

Abstract
The EphA2 receptor tyrosine kinase signals through two distinct mechanisms, one regulated by tyrosine phosphorylation and the other by serine/threonine phosphorylation. Serine 892 (S892) is one of the major serine/threonine phosphorylation sites in EphA2, but little is known about its regulation and function. S892 is located in the linker connecting the EphA2 kinase and SAM domains, and is part of a cluster of five phosphorylated residues that includes the well characterized S897. EphA2 can be phosphorylated on S897 by the RSK, AKT and PKA kinases to promote a non-canonical form of signaling that plays an important role in cancer malignancy. Here we show that the Protein Kinase C (PKC) family phosphorylates the EphA2 S892 motif in vitro and in cells. By using a newly developed phosphospecific antibody, we detected EphA2 S892 phosphorylation in a variety of cell lines. As expected for a PKC target site, the PKC activator 12-O-tetradecanoylphorbol-13-acetate (TPA) increases S892 phosphorylation whereas the broad-spectrum PKC inhibitor Go 6983 inhibits both basal and TPA-induced S892 phosphorylation. Besides phosphorylating S892, PKC can also increase EphA2 phosphorylation on S897 through the MEK kinase, which regulates the ERK-RSK signaling axis. We also found that S892 and S897 phosphorylation induced by PKC activation can be downregulated by ephrin ligand-induced EphA2 canonical signaling. Our data reveal that the PKC family contributes to the phosphorylation cluster in the EphA2 kinase-SAM linker, which regulates EphA2 non-canonical signaling and cancer malignancy.
Keywords: Eph receptor, receptor tyrosine kinase, serine/threonine phosphorylation, phosphorylation cluster, cancer
1. INTRODUCTION
EphA2 signals through an unusual dual mechanism. Similar to other receptor tyrosine kinases, EphA2 “canonical” signaling is induced by the binding of ephrin-A ligands to the ligand-binding domain in the receptor extracellular region, leading to EphA2 autophosphorylation on tyrosine residues and increased kinase activity [1]. A completely different, “non-canonical” signaling mechanism depends on EphA2 phosphorylation by serine/threonine kinases in the linker connecting the kinase and SAM domains [2, 3]. This linker (comprising ~20 amino acids, from T883 to E902 [4]) includes a cluster of five serine/threonine residues, all of which are highly phosphorylated (phosphosite.org) and likely act in concert to regulate EphA2 non-canonical signaling. Indeed, clusters of phosphorylated serine and threonine residues are common in disordered regions of proteins such as linkers, where different phosphosites are typically concomitantly regulated and functionally cooperate [5–7]. Multiple phosphorylation sites, for example, can achieve more robust functional responses than a single site, set a response threshold, and/or enable gradual effects.
Among the serine/threonine residues in the EphA2 kinase-SAM linker, only S897 has been extensively characterized. EphA2 can be phosphorylated on S897 by kinases of the AGC (PKA, PKG, PKC) group, including RSK, AKT and PKA [2, 8, 9]. By phosphorylating S897, these AGC kinases promote EphA2 pro-oncogenic effects such as cancer cell invasiveness and metastatic ability, cell cycle progression, cancer stem cell-like properties, and drug resistance through signaling pathways that are not well understood [3, 10, 11]. It is known that non-canonical signaling is independent of ligand binding and kinase activity [2]. The main EphA2 downstream effector involved in non-canonical signaling identified so far is the RHOG guanine nucleotide exchange factor Ephexin4 (ARHGEF16), which can promote RAC1 and PI3 kinase-AKT activation [3, 11–14].
We have previously shown that S897 phosphorylation leads to the phosphorylation of another residue in the phosphorylation cluster, S901 [9]. This is because S897 phosphorylation creates a S901 negatively charged substrate motif recognized by the constitutively active CK1 kinase [9]. However, the kinases responsible for phosphorylation of the other three residues (S892, T898 and S899) in the phosphorylation cluster of the EphA2 linker have remained unknown.
Here, we identify PKC as a major family of serine/threonine kinases that phosphorylate EphA2 on S892 and that can also indirectly increase S897 phosphorylation, thus contributing to the functionally important phosphorylation cluster in the EphA2 kinase-SAM linker. This represents a novel signaling interplay, where EphA2 serine phosphorylation may mediate activities of PKC family members in cancer cells.
2. MATERIALS AND METHODS
2.1. In vitro kinase reactions
The peptide substrates were synthesized by Kinexus (Vancouver, Canada) at >95% or > 98% purity. The recombinant protein kinases were cloned, expressed and purified using Kinexus proprietary methods and underwent routine quality control testing. The assay conditions were optimized to yield acceptable enzymatic activity and high signal-to-noise ratio. Protein kinase assays were performed in a final volume of 25 μl, including 5 μl diluted active kinase (~10–50 nM final concentration), 5 μl stock peptide substrate solution (500 μM final concentration), 10 μl kinase assay buffer, 5 μl [γ−33P]ATP (250 μM stock solution, 0.8 μCi). Assays were initiated by the addition of [γ−33P]ATP and the reaction mixture was incubated at room temperature for 30 min. The assay was terminated by spotting 10 μl of the reaction mixture onto a multiscreen phosphocellulose P81 plate, which was then washed 3 × 15 min in a 1% phosphoric acid solution. The radioactivity on the plate was measured in the presence of scintillation fluid in a Trilux scintillation counter.
2.2. Cell lines
The following cell lines were purchased from the American Type Culture Collection: PC3 prostate cancer (CRL-1435), HCC1937 triple negative breast cancer (CRL-2336), SKOV3 ovarian serous cystadenocarcinoma (HTB-77), SW626 colon adenocarcinoma (HTB-78) and H1648 lung adenocarcinoma (CRL-5882). The HOP62 lung adenocarcinoma cell line was purchased from the tumor/cell line repository of the Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute; the MEL-JUSO melanoma cell line from the DSMZ (ACC 74) and the HEK 293-AD human embryonic kidney cell line from Cell Biolabs (AD-100). The MDA-MB-231 breast cancer cell line was provided by J. Smith (Sanford Burnham Prebys Medical Discovery Institute); the BT549 breast cancer cell line by R. Maki (Sanford Burnham Prebys Medical Discovery Institute) and the PANC-1 cell line by F. Levine (Sanford Burnham Prebys Medical Discovery Institute). The MEL-JUSO and HEK293 cell lines were stably infected with the pLVX-IRES-Neo lentivirus encoding human EphA2 wild-type with an N-terminal FLAG tag [9]. In addition, HEK293 cells were transiently transfected with pLVX-IRES-Neo lentivirus encoding FLAG-tagged human EphA2 wild-type and the S892A and S897A mutants [9], FLAG-tagged human EphA1, or EGFP.
The PC3, MDA-MB-231, BT549, H2009, PANC-1 and MCF10A cell lines were authenticated by performing short tandem repeat analysis on isolated genomic DNA with the GenePrint® 10 System (Promega), and peaks were analyzed using GeneMarker HID (Softgenetics). Allele calls were searched against short tandem repeat databases [34].
PC3, HCC1937, HOP62, H1648, MEL-JUSO and BPH-1 cells were cultured in RPMI 1640 medium (ThermoFisher Scientific/Gibco 11875–093); MDA-MB-231, BT549, SW626, PANC-1, and HEK293-AD cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Corning, 10–013-CV), H2009 cells were cultured in DMEM/F12 medium (Corning, 10–092-CV) and SKOV3 cells were cultured in McCoy’s 5A medium (ThermoFisher Scientific, 16600082). MCF10A cells were cultured in DMEM/F12 medium supplemented with 5% horse serum, 100 ng/ml cholera toxin (Sigma-Aldrich), and Mammary Epithelial Cell Growth Medium SingleQuots supplements and growth factors (Lonza, CC-4136) but without including gentamycin/amphotericin. All culture media (except for the MCF10A medium) contained 10% fetal bovine serum as well as antimycotics and antibiotics (Corning, 30–004-Cl).
Cells were serum-starved for 1 hour and then treated with 1 μM Go 6983 (Selleck, S2911) for 60 min or 200 μM TPA (Cell Signaling Technology, 4174S) for 30 min. For combined treatments, cells were treated with Go 6983 for 60 min followed by TPA for an additional 30 min in the continued presence of Go 6983. Alternatively, cells were treated with TPA for 10 min followed by 0.5 μg/ml ephrin-A1 Fc (R&D Systems, 602-A1–200) for an additional 20 min in the continued presence of TPA. In some cases cells were serum-starved before the treatments for 1 hour (Fig. 3A), 2 hours (Fig. 2A and HEK293 in Fig. 6C), or overnight (Fig. 2B,C, Fig. 3B,C, and Fig. 6A).
Fig. 3. A new phosphospecific antibody recognizes EphA2 phosphorylated on S892.
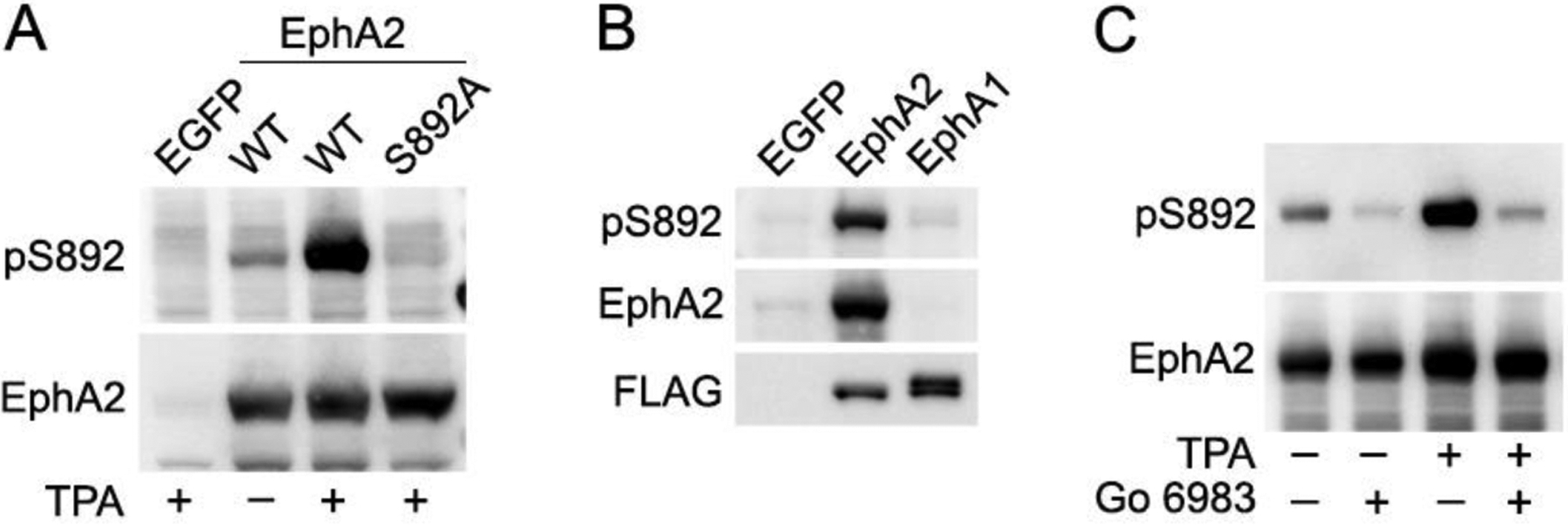
(A) Lysates from HEK293 cells transiently transfected with constructs encoding EGFP, EphA2 wild-type (WT) or the EphA2 S892A mutant were treated with the PKC activator TPA or left untreated. Lysates were probed with a newly generated antibody recognizing the phosphorylated S892 motif (pS892) and re-probed for EphA2. (B) Lysates from HEK293 cells transiently transfected with EGFP, FLAG-EphA2 or FLAG-EphA1 were probed for S892 phosphorylation and reprobed as indicated. (C) HEK293 cells transiently expressing EphA2 WT were treated with the PKC inhibitor Go 6983, TPA or both, and lysates were probed for S892 phosphorylation and reprobed for EphA2. Go 6983 inhibits both basal and TPA-induced S892 phosphorylation.
Fig. 2. PKC phosphorylates EphA2 on S892 in cells.
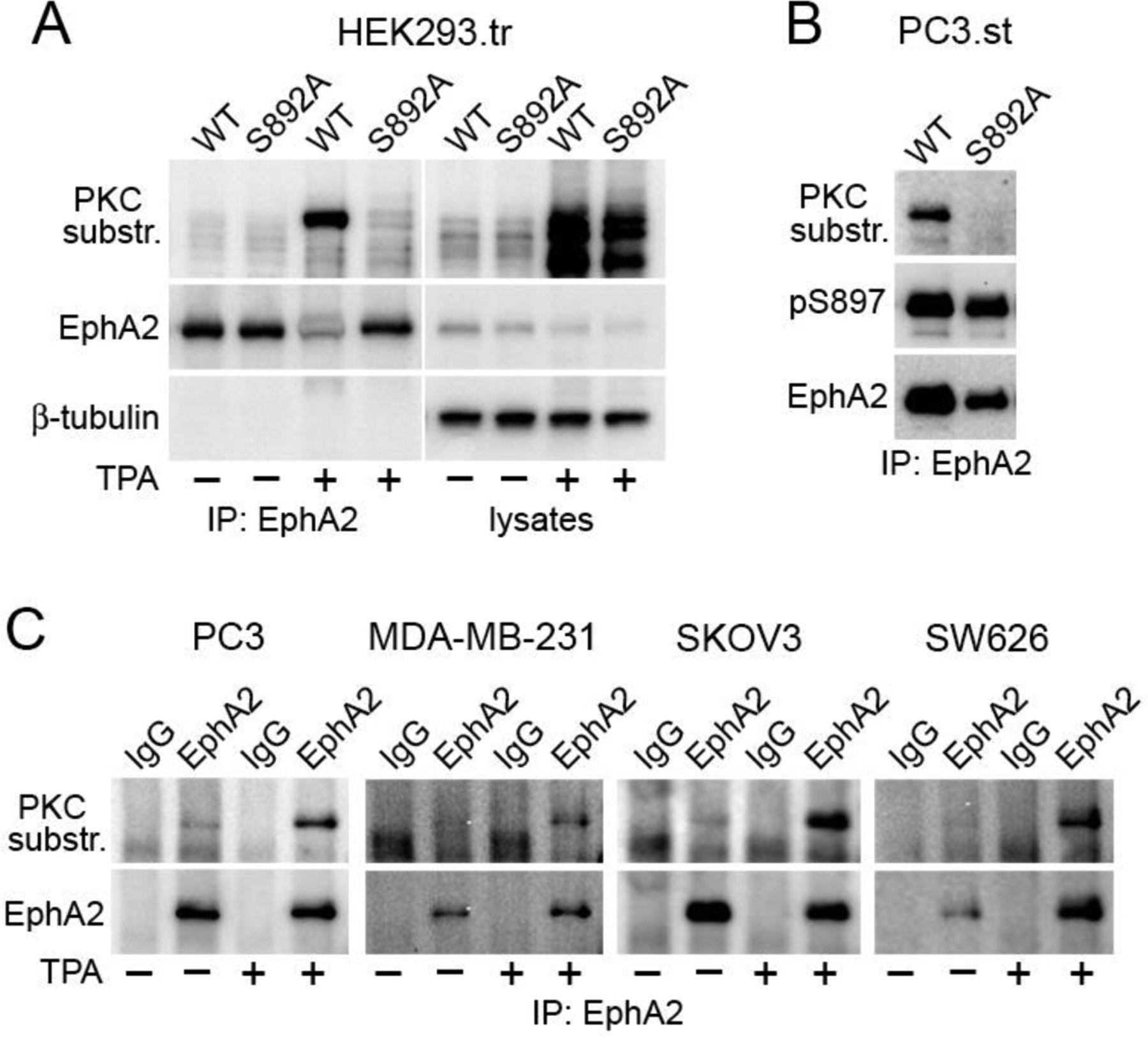
(A) HEK293 cells were transiently transfected (.tr) with FLAG-tagged EphA2 wild-type (WT) or the S892A mutant and stimulated with 200 nM TPA for 30 min (+) or left untreated (–). FLAG immunoprecipitates (IP) and lysates were probed with a phospho-(Ser) PKC substrate antibody and reprobed for EphA2 and β-tubulin. (B) FLAG immunoprecipitates from EphA2 knockdown PC3 cells stably expressing (.st) FLAG-tagged EphA2 WT or the S892A mutant were probed as indicated. (C) The indicated cancer cell lines were treated with TPA (+) or left untreated (–) and EphA2 immunoprecipitates were probed as indicated. Immunoprecipitates using non-immune IgGs serve as a control.
Fig. 6. Regulation of EphA2 S892 phosphorylation.
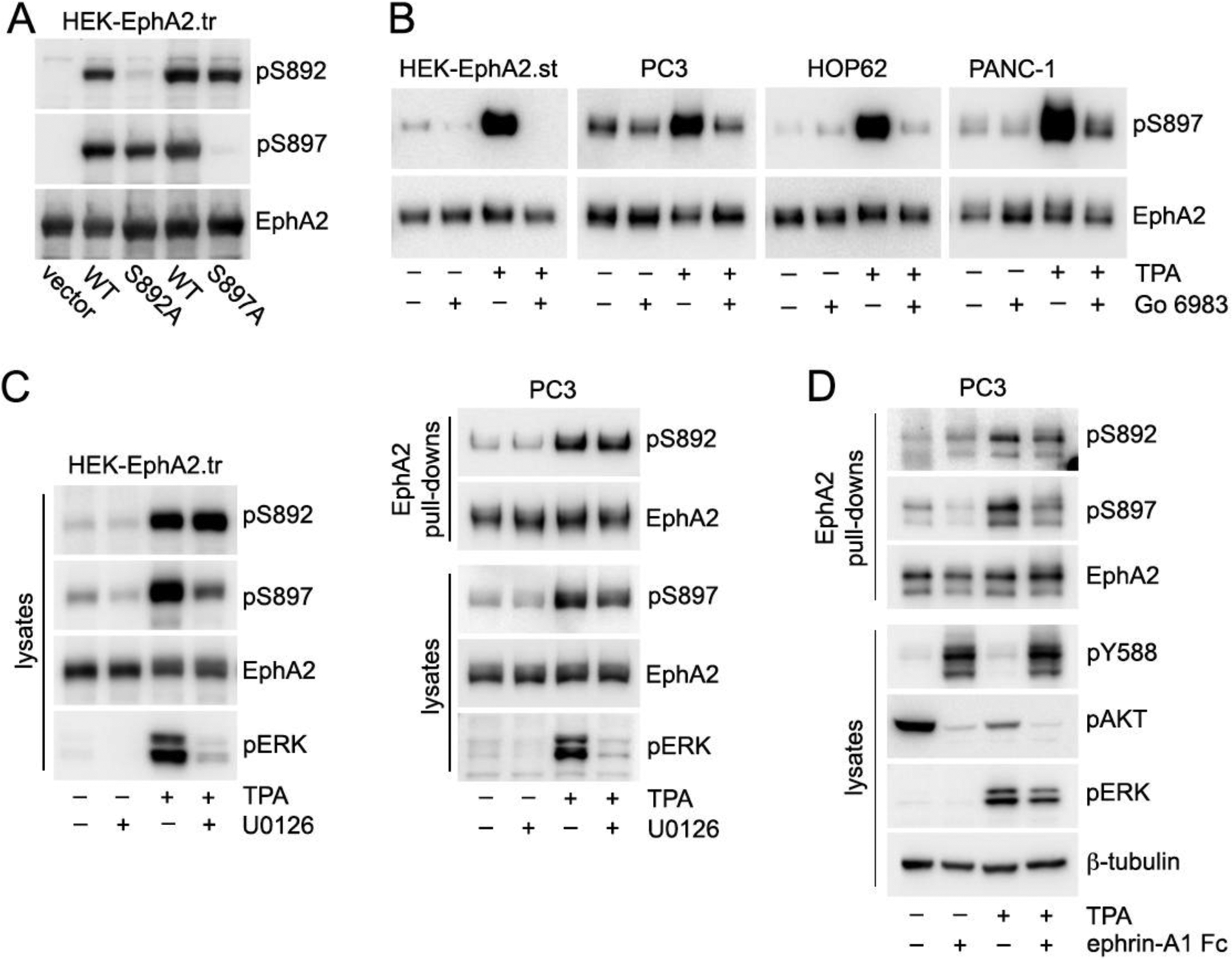
(A) HEK293 cells were transiently transfected (.tr) with constructs encoding EphA2 WT, S892A mutant or S897A mutant, or empty vector as a control. Lysates were probed with the indicated antibodies, showing that the EphA2 S897A mutant can be substantially phosphorylated on S892 and the S892A mutant can be substantially phosphorylated on S897. (B) HEK293 cells stably transfected (.st) with EphA2 WT and the indicated cancer cell lines expressing endogenous EphA2 were treated with the PKC inhibitor Go 6983 and/or with the PKC activator TPA. EphA2 was isolated by pull-down with a peptide that binds to the EphA2 ligand-binding domain. The EphA2 immunoblots for HEK-EphA2.st, HOP62 and PANC-1 cells are the same as in Fig. 5.They were stripped and then reprobed for EphA2 phosphorylation on S897. Different aliquots of the EphA2 pulldowns from PC3 cells were probed for pS897 and EphA2. TPA induces EphA2 phosphorylation on S897 and the PKC inhibitor strongly decreases the phosphorylation induced by TPA. (C) HEK293 cells transiently transfected (.tr) with EphA2 WT and PC3 cells were treated with the MEK inhibitor U0126 and/or TPA and lysates or peptide pull-downs were probed as indicated. EphA2 phosphorylation on both S892 and S897 is increased by TPA, but only the phosphorylation on S897 is inhibited by U0126. This is consistent with direct phosphorylation of S892 by PKC and indirect effects of PKC on S897 phosphorylation through the MEK-ERK-RSK pathway. (D) PC3 cells were treated with TPA and/or ephrin-A1 Fc ligand. EphA2 immunoprecipitates were probed as indicated. Ephrin-A1 Fc induces EphA2 phosphorylation on tyrosine residues such as Y588, but decreases EphA2 phosphorylation on S897 as well as S892, concomitant with inhibition of ERK and AKT phosphorylation. Notably, in PC3 cells TPA increases ERK phosphorylation but decreases AKT phosphorylation.
2.3. Immunoprecipitations
To obtain lysates for immunoprecipitations, cells were cultured until they reached ~80% confluence, rinsed once with ice cold PBS containing Ca+ and Mg+ (Lonza, 17–513F), and collected in modified RIPA buffer (1% TX-100, 0.5% sodium deoxycholate, 0.1% SDS, 2 mM EDTA in PBS, pH 7.5) with Halt Protease and Phosphatase inhibitor cocktail (ThermoFisher Scientific, 78443). Cell lysates were incubated for 5 min on ice with periodic mixing and then centrifuged for 10 min at 16,700 g at 4°C to remove insoluble material. The supernatant was precleared by incubation with Sepharose beads (Sigma-Aldrich, 4B200) for 15 min at 4°C on a rotator followed by centrifugation for 1 min at 4,000 g. Each FLAG immunoprecipitation was performed by incubating 20–25 μl anti-FLAG M2 affinity gel (Sigma-Aldrich, A2220) with cell lysates for 2 hours at 4°C on a rotator. The immunoprecipitates were washed 3 times with 1 ml modified RIPA buffer and once with PBS, and eluted by incubation at 95°C for 2 min in 25 μl SDS-containing sample buffer without β-mercaptoethanol (to avoid dissociating the chains of the FLAG antibody not directly linked to the beads). Following centrifugation for 30 sec at 4,000 g, the supernatant was collected and β-mercaptoethanol was added to a concentration of 2.5%.
For immunoprecipitations with a mouse monoclonal anti-EphA2 antibody (EMD Millipore, clone D7, 05-480), each immunoprecipitation was performed using 4–6 μg of antibody bound to 20–30 μl of GammaBind Plus Sepharose (GE Healthcare, 17-0886-02) for 2 hours at 4°C on a rotator. Immunoprecipitates were washed in the same way as the FLAG immunoprecipitates and eluted in SDS-containing sample buffer or Bolt LDS sample buffer (Life Technologies, B0007) containing 2.5% β-mercaptoethanol.
2.4. Peptide pull-downs
To obtain lysates for the peptide pull-downs, cells were cultured until they reached ~80% confluence, rinsed once with ice cold PBS containing Ca+ and Mg+ (Lonza, 17–513F), and collected in cold PBS containing 0.5% TX-100 with Halt Protease and Phosphatase inhibitor cocktail (Fisher Scientific, 78443). Cells were incubated for 10 min on ice with periodic mixing and then centrifuged at 16,700 g for 5 min at 4°C to remove insoluble material. For peptide pull-downs, we used the biotinylated βA-WLA-YRPKam-bio (20) peptide (97.5% purity, GenScript custom peptide), which binds to the EphA2 ligand-binding domain with high affinity [35]. Five μg peptide diluted in 100 μl PBS were bound to 25 μl streptavidin agarose (Thermo Fisher, 20349) by placing the streptavidin agarose beads in a spin column (G-Biosciences, 786–718) and running the peptide solution through the column twice by centrifuging at 500 g for 30 sec. The spin columns were washed once with PBS. Cell lysates were then run through the spin columns 3 times by centrifuging at 500 g for 30 sec each time. The spin columns were washed 3 times with 0.3 ml ice cold 0.5% TX-100 in PBS by centrifuging at 2,000 g for 30 sec each time. Bound proteins were eluted with 30 to 70 μl SDS-containing sample buffer or Bolt LSD sample buffer (Life Technologies, B0007) containing 2.5% β-mercaptoethanol by centrifuging the columns twice for 30 sec at 500 g followed by 15 sec at maximum speed each time.
2.5. Immunoblotting
To obtain lysates to be used only for immunoblotting, cells were rinsed once with ice cold PBS containing Ca+ and Mg+ (Lonza, 17–513F), and collected in SDS-containing sample buffer with 2.5% β-mercaptoethanol. Aliquots of lysates collected for immunoprecipitations or for pull-downs were also used for immunoblotting, after addition of sample buffer. Lysates and immunoprecipitated or pulled down samples were heated at 95° for 2 min, briefly sonicated (in the case of cell lysates) and run on either Novex Wedge Well 4–20% Tris-Glycine gels (Invitrogen, XP04205) or Bolt 4–12% Bis-Tris Plus gels (Invitrogen, NW04125). After semi-dry transfer using PVDF transfer packs (Bio-Rad, 1704272) with a Trans-Blot Turbo Transfer System (Bio-Rad Laboratories), the PVDF membranes were blocked for 1 hour at room temperature with 5% bovine serum albumin in 0.1% Tween-20 in TBS (150 mM NaCl, 50 mM TrisHCl, pH 7.5) and then incubated in the cold overnight in blocking buffer containing primary antibodies from Cell Signaling Technology recognizing EphA2 (#6997; 1:1,000 dilution), EphA2 pY588 (#12677; 1:2,000 dilution), EphA2 pS897 (#6347; 1:1,000 dilution), β-tubulin (#2128; 1:1,000 dilution), the FLAG tag (#8146; 1:1,000 dilution), phospho p44/42 MAPK (ERK1/2) (#9101; 1:1,000 dilution), phospho AKT S473 (#4060; 1:2,000 dilution) or phospho-(Ser) PKC substrate antibody (#2261; 1:1,000 dilution).
Antibodies recognizing EphA2 phosphorylated on S901 were generated in our laboratory, validated as described [9], and used at a concentration of 1 μg/ml. Antibodies recognizing EphA2 phosphorylated on S892 were generated using a similar protocol. Briefly, the peptide KDFDPRVpSIRLPST, corresponding to EphA2 residues 886–898 with phosphorylated S892 and an added N-terminal lysine, was coupled to BSA using glutaraldehyde and used to immunize rabbits. Antibodies recognizing the non-phosphorylated S892 motif were then removed from the immune serum by affinity purification using the non-phosphorylated peptide (KDFDPRVSIRLPST) coupled to an Affi-Gel 10 column (1536046; Bio-Rad Laboratories). The absorbed serum was then affinity purified using the phosphorylated peptide (KDFDPRVpSIRLPST) coupled to an Affi-Gel 10 column. The purified antibodies were used for immunoblotting at a concentration of 1 μg/ml. They specifically recognize EphA2 phosphorylated on S892, since they label EphA2 wild-type overexpressed by transient transfection in HEK293 cells, but not the EphA2 S892A mutant, which lacks the S892 phosphorylation site (Figs. 3A and 6A). The pS892 antibodies also do not recognize the closely related EphA1 (Fig. 3B). These data confirm the desired specificity of the antibodies for the phosphorylated S892 motif. Requests of small amounts of the pS892 antibodies should be directed to the corresponding author. Antibody transfers will be made with no more restrictive terms than required by the Sanford Burnham Prebys Medical Discovery Institute.
After washing, the membranes were incubated at room temperature for 1 hour with a horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibody (Invitrogen, A16110; 1:3,000 dilution) followed by ECL (GE Healthcare, RPN2106) or SuperSignal West Dura (ThermoFisher Scientific, 34076) chemiluminescence detection. The chemiluminescence signal was captured using the ChemiDoc Touch Imaging System (Bio-Rad), quantified using Image Lab (Bio-Rad), and analyzed using Prism software (GraphPad).
2.6. Heat map and hierarchical clustering
Each phosphorylation signal was normalized to the level of EphA2 in a cell line. For each phosphosite, the signal was then normalized to the signal in the cell line with highest signal. Heatmap and hierarchical clustering were built for the normalized phosphorylation signals using the R pheatmap package with default parameters. Bioinformatics analysis was performed by the Sanford Burnham Prebys Bioinformatics Core
3. RESULTS
3.1. The PKC family phosphorylates the EphA2 S892 motif
Seven PKC family members are among the top eight kinases predicted by the Kinexus PhosphoNET online resource (www.phosphonet.ca) to phosphorylate EphA2 on S892 (Fig. 1A). These include all conventional and novel PKCs while the two atypical PKCs are lower in the list but still among the top forty-five (Figs. 1A and Table S1). In contrast, none of the PKCs are among the top 40 kinases predicted to phosphorylate the other four residues in the EphA2 kinase-SAM linker (S897, T898, S899 and S901). Indeed, the RVS892IR motif in the EphA2 linker conforms to the PKC substrate consensus motif (R/K)-X-pS-Hyd-(R/K), where X indicates any residue and Hyd indicates a hydrophobic residue) [15, 16] (phosphonet.ca).
Fig. 1. PKC family kinases phosphorylate the EphA2 S892 motif in vitro.
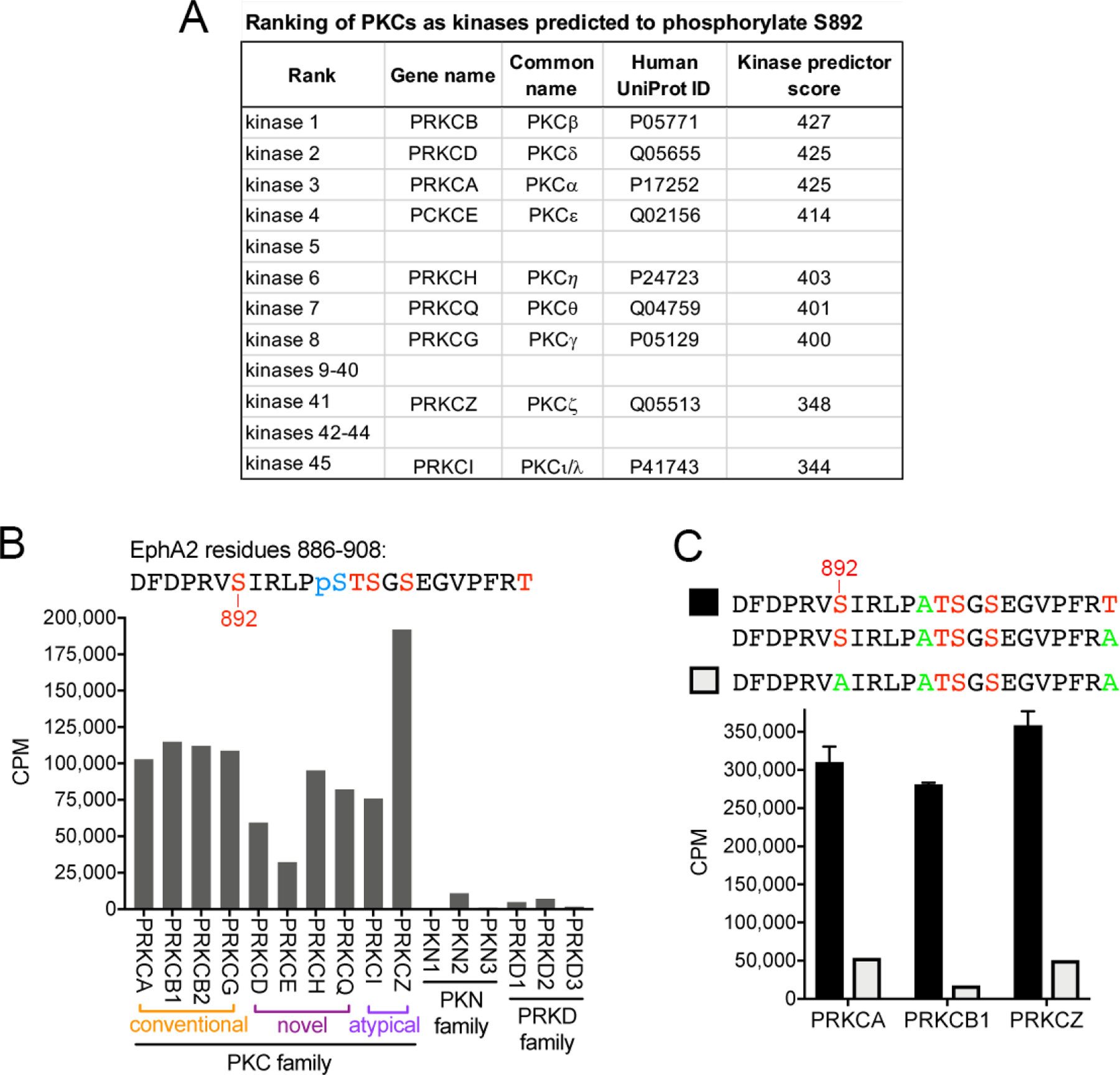
(A) Ranking of the nine PKC family members among the kinases predicted to phosphorylate the EphA2 S892 motif, based on the PhosphoNET kinase predictor score. (B) In vitro kinase reactions with radioactive ATP show that most PKC kinases (including PKCβ isoforms 1 and 2) phosphorylate at high levels a peptide substrate in which S892, T898, S899, S901 and T908 are potential phosphorylation sites (indicated in red). Phosphorylated S897 (pS897, blue) was incorporated during peptide synthesis, preventing incorporation of radioactive phosphate at this site during the in vitro kinase reaction. Other closely related kinases (PKN and PRKD families) do not or only minimally phosphorylate the peptide. CPM, counts per minute. (C) Three representative members of the PKC family phosphorylate peptide substrates containing S892, whereas phosphorylation is greatly reduced when S892 is mutated to Ala (green), which cannot be phosphorylated. This demonstrates that PKC kinases preferentially phosphorylate the S892 motif in the EphA2 kinase-SAM linker.
To experimentally verify that PKC family kinases can phosphorylate the S892 motif, in vitro kinase reactions were carried out with purified kinases and a peptide substrate corresponding to residues 886–908 in the EphA2 kinase-SAM linker. This peptide substrate contained already phosphorylated S897, preventing phosphate incorporation at this position. All PKC family members readily phosphorylated the peptide (Fig. 1B; Table S1). Different PKC family members catalyzed different levels of radioactive phosphate incorporation, which may reflect the suitability of the peptide as a substrate for a particular PKC family member and/or other factors, such as the intrinsic activity of each recombinant kinase. Interestingly the 3 PKN kinases, which represent the family most closely related to the PKC family [17], caused little or no phosphorylation of the peptide substrate (Fig. 1B). We also examined the 3 members of the PRKD family because like conventional and novel PKCs they are also regulated by diacylglycerol (Table S1), and have thus been considered similar to PKCs (with PRDK1 also known as PKCμ and PRDK3 also known as PKCν) [18, 19]. This revealed that PRKD kinases also essentially do not phosphorylate the peptide substrate (Fig. 1B). These results demonstrate the ability of the in vitro kinase assay to discriminate the substrate specificity of PKCs and related kinases.
To determine whether PKCs indeed phosphorylate S892 (rather than T898, S899 or S901), we compared phosphorylation of similar peptides either containing S892 or with S892 replaced by Ala to prevent S892 phosphorylation. These peptide substrates also contained alanine instead of S897. Replacement of S892 with greatly reduced peptide phosphorylation by PKCα, PKCβ and PKCζ used as representative PKC family members that highly phosphorylate the S892 motif (Fig. 1C), indicating that S892 is the main residue phosphorylated by the PKC family. These data show that the S892 motif is specifically phosphorylated in vitro by multiple PKC family members. In addition, the phosphorylation can occur regardless of whether S897 is phosphorylated or replaced by the non-phosphorylatable alanine (Fig. 1B,C).
3.2. PKC kinases phosphorylate EphA2 on S892 in cells
As a first step to determine whether EphA2 S892 is phosphorylated by PKC in cells, we treated HEK293 cells engineered to transiently express EphA2 with the phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA), which promotes PKC activation by mimicking the action of diacylglycerol, which is the natural activator of conventional and novel PKCs [20] (Table S1). A strong signal was obtained by labeling immunoprecipitated EphA2 with a commercial “phospho-(Ser) PKC substrate” antibody that recognizes the PKC substrate motif (R/K)-X-pS-Hyd-(R/K) (Fig. 2A). The phospho PKC substrate antibody did not label the EphA2 S892A mutant (Fig. 2A), and thus specifically recognizes the phosphorylated S892 motif in EphA2. Thus, EphA2 S892 phosphorylation in cells is enhanced by the PKC activator TPA and recognized by a phospho PKC substrate antibody. These data support a role for PKC family members in the phosphorylation of EphA2 on S892 in cells.
The phospho PKC substrate antibody also labels immunoprecipitated EphA2 stably expressed in engineered PC3 prostate cancer cells not treated with TPA and does not label the EphA2 S892A mutant (Fig. 2B). Furthermore, the antibody recognizes endogenous EphA2 immunoprecipitated from several cancer cell lines treated with TPA, including PC3 prostate cancer, MDA-MB-231 breast cancer, SKOV3 ovarian cancer and SW626 colon cancer cells (Fig. 2C). These data demonstrate that PKC kinases can phosphorylate EphA2 in cells. However, the phospho PKC substrate antibody recognizes a complex motif with multiple amino acids at each position, which might explain why it yields a very low to undetectable signal for S892 phosphorylation of endogenous EphA2 in the absence of TPA treatment.
We therefore generated a new antibody specifically recognizing the phosphorylated S892 motif. This phosphospecific antibody detects S892 phosphorylation in lysates of HEK293 cells transiently transfected with EphA2 wild-type and the signal is increased by TPA treatment for EphA2 wild-type but not the S892A mutant (Fig. 3A). We also verified that the antibody is specific for EphA2 and does not recognize EphA1, the only other Eph receptor that also contains the cluster of phosphorylation sites in the kinase-SAM linker, including T901 corresponding to EphA2 S892 (Fig. 3B). The specificity of the antibody is consistent with the differences in the phosphorylated motifs of the two receptors, which are DPRVpS892IRLP in EphA2 and DPRMpT901LRLP in EphA1(residues that are different in the two receptors are shown in bold). Thus, the new phosphospecific antibody we generated represents a valuable tool to detect EphA2 phosphorylation on S892 in cells.
Importantly, strong inhibition of both basal and TPA-induced S892 phosphorylation by the broad-spectrum PKC inhibitor Go 6983 (Fig. 3C) further supports the notion that PKC is the main kinase family that phosphorylates EphA2 on S892 in HEK293 cells.
3.3. EphA2 is phosphorylated on S892 in a variety of cancer cell lines
EphA2 phosphorylation on S892 has been detected by mass spectrometry in a wide variety of cancer cell lines, including many breast, gastric and lung cancer cell lines [21–24] (phosphosite.org), although in most cases the level of S892 phosphorylation was not determined. Immunoblotting analysis of EphA2 from a number of cell lines from different cancer types revealed widespread and variable EphA2 S892 phosphorylation (Fig. 4). Highest EphA2 phosphorylation on S892 was detected in the PC3 androgen-independent prostate cancer cell line, the HOP62 lung adenocarcinoma cell line, the PANC-1 pancreatic cancer cell line, and the MEL-JUSO melanoma cell line stably engineered to express EphA2 (Fig. 4B). We also detected substantial EphA2 S892 phosphorylation in the MDA-MB-231 and BT549 triple negative breast cancer cell lines, the NCI-H1648 lung adenocarcinoma cell line, and the AsPC-1 pancreatic cancer cell line. Phosphorylation on S892 appears to correlate less closely with S897 phosphorylation compared to S901 phosphorylation (Fig. 4A), suggesting differences in the regulation of S892 phosphorylation versus S897 and S901 phosphorylation.
Fig. 4. EphA2 is phosphorylated on S892 in different cancer cell lines.
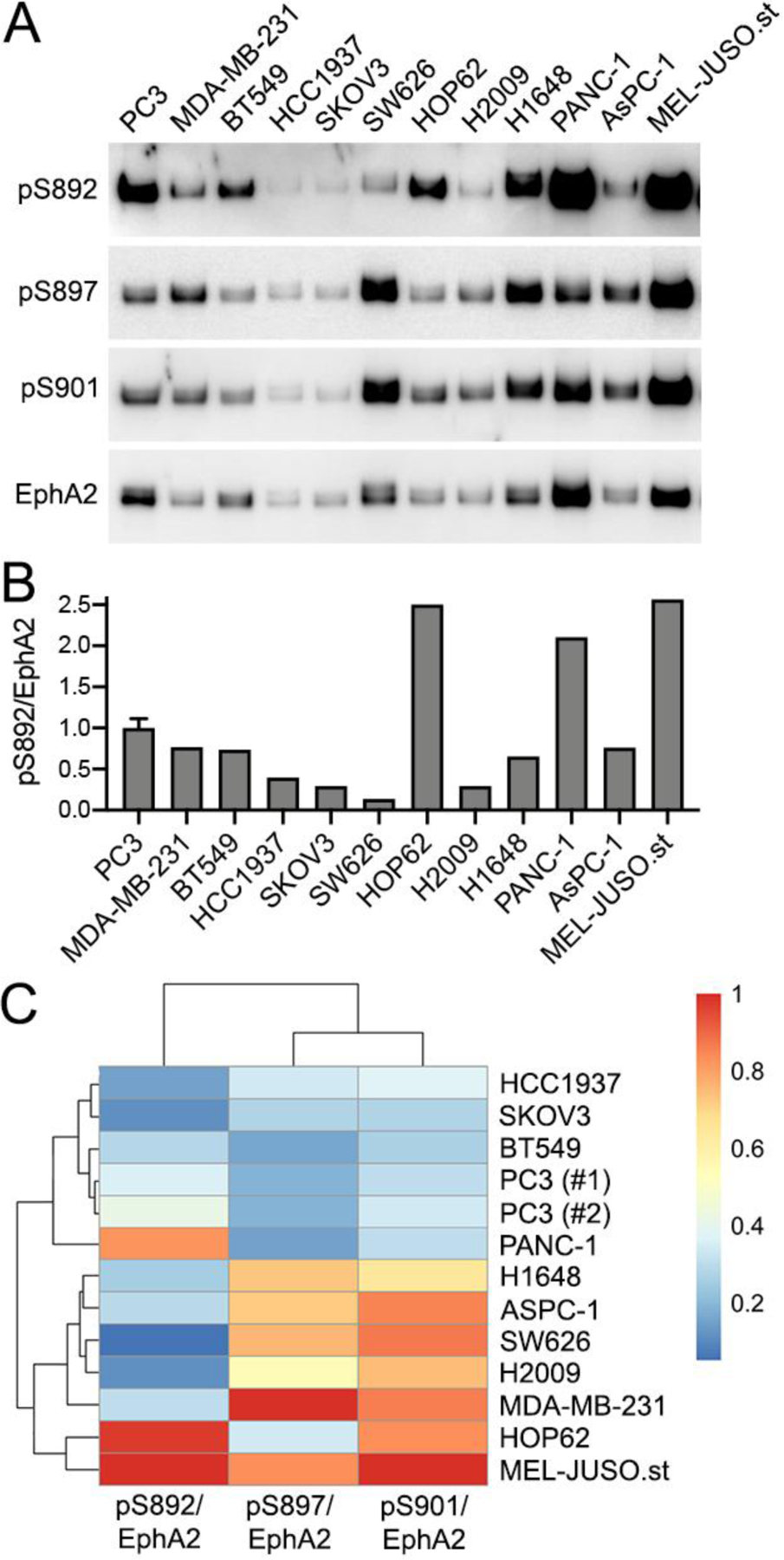
(A) EphA2 was pulled down with a peptide that binds to the EphA2 ligand-binding domain. Each pull-down was divided into four aliquots that were probed in separate immunoblots for EphA2 phosphorylation on S892, S897 and S901 as well as for total EphA2. (B) Quantification (from the blots in panel A) of the levels of S892 phosphorylation relative to the amount of pulled down EphA2. For PC3 cells, the histogram shows the mean and SD of the signals from two lanes from the same blot (only one is shown). The cell lines analyzed are: PC3 androgen-independent prostate cancer; MDA-MB-231, BT549 and HCC1937 triple negative breast cancer; SKOV3, ovarian serous cystadenocarcinoma; SW626, colon adenocarcinoma; HOP62, H2009, H1648 lung adenocarcinoma; PANC-1 and AsPC-1 pancreatic cancer; and MEL-JUSO.st, a melanoma cell line engineered to stably express EphA2 wild-type. (C) Heat map and hierarchical clustering for the phosphorylation signals shown in panel A, normalized to the total amount of EphA2. pS897 and pS901 cluster together and pS892 is more divergent (top). The cell lines are subdivided in two groups with low or high kinase-SAM linker phosphorylation. PC3 (#1) and PC3(#2) are the signals from two lanes in the same blot. The color key for the normalized phosphorylation signals is also shown.
3.4. The PKC family plays an important role in EphA2 S892 phosphorylation in cells
We found that S892 phosphorylation of endogenous EphA2 is inhibited by the broad spectrum PKC inhibitor Go 6983 in most cancer cell lines (Fig. 5A) and some non-transformed cell lines (Fig. 5B). This implies that one or more of the kinases targeted by Go 6983 (including conventional PKCs, PKCδ and PKCζ; Table S1) are responsible for physiological and pathological EphA2 phosphorylation on S892. Furthermore, in all the cell lines examined, TPA treatment dramatically increased EphA2 phosphorylation on S892 and Go 6983 strongly inhibited the TPA-induced phosphorylation (Fig. 5A,B). This suggests that PKC family members that are activated by TPA and inhibited by Go 6983 (which includes PKCα, β, γ and δ Table S1) can phosphorylate EphA2 on S892 in the cell lines examined. In non-transformed cell lines, we detected low EphA2 phosphorylation on S892 in BPH-1 benign prostatic hyperplasia epithelial cells and HEK293 human embryonic kidney cells and slightly higher S892 phosphorylation the MCF10 mammary epithelial cell line (Fig. 5B).
Fig. 5. PKC inhibition decreases EphA2 phosphorylation on S892.
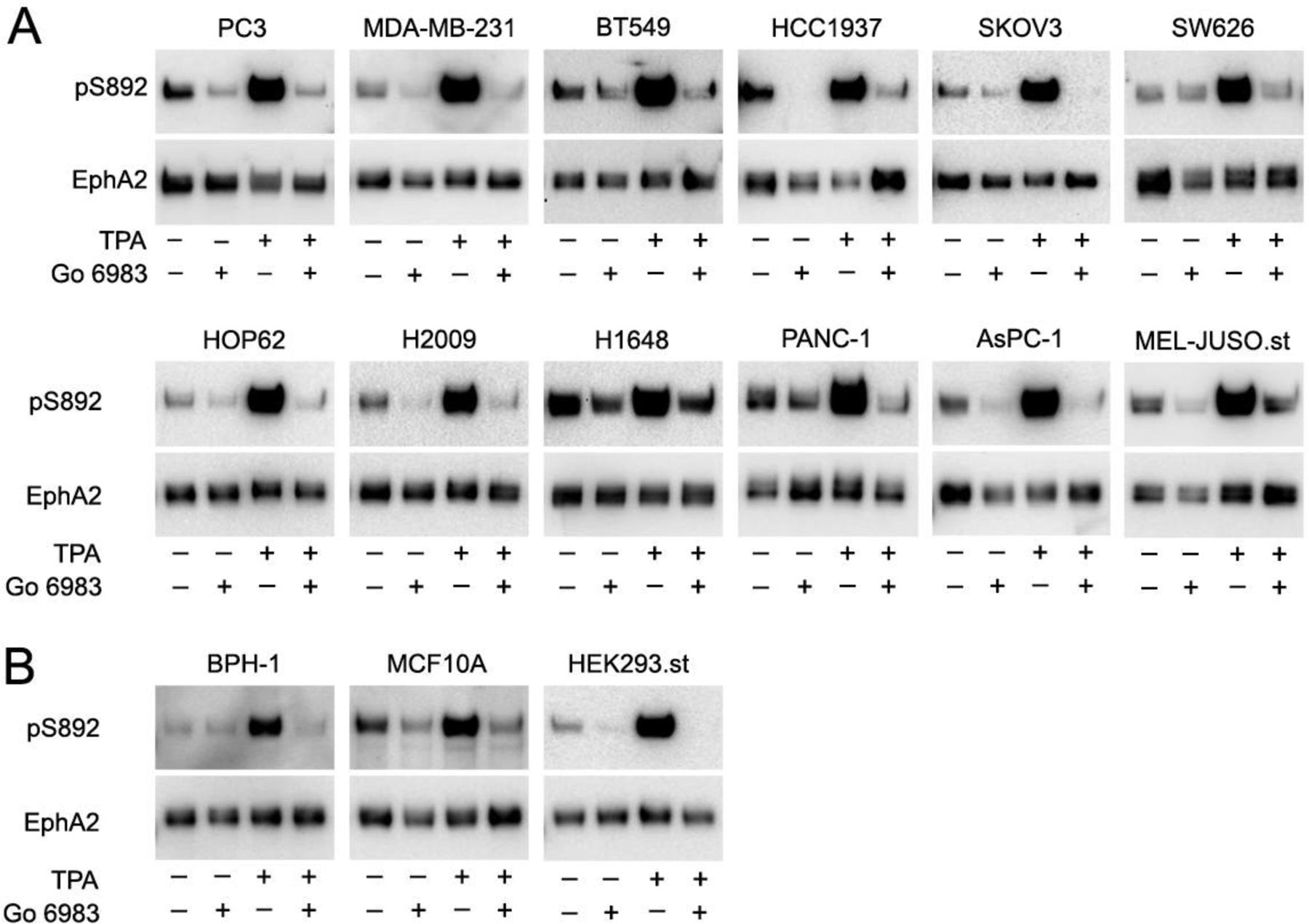
(A) The indicated cancer cells were treated with the PKC inhibitor Go 6983 and/or with the PKC activator TPA. EphA2 was pulled down with a peptide that binds to the EphA2 ligand-binding domain. Each pulldown was divided into two aliquots that were probed in separate immunoblots for EphA2 phosphorylation on S892 and for total EphA2. The cancer cell lines analyzed are the same as in Fig. 4. (B) Analysis as in panel A, using non-transformed cell lines from prostate (BPH-1) and mammary (MCF10A) epithelium as well as the HEK293 embryonic kidney cell line stably expressing EphA2. The exposures of the pS892 blots were chosen in order to obtain a similar (maximal) signal in the TPA-stimulated lanes.
3.5. Regulation of EphA2 S892 and S897 phosphorylation
We have previously shown that once S897 becomes phosphorylated, and thus negatively charged, the nearby S901 is phosphorylated by the acidophilic kinase CK1 [9]. In contrast, S892 can be readily phosphorylated by PKC in in vitro kinase reactions with a peptide substrate in which S897 is mutated to Ala and therefore not phosphorylated (Fig. 1C). Consistent with this, S892 can be readily phosphorylated in the EphA2 S897A mutant expressed in HEK293 cells (Figs. 2B and 6A). These data show that S892 phosphorylation is regulated differently from S901 phosphorylation. In addition, as we have previously shown [9], S897 can be phosphorylated in the EphA2 S892A mutant (Fig. 6A). Thus, the S892 and S897 residues can be independently phosphorylated.
Nevertheless, we found that TPA treatment can increase EphA2 phosphorylation on not only S892 but also S897 (Fig. 6B). The increase in S897 phosphorylation induced by TPA appears to depend on PKC, since it is inhibited by the Go 6983 inhibitor (Fig. 6B). However, our data suggest that PKC does not directly phosphorylate S897 because S897 phosphorylation induced by TPA is inhibited by the U0126 kinase inhibitor, which targets the ERK upstream kinase MEK (Fig. 6C). This is consistent with the known ability of PKC to activate ERK [25–27] and of the ERK downstream kinase RSK to phosphorylate EphA2 on S897 [3]. In contrast, EphA2 phosphorylation on S892 is not detectably affected by the MEK inhibitor, consistent with direct phosphorylation of S892 by PKC (Fig. 6C). These data suggest that PKC can regulate the phosphorylation of not only S892 but also S897, and thus presumably S901, in the EphA2 kinase-SAM linker.
It is known that the ephrin-A1 Fc ligand can downregulate EphA2 phosphorylation on S897 [2, 9]. This may be an effect of EphA2 kinase-dependent signaling, which can inhibit the AKT and ERK kinases [2, 9, 28, 29]. We found that ephrin-A1 Fc stimulation of PC3 cells treated with TPA causes a marked decrease of not only S897 phosphorylation but also S892 phosphorylation (Fig. 6D). Thus, EphA2 canonical signaling can inhibit phosphorylation of multiple residues in the EphA2 kinase-SAM linker. It will be interesting to investigate whether EphA2 activation induced by ephrin-A1 Fc negatively regulates S892 phosphorylation by inhibiting PKC or by activating a phosphatase.
4. CONCLUSION
We show here that S892 is a major EphA2 phosphorylation site, which can be regulated by PKC in cancer cells as well as in non-transformed cells, although EphA2 phosphorylated on S892 is likely more abundant in cancer cells due to their higher EphA2 expression. To our knowledge, this is the first report demonstrating phosphorylation of an Eph receptor by PKC. This regulation likely extends to EphA1, in which the RMT901LR motif (corresponding to the RVS892IR motif in EphA2) also conforms to a PKC substrate motif [15, 16] (phosphonet.ca). This PKC motif, and the other sites in the phosphorylation cluster of the EphA2 kinase-SAM linker, are not conserved in other Eph receptors. This suggests a distinctive PKC-dependent regulation of EphA2, and possibly EphA1, among the Eph receptors.
The effects of the Go 6983 inhibitor and TPA suggest that conventional PKCs, and possibly the novel PKCδ, can phosphorylate EphA2 on S892. Studies with additional more selective inhibitors, and knockdown or inactivation of individual PKC genes, will be needed to establish the role of specific PKC family members in EphA2 kinase-SAM linker phosphorylation in cells. It will also be important to determine how physiological stimuli regulate EphA2 phosphorylation by PKC. Available mass spectrometry data show that the activities of the EGF and MET receptor tyrosine kinases can affect EphA2 phosphorylation on S892 [22, 24], consistent with the known ability of these receptors to activate PKC [25, 30, 31].
We previously reported that phosphorylation of S901, another residue in the EphA2 kinase SAM linker, occurs coordinately with S897 phosphorylation [9]. In contrast, our data show that EphA2 phosphorylation on S892 can be regulated independently of S897 phosphorylation. Nevertheless, PKC activated by TPA increases the phosphorylation of both S892 and S897, suggesting that in some cellular context PKC may promote phosphorylation of the EphA2 kinase-SAM linker on multiple residues. In addition, depending on the cellular context and the serine/threonine kinases involved, non-canonical signaling (involving phosphorylation of the kinase-SAM linker) could occur concomitantly with canonical signaling (involving ligand-induced EphA2 activation and kinase activity) [9].
PKC-dependent phosphorylation of receptor tyrosine kinases belonging to other families has been shown to promote receptor internalization and degradation [32]. Phosphorylation by PKC can also inhibit EGF receptor activation through a negative feedback mechanism [33]. Additional studies are needed to establish the functional significance of the multiple phosphorylation sites in the EphA2 kinase-SAM linker. It is well established that EphA2 phosphorylation on S897 promotes the malignant properties of cancer cells [3, 10, 11], and that phosphorylation sites that are part of a cluster often reinforce each other’s function [5]. Thus, PKC and other serine/threonine kinases that phosphorylate residues in the EphA2 kinase-SAM linker may function together to promote EphA2 non-canonical signaling ability. If this is the case, S892 phosphorylation may be one of multiple phosphorylation events that control the structure and function of the EphA2 cytoplasmic region, and its functional effects may be only mild and hard to discern.
The phorbol ester TPA, which induces prolonged PKC activation, was initially defined as a tumor promoter [20]. However, the PKC family has complex roles in tumorigenesis and different PKCs have been shown to promote or inhibit cancer malignancy, depending on cellular context and the specific family member involved. It will be important in future studies to determine the role of EphA2 phosphorylation in the cellular activities of PKC. Nevertheless, uncovering the ability of PKC to phosphorylate EphA2 on S892 advances our knowledge of the complex EphA2 signaling mechanisms and their regulation by cellular signaling networks.
Supplementary Material
ACKNOWLEDGMENTS
We thank A. Conde-Perez for generating the MEL-JUSO cell line stably expressing FLAG-EphA2; J. Smith, R. Maki and F. Levine for providing cell lines, J. Yin for generating the heat map and hierarchical clustering, M. Gomez-Soler and M. Matsumoto for comments on the manuscript.
FUNDING
This work was supported by NIH grants R01GM131374 and institutional funding (to EBP) and NCI Cancer Center Support Grant P30CA030199, which funded SBP Core Facilities and a pilot project.
ABBREVIATIONS
- AGC, PKA, PKG, PKC
- MAPK
mitogen-activated protein kinase
- PDZ
postsynaptic density protein (PSD95), Drosophila disc large tumor suppressor (Dlg1), and zonula occludens-1 protein (ZO-1)
- PI3 kinase
phosphatidylinositol 3-kinase
- PKA
protein kinase A
- PKC
protein kinase C
- SAM
sterile-α-motif
- TPA
12-O-tetradecanoylphorbol-13-acetate
Footnotes
CONFLICT OF INTEREST
The authors declare no competing interests.
REFERENCES
- [1].Pasquale EB, Eph receptors and ephrins in cancer: bidirectional signalling and beyond, Nat Rev Cancer 10 (2010) 165–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, Basilion JP, Sedor J, Wu J, Danielpour D, Sloan AE, Cohen ML, Wang B, EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt, Cancer Cell 16 (2009) 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhou Y, Sakurai H, Emerging and Diverse Functions of the EphA2 Noncanonical Pathway in Cancer Progression, Biol Pharm Bull 40 (2017) 1616–1624. [DOI] [PubMed] [Google Scholar]
- [4].Seiradake E, Harlos K, Sutton G, Aricescu AR, Jones EY, An extracellular steric seeding mechanism for Eph-ephrin signaling platform assembly, Nat Struct Mol Biol 17 (2010) 398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schweiger R, Linial M, Cooperativity within proximal phosphorylation sites is revealed from large-scale proteomics data, Biol Direct 5 (2010) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Iakoucheva LM, Radivojac P, Brown CJ, O’Connor TR, Sikes JG, Obradovic Z, Dunker AK, The importance of intrinsic disorder for protein phosphorylation, Nucleic Acids Res 32 (2004) 1037–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Landry CR, Levy ED, Michnick SW, Weak functional constraints on phosphoproteomes, Trends Genet 25 (2009) 193–7. [DOI] [PubMed] [Google Scholar]
- [8].Zhou Y, Yamada N, Tanaka T, Hori T, Yokoyama S, Hayakawa Y, Yano S, Fukuoka J, Koizumi K, Saiki I, Sakurai H, Crucial roles of RSK in cell motility by catalysing serine phosphorylation of EphA2, Nat Commun 6 (2015) 7679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Barquilla A, Lamberto I, Noberini R, Heynen-Genel S, Brill LM, Pasquale EB, Protein kinase A can block EphA2 receptor-mediated cell repulsion by increasing EphA2 S897 phosphorylation, Mol Biol Cell 27 (2016) 2757–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Barquilla A, Pasquale EB, Eph receptors and ephrins: therapeutic opportunities, Annu Rev Pharmacol Toxicol 55 (2015) 465–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kaibori Y, Saito Y, Nakayama Y, EphA2 phosphorylation at Ser897 by the Cdk1/MEK/ERK/RSK pathway regulates M-phase progression via maintenance of cortical rigidity, FASEB J 33 (2019) 5334–5349. [DOI] [PubMed] [Google Scholar]
- [12].Hiramoto-Yamaki N, Takeuchi S, Ueda S, Harada K, Fujimoto S, Negishi M, Katoh H, Ephexin4 and EphA2 mediate cell migration through a RhoG-dependent mechanism, J Cell Biol 190 (2010) 461–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Harada K, Hiramoto-Yamaki N, Negishi M, Katoh H, Ephexin4 and EphA2 mediate resistance to anoikis through RhoG and phosphatidylinositol 3-kinase, Exp Cell Res 317 (2011) 1701–13. [DOI] [PubMed] [Google Scholar]
- [14].Kawai H, Kobayashi M, Hiramoto-Yamaki N, Harada K, Negishi M, Katoh H, Ephexin4-mediated promotion of cell migration and anoikis resistance is regulated by serine 897 phosphorylation of EphA2, FEBS Open Bio 3 (2013) 78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pearson RB, Kemp BE, Protein kinase phosphorylation site sequences and consensus specificity motifs: tabulations, Methods Enzymol 200 (1991) 62–81. [DOI] [PubMed] [Google Scholar]
- [16].Nishikawa K, Toker A, Johannes FJ, Songyang Z, Cantley LC, Determination of the specific substrate sequence motifs of protein kinase C isozymes, J Biol Chem 272 (1997) 952–60. [DOI] [PubMed] [Google Scholar]
- [17].Mellor H, Parker PJ, The extended protein kinase C superfamily, Biochem J 332 (1998) 281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Griner EM, Kazanietz MG, Protein kinase C and other diacylglycerol effectors in cancer, Nat Rev Cancer 7 (2007) 281–94. [DOI] [PubMed] [Google Scholar]
- [19].Antal CE, Newton AC, Tuning the signalling output of protein kinase C, Biochem Soc Trans 42 (2014) 1477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Isakov N, Protein kinase C (PKC) isoforms in cancer, tumor promotion and tumor suppression, Semin Cancer Biol 48 (2018) 36–52. [DOI] [PubMed] [Google Scholar]
- [21].Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP, A quantitative atlas of mitotic phosphorylation, Proc Natl Acad Sci U S A 105 (2008) 10762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Moritz A, Li Y, Guo A, Villen J, Wang Y, MacNeill J, Kornhauser J, Sprott K, Zhou J, Possemato A, Ren JM, Hornbeck P, Cantley LC, Gygi SP, Rush J, Comb MJ, Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases, Sci Signal 3 (2010) ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Klammer M, Kaminski M, Zedler A, Oppermann F, Blencke S, Marx S, Muller S, Tebbe A, Godl K, Schaab C, Phosphosignature predicts dasatinib response in non-small cell lung cancer, Mol Cell Proteomics 11 (2012) 651–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Huang H, Haar Petersen M, Ibanez-Vea M, Lassen PS, Larsen MR, Palmisano G, Simultaneous Enrichment of Cysteine-containing Peptides and Phosphopeptides Using a Cysteine-specific Phosphonate Adaptable Tag (CysPAT) in Combination with titanium dioxide (TiO2) Chromatography, Mol Cell Proteomics 15 (2016) 3282–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Adachi T, Nakashima S, Saji S, Nakamura T, Nozawa Y, Mitogen-activated protein kinase activation in hepatocyte growth factor-stimulated rat hepatocytes: involvement of protein tyrosine kinase and protein kinase C, Hepatology 23 (1996) 1244–53. [DOI] [PubMed] [Google Scholar]
- [26].Mackay HJ, Twelves CJ, Targeting the protein kinase C family: are we there yet?, Nat Rev Cancer 7 (2007) 554–62. [DOI] [PubMed] [Google Scholar]
- [27].Garg R, Benedetti LG, Abera MB, Wang H, Abba M, Kazanietz MG, Protein kinase C and cancer: what we know and what we do not, Oncogene 33 (2014) 5225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Miao H, Wei BR, Peehl DM, Li Q, Alexandrou T, Schelling JR, Rhim JS, Sedor JR, Burnett E, Wang BC, Activation of EphA receptor tyrosine kinase inhibits the Ras/MAPK pathway, Nat Cell Biol 3 (2001) 527–530. [DOI] [PubMed] [Google Scholar]
- [29].Yang NY, Fernandez C, Richter M, Xiao Z, Valencia F, Tice DA, Pasquale EB, Crosstalk of the EphA2 receptor with a serine/threonine phosphatase suppresses the Akt-mTORC1 pathway in cancer cells, Cell Signal 23 (2011) 201–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lemmon MA, Schlessinger J, Cell signaling by receptor tyrosine kinases, Cell 141 (2010) 1117–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Machide M, Kamitori K, Nakamura Y, Kohsaka S, Selective activation of phospholipase C gamma1 and distinct protein kinase C subspecies in intracellular signaling by hepatocyte growth factor/scatter factor in primary cultured rat neocortical cells, J Neurochem 71 (1998) 592–602. [DOI] [PubMed] [Google Scholar]
- [32].Seedorf K, Shearman M, Ullrich A, Rapid and long-term effects on protein kinase C on receptor tyrosine kinase phosphorylation and degradation, J Biol Chem 270 (1995) 18953–60. [DOI] [PubMed] [Google Scholar]
- [33].Welsh JB, Gill GN, Rosenfeld MG, Wells A, A negative feedback loop attenuates EGF-induced morphological changes, J Cell Biol 114 (1991) 533–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yu M, Selvaraj SK, Liang-Chu MM, Aghajani S, Busse M, Yuan J, Lee G, Peale F, Klijn C, Bourgon R, Kaminker JS, Neve RM, A resource for cell line authentication, annotation and quality control, Nature 520 (2015) 307–11. [DOI] [PubMed] [Google Scholar]
- [35].Gomez-Soler M, Petersen Gehring M, Lechtenberg BC, Zapata-Mercado E, Hristova K, Pasquale EB, Engineering nanomolar peptide ligands that differentially modulate EphA2 receptor signaling, J Biol Chem 294 (2019) 8791–8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.