

Abstract
Intratumoral hypoxia occurs in 90% of solid tumors and is associated with a poor prognosis for patients. Cancer cells respond to hypoxic microenvironments by activating the transcription factors hypoxia-inducible factor 1 (HIF-1) and HIF-2. Here we studied the unique gene expression patterns of 31 different breast cancer cell lines exposed to hypoxic conditions. The epidermal growth factor receptor (EGFR), a member of the ErbB (avian erythroblastosis oncogene B) family of receptors that play a role in cell proliferation, invasion, metastasis, and apoptosis, was induced in 7 of the 31 breast cancer cell lines by hypoxia. A functional hypoxia response element (HRE) was identified, which is activated upon HIF-1 binding to intron 18 of the EGFR gene in cell lines in which EGFR was induced by hypoxia. CpG methylation of the EGFR HRE prevented induction under hypoxic conditions. The HRE of EGFR was methylated in normal breast tissue and some breast cancer cell lines and could be reversed by treatment with DNA methyltransferase inhibitors. Induction of EGFR under hypoxia led to an increase in AKT, ERK, and Rb phosphorylation as well as increased levels of cyclin D1, A, B1, E2F, and repression of p21 in a HIF-1α-dependent manner, leading to cell proliferation and migration. Increased expression of EGFR sensitized cells to EGFR inhibitors. Collectively, our data suggest that patients with hypoxic breast tumors and hypomethylated EGFR status may benefit from EGFR inhibitors currently used in the clinic.
Graphical Abstract
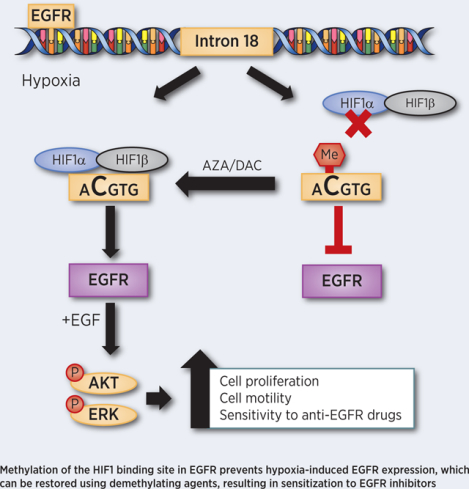
Introduction
Increased cell proliferation and oxygen consumption results in lower oxygen availability in solid tumors as compared to normal tissue (1,2). Intratumoral hypoxia has been associated with invasion, metastasis, treatment failure, and patient mortality (3,4). In murine models of metastasis, cells exposed to hypoxia in the primary tumor were able to metastasize five times more readily than their oxygenated counterpart(5). Cancer cells survive and adapt to hypoxic conditions, in part, through the activation of hypoxia-inducible factor 1 (HIF-1) and HIF-2, which induce the expression of gene products involved in angiogenesis, glucose utilization, invasion, and metastasis (6). HIF-1 is a heterodimeric protein composed of a constitutively expressed HIF-1β subunit and an O2-regulated HIF-1α subunit (7). Our recent work suggests that tumors may have a unique transcriptional response to hypoxia with a select number of conserved genes that are induced or repressed across 31 individual cell lines (8). We selected the epidermal growth factor receptor (EGFR), which is induced in 7 of 31 cell lines under hypoxic conditions in order to determine the mechanisms and potential clinical implications of the heterogeneity in the hypoxic response.
The EGFR is a member of the ErbB (avian erythroblastosis oncogene B) family of receptors and activates multiple signaling pathways, including mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinases (ERK) and phosphoinositide-3-kinase (PI3K)/V-AKT murine thymoma viral oncogene homolog (AKT) pathways (9,10). The activation of EGFR has many implications in tumor biology such as cell proliferation, invasion, metastasis, and apoptosis (11,12). EGFR is overexpressed in various human cancers, including lung cancer, breast cancer, colon cancer, glioblastoma, and is associated with tumor malignancy and poor prognosis (13,14). Approximately half of the cases of triple-negative breast cancer (TNBC) and inflammatory breast cancer (IBC) present with the overexpression of EGFR (11). Several studies have shown an inverse correlation between EGFR expression and disease-free and overall survival of breast cancer patients (14,15). Taken together, these findings have prompted the evaluation of EGFR inhibitors for the treatment of TNBC (16). However, the results of such studies in breast cancer treatment have been disappointing (16–18), partially due to the lack of biomarkers to predict which patients are most likely to respond to treatment with EGFR inhibitors (18).
Under normal circumstances, EGFR expression is primarily regulated by the abundance of its mRNA (19). EGFR gene amplification is a common mechanism of over-expression in high-grade gliomas (20), but it is less common in other solid tumors (21). A recent study of non-small cell lung cancer (NSCLC) found that only 6% of primary NSCLC tumors have gene amplification of EGFR (22). Epigenetic regulation is a biological mechanism by which gene expression is modulated through DNA methylation or histone modifications (23). DNA methylation of cytosine at CpG dinucleotides is an important and well-studied regulatory modification throughout the genome (24). Hypermethylation of the promoter region of EGFR has been described in several types of cancer and alters EGFR expression (25,26). Whether and how hypomethylation of EGFR can alter gene expression has not previously been considered.
Here, we demonstrate that EGFR is induced under hypoxic conditions. We uncover a functional hypoxia response element that is activated upon HIF-1 binding to an intron region of the EGFR gene. In normal breast tissue, intron 18 of EGFR is methylated, which prevents EGFR induction. The treatment of cells with a DNA methyltransferase inhibitor causes the demethylation of intron 18 of EGFR, thereby restoring the hypoxic regulation of EGFR. In cancer tissue and cancer cell lines with non-methylated EGFR, hypoxia leads to AKT, ERK, and Rb phosphorylation as well as induction of cyclin D1 and repression of p21 in a HIF-1α dependent manner resulting in cell proliferation and migration. On the other hand, increased levels of EGFR under hypoxia enhances the efficacy of EGFR inhibitors. Taken together, our data suggest that EGFR inhibitors, in combination with methyl transferase inhibitors or in a subset of patients with hypomethylated EGFR, may have a therapeutic benefit for patients with hypoxic tumors.
Materials & Methods
Cell Culture
All cell lines, with the exception of SUMs, were obtained from the ATCC. SUMs were purchased from Asterand Bioscience. Cells were cultured per company provided protocols. The MCF10A and MCF10A ER-expressing cells were a kind gift from Ben Ho Park and cultured as previously described (27). CRISPR edited MCF-7 HIF-1α, HIF-2α and control knockout cell lines were previously generated in our laboratory (8). All cell lines used in the study were authenticated by STR sequencing and confirmed to be mycoplasma free. Cells were maintained in a humidified environment at 37°C and 5% CO2 during culture and live-cell imaging. Hypoxic cells were maintained at 37°C in an invivo 200 hypoxia workstation equipped with a digitally controlled oxygen regulator and maintained at 1% O2, 5% CO2, and 94% N2. Live-cell microscopy experiments were conducted in a McCoy incubator maintained at 1% O2, 5% CO2, and 94% N2 and imaged with a Lionheart microscope (Biotek).
Reverse Transcription and qPCR
Total RNA was extracted from cells using a Direct-zol RNA Miniprep kit with DNase I treatment per the manufacturer’s instructions (Zymo Research). One microgram of total RNA was used for first-strand DNA synthesis with the iScript cDNA Synthesis System (Bio-Rad). qPCR was performed using human-specific primers and iTaq SYBR Green Universal Master Mix (Bio-Rad). The expression of each target mRNA relative to 18s rRNA was calculated on the basis of the threshold cycle (Ct) as 2−Δ(ΔCt), where ΔCt = Ct,target – Ct,18s and Δ(ΔCt) = ΔCt,test - ΔCt,control. Primer sequences are shown in Supplementary Table 1.
Immunoblot Assays
Aliquots of whole-cell lysates were prepared in NP-40 buffer (150 mmol/L NaCl, 1% NP-40, 50 mmol/L Tris-HCl, pH 9.0) and fractionated by 10% or 12.5% SDS-PAGE. Proteins were transferred from the gel to a nitrocellulose membrane for 15 min using a Trans-blot Turbo (Bio-Rad). The nitrocellulose membrane was blocked in 5% milk (w/v) in Tris-buffered saline and 0.1% Tween-20 (TBS-T) for 30 minutes. Antibodies against EGFR (1:1000, Proteintech 18986–1-AP), phosphorylated EGFR Y1068 (1:500 Sigma SAB4300063), HIF-1α (1:500, BD biosciences 610958), phosphorylated Erk T202/Y204 (1:1000, Cell Signaling 9106S), Erk (1:1000, Cell Signaling 4695S), phosphorylated Akt S473 (1:1000, Cell Signaling 9271L), Akt (1:1000, Cell Signaling 9272S), phosphorylated Rb S780 (1:500, Cell Signaling 9307S), Rb (1:500, Cell Signaling 9309S), Cyclin D1 (1:1000, Cell Signaling 2922S), Cyclin A (1:500, Santa Cruz Sc-271682), Cyclin E (1:500, Cell Signaling 4129S), Cyclin B1 (1:500, Santa Cruz Sc-245), C-myc (1:500, Cell Signaling 13987S), and p21 Waf1/Clip1 (1:500, Cell Signaling 2947S) were used with overnight incubation at 4°C with orbital shaking. Blots were washed three times with TBS-T. β-actin-HRP (1:10000, Proteintech HRP-60008), secondary anti-mouse-HRP (Azure AC2115), and anti-rabbit-HRP (Azure AC2114) were then utilized with 1.5 h incubation at room temperature with orbital shaking following by three additional TBS-T washes. Enhanced chemiluminescent substrate (Perkin Elmer) was utilized as the substrate for HRP-catalyzed detection and imaged using a c300 imager (Azure Biosystems).
Patient Data Analysis
The TCGA Breast cancer (BRCA) transcriptional data and clinical data were downloaded from the NIH GDC Data Portal. The transcriptional data were quantile normalized before analyses. The BRCA TCGA methylation (HumanMethylation 450k) data sets were downloaded from the website firebrowse (http://firebrowse.org). Statistical analysis on the TCGA data were performed with R software (version 3.6.0). All Mann-Whitney P values was calculated with the R function wilcox.test. The correlation plots and Pearson correlation statistics were calculated with the R function cor.test. The hypoxia score is the average of the z-score of each of the 42 genes in the hypoxia signature. The hypoxia signature was defined as presented in our prior publication (8) by comparing the transcriptional profile of 34 breast cancer cell lines exposed to hypoxia to identify genes with consistent regulation under hypoxic conditions.
Animal Studies
Animal research complied with all relevant ethical regulations according to protocols approved by the Johns Hopkins University Animal Care and Use Committee. Female 5 to 7-week-old NOD-SCID Gamma (NSG) mice were anesthetized and 2 × 106 BT-474 cells were injected into the mammary fat pad. Slow-release estradiol pellets (2 mg per pellet) were implanted subcutaneously three days prior to BT-474 cell injection. Tumors were excised when they reached 0.5 mm in diameter. Excised tumors were formalin-fixed (Sigma-Aldrich) and paraffin-embedded.
Immunohistochemistry Staining
Paraffin-embedded tissue sections were dewaxed with xylenes and hydrated with decreasing gradients of ethanol. Tissue sections were treated with Tris-EDTA buffer (10 mM Tris-Cl, 1 mM EDTA, pH 9.0) at near-boiling temperature for 20 minutes for antigen retrieval. Immunohistochemistry was conducted with the Vectastain® Elite® ABC HRP Kit (Vector Laboratories PK-7200) and DAB Peroxidase (HRP) Substrate Kit (Vector Laboratories SK-4100) according to the manufacturer’s instruction. Primary antibodies against EGFR (1:400, Proteintech 18986–1-AP) and HIF-1α (1:400, BD biosciences 610958) were diluted in 1% BSA in PBS and applied to slides for 1h at room temperature. Slides were imaged in bright field on a Cytation 5 Cell Imaging Multi-mode Reader (BioTek).
Cell Viability Assay
Cells (1–2 × 105) were seeded in 96-well plates and exposed to 20% or 1% O2 in the presence of Erlotinib (Selleck chemicals), Gefitinb (Selleck chemicals) or DMSO at the indicated dose. After 48 h, cells were washed and harvested in 0.5 mL of trypsin. An additional 0.5 mL of media was added, and cells were counted after 0.4% Trypan Blue (Gibco) staining using a Countess II FL Automated Cell Counter (Thermo Fisher). Alternatively, cells were treated with the aforementioned drugs for 2 days and then incubated with 10% AlamarBlue ™ Cell Viability Reagent (ThermoFisher) for 4 h. 100 μl of media from each well was collected and transferred to a black, clear-bottom 96-well plate. The fluorescence intensity was measured at an excitation wavelength of 560 nm and emission wavelength of 590 nm on a Cytation 5 Multi-Mode Reader (BioTek). The fluorescence intensity of AlamarBlue™ media in a control well with no cells was subtracted from the measurement of all experimental samples prior to analysis. After measuring the AlamarBlue intensity, the cells were fixed with 0.5% Crystal Violet (Sigma Aldrich) with methanol for 10 min. Then, Crystal Violet solution was removed and followed by 3–5 washes with PBS. The plate was left to dry overnight and imaged with Cytation 5 Cell Imaging Multi-Mode Reader (BioTek).
Propidium Iodide Staining
MCF-7 cells were plated in serum-free medium for 24 h. Cells were then exposed to hypoxia for 24 h and then treated with 100ng/ul EGF while being maintained under hypoxia for an additional 16 h. Cells were then pelleted, resuspended in water, and fixed by adding 100% ethanol drop-wise to a final concentration of 70%. Fixed cells were maintained on ice for 2 hours. Cells were then washed with PBS, Pelleted and incubated in staining buffer (PBS with 100ug/ml RNase A and 50 ug/ml propidium iodide) overnight at 4C in the dark. Flow cytometry for cell cycle analysis was performed on an LSR II (BD Biosciences). Data was analyzed with FlowJo V10 software (Tree Star Inc.).
Automated Analysis for Percentage of Cells Positive for Ki67
Ki67 and DAPI-stained cells were imaged in a 3 by 3 montage per well with a Cytation 5 Cell Imaging Multi-Mode Reader (BioTek). Using the Gen5 3.05 software (BioTek), the DAPI channel was used to count the number of cells in a field of view and to create an individual mask on the area of each nucleus. Ki67 intensity within each individual mask was then quantified. By visually observing cells, a threshold intensity of 8,000 was selected. Any object with an intensity greater or equal to the threshold was deemed positive, and all others were deemed negative. Percentage was determined for each well by dividing the number of positive cells over the total number of counted cells.
Chromatin Immunoprecipitation (ChIP) Assay
Cells were cross-linked with 1% formaldehyde for 10 min and quenched in 0.125 M glycine. Chromatin was sheared by sonication using a Covaris sonicator (settings: Power (W): 150, Duty Factor: 5%, Cycles: 200 Treatment time: 420 sec (7 min)). Sonicated lysates were precleared with salmon sperm DNA/protein A agarose slurry (Millipore). IgG (Santa Cruz) or primary antibodies against HIF-1α (Santa Cruz), HIF-2α (Novus Biologicals), or HIF-1β (Novus Biologicals) were added and incubated overnight with precleared lysates. The following day, salmon sperm DNA/protein A agarose beads were added for 4 h at 37°C. The agarose beads were collected and washed sequentially with: low- and high-salt immune complex wash buffers; LiCl immune complex wash buffer; and twice with TE buffer. The DNA was eluted from the agarose gel in 1% SDS/0.1 M NaHCO3 and crosslinks were reversed by addition of NaCl to a final concentration of 0.2 M. Proteinase K was added to degrade protein in the lysate. DNA was recovered by phenol-chloroform extraction followed by ethanol precipitation, treated with RNase, and analyzed by qPCR. Fold enrichment was calculated based on the cycle threshold (Ct) as 2−Δ(ΔCt), where ΔCt = Ct,IP − Ct,Input and Δ(ΔCt) = ΔCt,antibody − ΔCt,IgG.
Luciferase Reporter Plasmid Constructs
The EGFR, mutated EGFR or LDHA luciferase reporter vectors were generated by annealing 10 μM of the forward strand and 10 μM of reverse strand of 60bp oligonucleotide sequence of EGFR or LDHA containing the HIF-1α binding site. The 60bp fragment was cloned into a pENTR TOPO vector using a pENTR Directional TOPO Cloning Kit according to manufacturer’s protocol (Life Technologies). The pENTR plasmids were then recombined into into the pGL4.23-GW plasmid upstream of firefly luciferase using Gateway LR Clonase enzyme (Thermofisher Scientific). Oligo nucleotide sequences are listed in Supplementary Table 1. Plasmid constructs were then confirmed by Sanger sequencing.
Firefly Luminescence Assay
MCF-7 and 293T cells were seeded overnight in 24-well plates and co-transfected with 0.4 ug of the indicated vectors, 0.05 ug of psVmRL renilla luciferase vector, and 0.05 ug of pcDNA3-EGFP per well using PolyJet In Vitro DNA Transfection Reagent (SignaGen Laboratories). psVmRL renilla luciferase vector was used as an internal control. Approximately 16 h later, media was refreshed, and the transfected cells were exposed to 20% or 1% O2 for 24 h. Cells were analyzed for luciferase activity using a Dual-Luciferase Reporter Assay System per manufacturer’s instructions (Promega).
Bisulfite Modification.
200 ng of DNA from each of the indicated samples was treated with sodium bisulfite using the EpiTect® Bisulfite Kit (Qiagen, 59824) according to the manufacturer’s protocol. 20 ng of the bisulfite treated DNA was used Methylation-specific PCR or Sanger sequencing. PCR was used to amplify bisulfite treated DNA prior to Sanger sequencing. Primers used for PCR are provided in Supplementary Table 1. Sanger sequencing was performed by Johns Hopkins Genetic Resources Core Facility.
MS-HRM PCR (methylation-specific high-resolution melt PCR)
EGFR forward and reverse primers specific for the detection of only non-methylated bisulfite treated DNA are provided in Supplementary Table 1. The specificity of the primer pair was assessed using fully methylated or non-methylated synthetic DNA (Ultramer® DNA Oligonucleotides, IDT). The ultramers were mixed to achieve the following percentage of methylated DNA: 0%, 6.25%, 12.5%, 25%, 50%, 75%, and 100%. PCR was performed in a final volume of 20 μl, containing 10 μl of Precision Melt Supermix (BIO-RAD, 172–5110), 2μM of each primer and 20 ng of bisulfite modified DNA template and remaining volume of DNase-free water. Each reaction was performed in triplicate. All analyses were run according to the following conditions: 1 cycle of 95°C for 2 min, 43 cycles of 95°C for 10 s, Ta for 30 s and 60°C for 30s; followed by an HRM step of 95°C for 30 s and 60°C for 1 min, 65°C for 15 s, and continuous acquisition to 95°C at one acquisition per 0.2°C.
The percent methylation was calculated after normalizing to DAPIKI expression on the basis of the threshold cycle (Ct) as 2−Δ(ΔCt), where ΔCt = Ct,sample – Ct,DAPIKI and Δ(ΔCt) = ΔCt,test - ΔCt,control. DAPIKI primers were designed to detect both non-methylated and methylated DAPIKI. For samples with no signal detection, the Ct value was set to 43. Raw data from Methylation-specific PCR was analyzed utilizing web-based high-resolution DNA melting analysis software (uAnalyze 2.0), with normalized curves for comparison among samples (28).
Cell Migration
BT-474 cells (1 × 104) were plated in 6‐well plates coated with soluble rat tail type I collagen (Corning). Cells were incubated overnight, and phase contrast images were taken every 5 minutes for 23 h using a lionheart (Biotek). MetaMorph software was used to determine x and y coordinates at each time interval and to construct cell trajectory maps. The cell trajectories were fit using an anisotropic persistent random walk (APRW) model of cell motility to calculate Distance traveled from origin and total cell diffusivities (Dtot). APRW model analysis was performed as described in detail using MATLAB (29). Three‐dimensional cell trajectory data were used to statistically profile cell migration using the MSD, which can be obtained from (x[t], y[t]) coordinates of cells with time (t). MSD (τ) = (x[t + τ] − x[t]) + (y[t + τ] – y[t]) where τ = 5 min * frame number. Values of persistence and speed are obtained from APRW model fitting and expressed as speed (S) and persistence (P) of cells, which can be used to calculated total cell diffusivity (Dtot). Dtot = (Sp2Pp + Snp2Pnp)/4 where both speed (S) and persistence (P) are calculated along both the primary and nonprimary axes.
Statistics
All the values in text and figures are presented as mean ± SEM unless otherwise stated. Statistical significance was determined when appropriate by Student’s t-test or one/two-way ANOVA with Bonferonni post-test. P-values of <0.05 were considered significant.
Results
EGFR expression is induced by hypoxia in some but not all breast cancer cell lines.
In our previous work (8), we performed an RNA sequencing analysis of 31 breast cancer cell lines exposed to 20% or 1% O2 conditions for 24 h. Our results show that more than 1,000 genes are induced or repressed in each cell line in response to hypoxia; however, only 42 genes shared a conserved response to hypoxia. Intriguingly, EGFR was among the genes that showed induction under hypoxic conditions in some but not all breast cancer cell lines. To confirm this finding, we performed real-time quantitative PCR (RT-PCR) and verified that only seven of the 31 breast cancer cell lines had a two-fold or greater increase in EGFR expression upon exposure to hypoxia (Figure 1A). Subsequently, we determined the baseline and hypoxia-induced mRNA and protein expression of EGFR in luminal and basal cell lines (Figure 1B–D, Supplementary Figure S1A–B). One basal cell line, SUM149, showed increased EGFR expression under hypoxic conditions, whereas all of the luminal cell lines except MDA-MB-175 displayed increased EGFR expression (Figure 1B–D and Supplementary Figure S1A–B). EGFR is localized in hypoxic regions in orthotopic tumors derived from luminal, BT-474 cells (Supplementary Figure S1C).
Figure 1. EGFR expression is induced by hypoxia in some but not all breast cancer cell lines.
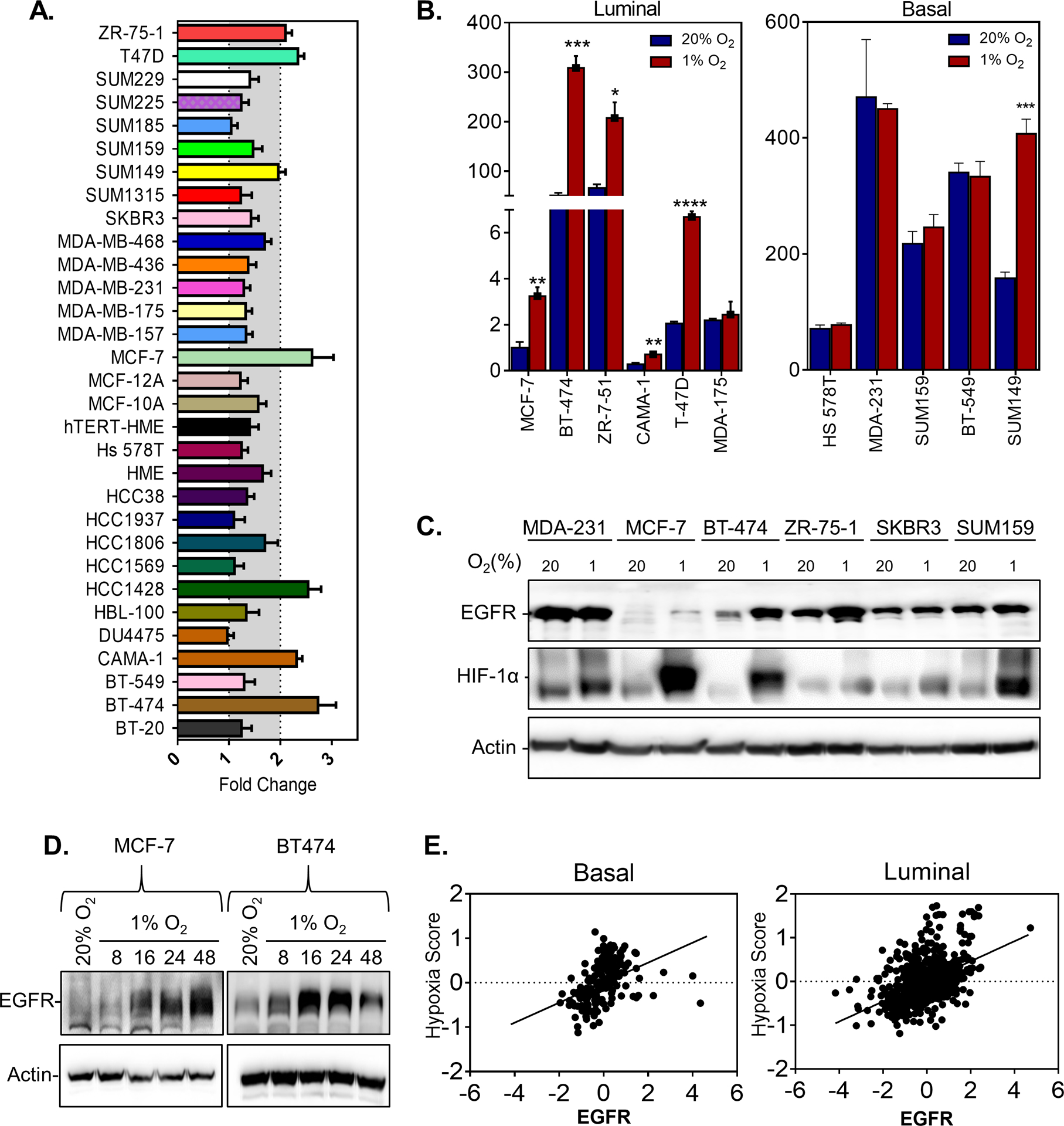
A. The fold change in EGFR mRNA expression in cells exposed to 1% compared to 20% O2 conditions as measured by qPCR in 31 breast cancer cell lines. B. EGFR mRNA levels in luminal or basal cell lines normalized by EGFR expression in MCF-7 cells cultured under 20% O2 conditions. n=3. Student’s t-test * P<0.05, ** P<0.01 *** P<0.001, **** P<0.0001. C-D. Immunoblot assays were performed to assess EGFR protein levels using lysates prepared from (C) MDA-MB-231 (MDA-231), MCF-7, BT-474, ZR-75–1, SKBR3 and SUM159 cells exposed to 20% or 1% O2 for 48 h or (D) MCF-7 and BT-474 cells, exposed to 20% or 1% O2 for 8, 16, 24, or 48 h. E. Scatterplot correlating the expression of a hypoxia score(8) and EGFR mRNA expression in breast cancer samples from patients with basal (n=152; Pearson R=0.38; P<0.0001) or luminal (n = 692; Pearson R=0.45; P<0.0001) breast cancer from the TCGA.
Given that the luminal cell lines express the estrogen receptor (ER), whereas basal cell lines do not, we reasoned that ER expression may play a role in EGFR induction upon exposure to hypoxia. To test this hypothesis, we inhibited ER activity by treating cells with 4-hydroxytamoxifen (OHT) or fulvestrant. Both treatments inhibited ER-regulated TFF1 induction but did not affect EGFR expression under hypoxic conditions in both BT474 and MCF-7 cells (Supplementary Figure S1D–F). We also utilized an MCF10A human breast epithelial cell line engineered to overexpress an ER cDNA (30) to determine whether ER expression would promote EGFR expression under hypoxic conditions. ER expression did not promote EGFR expression under hypoxic conditions (Supplementary Figure S1G–H). The results verify that hypoxia selectively induces EGFR expression in some breast cancer cell lines, but manipulating ER expression did not alter this response. . Using RNA expression data from The Cancer Genome Atlas (TCGA) we find that EGFR expression correlates with expression of our hypoxia score (8) in patients with either luminal (ER+) or basal (ER-) breast cancer samples (Figure 1E). However, it is important to note that both the hypoxia score (8) and EGFR expression (31) have been shown to be enriched in basal breast cancer and may play a role in the aforementioned result.
HIF-1α is required for EGFR induction under hypoxic conditions.
To determine whether hypoxia-inducible factors (HIFs) are required for EGFR induction under hypoxic conditions, we assessed the expression of EGFR in CRISPR-depleted HIF-1α or HIF-2α knockout MCF-7 subclones (Supplementary Figure S2A). The knockout of HIF-1α abrogated EGFR induction upon exposure to hypoxia at both the mRNA and protein levels whereas the knockout of HIF-2α did not, demonstrating that HIF-1α (but not HIF-2α) is required for EGFR induction under hypoxic conditions (Figure 2A–B). To determine whether the increase in EGFR levels under hypoxia is sufficient to activate the EGFR pathway, we stimulated cells with EGF. MCF-7, BT474, and HCC1428cells showed increased phosphorylation of AKT and ERK under both hypoxic and normal O2 conditions following 30 min of stimulation with EGF albeit the levels of pAKT and pERK induction under hypoxia varied between the cell lines with HCC1428 cells showing the most striking induction (Figure 2C, Supplementary Figure S2B–C). The knock-out of HIF-1α abrogated this effect (Figure 2D). Next, we stimulated MCF-7 and BT474 cells with EGF and exposed the cells to hypoxia in the presence of the EGFR inhibitor, Gefitinib. The robust increase in AKT and ERK phosphorylation in response to hypoxia was abrogated in a dose-dependent manner in response to gefitinib treatment (Figure 2E, Supplementary Figure S2D). Taken together, the results demonstrate that HIF-1α increases EGFR expression under hypoxic conditions leading to robust activation of the EGFR pathway in response to ligand (Figure 2F).
Figure 2. HIF-1α is required for EGFR induction under hypoxic conditions.
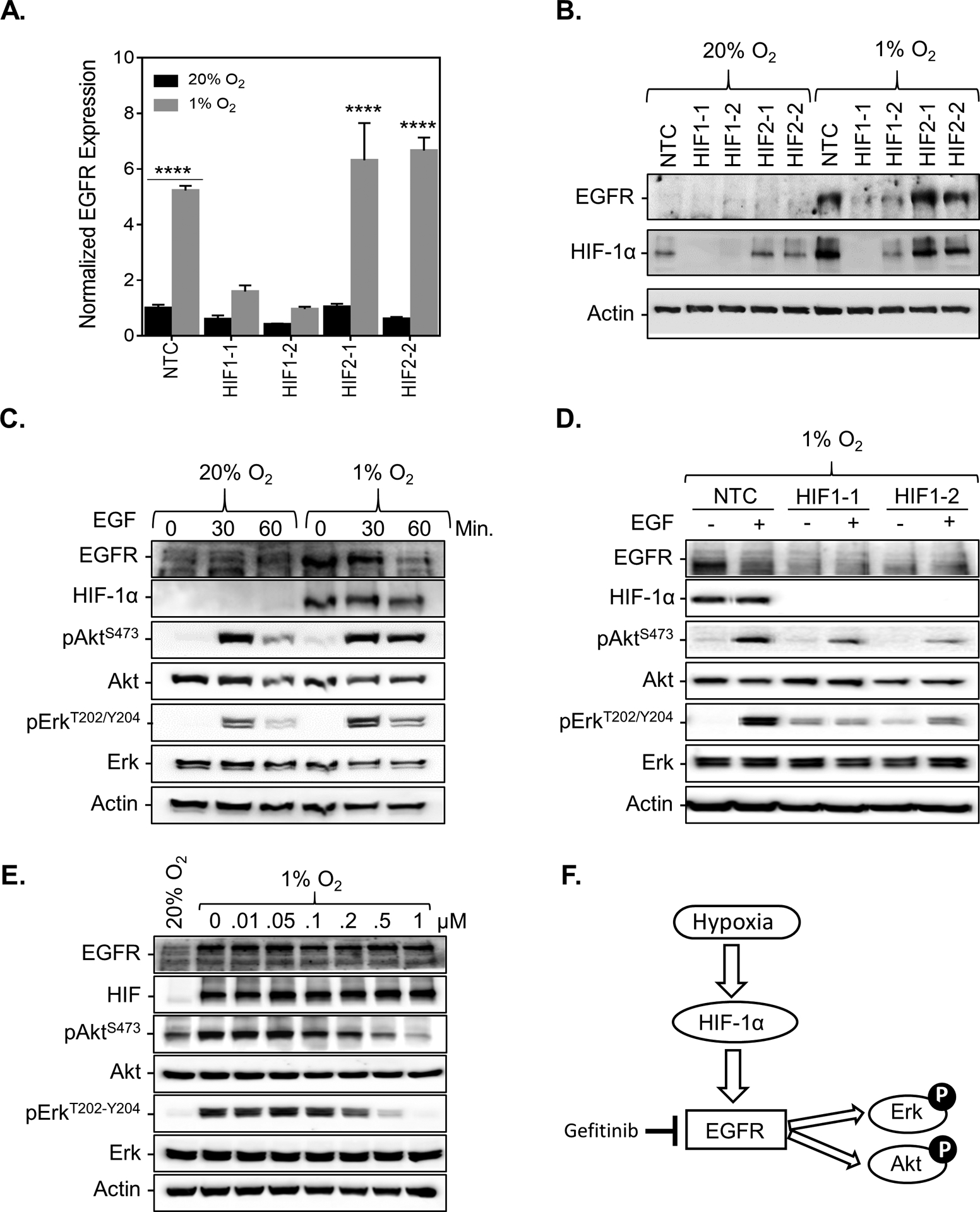
A. EGFR mRNA levels were analyzed by qPCR in MCF-7 subclones, which were stably transfected with a non-target control (NTC) CRISPR vector or vectors encoding sgRNA sequences that target either HIF-1α (HIF1–1, HIF1–2) or HIF-2α (HIF2–1, HIF2–2) and exposed to 20% or 1% O2 for 24 h (mean ± SEM, n = 3); **** P < 0.001 versus NTC at 20% O2 (two-way ANOVA with Bonferroni posttest). B-F. Immunoblot assays were performed using lysates prepared from (B) MCF7 subclones exposed to 20% or 1% O2 for 48 h. (C) MCF-7 cells exposed to 20% or 1% O2 for 24 h in the presence of 100ng/mL EGF for 0, 30, or 60 min. (D) MCF-7 subclones exposed to 1% O2 for 24 h with or without 100ng/mL EGF for 60 min. (E) MCF-7 cells exposed to 20% or 1% O2 for 30 h in the presence of 100ng/mL EGF for 1 h and treated with increasing doses of Gefitinib or an equivalent volume of DMSO for 6 h. (F) Proposed mechanism.
The EGFR gene contains a functional hypoxia response element.
To determine if HIF-1α is a direct transcriptional regulator of EGFR, we searched for putative HIF-1α binding sites within the EGFR gene. We also leveraged the results of a previous study which used high-resolution genome-wide mapping of HIF-binding sites in MCF-7 cells exposed to 0.5% O2 or 2mM of DMOG (32). A high-stringency HIF-1α-binding region was identified in intron 18 of the EGFR loci (Figure 3A). Using a ChIP assay, we confirmed that HIF-1α and HIF-1β but not HIF-2α were bound to this regionwhich contained three ACGTG sites with enrichment levels similar to LDHA binding (Figure 3B and Supplementary Figure S3A). On the other hand, HIF-1α did not bind to a nearby 2 different nearby regions of intron 17 in EGFR. Likewise, HIFs were not enriched in an intronic region of EGFR which contained 5 ACGTG binding sites (Supplementary Figures S3B–C). This demonstrates the specific recruitment of HIF-1α to Intron 18 of the EGFR gene under hypoxic conditions in BT474 cells.
Figure 3. The EGFR gene contains a functional hypoxia response element.
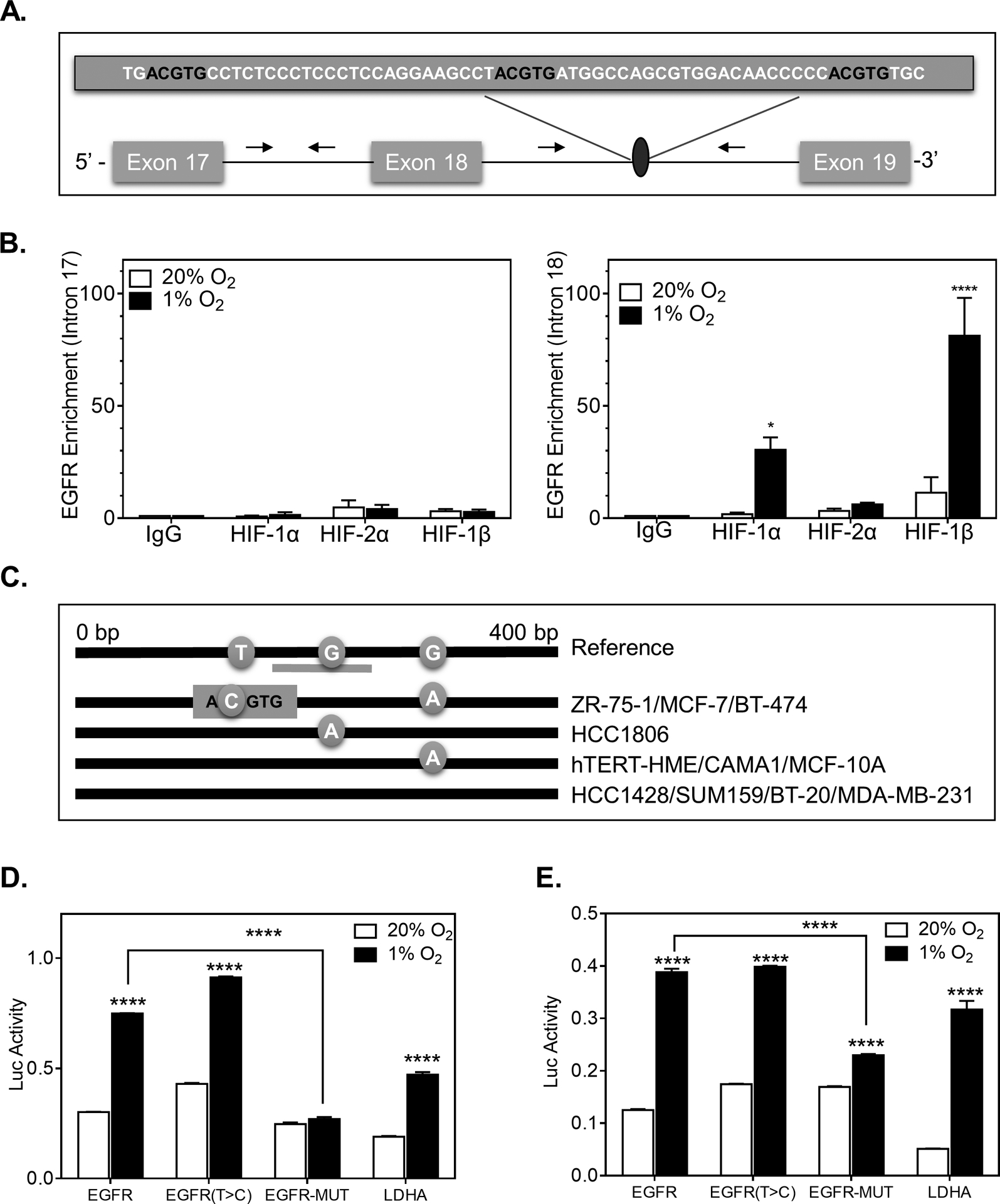
A. Candidate HIF-binding sites (ACGTG) were identified in the Intron region between Exon 18 and Exon 19. B. A ChIP assay was performed to assess the enrichment of HIF-binding in intron 17 (no putative HIF-binding sites) and intron 18 of EGFR in BT-474 cells exposed to 20% or 1% O2 for 4 h using IgG or antibodies against HIF-1α, HIF-2α, or HIF-1β. n=3 × N=3. Two-way ANOVA with Bonferroni post-test * P<0.05, **** P<0.001. C. Sanger sequencing of PCR-amplified genomic DNA isolated from the indicated cell lines. Reference genome sequence showing the position of T and G nucleotides variants. D-E. BT-474 (D) or 293T-cells (E) that were transiently transfected with a pGL4.23-GW -promoter construct containing a WT EGFR HRE, an HRE with a single nucleotide variant of EGFR (T>C), a fully mutated EGFR HRE or HRE from LDHA and co-transfected with a Renilla luciferase vector. Following transfection, the cells were exposed to 20% or 1% O2 for 24 h and luciferase reporter activity (Luc) was determined. The firefly to renilla ratio was calculated and normalized by the value for 20% O2. n=3. Student’s t-test **** P<0.0001.
Since only 7 of the 31 breast cancer cell lines displayed a significant induction of EGFR upon exposure to hypoxia (Figure 1A), we hypothesized that EGFR may contain one or more single nucleotide variants (SNV) in the HIF-1 binding region. To address this consideration, we isolated DNA from 10 cell lines and Sanger sequenced and amplified a 400-bp region of EGFR containing the HIF-DNA binding site (Figure 3C). The DNA isolated from ZR-75–1, MCF-7 and BT-474 displayed a unique point mutation (T > C) that generated an additional ACGTG site (Supplementary Table S2). To determine whether the SNV altered EGFR expression under hypoxia and to verify that we had identified a functional HRE, we utilized a luciferase reporter assay. We inserted a 60-bp sequence spanning the HIF binding sites in EGFR into the reporter plasmid, pGL4.23-GW-luciferase, in which a basal SV40 promoter drives firefly luciferase expression. Two additional constructs were also generated, one containing the single nucleotide (T > C) variant of EGFR and one in which all three HIF sites were mutated (EGFR-MUT). In MCF-7 and 293T cells, transfected with pGL4.23-EGFR and pGL4.23-EGFR (T > C), luciferase activity increased 2.5-fold on exposure to hypoxia, whereas in cells transfected with pGL4.23-EGFR-MUT, the hypoxic induction of luciferase expression was abrogated (Figure 3D–E). We also constructed an LDHA luciferase reporter construct as a positive control. Thus, the ChIP and luciferase reporter assays demonstrate that EGFR is a direct HIF-1 target gene. However, the single nucleotide variation that we identified did not alter luciferase expression nor did it provide evidence for the difference in EGFR regulation by hypoxia between cell lines.
HIF-binding sites in EGFR have altered methylation patterns in breast cancer cell lines.
Given that nucleotide variations did not predict for hypoxia-induced EGFR expression amongst breast cancer cell lines, we next questioned whether the methylation status of the ACGTG binding site might play a role. Methylation of promoter regions is a well-established mechanism for gene silencing (33). In order to assess the methylation status of the HIF-1 binding region, we isolated, bisulfite-treated, and PCR amplified a 400-bp region of EGFR containing the HIF-DNA binding site followed by Sanger sequencing of ten breast cancer cell lines (Figure 4A–B, Supplementary Figure S4A–B, Supplementary Table S3). The chromatogram analysis demonstrated that all cytosine residues in the amplified region of EGFR in MCF-7, CAMA1, HCC1428, and BT-474 cells were unmethylated. On the other hand, every cytosine residue in the HIF-binding region of EGFR in MDA-MB-231, hTERT-HME, MCF-10A, HCC1806, BT-20, and SUM159 was methylated.
Figure 4. HIF-binding sites are methylated in some breast cancer cell-lines.
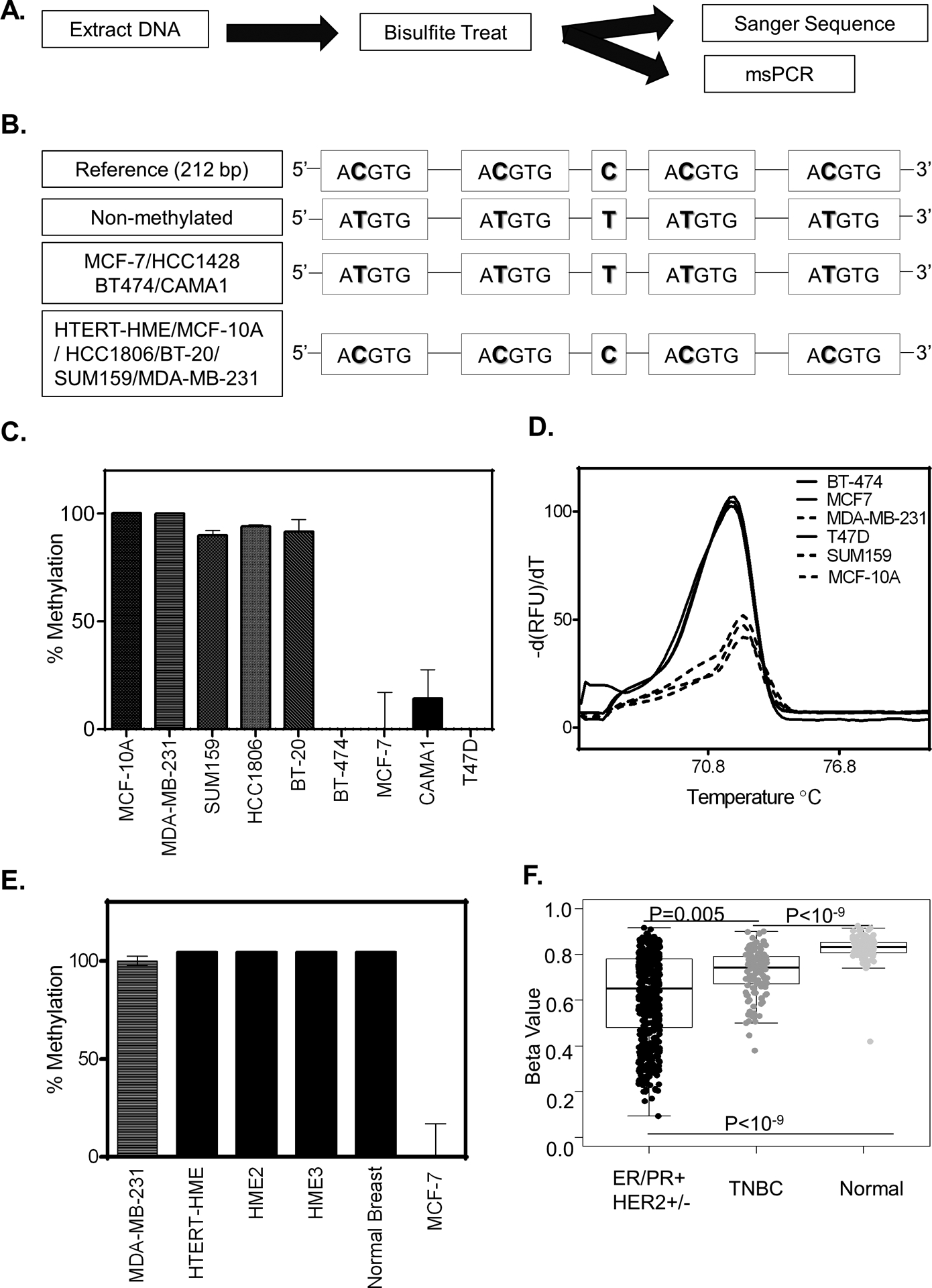
A. Schematic overview of experimental design to assess methylation status. B. Sanger sequence of PCR amplified bisulfite treated DNA for the cell lines indicated. C-E. Methylation status as measured by methylation-specific qPCR using a primer specific for the non-methylated HIF-binding region of EGFR for cell lines indicated. (C) The methylation percentage was calculated by first standardizing all expression values to MCF-7 and then normalizing the result to MDA-MB-231(MDA-231). (D) UAnalyze software was used to plot the melt curves from the methylation specific-high resolution melting (MS-HRM) analysis of the indicated cell lines. (E) Percent methylation as determined in normal breast tissue. F. Boxplots showing the methylation level in breast cancer patients based on cancer subtypes compared to normal breast tissue. (P-value is calculated by the wilcox.test, ER/PR+ HER2+/− n= 551, TNBC n=122 Normal n=97).
To confirm the results of Sanger Sequencing, we developed a methylation-specific PCR (MSP) assay paired with a high-resolution melt curve analysis (HRM). To test the ability of our assay to discriminate between non-methylated and methylated bisulfite-treated DNA we designed synthetic oligos that represent a PCR amplified bisulfite treated DNA sequence of a fully methylated or non-methylated HIF-1 binding region in the EGFR intron 18. The oligos were mixed at ratios from 0:1 to 1:0 (methylated: non-methylated) to show the specificity of the primers to detect only non-methylated DNA (Supplementary Figure S5A). Next, we used uAnalyze (28), a web-based high-resolution DNA melting analysis tool to determine the unique melt curve signature of the methylated and non-methylated DNA sequences (Supplementary Figure S5B–C). Oligonucleotide DNA had distinct melting curves of 70°C and 72°C for non-methylated and methylated DNA, respectively. Mixed ratios of non-methylated to methylated DNA had bimodal melting curves that reflect the input quantities of each oligo. Given that temperature plays a role in the helical twist of DNA, we also considered the percent helicity of DNA which ranged between −40% and 20% for methylated and non-methylated amplicons, respectively.
After verifying that our MSP paired HRM assay can successfully distinguish methylated and non-methylated EGFR amplicons, we used the assay to confirm the methylation status of the cell lines previously tested using Sanger Sequencing of bisulfite treated DNA (Figure 4C–D). To determine whether the HIF-1 binding site is methylated in normal epithelial cells, we tested cells from 4 different individuals and determined that they were 100% methylated (Figure 4E). Intrigued by the result, we assessed the 450K Methylation array data from the TCGA which contained probe cg20062492 to detect the methylation status of the EGFR region of interest (Supplementary Figure S6A). The results show that normal breast tissue displayed a higher level of methylation in HIF-binding region compared to breast cancer tissue (Figure 4F). The methylation levels are significantly lower for breast cancer compared to normal breast tissue. Together these results suggest that the methylation status of the HIF-1 binding site prevents EGFR induction under hypoxic conditions.
Demethylation of the HIF-binding site of EGFR restores EGFR induction under hypoxic conditions.
Our results demonstrate that the methylation status of the HIF binding region correlate with increased expression of EGFR under hypoxic conditions. To determine whether demethylation could restore EGFR induction under hypoxic conditions, we treated MDA-MB-231 and SUM-159 cells with 500nM azacytidine (AZA) or 100nM of decitabine (DAC) for 3 days followed by drug withdrawal for an additional 6 or 10 days. The MSP paired HRM (Figure 5A–B and Supplementary Figure S7A–B) showed that both AZA and DAC decreased methylation at this site by 80%. Using a ChIP assay, we also confirmed that DAC treatment enhances HIF-1α (but not HIF-2α) to the HRE of the EGFR gene under hypoxia (Figure 5C–D and Supplementary Figure S7C). Treatment with either AZA or DAC also restored EGFR mRNA and protein induction by hypoxia in MDA-MB-231 or SUM-159 cells (Figure 5E–H and Supplementary Figure S7D). The results demonstrated that cytosine methylation within the HIF-binding region prevents the induction of EGFR under hypoxia which can be restored by treatment with demethylating agents.
Figure 5. Treatment with demethylating agents restores EGFR induction under hypoxic conditions for cell lines with methylated HIF-binding regions of EGFR.
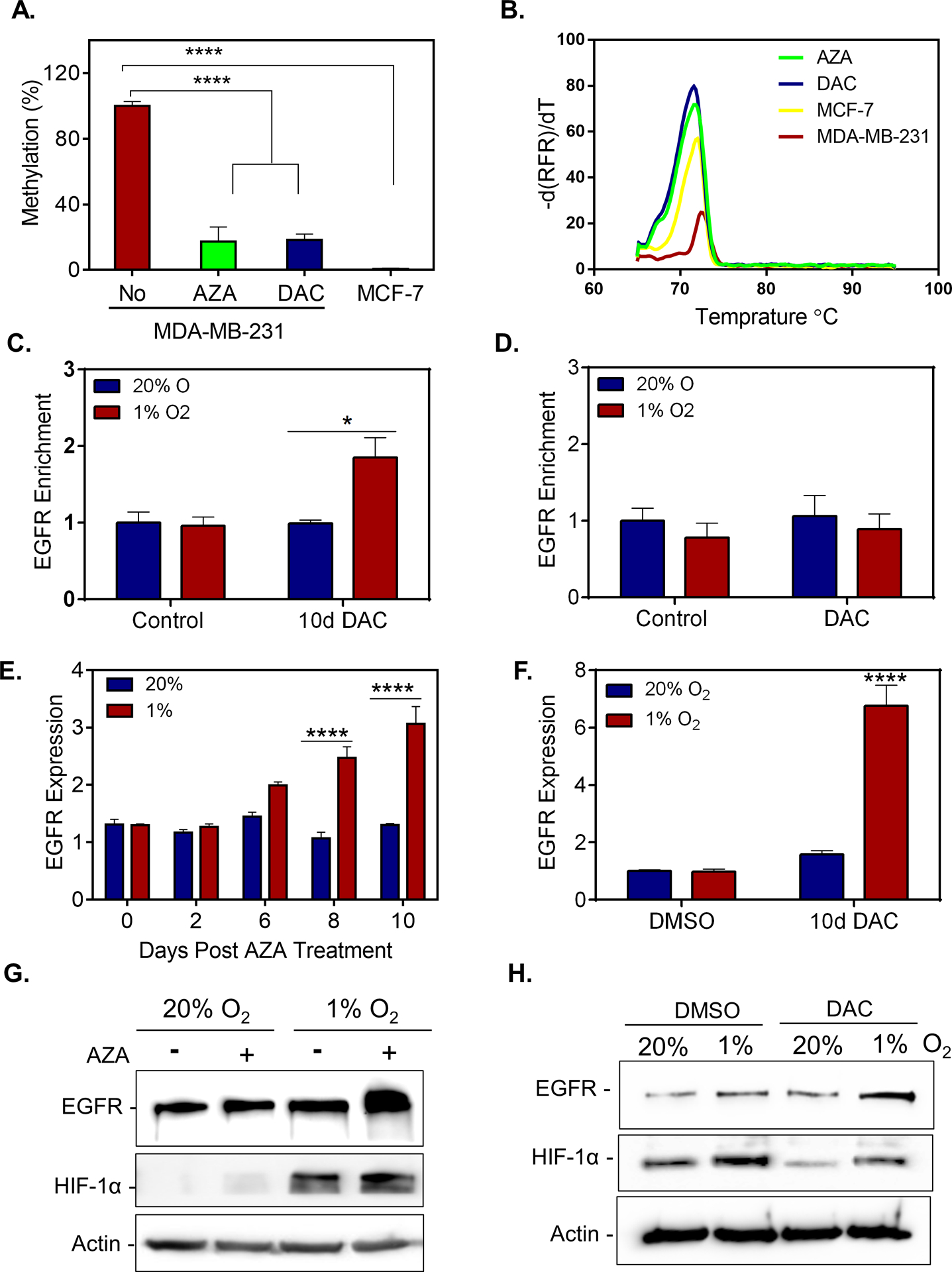
A-B. MS-HRM assay was used to evaluate the methylation status of MDA-MB-231 cells treated with 500nM AZA, 250nM DAC or DMSO for 3 consecutive days followed by 10 days of culture in the absence of drug. (A) Methylation percentage after standardizing to MCF-7 and normalizing by MDA-MB-231(MDA-231) levels. (B) MS-HRM analysis using UAnalyze software. C-D. A ChIP assay was performed to assess the enrichment of EGFR in MDA-MB-231 cells treated with 250nM DAC or DMSO for 3 consecutive days followed by 8 days of culture in the absence of drug and exposed to 20% or 1% O2 for 5 h using antibodies against (C) HIF-1α or (D) HIF-2α (mean ± SEM, n = 3); *P < 0.05 versus MDA-MB-231 cells treated with DMSO (control) at 20% O2 (two-way ANOVA with Bonferroni posttest).E-F. EGFR mRNA levels measured by qPCR in MDA-MB-231 (MDA-231) cells treated with (E) 500nM of AZA or (F) 250nM of DAC for 3 consecutive days followed by 6, 8, or 10 days of culture in the absence of drug. Cells were exposed to 20% or 1% O2 for the last 24 h of the experiment (mean ± SEM, n = 3); **** P < 0.0001 versus MDA-MB-231 cells treated with equivalent concentration of DMSO (control) at 20% O2 (two-way ANOVA with Bonferroni posttest). G-H. Immunoblot assays were performed using lysates prepared from MDA-MB-231 cells treated with 500nM AZA (G) or 250nM DAC (H) for 3 consecutive days followed by 10 days of culture in the absence of drug. Cells were exposed to 20% or 1% O2 for the last 48 h of the experiment.
Hypoxic cells are sensitive to EGFR inhibitors.
Mitogen activation stimulates the Ras/MEK/ERK and Ras/PI3K/AKT pathway leading to the induction of cyclins, c-myc and Rb phosphorylation (34–37). We find that hypoxia, EGF, or the combination of EGF and hypoxia led to an increase in the level of almost every cyclin tested, promoted Rb phosphorylation, and enhanced E2F levels while concomitantly decreasing p21 levels 20% O2 conditions (Figure 6A–B and Supplementary Figure S8A). The expression of cyclin D1, c-myc and phosphorylated RB are reduced in HIF-1α knockout cell lines (Supplementary Figure S8B).
Figure 6. Hypoxic cells are more sensitive to EGFR inhibitors.
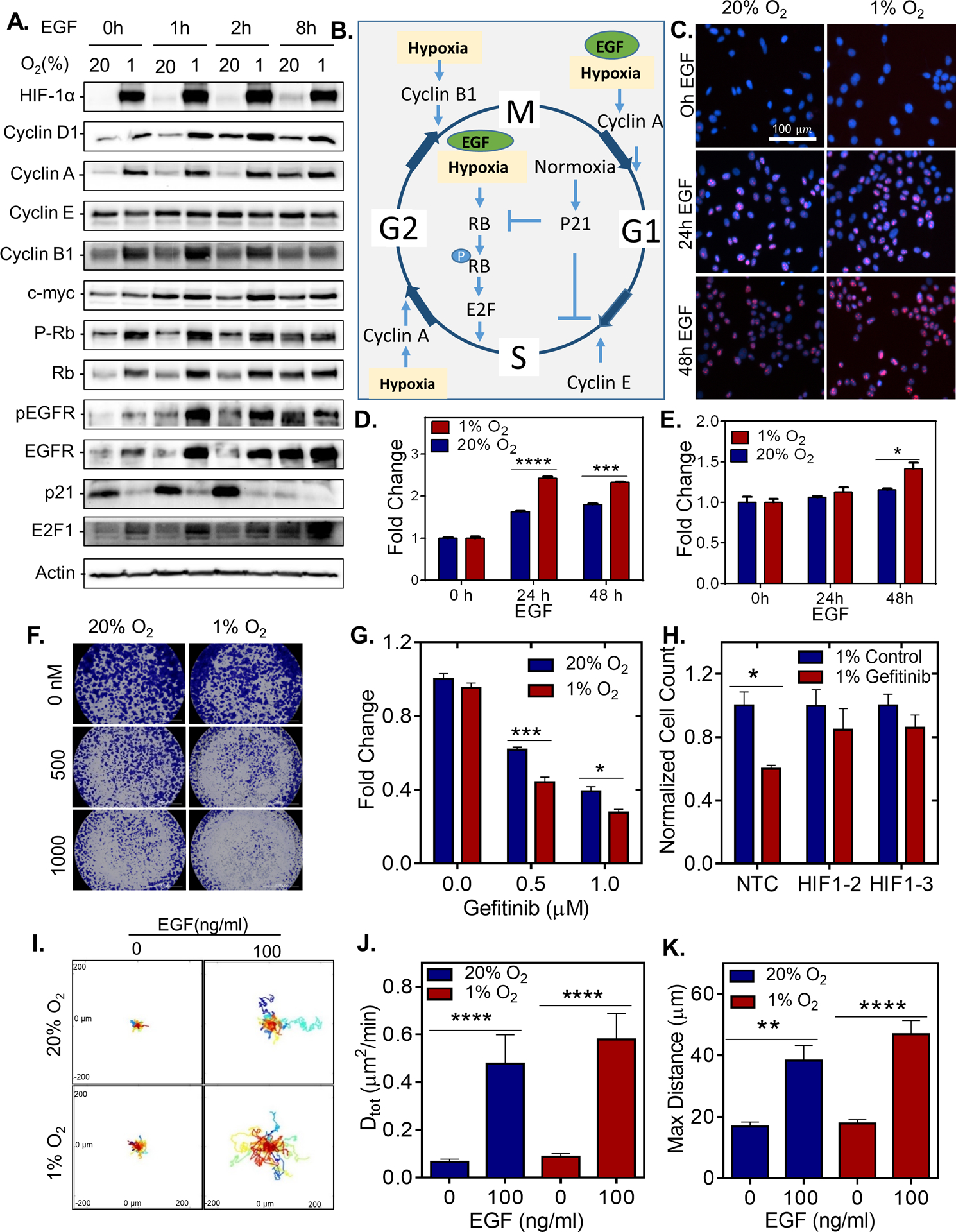
A. Immunoblot assays were performed using lysates prepared MCF7 cells serum starved for 48 h and exposed to 20% or 1% O2 for 48 h with or without 100ng/mL EGF treatment for 1, 2 or 8 h. B. Proposed mechanism; the effect of hypoxia and EGF stimulation on cell cycle. C-E. MCF-7 cells were serum starved for 72 h and exposed to 20% or 1% O2 for 48 h and treated with vehicle or with 100ng/mL EGF treatment for 24 or 72 h. (C) Representative images of Ki67- and DAPI-stained MCF7 cells. (D) The fold change in percentage of MCF-7 cells positive for ki67 as measured by KI67 staining (mean ± SEM; n = 3); ***, P < 0.001 ****, P < 0.0001 versus untreated (two-way ANOVA with Bonferroni posttest). (E) The fold change in cell viability of as measured by presto Blue Assay. (mean ± SEM; n = 3); *, P < 0.05, versus 20% untreated (two-way ANOVA with Bonferroni posttest). F. Crystal Violet staining of BT-474 cells treated with different concentrations of Gefitinib. G-H. Cell counts of (G), BT474 determined by AlamarBlue stain, and (H), MCF-7 subclones determined by manual counting, which were stably transfected with a non-target control (NTC) CRISPR vector or vectors encoding gDNA sequences that target either HIF-1α (1–1, 1–2) exposed to 20% or 1% O2 for 24 h and treated with Gefitinib for 48h. n=3. Two-way ANOVA with Bonferroni Post Test * P<0.05, ***P<0.001. I. Representative BT-474 cell trajectories plotted using x, y coordinates, obtained at 5 min intervals over a 23 h period for cells treated with or without 100ng/mL of EGF and incubated under 20% or 1% O2. J. the diffusivity (D tot) of each cell on collagen-coated plates. ** P < 0.01, **** P < 0.0001 (one-way ANOVA with Bonferroni posttest). K. The maximum distance of each cell from starting to ending coordinate.
To determine whether EGFR induction in hypoxic breast cancer cells promotes cell cycle progression, we stimulated MCF-7 cells with EGF for 0, 24, or 48h and assessed cell proliferation (Figure 6C–D and Supplementary Figure S8C). EGF treatment led to a robust increase in Ki-67 expression under hypoxia (Figure 6C–D) accompanied by increased proliferation (Figure 6E). EGF stimulation led to a 21% percent increase in the number of cells in S-phase when cultured under hypoxia as compared to a 12% increase in the number of cells in S-phase under normal O2 conditions (Supplementary Figure S8C).
Given the enhanced expression and activation of EGFR under hypoxic conditions, we reasoned that similar to cells with an EGFR amplification (38,39), cells exposed to hypoxia may be more sensitive to EGFR inhibitors, Gefitinib and Erlotinib. Under hypoxic conditions, MCF-7 and BT474 cells were more sensitive to both Erlotinib and Gefitinib (Figure 6F–G and Supplementary Figures S8D–E), whereas HIF-1α knockout sub-clones were resistant to gefitinib treatment under hypoxia (Figure 6H). Collectively, the results suggest that HIF-1α induction enhances EGFR expression to promote activation by ligand or suppression via EGFR targeted therapies.
In addition to stimulating cell proliferation, EGF stimulation (40,41) and hypoxia (42) independently promote 2D cell motility. To determine whether EGF and hypoxia would synergistically enhance cell motility, EGF-stimulated BT474 were exposed to hypoxia, and migration was monitored using time-lapse phase microscopy. The data demonstrated that cells exposed to hypoxia in the presence of EGF showed the highest increase in motility (Figure 6I–K).
Discussion
Although the HRE consensus sequence 5’-ACGTG-3’ contains a methylation-prone CpG dinucleotide (43,44), the extent and role of methylation on hypoxic gene regulation have been limited. Our results uncovered a functional HRE in an intron region of EGFR that is activated under hypoxic conditions in a HIF-dependent manner. The methylation status of the CpG dinucleotide within the HIF-binding region in intron-18 correlates to the ability for EGFR to be induced under hypoxic conditions in breast cancer. In cell lines with methylation of this region, treatment with a demethyltransferases restored EGFR regulation under hypoxia. Similar to our findings with EGFR, erythropoietin (EPO) is a well-known hypoxia-regulated gene whose expression correlates inversely with methylation (45,46). Direct methylation of the HRE sequence of the EPO gene has been shown to abrogate both HIF-1 DNA binding and hypoxic reporter gene activation (45). Likewise, a study of the MUC17 gene in pancreatic cancer demonstrated a robust hypoxic induction only in cell lines without methylated HRE regions (47,48). Further investigation is warranted to determine how globally the methylation status of DNA affects the expression of hypoxia-regulated genes and the mechanisms that are involved.
DNA derived from the normal human mammary epithelial cells of 4 donor patients indicate that the EGFR region is methylated in normal mammary epithelium. This finding is also supported by the high methylation beta values in this region of EGFR reported for more than 60 patients that donated normal tissue for the TCGA project. Therefore, the data from patient tissue and breast cancer cell lines suggest that hypomethylation of the region occurs in cancer and correlates with increased expression under hypoxia. Although DNA hypomethylation was the initial epigenetic abnormality recognized in human tumors (49), hypermethylation of promoters of genes that are silenced in cancers (e.g., tumor-suppressor genes) have been the most well studied to date. Recent high-resolution genome-wide studies confirm that DNA hypomethylation is as prevalent as DNA hypermethylation (50). One study found that in addition to global hypomethylation of repeat sequences, hypomethylation of certain genes in cancer, especially in genes linked with signaling pathways (e.g., BCR, LYN RAB8A, NFKBIB), chromatin modifications (e.g., CHD2, CHD3, SMARCB1), cell growth and development (e.g., EBF1, EGR1, EGFR, ERBB2, MYC), apoptosis inhibition (e.g., BCL2, TRAF1), and cell proliferation (e.g., CCND1, LYN, BCL3) can occur. It will be important to determine how hypomethylation of HREs occurs and whether hypomethylation correlates strictly within HREs found in intronic regions on the genome.
There are several functional implications for the activation of EGFR in response to hypoxia. First, cells exposed to hypoxia will be more sensitive to EGF-induced signaling with enhanced AKT and ERK phosphorylation that occurs in a HIF-dependent manner. We find that hypoxia promotes G1/S progression, progression from G2 to M phase, and also prevents apoptosis in the presence of EGF by reducing the levels of p21. Some tumor types produce EGF in excess, which would amplify the activation of the EGFR receptor under hypoxic conditions. On the other hand, hypoxia sensitizes cells to EGFR inhibition. To date, 6 phase II clinical trials to investigate the efficacy and safety of anti-EGFR mAbs in patients with TNBC have been reported (18). In breast cancer, the clinical trials of EGFR inhibitors have shown low response rates, however some patients have shown a meaningful response. Therefore, it may be necessary to stratify patients in order to allow those patients that may benefit from EGFR targeting agents to have access to agents currently used in the clinic. Our results suggest that patients with hypoxic tumors and hypomethylated EGFR gene may be candidates for the addition of EGFR inhibitors to their current treatment regimens.
Supplementary Material
Significance.
Hypoxia sensitizes breast cancer cells to EGFR inhibitors in a HIF-1α- and methylation-specific manner, suggesting patients with hypoxic tumors may benefit from EGFR inhibitors already available to the clinic.
Acknowledgements
We thank Julia Ju for helping with sample preparation for this work and Sara Sukumar and Mary Jo Fackler for helpful advice on methylation specific PCR assays and design. Work in the Gilkes lab is supported by U54-CA210173 (NCI), R00-CA181352 (NCI), Susan G. Komen Foundation (CCR17483484), The Jayne Koskinas Ted Giovanis Foundation for Health and Policy, The Emerson Collective, The Allegany Health Network, the NIH Predoctoral and Postdoctoral Training Program (T32 CA 153952), and the SKCCC Core Grant (P50CA006973 (NCI)).
Footnotes
Disclosure of Potential Conflicts of Interest
The authors have no potential conflicts to disclose.
References
- 1.Dewhirst MW. Intermittent hypoxia furthers the rationale for hypoxia-inducible factor-1 targeting. Cancer Res 2007;67(3):854–5. [DOI] [PubMed] [Google Scholar]
- 2.Brahimi-Horn MC, Chiche J, Pouyssegur J. Hypoxia and cancer. J Mol Med 2007;85(12):1301–7. [DOI] [PubMed] [Google Scholar]
- 3.Gillies RJ, Gatenby RA. Hypoxia and adaptive landscapes in the evolution of carcinogenesis. Cancer Metastasis Rev 2007;26(2):311–7. [DOI] [PubMed] [Google Scholar]
- 4.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 2003;3(10):721–32. [DOI] [PubMed] [Google Scholar]
- 5.Godet I, Shin YJ, Ju JA, Ye IC, Wang G, Gilkes DM. Fate-mapping post-hypoxic tumor cells reveals a ROS-resistant phenotype that promotes metastasis. Nature communications 2019;10(1):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010;29(5):625–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A 1995;92(12):5510–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ye IC, Fertig EJ, DiGiacomo JW, Considine M, Godet I, Gilkes DM. Molecular Portrait of Hypoxia in Breast Cancer: A Prognostic Signature and Novel HIF-Regulated Genes. Molecular cancer research : MCR 2018;16(12):1889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R, et al. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest 2007;117(8):2051–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.She QB, Solit DB, Ye Q, O’Reilly KE, Lobo J, Rosen N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell 2005;8(4):287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masuda H, Zhang D, Bartholomeusz C, Doihara H, Hortobagyi GN, Ueno NT. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat 2012;136(2):331–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitsudomi T, Yatabe Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. The FEBS journal 2010;277(2):301–8. [DOI] [PubMed] [Google Scholar]
- 13.Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008;27(34):4702–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rimawi MF, Shetty PB, Weiss HL, Schiff R, Osborne CK, Chamness GC, et al. Epidermal growth factor receptor expression in breast cancer association with biologic phenotype and clinical outcomes. Cancer 2010;116(5):1234–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Magkou C, Nakopoulou L, Zoubouli C, Karali K, Theohari I, Bakarakos P, et al. Expression of the epidermal growth factor receptor (EGFR) and the phosphorylated EGFR in invasive breast carcinomas. Breast cancer research : BCR 2008;10(3):R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Laurentiis M, Cianniello D, Caputo R, Stanzione B, Arpino G, Cinieri S, et al. Treatment of triple negative breast cancer (TNBC): current options and future perspectives. Cancer treatment reviews 2010;36 Suppl 3:S80–6. [DOI] [PubMed] [Google Scholar]
- 17.Burness ML, Grushko TA, Olopade OI. Epidermal growth factor receptor in triple-negative and basal-like breast cancer: promising clinical target or only a marker? Cancer J 2010;16(1):23–32. [DOI] [PubMed] [Google Scholar]
- 18.Nakai K, Hung MC, Yamaguchi H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. American journal of cancer research 2016;6(8):1609–23. [PMC free article] [PubMed] [Google Scholar]
- 19.Xu YH, Richert N, Ito S, Merlino GT, Pastan I. Characterization of epidermal growth factor receptor gene expression in malignant and normal human cell lines. Proceedings of the National Academy of Sciences of the United States of America 1984;81(23):7308–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nathoo N, Goldlust S, Vogelbaum MA. Epidermal growth factor receptor antagonists: novel therapy for the treatment of high-grade gliomas. Neurosurgery 2004;54(6):1480–8; discussion 8–9. [DOI] [PubMed] [Google Scholar]
- 21.Spaulding DC, Spaulding BO. Epidermal growth factor receptor expression and measurement in solid tumors. Seminars in oncology 2002;29(5 Suppl 14):45–54. [DOI] [PubMed] [Google Scholar]
- 22.Hirsch FR, Varella-Garcia M, Bunn PA Jr., Di Maria MV, Veve R, Bremmes RM, et al. Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2003;21(20):3798–807. [DOI] [PubMed] [Google Scholar]
- 23.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature genetics 2003;33 Suppl:245–54. [DOI] [PubMed] [Google Scholar]
- 24.Ziller MJ, Gu H, Muller F, Donaghey J, Tsai LT, Kohlbacher O, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013;500(7463):477–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gomori E, Pal J, Kovacs B, Doczi T. Concurrent hypermethylation of DNMT1, MGMT and EGFR genes in progression of gliomas. Diagnostic pathology 2012;7:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weng X, Zhang H, Ye J, Kan M, Liu F, Wang T, et al. Hypermethylated Epidermal growth factor receptor (EGFR) promoter is associated with gastric cancer. Scientific reports 2015;5:10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karakas B, Weeraratna A, Abukhdeir A, Blair BG, Konishi H, Arena S, et al. Interleukin-1 alpha mediates the growth proliferative effects of transforming growth factor-beta in p21 null MCF-10A human mammary epithelial cells. Oncogene 2006;25(40):5561–9. [DOI] [PubMed] [Google Scholar]
- 28.Dwight ZL, Palais R, Wittwer CT. uAnalyze: web-based high-resolution DNA melting analysis with comparison to thermodynamic predictions. IEEE/ACM Trans Comput Biol Bioinform 2012;9(6):1805–11. [DOI] [PubMed] [Google Scholar]
- 29.Wu PH, Giri A, Wirtz D. Statistical analysis of cell migration in 3D using the anisotropic persistent random walk model. Nat Protoc 2015;10(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abukhdeir AM, Blair BG, Brenner K, Karakas B, Konishi H, Lim J, et al. Physiologic estrogen receptor alpha signaling in non-tumorigenic human mammary epithelial cells. Breast Cancer Res Treat 2006;99(1):23–33. [DOI] [PubMed] [Google Scholar]
- 31.Changavi AA, Shashikala A, Ramji AS. Epidermal Growth Factor Receptor Expression in Triple Negative and Nontriple Negative Breast Carcinomas. J Lab Physicians 2015;7(2):79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schodel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011;117(23):e207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Newell-Price J, Clark AJ, King P. DNA methylation and silencing of gene expression. Trends Endocrinol Metab 2000;11(4):142–8. [DOI] [PubMed] [Google Scholar]
- 34.Chen YF, Chiu HH, Wu CH, Wang JY, Chen FM, Tzou WH, et al. Retinoblastoma protein (pRB) was significantly phosphorylated through a Ras-to-MAPK pathway in mutant K-ras stably transfected human adrenocortical cells. DNA Cell Biol 2003;22(10):657–64. [DOI] [PubMed] [Google Scholar]
- 35.Halilovic E, She QB, Ye Q, Pagliarini R, Sellers WR, Solit DB, et al. PIK3CA mutation uncouples tumor growth and cyclin D1 regulation from MEK/ERK and mutant KRAS signaling. Cancer Res 2010;70(17):6804–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lavoie JN, LAllemain G, Brunet A, Muller R, Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44(MAPK) and negatively by the p38/HOG(MAPK) pathway. J Biol Chem 1996;271(34):20608–16. [DOI] [PubMed] [Google Scholar]
- 37.Tsai WB, Aiba I, Long Y, Lin HK, Feun L, Savaraj N, et al. Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res 2012;72(10):2622–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amann J, Kalyankrishna S, Massion PP, Ohm JE, Girard L, Shigematsu H, et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Research 2005;65(1):226–35. [PubMed] [Google Scholar]
- 39.Shepherd FA, Rosell R. Weighing tumor biology in treatment decisions for patients with non-small cell lung cancer. J Thorac Oncol 2007;2(6):S68–S76. [DOI] [PubMed] [Google Scholar]
- 40.Saadi W, Wang SJ, Lin F, Jeon NL. A parallel-gradient microfluidic chamber for quantitative analysis of breast cancer cell chemotaxis. Biomed Microdevices 2006;8(2):109–18. [DOI] [PubMed] [Google Scholar]
- 41.Wang SJ, Saadi W, Lin F, Minh-Canh Nguyen C, Li Jeon N. Differential effects of EGF gradient profiles on MDA-MB-231 breast cancer cell chemotaxis. Exp Cell Res 2004;300(1):180–9. [DOI] [PubMed] [Google Scholar]
- 42.Ju JA, Godet I, Ye IC, Byun J, Jayatilaka H, Lee SJ, et al. Hypoxia Selectively Enhances Integrin alpha5beta1 Receptor Expression in Breast Cancer to Promote Metastasis. Mol Cancer Res 2017;15(6):723–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nanduri J, Semenza GL, Prabhakar NR. Epigenetic changes by DNA methylation in chronic and intermittent hypoxia. American journal of physiology Lung cellular and molecular physiology 2017;313(6):L1096–L100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Science’s STKE : signal transduction knowledge environment 2005;2005(306):re12. [DOI] [PubMed] [Google Scholar]
- 45.Wenger RH, Kvietikova I, Rolfs A, Camenisch G, Gassmann M. Oxygen-regulated erythropoietin gene expression is dependent on a CpG methylation-free hypoxia-inducible factor-1 DNA-binding site. European journal of biochemistry 1998;253(3):771–7. [DOI] [PubMed] [Google Scholar]
- 46.Yin H, Blanchard KL. DNA methylation represses the expression of the human erythropoietin gene by two different mechanisms. Blood 2000;95(1):111–9. [PubMed] [Google Scholar]
- 47.Kitamoto S, Yokoyama S, Higashi M, Yamada N, Matsubara S, Takao S, et al. Expression of MUC17 is regulated by HIF1alpha-mediated hypoxic responses and requires a methylation-free hypoxia responsible element in pancreatic cancer. PloS one 2012;7(9):e44108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kitamoto S, Yamada N, Yokoyama S, Houjou I, Higashi M, Goto M, et al. DNA methylation and histone H3-K9 modifications contribute to MUC17 expression. Glycobiology 2011;21(2):247–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983;301(5895):89–92. [DOI] [PubMed] [Google Scholar]
- 50.Kushwaha G, Dozmorov M, Wren JD, Qiu J, Shi H, Xu D. Hypomethylation coordinates antagonistically with hypermethylation in cancer development: a case study of leukemia. Human genomics 2016;10 Suppl 2:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.