

Abstract
Osteoclasts are bone-resorbing cells that play an essential role in the remodeling of bone under physiological conditions and numerous pathological conditions, such as osteoporosis, bone metastasis, and inflammatory bone erosion. Nuclear receptors are crucial to various physiological processes, including metabolism, development and inflammation, and function as transcription factors to activate target genes. Synthetic ligands of nuclear receptors are also available for the treatment of metabolic and inflammatory diseases. However, dysregulated bone phenotypes have been documented in patients who take synthetic nuclear receptor ligands as a therapy. Therefore, the effect of nuclear receptors on bone cells has become an important area of exploration; additionally, the molecular mechanisms underlying the action of nuclear receptors in osteoclasts have not been completely understood. Here, we cover the recent progress in our understanding of the roles of nuclear receptors in osteoclasts.
Introduction
Nuclear receptors are a protein family of transcription factors, activated by ligand binding [1–3]. The body expresses more than 48 different nuclear receptors (NRs), officially divided into six groups based on phylogeny [4,5]. Group 1 includes the receptors TRs, RARs, VDR, and PPARs, RORs, Rev-erbs, CAR, PXR, LXRs, and others. Group 2 contains RXRs, COUP-TF, and HNF-4. Group 3 includes the steroid receptors (ERs, GRs, PRs, and ARs) as well as the ERRs. Group 4 contains the nerve growth factor-induced clone B group of orphan receptors including NGFI-B, NURR1, and NOR1. Group 5 includes the steroidogenic factor 1and the receptors related to the Drosophila FTZ-F1. Group 6 contains only the GCNF1 receptor, which does not fit well into any other subfamilies. NRs signal in both genomic and non-genomic ways (Figure 1). Ligand binding to NRs facilitates conformational changes promoting DNA binding to NR/ligand complexes and allowing for interactions with transcription factors. Ultimately, NRs regulate target gene expression through a consensus DNA binding site. However, recent findings suggest that NRs bind to other genomic sequences by interacting with additional transcription factors [6]. NRs also function via non-genomic effects, including regulation of protein activity, modulation of ion channels and intracellular calcium levels, and production of secondary messengers [7].
Figure 1. Nuclear receptor (NR) signaling.
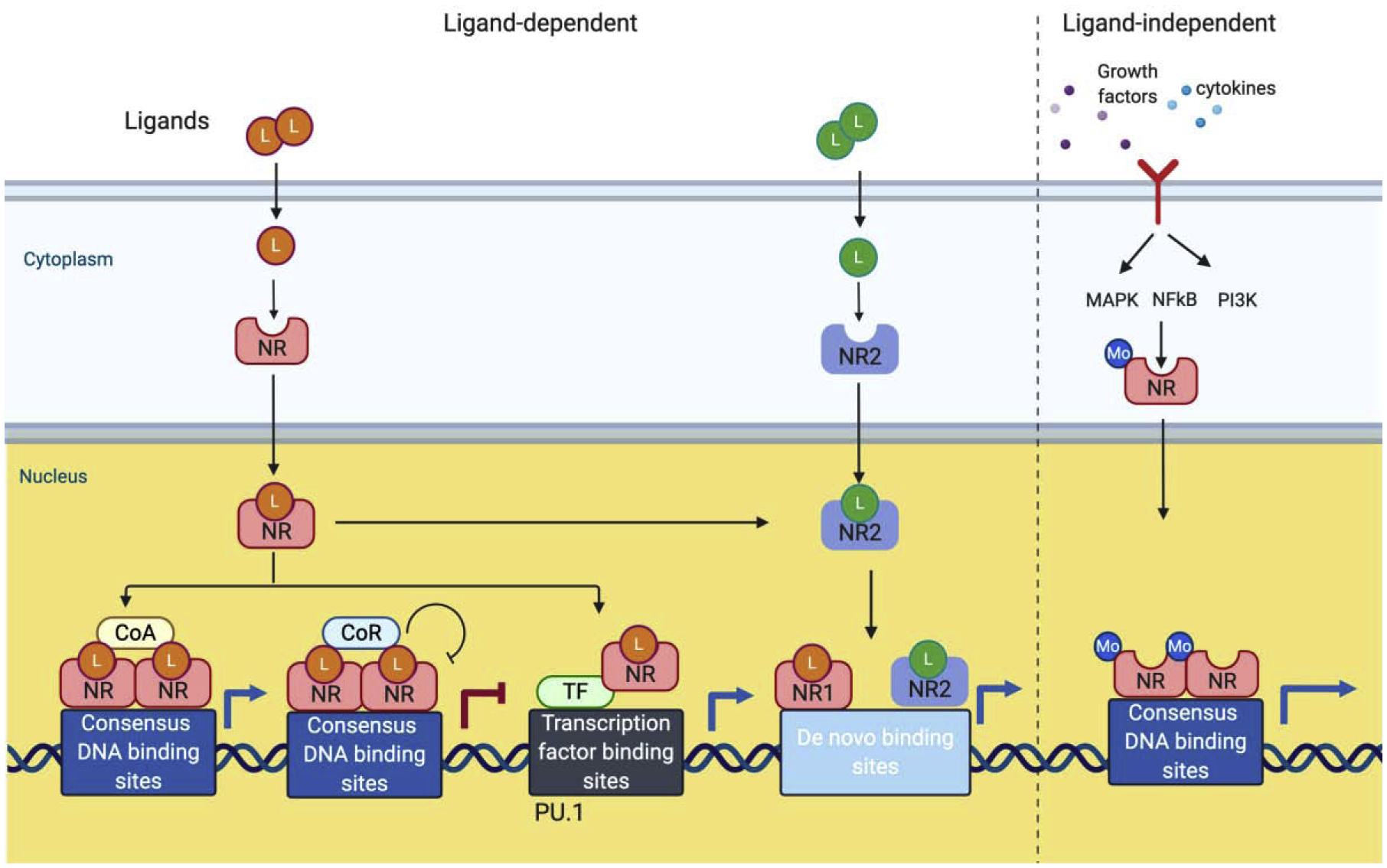
Upon binding on ligands (agonist or antagonist), cytoplasmic NRs undergo the conformational changes and translocate into the nucleus, where they can signal in genomic ways. Activated NRs also exert their action in non-genomic ways (not shown). In the nucleus, ligand-bound NRs directly bind to consensus DNA binding sites of NRs and activate or suppress target gene expression by interacting with co-activator (CoA) or co-repressor (CoR), respectively. Activated NRs also activate target by binding to other transcription factors (TF). Co-binding of NRs with pioneer factors such as PU.1 and osteoclast-specific TFs including NFATc1 need to be determined. Interplay between two different NRs open up de novo binding sites in chromosome and can regulate gene expression. Some NRs can be activated by different modification (Mo) including phosphorylation through other stimulus such as growth factors or cytokines.
Nuclear receptors show notable structural similarity [1–3]. Most members of the nuclear receptor group share a general structure (Figure 2). A-B is the N-terminal domain containing activation function-1 (AF-1) and is variable in size and sequence. C has the highly conserved DNA binding domain (DBD), and D is the hinge domain. E has the Ligand binding domain (LBD) that contains AF-2, which is important for the binding of coactivators. Ligand binding to LBD causes the conformational change of NRs. F is the highly variable C-terminal domain. The function of AF-1 is independent of ligand binding and AF-1 alone leads to a weak transcriptional activation. In contrast, the action of AF-2 is dependent on ligands and co-activation of AF-1 with AF-2 promotes a strong transcriptional activation of target genes. In the absence of ligand, nuclear receptor corepressors such as SMRT and NcoR bind to NRs to repress the basal transcription of the target genes. Although corepressors bind to corepressor NR boxes in the LBD, their binding is independent of AF-2. Ligand binding facilitates the conformational changes of AF-2 to trigger the release of corepressor and to induce the subsequent interaction with nuclear receptor coactivators such as SRC1 and TIF2. The deletion of AF-2 helix enhances the binding of corepressors to some NRs such as VDR. Moreover, AF-2 dependent recruitment of coactivators enhances the binding of transcriptional machinery to NRs and facilitates transcriptional activation of the target genes.
Figure 2. The structure of nuclear receptors.
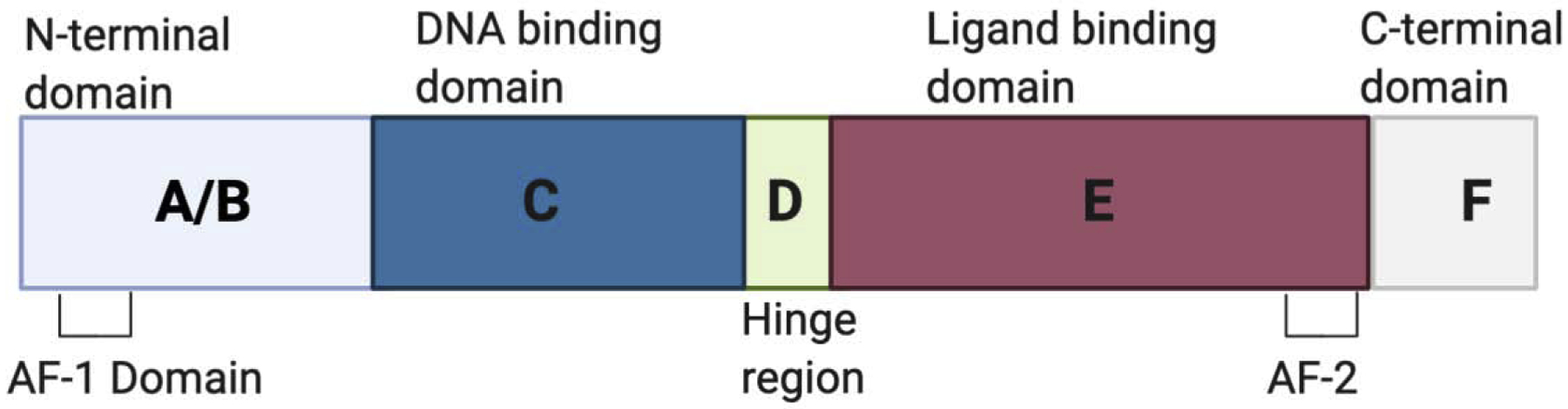
Nuclear receptors share structural homology. A-B is the N-terminal domain containing ligand-independent activation function −1 (AF-1) domain. C has the highly conserved DNA binding domain (DBD), and D is the hinge domain linking DBD to LBD. E has the Ligand binding domain (LBD) that contains AF-2, which is important for the binding of coactivators. Ligand binding to LBD causes the conformational change of nuclear receptors. F is the less conserved C-terminal domain.
Osteoclasts are multinucleated, bone-resorbing cells derived from myeloid lineage cells [8–10]. Osteoclastogenesis progresses in three different phases; commitment, fusion/maturation, and resorption [8] (Figure 3). Macrophage-colony stimulatory factor (M-CSF) and receptor activator of nuclear factor κB ligand (RANKL) are essential cytokines for osteoclast differentiation. M-CSF is a crucial factor for function, survival, and differentiation of monocytes/ macrophages and osteoclasts [11,12]. Engagement of RANKL to its receptor, RANK, activates multiple signaling pathways, including p38, ERK, JNK, AKT, and NF-κB [13], and this induces expression of osteoclast transcription factors, such as c-FOS and MYC [8]. With ITAM-mediated signals, RANKL induces and activates NFATc1, initiating osteoclast differentiation and promoting cell-cell fusion to generate multinuclear osteoclasts by induction of the membrane fusion promoter, DC-STAMP, and other fusion genes. During osteoclastogenesis, osteoclasts also undergo cytoskeletal remodeling to migrate and attach to the bone surface via the PI3K dependent mechanism [14]. In early phase of osteoclastogenesis, columnar actin puncta, known as podosomes, starts to form and is organized as actin ring as osteoclastogenesis progresses [15]. Fusion of mononuclear preosteoclasts occurs in a site-specific manner [16]. Mature osteoclasts are able to resorb bone by secreting bone resorbing factors. Osteoclastogenesis is regulated by RANKL and various factors including inflammatory mediators, PTH, and ligands of NRs such as hormones and vitamin D. Ostoprotegrin (OPG) is a decoy receptor for RANKL and is secreted from stromal cells and osteoblasts [17]. OPG prevents RANKL and RANK interactions and the ratio of RANKL-to OPG correlates with in vivo osteoclastogenesis and fine-tune osteoclast differentiation and bone remodeling.
Figure 3. Osteoclastogenesis.
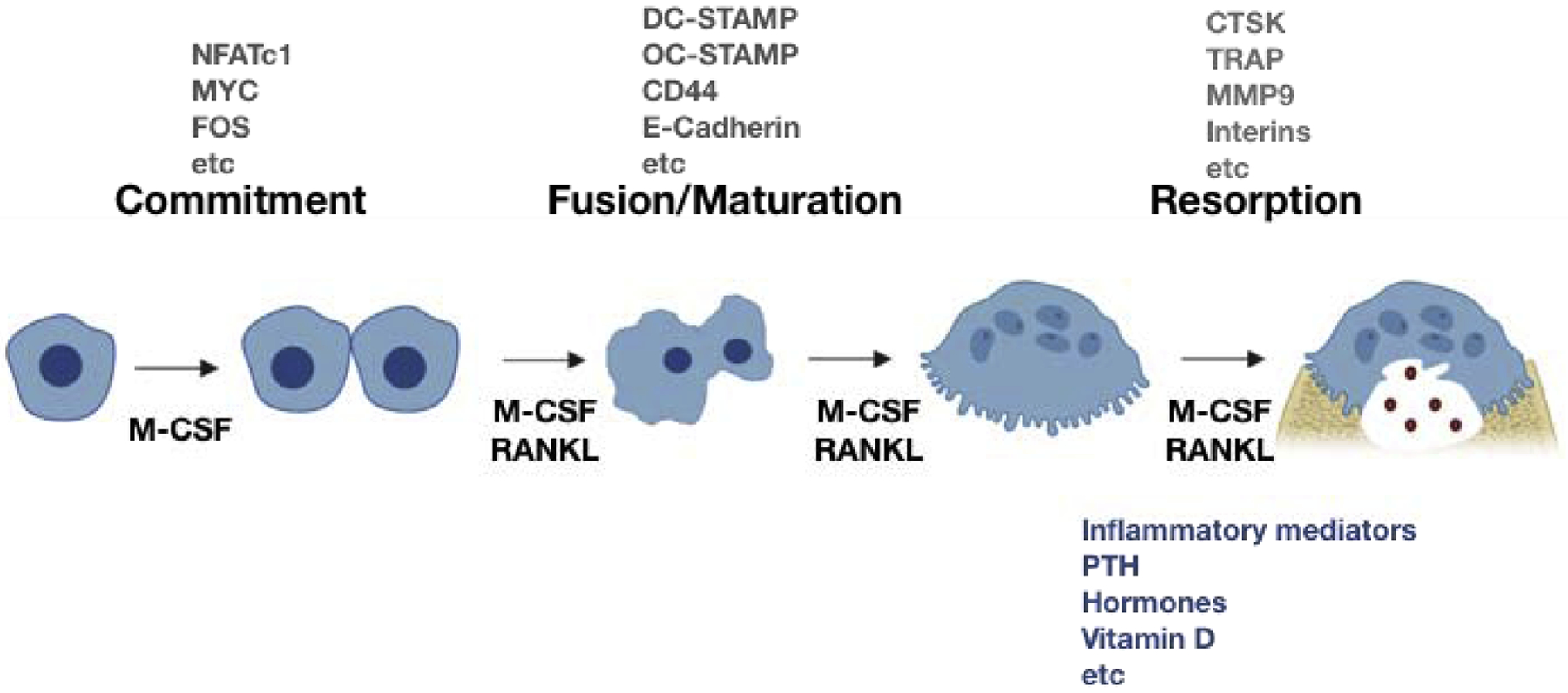
Osteoclastogenesis progresses in three different phases; commitment, fusion/maturation, and resorption. Osteoclasts differentiate from multipotential, myeloid lineage precursor cells under the influence of a variety of cytokines and local factors. M-CSF and RANKL are essential factors for osteoclastogenesis and induce various transcription osteoclasts. Cell-cell adhesion is initiated by E-cadherin, integrins, and cytoskeletal rearrangement, resulting in the podosome formation. Subsequently, cell-cell fusion is mediated by induction of fusion related genes such a DC-STAMP and OC-STAMP. Osteoclasts tightly attach to bone via the sealing zone and form the resorption pit. To break bone, osteoclasts resorb bone by releasing several enzymes, including CTSK, TRAP, and MMP9 and ions such as H+ and Cl−. The microenvironment surrounding osteoclasts such as inflammatory mediators, PTH, hormones, vitamin D greatly affects osteoclast differentiation and function.
Multiple NRs are expressed in osteoclasts [18,19] (Table1). It is established that NRs play a critical role in bone homeostasis and remodeling [18,19]. NRs affect skeleton size and shape during development and maintain skeletal homeostasis through both direct and indirect regulation of osteoclast differentiation and activity. Activation of NRs directly regulates the expression of target genes in osteoclasts. NRs indirectly influence osteoclastogenesis by modulating the RANKL/OPG ratio. Ligands for many NRs have not been discovered to date, and these NRs are often named orphan NRs based on their structural domain properties. Adopted NRs emerge when their endogenous ligands are characterized[20]. Many synthetic ligands for NRs have been developed, and pharmacological activation of NR with specific agonists has been used for the treatment of metabolic and inflammatory diseases. However, they are often associated with abnormal skeletal phenotypes and increased risk of fracture [21], and thus, it is important to understand the underlying mechanism of how NRs regulate osteoclast formation and function. In this review, we provide an update on six selected NRs with profound effects on osteoclasts and whose synthetic ligands are used in clinical treatments; additionally, the effects of other NRs on osteoclasts are reviewed in [18,19,22].
Table 1.
The function of NRs in osteoclasts
| Type | NRs | Natural ligands | Synthetic ligands | Effect on osteoclasts | Mechanisms in osteoclasts |
|---|---|---|---|---|---|
| RXRα | Retinoicacid, Retinol | Bexarotene | Regulate the proliferation of osteoclasts |
|
|
| RXRβ | |||||
| RXRγ | No expression in osteoclasts | N/A | |||
| PPARα | Polysatu rated fatty acid | Fenofibrate, statin | Ligand-induced suppression of osteoclastogen esis |
|
|
| PPARβ/δ | Unsaturated/saturated fatty acid | GW0742, L-165041 | Ligand-induced suppression of osteoclastogen esis |
|
|
| PPARγ | 15-deoxy-12, 14 prostaglandin J2 (15d-PGJ2) | Thiazolidinedione (rosiglitazone, ciglitazone, pioglitazone) | Ligand-induced stimulation of osteoclastogen esis in vivo: contradicting results |
|
|
| LXRα | Oxysterols (25OHC, 27OH, 24OHC, etc) | GW3965, T0901317 | Activation of LXRs by agonists suppresses osteoclastogen esis and apoptosis of enhances mature osteoclasts |
|
|
| LXRβ | |||||
| ERα | Oestrogen (ostetrone (E1), ostetradiol (E2), and oestriol (E3) | SERMs Synthetic estrogen (ethinyl estradiol (EE2), diethylstilbe strol(DES) | Negative regulation of osteoclastogenesis Induction of apoptosis in mature osteoclasts |
|
|
| ERβ | N/A |
|
|||
| GR | Glucocorticoid | Synthetic glucocorticoid (Dexamethasone, Prednisolone RU38486, A348441) | Suppress or increase osteoclast activity in vitro Prolonged life span of osteoclasts |
|
|
| ERRβ | N/A | 4-methylensterols DY131 (GSK9089) GSK4716 | N/A |
|
|
| ERRγ | N/A | Bisphenol A | Overexpressio n of ERRγ suppresses osteoclastogenesis |
|
|
| ERRα | Cholesterol | inverse agonist, XCT790, Isoflavones, flavone |
|
Estrogen receptor (ER)
Estrogen plays an important role in reproductive organ development, energy metabolism and bone homeostasis [23,24]. In post-menopausal women, estrogen is diminished, and estrogen treatment prevents bone loss [25]. SERM (selective estrogen receptor modulators) are FDA approved synthetic estrogen analogs that are also used to prevent bone loss [26]. ERs are major mediators for estrogen actions and have two isoforms; ERα and ERβ. ERα regulates gene expression in a tissue and cell typespecific manner through the coordinated actions of transcription factors and co-factors. Following estrogen binding, activated nuclear ERs bind to genomic DNA, containing an estrogen response element (ERE), and co-regulators are recruited to modulate ER-mediated gene transcription.
Estrogen-deficiency in postmenopausal women, ovariectomized women, and ERα-deficient men all have diminished cortical and trabecular bone mineral density and enhanced osteoclast-mediated bone resorption [27,28]. In contrast, female ERα-deficient mice show increased bone mass with low bone turnover, and ERβ-deficient mice exhibit a milder bone phenotype compared to ERα-deficient mice [29,30]. In addition, ovariectomized female ERα-deficient mice show decreased bone mass. The discrepancy in bone phenotype for female global ERα-deficient mice can be explained through elevated circulating testosterone and androgen signaling. However, ERαf/f- LysozymeM cre mice, carrying a conditional deletion of ERα in osteoclast precursor cells, show increased osteoclast number and decreased trabecular bone mass [31]. ERαf/f- CathepsinK cre mice, resulting in the deletion of ERα in mature osteoclasts, exhibit decreased trabecular bone mass only in females [32]. These results suggest that activation of ERs in osteoclasts negatively regulates osteoclast differentiation and bone resorption.
Estrogen promotes osteoclast apoptosis and regulates osteoclast lifespan by inducing expression of FAS ligands (FASL)[32]. Activation of ERα also induces the destabilization of HIF1α (hypoxia inducible factor) in osteoclasts [33]. The enhanced osteoclastogenesis in cells from ERαf/f-CathepsinK cre mice is reversed by HIF1α- deficiency. HIF1α is stabilized and accumulated during osteoclastogenesis, positively regulating osteoclast differentiation[34]. Together, estrogen and SERM suppress the accumulation of HIF1α in osteoclasts [35]. Estrogen indirectly regulates osteoclastogenesis by inducing FASL in osteoblasts and regulating the RANKL/OPG ratio [36]. ERα regulates gene expression in a ligand-independent manner. ERα regulates CREB phosphorylation by activating other kinases, such as mitogen activated protein kinase (MAPK), and controlling CREB-mediated gene regulation[37]. ERα also undergoes posttranslational modification through signaling pathways independent of ligands [38]. The action of ERα is regulated by growth factors and cytokines [39] and by competing with ERE-BP (estrogen response element-binding protein) for occupancy of ERRs. ERE-BP overexpression causes estrogen resistance [40] and enhances osteoclast differentiation[41]. Finally, myeloid-specific deletion of Sirt6 decreases ERα protein levels and apoptosis in pre-osteoclasts. Sirt6 deacetylates ERα protein, preventing further proteasome degradation [42]. The effect of ERβ on osteoclasts has not been characterized. Taken together, estrogen therapy would be beneficial for treating bone loss by suppressing osteoclasts and promoting osteoblast apoptosis. However, other cells also express ERs and estrogen therapy shows the adverse effect on other organs such as uterine and breast [43,44]. Importantly, SERMs such as roxifene is used to prevent or treat bone loss in women after menopause and functions as an estrogen blocker in uterine and breast to diminish the side effects of estrogen therapy.
Estrogen related receptor (ERR)
ERRs are important in energy metabolism and cancer development and express in tissues with high-energy demands [45]. ERRs were discovered from their sequence similarity to DNA binding domains of ERα [46]. However, ERRs do not bind to estrogen. They contain three members; ERRα, ERRβ, and ERRγ [46]. Estrogen-related receptors (ERRs) bind to the ERR response element (ERRE) and the ERE [46]. The ligands that interact with ERRs are unclear, so they are orphan NRs [47]. However, a recent study identified cholesterol as a potential endogenous ligand for ERRα [45].
ERRα regulates many key functions in osteoclasts. It is a positive regulator of osteoclast differentiation by mediating metabolic reprogramming [48,49]. ERRα also regulates metabolic genes controlling mitochondrial respiration in osteoclasts [49]. During osteoclastogenesis, MYC induces ERRα to regulate oxidative phosphorylation [49]. Several cofactors interact with ERRs, including peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1-α) and PGC1-β [50]. In osteoclasts, ERRα complexes with PGC1β upon RANKL stimulation to regulate target genes such as Ndg2, Aco2, and IDH3 [48]. Beyond regulating osteoclast functions, ERRα also affects pathological bone loss. Deletion or pharmacological inhibition of ERRα attenuates ovariectomy-induced bone loss in mouse models of osteoporosis [49,51]. In addition, cholesterol, a natural ERRα ligand, promotes osteoclastogenesis, and its effect on osteoclast differentiation was diminished in ERRα-deficient mice [45]. ERRα is a potential therapeutic target in pathologic bone loss. In addition to ERRα agonists and antagonists, cholesterol level modulation can regulate ERRα activity. The physiological role of ERRα in osteoclasts is not conclusive due to contradicting reports using global ERRα-deficient mice. While an increase in osteoblast number, and a decrease in osteoclast number are reported in ERRα-deficient mice by Wei et al[48], other studies only detect an increase in osteoblast number with no change in osteoclast number[51,52]. Bisphosphonates are an anti-resorptive drug and are widely used for suppressing osteoclast-mediated bone resorption[53]. Bisphosphonates inhibit two enzymes in the cholesterol biosynthesis pathway. Although the linking of the action of bisphosphonates to ERRα is not well established, pharmacological inhibition of ERRα can be useful treatment for pathological bone loss due to the dual effect of ERRα on bone cells and on cholesterol-mediated actions in bone.
Compared to ERRα less is known about ERRβ and ERRγ in osteoclasts. To date, no study addresses ERRβ’s role in osteoclasts. ERRγ is high in osteoclast precursor cells and decreased in osteoclasts. Overexpression of ERRγ suppresses osteoclast differentiation by inhibiting RANKL-induced NF-κB pathway [54], suggesting that ERRγ is a negative regulator of osteoclastogenesis. Administration of ERRγ-agonists also protects mice from pathological bone loss[54]. However, ERRγ-deficiency increases bone mass with no changes in osteoclasts [55], suggesting that the effect of ERRγ on bone remodeling is attributed to regulating osteoblast function under physiological conditions.
Glucocorticoid receptor (GR)
Endogenous glucocorticoids (GCs) are essential for physiological and developmental processes [56]. Moreover, synthetic GCs are an effective therapy for autoimmune and inflammatory diseases. Both endogenous and synthetic GCs bind to GRs to regulate transcription of targets [57]. Human GRs are endocrine NRs, containing GRα and GRβ. Human GRα is present in the cytoplasm of almost every cell. It is a ligand-dependent transcription factor, mediating many known actions of glucocorticoids. In contrast to GRα, human GRβ is in the nucleus and does not bind to GCs or activate GC-response genes [58]. GRβ is a dominant negative factor for GRα and confers glucocorticoid resistance in inflammatory diseases [58]. The cellular response to GCs varies in sensitivity and specificity among individuals and within tissues of the same individual [59]. GR binds to the GRE (GC-responsive element) of targets to regulate gene expression by interacting with transcription factors [60].
Therapeutic use of GCs come with serious side effects, such as glucocorticoid-induced osteoporosis (GIO) [61,62]. GIO has been studied, focusing on the effects of GCs on osteogenesis. GCs block osteoblast differentiation and accelerate apoptosis of osteoblasts and osteocytes [63]. Several different mechanisms underlie the impaired osteogenesis by GCs have been suggested. GR-mediated suppression on osteogenic genes via regulation of chromatin remodeling complexes, BRG1 and BRM [64], suppressing IL-11 [65], and inducing WNT antagonists such as DKK1, sFRP1, and WIF [66–68]. In addition, GCs reduce OPG and increase RANKL in osteocytes/osteoblasts, and an altered RANKL to OPG ratio by GCs promote osteoclast-mediated cortical bone resorption [69]. Although the indirect role of GCs in osteoclasts are well established, GCs’ direct effect on osteoclasts is inconclusive [70]. GCs enhance osteoclast activity and differentiation in vitro, especially, when precursor cells were seeded in bone. These promoting effects of GCs decrease in dimerization-defective GRdim osteoclasts [71], showing that GC-mediated enhancement of osteoclastogenesis is GR-dependent. Other studies show that the administration of GCs suppresses osteoclast precursors, but osteoclast numbers remain constant due to their prolonged life span [72]. However, treatment with dexamethasone (DEX) suppresses osteoclast-spreading and bone resorption. DEX-mediated suppression of osteoclastogenesis is undetectable in GRαf/f- LysozymeM cre mice, a conditional deletion of GRα in osteoclast precursor cells [73]. Although one study showed that GCs regulate osteoclast activity by PKCIIβ-mediated TRPV1 and CB2 regulation [74], the mechanisms underlying GR’s activity under physiological and pathological conditions are not fully characterized. Glucocorticoid-induced bone loss is biphasic with a rapid early phase of bone loss (5~15% per year) followed by a constant low rate of bone loss (2% per year) [75]. Despite the controversial effects of GCs on osteoclasts, the timing and dosage of GCs generate the differential effect on osteoclastogenesis (Kaneko et al., unpublished observations), and the combinational effects of GCs that increase apoptosis of osteoblasts and osteocytes, increase adipocyte hypertrophy, decrease bone vasculature, and decrease osteoclast apoptosis might explain the clinical effects of GCs.
Peroxisome Proliferator-Activator receptor (PPARγ)
The peroxisome proliferator-activated receptor (PPAR) family of nuclear receptors regulates lipid-related genes and functions as a metabolic sensor [76]. PPARs are activated by natural ligands, such as fatty acids [77]. They contain three members, PPARα, PPARβ/δ, and PPARγ. PPARδ and PPARγ express in human and murine osteoclasts, but PPARα expresses only in human osteoclasts. PPARs form heterodimeric complexes with RXR (retinoid X receptor), and PPAR/RXR complexes bind to peroxisome proliferator response elements (PPRE) in the genome with different cofactors. PPARγ has been involved in regulation of the bone metabolism [78–80], but the role of other PPARs in osteoclast differentiation and function is not well characterized. PPARγ is originally identified as the master regulator of adipogenic differentiation [81]. Ligands of PPARγ are naturally occurring lipophilic ligands, such as polyunsaturated fatty acids and oxidized prostaglandin J2, as well as the synthetic agonist, TZDs [81,82]. PPARα-deficient mice have no bone phenotype, but treatment with fenofibrate, a synthetic ligand for PPARα, decreases osteoclasts, increases osteoblast differentiation [83], and it protects from OVX-induced bone loss in rats [84]. PPARδ is a key regulator of fatty acid metabolism in muscle [85] and has been implicated as a regulator of the crosstalk between osteoblasts and osteoclasts [86]. The treatment of GW501516, an agonist of PPARβ/δ, promotes Wnt-dependent and β-catenin-dependent signaling in osteoblasts and the differentiation of osteoblasts [86]. Activation of PPARβ/δ by GW501516 suppresses osteoclastogenesis in a osteoblast dependent manner and also protects mice from OVX-induced bone loss by altering the OPG/RANKL ratio [86]. Conversely, PPARδ-Sox2 cre mice exhibit osteopenic phenotype with diminished osteoclast number in vivo. Although bone formation in vivo is not altered in PPARδ-Sox2 cre mice, osteoblasts increases the expression of OPG to attenuate osteoclastogenesis. Therefore, PPARδ regulates osteoclasts by controlling the crosstalk between osteoclasts and osteoblasts.
The thiazolidinedione (TZD) family is a synthetic agonist for PPARγ used to treat insulin-resistant type II diabetes, and both pioglitazone and rosiglitazone are the currently approved for clinical use. However, it has been shown that long-term usage of TZD increases fracture risk and bone loss [87,88] with higher incidence in women than men [89]. TZDs regulate bone metabolisms by supporting osteoclastogenesis [48,80] and suppressing osteoblastogenesis [78]. Rosiglitazone-fed mice show significant decrease in BMD, lower cortical thickness and trabecular bone volume [90,91]. TZDs change marrow structure and function showing a decrease in osteoblast number, an increase in marrow fat cells and osteoclast number [92]. Although TZDs decrease osteoblasts and induce osteocyte apoptosis, in aged animal, TZD-mediated bone loss is dominantly associated with an increased osteoclastogenesis by increased RANKL expression [92]. PGC1β is required for TZD-enhanced osteoclastogenesis and is induced by β-catenin in cells treated with rosiglitazone [48]. PGC1β is a co-activator for PPARγ activation of osteoclastogenesis and regulates osteoclastogenesis [93]. Both PGC1β and PPARγ bind to ERRα’s promoter to induce ERRα [48], suggesting several NRs cooperatively regulate osteoclast function. However, the physiological role of PPARγ in osteoclasts is controversial because the opposite bone phenotype of PPARγ- deficient mice has been observed from two different laboratories. Wan et al show that osteoclastogenesis and bone mass of PPARγ-Tie2cre mice decreased compared to control mice, and PPARγ promoted osteoclastogenesis by enhancing c-FOS expression [80]. In contrast, the Teitalbum group demonstrated that various osteoclast-lineage cell specific deletion of PPARγ using Tie2cre, LysozymeM cre, Vav1 cre, and RANK cre mice (PPARγ cKO mice) all exhibits no changes in in vitro osteoclastogenesis and unaltered bone mass [94], and PPARγ cKO mice express similar levels of osteoclast-specific proteins including c-FOS, cathepsin K, and integrin β3 compared to wild type mice. Furthermore, PPARγ deficiency cannot protect mice from OVX-induced bone loss [94], suggesting that PPARγ does not regulate physiological and pathological osteoclastogenesis. In other studies, ASXL2-deficient mice exhibit osteopenic phenotype with diminished osteoclast activity [95]. As ASXL2 is a coactivator of PPARγ, this data may also suggest the physiological role of PPARγ in osteoclastogenesis. Although these contradicting results argue that basal activation of PPARγ in early stages of osteoclast differentiation, it is clear that drug-induced activation of PPARγ promotes osteoclastogenesis, and thus, osteoclasts likely contribute to the increased fracture risk by TZDs. Therefore, it is important to understand the mechanisms how TZDs regulate osteoclast differentiation and activity.
Liver X receptors (LXRs)
LXRα and LXRβ are important regulators of cholesterol, glucose metabolism, and inflammatory response [96]. While LXRβ is ubiquitously expressed, LXRα expression is limited to few tissues including liver, adipose tissue, intestines, lungs, and macrophages. LXRα and LXRβ proteins have considerable sequence homology and respond to the same ligands. The endogenous ligands for LXR are sterol metabolites, including oxysterols [97,98]. LXRs form heterodimers with Retinoid X Receptor (RXR) and bind to LXR response elements (LXREs). Synthetic agonists including GW3965 and T0901317, inverse agonists such as SR9238, and antagonists such as GSK2033 for LXRs have been developed. Although bone mass is not altered in LXRα-, LXRβ-, or LXRαβ-deficient mice, some bone parameters are changed in LXR-deficient mice-LXRα-deficient female mice that significantly increase total BMD and femur length at 16 weeks, and LXRβ-deficient male mice significantly decrease trabecular BMD at 16 weeks [99]. In addition, bone mass is not altered by the treatment with the synthetic LXR ligands GW3965 or T0901317 in mice [100]. However, activation of LXRs by GW3965 regulates both osteoclasts and osteoblasts, and it is well documented that LXR agonists are important players for osteoclastogenesis. Activation of LXRs inhibits osteoclast differentiation and activity by regulating MITF (microphthalmia-associated transcription factor)-NFATc1 expression via an LXRβ-dependent mechanism [101]. The treatment of GW3965 or T0901317 protects mice from OVX-induced bone loss and PTH-induced bone loss by decreasing the RANKL-to OPG ratio [102]. GW3965 treatment block osteoblast-induced osteoclast formation in osteoclast/osteoblast co-culture by regulating the RANKL to OPG ratio [102]. GW3965 also suppresses LPS-induced osteoclastogenesis by regulating Akt pathway via an LXRβ-dependent mechanism [103]. LXR agonists inhibit RANKL-induced c-Fos and NFATc1 expression and accelerate apoptosis in mature osteoclasts by the induction of caspase-3 and −9 activity and Bim expression [104]. LXR agonists have a minimal effect on physiological bone homeostasis, but activation of LXRs is useful for estrogen deficiency-induced bone loss or inflammatory bone loss. These results suggest that activation of LXRs would be beneficial for the treatment of pathological bone loss. However, in animal models, LXR agonists also increase fatty acid and triglyceride (TG) synthesis, which also affect osteoclast activity and function. Thus, both beneficial and adverse effects of LXR agonists in other cells need to be considered for the use of LXR agonists for bone diseases.
Retinoic X receptor (RXR)
Retinoid X receptor (RXR) controls a wide variety of cellular processes such as lipid and glucose metabolism. RXRs consist of three subtypes- RXRα, RXRβ, and RXRγ. RXRα and RXRβ express in osteoclasts and myeloid cells [105]. RXRs form either homodimers or heterodimers with other nuclear receptors, including LXRs, VDR, or PPARs [106,107]. RXRαβ-Mx1cre mice (RXR KO), which have deletion of RXR in any IFN-responsible cells including osteoclasts and osteoblasts, exhibit increased cortical and trabecular bone mass, but the number of osteoclasts remains the same [108]. RXR-deficient osteoclasts form bigger osteoclasts with low bone resorption activity than wild type osteoclasts and express low MAFB, resulting in altered proliferation of osteoclasts. Paradoxically, both RXR-deficiency as well as pharmacological activation of RXR by bexarotene protect mice from OVX-induced bone loss. This protective role of RXR activation in OVX-induced bone loss can be explained by altered MAFB expression. While RXR homodimers can directly bind to the promoter of Mafb to induce MAFB expression, pharmacological activation of RXR leads to the formation of RXR/LXR heterodimers that indirectly increase MAFB by inducing SREBP1c expression. Bexarotene is the only approved RXR agonist for the clinical use and has been used as a treatment for cutaneous T cell lymphoma[109]. Bexarotene has no effect on physiological bone remodeling in mice [108]. Although RXR activation protects mice from OVX-induced bone loss [108], the bone mass is significantly but marginally reversed by bexarotene. Therefore, it further needs to be evaluated the effect of RXR activation on physiological and pathological bone remodeling.
Conclusions
Despite the importance of NRs, many aspects of NR’s skeletal actions are not fully characterized. Studies have reported the alteration of osteoclast differentiation and activity by synthetic ligands of NRs (Figure 4); however, knowledge of the mechanisms of NR’s action in osteoclasts is limited. Several factors should be considered. First, genome-wide screening of NRs has identified distinct cell-type specific NR binding sites, their consensus DNA binding sites, and cofactors. For example, genomic mapping of ERα identifies co-regulators of ERα, including FOXA1 in breast cancer cells [110]. However, there are no genomic mapping studies of NRs in osteoclasts. It has been shown that nuclear receptor binding profiles are tissue-specific and species-specific. In addition, in macrophages the co-binding of PU.1, a key factor for myeloid and B cell development, with PPARγ has been found[111], suggesting the co-binding between NRs and key transcription factors facilitate the function of cells. Thus, identifying the osteoclast-specific binding sites of NRs and other co-factors, and mapping the interaction of NRs with the osteoclast-specific transcription factors such as NFATc1 and IRF8 needs to be determined [112]. Secondly, as NRs are regulated by other signals, cross-regulation of NRs with important signals in osteoclasts needs to be determined. Third, multiple NRs co-exist in osteoclasts, and the interplay between NRs reprograms chromatin to open sites in addition to NR’s designated binding sites. The crosstalk between NRs needs to be characterized during osteoclastogenesis. Fourth, alternative translation and post-translation modification of NRs generate multiple functional isoforms, and the exact isoform relevant to osteoclast activity needs to be characterized. Fifth, NRs have profound effects on osteoblasts, bone-forming cells. Since the effect comes from global knock mice, it is difficult to differentiate contributions to osteoclasts and osteoblasts. Therefore, osteoclast-specific NR-deficient mice are needed to understand the contributions to osteoclasts. Since NRs play an important role in skeletal biology, further studies to uncover transcriptional networks driven by NRs, binding sites in osteoclasts and evidence that they will affect bone under physiological and pathological conditions will provide a better method of targeting NRs without creating skeletal abnormalities.
Figure 4. Nuclear receptor (NR) signaling in osteoclasts.
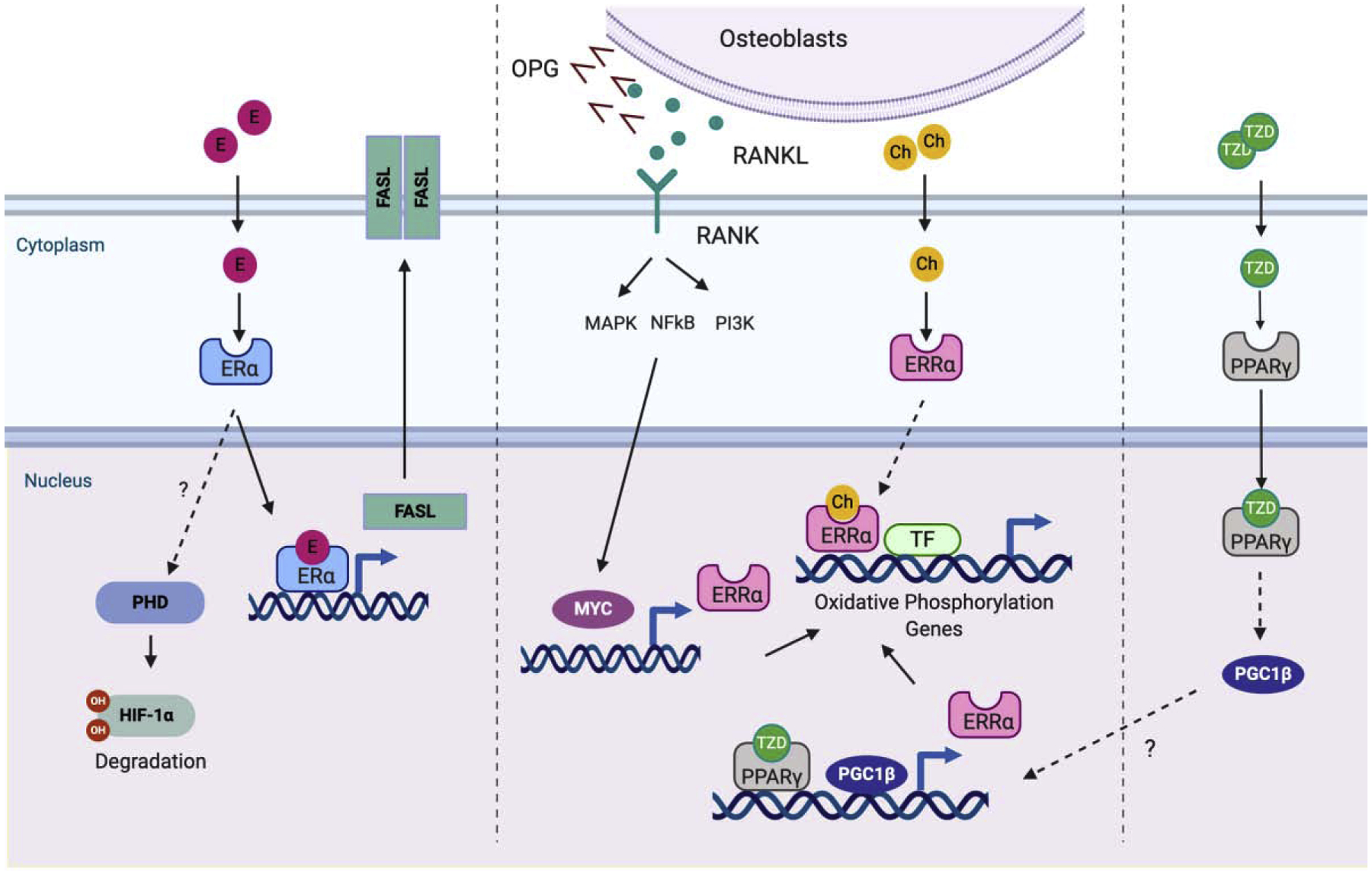
On binding of RANKL to RANK, MYC is induced. MYC binds to the promoter of ERRα to induce the expression of ERRα. ERRα subsequently activates genes related to oxidative phosphorylation which is a key metabolic mechanism of osteoclast differentiation. The MYC-ERRα axis plays an important role in osteoclast differentiation. The thiazolidinedione (TZD) family binds to and activates PPARγ. PPARγ indirectly induces PGC1β. Both PPARγ and PGC1β bind to the promoter of ERRα. Estrogen negatively regulates osteoclastogenesis. Estrogen binds to and activates ERα. Activated ERα induces HIF1α degradation and FASL expression to enhance apoptosis of osteoclasts. Osteoblasts/osteocytes secrete OPG and RANKL. The activation of NRs in osteoblasts regulates the ratio of RANKL to OPG to modify RANKL-induced osteoclastogenesis. factors to initiate the osteoclastogenenic program. Then committed cells (osteoclast precursor cells) fuse to each other and generate multinucleated
Acknowledgements
This work was supported by the National Institutes of Health 5R01 AR069562; 5R01 AR073156 (to P.M.K.H.). Figures were generated by www.biorender.com
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
Nothing declared.
References
Papers of particular interest have been highlighted as:
*of special interest
**of outstanding interest
- 1.Dhiman VK, Bolt MJ, White KP: Nuclear receptors in cancer - uncovering new and evolving roles through genomic analysis. Nat Rev Genet 2018, 19:160–174. [DOI] [PubMed] [Google Scholar]
- 2.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, et al. : The nuclear receptor superfamily: the second decade. Cell 1995, 83:835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao L, Hu H, Gustafsson JA, Zhou S: Nuclear Receptors in Cancer Inflammation and Immunity. Trends Immunol 2020, 41:172–185. [DOI] [PubMed] [Google Scholar]
- 4.Nuclear Receptors Nomenclature C: A unified nomenclature system for the nuclear receptor superfamily. Cell 1999, 97:161–163. [DOI] [PubMed] [Google Scholar]
- 5.Germain P, Staels B, Dacquet C, Spedding M, Laudet V: Overview of nomenclature of nuclear receptors. Pharmacol Rev 2006, 58:685–704. [DOI] [PubMed] [Google Scholar]
- 6.Kato S, Yokoyama A, Fujiki R: Nuclear receptor coregulators merge transcriptional coregulation with epigenetic regulation. Trends Biochem Sci 2011, 36:272–281. [DOI] [PubMed] [Google Scholar]
- 7.Unsworth AJ, Flora GD, Gibbins JM: Non-genomic effects of nuclear receptors: insights from the anucleate platelet. Cardiovasc Res 2018, 114:645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park-Min KH: Mechanisms involved in normal and pathological osteoclastogenesis. Cell Mol Life Sci 2018, 75:2519–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lorenzo J, Horowitz M, Choi Y: Osteoimmunology: interactions of the bone and immune system. Endocr Rev 2008, 29:403–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsukasaki M, Takayanagi H: Osteoimmunology: evolving concepts in bone immune interactions in health and disease. Nat Rev Immunol 2019, 19:626–642. [DOI] [PubMed] [Google Scholar]
- 11.Hamilton JA: Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 2008, 8:533–544. [DOI] [PubMed] [Google Scholar]
- 12.Arai F, Miyamoto T, Ohneda O, Inada T, Sudo T, Brasel K, Miyata T, Anderson DM, Suda Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-fms and receptor activator of nuclear factor kB (RANK) receptors. J Experimental Medicine 1999, 190:1741–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka S, Nakamura K, Takahasi N, Suda T: Role of RANKL in physiological and pathological bone resorption and therapeutics targeting the RANKL-RANK signaling system. Immunol Rev 2005, 208:30–49. [DOI] [PubMed] [Google Scholar]
- 14.Oikawa T, Kuroda Y, Matsuo K: Regulation of osteoclasts by membrane-derived lipid mediators. Cell Mol Life Sci 2013, 70:3341–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jurdic P, Saltel F, Chabadel A, Destaing O: Podosome and sealing zone: specificity of the osteoclast model. Eur J Cell Biol 2006, 85:195–202. [DOI] [PubMed] [Google Scholar]
- 16.Soe K, Andersen TL, Hinge M, Rolighed L, Marcussen N, Delaisse JM: Coordination of Fusion and Trafficking of Pre-osteoclasts at the Marrow- Bone Interface. Calcif Tissue Int 2019, 105:430–445. [DOI] [PubMed] [Google Scholar]
- 17.Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et al. : Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 1997, 89:309–319. [DOI] [PubMed] [Google Scholar]
- 18.*.Imai Y, Youn MY, Inoue K, Takada I, Kouzmenko A, Kato S: Nuclear receptors in bone physiology and diseases. Physiol Rev 2013, 93:481–523. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review provides an overview of the role of nuclear receptors in bone cells.
- 19.*.Jin Z, Li X, Wan Y: Minireview: nuclear receptor regulation of osteoclast and bone remodeling. Mol Endocrinol 2015, 29:172–186. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review provides an overview of the role of nuclear receptors in osteoclasts.
- 20.Mullican SE, Dispirito JR, Lazar MA: The orphan nuclear receptors at their 25-year reunion. J Mol Endocrinol 2013, 51:T115–140.24096517 [Google Scholar]
- 21.Nelson HD, Humphrey LL, Nygren P, Teutsch SM, Allan JD: Postmenopausal hormone replacement therapy: scientific review. JAMA 2002, 288:872–881. [DOI] [PubMed] [Google Scholar]
- 22.Zuo H, Wan Y: Nuclear Receptors in Skeletal Homeostasis. Curr Top Dev Biol 2017, 125:71–107. [DOI] [PubMed] [Google Scholar]
- 23.Mauvais-Jarvis F, Clegg DJ, Hevener AL: The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev 2013, 34:309–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khosla S, Oursler MJ, Monroe DG: Estrogen and the skeleton. Trends Endocrinol Metab 2012, 23:576–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harman SM: Estrogen replacement in menopausal women: recent and current prospective studies, the WHI and the KEEPS. Gend Med 2006, 3:254–269. [DOI] [PubMed] [Google Scholar]
- 26.Pinkerton JV, Thomas S: Use of SERMs for treatment in postmenopausal women. J Steroid Biochem Mol Biol 2014, 142:142–154. [DOI] [PubMed] [Google Scholar]
- 27.Khosla S, Melton LJ 3rd, Atkinson EJ, O’Fallon WM, Klee GG, Riggs BL: Relationship of serum sex steroid levels and bone turnover markers with bone mineral density in men and women: a key role for bioavailable estrogen. J Clin Endocrinol Metab 1998, 83:2266–2274. [DOI] [PubMed] [Google Scholar]
- 28.Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, Williams TC, Lubahn DB, Korach KS: Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med 1994, 331:1056–1061. [DOI] [PubMed] [Google Scholar]
- 29.Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M: Effect of single and compound knockouts of estrogen receptors alpha (ERalpha) and beta (ERbeta) on mouse reproductive phenotypes. Development 2000, 127:4277–4291. [DOI] [PubMed] [Google Scholar]
- 30.Sims NA, Dupont S, Krust A, Clement-Lacroix P, Minet D, Resche-Rigon M, Gaillard-Kelly M, Baron R: Deletion of estrogen receptors reveals a regulatory role for estrogen receptors-beta in bone remodeling in females but not in males. Bone 2002, 30:18–25. [DOI] [PubMed] [Google Scholar]
- 31.Martin-Millan M, Almeida M, Ambrogini E, Han L, Zhao H, Weinstein RS, Jilka RL, O’Brien CA, Manolagas SC: The estrogen receptor-alpha in osteoclasts mediates the protective effects of estrogens on cancellous but not cortical bone. Mol Endocrinol 2010, 24:323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.**.Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, Harada Y, Azuma Y, Krust A, Yamamoto Y, et al. : Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell 2007, 130:811–823. [DOI] [PubMed] [Google Scholar]; This paper is the first paper to provide the molecular basis for how estrogen attenuates bone resorption, using osteoclast-specific estrogen receptor deficient mice.
- 33.**.Miyauchi Y, Sato Y, Kobayashi T, Yoshida S, Mori T, Kanagawa H, Katsuyama E, Fujie A, Hao W, Miyamoto K, et al. : HIF1alpha is required for osteoclast activation by estrogen deficiency in postmenopausal osteoporosis. Proc Natl Acad Sci U S A 2013, 110:16568–16573. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study links the estrogen-dependent destabilization of HIF1α to the promotion of bone loss in estrogen-deficient conditions. HIF1α is stabilized under hypoxic conditions. However, when estrogen is present, HIF1α in osteoclasts is unstable even in hypoxic conditions. Therefore, estrogen deficiency stabilizes HIF1α in osteoclasts to accelerate osteoclast-mediated bone resorption.
- 34.Knowles HJ: Hypoxic regulation of osteoclast differentiation and bone resorption activity. Hypoxia (Auckl) 2015, 3:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morita M, Sato Y, Iwasaki R, Kobayashi T, Watanabe R, Oike T, Miyamoto K, Toyama Y, Matsumoto M, Nakamura M, et al. : Selective Estrogen Receptor Modulators Suppress Hif1alpha Protein Accumulation in Mouse Osteoclasts. PLoS One 2016, 11:e0165922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bord S, Ireland DC, Beavan SR, Compston JE: The effects of estrogen on osteoprotegerin, RANKL, and estrogen receptor expression in human osteoblasts. Bone 2003, 32:136–141. [DOI] [PubMed] [Google Scholar]
- 37.Dyson HJ, Wright PE: Role of Intrinsic Protein Disorder in the Function and Interactions of the Transcriptional Coactivators CREB-binding Protein (CBP) and p300. J Biol Chem 2016, 291:6714–6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bennesch MA, Picard D: Minireview: Tipping the balance: ligand-independent activation of steroid receptors. Mol Endocrinol 2015, 29:349–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weitzmann MN, Pacifici R: Estrogen deficiency and bone loss: an inflammatory tale. J Clin Invest 2006, 116:1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen H, Stuart W, Hu B, Nguyen L, Huang G, Clemens TL, Adams JS: Creation of estrogen resistance in vivo by transgenic overexpression of the heterogeneous nuclear ribonucleoprotein-related estrogen response element binding protein. Endocrinology 2005, 146:4266–4273. [DOI] [PubMed] [Google Scholar]
- 41.Chen H, Gilbert LC, Lu X, Liu Z, You S, Weitzmann MN, Nanes MS, Adams J: A new regulator of osteoclastogenesis: estrogen response element-binding protein in bone. J Bone Miner Res 2011, 26:2537–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moon YJ, Zhang Z, Bang IH, Kwon OK, Yoon SJ, Kim JR, Lee S, Bae EJ, Park BH: Sirtuin 6 in preosteoclasts suppresses age- and estrogen deficiency-related bone loss by stabilizing estrogen receptor alpha. Cell Death Differ 2019, 26:2358–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clemons M, Goss P: Estrogen and the risk of breast cancer. N Engl J Med 2001, 344:276–285. [DOI] [PubMed] [Google Scholar]
- 44.Yager JD, Davidson NE: Estrogen carcinogenesis in breast cancer. N Engl J Med 2006, 354:270–282. [DOI] [PubMed] [Google Scholar]
- 45.**.Wei W, Schwaid AG, Wang X, Wang X, Chen S, Chu Q, Saghatelian A, Wan Y: Ligand Activation of ERRalpha by Cholesterol Mediates Statin and Bisphosphonate Effects. Cell Metab 2016, 23:479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]; Despite the intensive search, the ligand of an orphan receptor ERRα has not been found. The authors discover the endogenous ligand for ERRα and provide the cholesterol-ERRα axis as a novel ERRα pathways for therapeutic interventions.
- 46.Giguere V, Yang N, Segui P, Evans RM: Identification of a new class of steroid hormone receptors. Nature 1988, 331:91–94. [DOI] [PubMed] [Google Scholar]
- 47.Huss JM, Garbacz WG, Xie W: Constitutive activities of estrogen-related receptors: Transcriptional regulation of metabolism by the ERR pathways in health and disease. Biochim Biophys Acta 2015, 1852:1912–1927. [DOI] [PubMed] [Google Scholar]
- 48.**.Wei W, Wang X, Yang M, Smith LC, Dechow PC, Sonoda J, Evans RM, Wan Y: PGC1beta mediates PPARgamma activation of osteoclastogenesis and rosiglitazone-induced bone loss. Cell Metab 2010, 11:503–516. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that PGC-1β is required for the rosiglitazone-induced bone loss. PGC1β is a coactivator of PPARγ and is involved in enhanced osteoclastogenesis by PPARγ activation by inducing FOS expression and mitochondrial biogenesis. Of note, PGC1β itself is an important regulator of osteoclastogenesis and physiological bone remodeling.
- 49.**.Bae S, Lee MJ, Mun SH, Giannopoulou EG, Yong-Gonzalez V, Cross JR, Murata K, Giguere V, van der Meulen M, Park-Min KH: MYC-dependent oxidative metabolism regulates osteoclastogenesis via nuclear receptor ERRalpha. J Clin Invest 2017, 127:2555–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows for the first time that MYC governs osteoclastogenic mitochondrial respiration via ERRα. The inhibition of the MYC-ERRα axis protects mice from OVX-induced bone loss.
- 50.Deblois G, Giguere V: Oestrogen-related receptors in breast cancer: control of cellular metabolism and beyond. Nat Rev Cancer 2013, 13:27–36. [DOI] [PubMed] [Google Scholar]
- 51.Teyssier C, Gallet M, Rabier B, Monfoulet L, Dine J, Macari C, Espallergues J, Horard B, Giguere V, Cohen-Solal M, et al. : Absence of ERRalpha in female mice confers resistance to bone loss induced by age or estrogen-deficiency. PLoS One 2009, 4:e7942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delhon I, Gutzwiller S, Morvan F, Rangwala S, Wyder L, Evans G, Studer A, Kneissel M, Fournier B: Absence of estrogen receptor-related-alpha increases osteoblastic differentiation and cancellous bone mineral density. Endocrinology 2009, 150:4463–4472. [DOI] [PubMed] [Google Scholar]
- 53.Wilson C: Bone: Bisphosphonate use and atypical femur fractures. Nat Rev Endocrinol 2010, 6:420. [DOI] [PubMed] [Google Scholar]
- 54.Kim HJ, Kim BK, Ohk B, Yoon HJ, Kang WY, Cho S, Seong SJ, Lee HW, Yoon YR: Estrogen-related receptor gamma negatively regulates osteoclastogenesis and protects against inflammatory bone loss. J Cell Physiol 2019, 234:1659–1670. [DOI] [PubMed] [Google Scholar]
- 55.Cardelli M, Aubin JE: ERRgamma is not required for skeletal development but is a RUNX2-dependent negative regulator of postnatal bone formation in male mice. PLoS One 2014, 9:e109592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cain DW, Cidlowski JA: Immune regulation by glucocorticoids. Nat Rev Immunol 2017, 17:233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heitzer MD, Wolf IM, Sanchez ER, Witchel SF, DeFranco DB: Glucocorticoid receptor physiology. Rev Endocr Metab Disord 2007, 8:321–330. [DOI] [PubMed] [Google Scholar]
- 58.Lewis-Tuffin LJ, Cidlowski JA: The physiology of human glucocorticoid receptor beta (hGRbeta) and glucocorticoid resistance. Ann N Y Acad Sci 2006, 1069:1–9. [DOI] [PubMed] [Google Scholar]
- 59.Lu NZ, Cidlowski JA: Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell 2005, 18:331–342. [DOI] [PubMed] [Google Scholar]
- 60.Kassel O, Herrlich P: Crosstalk between the glucocorticoid receptor and other transcription factors: molecular aspects. Mol Cell Endocrinol 2007, 275:13–29. [DOI] [PubMed] [Google Scholar]
- 61.*.Buckley L, Humphrey MB: Glucocorticoid-Induced Osteoporosis. N Engl J Med 2018, 379:2547–2556. [DOI] [PubMed] [Google Scholar]
- 62.Henneicke H, Gasparini SJ, Brennan-Speranza TC, Zhou H, Seibel MJ: Glucocorticoids and bone: local effects and systemic implications. Trends Endocrinol Metab 2014, 25:197–211. [DOI] [PubMed] [Google Scholar]
- 63.Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC: Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest 1998, 102:274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pico MJ, Hashemi S, Xu F, Nguyen KH, Donnelly R, Moran E, Flowers S: Glucocorticoid receptor-mediated cis-repression of osteogenic genes requires BRM-SWI/SNF. Bone Rep 2016, 5:222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rauch A, Seitz S, Baschant U, Schilling AF, Illing A, Stride B, Kirilov M, Mandic V, Takacz A, Schmidt-Ullrich R, et al. : Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab 2010, 11:517–531. [DOI] [PubMed] [Google Scholar]
- 66.Butler JS, Queally JM, Devitt BM, Murray DW, Doran PP, O’Byrne JM: Silencing Dkk1 expression rescues dexamethasone-induced suppression of primary human osteoblast differentiation. BMC Musculoskelet Disord 2010, 11:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang FS, Lin CL, Chen YJ, Wang CJ, Yang KD, Huang YT, Sun YC, Huang HC: Secreted frizzled-related protein 1 modulates glucocorticoid attenuation of osteogenic activities and bone mass. Endocrinology 2005, 146:2415–2423. [DOI] [PubMed] [Google Scholar]
- 68.Morimoto E, Li M, Khalid AB, Krum SA, Chimge NO, Frenkel B: Glucocorticoids Hijack Runx2 to Stimulate Wif1 for Suppression of Osteoblast Growth and Differentiation. J Cell Physiol 2017, 232:145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Piemontese M, Xiong J, Fujiwara Y, Thostenson JD, O’Brien CA: Cortical bone loss caused by glucocorticoid excess requires RANKL production by osteocytes and is associated with reduced OPG expression in mice. Am J Physiol Endocrinol Metab 2016, 311:E587–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.*.Teitelbaum SL: Glucocorticoids and the osteoclast. Clin Exp Rheumatol 2015, 33:S37–39. [PubMed] [Google Scholar]
- 71.Conaway HH, Henning P, Lie A, Tuckermann J, Lerner UH: Activation of dimeric glucocorticoid receptors in osteoclast progenitors potentiates RANKL induced mature osteoclast bone resorbing activity. Bone 2016, 93:43–54. [DOI] [PubMed] [Google Scholar]
- 72.Jia D, O’Brien CA, Stewart SA, Manolagas SC, Weinstein RS: Glucocorticoids act directly on osteoclasts to increase their life span and reduce bone density. Endocrinology 2006, 147:5592–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim HJ, Zhao H, Kitaura H, Bhattacharyya S, Brewer JA, Muglia LJ, Ross FP, Teitelbaum SL: Glucocorticoids suppress bone formation via the osteoclast. J Clin Invest 2006, 116:2152–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bellini G, Torella M, Manzo I, Tortora C, Luongo L, Punzo F, Colacurci N, Nobili B, Maione S, Rossi F: PKCbetaII-mediated cross-talk of TRPV1/CB2 modulates the glucocorticoid-induced osteoclast overactivity. Pharmacol Res 2017, 115:267–274. [DOI] [PubMed] [Google Scholar]
- 75.Bouvard B, Audran M, Legrand E, Chappard D: Ultrastructural characteristics of glucocorticoid-induced osteoporosis. Osteoporos Int 2009, 20:1089–1092. [DOI] [PubMed] [Google Scholar]
- 76.Evans RM, Barish GD, Wang YX: PPARs and the complex journey to obesity. Nat Med 2004, 10:355–361. [DOI] [PubMed] [Google Scholar]
- 77.Grygiel-Gorniak B: Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications--a review. Nutr J 2014, 13:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kawai M, Rosen CJ: PPARgamma: a circadian transcription factor in adipogenesis and osteogenesis. Nat Rev Endocrinol 2010, 6:629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Viccica G, Francucci CM, Marcocci C: The role of PPARgamma for the osteoblastic differentiation. J Endocrinol Invest 2010, 33:9–12. [PubMed] [Google Scholar]
- 80.**.Wan Y, Chong LW, Evans RM: PPAR-gamma regulates osteoclastogenesis in mice. Nat Med 2007, 13:1496–1503. [DOI] [PubMed] [Google Scholar]; This study shows that PPARγ deficiency suppresses osteoclastogenesis using PPARγ-tie2 cre mice.
- 81.Tontonoz P, Spiegelman BM: Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem 2008, 77:289–312. [DOI] [PubMed] [Google Scholar]
- 82.Lecka-Czernik B, Moerman EJ, Grant DF, Lehmann JM, Manolagas SC, Jilka RL: Divergent effects of selective peroxisome proliferator-activated receptor-gamma 2 ligands on adipocyte versus osteoblast differentiation. Endocrinology 2002, 143:2376–2384. [DOI] [PubMed] [Google Scholar]
- 83.Still K, Grabowski P, Mackie I, Perry M, Bishop N: The peroxisome proliferator activator receptor alpha/delta agonists linoleic acid and bezafibrate upregulate osteoblast differentiation and induce periosteal bone formation in vivo. Calcif Tissue Int 2008, 83:285–292. [DOI] [PubMed] [Google Scholar]
- 84.Stunes AK, Westbroek I, Gustafsson BI, Fossmark R, Waarsing JH, Eriksen EF, Petzold C, Reseland JE, Syversen U: The peroxisome proliferator-activated receptor (PPAR) alpha agonist fenofibrate maintains bone mass, while the PPAR gamma agonist pioglitazone exaggerates bone loss, in ovariectomized rats. BMC Endocr Disord 2011, 11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kleiner S, Nguyen-Tran V, Bare O, Huang X, Spiegelman B, Wu Z: PPAR{delta} agonism activates fatty acid oxidation via PGC-1{alpha} but does not increase mitochondrial gene expression and function. J Biol Chem 2009, 284:18624–18633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.**.Scholtysek C, Katzenbeisser J, Fu H, Uderhardt S, Ipseiz N, Stoll C, Zaiss MM, Stock M, Donhauser L, Bohm C, et al. : PPARbeta/delta governs Wnt signaling and bone turnover. Nat Med 2013, 19:608–613. [DOI] [PubMed] [Google Scholar]; This study shows that the activation of PPARδ regulates osteoclastogenesis by a osteoblast-dependent mechanisms.
- 87.Schwartz AV: TZDs and Bone: A Review of the Recent Clinical Evidence. PPAR Res 2008, 2008:297893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Grey A: Skeletal consequences of thiazolidinedione therapy. Osteoporos Int 2008, 19:129–137. [DOI] [PubMed] [Google Scholar]
- 89.Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, Kravitz BG, Lachin JM, O’Neill MC, Zinman B, et al. : Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med 2006, 355:2427–2443. [DOI] [PubMed] [Google Scholar]
- 90.Rzonca SO, Suva LJ, Gaddy D, Montague DC, Lecka-Czernik B: Bone is a target for the antidiabetic compound rosiglitazone. Endocrinology 2004, 145:401–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Soroceanu MA, Miao D, Bai XY, Su H, Goltzman D, Karaplis AC: Rosiglitazone impacts negatively on bone by promoting osteoblast/osteocyte apoptosis. J Endocrinol 2004, 183:203–216. [DOI] [PubMed] [Google Scholar]
- 92.Lazarenko OP, Rzonca SO, Hogue WR, Swain FL, Suva LJ, Lecka-Czernik B: Rosiglitazone induces decreases in bone mass and strength that are reminiscent of aged bone. Endocrinology 2007, 148:2669–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.**.Ishii KA, Fumoto T, Iwai K, Takeshita S, Ito M, Shimohata N, Aburatani H, Taketani S, Lelliott CJ, Vidal-Puig A, et al. : Coordination of PGC-1beta and iron uptake in mitochondrial biogenesis and osteoclast activation. Nat Med 2009, 15:259–266. [DOI] [PubMed] [Google Scholar]; This study shows that PGC1β is induced by a CREB-dependent pathway and a positive regulator of osteoclastogenesis. Intriguingly, the authors also show that iron uptake activates PGC1β and osteoclastic bone resorption. Thus, this study establishes the PGC1β-mediated mitochondrial biogenesis is coupled with iron uptake via TfR1, resulting in the regulation of osteoclastogenesis.
- 94.**.Zou W, Rohatgi N, Chen TH, Schilling J, Abu-Amer Y, Teitelbaum SL: PPAR-gamma regulates pharmacological but not physiological or pathological osteoclast formation. Nat Med 2016, 22:1203–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows the contradicting results about the physiological role of PPARγ in osteoclasts. Using all the currently available cre mice in osteoclast lineage cells, PPARγ is deleted in osteoclast lineage cells. The authors demonstrate that PPARγ has no apparent role in physiological and pathological bone resorption. However, the accelerating osteoclastogenesis by TZDs, PPARγ agonists, is in agreement among the studies.
- 95.Izawa T, Rohatgi N, Fukunaga T, Wang QT, Silva MJ, Gardner MJ, McDaniel ML, Abumrad NA, Semenkovich CF, Teitelbaum SL, et al. : ASXL2 Regulates Glucose, Lipid, and Skeletal Homeostasis. Cell Rep 2015, 11:1625–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zelcer N, Tontonoz P: Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest 2006, 116:607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ: An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 1996, 383:728–731. [DOI] [PubMed] [Google Scholar]
- 98.Fu X, Menke JG, Chen Y, Zhou G, MacNaul KL, Wright SD, Sparrow CP, Lund EG: 27-hydroxycholesterol is an endogenous ligand for liver X receptor in cholesterol-loaded cells. J Biol Chem 2001, 276:38378–38387. [DOI] [PubMed] [Google Scholar]
- 99.Robertson KM, Norgard M, Windahl SH, Hultenby K, Ohlsson C, Andersson G, Gustafsson JA: Cholesterol-sensing receptors, liver X receptor alpha and beta, have novel and distinct roles in osteoclast differentiation and activation. J Bone Miner Res 2006, 21:1276–1287. [DOI] [PubMed] [Google Scholar]
- 100.Prawitt J, Beil FT, Marshall RP, Bartelt A, Ruether W, Heeren J, Amling M, Staels B, Niemeier A: Short-term activation of liver X receptors inhibits osteoblasts but long-term activation does not have an impact on murine bone in vivo. Bone 2011, 48:339–346. [DOI] [PubMed] [Google Scholar]
- 101.Remen KM, Henning P, Lerner UH, Gustafsson JA, Andersson G: Activation of liver X receptor (LXR) inhibits receptor activator of nuclear factor kappaB ligand (RANKL)-induced osteoclast differentiation in an LXRbeta-dependent mechanism. J Biol Chem 2011, 286:33084–33094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.**.Kleyer A, Scholtysek C, Bottesch E, Hillienhof U, Beyer C, Distler JH, Tuckermann JP, Schett G, Kronke G: Liver X receptors orchestrate osteoblast/osteoclast crosstalk and counteract pathologic bone loss. J Bone Miner Res 2012, 27:2442–2451. [DOI] [PubMed] [Google Scholar]; This study shows that the activation of LXRs protects mice from pathological bone loss by decreasing the RANKL to OPG ratio, suggesting the role of LXRs in the crosstalk between osteoclasts and osteoblasts.
- 103.Robertson Remen KM, Lerner UH, Gustafsson JA, Andersson G: Activation of the liver X receptor-beta potently inhibits osteoclastogenesis from lipopolysaccharide-exposed bone marrow-derived macrophages. J Leukoc Biol 2013, 93:71–82. [DOI] [PubMed] [Google Scholar]
- 104.Kim HJ, Yoon KA, Yoon HJ, Hong JM, Lee MJ, Lee IK, Kim SY: Liver X receptor activation inhibits osteoclastogenesis by suppressing NF-kappaB activity and c-Fos induction and prevents inflammatory bone loss in mice. J Leukoc Biol 2013, 94:99–107. [DOI] [PubMed] [Google Scholar]
- 105.Roszer T, Menendez-Gutierrez MP, Cedenilla M, Ricote M: Retinoid X receptors in macrophage biology. Trends Endocrinol Metab 2013, 24:460–468. [DOI] [PubMed] [Google Scholar]
- 106.Zhang XK, Lehmann J, Hoffmann B, Dawson MI, Cameron J, Graupner G, Hermann T, Tran P, Pfahl M: Homodimer formation of retinoid X receptor induced by 9-cis retinoic acid. Nature 1992, 358:587–591. [DOI] [PubMed] [Google Scholar]
- 107.Schierle S, Merk D: Therapeutic modulation of retinoid X receptors - SAR and therapeutic potential of RXR ligands and recent patents. Expert Opin Ther Pat 2019, 29:605–621. [DOI] [PubMed] [Google Scholar]
- 108.**.Menendez-Gutierrez MP, Roszer T, Fuentes L, Nunez V, Escolano A, Redondo JM, De Clerck N, Metzger D, Valledor AF, Ricote M: Retinoid X receptors orchestrate osteoclast differentiation and postnatal bone remodeling. J Clin Invest 2015, 125:809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows the role of RXRs in osteoclastogenesis. RXR deficiency results in the generation of giant and non-resolving osteoclasts. The activation of RXRs suppresses osteoclastogenesis by inducing the expression of MAFB, a negative regulator of osteoclastogenesis, and it also protects mice from OVX-induced bone loss.
- 109.Querfeld C, Nagelli LV, Rosen ST, Kuzel TM, Guitart J: Bexarotene in the treatment of cutaneous T-cell lymphoma. Expert Opin Pharmacother 2006, 7:907–915. [DOI] [PubMed] [Google Scholar]
- 110.Jozwik KM, Carroll JS: Pioneer factors in hormone-dependent cancers. Nat Rev Cancer 2012, 12:381–385. [DOI] [PubMed] [Google Scholar]
- 111.Pott S, Kamrani NK, Bourque G, Pettersson S, Liu ET: PPARG binding landscapes in macrophages suggest a genome-wide contribution of PU.1 to divergent PPARG binding in human and mouse. PLoS One 2012, 7:e48102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Izawa N, Kurotaki D, Nomura S, Fujita T, Omata Y, Yasui T, Hirose J, Matsumoto T, Saito T, Kadono Y, et al. : Cooperation of PU.1 With IRF8 and NFATc1 Defines Chromatin Landscapes During RANKL-Induced Osteoclastogenesis. J Bone Miner Res 2019, 34:1143–1154. [DOI] [PubMed] [Google Scholar]