

1. INTRODUCTION
One in three adults in America lives with chronic pain [1; 23]. In the majority of cases, pain is reported in multiple body locations and is associated with other non-painful bodily symptoms. These co-morbid disorders are often idiopathic, as such no identifiable structural pathology or biochemical aberration can be associated with the reported pain, and are commonly accompanied with dysregulation of the central, peripheral and/or enteric nervous systems [12; 13].
Recently, mitochondrial dysfunction has been shown to contribute to pain perception and chronic pain conditions [17; 49; 59]. This organelle is found in most eukaryotic cells, including most human cells. Copy numbers for mitochondria vary greatly among cell types, with each mitochondrion containing a few to thousands of copies of its own mitochondrial DNA, (mtDNA) [42; 67]. Mitochondria fuse and divide in an ongoing, dynamic process in response to various cell stimuli and needs [52]. They are often referred to as the “energy powerhouses of the cell” as they generate most of the cell’s chemical energy in the form of adenosine triphosphate (ATP) [56]. The mitochondrion’s genome (mtDNA) is haploid and is exclusively inherited from the maternal line [19]. Mitochondria play key roles in neuronal transmission and plasticity [25; 58], immune function [41; 68], and the ability to modulate a cell’s fate [24; 55; 70]. Mitochondria-related diseases generally result in abnormalities in tissues of neuronal and muscular origin, likely because these tissues have high and fluctuating energy requirements.
Several studies have shown a link between energy metabolism and chronic pain [17; 49; 59], suggesting several pathways through which mitochondrial dysfunction can increase or inhibit neuropathic and inflammatory pain. These include the mitochondria’s critical functions such as energy metabolism and metabolism of reactive oxygen species. Furthermore, mtDNA may drive some of these dysfunctions. Despite comprising only 37 genes, mtDNA has both a high frequency inherited polymorphisms and occurrences of new mtDNA mutations. Yet, due to several unique characteristics of mitochondrial genetics, each polymorphism can be either heteroplasmic (abnormal and wild type coexist in the same cell) or homoplasmic (in which all mtDNA is affected). Furthermore, mtDNA mutations generally demonstrate marked variability in terms of clinical expression, organ systems affected, severity, age of onset, and natural history of disease. This complicates studying the role of mtDNA in the context of chronic diseases. Despite these challenges, several preliminary studies have found associations of candidate polymorphisms in mitochondrial DNA with irritable bowel syndrome, non-specific abdominal pain, migraine and cyclic vomiting syndrome [8; 35; 61; 64; 72].
These previous studies, however, were often small, targeted a specific genetic group of people (often mtDNA haplotype H) as well as specific pain conditions (e.g., only patients with irritable bowel syndrome), and focused on a limited number of polymorphisms. This current study aimed to examine the full mitochondrial genetic makeup from a wide variety of people affected by chronic pain in the largest sample reported to date.
2. MATERIALS AND METHODS
2.1. Study approval
The primary study, Complex Persistent Pain Conditions (CPPC): Unique and Shared Pathways of Vulnerability, was approved by the Institutional Review Boards (IRBs) of the University of North Carolina and McGill University. The replication study, conducted using data and samples from the Orofacial Pain: Prospective Evaluation and Risk Assessment (OPPERA) cohort, was approved by IRBs of the four recruitment sites (the University of Florida, the University of North Carolina at Chapel Hill, the University of Maryland, and the University at Buffalo), the data coordinating center at Battelle Memorial Institute, and by McGill University.
2.2. Discovery cohort
The CPPC cohort [57] included participants enrolled in a cross-sectional study of overlapping pain conditions conducted at the University of North Carolina at Chapel Hill. A total of 848 study participants were enrolled, of which 752 had high quality DNA to perform mitochondria deep-sequencing. After quality controls filters were applied, 609 participants had simultaneous clinically assessed phenotypes with genomic data available for association studies (Table 1). Subjects were aged 18 to 64 years old, and included both sexes (86% female) and major ethnic and racial groups (69% Caucasian as determined by participant self-report). Subjects had at least one of five index CPPCs (episodic migraine [EM, 263 subjects], irritable bowel syndrome [IBS, 221 subjects], fibromyalgia [FM, 96 subjects], vulvar vestibulitis [VVS, 100 subjects], or temporomandibular disorders [TMD, 172 subjects], or were otherwise healthy controls with none of these conditions (237 subjects). Each pain condition was classified by study clinicians using validated protocols: EM was classified following an examination with a neurologist, IBS was classified according to ROME-II criteria [37], FM was classified using ACR-1990 criteria [69], and TMD was classified using RDC-TMD criteria [15]. Women were classified as VVS cases if they reported provoked pain on contact in the genital region; or having been told by a gynecologist that they have VVS; or both. Women who endorsed generalized pain and/ or itching in the genital area for 3 months or more were excluded from VVS cases. We performed Principal Component Analyses of the phenotype matrix (1=unaffected, 2=affected) using the R statistical package’s function “prcomp”.
Table 1.
CPPC study demographic and patient characteristics.
| Males [N] | Females [N] | Total [N] | F/M | P-value | |
|---|---|---|---|---|---|
| CPPC | |||||
| EM | 18 | 245 | 263 | 2.3x | < 0.01 |
| TMD | 10 | 162 | 172 | 2.7x | < 0.01 |
| IBS | 20 | 201 | 221 | 1.7x | 0.02 |
| FM | 2 | 94 | 96 | 7.8x | < 0.01 |
| VVS | 0 | 100 | 100 | N/A | N/A |
| ANY | 30 | 342 | 372 | 1.9x | < 0.01 |
| CTL | 57 | 180 | 237 | 0.5x | < 0.01 |
| # CPPCs | |||||
| 0 | 57 | 180 | 237 | 0.5x | < 0.01 |
| 1 | 19 | 123 | 142 | 1.1x | 0.90 |
| 2 | 3 | 49 | 52 | 2.7x | 0.10 |
| 3 | 7 | 115 | 122 | 2.7x | < 0.01 |
| 4 | 1 | 39 | 40 | 6.5x | 0.04 |
| 5 | 0 | 16 | 16 | N/A | N/A |
| Ancestry | |||||
| Cauc | 55 | 366 | 421 | 1.1x | 0.53 |
| Af-Am | 25 | 117 | 142 | 0.8x | 0.28 |
| Other | 7 | 39 | 46 | 0.9x | 0.83 |
| Age [y] | 34.6 (±11.9) | 36.2 (±11.6) | 36.0 (±11.6) | 1.05x | 0.61 |
| min/max | 18/61 | 18/64 | 18/64 | ||
| Counts | 87 | 522 | 609 |
Distribution of CPPCs and characteristics in males and in females. Participants can report more than one chronic pain conditions, therefore the counts in each sex is higher than the total number of individuals in the cohort. All P-values obtained using exact Fisher test, except for age, using Welch’s two-sample unequal variance. Complex persistent pain conditions were: episodic migraine, EM; temporomandibular disorders, TMD; irritable bowel syndrome, IBS; fibromyalgia, FM; vulvar vestibulitis, VVS; any of preceding, ANY; or controls, CTL. Number of CPPCs (#) in any study participant range from 0 to 5, inclusively. Ancestries were: Caucasians, Cauc; African-Americans, Af-Am; and others.
2.3. Sequencing of mitochondrial DNA
Whole blood was collected by venipuncture and genomic DNA was extracted using the NucleoSpin® Tissue kit (Macherey & Nagel, Dueren, Germany), diluted to 20 ng/μl, and aliquoted to 25 μl per sample. High-coverage (>100x) mitochondria DNA sequencing (mtSeq) from whole blood fractions was performed to determine allele content and assess heteroplasmy levels at each genomic position. The high coverage was made possible using the Ovation® Human Mitochondrion Target Enrichment System (NuGEN, San Carlos, CA). Deep-sequencing was performed at the University of Toronto on an Illumina 2500 instrument (Illumina, San Diego, CA), with data converted to FASTQ using Illumina’s CASAVA software. Length of reads was 101 nucleotides, single-ended.
2.4. Bioinformatics
Deep-sequencing reads were trimmed using Trimmomatic v0.32 [6], using aggressive trimming command-line option: “-phred33 ILLUMINACLIP:adapters.fa:1:30:6 LEADING:20 TRAILING:20 SLIDINGWINDOW:5:20 MINLEN:22”. Reads were then mapped to the human genome version GRCh38/hg38, which consists of the revised Cambridge Reference Sequence (rCRS) [3]. The alignment of reads was done using the Bowtie v2 aligner [30], chosen for its ability to perform local alignments (in contrast to Bowtie v1 which performs end-to-end global alignment only), with the following command-line arguments: “--very-sensitive-local -k 10”. The circular nature of mitochondrial DNA was not an issue because of the very deep sequencing coverage. PCR-duplicates were removed using scripts provided by NuGEN (NuGEN, San Carlos, CA). BAM flags for secondary alignments were converted into primary alignments since Bowtie randomly assigns one of the equally-scored alignment as primary, and downstream bioinformatics tool only consider primary alignments. BAM files were analyzed using MitoSeek [22], which provided counts of alleles for each genomic position. Sequencing quality controls included: PHRED scores ≥ 30, ≥ 10 counts, ≥ 95% same allele (sequencing errors and low levels of heteroplasmy were tolerated). Sequencing data was transformed into genotyping data with allele counts provided by MitoSeek. Genotyping quality controls included: Hardy-Weinberg equilibrium P-value ≥ 1×10−4 (i.e. not peculiar), genotyping rate per SNP ≥ 98%, genotyping rate per individual ≥ 98%, minor allele frequency ≥ 5%, and required that in an individual, a position had to feature at least 95% of the same nucleotide, thus allowing for parsimonious amounts of heteroplasmy levels.
A total of 752 samples were deep sequenced at a read length of 101 nucleotides. The total number of sequenced reads per sample was: minimum 50K, maximum 11.9M, mean 2.0M, standard deviation (s.d.) 1.1M. After PCR duplicates removal, the ratio of number of mapped reads to reads sequenced was: mean 55.2%, s.d. 7.8%. The ratio of number of reads mapped to mitochondria versus total number mapped, including nuclear DNA: mean 81.4%, s.d. 2.8%. No single read aligned on mitochondrial DNA were reported aligned elsewhere on nuclear DNA, indicating perfect specificity of sequenced reads for mitochondria studies. Mapped alignment length in nucleotides on mitochondria (CIGAR ‘M’ symbol): mean 81.0, s.d. 25.6. The average nucleotide coverage was (Figure S1A): mean 3913x, s.d. 3383x; in the 100 nucleotides from 5’ and 3’ ends of the chromosome (Figure S1B): mean 799x, s.d. 595x. We employed the Bowtie2 program that was able to analyze the circular configuration of the mitochondrial chromosome with the help of long reads, deep sequencing of mtDNA-enriched samples, and the ability to perform a local alignment (bowtie1 performs end-to-end alignment only). Reads spanning the control region were either mapped at the 5’ or the 3’ end of the linearized chromosome sequence, whichever end yielded better alignment scores. The coverage is on par in quality with previous studies on mitochondria [65; 71].
2.5. Statistics
Mitochondria-wide association analyses were conducted using PLINK v1.9 [47], with CPPCs as phenotypes, and sex, age, as well as the first two principal genetic components as co-variables. The mitochondrial control region (rCRS nucleotides 1 to 576 and 16024 to 16579), known to be hyper-variable, was excluded from association analyses. Sites with less than 95% of the same allele were coded as ‘0’ (“undefined genotype”) for input to PLINK. The chromosome designation ‘26’ for mitochondria was also used to instruct PLINK that the genotyping data are haploid-based. Separate association tests were conducted for each CPPC using logistic regression models in which cases were defined as subjects with the relevant CPPC and controls were subjects who did not have that CPPC. Here, we followed recommendations to object to rely on “super-controls” for association testing, as people considered cases for one CPPC can also be subjects to other CPPCs, just as the control subjects [46]. An additional linear regression model tested for associations with the total number of CPPCs (i.e., ranging from zero to five). A position was tested if its minor allele frequency was at least 5% (i.e. not a rare variant). A sex-stratified analysis was done for each comparison. A principal component analysis (PCA)-based approach that considered correlated SNP alleles in linkage disequilibrium was used for determining statistical significance, then Bonferroni correction was applied for multiple testing based on the estimated number of effective SNPs. The Genetic Type I error calculator (GEC) was used to estimate the effective number of SNPs [33] as an alternative to mitochondrial haplogroup assignment.
Haplogroup assignments were determined from the deep sequencing by reconstructing each individual’s mitochondrial DNA sequence, in which the original rCRS sequence was adjusted to MitoSeek’s major allele base call. Haplogroup assignments were performed using HAPLOFIND [63] and HaploGrep 2 [66], and results were congruent between the two methods. In African-Americans, 71.9% were classified as haplogroup L, 7.8% as H and 5.9% as U. In Caucasians, 38.8% were in H, 16.0% in U and 10.2% in T. In the ‘other’ ancestry group, haplogroups B (22.2%), A (18.5%) and D (14.8%) were most common. Results were consistent with known high-levels of Caucasian admixture among African Americans, and Native Americans among Hispanics, who constituted much of the ‘other’ group. These proportions are in line with the findings from the 1,000 Genomes Project about the distribution of mitochondrial haplogroups in the US population [50]. We performed haplogroup-based association tests with CPPC by comparing individuals of one haplogroup to those of all other haplogroups. Haplogroups tested featured at least 30 individuals, and were: H (n=177), L (n=137), U (n=79), J (n=51), T (n=47), and K (n=32). Age and sex were used as co-variables.
Rare variant association tests were performed using the SKAT-O approach [32]. SNPs were pooled by genes or by pathways. Tested genes comprised all 13 mitochondrial protein-coding genes, 22 transfer RNAs, and small (12S rRNA) and large (16S rRNA) ribosomal subunits. Tested pathways pertained to the oxidative phosphorylation complexes, and were [34]: Complex I = [MT-ND1, MT-ND2, MT-ND3, MT-ND4, MT-ND4L, MT-ND5, MT-ND6], Complex III = [MT-CYB], Complex IV = [MT-CO1, MT-CO2, MT-CO3], and Complex V = [MT-ATP6, MT-ATP8]. Age, sex, and the first two principal genetic components were used to define the SKAT test null model.
Association tests with heteroplasmy levels were conducted as follows: for each genomic position, the distribution of heteroplasmy odds ratio in subjects with a CPPC was contrasted against the distribution of those without. In each individual, the heteroplasmy odds ratio was established from a 2×2 Fisher table composed of the observed major and minor allele counts, and estimated counts of major and minor alleles from 0.1% heteroplasmy baseline levels, which could be attributed to sequencing error, deep-sequencing read misalignment, etc. The odds ratio was calculated using the observed minor allele count to that expected, given the background of observed and estimated major allele counts. Sequencing depth modulated the statistical significance of the odds ratios, but here we performed the tests based on effect size only, while making sure that genomic positions with marked differences in heteroplasmy levels would correspond to deeply sequenced positions, i.e. with several thousand-fold coverage on average. We performed logistic tests for CPPC as a function of heteroplasmy odds ratios, using age, sex, and first two genetic principal components as co-variables. Because the odds ratio is self-normalized there was no need to account for sample size factors (sample-wide sequencing depth).
2.6. Visualization
Graphics were plotted using the R statistical package, version 3.5.2 (2018-12-20) [48].
2.7. Replication cohort
The replication case–control cohort included 1,754 female subjects selected from the OPPERA study [39], of whom 53 were fibromyalgia cases while 1,701 were non-cases based on self-reported item in the Medical History questionnaire in OPPERA (Fibromyalgia/Chronic Fatigue Syndrome). Cases were defined as those that answered “yes” to the question: “did you have this fibromyalgia in the past or have it now?”. Cases and controls were not excluded if they had other CPPCs. (Supplementary Table S1). Genotyping was performed by the Center for Inherited Disease Research (Baltimore, MD) using the Illumina HumanOmni 2.5 Exome Bead Chip platform (Illumina, Inc, San Diego, CA). Genetic data cleaning was accomplished by the Genetic Analysis Center at the University of Washington following their established pipeline [31]. The genotyping array included SNP m.2352T>C, probed via exm2216242 [57]. Association tests used as co-variables age, sex, and the first two genetic principal components to account for population stratification.
2.8. Cell culture
Twenty B lymphoblast cell lines were obtained from the NHGRI sample repository for Human Genetic Research through the Coriell Institute (Camden, NJ). Of these cell lines, 10 had the major allele at m.2352T>C while 10 had the minor allele at this position. All cell lines were derived from women of African-American or Western African ancestries; 16 females were of African-American ancestry living in the U.S. (1,000 Genomes Project population code ASW), while 4 were from Western Africa (YRI). These ancestries were selected over other ancestries because they carry significantly higher minor allele content at m.2352T>C and women were selected over men because they showed stronger association with pain phenotypes. The cell lines were maintained according to the supplier’s protocol. Briefly, cells were cultured in maintenance medium of RPMI 1640 (GIBCO®; Thermo Fisher Scientific, Waltham) supplemented with 15% fetal bovine serum (GE Healthcare Bio-Sciences, Marlborough), and 200mM glutamine (GIBCO®). B lymphoblast density was kept between 0.2 and 1 × 106 cells/ml.
2.9. JC-10 staining
Each B lymphoblast line was washed and re-suspended at 0.5 × 106 cells/ml in either RPMI 1640 without glucose supplemented with 4.5g/L glucose (Sigma-Aldrich, St-Louis), 15% dialyzed fetal bovine serum (GIBCO) and 200mM glutamine, or RPMI 1640 without glucose supplemented with 4.5g/L galactose (Sigma-Aldrich), 15% dialyzed fetal bovine serum and 200mM glutamine. Cells were incubated for 24 hours at 37°C with 5% CO2 in a humidified chamber. Based on optimization experiments, 1 × 105 cells were stained in 200 μl JC-10 staining solution (AbCam, Cambridge) for 30 minutes at 37°C according to manufacturer’s instructions. Control samples were depleted for JC-10 aggregates by a mitochondrial uncoupling agent: 10 μM carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) (Sigma-Aldrich). 3×104 events per sample were acquired on an LSR-Fortessa SORP (BD BioSciences, Franklin Lakes) using excitation at 488nm and 530/30nm detection filter for the JC-10 monomers, and excitation at 561nm and 582/15nm detection filter for the JC-10 aggregates.
2.10. Flow cytometry data analysis
Preliminary cleaning of flow cytometry data was done using a time-gate to exclude anomalies from abrupt changes in the flow rate, followed by exclusion of debris and doublets based on forward and side scatter parameters. Approximately 5000 live B-lymphoblasts were selected per sample for unsupervised clustering analysis. Mean fluorescent intensity (MFI) signals from JC-10 monomers and polymers were normalized (mean = 0, standard deviation = 1) across all the 40 samples (two conditions x (ten samples with the reference allele + ten samples with the alternative allele)). Density-based spatial clustering of applications with noise was performed using the DBSCAN R-package [16]. Two clusters were identified: cluster A (high in JC-10 aggregate signal) and cluster B (high in JC-10 monomer signal). Linear modelling was performed with the ratio of cluster B to cluster A as the dependent variable, and the presence of minor allele and culture media (glucose or galactose) as independent variables. The cluster ratio served as a marker for the number of cells with low mitochondrial membrane potential (Δψm) compared to healthy cells. FlowJo™ (FlowJo, LLC, Ashland) and R version 3.6.0 were used for the analyses.
3. RESULTS
3.1. Discovery cohort sample characteristics
A total of 848 participants were enrolled in the Complex Persistent Pain Condition (CPPC) cohort. The pairing of samples with epidemiological data with those with deep-sequencing of mitochondria data resulted in 609 matched samples (Table 1). The cohort was comprised of individuals with at least one CPPC (61.1%; controls 38.9%), and most were female (85.7%), of predominantly Caucasian (69.1%) or African-American (23.3%) ancestries, and aged between 18 to 64 years (mean 36.0 ±11.6).
Complex persistent pain conditions were: episodic migraine (EM, 43.2%), temporomandibular disorders (TMD, 28.2%), irritable bowel disorder (IBS, 36.3%), fibromyalgia (FM, 15.8%), and vulvar vestibulitis (VVS, 16.4%). Women were more likely to have each CPPC compared to men; from 1.7x for IBS to 7.8x for FM (VVS, by definition, affects women only). Most individuals with one or more CPPC had co-morbid conditions: 142 (23.3%) individuals had only one CPPC, while 230 (37.8%) had two or more CPPCs. Principal component analysis of the phenotype matrix distinguished, as expected, the health status against the chronic pain conditions (Figure 1). This major axis (PC 1) contributed to as much as 49% of the variance in the matrix. The second major axis (PC 2) explained 15%; at opposite ends of the spectrum were fibromyalgia and irritable bowel syndrome. The principal component eigenvectors extracted from the phenotype matrix were useful surrogates for an association study with a simplified endophenotype underlying all CPPCs [4], while enabled consideration of all CPPCs into one association study, thus circumventing the need for sample overlap correction in a risk meta-analysis.
Figure 1.
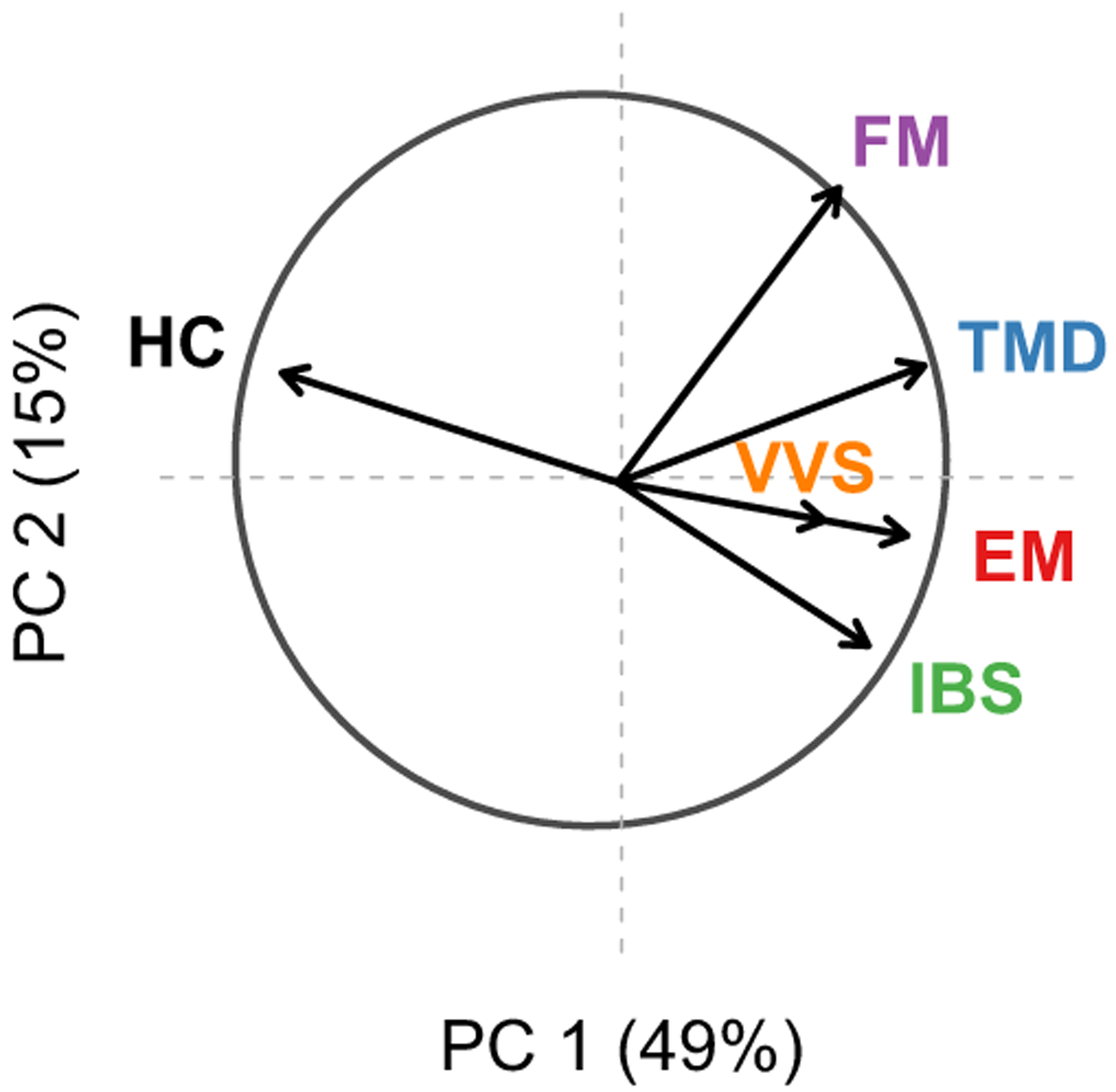
Correlation wheel of CPPCs with the first two principal components of the phenotype matrix. Complex persistent pain conditions are: episodic migraine, EM; temporomandibular disorders, TMD; irritable bowel syndrome, IBS; fibromyalgia, FM; vulvar vestibulitis, VVS. HC stands for healthy controls. Percent variance explained by each principal component shown in parentheses.
3.2. Association with complex persistent pain conditions
First, we performed association tests between each of the five CPPCs and allelic polymorphic content along the mitochondrial chromosome (Figure 2). We also tested for association with any CPPC, and the number of CPPCs.
Figure 2.
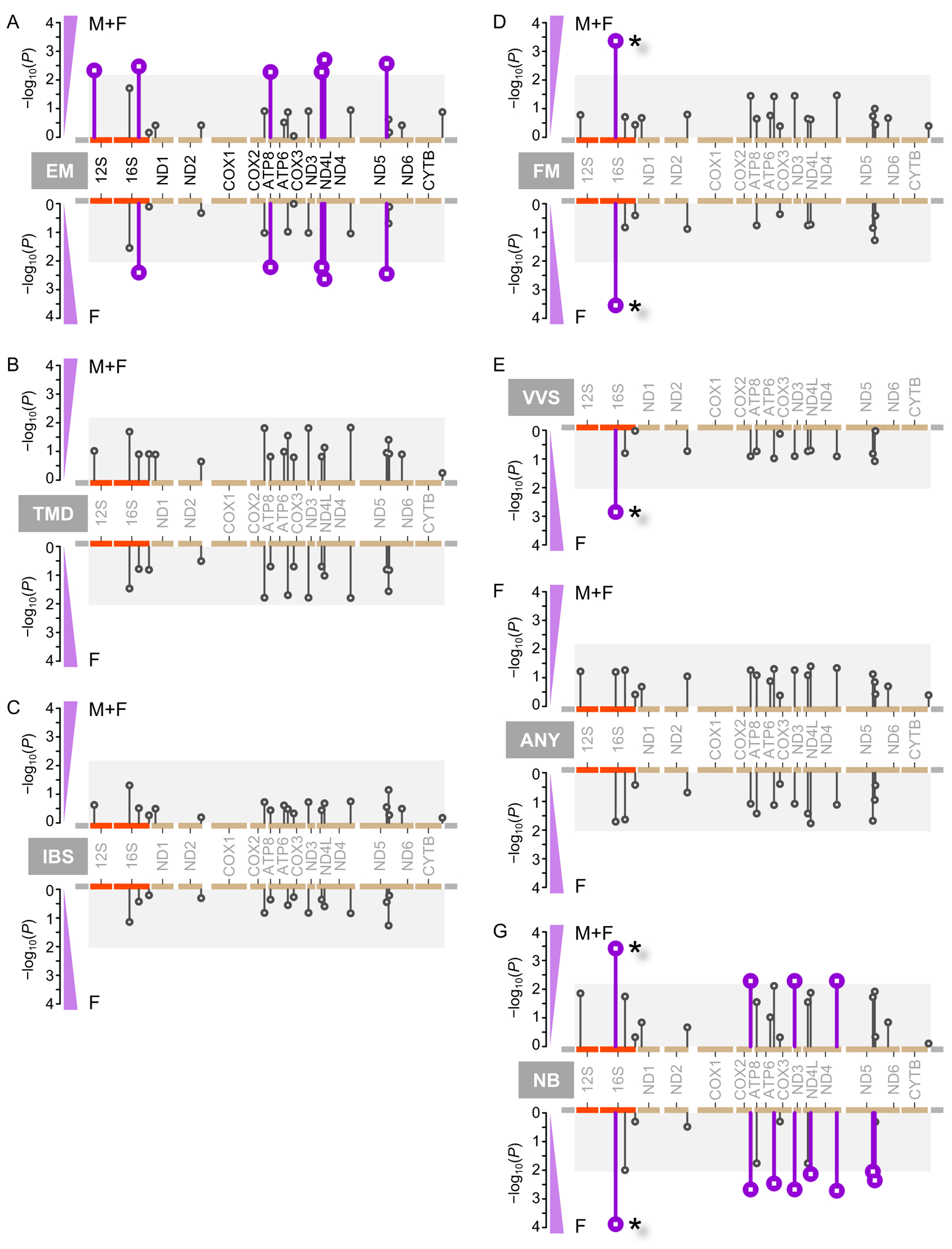
Mitochondria-wide association studies of CPPCs. Shown are Manhattan plots, tracking association P-value (P) along the mitochondrial chromosome, in everyone (top) and in females only (bottom). The mitochondrial chromosome shown is the linearized version with annotated genomic features: rRNA (orange), protein coding genes (tan), control region or D-loop (grey). Vertical bars indicate tested positions, with minor allele frequency ≥5%. Grey boxes indicate areas outside of statistical significance, while vertical purple bars highlight significance. Significant associations with position m.2352T>C marked with a star (*). (A) Episodic migraine (EM). (B) Temporomandibular disorders (TMD). (C) Irritable bowel syndrome (IBS). (D) Fibromyalgia (FM). (E) Vulvar vestibulitis (VVS). (F) Presence of any CPPC (ANY). (G) Number of CPPCs (NB).
The most significantly associated mitochondrial position with a CPPC after correction for effective number of SNPs was position 2352 with fibromyalgia (OR=5.1, P=2.8×10−4, F only; OR=4.62, P=4.3×10−4, F+M) (Figure 2D; Supplementary Table S2). This position corresponds to SNP m.2352T>C (rs28358579) that is located in the large mitochondrial ribosomal subunit (16S rRNA) encoded by the MT-RNR2 gene. The 16S rRNA locus also hosts a peptide-coding gene, humanin, found to have neuroprotective [60] and anti-apoptosis [21] properties, and in which m.2352T>C lies in its 5’UTR. That SNP was also significantly associated with vulvar vestibulitis (OR=4.6, P=1.4×10−3) (Figure 2E) and with the number of CPPCs (beta=+0.83, P=1.3×10−4, F only; beta=+0.68, P=3.8×10−4; F+M) (Figure 2G). Consistently, the presence of the C allele increased risk with almost a unit increase in number of CPPCs with the risk allele (beta close to +1). The latter associations were slightly stronger in female only populations.
A detailed account of the relationships between SNP m.2352T>C, fibromyalgia status and ancestry is presented in Figure 3. The percentage of subjects with fibromyalgia was similar in Caucasians (15.9%) and African-Americans (17.6%) (Figure 3A). The C minor allele was present in 32.5% of cases, whereas the T major allele was present in only 14.8% (Figure 3B). Only 9 (<1.5%) individuals featured heteroplasmy levels above 5% for SNP m.2352T>C (Figure 3B; allele ‘0’). The C minor allele was predominantly found in participants of African-American ancestry (Figure 3C): about 24% in African-Americans while only about 1% in Caucasians.
Figure 3.
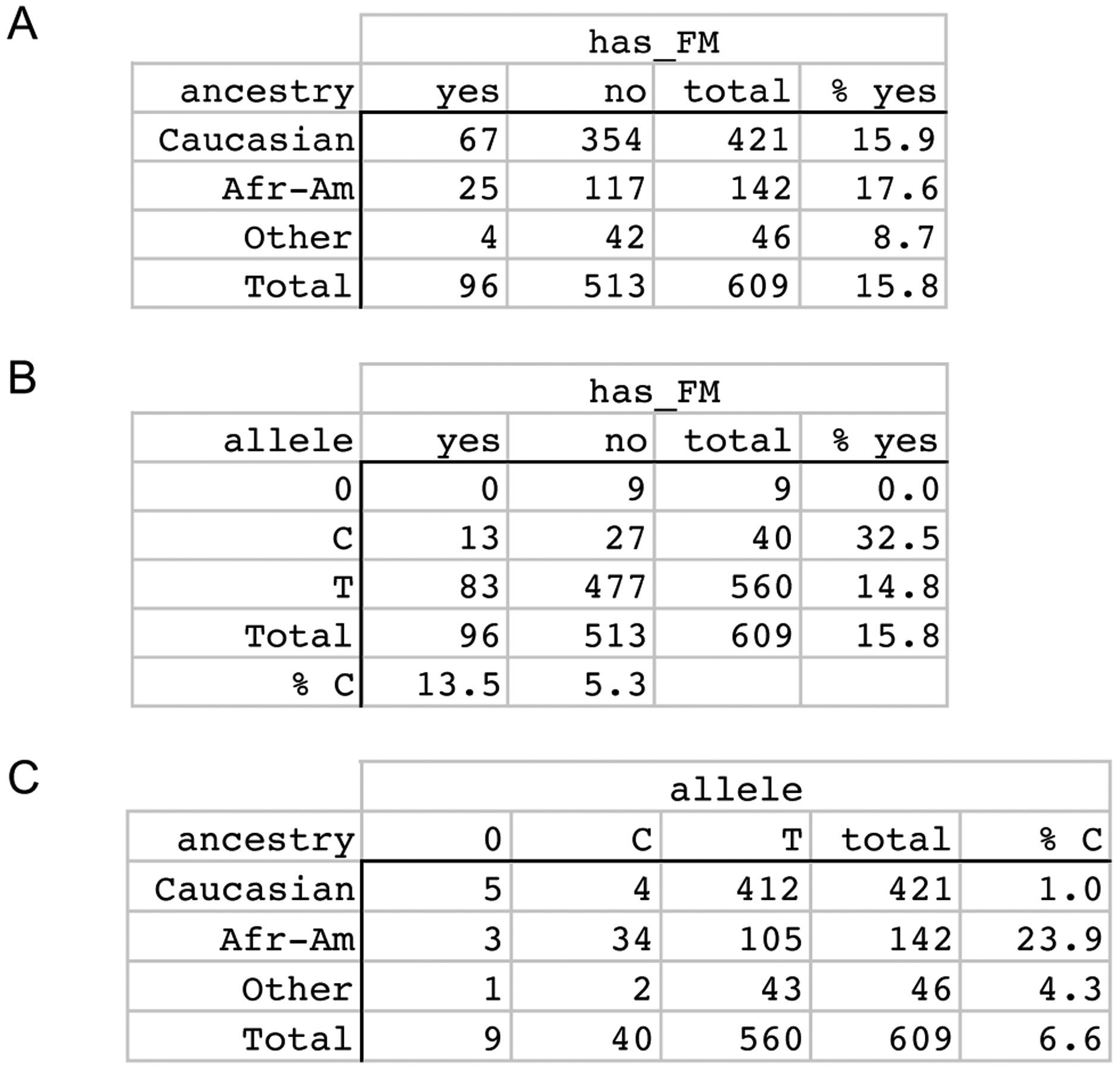
Relationship between fibromyalgia, ancestry, and allele content at SNP m.2352T>C in females. (A) Fibromyalgia in different ancestral groups. (B) Allelic distribution in fibromyalgia patients. (C) Allelic distribution in different ancestral groups. Allele ‘0’ stands for discarded samples due to heteroplasmy.
We next examined each CPPC for the effect of SNP m.2352T>C as secondary analyses (Figure 4; Supplementary Table S3). First, we recapitulated the initial findings for FM in both CPPC and OPPERA cohorts in a forest plot (Figure 4A). Then, we observed a consistent, nominally significant (P<0.05) increased risk for all other CPPC with the presence of the C allele. Notably, for episodic migraine (EM: OR=2.5, P=1.9×10−2) (Figure 4B), for temporomandibular disorders (TMD: OR=2.6, P=2.1×10−2) (Figure 4C), and with number of CPPC (NB: beta=+0.7, P=3.8×10−4) (Figure 4D). The association with any CPPC was also nominally significant in females only (ANY: OR=3.0, P=2.0×10−2) (Figure 4E), but highly significant in vulvar vestibulitis (VVS: OR=4.6, P=1.4×10−3) (Figure 4F).
Figure 4.
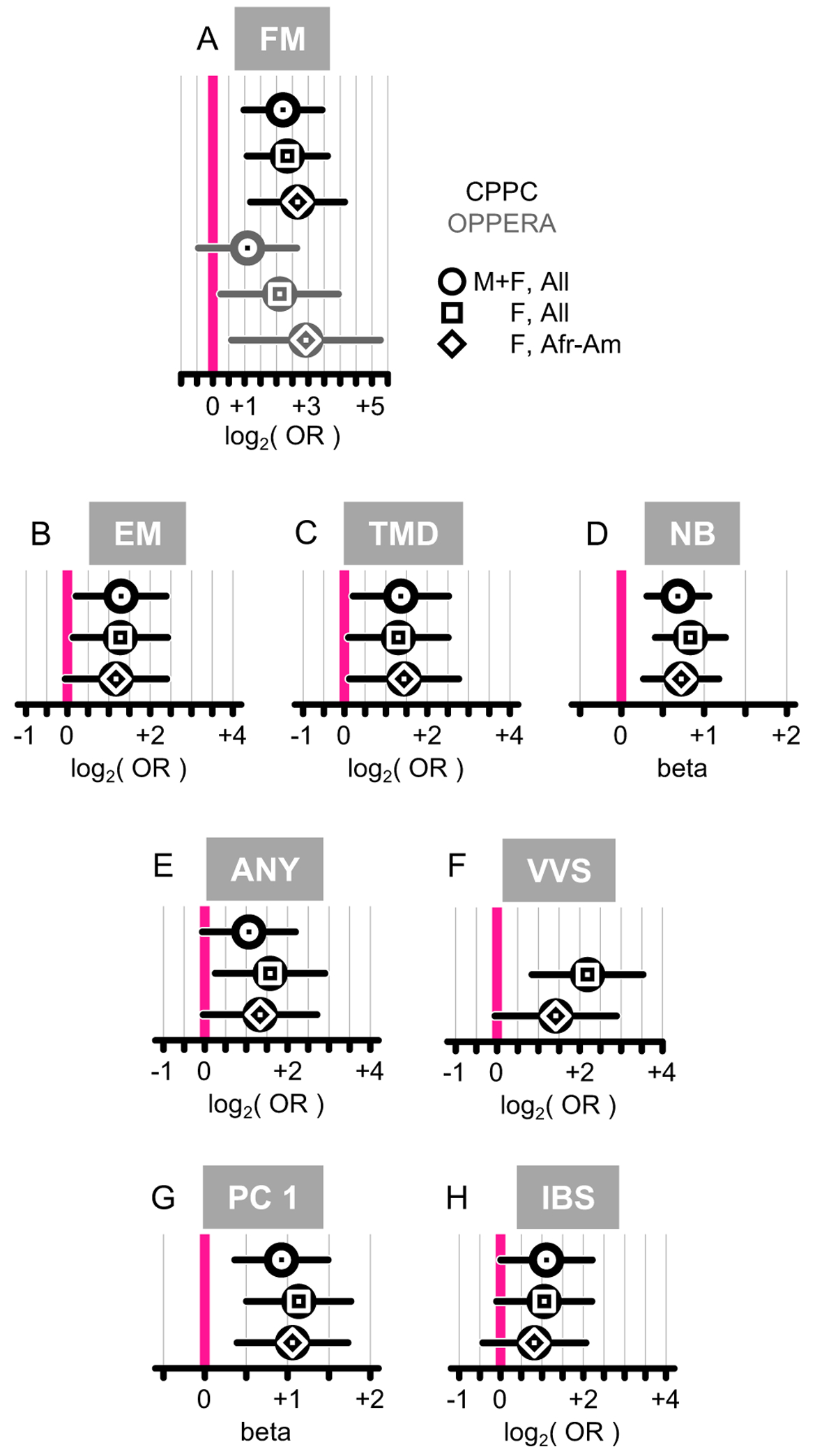
Forest plots for the association of m.2352T>C with CPPCs. Odd ratios (log2 scale) or betas with 95% confidence intervals shown. CPPC discovery data in black, while OPPERA replication data in grey. Population stratifications are: everyone (circle), females only (square) or African-American females (lozenge). (A) Fibromyalgia (FM). (B) Episodic migraine (EM). (C) Temporomandibular disorders (TMD). (D) Number of CPPCs (NB). (E) Presence of any CPPC (ANY). (F) Vulvar vestibulitis (VVS). (G) Principal component 1 (PC 1). (H) Irritable bowel syndrome (IBS).
We next used principal component analysis (PCA) of the phenotype matrix, which provided for an eigenvector associated with the largest eigenvalue (Figure 1). The eigenvector PC 1 was used as a quantitative phenotype for association with m.2352T>C, to estimate the contribution of the polymorphism to chronic pain states at large. We found that the SNP’s effect (beta) was positive, implying that the minor allele conferred significantly increased risk for the presence of pain (PC 1: beta=+0.93, P=1.4×10−3; Figure 4G). Again, this effect was even stronger in women (PC 1: beta=+1.14, P=4.7×10−4; Figure 4G).
Haplotype-based tests were performed to explore association between CPPC and maternal-lineage ancestry. We tested the most represented haplotypes H, L, U, J, T, and K. No association results were deemed significant at the FDR 10% level (Supplementary Table S4). We also performed rare variant-based tests with SKAT-O. SNPs were pooled by genes or by oxidative phosphorylation protein complexes comprising one or multiple genes. Again, no association results were significant at the FDR 10% level (Supplementary Table S5).
Finally, we capitalized on the very deep mitochondrial DNA sequencing to assess impact of heteroplasmy levels with respect to CPPC status (Supplementary Table S6). Each time, differences in heteroplasmy levels between cases and controls were minimal (FDR > 10%). Overall, heteroplasmy levels were significantly associated with the presence of all CPPC at multiple positions, with higher levels in control subjects than in cases. Notably, in association with any CPPCs (ANY) position m.6412A>G in the MT-CO1 gene (beta=−0.84, P=2.1×10−7) is a non-synonymous mutation AAU to AGU, coding a change from asparagine to serine.
3.3. Replication of m.2352T>C in the OPPERA cohort
We next tested our finding for replication in an independent cohort: Orofacial Pain: Prospective Evaluation and Risk Assessment Study – The OPPERA Study [39]. Even though the cohort was focused on the study of TMD, it contained self-reported data on fibromyalgia status and other CPPCs. Furthermore, the ancestry structure of OPPERA was similar to the discovery cohort. Female subjects were predominantly of Caucasian (61.3%) and African-American (23.2%) ancestries and aged between 18 to 44 years (mean 27.7 ±7.7). They were partitioned into 52 (3%) cases for fibromyalgia and 1660 controls (non-fibromyalgia), for a total of 1712 individuals. Women of African-American ancestry comprised 8 (2%) cases and 402 controls (Supplementary Table S1).
We found that fibromyalgia status was associated significantly with SNP m.2352T>C in women (OR=4.3, P=2.6×10−2), thus replicating our initial finding. The polymorphism had even stronger effect size in a smaller population of African-American women (OR=7.6, P=1.4×10−2; Figure 4A; Supplementary Table S7). Thus, overall, we found that m.2352T>C consistently increased risk for fibromyalgia in both the discovery and replication cohorts (Figures 4A; Supplementary Table S7) with very robust effect size.
3.4. Replication of previous work
Several previous studies have suggested a role for mtDNA polymorphisms, such as m.16519T>C and m.3010G>A in various chronic pain-related disorders such as IBS, migraine and cyclic vomiting [5; 8; 61; 62; 72]. In our study, these two SNPs exhibited heteroplasmy levels greater than 5% in about 5% of samples, and so were excluded from primary and secondary analyses after quality control for genotyping rate. Heteroplasmy results showed nominal association between dosage of A at position 3010 with migraine (P=0.03) or presence of any CPPC (P=0.04), with controls displaying greater levels of heteroplasmy. At position 16519, dosage of C was nominally associated with TMD (P=0.02), IBS (P=0.03) or presence of any CPPC (P=0.05), with controls displaying greater levels of heteroplasmy here too.
3.5. Effect of m.2352T>C on mitochondrial membrane potential
The mitochondrial genome encodes 37 genes, including 2 rRNAs, 22 tRNAs and 13 polypeptides, which are required for oxidative phosphorylation as part of the electron transport chain (ETC). The mitochondrial rRNAs and their assembled ribosomes are solely responsible for translating these electron transport chain proteins. Because the m.2352T>C polymorphism is situated in the mitochondria’s large ribosomal subunit 16S rRNA it could potentially impact the translation of ETC transcripts, and consequently oxidative phosphorylation.
To test the functional effects of the m.2352T>C polymorphism, we employed B lymphoblast cell lines with known genotypes from female individuals of African-American or West African ancestry from the NHGRI sample repository. The obtained cell lines were from populations enrolled in the 1000 Genome Project, with available genotypes, sex, age, and ancestry, but no pain phenotypes. We identified 10 individuals that carried the T (major) allele, and 10 other individuals with the C (minor) allele at m.2352, for a total of 20.
We tested the effect of the SNP on oxidative phosphorylation in B lymphoblasts by growing cells in either glucose-containing media that allowed energy production by glycolysis and oxidative phosphorylation or in galactose-containing media which forces the cells to use oxidative phosphorylation for ATP production [14; 51]. Increased oxidative phosphorylation results in a greater mitochondrial membrane potential (Δψm) due to a corresponding increase in the transport of protons across the inner mitochondrial membrane due to the ETC [45; 73]. Thus, Δψm is correlated with oxidative phosphorylation. The staining of both genotypic variants of cell lines with JC-10 dye was used to assess mitochondrial membrane potential by measuring fluorescence. The JC-10 dye is present as either a J-monomer in cells with low Δψm or as aggregates in mitochondria when Δψm is increased. These two forms of the dye have different excitation and emission spectra, and the ratio of fluorescence was used as a proxy for Δψm. Furthermore, FCCP reagent, which uncouples the electron transport chain and therefore decreases Δψm, was used as a negative control to specifically deplete JC-10 aggregate fluorescent signal (561nm excitation laser, 582/15nm detection filter) without significantly affecting JC-10 monomer staining (488nm excitation, 530/30nm detection filter). As expected, there was much lower JC-10 aggregate staining in FCCP treated cells (Figure 5A, FCCP-treated cells).
Figure 5.
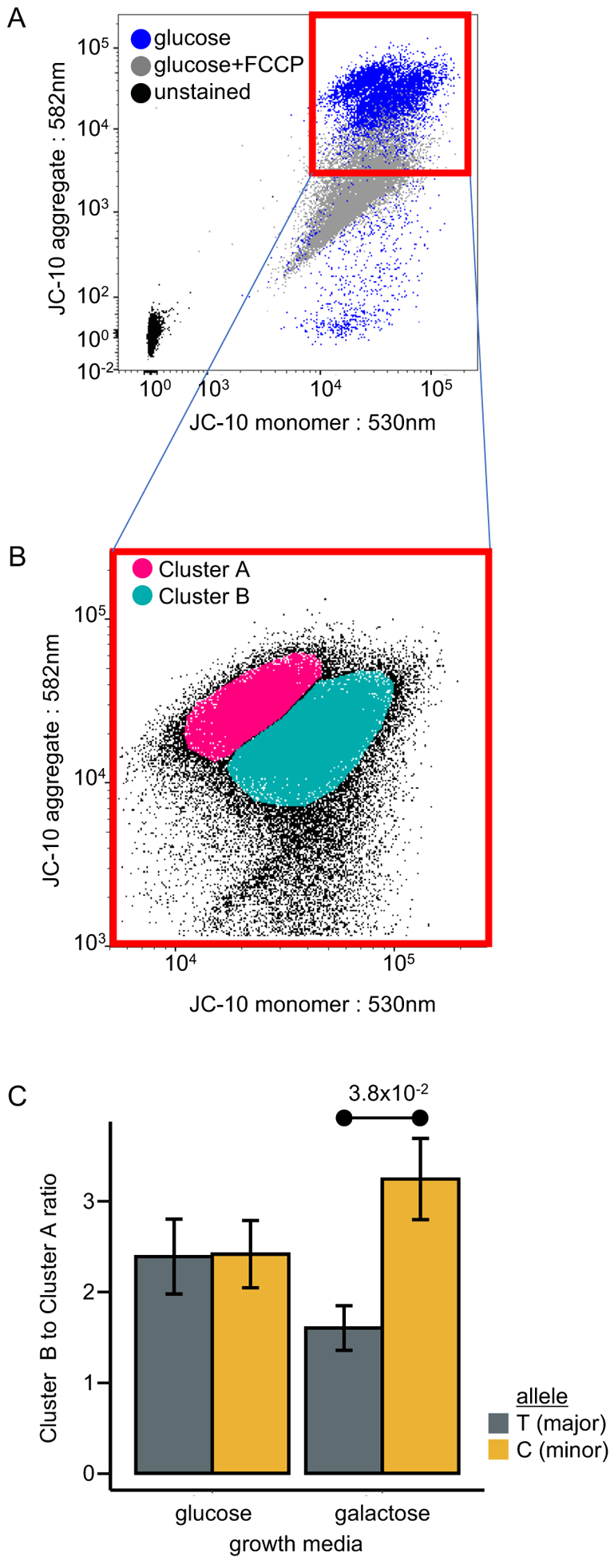
Cellular mitochondrial assay for functional characterization of SNP m.2352T>C using JC-10 staining. (A) Fluorescence scatter plot showing JC-10 aggregate intensity (582nm channel) as a function of JC-10 monomer intensity (530nm channel). Plotted data from JC-10 stained B lymphoblast cells either without treatment (blue) or treated with mitochondrial uncoupling agent, FCCP (10 μM) (grey). Background signal shown by cells left unstained for JC-10 (black). (B) Representation of the two cell clusters of live B lymphoblasts, A and B, generated by density-based spatial clustering of applications with noise (DBSCAN) on a fluorescence scatter plot showing JC-10 aggregate intensity (582nm channel) as a function of JC-10 monomer intensity (530nm channel). (C) The ratio of cells in cluster B, which have lower ΔΨm, to cluster A, which have higher ΔΨm. Ratio measurements performed in glucose and galactose-containing growth media. In each medium, ratio measurements were performed for B lymphoblasts with the T- (major) and C- (minor) alleles. Error bars represent mean ± SEM. 10 biological replicates for each treatment group.
For the experimental samples, the vast majority of the lymphoblasts formed two discrete clusters when JC-10 monomer was plotted against JC-10 aggregate signal (Figure 5A). Both of these clusters had more JC-10 aggregate staining than the FCCP-treated control cells, indicating that our assay captured gross changes in Δψm. Also, the presence of JC-10 aggregate staining in untreated lymphoblasts indicated that they were largely a viable population since a collapse of Δψm is recognized as an early hallmark of apoptosis. Our assay also captured more subtle changes in Δψm since cells that were grown in galactose-containing media showed an increase in JC-10 aggregate staining compared to cells grown in glucose (P=1.0×10−2)(data not shown). The increase in Δψm in galactose-containing media was expected since the cells were reliant on oxidative phosphorylation.
As the overlap in distributions on both axes did not allow for adequate conventional flow cytometric analysis, an unbiased cluster analysis was performed to determine that the population of live cells contained subpopulations, cluster A (high Δψm) and cluster B (low Δψm) (Figure 5B). There were no obvious differences in cluster A or cluster B between B lymphoblasts with the minor or major alleles when grown in glucose-containing media. However, when grown in galactose-containing media, the B lymphoblasts with the minor allele showed a significant increase in the number of cells with a higher JC-10 monomer signal and a lower JC-10 aggregate signal (cluster B, low Δψm) than B lymphoblasts with the major allele (P=3.8×10−2) (Figure 5C). This indicated that the cells with the minor allele had decreased Δψm under conditions where oxidative phosphorylation was required.
4. DISCUSSION
Although previous studies have unmasked the role of mitochondria in chronic pain, our analysis is the first to examine the full mitochondrial genetic makeup of people affected by a panoply of chronic pain conditions. It is also the largest sample size reported in the literature to date involving mitochondrial genetics and chronic pain. Because we generated mitochondrial sequencing data of a very high density in the CPPC cohort, we were able to test different modalities of the mitochondrial genetic contribution to chronic pain, such as rare mutations, common variants, haplogroups, and heteroplasmy.
The most robust association results have been obtained for the common polymorphic variants. We found that SNP m.2352T>C was associated with an increased risk for fibromyalgia in the presence of the alternative C allele. The replicated genetic effect size of the C allele on the disease risk (OR 5.1 and 4.3 in discovery and replication cohort, respectively) is impressive and has little precedence within the field of common diseases. The relatively high minor allelic frequency of the associated allele makes our result even more unique, as an inverse relationship between a SNP’s frequency and disease odds ratio is predicted by population genetics and is observed in a daily fashion with the outpouring of GWAS results [18; 44]. In the sex-specific stratified analysis the association was significant only in women but not in men, although it is difficult to be sure that the identified effect is truly sex-specific because of the limited number of male fibromyalgia patients. Furthermore, the minor allele of this SNP was abundant in participants of African American ancestry and much rarer in other ancestry groups. However, for the majority of pain phenotypes the effect size of the C allele was stronger in a mixed population than in the African American population (Figures 4F–H), suggesting that the effect of the C allele is not race-specific.
Although our primary screen identified SNP m.2352T>C most significantly associated with FM, we observed other significant associations with VVS and number of CPPCs, and noticed nominal associations with all other CPPCs in secondary analyses, in a consistent risk-associated fashion. The association was observed in episodic migraine, temporomandibular disorders, IBS, and number of CPPCs. Association with the global pain phenotype via first principal component was also significant.
This SNP was not previously documented to be associated with a disease or trait in the Online Mendelian Inheritance in Man (OMIM®) catalog [2], nor is its clinical significance reported in the ClinVar resource [29]. This might be because genome-wide association studies are mainly conducted in people of European ancestry, though this shortcoming is now being addressed [43]. Furthermore, the m.2352T>C SNP isn’t routinely assessed as it is missing from popular genotyping arrays, including those used by large genetic studies such as the UK Biobank project and 23AndMe. We attempted, but failed to unambiguously impute the SNP using the large database of complete human mitochondrial sequences (MITOMAP [38]), indicating low linkage disequilibrium with neighboring genotyped SNPs. Thus, our results suggest that including the m.2352T>C SNP into future genotyping platforms will benefit the research field of mitochondrial genetics.
The m.2352T>C SNP is situated in mitochondria’s 16S rRNA gene, the large subunit of the ribosome [26] (gene MT-RNR2). It’s also situated in the 5’UTR of the humanin gene, a peptide with anti-apoptotic and neuroprotective properties, although, there is uncertainty whether this is a transcribed protein-coding gene, or if it is a nuclear pseudogene of the mitochondrial MT-RNR2 gene [21; 36; 40]. The alternative allele may affect the stability of the transcript or its secondary structure of one or both corresponding RNAs, as well as the translation level of humanin. It is also possible that m.2352T>C polymorphism alters ribosomal translation speed or decoding fidelity. Unfortunately, the crystal structure of the large ribosomal subunit of human mitochondria does not display enough electron density to unambiguously resolve the position of m.2352 (chain A of PDB code 3J7Y [7]), hampering the deciphering of its role.
To test if the polymorphic variant affects overall cellular function, we conducted an assay in which mitochondria were presented with alternative energy sources while monitoring the mitochondrial membrane potential. The mitochondrial membrane potential is a consequence of electron transport in mitochondria, which is necessary for ATP synthesis. It also has non-ATP producing functions such as cell viability [73]. Although limited variation in the mitochondrial membrane potential is common, prolonged changes can affect mitochondrial function [73]. We found that cells carrying the minor allele compared to the major allele were more likely to be in the cell cluster with increased JC-10 monomer staining when grown in galactose, rather than in glucose. This result suggests that the presence of the minor allele decreased the mitochondrial membrane potential under conditions where oxidative phosphorylation is required. Although the exact mechanism is unknown, our results indicate that SNP m.2352T>C impacts oxidative phosphorylation, thus potentially linking oxidative phosphorylation with the development of chronic pain conditions. Our results are concordant with previous findings on an association between impaired mitochondrial metabolism and fibromyalgia [10] and a decreased level of coenzyme Q10, an essential electron carrier in the mitochondrial respiratory chain, in the blood of fibromyalgia patients [11]. These relationships are of particular interest since they are indicative of potential therapeutic targets [9].
To our surprise, we did not find any significant associations when we tested the contribution of rare variants to pain states using the SKAT-O approach, neither by combining their effects on genes nor pathways. In contrast, such associations have been found for Complex I of the OXPHOS pathway and cancer [34], mitochondria-wide rare variants and schizophrenia [20], or specific, combined gene-based analyses with various metabolic traits [28]. Furthermore, haplogroup-based tests didn’t produce any significant results with pain-related phenotypes.
When we tested heteroplasmy levels, the coexistence of multiple alleles at the same genomic locus in a given individual, we found overall small but significantly elevated heteroplasmy levels at defined mitochondrial genomic loci in control subjects compared to those with any one of the CPPC. This indicated that nucleotide diversity might be beneficial regarding protection from painful conditions. We could not find a cohort to test this finding for replication. However, our results are in line with previous findings that have shown that mitochondrial heteroplasmy is widespread and tolerated in healthy subjects despite its pathogenicity under specific circumstances [71]. Moreover, the relationship between heteroplasmy level and pathogenicity has been demonstrated to display non-linear behavior, as for the case with m.3243A>G [27], in which distinct cellular consequences can be observed dependent on increasing minor allele dosage. The diversity hypothesis has been shown fruitful for cell survival in other cellular contexts [53; 54].
This study has many strengths, including (1) Deep-sequencing of mitochondrial-enriched DNA fragments to determine with high accuracy the complete mitochondrial genetic makeup of people affected by chronic pain. (2) A large sample size compared to other studies of the mitochondria’s genetic role in pain. (3) Inclusion of human subjects with diverse mtDNA haplogroups, in particular H and L. (4) Inclusion of several pain conditions previously unstudied with respect to mtDNA. (5) Assessment of all participants by medical experts for each of the five conditions providing reliable, high-quality phenotyping. It also has several weaknesses. (1) Even though the sample size of our study is the largest published yet, the results indicated that the cohort is underpowered to detect low effect size for common SNPs, and combined effects for grouped rare variants. (2) We have assessed the association between heteroplasmy and CPPCs, but couldn’t find an appropriate cohort to replicate our findings. (3) We employed B lymphoblast cell lines for our functional assays to test the allelic effect on oxidative phosphorylation. The B lymphoblasts were used as a cell model for measuring allelic-dependent mitochondrial membrane potential, as these were the only cell lines with fully characterized genotypes available; the cells contributing to pathophysiology of pain states, like neurons, would be better choice (4) Great care has been put into selecting individuals from the NHGRI sample repository such that the subjects with both the T and C alleles of m.2352 were randomized, but we cannot rule out the possibility that other SNPs in high linkage disequilibrium are the true effectors of the observed modulation of the mitochondria membrane potential.
In conclusion, our results suggest that the m.2352T>C polymorphism has a strong clinical effect on the risk of fibromyalgia and possibly other chronic pain conditions. Prevalence of the SNP was elevated in participants of African-American ancestry, while almost absent in those of Caucasian ancestry. Using a cellular assay, we identified differences in mitochondrial functions in B lymphoblast cells from individuals with defined allelic variants at that SNP position. We show that the SNP allele is associated with lower mitochondrial inner membrane potential during oxidative phosphorylation. This implies that decreased cellular energy metabolism may contribute to chronic pain, although the exact mechanisms still need to be identified. Taken together, our findings suggest a novel pathway for the development of treatments for chronic pain patients directed at detecting and restoring mitochondrial dysfunction.
Supplementary Material
Supplementary Table S1. Distribution of CPPCs and characteristics in males and in females. Participants can report more than one chronic pain condition, therefore the counts in each sex are higher than the total number of individuals in the cohort. All P-values were obtained using exact Fisher test, except for age, using Welch's two-sample unequal variance. Complex persistent pain conditions were: episodic migraine, EM; temporomandibular disorders, TMD; irritable bowel syndrome, IBS; fibromyalgia, FM; vulvar vestibulitis, VVS; any of preceding, ANY; or controls, CTL. Number of CPPCs (#CPPCs) in any study participant range from 0 to 5, inclusively. Ancestries were: Caucasians, Cauc; African-Americans, Af-Am; and Others.
Supplementary Table S2. Mitochondria-wide association studies with CPPCs. Association tests were performed within various strata: by ancestry (ANC) and by sex (SEX). Ancestries were: Caucasians (CAU), African-Americans (AA), or others (OTH). For each stratum, only the top three strongest associated positions were reported: shown are the odds ratios (OR), standard error (SE), 95% confidence intervals for odds ratios (L95, U95), and P-values (P). Threshold for statistical significance after Bonferroni correction for effective number of SNPs indicated for each stratum (SIGNIF). NMISS is the number of samples with non-missing genotyping, phenotyping, and co-variables information. (A) Episodic migraine (EM). (B) Temporomandibular disorders (TMD). (C) Irritable bowel syndrome (IBS). (D) Fibromyalgia (FM). (E) Vulvar vestibulitis (VVS). (F) Presence of any CPPC. (G) Number of CPPCs.
Supplementary Table S3. Secondary analyses of association of CPPCs status with SNP m.2352T>C. CPPC include: episodic migraine (EM), temporomandibular disorders (TMD), irritable bowel syndrome (IBS), fibromyalgia (FM), vulvar vestibulitis (VVS), presence of any CPPC (ANY), number of CPPCs (NB), and principal component 1 (PC 1). Columns like Supplementary Table S2.
Supplementary Table S4. Haplotype-based association tests with CPPCs. CPPCs (pheno) were: episodic migraine (EM), temporomandibular disorders (TMD), irritable bowel syndrome (IBS), fibromyalgia (FM), vulvar vestibulitis (VVS), and presence of any CPPC (ANY). Tests were conducted in a sex-stratified fashion (sex): females-only (F), males-only (M), and combined (M+F). Tested mitochondrial haplotypes (haplo) were: H, L, U, T and J. Effect sizes (beta) and association P-values (pval) reported. Correction for multiple testing across all tests using FDR. For each haplotype, the number of cases (nb.cases.H) and controls (nb.ctrls.H) were shown alongside numbers for cases (nb.cases.X) and controls (nb.cases.X) not of that haplotype.
Supplementary Table S5. Rare variants association tests with CPPCs. Rare variants were pooled by genes or by oxidative phosphorylation complexes (OXPHOS) (SetID). Tests were conducted in a sex-stratified fashion: females-only (F), males-only (M), and combined (M+F). CPPCs were: episodic migraine (EM), temporomandibular disorders (TMD), irritable bowel syndrome (IBS), fibromyalgia (FM), vulvar vestibulitis (VVS), and presence of any CPPC (ANY). Correction for multiple testing using FDR within each combination of SNP pooling, sex stratification and CPPC. Also shown are: total number of markers in the SNP set (N.Marker.All), number of markers tested (N.Marker.Test), total minor allele count (MAC), number of individuals with the minor allele (m), the SKAT-O method (Method.bin), and minimum possible P-value (MAP).
Supplementary Table S6. Heteroplasmy association tests with CPPCs. CPPCs were: episodic migraine (EM), temporomandibular disorders (TMD), irritable bowel syndrome (IBS), fibromyalgia (FM), vulvar vestibulitis (VVS), and presence of any CPPC (ANY). Correction for multiple testing using FDR within each combination of sex stratification and CPPC, mitochondria-wide. Also shown are: average heteroplasmy levels in cases (avg_cases) as well as in controls (avg_ctrls), with the number of cases (nb_cases) and controls (nb_ctrls), and the average deep-sequencing depth at that position across all samples. Only the top 100 best P-values are shown.
Supplementary Table S7. Replication of association of SNP m.2352T>C in the OPPERA cohort. Both CPPC and OPPERA cohort results are shown. (A) Fibromyalgia. (B) All CPPCs, including episodic migraine (EM); temporomandibular disorders (TMD); irritable bowel syndrome (IBS); fibromyalgia (FM); vulvar vestibulitis (VVS); presence of any CPPC (ANY); number of CPPCs (NB), and principal component 1 (PC 1). Columns like Supplementary Table S2.
Supplementary Figure S1. mtDNA-Seq sequencing depth. (A) Shown is the linearized version of the mitochondrial chromosome, with annotated genomic features: rRNA (orange), protein coding genes (tan), control region or D-loop (grey). Depth of sequencing (D) displayed on the log10 scale, for mean sequencing (black), first (red) and third (green) quartiles of depth. (B) Zoom into the first and last 100 nucleotide regions.
Acknowledgments:
We would like to thank all participants enrolled in this study. We also thank Ashesh Saraiya and Greg Dijkman for performing mtDNA sequencing, as well as Drs. Essam Zaki and Eric Shoubridge for helpful discussions. Flow cytometry work was performed in the McGill University Flow Cytometry Core Facility of the Life Science Complex and supported by funding from the Canadian Foundation for Innovation.
Funding: The Complex Persistent Pain Conditions: Unique and Shared Pathways of Vulnerability Program Project were supported by NIH/National Institute of Neurological Disorders and Stroke (NINDS; https://www.ninds.nih.gov) grant NS045685 to the University of North Carolina at Chapel Hill. OPPERA was supported by the National Institute of Dental and Craniofacial Research (NIDCR; https://www.nidcr.nih.gov/) grant number U01DE017018. Funding for this work was also kindly provided by the Canadian Excellence Research Chairs (CERC) Program (www.cerc.gc.ca) grant CERC09 (to LD).
Footnotes
Competing interests: The authors declare no competing financial interests.
REFERENCES
- [1].Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. Washington (DC), 2011. [PubMed] [Google Scholar]
- [2].Amberger JS, Bocchini CA, Scott AF, Hamosh A. OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res 2019;47(D1):D1038–D1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 1999;23(2):147. [DOI] [PubMed] [Google Scholar]
- [4].Aschard H, Vilhjalmsson BJ, Greliche N, Morange PE, Tregouet DA, Kraft P. Maximizing the power of principal-component analysis of correlated phenotypes in genome-wide association studies. Am J Hum Genet 2014;94(5):662–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Boles RG, Zaki EA, Lavenbarg T, Hejazi R, Foran P, Freeborn J, Trilokekar S, McCallum R. Are pediatric and adult-onset cyclic vomiting syndrome (CVS) biologically different conditions? Relationship of adult-onset CVS with the migraine and pediatric CVS-associated common mtDNA polymorphisms 16519T and 3010A. Neurogastroenterol Motil 2009;21(9):936–e972. [DOI] [PubMed] [Google Scholar]
- [6].Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Brown A, Amunts A, Bai XC, Sugimoto Y, Edwards PC, Murshudov G, Scheres SHW, Ramakrishnan V. Structure of the large ribosomal subunit from human mitochondria. Science 2014;346(6210):718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Camilleri M, Carlson P, Zinsmeister AR, McKinzie S, Busciglio I, Burton D, Zaki EA, Boles RG. Mitochondrial DNA and gastrointestinal motor and sensory functions in health and functional gastrointestinal disorders. Am J Physiol Gastrointest Liver Physiol 2009;296(3):G510–516. [DOI] [PubMed] [Google Scholar]
- [9].Cordero MD, Cotan D, del-Pozo-Martin Y, Carrion AM, de Miguel M, Bullon P, Sanchez-Alcazar JA. Oral coenzyme Q10 supplementation improves clinical symptoms and recovers pathologic alterations in blood mononuclear cells in a fibromyalgia patient. Nutrition 2012;28(11–12):1200–1203. [DOI] [PubMed] [Google Scholar]
- [10].Cordero MD, De Miguel M, Moreno Fernandez AM, Carmona Lopez IM, Garrido Maraver J, Cotan D, Gomez Izquierdo L, Bonal P, Campa F, Bullon P, Navas P, Sanchez Alcazar JA. Mitochondrial dysfunction and mitophagy activation in blood mononuclear cells of fibromyalgia patients: implications in the pathogenesis of the disease. Arthritis Res Ther 2010;12(1):R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cordero MD, Moreno-Fernandez AM, deMiguel M, Bonal P, Campa F, Jimenez-Jimenez LM, Ruiz-Losada A, Sanchez-Dominguez B, Sanchez Alcazar JA, Salviati L, Navas P. Coenzyme Q10 distribution in blood is altered in patients with fibromyalgia. Clin Biochem 2009;42(7–8):732–735. [DOI] [PubMed] [Google Scholar]
- [12].Diatchenko L, Fillingim RB, Smith SB, Maixner W. The phenotypic and genetic signatures of common musculoskeletal pain conditions. Nat Rev Rheumatol 2013;9(6):340–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Diatchenko L, Nackley AG, Slade GD, Fillingim RB, Maixner W. Idiopathic pain disorders--pathways of vulnerability. Pain 2006;123(3):226–230. [DOI] [PubMed] [Google Scholar]
- [14].Dott W, Mistry P, Wright J, Cain K, Herbert KE. Modulation of mitochondrial bioenergetics in a skeletal muscle cell line model of mitochondrial toxicity. Redox Biol 2014;2:224–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dworkin SF, LeResche L. Research diagnostic criteria for temporomandibular disorders: review, criteria, examinations and specifications, critique. J Craniomandib Disord 1992;6(4):301–355. [PubMed] [Google Scholar]
- [16].Ester M, Kriegel H, Sander J, Xu X. A density-based algorithm for discovering clusters a density-based algorithm for discovering clusters in large spatial databases with noise Proceedings of the Second International Conference on Knowledge Discovery and Data Mining. Portland, Oregon: AAAI Press, 1996. pp. 226–231. [Google Scholar]
- [17].Flatters SJ. The contribution of mitochondria to sensory processing and pain. Prog Mol Biol Transl Sci 2015;131:119–146. [DOI] [PubMed] [Google Scholar]
- [18].Rare Gibson G. and common variants: twenty arguments. Nat Rev Genet 2012;13(2):135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Giles RE, Blanc H, Cann HM, Wallace DC. Maternal inheritance of human mitochondrial DNA. Proc Natl Acad Sci U S A 1980;77(11):6715–6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Goncalves VF, Giamberardino SN, Crowley JJ, Vawter MP, Saxena R, Bulik CM, Yilmaz Z, Hultman CM, Sklar P, Kennedy JL, Sullivan PF, Knight J. Examining the role of common and rare mitochondrial variants in schizophrenia. PLoS One 2018;13(1):e0191153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, Reed JC. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 2003;423(6938):456–461. [DOI] [PubMed] [Google Scholar]
- [22].Guo Y, Li J, Li CI, Shyr Y, Samuels DC. MitoSeek: extracting mitochondria information and performing high-throughput mitochondria sequencing analysis. Bioinformatics 2013;29(9):1210–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Johannes CB, Le TK, Zhou X, Johnston JA, Dworkin RH. The prevalence of chronic pain in United States adults: results of an Internet-based survey. J Pain 2010;11(11):1230–1239. [DOI] [PubMed] [Google Scholar]
- [24].Kallergi E, Kalef-Ezra E, Karagouni-Dalakoura K, Tokatlidis K. Common players in mitochondria biogenesis and neuronal protection against stress-induced apoptosis. Neurochem Res 2014;39(3):546–555. [DOI] [PubMed] [Google Scholar]
- [25].Kann O, Kovacs R. Mitochondria and neuronal activity. Am J Physiol Cell Physiol 2007;292(2):C641–657. [DOI] [PubMed] [Google Scholar]
- [26].Kearsey SE, Craig IW. Altered ribosomal RNA genes in mitochondria from mammalian cells with chloramphenicol resistance. Nature 1981;290(5807):607–608. [DOI] [PubMed] [Google Scholar]
- [27].Kopinski PK, Janssen KA, Schaefer PM, Trefely S, Perry CE, Potluri P, Tintos-Hernandez JA, Singh LN, Karch KR, Campbell SL, Doan MT, Jiang H, Nissim I, Nakamaru-Ogiso E, Wellen KE, Snyder NW, Garcia BA, Wallace DC. Regulation of nuclear epigenome by mitochondrial DNA heteroplasmy. Proc Natl Acad Sci U S A 2019;116(32):16028–16035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kraja AT, Liu C, Fetterman JL, Graff M, Have CT, Gu C, Yanek LR, Feitosa MF, Arking DE, Chasman DI, Young K, Ligthart S, Hill WD, Weiss S, Luan J, Giulianini F, Li-Gao R, Hartwig FP, Lin SJ, Wang L, Richardson TG, Yao J, Fernandez EP, Ghanbari M, Wojczynski MK, Lee WJ, Argos M, Armasu SM, Barve RA, Ryan KA, An P, Baranski TJ, Bielinski SJ, Bowden DW, Broeckel U, Christensen K, Chu AY, Corley J, Cox SR, Uitterlinden AG, Rivadeneira F, Cropp CD, Daw EW, van Heemst D, de Las Fuentes L, Gao H, Tzoulaki I, Ahluwalia TS, de Mutsert R, Emery LS, Erzurumluoglu AM, Perry JA, Fu M, Forouhi NG, Gu Z, Hai Y, Harris SE, Hemani G, Hunt SC, Irvin MR, Jonsson AE, Justice AE, Kerrison ND, Larson NB, Lin KH, Love-Gregory LD, Mathias RA, Lee JH, Nauck M, Noordam R, Ong KK, Pankow J, Patki A, Pattie A, Petersmann A, Qi Q, Ribel-Madsen R, Rohde R, Sandow K, Schnurr TM, Sofer T, Starr JM, Taylor AM, Teumer A, Timpson NJ, de Haan HG, Wang Y, Weeke PE, Williams C, Wu H, Yang W, Zeng D, Witte DR, Weir BS, Wareham NJ, Vestergaard H, Turner ST, Torp-Pedersen C, Stergiakouli E, Sheu WH, Rosendaal FR, Ikram MA, Franco OH, Ridker PM, Perls TT, Pedersen O, Nohr EA, Newman AB, Linneberg A, Langenberg C, Kilpelainen TO, Kardia SLR, Jorgensen ME, Jorgensen T, Sorensen TIA, Homuth G, Hansen T, Goodarzi MO, Deary IJ, Christensen C, Chen YI, Chakravarti A, Brandslund I, Bonnelykke K, Taylor KD, Wilson JG, Rodriguez S, Davies G, Horta BL, Thyagarajan B, Rao DC, Grarup N, Davila-Roman VG, Hudson G, Guo X, Arnett DK, Hayward C, Vaidya D, Mook-Kanamori DO, Tiwari HK, Levy D, Loos RJF, Dehghan A, Elliott P, Malik AN, Scott RA, Becker DM, de Andrade M, Province MA, Meigs JB, Rotter JI, North KE. Associations of Mitochondrial and Nuclear Mitochondrial Variants and Genes with Seven Metabolic Traits. Am J Hum Genet 2019;104(1):112–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, Karapetyan K, Katz K, Liu C, Maddipatla Z, Malheiro A, McDaniel K, Ovetsky M, Riley G, Zhou G, Holmes JB, Kattman BL, Maglott DR. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 2018;46(D1):D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9(4):357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Laurie CC, Doheny KF, Mirel DB, Pugh EW, Bierut LJ, Bhangale T, Boehm F, Caporaso NE, Cornelis MC, Edenberg HJ, Gabriel SB, Harris EL, Hu FB, Jacobs KB, Kraft P, Landi MT, Lumley T, Manolio TA, McHugh C, Painter I, Paschall J, Rice JP, Rice KM, Zheng X, Weir BS, Investigators G. Quality control and quality assurance in genotypic data for genome-wide association studies. Genet Epidemiol 2010;34(6):591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, Team NGESP-ELP, Christiani DC, Wurfel MM, Lin X. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet 2012;91(2):224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Li MX, Yeung JM, Cherny SS, Sham PC. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum Genet 2012;131(5):747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li Y, Beckman KB, Caberto C, Kazma R, Lum-Jones A, Haiman CA, Le Marchand L, Stram DO, Saxena R, Cheng I. Association of Genes, Pathways, and Haplogroups of the Mitochondrial Genome with the Risk of Colorectal Cancer: The Multiethnic Cohort. PLoS One 2015;10(9):e0136796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu ZY, Gua XX, Zhang RG, Wang XX, Ai J, Wang WF, Yang YS. Association of mitochondrial displacement loop polymorphisms with diarrhea-predominant irritable bowel syndrome: A preliminary study. J Dig Dis 2018;19(5):295–300. [DOI] [PubMed] [Google Scholar]
- [36].Logan IS. Pseudogenization of the Humanin gene is common in the mitochondrial DNA of many vertebrates. Zool Res 2017;38(4):198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Longstreth GF, Thompson WG, Chey WD, Houghton LA, Mearin F, Spiller RC. Functional bowel disorders. Gastroenterology 2006;130(5):1480–1491. [DOI] [PubMed] [Google Scholar]
- [38].Lott MT, Leipzig JN, Derbeneva O, Xie HM, Chalkia D, Sarmady M, Procaccio V, Wallace DC. mtDNA Variation and Analysis Using Mitomap and Mitomaster. Curr Protoc Bioinformatics 2013;44:1 23 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Maixner W, Diatchenko L, Dubner R, Fillingim RB, Greenspan JD, Knott C, Ohrbach R, Weir B, Slade GD. Orofacial pain prospective evaluation and risk assessment study--the OPPERA study. J Pain 2011;12(11 Suppl):T4–11 e11–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Maximov V, Martynenko A, Hunsmann G, Tarantul V. Mitochondrial 16S rRNA gene encodes a functional peptide, a potential drug for Alzheimer’s disease and target for cancer therapy. Med Hypotheses 2002;59(6):670–673. [DOI] [PubMed] [Google Scholar]
- [41].Meyer A, Laverny G, Bernardi L, Charles AL, Alsaleh G, Pottecher J, Sibilia J, Geny B. Mitochondria: An Organelle of Bacterial Origin Controlling Inflammation. Front Immunol 2018;9:536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Miller FJ, Rosenfeldt FL, Zhang C, Linnane AW, Nagley P. Precise determination of mitochondrial DNA copy number in human skeletal and cardiac muscle by a PCR-based assay: lack of change of copy number with age. Nucleic Acids Res 2003;31(11):e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mills MC, Rahal C. A scientometric review of genome-wide association studies. Commun Biol 2019;2:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Park JH, Gail MH, Weinberg CR, Carroll RJ, Chung CC, Wang Z, Chanock SJ, Fraumeni JF, Jr., Chatterjee N. Distribution of allele frequencies and effect sizes and their interrelationships for common genetic susceptibility variants. Proc Natl Acad Sci U S A 2011;108(44):18026–18031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Perry SW, Norman JP, Barbieri J, Brown EB, Gelbard HA. Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques 2011;50(2):98–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Poole C Controls who experienced hypothetical causal intermediates should not be excluded from case-control studies. Am J Epidemiol 1999;150(6):547–551. [DOI] [PubMed] [Google Scholar]
- [47].Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81(3):559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing, 2018. [Google Scholar]
- [49].Reichling DB, Green PG, Levine JD. The fundamental unit of pain is the cell. Pain 2013;154 Suppl 1:S2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Rishishwar L, Jordan IK. Implications of human evolution and admixture for mitochondrial replacement therapy. BMC Genomics 2017;18(1):140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res 2004;64(3):985–993. [DOI] [PubMed] [Google Scholar]
- [52].Sanchis-Gomar F, Garcia-Gimenez JL, Gomez-Cabrera MC, Pallardo FV. Mitochondrial biogenesis in health and disease. Molecular and therapeutic approaches. Curr Pharm Des 2014;20(35):5619–5633. [DOI] [PubMed] [Google Scholar]
- [53].Schwartz MH, Pan T. Function and origin of mistranslation in distinct cellular contexts. Crit Rev Biochem Mol Biol 2017;52(2):205–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Schwartz MH, Waldbauer JR, Zhang L, Pan T. Global tRNA misacylation induced by anaerobiosis and antibiotic exposure broadly increases stress resistance in Escherichia coli. Nucleic Acids Res 2016;44(21):10292–10303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Shin EJ, Tran HQ, Nguyen PT, Jeong JH, Nah SY, Jang CG, Nabeshima T, Kim HC. Role of Mitochondria in Methamphetamine-Induced Dopaminergic Neurotoxicity: Involvement in Oxidative Stress, Neuroinflammation, and Pro-apoptosis-A Review. Neurochem Res 2018;43(1):66–78. [DOI] [PubMed] [Google Scholar]
- [56].Siekevitz P Powerhouse of the cell. Scientific American 1957;197(1):131–140. [Google Scholar]
- [57].Smith SB, Parisien M, Bair E, Belfer I, Chabot-Dore AJ, Gris P, Khoury S, Tansley S, Torosyan Y, Zaykin DV, Bernhardt O, de Oliveira Serrano P, Gracely RH, Jain D, Jarvelin MR, Kaste LM, Kerr KF, Kocher T, Lahdesmaki R, Laniado N, Laurie CC, Laurie CA, Mannikko M, Meloto CB, Nackley AG, Nelson SC, Pesonen P, Ribeiro-Dasilva MC, Rizzatti-Barbosa CM, Sanders AE, Schwahn C, Sipila K, Sofer T, Teumer A, Mogil JS, Fillingim RB, Greenspan JD, Ohrbach R, Slade GD, Maixner W, Diatchenko L. Genome-wide association reveals contribution of MRAS to painful temporomandibular disorder in males. Pain 2019;160(3):579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Su B, Wang X, Zheng L, Perry G, Smith MA, Zhu X. Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochim Biophys Acta 2010;1802(1):135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sui BD, Xu TQ, Liu JW, Wei W, Zheng CX, Guo BL, Wang YY, Yang YL. Understanding the role of mitochondria in the pathogenesis of chronic pain. Postgrad Med J 2013;89(1058):709–714. [DOI] [PubMed] [Google Scholar]
- [60].Tajima H, Niikura T, Hashimoto Y, Ito Y, Kita Y, Terashita K, Yamazaki K, Koto A, Aiso S, Nishimoto I. Evidence for in vivo production of Humanin peptide, a neuroprotective factor against Alzheimer’s disease-related insults. Neurosci Lett 2002;324(3):227–231. [DOI] [PubMed] [Google Scholar]
- [61].van Tilburg MA, Zaki EA, Venkatesan T, Boles RG. Irritable bowel syndrome may be associated with maternal inheritance and mitochondrial DNA control region sequence variants. Dig Dis Sci 2014;59(7):1392–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Venkatesan T, Zaki EA, Kumar N, Sengupta J, Ali M, Malik B, Szabo A, van Tilburg MA, Boles RG. Quantitative pedigree analysis and mitochondrial DNA sequence variants in adults with cyclic vomiting syndrome. BMC Gastroenterol 2014;14:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Vianello D, Sevini F, Castellani G, Lomartire L, Capri M, Franceschi C. HAPLOFIND: a new method for high-throughput mtDNA haplogroup assignment. Hum Mutat 2013;34(9):1189–1194. [DOI] [PubMed] [Google Scholar]
- [64].Wang WF, Li X, Guo MZ, Chen JD, Yang YS, Peng LH, Wang YH, Zhang CY, Li HH. Mitochondrial ATP 6 and 8 polymorphisms in irritable bowel syndrome with diarrhea. World J Gastroenterol 2013;19(24):3847–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wei W, Tuna S, Keogh MJ, Smith KR, Aitman TJ, Beales PL, Bennett DL, Gale DP, Bitner-Glindzicz MAK, Black GC, Brennan P, Elliott P, Flinter FA, Floto RA, Houlden H, Irving M, Koziell A, Maher ER, Markus HS, Morrell NW, Newman WG, Roberts I, Sayer JA, Smith KGC, Taylor JC, Watkins H, Webster AR, Wilkie AOM, Williamson C, Diseases NB-R, Pilot GP-RD, Ashford S, Penkett CJ, Stirrups KE, Rendon A, Ouwehand WH, Bradley JR, Raymond FL, Caulfield M, Turro E, Chinnery PF. Germline selection shapes human mitochondrial DNA diversity. Science 2019;364(6442). [DOI] [PubMed] [Google Scholar]
- [66].Weissensteiner H, Pacher D, Kloss-Brandstatter A, Forer L, Specht G, Bandelt HJ, Kronenberg F, Salas A, Schonherr S. HaploGrep 2: mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res 2016;44(W1):W58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wiesner RJ, Ruegg JC, Morano I. Counting target molecules by exponential polymerase chain reaction: copy number of mitochondrial DNA in rat tissues. Biochem Biophys Res Commun 1992;183(2):553–559. [DOI] [PubMed] [Google Scholar]
- [68].Wilkins HM, Swerdlow RH. Relationships Between Mitochondria and Neuroinflammation: Implications for Alzheimer’s Disease. Curr Top Med Chem 2016;16(8):849–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wolfe F, Smythe HA, Yunus MB, Bennett RM, Bombardier C, Goldenberg DL, Tugwell P, Campbell SM, Abeles M, Clark P, et al. The American College of Rheumatology 1990 Criteria for the Classification of Fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum 1990;33(2):160–172. [DOI] [PubMed] [Google Scholar]
- [70].Wu Y, Chen M, Jiang J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019;49:35–45. [DOI] [PubMed] [Google Scholar]
- [71].Ye K, Lu J, Ma F, Keinan A, Gu Z. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc Natl Acad Sci U S A 2014;111(29):10654–10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Zaki EA, Freilinger T, Klopstock T, Baldwin EE, Heisner KR, Adams K, Dichgans M, Wagler S, Boles RG. Two common mitochondrial DNA polymorphisms are highly associated with migraine headache and cyclic vomiting syndrome. Cephalalgia 2009;29(7):719–728. [DOI] [PubMed] [Google Scholar]
- [73].Zorova LD, Popkov VA, Plotnikov EY, Silachev DN, Pevzner IB, Jankauskas SS, Babenko VA, Zorov SD, Balakireva AV, Juhaszova M, Sollott SJ, Zorov DB. Mitochondrial membrane potential. Anal Biochem 2018;552:50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Distribution of CPPCs and characteristics in males and in females. Participants can report more than one chronic pain condition, therefore the counts in each sex are higher than the total number of individuals in the cohort. All P-values were obtained using exact Fisher test, except for age, using Welch's two-sample unequal variance. Complex persistent pain conditions were: episodic migraine, EM; temporomandibular disorders, TMD; irritable bowel syndrome, IBS; fibromyalgia, FM; vulvar vestibulitis, VVS; any of preceding, ANY; or controls, CTL. Number of CPPCs (#CPPCs) in any study participant range from 0 to 5, inclusively. Ancestries were: Caucasians, Cauc; African-Americans, Af-Am; and Others.
Supplementary Table S2. Mitochondria-wide association studies with CPPCs. Association tests were performed within various strata: by ancestry (ANC) and by sex (SEX). Ancestries were: Caucasians (CAU), African-Americans (AA), or others (OTH). For each stratum, only the top three strongest associated positions were reported: shown are the odds ratios (OR), standard error (SE), 95% confidence intervals for odds ratios (L95, U95), and P-values (P). Threshold for statistical significance after Bonferroni correction for effective number of SNPs indicated for each stratum (SIGNIF). NMISS is the number of samples with non-missing genotyping, phenotyping, and co-variables information. (A) Episodic migraine (EM). (B) Temporomandibular disorders (TMD). (C) Irritable bowel syndrome (IBS). (D) Fibromyalgia (FM). (E) Vulvar vestibulitis (VVS). (F) Presence of any CPPC. (G) Number of CPPCs.
Supplementary Table S3. Secondary analyses of association of CPPCs status with SNP m.2352T>C. CPPC include: episodic migraine (EM), temporomandibular disorders (TMD), irritable bowel syndrome (IBS), fibromyalgia (FM), vulvar vestibulitis (VVS), presence of any CPPC (ANY), number of CPPCs (NB), and principal component 1 (PC 1). Columns like Supplementary Table S2.
Supplementary Table S4. Haplotype-based association tests with CPPCs. CPPCs (pheno) were: episodic migraine (EM), temporomandibular disorders (TMD), irritable bowel syndrome (IBS), fibromyalgia (FM), vulvar vestibulitis (VVS), and presence of any CPPC (ANY). Tests were conducted in a sex-stratified fashion (sex): females-only (F), males-only (M), and combined (M+F). Tested mitochondrial haplotypes (haplo) were: H, L, U, T and J. Effect sizes (beta) and association P-values (pval) reported. Correction for multiple testing across all tests using FDR. For each haplotype, the number of cases (nb.cases.H) and controls (nb.ctrls.H) were shown alongside numbers for cases (nb.cases.X) and controls (nb.cases.X) not of that haplotype.
Supplementary Table S5. Rare variants association tests with CPPCs. Rare variants were pooled by genes or by oxidative phosphorylation complexes (OXPHOS) (SetID). Tests were conducted in a sex-stratified fashion: females-only (F), males-only (M), and combined (M+F). CPPCs were: episodic migraine (EM), temporomandibular disorders (TMD), irritable bowel syndrome (IBS), fibromyalgia (FM), vulvar vestibulitis (VVS), and presence of any CPPC (ANY). Correction for multiple testing using FDR within each combination of SNP pooling, sex stratification and CPPC. Also shown are: total number of markers in the SNP set (N.Marker.All), number of markers tested (N.Marker.Test), total minor allele count (MAC), number of individuals with the minor allele (m), the SKAT-O method (Method.bin), and minimum possible P-value (MAP).
Supplementary Table S6. Heteroplasmy association tests with CPPCs. CPPCs were: episodic migraine (EM), temporomandibular disorders (TMD), irritable bowel syndrome (IBS), fibromyalgia (FM), vulvar vestibulitis (VVS), and presence of any CPPC (ANY). Correction for multiple testing using FDR within each combination of sex stratification and CPPC, mitochondria-wide. Also shown are: average heteroplasmy levels in cases (avg_cases) as well as in controls (avg_ctrls), with the number of cases (nb_cases) and controls (nb_ctrls), and the average deep-sequencing depth at that position across all samples. Only the top 100 best P-values are shown.
Supplementary Table S7. Replication of association of SNP m.2352T>C in the OPPERA cohort. Both CPPC and OPPERA cohort results are shown. (A) Fibromyalgia. (B) All CPPCs, including episodic migraine (EM); temporomandibular disorders (TMD); irritable bowel syndrome (IBS); fibromyalgia (FM); vulvar vestibulitis (VVS); presence of any CPPC (ANY); number of CPPCs (NB), and principal component 1 (PC 1). Columns like Supplementary Table S2.
Supplementary Figure S1. mtDNA-Seq sequencing depth. (A) Shown is the linearized version of the mitochondrial chromosome, with annotated genomic features: rRNA (orange), protein coding genes (tan), control region or D-loop (grey). Depth of sequencing (D) displayed on the log10 scale, for mean sequencing (black), first (red) and third (green) quartiles of depth. (B) Zoom into the first and last 100 nucleotide regions.