both base excision and ICL unhooking activity in vitro. Based on the nanomolar ssDNA binding affinity of the tandem GRFs and the requirement for ssDNA in NEIL3 substrates (8, 17, 27), we hypothesize that the GRF and glycosylase domains compete for the ssDNA substrate. Indeed, when we introduced point mutations that ablate GRF–ssDNA binding, we rescued glycosylase activity. An alternative possibility is that the GRF12 domain interacts in cis with the NEIL3–GD to modulate its activity. Indeed, protein structures that mimic ssDNA binding to APE2 have been observed in the X-ray structure of the APE2 helix–GRF domain region, suggesting that GRF–ssDNA binding could be regulated by GRF protein–protein interactions (39, 41–43). We propose that the GRF domain endows NEIL3 with a specificity for ICL-stalled replication forks and a variety of other complex environments through its interactions with DNA and other macromolecules.
The C-terminal ZF domains of NEIL3 aid in the recruitment of NEIL3 to replication forks that have converged at an ICL (28). The NZF domain interacts with ubiquitinylated CMG helicase, and the GRF domain enhances the interaction, likely by interacting with ssDNA on the lagging-strand template (28). In those studies, disrupting the GRF–ssDNA interaction via mutagenesis resulted in loss of ICL unhooking by NEIL3 in Xenopus egg extracts, but not in vitro, consistent with the GRF–DNA interaction being important for the recruitment of NEIL3 to converged forks. Because we now find that disruption of the GRF–ssDNA interaction increases glycosylase activity on a simple ICL fork substrate in vitro, it is likely that at converged forks in the cell, the GRF domain is bound to ssDNA away from the lesion so it does not interfere with glycosylase domain access. The other interactions between NEIL3 and the replisome, such as NZF and ubiquitylated CMG, would direct the manner in which NEIL3 is positioned. The autoinhibition of NEIL3 activity in vitro also suggests that the GRF domain may regulate catalytic activity prior to recruitment to converged forks, as a means of avoiding spurious strand cleavage on ssDNA present during replication.
We find that two tandem GRF motifs provide enhanced and more specific binding to ssDNA than each individual GRF motif alone. In addition, the different relative configurations of GRF1–GRF2 motifs observed between the two crystallographically unique molecules in the structure suggest an overall flexibility of the tandem GRF12 domain that may promote binding to ssDNA in different contexts or broaden the conformations that can be sampled upon binding. For example, the C-terminus of NEIL3 plays a role in recruitment of the protein to telomeric damage (20), which is a different DNA environment from converged replisomes. This flexibility may also provide opportunities for the GRF to interact with protein partners. Interestingly, GRF2 contains putative ubiquitylation sites that may aid in such protein–protein interactions. NEIL3 interactions with PCNA, FEN1, TRF1, and CMG have been reported (20, 28). There are likely others, especially given that NEIL3 has been associated with roles outside of replication, including cell signaling, immunity, pulmonary function, myocardial infarction, and ischemic stroke (1, 47–60).
Experimental procedures
Protein purification
The mNEIL3 gene in pET30a was provided by S. Doublié (University of Vermont) (12). The C-terminal His6 tag was replaced with a FLAG tag by a Q5 site-directed mutagenesis kit. Protein was expressed in E. coli BL21 RIL cells in LB medium supplemented with 100 μm ZnSO4 by induction with 250 μm IPTG overnight at 16 °C. The cells were lysed in 20 mm Tris-HCl, pH 8.0, 300 mm NaCl, 15% glycerol, 0.1% NP-40, 1 mm phenylmethylsulfonyl fluoride, 1 mm leupeptin, and 5 mm β-mercaptoethanol (βME). Lysate was centrifuged at 20,000 × g for 30 min, and supernatant was collected. Anti-FLAG beads were incubated with the supernatant for 2–4 h at 4 °C. The beads were isolated by centrifugation and washed three times with 20 mm Tris-HCl, pH 8.0, 600 mm NaCl, 15% glycerol, 0.05 mm TCEP, and 0.1% NP-40. Protein was eluted with 134 μm FLAG peptide in 20 mm Tris-HCl, pH 8.0, 300 mm NaCl, 15% glycerol, 0.05 mm TCEP, and 0.1% NP-40 for 15 min at 4 °C.
Nucleotides encoding mNEIL3–GD (residues 1–282) and containing a C-terminal His6 tag was expressed from pET30a in E. coli BL21(DE3) Star cells by autoinduction as previously described (12). The cells were lysed in buffer A (20 mm Tris-HCl, pH 8.0, 300 mm NaCl, 10% glycerol, 5 mm βME) containing 20 mm imidazole, 1 mm leupeptin, and 1 mm pepstatin A. Lysate was incubated with Ni-NTA (Thermo Scientific) beads for 30 min at 4 °C. Protein was eluted using a 20–500 mm imidazole gradient in Buffer A. Fractions were pooled, diluted to 75 mm NaCl in buffer B (40 mm HEPES-NaOH, pH 7.0, 10% glycerol, and 5 mm βME), and dialyzed at 4 °C overnight. Protein was loaded onto a heparin–Sepharose (GE Healthcare) column, washed, and eluted with an NaCl gradient (0–1 m) in Buffer B. The fractions were pooled, diluted to 500 mm NaCl in buffer B, and loaded onto a HiLoad 26/600 Superdex 200 size-exclusion column (GE Healthcare). The protein was eluted in 20 mm HEPES-NaOH, pH 7.0, 100 mm NaCl, 1 mm TCEP, concentrated, and frozen.
Nucleotides encoding mNEIL3 GRF1 (residues 506–549), GRF2 (residues 550–595), tandem GRF12 (residues 506–595), and NZF (residues 319–353) proteins were cloned into pBG101 (Vanderbilt Center for Structural Biology), which produces an N-terminal His6–GST fusion protein that can be cleaved by rhinovirus 3C protease. Proteins were expressed in E. coli BL21 RIL cells in LB medium containing 10 μm ZnSO4 by induction with 250 mm IPTG overnight at 16 °C. The cells were lysed in buffer containing 50 mm Tris-HCl (pH 8.0 for GRF1, pH 7.0 for GRF2, or pH 7.5 for GRF12 and NZF), 500 mm NaCl, 15% glycerol, 0.5 mm TCEP, 0.02% NP-40, 1 μm leupeptin, and 1 μm pepstatin A. Lysate was incubated with Ni-NTA (Thermo Scientific) beads for 30 min at 4 °C. The beads were washed with 20 mm imidazole, and protein was eluted with 500 mm imidazole in buffer C (50 mm Tris-HCl, 500 mm NaCl, 15% glycerol, 0.5 mm TCEP) supplemented with 0.02% NP-40. Imidazole was diluted to less than 200 mm, and protein was incubated overnight at 4 °C with GSH–Sepharose resin (Thermo Scientific) in buffer C with 0.02% NP-40. GSH beads were washed with buffer C and 0.02% NP-40, and protein was eluted with 10 mm reduced GSH in buffer C with 0.01% NP-40. The protein fractions were pooled and concentrated. At this stage the purified His6–GST fusions were either stored for use in EMSAs or dialyzed into buffer C and cleaved by 3C protease. The His6–GST tag was removed with Ni-NTA in buffer C containing 15–20 mm imidazole. GRF protein was concentrated and stored in buffer C. GRF1-R518E, GRF1-K522E, GRF2-K568E, and GRF12-K522E-K568E (GRFmut) point mutants were generated using a QuikChange (Agilent) mutagenesis kit and purified the same as the WT proteins.
Human NEIL3 GRF12 for crystallization (residues 501–605) was cloned into pMCSG9 by ligation-independent cloning to produce a His6–MBP–TEV–GRF construct. For expression, plasmid was transformed into BL21AI cells and grown in terrific broth to an A600 of 0.8. The cells were induced with 20% l-arabinose, 50 mm ZnSO4, and 100 mm IPTG and grown overnight at 16 °C. For purification, the cells were resuspended in 50 mm Tris, pH 8.0, 500 mm NaCl, 1 mm TCEP, 0.1 g of lysozyme, and protease inhibitor tablet (Roche). The cells were lysed and centrifuged. Lysate was added to amylose resin, flow-through was collected, and resin was washed with the above buffer followed by 50 mm Tris, pH 8.0, 1 m NaCl, and 1 mm TCEP. Salt was dropped to 150 mm NaCl, and protein was eluted with 50 mm Tris, pH 8.0, 150 mm NaCl, 1 mm TCEP, and 10 mm maltose. Elutions were collected, incubated with TEV protease, and subjected to heparin–Sepharose chromatography with a 0.05–1.0 m NaCl gradient in 20 mm Tris, pH 8, and 1 mm TCEP. Fractions were pooled and run over a HiLoad 16/600 Superdex 75 (GE Healthcare) size-exclusion column in 10 mm Tris pH 8, 100 mm NaCl, and 1 mm TCEP, and protein was concentrated to 10 mg/ml.
X-ray crystallography
Crystals of hNEIL3 GRF12 were obtained by sitting-drop vapor diffusion at 20 °C by mixing the protein solution with 0.1 m Tris, pH 8.5, and 25% PEG 6000. The crystals were cryoprotected in 20% ethylene glycol. X-ray diffraction data from a single crystal were collected at 105 K on Beamline 22-ID of the Advanced Photon Source and processed with HKL2000 (61). Phasing and refinement was carried out using the PHENIX suite of programs (62). The structure was determined by SAD phasing from 2.8 Å anomalous data collected at the zinc edge (1.27059 Å) using AutoSol and Phaser. An initial model was built using side chain–truncated APE2 GRF residues 461–508 from PDB 5U6Z as a guide and the Zn-SAD electron density, followed by manual building of the remainder of the model in Coot (63). The model was refined against 2.6 Å native data collected at 1.0000 Å wavelength. The final model was validated with MolProbity (64) and contained no residues in disallowed regions of the Ramachandran plot. Refinement and validation statistics are shown in Table 1. Structures were analyzed and figures made using PyMOL (Schrödinger). All software was curated by SBGrid (65).
Electrophoretic mobility shift assays
Oligonucleotides used in EMSAs are shown in Table S1. His-GST fusions of mNEIL3 GRF and NZF constructs (0–10 μm) were incubated with 10 nm 6-carboxyfluorescein (FAM)–labeled DNA for 30 min at 21 °C in 20 mm HEPES, pH 6.6, 100 mm NaCl, 5 mm MgCl2, 3% (v/v) glycerol, 0.2% (v/v) NP-40, and 0.5 mm TCEP. The samples were separated by electrophoresis on a 5% polyacrylamide/0.5× TBE gel at 200 V for 1 h. The gels were imaged using a Typhoon Trio variable mode imager (GE Healthcare) at 532-nm excitation and 526-nm emission wavelengths. Band intensities were quantified using Gel Analyzer and plotted using GraphPad Prism 8.
Fluorescence anisotropy
DNA binding of WT and mutant GRF constructs were monitored by fluorescence anisotropy. For Kd determination, proteins at varying concentration were incubated with 25 nm FAM-labeled ssDNA (Table S1) in 20 mm HEPES, pH 7.0, 100 mm NaCl, 5 mm MgCl2, 3% glycerol, and 0.5 mm TCEP at 4 °C in the dark for 30 min. To determine stoichiometry of binding, 50 nm FAM-labeled and 5 μm unlabeled 40-mer ssDNA was used so that the total DNA concentration was over 20-fold excess of the Kd. Fluorescence anisotropy data at excitation and emission wavelengths of 485 and 528 nm were collected at room temperature in 96-well plates using a BioTek Synergy H1 plate reader. The data were fit to a two-state binding model using GraphPad Prism 8.
Glycosylase activity assays
DNA substrates containing an AP-ICL or DHT were prepared as previously described (17). Both AP-ICL unhooking and DHT excision assays were performed under single-turnover conditions, which was verified by confirming that the reaction rates remained constant at higher enzyme:DNA ratios. AP-ICL unhooking reactions were performed at 25 °C and contained 250 nm full-length mNEIL3 or deletion mutants and 25 nm FAM-labeled DNA in glycosylase buffer (20 mm HEPES-NaOH, pH 7.0, 100 mm NaCl, 5% glycerol, 1 mm DTT, and 10 µg/ml BSA). The reactions were stopped by the addition of 10 mm EDTA, 80% (v/v) formamide at 70 °C for 5 min and run on precast 10% TBE/urea gels (Invitrogen) at 180 V in 0.5× TBE buffer. Base excision activity of mNEIL3–GD in the presence of purified GRF12 proteins was carried out by mixing 10 μm mNEIL3–GD with 10 μm or either GRF12 or GRF12mut, followed by incubation at 25 °C with 50 nm FAM-labeled DHT-containing DNA in glycosylase buffer. Reactions were stopped by the addition of 0.1 n NaOH and 10 mm EDTA, 80% (v/v) formamide and heated at 70 °C for 5 min. Band intensities were quantified using gel analyzer and plotted with one-phase association exponential fit using GraphPad Prism 8.
Data availability
Atomic coordinates and structure factors for the hNEIL3-GRF12 crystal structure have been deposited in the Protein Data Bank under accession code 7JL5. All other data are contained within the article.
Supplementary Material
Acknowledgments
We thank Sylvie Doublié for the NEIL3/pET-30a vector. We thank Lars Pedersen of the NIEHS, National Institutes of Health collaborative crystallography group, and the Advanced Photon Source SE Regional Collaborative Access Team staff for assistance with crystallographic data collection. We thank Lars Pedersen and Bill Beard for comments. The X-ray diffraction data were collected at the Southeast Regional Collaborative Access Team 22-ID Beamline at the Advanced Photon Source at the Argonne National Laboratory. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract W-31-109-Eng-38.
This article contains supporting information.
Author contributions—A. A. R., J. L. W., and B. H. G. data curation; A. A. R., J. L. W., B. H. G., R. S. W., and B. F. E. formal analysis; A. A. R., K. S. G., R. S. W., and B. F. E. funding acquisition; A. A. R. investigation; A. A. R., J. L. W., and B. H. G. writing-original draft; A. A. R., J. L. W., B. H. G., K. S. G., R. S. W., and B. F. E. writing-review and editing; T. H., K. S. G., R. S. W., and B. F. E. resources; R. S. W. and B. F. E. supervision; A. A. R., J. L. W., R. S. W., and B. F. E. validation; B. F. E. conceptualization; B. F. E. visualization; B. F. E. project administration.
Funding and additional information—This work was supported by National Institutes of Health Grants R01GM131071 and P01CA092584 (to B. F. E.) and NIEHS, National Institute of Health Intramural Program Grant 1Z01ES102765 (to R. S. W.). A. A. R. was supported by Vanderbilt Molecular Biophysics Training Program Grant T32GM08320 and National Science Foundation Graduate Research Fellowship DGE-1445197. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the National Science Foundation.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- ssDNA
- single-stranded DNA
- ICL
- interstrand cross-link
- RMSD
- root-mean-square deviation
- DHT
- 5,6-dihydrothymine
- AP
- apurinic/apyrimidinic
- GD
- glycosylase domain
- ZF
- zinc finger
- NZF
- RanBP/NPL4-type ZF
- APE
- AP endonuclease
- CMG
- Cdc45/Mcm2-7/GINS
- m
- M. musculus
- EMSA
- electrophoretic mobility shift assay
- x
- X. laevis
- h
- human
- ZR
- zinc ribbon
- TopI
- topoisomerase I
- IPTG
- isopropyl β-d-thiogalactopyranoside
- TCEP
- tris(2-carboxyethyl)phosphine
- NP-40
- Nonidet P-40
- βME
- β-mercaptoethanol
- Ni-NTA
- nickel–nitrilotriacetic acid
- FAM
- 6-carboxyfluorescein.
References
- 1. Liu M., Doublié S., and Wallace S. S. (2013) Neil3, the final frontier for the DNA glycosylases that recognize oxidative damage. Mutat. Res. 743–744, 4–11 10.1016/j.mrfmmm.2012.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hazra T. K., Izumi T., Boldogh I., Imhoff B., Kow Y. W., Jaruga P., Dizdaroglu M., and Mitra S. (2002) Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. U.S.A. 99, 3523–3528 10.1073/pnas.062053799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Das A., Rajagopalan L., Mathura V. S., Rigby S. J., Mitra S., and Hazra T. K. (2004) Identification of a zinc finger domain in the human NEIL2 (Nei-like-2) protein. J. Biol. Chem. 279, 47132–47138 10.1074/jbc.M406224200 [DOI] [PubMed] [Google Scholar]
- 4. Albelazi M. S., Martin P. R., Mohammed S., Mutti L., Parsons J. L., and Elder R. H. (2019) The biochemical role of the human NEIL1 and NEIL3 DNA glycosylases on model DNA replication forks. Genes (Basel) 10, 315 10.3390/genes10040315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mullins E. A., Rodriguez A. A., Bradley N. P., and Eichman B. F. (2019) Emerging roles of DNA glycosylases and the base excision repair pathway. Trends Biochem. Sci. 44, 765–781 10.1016/j.tibs.2019.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grin I. R., and Zharkov D. O. (2011) Eukaryotic endonuclease VIII–like proteins: new components of the base excision DNA repair system. Biochemistry (Mosc.) 76, 80–93 10.1134/s000629791101010x [DOI] [PubMed] [Google Scholar]
- 7. Krokeide S. Z., Laerdahl J. K., Salah M., Luna L., Cederkvist F. H., Fleming A. M., Burrows C. J., Dalhus B., and Bjørås M. (2013) Human NEIL3 is mainly a monofunctional DNA glycosylase removing spiroimindiohydantoin and guanidinohydantoin. DNA Repair (Amst.) 12, 1159–1164 10.1016/j.dnarep.2013.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu M., Bandaru V., Bond J. P., Jaruga P., Zhao X., Christov P. P., Burrows C. J., Rizzo C. J., Dizdaroglu M., and Wallace S. S. (2010) The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 107, 4925–4930 10.1073/pnas.0908307107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu M., Imamura K., Averill A. M., Wallace S. S., and Doublié S. (2013) Structural characterization of a mouse ortholog of human NEIL3 with a marked preference for single-stranded DNA. Structure 21, 247–256 10.1016/j.str.2012.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krokeide S. Z., Bolstad N., Laerdahl J. K., Bjørås M., and Luna L. (2009) Expression and purification of NEIL3, a human DNA glycosylase homolog. Protein Expr. Purif. 65, 160–164 10.1016/j.pep.2008.11.014 [DOI] [PubMed] [Google Scholar]
- 11. Takao M., Oohata Y., Kitadokoro K., Kobayashi K., Iwai S., Yasui A., Yonei S., and Zhang Q. M. (2009) Human Nei-like protein NEIL3 has AP lyase activity specific for single-stranded DNA and confers oxidative stress resistance in Escherichia coli mutant. Genes Cells 14, 261–270 10.1111/j.1365-2443.2008.01271.x [DOI] [PubMed] [Google Scholar]
- 12. Liu M., Bandaru V., Holmes A., Averill A. M., Cannan W., and Wallace S. S. (2012) Expression and purification of active mouse and human NEIL3 proteins. Protein Expr. Purif. 84, 130–139 10.1016/j.pep.2012.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Neurauter C. G., Luna L., and Bjørås M. (2012) Release from quiescence stimulates the expression of human NEIL3 under the control of the Ras dependent ERK-MAP kinase pathway. DNA Repair (Amst.) 11, 401–409 10.1016/j.dnarep.2012.01.007 [DOI] [PubMed] [Google Scholar]
- 14. Regnell C. E., Hildrestrand G. A., Sejersted Y., Medin T., Moldestad O., Rolseth V., Krokeide S. Z., Suganthan R., Luna L., Bjørås M., and Bergersen L. H. (2012) Hippocampal adult neurogenesis is maintained by Neil3-dependent repair of oxidative DNA lesions in neural progenitor cells. Cell Rep. 2, 503–510 10.1016/j.celrep.2012.08.008 [DOI] [PubMed] [Google Scholar]
- 15. Sejersted Y., Hildrestrand G. A., Kunke D., Rolseth V., Krokeide S. Z., Neurauter C. G., Suganthan R., Atneosen-Åsegg M., Fleming A. M., Saugstad O. D., Burrows C. J., Luna L., and Bjørås M. (2011) Endonuclease VIII–like 3 (Neil3) DNA glycosylase promotes neurogenesis induced by hypoxia-ischemia. Proc. Natl. Acad. Sci. U.S.A. 108, 18802–18807 10.1073/pnas.1106880108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morland I., Rolseth V., Luna L., Rognes T., Bjørås M., and Seeberg E. (2002) Human DNA glycosylases of the bacterial Fpg/MutM superfamily: an alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res. 30, 4926–4936 10.1093/nar/gkf618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Imani Nejad M., Housh K., Rodriguez A. A., Haldar T., Kathe S., Wallace S. S., Eichman B. F., and Gates K. S. (2020) Unhooking of an interstrand cross-link at DNA fork structures by the DNA glycosylase NEIL3. DNA Repair (Amst.) 86, 102752 10.1016/j.dnarep.2019.102752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou J., Liu M., Fleming A. M., Burrows C. J., and Wallace S. S. (2013) Neil3 and NEIL1 DNA glycosylases remove oxidative damages from quadruplex DNA and exhibit preferences for lesions in the telomeric sequence context. J. Biol. Chem. 288, 27263–27272 10.1074/jbc.M113.479055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou J., Fleming A. M., Averill A. M., Burrows C. J., and Wallace S. S. (2015) The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res. 43, 4039–4054 10.1093/nar/gkv252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou J., Chan J., Lambelé M., Yusufzai T., Stumpff J., Opresko P. L., Thali M., and Wallace S. S. (2017) NEIL3 repairs telomere damage during S phase to secure chromosome segregation at mitosis. Cell Rep. 20, 2044–2056 10.1016/j.celrep.2017.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hildrestrand G. A., Neurauter C. G., Diep D. B., Castellanos C. G., Krauss S., Bjørås M., and Luna L. (2009) Expression patterns of Neil3 during embryonic brain development and neoplasia. BMC Neurosci. 10, 45 10.1186/1471-2202-10-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kauffmann A., Rosselli F., Lazar V., Winnepenninckx V., Mansuet-Lupo A., Dessen P., van den Oord J. J., Spatz A., and Sarasin A. (2008) High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene 27, 565–573 10.1038/sj.onc.1210700 [DOI] [PubMed] [Google Scholar]
- 23. Zhang H., Ma H., Wang Q., Chen M., Weng D., Wang H., Zhou J., Li Y., Sun J., Chen Y., Liang X., Zhao J., Pan K., Wang H., and Xia J. (2010) Analysis of loss of heterozygosity on chromosome 4q in hepatocellular carcinoma using high-throughput SNP array. Oncol. Rep. 23, 445–455 10.3892/or_00000654 [DOI] [PubMed] [Google Scholar]
- 24. Farkas S. A., Vymetalkova V., Vodickova L., Vodicka P., and Nilsson T. K. (2014) DNA methylation changes in genes frequently mutated in sporadic colorectal cancer and in the DNA repair and Wnt/β-catenin signaling pathway genes. Epigenomics 6, 179–191 10.2217/epi.14.7 [DOI] [PubMed] [Google Scholar]
- 25. Mumbrekar K. D., Bola Sadashiva S. R., Kabekkodu S. P., Fernandes D. J., Vadhiraja B. M., Suga T., Shoji Y., Nakayama F., Imai T., and Satyamoorthy K. (2017) Genetic variants in CD44 and MAT1A confer susceptibility to acute skin reaction in breast cancer patients undergoing radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 97, 118–127 10.1016/j.ijrobp.2016.09.017 [DOI] [PubMed] [Google Scholar]
- 26. Klattenhoff A. W., Thakur M., Chu C. S., Ray D., Habib S. L., and Kidane D. (2017) Loss of NEIL3 DNA glycosylase markedly increases replication associated double strand breaks and enhances sensitivity to ATR inhibitor in glioblastoma cells. Oncotarget 8, 112942–112958 10.18632/oncotarget.22896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Semlow D. R., Zhang J., Budzowska M., Drohat A. C., and Walter J. C. (2016) Replication-dependent unhooking of DNA interstrand cross-links by the NEIL3 glycosylase. Cell 167, 498–511 10.1016/j.cell.2016.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu R. A., Semlow D. R., Kamimae-Lanning A. N., Kochenova O. V., Chistol G., Hodskinson M. R., Amunugama R., Sparks J. L., Wang M., Deng L., Mimoso C. A., Low E., Patel K. J., and Walter J. C. (2019) TRAIP is a master regulator of DNA interstrand crosslink repair. Nature 567, 267–272 10.1038/s41586-019-1002-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li N., Wang J., Wallace S. S., Chen J., Zhou J., and D'Andrea A. D. (2020) Cooperation of the NEIL3 and Fanconi anemia/BRCA pathways in interstrand crosslink repair. Nucleic Acids Res. 48, 3014–3028 10.1093/nar/gkaa038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Räschle M., Knipscheer P., Knipsheer P., Enoiu M., Angelov T., Sun J., Griffith J. D., Ellenberger T. E., Schärer O. D., and Walter J. C. (2008) Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 134, 969–980 10.1016/j.cell.2008.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Clauson C., Schärer O. D., and Niedernhofer L. (2013) Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb. Perspect. Biol. 5, a012732 10.1101/cshperspect.a012732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Price N. E., Johnson K. M., Wang J., Fekry M. I., Wang Y., and Gates K. S. (2014) Interstrand DNA-DNA cross-link formation between adenine residues and abasic sites in duplex DNA. J. Am. Chem. Soc. 136, 3483–3490 10.1021/ja410969x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Catalano M. J., Liu S., Andersen N., Yang Z., Johnson K. M., Price N. E., Wang Y., and Gates K. S. (2015) Chemical structure and properties of interstrand cross-links formed by reaction of guanine residues with abasic sites in duplex DNA. J. Am. Chem. Soc. 137, 3933–3945 10.1021/jacs.5b00669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Price N. E., Catalano M. J., Liu S., Wang Y., and Gates K. S. (2015) Chemical and structural characterization of interstrand cross-links formed between abasic sites and adenine residue in duplex DNA. Nucleic Acids Res. 43, 3434–3441 10.1093/nar/gkv174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang Z., Price N. E., Johnson K. M., Wang Y., and Gates K. S. (2017) Interstrand cross-links arising from strand breaks at true abasic sites in duplex DNA. Nucleic Acids Res. 45, 6275–6283 10.1093/nar/gkx394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang S., Liu K., Xiao L., Yang L., Li H., Zhang F., Lei L., Li S., Feng X., Li A., and He J. (2016) Characterization of a novel DNA glycosylase from S. sahachiroi involved in the reduction and repair of azinomycin B induced DNA damage. Nucleic Acids Res. 44, 187–197 10.1093/nar/gkv949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mullins E. A., Warren G. M., Bradley N. P., and Eichman B. F. (2017) Structure of a DNA glycosylase that unhooks interstrand cross-links. Proc. Natl. Acad. Sci. U.S.A. 114, 4400–4405 10.1073/pnas.1703066114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bradley N. P., Washburn L. A., Christov P. P., Watanabe C. M. H., and Eichman B. F. (2020) Escherichia coli YcaQ is a DNA glycosylase that unhooks DNA interstrand crosslinks. Nucleic Acids Res. 48, 7005–7017 10.1093/nar/gkaa346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wallace B. D., Berman Z., Mueller G. A., Lin Y., Chang T., Andres S. N., Wojtaszek J. L., DeRose E. F., Appel C. D., London R. E., Yan S., and Williams R. S. (2017) APE2 Zf-GRF facilitates 3'–5' resection of DNA damage following oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 114, 304–309 10.1073/pnas.1610011114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Grishin N. V. (2000) C-terminal domains of Escherichia coli topoisomerase I belong to the zinc-ribbon superfamily. J. Mol. Biol. 299, 1165–1177 10.1006/jmbi.2000.3841 [DOI] [PubMed] [Google Scholar]
- 41. Chen S. H., Wu C. H., Plank J. L., and Hsieh T. S. (2012) Essential functions of C terminus of Drosophila topoisomerase IIIα in double holliday junction dissolution. J. Biol. Chem. 287, 19346–19353 10.1074/jbc.M112.363044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tse-Dinh Y. C. (1991) Zinc (II) coordination in Escherichia coli DNA topoisomerase I is required for cleavable complex formation with DNA. J. Biol. Chem. 266, 14317–14320 [PubMed] [Google Scholar]
- 43. Zhang H. L., Malpure S., Li Z., Hiasa H., and DiGate R. J. (1996) The role of the carboxyl-terminal amino acid residues in Escherichia coli DNA topoisomerase III-mediated catalysis. J. Biol. Chem. 271, 9039–9045 10.1074/jbc.271.15.9039 [DOI] [PubMed] [Google Scholar]
- 44. Álvarez-Quilón A., Wojtaszek J. L., Mathieu M. C., Patel T., Appel C. D., Hustedt N., Rossi S. E., Wallace B. D., Setiaputra D., Adam S., Ohashi Y., Melo H., Cho T., Gervais C., Muñoz I. M., et al. (2020) Endogenous DNA 3' blocks are vulnerabilities for BRCA1 and BRCA2 deficiency and are reversed by the APE2 nuclease. Mol. Cell 78, 1152–1165.e8 10.1016/j.molcel.2020.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tan K., Zhou Q., Cheng B., Zhang Z., Joachimiak A., and Tse-Dinh Y. C. (2015) Structural basis for suppression of hypernegative DNA supercoiling by E. coli topoisomerase I. Nucleic Acids Res. 43, 11031–11046 10.1093/nar/gkv1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yu L., Zhu C. X., Tse-Dinh Y. C., and Fesik S. W. (1995) Solution structure of the C-terminal single-stranded DNA-binding domain of Escherichia coli topoisomerase I. Biochemistry 34, 7622–7628 10.1021/bi00023a008 [DOI] [PubMed] [Google Scholar]
- 47. Olsen M. B., Hildrestrand G. A., Scheffler K., Vinge L. E., Alfsnes K., Palibrk V., Wang J., Neurauter C. G., Luna L., Johansen J., Øgaard J. D. S., Ohm I. K., Slupphaug G., Kuśnierczyk A., Fiane A. E., et al. (2017) NEIL3-dependent regulation of cardiac fibroblast proliferation prevents myocardial rupture. Cell Rep. 18, 82–92 10.1016/j.celrep.2016.12.009 [DOI] [PubMed] [Google Scholar]
- 48. Rumsey W. L., Bolognese B., Davis A. B., Flamberg P. L., Foley J. P., Katchur S. R., Kotzer C. J., Osborn R. R., and Podolin P. L. (2017) Effects of airborne toxicants on pulmonary function and mitochondrial DNA damage in rodent lungs. Mutagenesis 32, 343–353 10.1093/mutage/gew063 [DOI] [PubMed] [Google Scholar]
- 49. Li P., Stetler R. A., Leak R. K., Shi Y., Li Y., Yu W., Bennett M. V. L., and Chen J. (2018) Oxidative stress and DNA damage after cerebral ischemia: Potential therapeutic targets to repair the genome and improve stroke recovery. Neuropharmacology 134, 208–217 10.1016/j.neuropharm.2017.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. de Sousa J. F., Torrieri R., Serafim R. B., Di Cristofaro L. F., Escanfella F. D., Ribeiro R., Zanette D. L., Paçó-Larson M. L., da Silva W. A. Jr., Tirapelli D. P., Neder L., Carlotti C. G. Jr., and Valente V. (2017) Expression signatures of DNA repair genes correlate with survival prognosis of astrocytoma patients. Tumour Biol. 39, 1010428317694552 10.1177/1010428317694552 [DOI] [PubMed] [Google Scholar]
- 51. Yang L. X., Zhang X., and Zhao G. (2016) Ginsenoside Rd attenuates DNA damage by increasing expression of DNA glycosylase endonuclease VIII–like proteins after focal cerebral ischemia. Chin. Med. J. (Engl.) 129, 1955–1962 10.4103/0366-6999.187851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tangye S. G. (2016) Genetic cause of immune dysregulation: one gene or two? J. Clin. Invest. 126, 4065–4067 10.1172/JCI90831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Skarpengland T., Holm S., Scheffler K., Gregersen I., Dahl T. B., Suganthan R., Segers F. M., Ostlie I., Otten J. J., Luna L., Ketelhuth D. F., Lundberg A. M., Neurauter C. G., Hildrestrand G., Skjelland M., et al. (2016) Neil3-dependent base excision repair regulates lipid metabolism and prevents atherosclerosis in Apoe-deficient mice. Sci. Rep. 6, 28337 10.1038/srep28337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Massaad M. J., Zhou J., Tsuchimoto D., Chou J., Jabara H., Janssen E., Glauzy S., Olson B. G., Morbach H., Ohsumi T. K., Schmitz K., Kyriacos M., Kane J., Torisu K., Nakabeppu Y., et al. (2016) Deficiency of base excision repair enzyme NEIL3 drives increased predisposition to autoimmunity. J. Clin. Invest. 126, 4219–4236 10.1172/JCI85647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jalland C. M., Scheffler K., Benestad S. L., Moldal T., Ersdal C., Gunnes G., Suganthan R., Bjørås M., and Tranulis M. A. (2016) Neil3 induced neurogenesis protects against prion disease during the clinical phase. Sci. Rep. 6, 37844 10.1038/srep37844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. He W., Huang P., Liu D., Zhong L., Yu R., and Li J. (2016) Polymorphism of the XRCC1 gene is associated with susceptibility and short-term recovery of ischemic stroke. Int. J. Environ. Res. Public Health 13, 1016 10.3390/ijerph13101016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ehlers C. L., Gizer I. R., Bizon C., Slutske W., Peng Q., Schork N. J., and Wilhelmsen K. C. (2016) Single nucleotide polymorphisms in the REG-CTNNA2 region of chromosome 2 and NEIL3 associated with impulsivity in a Native American sample. Genes Brain Behav. 15, 568–577 10.1111/gbb.12297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Skarpengland T., Laugsand L. E., Janszky I., Luna L., Halvorsen B., Platou C. G., Wang W., Vatten L. J., Damås J. K., Aukrust P., Bjørås M., and Åsvold B. O. (2015) Genetic variants in the DNA repair gene NEIL3 and the risk of myocardial infarction in a nested case-control study. The HUNT Study. DNA Repair (Amst.) 28, 21–27 10.1016/j.dnarep.2015.01.013 [DOI] [PubMed] [Google Scholar]
- 59. Rognlien A. G., Wollen E. J., Atneosen-Åsegg M., and Saugstad O. D. (2015) Increased expression of inflammatory genes in the neonatal mouse brain after hyperoxic reoxygenation. Pediatr. Res. 77, 326–333 10.1038/pr.2014.193 [DOI] [PubMed] [Google Scholar]
- 60. Chakraborty A., Wakamiya M., Venkova-Canova T., Pandita R. K., Aguilera-Aguirre L., Sarker A. H., Singh D. K., Hosoki K., Wood T. G., Sharma G., Cardenas V., Sarkar P. S., Sur S., Pandita T. K., Boldogh I., et al. (2015) Neil2-null mice accumulate oxidized DNA bases in the transcriptionally active sequences of the genome and are susceptible to innate inflammation. J. Biol. Chem. 290, 24636–24648 10.1074/jbc.M115.658146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 62. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Emsley P., Lohkamp B., Scott W., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., Snoeyink J., Richardson J. S., and Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 10.1093/nar/gkm216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Morin A., Eisenbraun B., Key J., Sanschagrin P. C., Timony M. A., Ottaviano M., and Sliz P. (2013) Collaboration gets the most out of software. eLife 2, e01456 10.7554/eLife.01456 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the hNEIL3-GRF12 crystal structure have been deposited in the Protein Data Bank under accession code 7JL5. All other data are contained within the article.