

Abstract
Hypoxia presents a challenge to anticancer therapy, reducing the efficacy of many available treatments. Photodynamic therapy (PDT) is particularly susceptible to hypoxia given that its mechanism relies on oxygen. Herein, we introduce two new osmium-based polypyridyl photosensitizers that are active in hypoxia. The lead compounds emerged from a systematic study of two Os(II) polypyridyl families derived from 2,2′-bipyridine (bpy) or 4,4′-dimethyl-2,2′-bipyridine (dmb) as coligands combined with imidazo[4,5-f][1,10]phenanthroline ligands tethered to n=0–4 thiophenes (IP-nT). The compounds were characterized and investigated for their spectroscopic and (photo)biological activities. The two hypoxia-active Os(II) photosensitizers had n=4 thiophenes, with the bpy analog 1–4T being the most potent. In normoxia, 1–4T had low nanomolar activity (EC50=1–13 nM) with phototherapeutic indices (PI) ranging from 5,500 to 55,000 with red and visible light, respectively. Submicromolar potency was maintained even in hypoxia (1% O2), with light EC50 and PI values of 732–812 nM and 68–76, respectively — among the largest PIs to date for hypoxic photoactivity. This high degree of activity coincided with a low-energy, long-lived (0.98–3.6 μs) mixed-character intraligand-charge-transfer (3ILCT)/ligand-to-ligand charge transfer (3LLCT) state only accessible in quaterthiophene complexes 1–4T and 2–4T. The coligand identity strongly influenced the photophysical and photobiological results in this study, whereby the bpy coligand led to longer lifetimes (3.6 μs) and more potent photocytotoxicity relative to dmb. The unactivated compounds were relatively nontoxic both in vitro and in vivo. The maximum tolerated dose (MTD) for 1–4T and 2–4T in mice was ≥ 200 mg kg−1, an excellent starting point for future in vivo validation.
Graphical Abstract
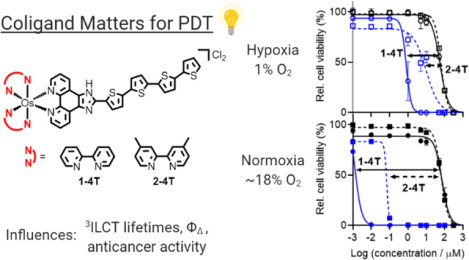
TOC-graphic 1. Light-activated osmium quaterthiophene complexes can be used with high potency against a human melanoma cell line. Choice of coligand dictates overall potency in both hypoxia (1% O2) and normoxia (~18.5% O2).
1. INTRODUCTION
Cancer is the second-leading cause of death worldwide (after cardiovascular disease), with around 17 million new cases annually.1 There are ongoing efforts to introduce alternatives or adjuvants where conventional approaches (e.g., surgery, radiation, chemotherapy) fail. Notwithstanding some remarkable breakthroughs over the past 10–30 years, particularly in the areas of immunotherapy and targeted medicine,2 the problems of resistance and metastasis endure, where populations of cancerous cells evade treatment and proliferate, particularly under favorable physiological conditions. Resistance can arise through many mechanisms; hypoxia, for example, has been shown to drive mutator phenotypes, and ensuing selection for mutant variants augments the capacity of cancer cells to adapt and defeat therapy.3–7
Light-activated treatments such as photodynamic therapy (PDT) and photochemotherapy (PCT) provide temporal and spatial selectivity by exploiting differences in toxicity in the light versus in the dark exhibited by prodrugs designed to elicit a phototoxic response. PDT, which has been known for over a century and is currently approved for clinical use,8–20 uses a relatively non-toxic prodrug — the photosensitizer (PS) — that generates cytotoxic 1O2 and other reactive oxygen species (ROS) when exposed to light. The multimodal activity of PDT-generated ROS implies that cancerous cells would be less likely to develop resistance to this mechanism.21 On the other hand, PDT’s reliance on oxygen can be problematic in the hypoxic environment that exists in some of the most aggressive tumors.22
PCT emerged, at least in part, to address the issue of hypoxia with PDT, but no PCT agents have advanced to clinical trials.23,24 PCT employs a light-responsive molecule that initiates oxygen-independent photoreactions that are ultimately cytotoxic.23–25 The process is stoichiometric and as a consequence is less potent than the catalytic generation of ROS in the PDT mechanism. While in theory PCT agents do not rely on oxygen for their photocytotoxic effects, most have been tested in normoxia. In fact, the mechanism most commonly exploited in the design of metal complex PCT agents, photoinduced ligand dissociation followed by covalent modification of biological targets, is rarely investigated in hypoxia, even in cell-free conditions.26–30 Of the PCT agents that have been evaluated in cellular hypoxia, very few have shown efficacy.30,31
The largest reported phototherapeutic indices (PIs) for PCT agents, including those that release cytotoxic ligands, are generally less than 20.27,30–33 This relatively low amplification of cytotoxicity with light is exemplified by a compound we reported with Glazer and coworkers, [Ru(6,6′-dmb)2(1-NIP)]Cl2, with a PI of 15 at 1% O2, the largest reported PI in hypoxia at the time of its publication in late 2019.31 This metal-based PCT agent, like most others, is preferentially activated with visible light rather than the clinically approved red light (630 nm) or the near infrared (NIR) light (>700 nm) that is highly desirable for certain tumors.
The recognition of this and other potential deficiencies of both PDT and PCT in clinical applications has motivated our lab to consider the photosensitizing prodrug from the perspective of the tumor characteristics,20 and to refine the parameters that might optimize the performance of a PS for particular conditions, e.g., hypoxic response or activation by tissue-penetrating long-wavelength light.31,34–42 The strategy has been promising, with one of our compounds, TLD1433,20 currently in phase II human clinical trials (Clinicaltrials.gov identifier: NCT03945162).
In this vein, we recently published a variety of Ru(II) polypyridyl families based on the imidazo[4,5-f][1,10]phenanthroline-oligothiophene (IP-nT) ligand motif that proved successful in TLD1433.20,36,40–44 Certain π-expansive ligands, and IP-nT in particular, can trap photoexcited energy in long-lived, ligand-localized triplet excited state reservoirs that can sensitize 1O2 with extremely high efficiency.45 The nature of this state in complexes incorporating the IP-nT ligand is an intraligand charge transfer (3ILCT) triplet excited state that is lower in energy than the triplet metal-to-ligand charge transfer (3MLCT) excited states that typically dominate Ru(II) polypyridyl photophysics.46 The thienyl-based CT character in particular allows for 1O2 sensitization at low oxygen tension and provides access to other important cytotoxic reactions, leading to usually high photocytotoxicity and unprecedented PIs. This high potency is best realized with visible wavelengths of light in the Ru(II) systems.
Access to reactive 3ILCT states afforded by the IP-nT ligand with red and NIR light could be realized in Os(II) polypyridyl systems with appropriate 3MLCT-3ILCT energy gaps, where one photon absorption populates 1MLCT states that ultimately decay to lowest energy 3ILCT states. The corresponding Os(II) complexes have less energetic MLCT excited states relative to their Ru(II) counterparts, which results in light absorption at longer wavelengths, possibly reaching into the PDT window (not clearly defined, but in the vicinity of 650–900 nm) where light penetrates tissue best.
Here we explore this possibility as well as the influence of the coligands on the energetics and dynamics of a series of Os(II) complexes (Chart 1) in their photoexcited states, and the ensuing effects on normoxic and hypoxic photocytotoxicity toward cancer cells. We show that the coligands influence the energy of the 3MLCT state, while not affecting the ligand-based 3ILCT energy, and as such, allow the manipulation of relaxation pathways. This led to remarkable differences in the photocytotoxicity between coligand families with strong contrasts in their normoxic and hypoxic activities. Subtle modification of the coligand alone was adequate for enhancing potency by roughly an order of magnitude in cells. The coligand is not innocent — it fine tunes the complex’s chemical, (photo)physical and (photo)biological properties.
Chart 1.
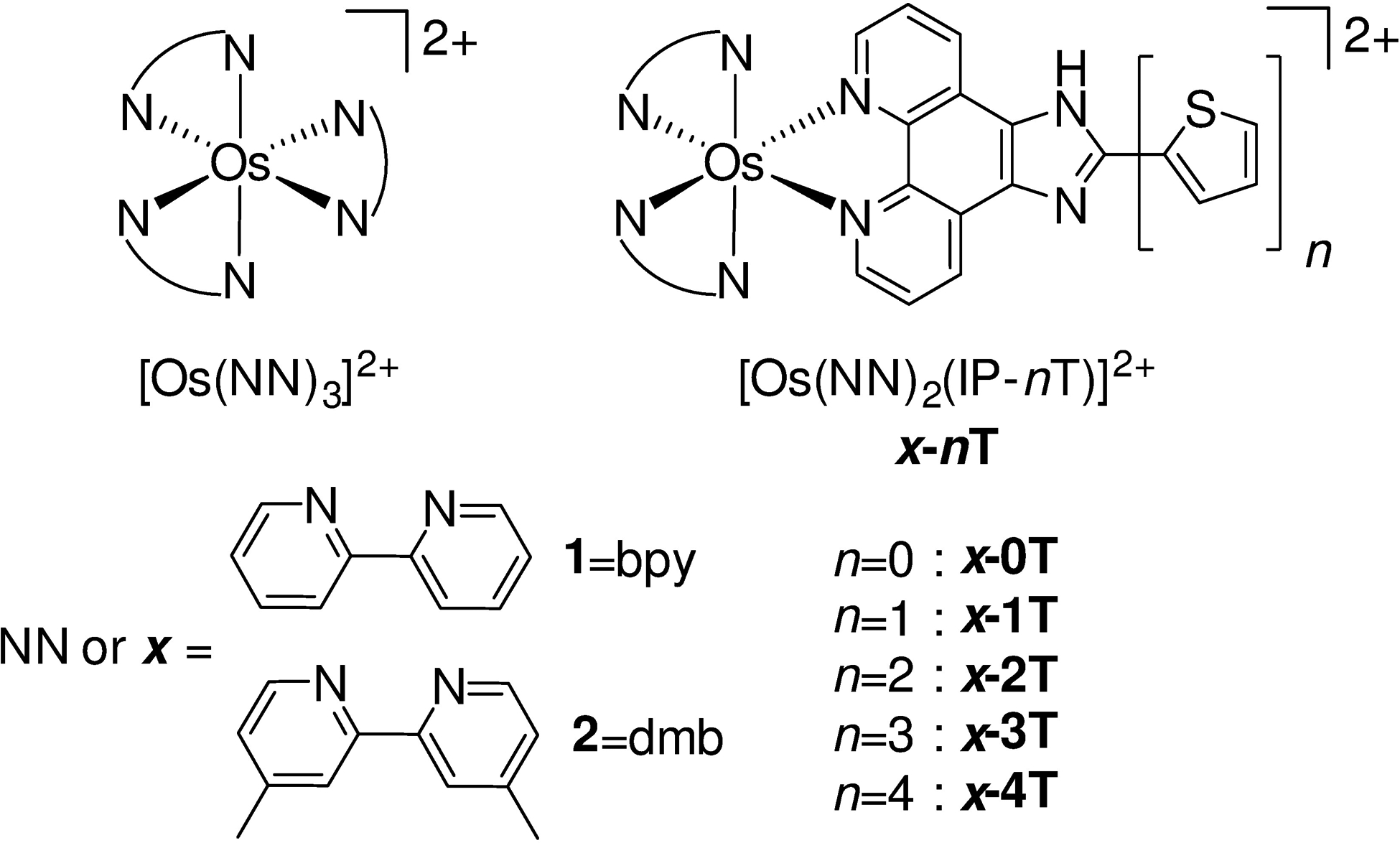
Molecular structures of the compounds used in this study. Chloride salts were used unless otherwise specified. The complexes were racemic mixtures of the Δ and Λ isomers.
2. MATERIALS AND METHODS
2.1. Materials
Unless otherwise specified, all reagents and solvents were purchased from commercial sources and used without further purification. Ultra-pure water (type I; ≥ 18.2 MΩ) was used for all biological experiments with a Barnstead or Milli-Q filtration system. Saturated solvents for log D experiments were prepared in-house using 10 mM phosphate buffer solution (saline-free PBS at pH=7.4) and 1-octanol (99.9%). Buffers were checked for an accurate pH against a two-point calibrated VWR B10P pH meter (pHref. = 4.00, 7.00; Fisher Science Education, S25849A/B).
2.2. Instrumentation
Microwave reactions were performed in a CEM Discover microwave reactor. Flash chromatography relied on the Teledyne Isco CombiFlash® EZ Prep system with Silicycle SiliaSep silica flash cartridges (FLH-R10030B-ISO25). Size-exclusion chromatography was performed on a manual column packed with Sephadex® LH-20. NMR spectra were collected using a JEOL ECA 500 NMR spectrometer (1H, 1H–1H COSY) at the NMR facility at the University of North Carolina at Greensboro (UNCG) and Agilent 700 MHz NMR spectrometer (13C, 13C–1H HSQC, 13C–1H HMBC) at the Joint School of Nanoscience and Nanoengineering at Greensboro (JSNN). The chemical shifts are reported in parts per million (ppm) and were referenced to the residual solvent peaks. ESI+ mass spectra were obtained using a Thermo Fisher LTQ Orbitrap XL coupled to a Water’s Acquity Ultra Performance Liquid Chromatography (UPLC) stack using a BEH C18 column at UNCG’s Triad Mass Spectrometry facility. High Performance Liquid Chromatography (HPLC) analyses were carried out on an Agilent/Hewlett Packard 1100 series instrument (ChemStation Rev. A. 10.02 software) using a Hypersil GOLD C18 column (Thermo 25005–254630, guard 25003–014001) with an A–B gradient (40 min run; 98% → 5% A; A=0.1% formic acid in water, B=0.1% formic acid in acetonitrile (MeCN). Reported retention times are accurate to within ± 0.1 min.
2.3. Synthesis
Os(bpy)2Cl2 ∙2H2O and Os(dmb)2Cl2 ∙2H2O intermediates47 (bpy = 2,2′-bipyridine, dmb = 4,4′-dimethyl-2,2′-bipyridine) and imidazo[4,5-f][1,10]phenanthroline (IP) based ligands48 were synthesized according to adapted literature protocols. The synthesis of IP-based ligands follows the procedure for the synthesis of IP-4T that is described below. [2,2′:5′,2″:5″,2‴-quaterthiophene]-5-carbaldehyde (4T-CHO) was prepared as previously described40 via the coupling of 5-bromo-5″-formyl-2,2′:5′,2″-terthiophene with 2-(tributylstannyl)thiophene, which were purchased from Alfa Aesar and Fisher Scientific, respectively.
Complexes 1-nT and 2-nT have not previously been reported and were isolated as their PF6− salts. Their Cl− salts were obtained from corresponding PF6− salts via anion metathesis on HCl-treated Amberlite IRA-410 resin (Alfa-Aesar, A1773436) with methanol as the eluent. Biological studies were carried out on Cl− salts and photophysical measurements were carried out on PF6− salts. Final complexes were a racemic mixture of Δ/Λ isomers. Reference compounds [Os(bpy)3]2+ and [Os(dmb)3]2+ and complexes 1-nT and 2-nT were characterized by TLC, 1H NMR (Figures S1–S7, S23–S29), high-resolution ESI+–MS (Figures S11–S16, S33–S38), and HPLC (Figures S17–S22, S39–S44). All complexes 1-nT and 2-nT were characterized by 1H–1H COSY NMR, and compounds 1–4T and 2–4T were additionally characterized by 13C, 13C–1H HSQC, and 13C–1H HMBC NMR for the full assignment of the 1H NMR and 13C NMR signals (Figures S8–S10, S30–S32). 1H NMR assignments were made in consultation with literature sources.49,50
IP-4T.
1,10-phenanthroline-5,6-dione (175 mg, 0.83 mmol), 4T-CHO (200 mg, 0.56 mmol), and ammonium acetate (1.38 g, 18 mmol) were added to a 250 mL round-bottom flask with glacial acetic acid (100 mL). The orange mixture was heated at 100°C for 96 hours. Once cooled, the reddish-brown mixture was neutralized with NH4OH. The precipitate was vacuum filtered using a Bϋchner funnel and washed with cold deionized water (50 mL) and cold ether (100 mL) to obtain the desired product as a brown solid (279 mg, 91%). 1H NMR (500 MHz, DMSO-d6, ppm): δ 9.05 (dd, J = 4.2, 1.7 Hz, 2H), 8.85 (d, J = 7.9 Hz, 2H), 7.88–7.81 (m, 3H), 7.57 (d, J = 4.7 Hz, 1H), 7.52 (d, J = 3.8 Hz, 1H), 7.48 (d, J = 3.9 Hz, 1H), 7.41–7.36 (m, 3H), 7.32 (d, J = 3.7 Hz, 1H), 7.13 (dd, J = 5.1, 3.5 Hz, 1H).
[Os(bpy)3]2+.
Os(bpy)2Cl2 ∙2H2O (98 mg, 0.16 mmol) and bpy (25 mg, 0.16 mmol) were added to a microwave vessel containing argon-purged ethylene glycol (2.5 mL) and subjected to microwave irradiation at 180°C for 15 minutes. The resulting black mixture was transferred to a separatory funnel with deionized water (20 mL) and CH2Cl2 (30 mL). After gentle mixing, the CH2Cl2 was drained and the remaining aqueous layer was washed with CH2Cl2 (30 mL portions) until the CH2Cl2 was colorless. At that point, another 30 mL of CH2Cl2 was added and allowed to settle to the bottom of the separatory funnel. Then, saturated aqueous KPF6 (5 mL) was added, and the mixture shaken gently and allowed to settle over time to facilitate transfer of the product from the aqueous layer to the CH2Cl2 layer, which was concentrated under reduced pressure. The crude product was purified by silica gel flash column chromatography with a gradient of MeCN, 10% water in MeCN, followed by 7.5% water in MeCN with 0.5% KNO3 to obtain [Os(bpy)3](PF6)2 as a black solid (67 mg, 44%). A portion of the PF6− salt (25 mg) was converted to its corresponding Cl− salt [Os(bpy)3]Cl2 in quantitative yield using Amberlite IRA-410 with MeOH as the eluent. Rf = 0.9 (0.5% KNO3, 7.5% H2O, 92% MeCN). 1H NMR (MeOD-d3, 500 MHz): δ 8.70 (d, J = 8.0 Hz, 6H; 3,3′), 7.94 (td, J = 8.0, 1.5 Hz, 6H; 4,4′), 7.73 (d, J = 6.0 Hz, 6H; 6,6′), 7.40 (td, J = 7.0, 1.5 Hz, 6H; 5,5′) (for hydrogen labels, see Chart 2). HRMS (ESI+) m/z: [M-2Cl]2+ Calcd for C30H24N6Os 330.0833; Found: 330.0825. HPLC retention time: 8.59 min (99% purity by peak area).
Chart 2:
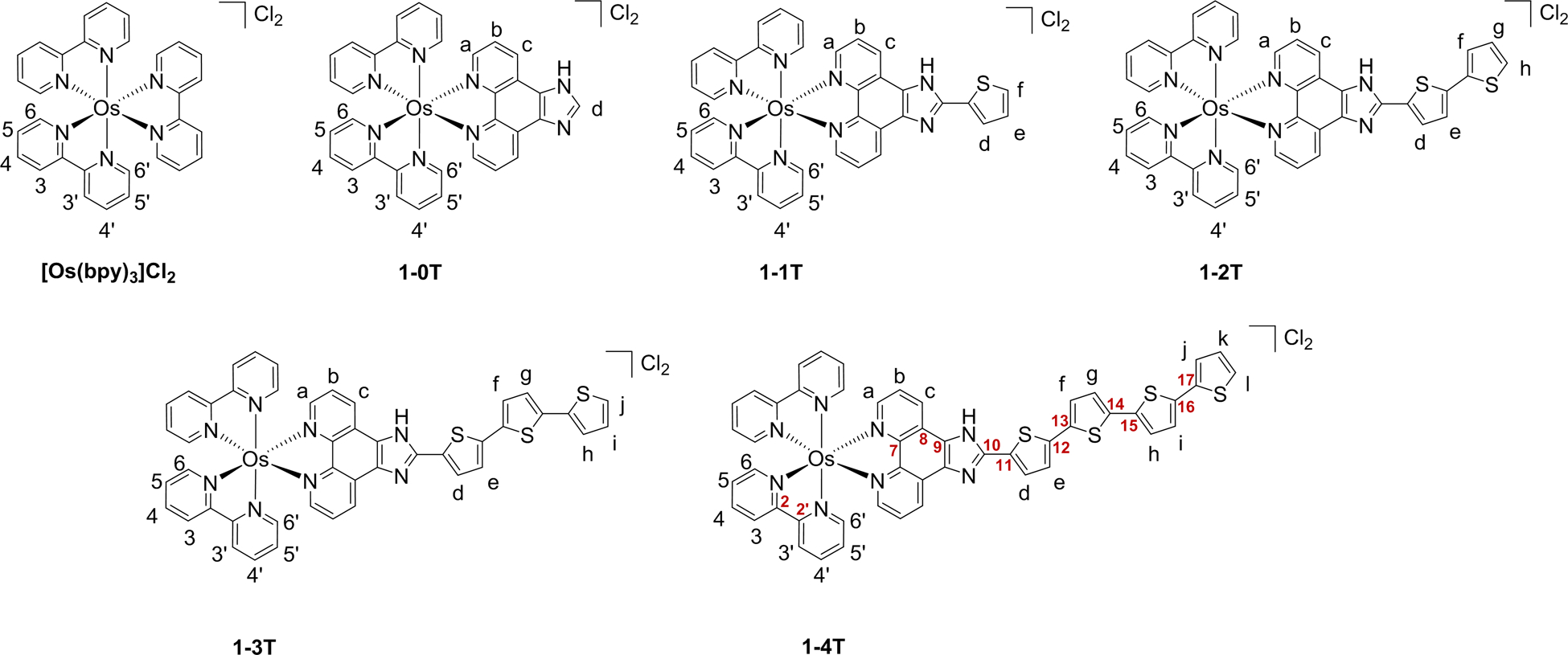
Hydrogen labels and selected carbon labels used in 1H NMR assignments of reference compound [Os(bpy)3]Cl2 and 1-nT.
[Os(bpy)2(IP)]Cl2 (1–0T).
Os(bpy)2Cl2 ∙2H2O (115 mg, 0.19 mmol) and IP (44 mg, 0.20 mmol) were combined and treated according to the procedure described for [Os(bpy)3]2+ to yield [Os(bpy)2(IP)](PF6)2 as a black solid (58 mg, 29%). The PF6− salt was converted to its corresponding Cl− salt [Os(bpy)2(IP)](Cl)2 using Amberlite IRA-410 with MeOH as the eluent (97% recovery). Rf = 0.06 (0.5% KNO3, 7.5% H2O, 92% MeCN). 1H NMR (MeOD-d3, 500 MHz): δ 8.81 (d, J = 8.0 Hz, 2H; c), 8.74 (d, J = 8.0 Hz, 2H; 3), 8.73 (s, 1H; d), 8.70 (d, J = 8.0 Hz, 2H; 3′), 8.06 (dd, J = 5.5, 1.0 Hz, 2H; a), 7.98 (td, J = 8.0, 1.0 Hz, 2H; 4), 7.87 (td, J = 8.0, 1.0 Hz, 2H; 4′), 7.86 (d, J = 5.5 Hz, 2H; 6), 7.81 (dd, J = 8.5, 5.5 Hz, 2H; b), 7.56 (d, J = 5.5 Hz, 2H; 6′), 7.46 (td, J = 7.0, 1.0 Hz, 2H; 5), 7.21 (td, J = 7.0, 1.0 Hz, 2H; 5′) (for hydrogen labels, see Chart 2). HRMS (ESI+) m/z: [M-2Cl]2+ Calcd for C33H24N8Os 362.0864; Found: 362.0858. [M-2Cl-H]+ Calcd for C33H23N8Os 723.1655; Found: 723.1646. HPLC retention time: 8.61 min (98% purity by peak area).
[Os(bpy)2(IP-1T)]Cl2 (1–1T).
Os(bpy)2Cl2∙2H2O (103 mg, 0.17 mmol) and IP-1T (54 mg, 0.18 mmol) were combined and treated according to the procedure described for [Os(bpy)3]2+ to yield [Os(bpy)2(IP-1T)](PF6)2 as a black solid (51 mg, 26%). A portion of the PF6− salt (44 mg) was converted to its corresponding Cl− salt [Os(bpy)2(IP-1T)]Cl2 using Amberlite IRA-410 with MeOH as the eluent (36 mg, 98% recovery). Rf = 0.13 (0.5% KNO3, 7.5% H2O, 92% MeCN). 1H NMR (MeOD-d3, 500 MHz): δ 8.90 (d, J = 8.5 Hz, 2H; c), 8.75 (d, J = 8.0 Hz, 2H; 3), 8.70 (d, J = 8.5 Hz, 2H; 3′), 8.03 (d, J = 5.5 Hz, 2H; a), 8.02 (m, 1H; d), 7.99 (td, J = 8.0, 1.0 Hz, 2H; 4), 7.87 (td, J = 8.5, 1.0 Hz, 2H; 4′), 7.86 (d, J = 6.5, 2H; 6), 7.79 (dd, J = 8.5, 5.5 Hz, 2H; b), 7.74 (dd, J = 4.5, 1.0 Hz, 1H; f), 7.58 (d, J = 6.5 Hz, 2H; 6′), 7.46 (td, J =7.0, 1.0 Hz, 2H; 5), 7.30 (dd, J = 4.5, 3.5 Hz, 1H; e), 7.23 (td, J =7.0, 1.0 Hz, 2H; 5′) (for hydrogen labels, see Chart 2). HRMS (ESI+) m/z: [M-2Cl]2+ Calcd for C37H26N8OsS 403.0802; Found: 403.0789. [M-2Cl-H]+ Calcd for C37H25N8OsS 805.1532; Found: 805.1517. HPLC retention time: 9.74 min (95% purity by peak area).
[Os(bpy)2(IP-2T)]Cl2 (1–2T).
Os(bpy)2Cl2∙2H2O (98 mg, 0.16 mmol) and IP-2T (65 mg, 0.17 mmol) were combined and treated according to the procedure described for [Os(bpy)3]2+ to yield [Os(bpy)2(IP-2T)](PF6)2 as a black solid (36 mg, 18%). The PF6− salt was converted to its corresponding Cl− salt [Os(bpy)2(IP-2T)]Cl2 using Amberlite IRA-410 with MeOH as the eluent (99% recovery). 1H NMR (MeOD-d3, 500 MHz): δ 8.80 (d, J = 8.5 Hz, 2H; c), 8.75 (d, J = 8.5 Hz, 2H; 3), 8.72 (d, J = 8.5 Hz, 2H; 3′), 8.01 (dd, J = 5.5, 1.0 Hz, 2H; a), 7.99 (td, J = 8.0, 1.0 Hz, 2H; 4), 7.92 (d, J = 4.0 Hz, 1H; d), 7.88 (td, J = 8.0, 1.0 Hz, 2H; 4′), 7.87 (d, J = 6.0 Hz, 2H; 6), 7.74 (dd, J = 8.5, 5.5 Hz, 2H; b), 7.63 (d, J = 6.0 Hz, 2H; 6′), 7.47 (td, J = 7.0, 1.0 Hz, 2H; 5), 7.45 (d, J = 5.0 Hz, 1H; h), 7.36 (dd, J = 3.5, 1.0 Hz, 1H; f), 7.33 (d, J = 4.0 Hz, 1H; e), 7.26 (td, J = 6.5, 1.0 Hz, 2H; 5′), 7.12 (dd, J = 5.0, 3.5 Hz, 1H; g) (for hydrogen labels, see Chart 2). HRMS (ESI+) m/z: [M-2Cl]2+ Calcd for C41H28N8OsS2 444.0741; Found: 444.0726. [M-2Cl-H]+ Calcd for C41H27N8OsS2 887.1409; Found: 887.1397. HPLC retention time: 20.38 min (95% purity by peak area).
[Os(bpy)2(IP-3T)]Cl2 (1–3T).
Os(bpy)2Cl2 ∙2H2O (92 mg, 0.15 mmol) and IP-3T (75 mg, 0.16 mmol) were combined and treated according to the procedure described for [Os(bpy)3]2+ to yield [Os(bpy)2(IP-3T)](PF6)2 as a black solid (60 mg, 30%). A portion of the PF6− salt (40 mg) was converted to its corresponding Cl− salt [Os(bpy)2(IP-3T)]Cl2 using Amberlite IRA-410 with MeOH as the eluent (91% recovery). 1H NMR (MeOD-d3, 500 MHz): δ 8.81 (d, J = 8.0 Hz, 2H; c), 8.75 (d, J = 8.5 Hz, 2H; 3), 8.73 (d, J = 8.5 Hz, 2H; 3′), 8.03 (dd, J = 5.5, 1.0 Hz, 2H; a), 7.98 (td, J = 8.0, 1.0 Hz, 2H; 4), 7.94 (d, J = 3.5 Hz, 1H; d), 7.90 (td, J = 8.0, 1.0 Hz, 2H; 4′), 7.86 (d, J = 6.0 Hz, 2H; 6), 7.74 (dd, J = 8.0, 5.5 Hz, 2H; b), 7.64 (d, J = 5.5 Hz, 2H; 6′), 7.47 (td, J = 6.5, 1.0 Hz, 2H; 5), 7.37 (d, J = 5.0 Hz, 1H; j), 7.28 (td, J = 6.5, 1.0 Hz, 2H; 5′), 7.26 (m, 3H; h,e,f), 7.15 (d, J = 4.0 Hz, 1H; g), 7.06 (dd, J = 5.0, 3.5 Hz, 1H; i) (for hydrogen labels, see Chart 2). HRMS (ESI+) m/z: [M-2Cl]2+ Calcd for C45H30N8OsS3 485.0680; Found: 485.0664. [M-2Cl-H]+ Calcd for C45H29N8OsS3 969.1287; Found: 969.1282. HPLC retention time: 22.37 min (92% purity by peak area).
[Os(bpy)2(IP-4T)]Cl2 (1–4T).
Os(bpy)2Cl2∙2H2O (117 mg, 0.19 mmol), and IP-4T (93 mg, 0.17 mmol) were combined and treated according to the procedure described for [Os(bpy)3]2+ to yield [Os(bpy)2(IP-4T)](PF6)2 as a black solid (29 mg, 13%). The PF6− salt was converted to its corresponding Cl− salt [Os(bpy)2(IP-4T)]Cl2 using Amberlite IRA-410 with MeOH as the eluent (93% recovery). 1H NMR (MeOD-d3, 700 MHz): δ 9.01–8.76 (bs, 2H; c), 8.74 (d, J = 8.4 Hz, 2H; 3), 8.71 (d, J = 8.4 Hz, 2H; 3′), 8.04 (d, J = 5.6 Hz, 2H; a), 7.99 (td, J = 7.7, 0.7 Hz, 2H; 4), 7.93 (d, J = 3.5 Hz, 1H; d), 7.88 (td, J = 8.4, 0.7 Hz, 2H; 4′), 7.86 (d, J = 6.3 Hz, 2H; 6), 7.79 (m, 2H; b), 7.59 (d, J = 6.3 Hz, 2H; 6′), 7.47 (td, J = 6.3, 0.7 Hz, 2H; 5), 7.41 (d, J = 3.5 Hz, 1H; e), 7.36 (d, J = 4.9 Hz, 1H; l), 7.35 (d, J = 3.5 Hz, 1H; f), 7.25 (m, 1H; j), 7.24 (td, J = 6.3, 0.7 Hz, 2H; 5′), 7.24 (d, J = 3.5 Hz, 2H; g,h), 7.15 (d, J = 3.5 Hz, 1H; i), 7.06 (dd, J = 4.9, 3.5 Hz; 1H; k) (for hydrogen labels, see Chart 2). 13C NMR (MeOH-d3, 175 MHz): δ 160.70 (2), 160.50 (2′), 151.96 (6), 151.91 (6′), 150.52 (a), 150.13 (10), 149.07–148.99 (7,9), 141.64 (12), 138.67 (4), 138.54 (4′), 138.48 (14), 138.36 (16), 137.84 (17), 136.41 (15), 136.21 (13), 131.82 (11), 131.67 (c), 131.12 (8), 129.71 (d), 129.40 (5), 129.33 (5′), 129.13 (k), 127.58 (b), 126.98 (f), 126.10 (l,h), 125.93 (e), 125.82 (3,g), 125.79 (3′), 125.59 (i), 125.13 (j) (for carbon labels, see Chart 2). HRMS (ESI+) m/z: [M-2Cl]2+ Calcd for C49H32N8OsS4 526.0618; Found: 526.0597. [M-2Cl-H]+ Calcd for C49H31N8OsS4 1051.1164; Found: 1051.1147. HPLC retention time: 23.94 min (97% purity by peak area).
[Os(dmb)3]2+.
Os(dmb)2Cl2∙2H2O (107 mg, 0.16 mmol) and 4,4′-dmb (29 mg, 0.16 mmol) were added to a microwave vessel containing argon-purged ethylene glycol (2.5 mL) and subjected to microwave irradiation at 180°C for 15 minutes. The resulting black mixture was transferred to a separatory funnel with deionized water (20 mL) and CH2Cl2 (30 mL). After gentle mixing, the CH2Cl2 was drained and the remaining aqueous layer was washed with CH2Cl2 (30 mL portions) until the CH2Cl2 was colorless. At that point, another 30 mL of CH2Cl2 was added and allowed to settle to the bottom of the separatory funnel. Then, saturated aqueous KPF6 (5 mL) was added, and the mixture shaken gently and allowed to settle over time to facilitate transfer of the product from the aqueous layer to the CH2Cl2 layer, which was concentrated under reduced pressure. The crude product was purified by silica gel flash column chromatography with a gradient of MeCN, 10% water in MeCN, followed by 7.5% water in MeCN with 0.5% KNO3 to obtain [Os(dmb)3](PF6)2 as a black solid (69 mg, 42%). The PF6− salt was converted to its corresponding Cl− salt [Os(dmb)3](Cl)2 using Amberlite IRA-410 with MeOH as the eluent (92% recovery). The Cl− salt (26 mg) was further purified on Sephadex LH-20 with MeOH as the eluent (17 mg, 65% recovery). Rf = 0.25 (0.5% KNO3, 7.5% H2O, 92% MeCN). 1H NMR (MeOD-d3, 500 MHz): δ 8.52 (d, J = 1.0 Hz, 6H; 3,3′), 7.51 (d, J = 6.0 Hz, 6H; 6,6′), 7.21 (dd, J = 6.0, 1.0 Hz, 6H; 5,5′), 2.64 (s, 18H; 4,4′-Me) (for hydrogen labels, see Chart S2). HRMS (ESI+) m/z: [M-2Cl]2+ Calcd for C36H36N6Os 372.1303; Found: 372.1292. HPLC retention time: 10.03 min (99% purity by peak area).
[Os(dmb)2(IP)]Cl2 (2–0T).
Os(dmb)2Cl2∙2H2O (100 mg, 0.15 mmol) and IP (35 mg, 0.16 mmol) were combined and treated according to the procedure for [Os(dmb)3]2+ to yield [Os(dmb)2(IP)](PF6)2 as a black solid (98 mg, 79%). A portion of the PF6− salt was converted to its corresponding Cl− salt [Os(dmb)2(IP)]Cl2 using Amberlite IRA-410 with MeOH as the eluent. Rf = 0.11 (0.5% KNO3, 7.5% H2O, 92% MeCN). 1H NMR (MeOD-d3, 500 MHz): δ 8.74 (d, J = 8.5 Hz, 2H; c), 8.70 (s, 1H; d), 8.59 (d, J = 2.0 Hz, 2H; 3), 8.54 (d, J = 2.0 Hz, 2H; 3′), 8.06 (dd, J = 5.0, 1.0 Hz, 2H; a), 7.76 (dd, J = 8.5, 5.0 Hz, 2H; b), 7.64 (d, J = 6.0 Hz, 2H; 6), 7.31 (d, J = 6.0 Hz, 2H; 6′), 7.30 (dd, J = 6.0, 1.5 Hz, 2H; 5), 7.03 (dd, J = 6.0, 1.5 Hz, 2H; 5′), 2.70 (s, 6H; 4-Me), 2.58 (s, 6H, 4′-Me) (for hydrogen labels, see Chart S2). HRMS (ESI+) m/z: [M-2Cl]2+ Calcd for C37H32N8Os 390.1177; Found: 390.1163. [M-2Cl-H]+ Calcd for C37H31N8Os 779.2281; Found: 779.2260. HPLC retention time: 9.81 min (99% purity by peak area).
[Os(dmb)2(IP-1T)]Cl2 (2–1T).
Os(dmb)2Cl2∙2H2O (67 mg, 0.10 mmol) and IP-1T (30 mg, 0.10 mmol) were combined and treated according to the procedure for [Os(dmb)3]2+ to yield [Os(dmb)2(IP-1T)](PF6)2 as a black solid (111 mg, 97%). A portion of the PF6− salt (80 mg) was converted to its corresponding Cl− salt [Os(dmb)2(IP-1T)]Cl2 using Amberlite IRA-410 with MeOH as the eluent (61 mg, 94% recovery). The solids were then further purified using Sephadex LH-20 (38 mg, 65%). Rf = 0.23 (0.5% KNO3, 7.5% H2O, 92% MeCN). 1H NMR (MeOD-d3, 500 MHz): δ 8.84 (broad d, J = 7.0 Hz, 2H; c), 8.59 (d, J = 1.0 Hz, 2H; 3), 8.54 (d, J = 1.0 Hz, 2H; 3′), 8.03 (d, J = 5.0 Hz, 2H; a), 8.00 (d, J = 4.0 Hz, 1H; d), 7.75 (dd, J = 8.0, 5.5 Hz, 2H; b), 7.74 (d, J = 5.0 Hz, 1H; f), 7.64 (d, J = 6.0 Hz, 2H; 6), 7.34 (d, J = 6.0 Hz, 2H, 6′), 7.30 (m, 3H; 5,e), 7.04 (dd, J = 6.0, 1.0 Hz, 2H; 5′), 2.70 (s, 6H; 4-Me), 2.58 (s, 6H, 4′-Me) (for hydrogen labels, see Chart S2). HRMS (ESI+) m/z: [M-2Cl]2+ Calcd for C41H34N8OsS 431.1115; Found: 431.1108. [M-2Cl-H]+ Calcd for C41H33N8OsS 861.2158; Found: 861.2158. HPLC retention time: 9.66 min (99% purity by peak area).
[Os(dmb)2(IP-2T)]Cl2 (2–2T).
Os(dmb)2Cl2∙2H2O (67 mg, 0.10 mmol) and IP-2T (39 mg, 0.10 mmol) were combined and treated according to the procedure for [Os(dmb)3]2+ to yield [Os(dmb)2(IP-2T)](PF6)2 as a black solid (115 mg, 93%). A portion of the PF6− salt (89 mg) was converted to its corresponding Cl− salt [Os(dmb)2(IP-2T)]Cl2 using Amberlite IRA-410 with MeOH as the eluent (63 mg, 86% recovery). The solids were then further purified using Sephadex LH-20 (38 mg, 61%). Rf = 0.23 (0.5% KNO3, 7.5% H2O, 92% MeCN). 1H NMR (MeOD-d3, 500 MHz): δ 8.73 (d, J = 8.5 Hz, 2H; c), 8.59 (d, J = 1.0 Hz, 2H; 3), 8.55 (d, J = 1.0 Hz, 2H; 3′), 8.01 (dd, J = 5.5, 1.0 Hz, 2H; a), 7.91 (d, J = 4.0 Hz, 1H; d), 7.70 (dd, J = 8.5, 5.5 Hz, 2H; b), 7.64 (d, J = 5.5 Hz, 2H; 6), 7.45 (dd, J = 5.0, 1.0 Hz, 1H; h), 7.37 (d, J = 6.0 Hz, 2H; 6′), 7.36 (dd, J = 3.5, 1.0 Hz, 1H; f), 7.34 (d, J = 4.0 Hz, 1H; e), 7.30 (dd, J = 6.0, 1.0 Hz, 2H; 5), 7.11 (dd, J = 5.0, 3.5 Hz, 1H; g), 7.06 (dd, J = 6.0, 1.0 Hz, 2H; 5′), 2.70 (s, 6H; 4-Me), 2.59 (s, 6H; 4′-Me) (for hydrogen labels, see Chart S2). HRMS (ESI+) m/z: [M-2Cl]2+ Calcd for C45H36N8OsS2 472.1054; Found: 472.1042. [M-2Cl-H]+ Calcd for C45H35N8OsS2 943.2035; Found: 943.2033. HPLC retention time: 22.05 min (97% purity by peak area).
[Os(dmb)2(IP-3T)]Cl2 (2–3T).
Os(dmb)2Cl2∙2H2O (99 mg, 0.15 mmol) and IP-3T (73 mg, 0.16 mmol) were combined and treated according to the procedure described for [Os(dmb)3]2+ to yield [Os(dmb)2(IP-3T)](PF6)2 as a black solid (111 mg, 54%). The PF6− salt was converted to its corresponding Cl− salt [Os(dmb)2(IP-3T)]Cl2 using Amberlite IRA-410 with MeOH as the eluent. A portion of the solids (12 mg) were further purified using Sephadex LH-20 (10 mg, 83%). Rf = 0.26 (0.5% KNO3, 7.5% H2O, 92% MeCN). 1H NMR (MeOD-d3, 500 MHz): δ 8.72 (d, J = 8.5 Hz, 2H; c), 8.59 (d, J = 1.0 Hz, 2H; 3), 8.56 (d, J = 1.0 Hz, 2H; 3′), 8.01 (dd, J = 5.0, 1.0 Hz, 2H; a), 7.90 (d, J = 3.5 Hz, 1H; d), 7.68 (dd, J = 8.5, 5.5 Hz, 2H; b), 7.65 (d, J = 6.0 Hz, 2H; 6), 7.40 (d, J = 6.0 Hz, 2H; 6′), 7.38 (dd, J = 5.0, 1.0 Hz, 1H; j), 7.31 (dd, J = 6.0, 1.0 Hz, 2H; 5), 7.26 (d, J = 3.5 Hz, 1H; h), 7.26 (d, J = 4.0 Hz, 1H; e), 7.24 (d, J = 4.0 Hz, 1H; f), 7.15 (d, J = 4.0 Hz, 1H; g), 7.10 (dd, J = 6.0, 1.0 Hz, 2H; 5′), 7.07 (dd, J = 5.0, 3.5 Hz, 1H; i), 2.70 (s, 6H; 4-Me), 2.60 (s, 6H; 4′-Me) (for hydrogen labels, see Chart S2). HRMS (ESI+) m/z: [M-2Cl]2+ Calcd for C49H38N8OsS3 513.0993; Found: 513.0978. [M-2Cl-H]+ Calcd for C49H37N8OsS3 1025.1913; Found: 1025.1914. HPLC retention time: 23.32 min (95% purity by peak area).
[Os(dmb)2(IP-4T)]Cl2 (2–4T).
Os(dmb)2Cl2∙2H2O (126 mg, 0.19 mmol) and IP-4T (90 mg, 0.16 mmol) were added to a microwave vial with 99.5% ethanol (3 mL) and subjected to microwave irradiation at 130°C for 20 minutes. Saturated aqueous KPF6 (2 mL) was added to the vial and the product was obtained by filtration with a fine frit and washed with cold water (6 mL) and cold diethyl ether (50 mL). The crude product was purified by silica gel flash column chromatography with a gradient of MeCN, 10% water in MeCN, followed by 7.5% water in MeCN with 0.5% KNO3 to obtain [Os(dmb)2(IP-4T)](PF6)2 as a black solid (111 mg, 48%). A portion of the PF6− salt (52 mg) was converted to its corresponding Cl− salt [Os(dmb)2(IP-4T)]Cl2 using Amberlite IRA-410 with MeOH as the eluent (39 mg, 89% recovery). Rf = 0.39 (0.5% KNO3, 7.5% H2O, 92% MeCN). 1H NMR (MeOD-d3, 700 MHz): δ 8.81 (bs, 2H; c), 8.59 (d, J = 0.7 Hz, 2H; 3), 8.55 (d, J = 0.7 Hz, 2H; 3′), 8.04 (dd, J = 4.9, 0.7 Hz, 2H; a), 7.92 (d, J = 4.2 Hz, 1H; d), 7.74 (dd, J = 7.7, 5.6 Hz, 2H; b), 7.65 (d, J = 5.6 Hz, 2H; 6), 7.39 (d, J = 3.5 Hz, 1H; e), 7.36 (d, J = 5.6 Hz, 2H; 6′), 7.36 (m, 1H; l), 7.33 (d, J = 3.5 Hz, 1H; f), 7.31 (dd, J = 5.6, 0.7 Hz, 2H; 5), 7.25 (dd, J = 3.5, 0.7 Hz, 1H; j), 7.20 (d, J = 3.5 Hz, 1H; h), 7.19 (d, J = 3.5 Hz, 1H; g), 7.14 (d, J = 4.2 Hz, 1H; i), 7.07 (dd, J = 5.6, 0.7 Hz, 2H; 5′), 7.05 (dd, J = 4.9, 3.5 Hz, 1H; k), 2.70 (s, 6H; 4-Me), 2.59 (s, 6H; 4′-Me) (for hydrogen labels, see Chart S2). 13C NMR (MeOH-d3, 175 MHz): δ 160.14 (2), 160.12 (2′), 151.26 (4), 151.15 (4′), 151.06 (6′), 150.88 (6), 150.48–150.46 (a), 150.06 (10), 149.57 (7,9), 141.53 (12), 138.41 (14), 138.22 (16), 137.83 (17), 136.41 (15), 136.24 (13), 131.91 (11), 131.70 (8), 130.88 (c), 130.02 (5), 129.97 (5′), 129.62 (d), 129.14 (k), 127.45 (b), 126.92 (f), 126.41–126.40 (3,3′), 126.11 (l), 126.08 (h), 125.90 (e), 125.80 (g), 125.58 (i), 125.13 (j), 21.03 (4-Me), 20.97 (4′-Me) (for carbon labels, see Chart S2). HRMS (ESI+) m/z: [M-2Cl]2+ Calcd for C53H40N8OsS4 554.0931; Found: 554.0919. [M-2Cl-H]+ Calcd for C53H39N8OsS4 1107.1790; Found: 1107.1790. HPLC retention time: 24.74 min (94% purity by peak area).
2.4. Lipophilicity
The relative lipophilicities (pH=7.4), or distribution coefficients (log D), of the complexes were determined using a modified “shake flask” method as described in previous work.40 Saturated solutions of (a) 1-octanol with phosphate buffer (1:4) and (b) buffer with 1-octanol (1:4) were prepared by shaking solutions at 230 rpm for 24 h (~20°C) using a New Brunswick Classic C26KC Incubator Shaker. In both solutions, excess solvent from saturation was removed via syringe. Chloride salts of metal complexes were prepared as 50 μM solutions first in saturated 1-octanol (500 μL) followed by saturated buffer (500 μL) for total 1 mL volume. Complex mixtures were shaken 200 times before being centrifuged at ~10,000 ×g for 2 min with a BioRad Model 16K Microcentrifuge. The isolated partitions of 1-octanol and phosphate buffer were removed by syringe and subsequently measured against a standard curve in their respective solvent (saturated 1-octanol or buffer). The longest wavelength peak maximum (400–600 nm) was used for endpoint-based absorption measurements on a SpectraMax M2e plate reader. The distribution coefficient with pH = 7.4, or log Do/w, was determined as the log transformed ratio of sample concentration in saturated 1-octanol to saturated buffer.
2.5. Computational
Computation was carried out using density functional theory (DFT) and its time-dependent extension (TDDFT),51 as implemented in Gaussian 09.52 This approach allows the exploration of the ground and excited states of metal complexes with reasonable reliability, and it is also a useful tool to predict the occurrence of Type II photoreactions in PDT.46,53–56 Ground state singlet and excited state triplet geometry optimization were performed in water without constraints by using PBE0 exchange-correlation functional (XC),57 in conjunction with the 6–31+G(d,p) basis set to describe all atoms except Os(II), which was modelled with the quasi-relativistic Stuttgart-Dresden pseudopotential.58 To simulate the effects of the solvent environment, the IEFPCM model (integral equation formalism polarizable continuum model)59,60 was adopted, setting a dielectric constant ɛ=80. Absorption spectra were also modelled in water on the ground state equilibrium structures, using the same basis set as for the optimizations by employing the M06 XC-functional,61 the performance of which has been widely tested and previously verified46,62–65 in modelling the photophysical properties of metallic complexes. This protocol allows a direct comparison with results previously obtained for analogous Ru(II)46 and Os(II)66 compounds.
2.6. Spectroscopy
2.6.1. General
Spectroscopic measurements were performed with dilute (5–20 μM) solutions of the PF6− salts of the osmium complexes in spectroscopic grade acetonitrile that had been further purified by distillation over calcium hydride under nitrogen. Solutions were deaerated for transient absorption by five freeze-pump-thaw cycles in custom Schlenk-style cuvettes, and by argon-sparging in septum-capped cuvettes for emission studies. The solutions were air-saturated for singlet oxygen measurements.
2.6.2. UV-Visible spectroscopy
A Jasco V730 spectrometer was used to collect UV-Vis absorption spectra, which were analyzed at local peak maxima by a regression of absorption vs. concentration for five dilutions in acetonitrile at room temperature.
2.6.3. Emission spectroscopy
Steady-state emission spectra were measured on a PTI Quantamaster spectrometer. The detectors used were a K170B PMT for UV to NIR (max ≈ 800 nm) wavelengths, and a Hamamatsu R5509–42 NIR PMT for longer wavelengths (600–1400 nm). The instrument internally corrected for wavelength-dependent nonlinearities in lamp output and detector sensitivities. Generally, the most intense and longest-wavelength peak in the excitation spectrum was chosen for λex.
2.6.4. Singlet oxygen
Singlet oxygen sensitization quantum yields (ΦΔ) were calculated from the intensity of the 1O2 emission band, centered around 1276 nm. [Ru(bpy)3]2+ was used as a standard (ΦΔ = 0.56 in aerated MeCN67) for the actinometric method shown in Equation 1, where I denotes the emission integration, A is the UV-Vis absorption of the solution at the excitation wavelength, and η is the solvent’s refractive index (η2/ηS2=1 here, since MeCN was used for both). The [Ru(bpy)3]2+ standard is indicated by the subscript S.
| Equation 1 |
The quantum yield experiments were undertaken as solutions of the PF6− salts in MeCN because water quenches the 1O2 state, plus MeCN/PF6− is commonly used in the literature, facilitating comparison. The longest wavelength in the excitation spectrum that maximized emission at 1276 nm was selected for the excitation wavelength. The emission was collected between 1200–1350 nm using a 1000 nm long-pass filter, and baseline corrected. Values were generally reproducible within ± 5%.
2.6.5. Transient absorption (TA)
Transient absorption lifetimes and differential excited state absorption (ESA) spectra were recorded on an Edinburgh Instruments LP-980 equipped with the PMT-LP detector. A Continuum Minilite Nd:YAG laser provided excitation pulses at 355 nm (1 Hz, ≈5 ns pulse width, ≈7–9 mJ per pulse). ESA spectra were acquired in 10 nm intervals, and TA lifetime measurements at single wavelengths bandwidth optimized for maximum detector response. Signals were deconvoluted from the excitation pulse when necessary. This configuration was also used to measure emission lifetimes.
2.7. Cellular Assays
2.7.1. Metal complex solutions
Stock solutions of 1-nT and 2-nT (n=0–4) along with reference tris homoleptics [Os(bpy)3]Cl2 and [Os(dmb)3]Cl2 were generally prepared at 5 mM in 10% v/v DMSO:H2O. Due to partial insolubility in the former solvent, 1–4T was prepared at 25 mM in DMSO. All stock solutions were stored at −20°C prior to use. Working solutions were prepared as dilutions in 1× Dulbecco’s Phosphate-Buffered Saline without Ca2+ or Mg2+ (DPBS; diluted and sterifiltered from 10× DPBS, Corning 20–031-CV). Cellular assays involved less than 1.2% v/v DMSO at the highest complex concentration.
2.7.2. Cytotoxicity and photocytotoxicity
All cell culture was performed as previously described for SK-MEL-28 malignant melanoma cells (ATCC HTB-72).31 The cell line was generally assayed within 5 passages or 10–15 passages from receipt of seed stock. SK-MEL-28 cells were seeded at 10,000 cells well−1 in 96-well plates for 3-day long experiments.
2.7.2.1. Normoxia
Cell viability experiments in normoxia (~18.5–21% O2)68 were performed and data analyzed as previously described.20,31 The light treatments used for full dose-response included red (633 nm, 20 mW cm−2) green (523 nm, 18 mW cm−2), and cool white visible (400–700 nm, maximum ≈450 nm, 21 mW cm−2) with a total fluence of 100 J cm−2. Compounds were screened by coligand (bpy, dmb) and the drug to light interval (DLI) was 16–21 h in both cases. Plates were read with a Molecular Devices M2e plate reader (bottom-read, λexc=530 nm, long-pass 570 nm, λem=620 nm).
The spectral profiles of the light sources are shown in Figure S45. Spectral output was monitored using the Luzchem SPR fiber optic detector in tandem with an Ocean Optics USB4000 spectrophotometer and an Ocean Optics UV-Vis XSR fiber optic of 230 μm diameter. Irradiance was measured using a Thorlabs Optical Power Meter PM100D and their corresponding thermal power sensor S310C. Irradiance was generally within 5% across the entire plate area for full-plate illumination.
2.7.2.2. Hypoxia
Cell culture and compound evaluation in hypoxia were performed as previously reported for SK-MEL-28 cells and included a compound known to be highly oxygen-dependent for its phototoxicity, [Ru(bpy)2(dppn)]Cl2.31 Hypoxia experiments (1% O2) were conducted in parallel to normoxic experiments with SK-MEL-28 cells and full dose-response experiments (EC50 determination) for better comparison. Briefly, cells were seeded at the same time as normoxic plates, pre-incubated for 2–3 h at 1% O2 (5% CO2, ~80% RH, 37°C) to facilitate cell attachment, dosed with compound dilutions (1 nM to 300 μM), incubated overnight for 21–26 h at low oxygen, sealed with qPCR films (VWR, 89134–428) to maintain low oxygen media, dark plates excluding – given light treatment (after normoxic plates), films were then removed, all plates returned to normoxia for post-treatment incubation over 2 days (~18.5% O2, 5% CO2, ≥90% RH, 37 °C), and tested for relative viability with a 4 h incubation using resazurin indicator (~60 μM final).
Data Manipulation and Statistics.
All results from the endpoint-based resazurin assay were background subtracted with negative controls (cell-free) and normalized relative to positive cell controls (PS-free). Any negative values were assumed to be a mismatch of background (i.e., fluorescence quenching) and assigned as zero values. Likewise, background fluorescence and/or quenching was observed at high concentration. Zero values were assigned for these cases when indicated by several consecutive concentrations for a given treatment (dark or light). Additional verification was conducted via light microscopy before finalizing data corrections. Further discussion of assay limits for these types of compounds is provided in a recent review.20
The normalized resazurin data over a wide concentration range was fit to both a three-parameter log-logistic and logistic models using GraphPad Prism 8.4.0 according to Equation 2 and Equation 3 (four-parameter shown) where bottom is constrained to equal zero and X is equal to concentration.
| Equation 2 |
| Equation 3 |
Experiments were done in triplicate with replicates plotted ± standard deviation (SD) on any dose-response figure. The effective concentration to reduce relative cell viability by 50% of the fitted curve (EC50) was used to report compound effectiveness; reported EC50 values were ± SEM (=standard error of the mean) for a given experiment. Steep hill slopes with ambiguous confidence intervals are unable to determine the SEM; therefore, error was labelled as not determined (n.d.) in those cases. Phototherapeutic indices (PI) are reported as the ratio of dark to light EC50 values and used as a measure of light-induced potency. Summary activity plots used for quickly comparing compound potency (Log EC50, PI, and Log PI) include SEM from log-logistic fits where applicable (Log EC50).
Correlation analyses for PI, lipophilicity, and ΦΔ were conducted using Pearson’s correlation coefficients and two-tailed t-tests for discerning significance at α = 0.05.
2.8. Maximum Tolerated Dose (MTD) in Mice
An 8-week old litter of female C57BL/6J mice, averaging 20 g per mouse, were treated by intraperitoneal injection of 1–4T and 2–4T in accordance with protocol A20–006 (approved by WFU Animal Care and Use Committee). Mice were dosed from 25–200 mg kg−1 with 200 μL injections of compound dissolved in 0.9% saline containing 10% v/v DMSO as the vehicle. Metal complex solutions were prepared immediately with sonication before injection. Mice were dosed by slow intraperitoneal injection (the lower right abdominal quadrant) only after visible confirmation of complete compound dissolution. Animals were continuously monitored for 2 hours, frequently over the next 6 hours, and periodically for up to 2 weeks before being sacrificed. Mice were accordingly euthanized if (a) a combination of moderate severity signs appeared, (b) a single severe sign appeared, or (c) the study period was complete, 2 weeks post-injection. The maximum tolerated dose (MTD) was defined as the dose that produces moderate signs of clinical toxicity in the final tested animal.
3. RESULTS AND DISCUSSION
3.1. Synthesis and Characterization
Complexes 1-nT and 2-nT were synthesized following adapted literature procedures.43,69,70 The complexes were isolated as their PF6− salts and purified with flash chromatography on silica, affording products in 13–30% yields for 1-nT and in 48–97% yields for 2-nT. The lower yields for the 1-nT complexes were due to poor resolution of product bands during LC purification, which was caused by the decreased solubility in the mobile phase (e.g., MeCN:H2O). The PF6− salts were converted to their corresponding Cl− salts in quantitative yields via anion metathesis using Amberlite IRA-410 and were further purified using size-exclusion chromatography on Sephadex LH-20, if needed.
The purities of complexes 1-nT and 2-nT were confirmed by HPLC analysis to be 95% or higher (Figures S17–S22, S39–S44), with the exception of 1–3T (92%) and 2–4T (94%). The structures of complexes 1-nT and 2-nT were confirmed with the help of high resolution ESI+ mass spectrometry (Figures S11–S16, S33–S38) and a detailed analysis of their 1D 1H and 2D 1H–1H COSY NMR spectra (Figures S1–S7, S23–S29). Assignments were made based on the connectivity observed in NMR and in consultation with literature sources.49,50 Compounds 1–4T and 2–4T required additional 13C, 13C–1H HSQC, and 13C–1H HMBC NMR experiments for full assignment of the 1H NMR signals of the quaterthiophene group (Figures S8–S10, S30–S32). It should be noted that due to the propensities of oligothienyl chains to aggregate, chemical shifts and signal resolution for the thiophene rings exhibited a concentration dependence, especially in complexes with longer thiophene chains. We previously found that the best resolution for all of the thiophene signals was obtained using ~1.5 mg of the compound in 0.8 mL of MeOD-d3 (~1.7 mM).
3.1.1. Determining NMR assignments for complexes Os-0T–Os-4T:
Hydrogen labels used in 1H NMR assignments of reference compound [Os(bpy)3]Cl2 and complexes 1-nT (n=0–4) are shown in Chart 2 with stacked 1H NMR spectra for comparison in Figure 1. Identical labels were used for 2-nT series (Figure S23), except that 4-Me and 4′-Me groups replace the 4 and 4′ hydrogen atoms. Tris-homoleptic compound [Os(bpy)3]Cl2, which was characterized in detail by Pazderski et al.,49 was used to establish the positions of signals 3–6 in complexes x-nT. Hydrogens from bipyridine ligands followed the pattern 3,3′ > 4,4′ > 6,6′ > 5,5′ (where “>” indicates further downfield).49 In each pair, the chemical shifts of primed and non-primed hydrogens were distinctly different. This difference in chemical shifts is dictated by the strength of a shielding effect experienced by these hydrogens caused by the spatial proximity of the π-system of a neighboring ligand. Non-primed hydrogens are shielded by the π-system of a neighboring bpy ligand in the 1-nT series (or dmb ligand in the 2-nT series), primed hydrogens are shielded by the π-system of the neighboring IP-nT ligand. Due to the coplanar and rigid phenanthroline core of the IP-nT ligand, it causes a stronger shielding effect than the more flexible bpy or dmb ligands, which resulted in primed hydrogens appearing significantly more upfield than their non-primed counterparts.50 Similar examples could be found in the literature.71–73 The difference in chemical shifts between primed and non-primed positions is most pronounced for the hydrogens that are the closest in space to the IP-nT ligand (the largest difference is observed for position 6/6′). Hydrogens 3 and 3′, which are oriented away from the IP-nT, are affected the least. Spin system a-b-c was assigned next, in the following order of decreasing chemical shift: c > a > b. Hydrogen b appeared most upfield, which is typical for meta-positioned hydrogens in complexed phenanthroline ligands. Hydrogen a was shifted upfield relative to c due to its proximity to the Os(II) center, which caused a shielding effect on the nearest hydrogen (hydrogen a, ortho-position).49 Hydrogen c (para-position) was too far away to be influenced by this effect. Additionally, hydrogen c is near the non-coordinated nitrogens of the IP-nT ligand, which causes a pronounced deshielding effect. It should be noted that while all other hydrogens appear on the spectrum as sharp signals, hydrogen c sometimes appears broadened. Observed broadening of its signal is attributed to the proximity of the nitrogens of the -N=-NH- fragment from IP-nT ligand. It should also be noted that due to the quick exchange between the two nitrogens of the imidazole ring in solution, and the quick deuterium exchange with the solvent, the signal for the imidazole-NH hydrogen was not observed, similarly to literature examples.71,74,75
Figure 1:
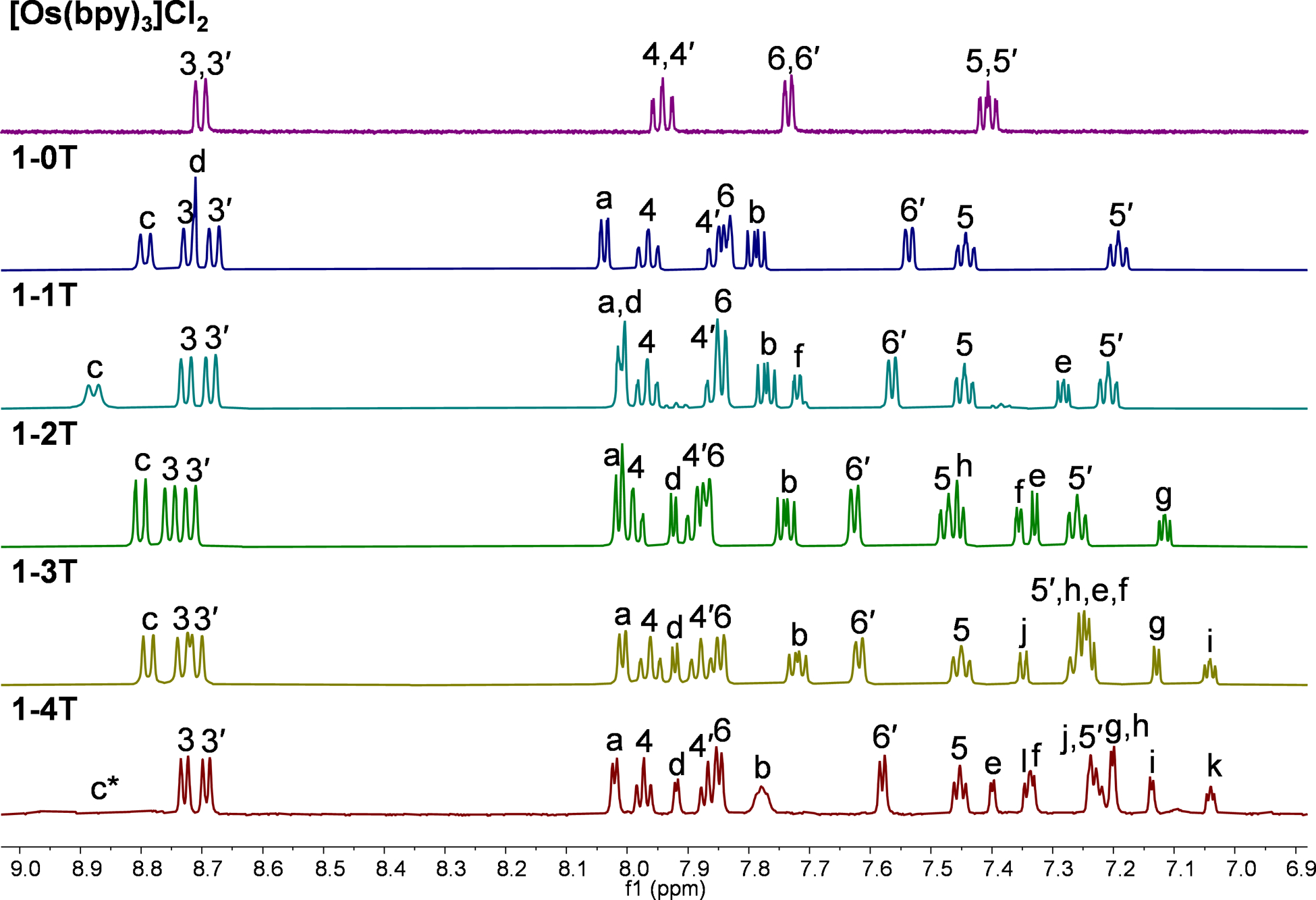
1H NMR spectra of reference compound [Os(bpy)3]Cl2 and 1–0T–1–4T (Cl− salts) in MeOD-d3 at 298 K, aromatic region. All data collected with 500 MHz NMR data, except for 1–4T, for which data was collected with 700 MHz NMR (* = c appears as a broad signal that is not visible at this intensity).
Among the hydrogens associated with the thiophene rings of complexes x-1T–x-4T, three hydrogen signals were the most diagnostic. Hydrogen d appeared the most downfield of all thiophene hydrogens due to the strong deshielding effect of the neighboring imidazo nitrogens. The hydrogen on the most distal ring positioned closest to the sulfur was also strongly deshielded (hydrogen f in x-1T, h in x-2T, j in x-3T, and l in x-4T). The most shielded hydrogen appeared as a distinct doublet of doublets and corresponded to the middle hydrogen of the most distal thiophene ring (hydrogen e in x-1T, g in x-2T, i in x-3T, and k in x-4T). Using these characteristic signals as starting points, the rest of the thiophene signals from the proximal and the most distal (relative to the imidazo group) thiophene ring were assigned via the observed 1H–1H COSY correlations. Assigning the signals for the internal thiophenes (those flanked on both sides by other thiophene rings) in complexes 1–4T and 2–4T required 13C–1H HSQC and 13C–1H HMBC NMR experiments (Figures S9–S10 and S31–S32). Firstly, two internal spin systems f-g and h-i were established using 1H–1H COSY correlations. Next, 13C–1H HSQC data was used to identify which 13C NMR signals corresponded to the thiophene hydrogens d–l. Then, 13C–1H HMBC data was used to establish diagnostic correlations, starting with hydrogens d and e. Both hydrogens d and e correlated to the same two 13C peaks, which were assumed to be carbons 11 and 12. The signal that hydrogen d correlated to more strongly was assigned as carbon 11 (around 131.8 ppm), and the signal that hydrogen e correlated to more strongly was assigned as carbon 12 (around 141.6 ppm). In addition to correlations with d and e, carbon 12 correlated with an additional hydrogen, which led to the assignment of this hydrogen as f. Hydrogen g was then readily identified via correlation to f observed in 1H–1H COSY NMR. Next, a diagnostic 13C signal around 137.7 ppm exhibited correlations to hydrogens l, j, and one more 1H signal that was assigned as hydrogen i. This 13C signal was assigned as carbon 17 due to the high probability of both l and j showing correlation to it. Lastly, hydrogen h was then readily identified via correlation to i observed in 1H–1H COSY NMR.
3.2. Lipophilicity
Complexes 1-nT and 2-nT (n=0–4) were evaluated for their lipophilicities at physiological pH (7.4) in phosphate buffer and 1-octanol using a modified shake-flask method, where positive log Do/w values indicate a preference for 1-octanol (higher lipophilicity) and negative values indicate aqueous solubility (lower lipophilicity). Results are shown in Figure 2 with supplemental data included in Table S1. The bpy complexes were approximately ten-fold more hydrophilic than their dmb counterparts regardless of the number of thiophene rings. The complexes without thiophenes (n=0) were the most hydrophilic, with lipophilicity increasing with n in both series. All of the bpy complexes had negative log Do/w values, whereas the dmb complexes with ≥2 thiophene rings had positive values. While 1–4T could not be evaluated due to precipitation, the installation of a fourth thiophene would be expected to result in a positive value for log Do/w given that 1–3T was amphiphilic. Surprisingly, the increased hydrophilic character in the 1-nT series did not correlate with better solubility in aqueous media for the most extended complexes (n=3–4). The dmb analogs, 2–3T and 2–4T, were fully soluble in high ionic strength aqueous media (≥1.2 mM) whereas their 1–3T and 1–4T counterparts were less soluble.
Figure 2:
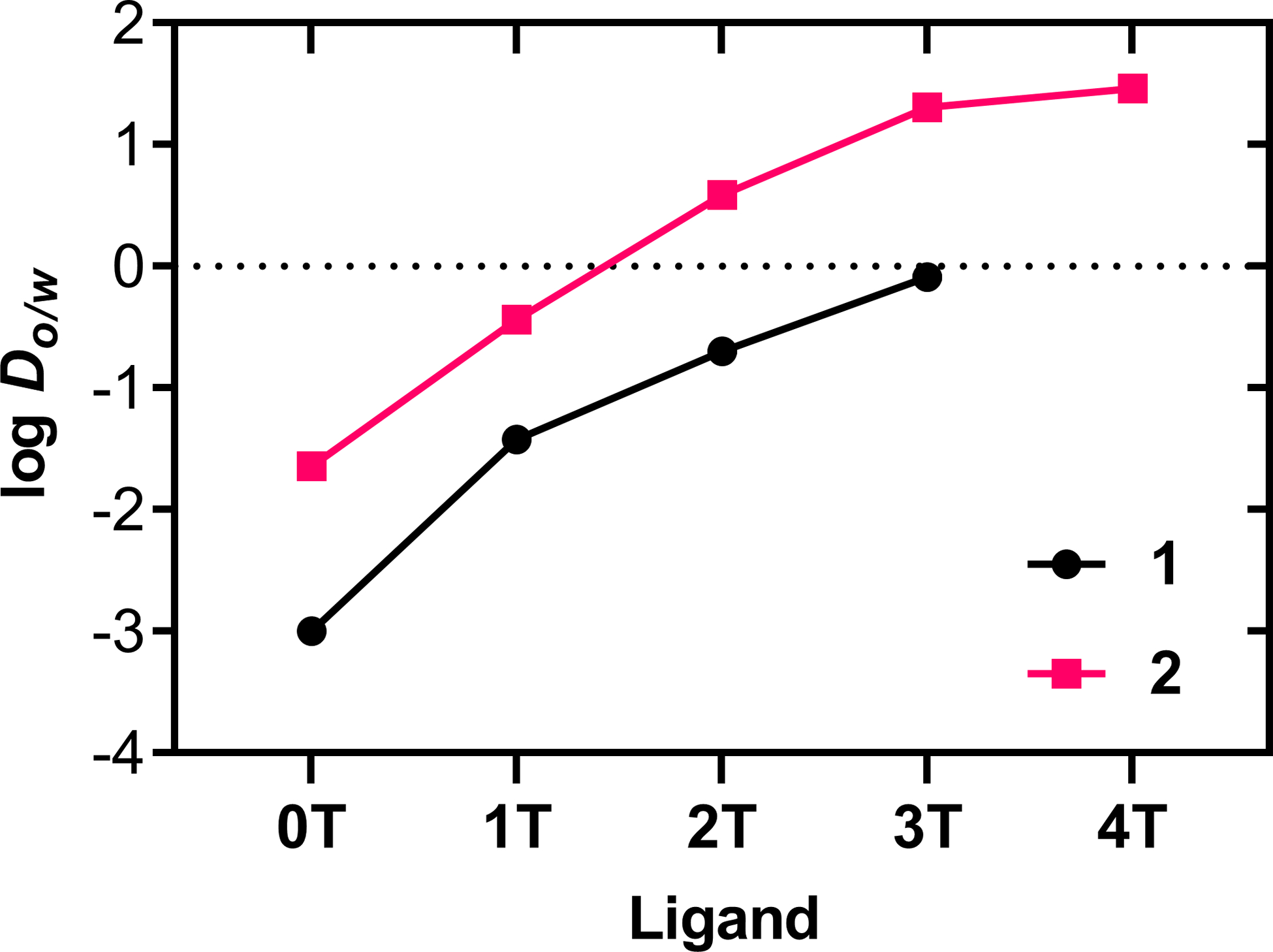
Lipophilicities of complexes 1-nT with black circles and 2-nT with fuchsia squares (n=0–4). In the case of 1–4T, a log Do/w value could not be estimated due to precipitation at the interface between saturated 1-octanol and phosphate buffer.
3.3. Computational Studies
3.3.1. Ground state configuration and UV-Vis spectroscopy
Computational studies of both series in water were conducted using density functional theory (DFT) and time-dependent DFT (TDDFT) calculations. Optimized geometries (PBE0/6–31+G(d,p)/SDD) show the pseudo-octahedral nature of the complexes, with the thiophene rings adopting a quasi-planar trans configuration (Figure 3). The rigid chelating IP ligand is coplanar with the first electron-donating thiophene ring, with subsequent rings twisted slightly out of coplanarity, up to around 16° for the fourth rings in 1–4T and 2–4T. The dihedral angle of the thiophene rings in the ground state were similar for the corresponding members of each series, however the out-of-plane angle was slightly greater in the 2-nT series (Table S2).
Figure 3:
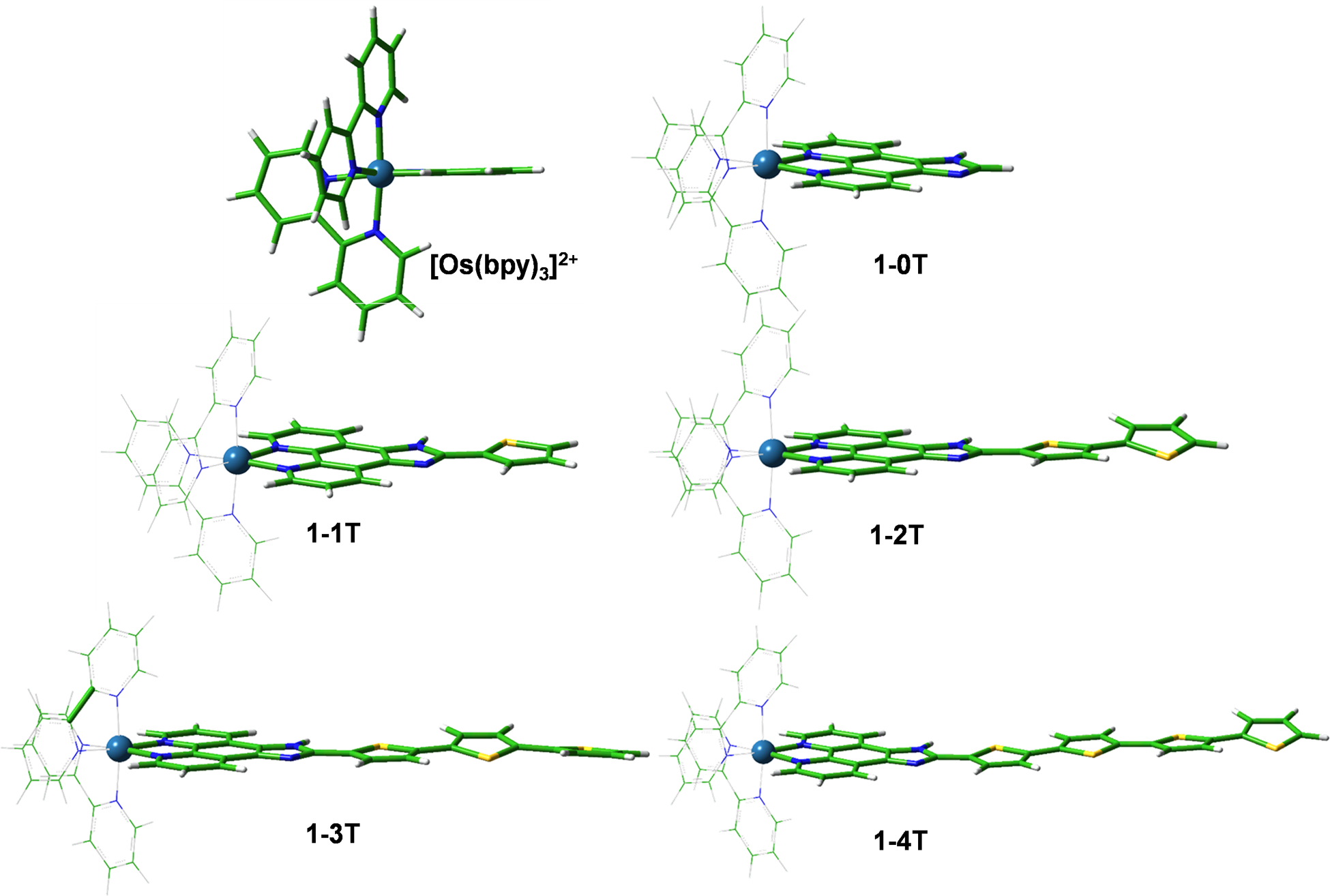
Optimized geometries of [Os(bpy)3]2+ and 1-nT (n=0–4) in a water environment at the PBE0/6–31+G(d,p)/SDD/ level of theory. The corresponding structures for the dmb series are in Figure S46.
The ground state electronic configurations for the bpy series are illustrated by the frontier orbitals depicted in Figure S47 and their energies in Figure 4. The HOMOs of [Os(bpy)3]2+, 1–0T, and 1–1T are largely centered on the metal ≈30% (Table 1), while in 1–2T–1–4T, the Os-d character has largely vanished with the HOMOs extending over the thiophene rings of the IP-nT ligand, constituting 68% of the HOMO in 1–4T that is predominantly organic in character. This illustrates how increasing the conjugation of the IP-nT ligand can allow it to take on more of the HOMO character. Figure 4a shows how the electronic energy of the organic ligand HOMO increases with added thiophene rings, eventually surpassing the relatively unchanging HOMO−1 orbitals that are comprised mainly of Os-d, bpy, and IP contributions. The crossover occurs around 1–2T, where the HOMO begins to be dominated by the thienyl chain. The composition of the HOMO−1 orbitals does not vary as much through the series, remaining localized to the Os with a significant degree of mixing with bpy and IP regions, but no contribution from the thienyl chain.
Figure 4:
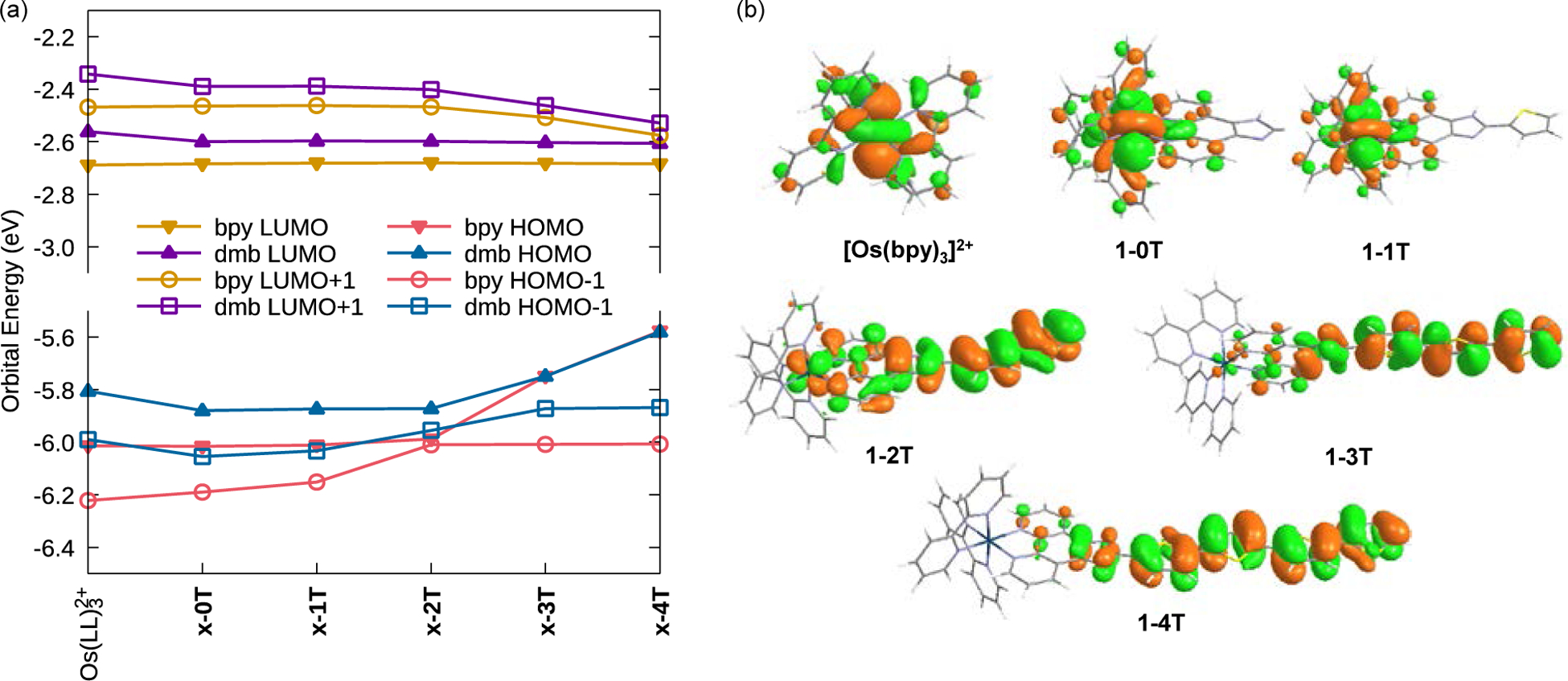
(a) Calculated frontier orbital energies. Data is tabulated in Tables S3 and S4. (b) Os(II)-based HOMO for [Os(bpy)3]2+, 1–0T, 1–1T and thienyl-based HOMOs for 1-nT with n=2–4, calculated at the M06/6–31+G(d,p)/SDD level of theory in water. See Figures S47 and S48 for additional plots.
Table 1:
Calculated orbital composition for the bpy-series singlet ground state. The corresponding dmb composition is in Table S5.
| HOMO−1 | HOMO | LUMO | LUMO+1 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % | Os | bpy | IP | thienyl | Os | bpy | IP | thienyl | Os | bpy | IP | thienyl | Os | bpy | IP | thienyl |
| Os(bpy)3 | 41 | 59 | — | — | 29 | 71 | — | — | 2 | 98 | — | — | 7 | 93 | — | — |
| 1–0T | 38 | 25 | 36 | 0 | 30 | 50 | 20 | 0 | 6 | 58 | 36 | 0 | 10 | 67 | 23 | 0 |
| 1–1T | 28 | 28 | 42 | 2 | 30 | 42 | 28 | 0 | 6 | 56 | 38 | 0 | 11 | 65 | 2 | 0 |
| 1–2T | 29 | 40 | 30 | 1 | 5 | 29 | 27 | 39 | 5 | 53 | 39 | 3 | 6 | 65 | 19 | 10 |
| 1–3T | 30 | 46 | 24 | 0 | 1 | 18 | 15 | 66 | 6 | 53 | 38 | 4 | 9 | 43 | 19 | 29 |
| 1–4T | 30 | 47 | 22 | 0 | 1 | 18 | 12 | 68 | 5 | 52 | 39 | 4 | 10 | 40 | 23 | 27 |
The dmb series follows a similar trend (Figure S47 and Table S5) but with 2–2T showing important differences. Unlike 1–2T, the HOMO of this complex remains mostly centered on the metal, with almost no contribution by the thiophene rings. Furthermore, the HOMO−1 has a large thiophene component, unlike 1–2T. This underlines how a seemingly minor modification to the ancillary ligand can have a profound effect on the molecular orbital structures of these complexes. Notwithstanding, the terthiophene and quaterthiophene units are so dominant that the HOMOs of 1–3T and 2–3T are essentially isoenergetic, as they are for 1–4T and 2–4T.
In contrast, the LUMO orbitals of both series remain nearly unaffected by the thiophene chain, being centered almost entirely on the ancillary ligands and IP residues. Consequently, the LUMO energies are influenced somewhat by the ancillary ligand, but not by the length of the thiophene chains. On the other hand, the LUMO+1 orbitals in both series are influenced by the number of thiophene rings, dropping to lower energy when n>2. Thus, the HOMO–LUMO gaps within a series (i.e., x-nT for changing n) are nearly entirely determined by the energy of the HOMO, which is influenced by the number of thiophene rings. The added methyl groups in the dmb ligands serve to elevate the energy of orbitals that encompass the ancillary ligands. Hence, the HOMO–LUMO gap between series (i.e., x-nT for x=1 vs x=2 for a given n) is influenced by the LUMO for n≥3, and the HOMO and LUMO for n≤2. The energetics of these frontier orbitals has an important influence over the UV-Vis spectra of the complexes.
Computed UV-Vis spectra provide useful insights into the low energy, spin-allowed singlet-singlet transitions observed experimentally in water and their MO origins. The calculated spectra are shown in Figure S49 (cf. Figure 5), and the transition descriptors are in Table 2 (bpy series) and Table S6 (dmb series). The models show that the lowest energy electronic configuration changes in nature with the incorporation of more than two thiophene rings. x-0T, x-1T, and x-2T have predominantly Os(d)→bpy(dmb) MLCT character, consistent with the metal-based nature of the occupied orbitals involved in the transitions (Figure S47). The computed transitions for x-3T and x-4T, however, are progressively more IL/ILCT in nature, with the involved HOMO orbital exclusively localized on the IP and thienyl-rings (Figure 4b and S47). This change is accompanied by a red-shift of the calculated UV-Vis absorption bands ascribed to these lowest energy singlet-singlet transitions, confirming the experimental trend. These findings are consistent with the calculated trends for the Ru(II) analogs46 and the related Os(II) phen66 series.
Figure 5:
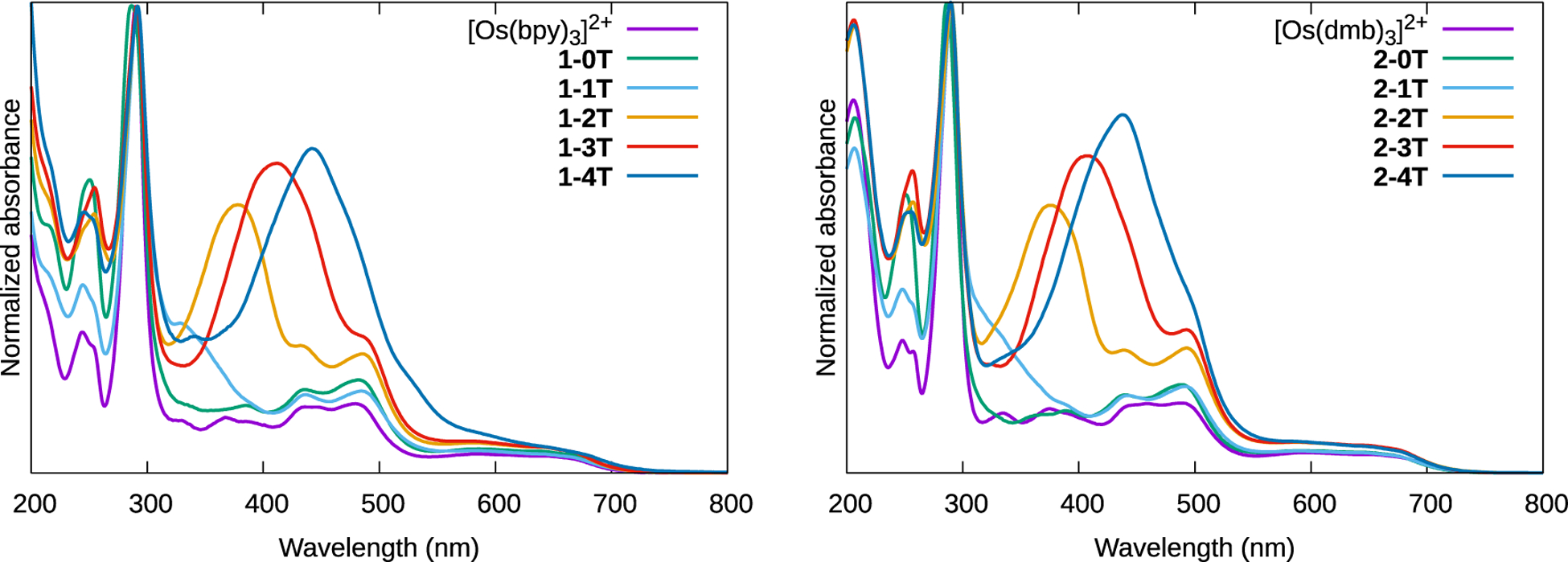
Normalized UV-Vis spectra of reference compounds [Os(bpy)3]2+ and [Os(dmb)3]2+ and complexes 1-0T–1-4T and 2-0T–2-4T as dilute (≈20 μM) PF6− solutions in acetonitrile at room temperature.
Table 2:
Calculated absorption wavelengths (λtheor>450 nm), configuration, experimental λmax in water, and theoretical peak assignments for the 1-nT series. The corresponding data for the 2-nT series is in Table S6. The dominant transition(s) at each wavelength are indicated, along with their fractional contribution to the signal intensity.
| cmpd | λtheor | f | λ expt | Transitions | Predominant Assignment |
|---|---|---|---|---|---|
| [Os(bpy)3]2+ | 461 | 0.138 | 479 | H−2⟶L+1 (26%); H−1⟶L+2 (25%); H−2⟶L+2 (20%) | MLCT |
| 1–0T | 463 460 |
0.138 0.139 |
480 | H−1 ⟶ L+2 (49%) H−2 ⟶ L+2 (51%) |
MLCT |
| 1–1T | 466 461 |
0.184 0.137 |
483 | H−1 ⟶ L+2 (51%) H-2⟶ L+2 (52%) |
MLCT |
| 1–2T | 487 | ||||
| 471 461 |
0.331 0.137 |
H⟶ L+1 (30%); H-2⟶L+2 (30%) H−2⟶L+1 (49%); H-3⟶L+2(27%) |
MLCT/ILCT MLCT |
||
| 1–3T | 492, 405 |
||||
| 487 466 |
0.950 0.394 |
H ⟶ L+1 (47%) H⟶ L+3 (42%) |
IL/ILCT IL/ILCT |
||
| 1–4T | 490, 444 | ||||
| 462 | 0.132 | H−3 ⟶ L+3 (40%) | MLCT |
In summary, the frontier orbital calculations indicate that the number of thiophene rings n influences the IL/ILCT energies while the ancillary ligands mainly impact the MLCT energies. Overall, the influence of π-conjugation on the IL/ILCT transitions with significant organic character is more pronounced than the ancillary ligand influence on the lowest energy transitions involving the metal ion. As expected, the increased electron density afforded by the methyl groups of the dmb ligands in the 2-nT series raises orbital energies in general. This can be seen in Figure 4a where for a given n, the HOMO, HOMO−1, LUMO, and LUMO+1 are slightly higher in energy for the 2-nT (dmb) series relative to the 1-nT (bpy) series.
3.3.2. Triplet states
The optimized geometries of the lowest energy triplet states reveal a flattening of the thiophene rings (Table S2) while preserving the pseudo-octahedral configuration at the metal with no changes in the Os-N bond lengths. The coplanarization mirrors the behavior of free oligothiophenes that are quinoidal and effectively coplanar, with maximal π-conjugation, in the photoexcited state.76,77 The most dramatic differences between the ground and excited state geometries occurs for n=3 and 4, where the terminal thienyl ring is twisted up to 16° out of plane in the ground state.
Table 3 summarizes the calculated T1 energies of the complexes, where those with n<2 are nearly identical, while 1–4T and 2–4T are as low as 1.43 and 1.46 eV, respectively. The orbital compositions as well as the energies and configurations of upper Tn states, mostly 3MLCT for all of the complexes, are compiled in Figure S51 and Table S7. Inspection of the donor and acceptor orbitals reported in Table S7 allows characterization of the nature of the lowest energy triplet state (T1) for each compound, particularly with regard to the extent of metal ion involvement in these transitions. The drop in T1 energy for x-nT with n≥2 corresponds to a change in its nature, from 3MLCT for x-1T to primarily involving the ligands for the compounds with longer thienyl chains. Both the donor and acceptor orbitals are localized mainly on the IP-nT ligand for n=2 and are predominantly 3ILCT/3IL. For n>2, T1 is 3ILCT/3LLCT in nature.
Table 3:
Calculated T1 energy (eV) for both series. MSD = Mulliken Spin Density on the Os metal center (no metal involvement was found for n>2).
| cmpd | T1 Energy (eV) | Type | MSD | cmpd | T1 Energy (eV) | Type | MSD |
|---|---|---|---|---|---|---|---|
| [Os(bpy)3]2+ | 2.07 | 3MLCT | 0.80 | [Os(dmb)3]2+ | 2.02 | 3MLCT | 0.82 |
| 1–0T | 2.08 | 3MLCT | 0.82 | 2–0T | 2.01 | 3MLCT | 0.80 |
| 1–1T | 2.07 | 3MLCT | 0.81 | 2–1T | 2.00 | 3MLCT | 0.81 |
| 1–2T | 1.91 | 3IL/3ILCT | ‒ | 2–2T | 1.92 | 3IL/3ILCT | ‒ |
| 1–3T | 1.62 | 3ILCT/3LLCT | ‒ | 2–3T | 1.64 | 3ILCT/3LLCT | ‒ |
| 1–4T | 1.43 | 3ILCT/3LLCT | ‒ | 2–4T | 1.46 | 3ILCT/3LLCT | ‒ |
Mulliken spin densities (MSDs) on the Os(II) centre corroborate this finding, being nearly one for n<2, indicative of a single unpaired electron on the metal center, as would be expected for the 3MLCT state. The lack of MSD on the metal for n≥2 supports the assignment of these lowest-energy triplet states as being predominantly ligand-based. The ancillary ligand has an effect on the 3MLCT T1 energies (around 0.06±0.01 eV greater in the bpy series) but not on the other T1 energies. In sharp contrast, the number of thiophenes is inversely related to the 3ILCT/3LLCT energies, but has no effect on the 3MLCT energies. The decrease in 3ILCT/3LLCT energy at n=4 is notable, but the triplet energies relative to the singlet ground state (singlet-triplet energy gaps, ΔS−T) show they remain sufficiently energetic to sensitize singlet oxygen (Figure S50).
These relationships show how judicious design of these complexes allows the manipulation of the triplet excited state energies and characters that may in turn control key photophysical – and hence, photobiological – behaviour.
3.4. Spectroscopy and Photophysics
3.4.1. UV-Visible spectroscopy
The normalized electronic absorption spectra of the compounds are shown in Figure 5. Qualitatively, the spectra show three key features. First, a pair of absorption bands between 400 and 500 nm (somewhat obscured in the x-3T and x-4T complexes) and a much broader and weaker band extending past 700 nm correspond to the Os2+(dπ) ⟶LL(π*) MLCT transitions for LL=bpy and dmb, with the lower energy transition being associated with a formally spin-forbidden absorption to the triplet state.78 The energies of these transitions were unaffected by the presence of IP-nT ligands, and the spectra of [Os(bpy)3]2+ and [Os(dmb)3]2+ replicate literature examples, for which these assignments had been made previously.79 Second, the intense signals near 290 nm correspond to the IL (1ππ*) transition localized on the ancillary LL ligands and were also largely unaffected by the presence of IP-nT ligands. Third, a peak appears in the 300–500 nm window only for complexes with thiophene units. This peak increases in intensity and decreases in energy as the number of conjugated thiophene rings increases and is therefore assigned to the thienyl-localized IL (1ππ*) transition of the IP-nT ligand, which is 1ILCT for n≥2, consistent with the free ligands and thienyl chains,40,41 related complexes,46,66 and the computational results described in this work. The spectra in water were qualitatively similar (Figure S52) but displayed the minor differences in the bands with charge transfer character that would be expected on altering the solvent polarity.
This region of the 1–4T spectrum shows variations compared to the rest of both series, including 2–4T: the unnormalized peak intensity is slightly attenuated compared to 2–4T, and there is a shoulder in the 500–550 nm region that does not appear in any other complex. These differences demonstrate that the identities of the coligands (bpy versus dmb) influence the nature of the singlet-singlet transitions that can be photoexcited. Such subtle yet, important differences could in turn lead to differences in the excited state dynamics of 1–4T and 2–4T, but were not suggested by the computational models. In addition, the aqueous experimental spectrum for 1–4T was also different from 2–4T and the other complexes, with bands that were broadened, red-shifted, and increased in intensity (Figure S52. The turbidity of the aqueous solution and additional scatter at the longer wavelengths in the experimental absorption spectrum of 1–4T suggest that this particular complex is more prone to agglomeration compared to the other compounds, and this was also not reflected in the computational studies.
3.4.2. Emission spectroscopy
In Ar-sparged room-temperature MeCN, both series exhibited broad, featureless steady-state emission spectra with maxima between 745–770 nm for the bpy series, and 768–774 nm for the dmb series (Table 4 and Figure S53), shifting to slightly longer wavelengths as the degree of conjugation increased. The excitation maxima did not vary meaningfully within or between families (≈470 nm), and were the same as had been observed previously for phenanthroline-based Os complexes.66 This indicates that the emission comes from predominantly 3MLCT state80 based on the ancillary ligand and/or the proximal phen portion of the IP-nT ligand, and that the π* acceptor orbital is similar in all of these complexes. The longer emission wavelength maxima in the 2-nT series indicates that the 3MLCT state is less energetic in these complexes compared to their 1-nT counterparts, consistent with computational findings, and this highlights how even minor structural modification of the ancillary ligands can manipulate the excited state energies.
Table 4:
Photophysical properties of the of the reference compounds [Os(bpy)3]2+ and [Os(dmb)3]2+ and complexes 1-0T–1-4T and 2-0T–2-4T collected on dilute (≈20 μM) PF6− solutions in deaerated acetonitrile at room temperature. Emission maxima, emission quantum yields (λex / nm in parentheses), emission lifetimes (excitation by a 355 nm laser pulse), and TA lifetime (λobs / nm, where a=ESA and b=bleach, and relative amplitudes from biexponential fits in parentheses).
| LL=bpy; x=1 | LL=dmb; x=2 | |||||||
|---|---|---|---|---|---|---|---|---|
| cmpd | λem/nm | Φem | τem/ns | τTA/ns | λem/nm | Φem | τem/ns | τTA/ns |
| [Os(LL)3]2+ | 745 | 5.6×10−3 (470) |
56 | 59 (480, b) | 768 (471) | 2.9×10−3 | 32 | 32 (370, a; 490, b) |
| x-0T | 745 | 6.2×10−3 (471) |
66 | 66 (370, a; 480, b) | 768 (470) | 7.8×10−3 | 76 | 78 (480, b) |
| x-1T | 754 | 4.1×10−3 (471) |
68 | 47 (370, a) 59 (480, b) |
768 (471) | 5.4×10−3 | 75 | 79 (490, b) |
| x-2T | 758 | 5.5×10−3 (470) |
68 | 52 (370, a) 60 (490, b) |
770 (470) | 6.9×10−3 | 75 | 66 (380, a) 78 (490, b) |
| x-3T | 765 | 4.1×10−3 (466) |
100 | 110 (440, b; 600, a) | 770 (470) | 4.0×10−3 | 76 | 83 (490, b) 73, 540 (2.3:1; 630, a) |
| x-4T | 770 | 2.8×10−3 (469) |
76–110 420–520 (1:2.2) |
480, 3000 (9.4:1; 440, b; 660, a) 460, 3600 (9.3:1; 680, a) |
774 (470) | 5.2×10−3 | 650 | 380–450, 980–1200 (2.1:1; 440, b) 430–580, 940–1100 (1.5:1; 660, a) |
There was no indication of emission from the 3ILCT state in the complexes, although 2–4T was the only complex in both series that showed 1ILCT-based fluorescence (and this was not due to free ligand impurity). The absence of fluorescence from the 1ILCT state in 1–4T suggests that the 1MLCT state is preferentially populated in 1–4T at the excitation wavelengths used for the measurement despite overlap of the 1ILCT and 1MLCT bands in the steady state absorption spectra and that the coligands have the ability to influence this preference.
Emission lifetimes were measured following excitation by a pulse (≈5 ns width) from a 355 nm laser, and the observed lifetimes of the two tris homoleptic complexes (τ=<100 ns) agree well with published data for 3MLCT emission.45,81 The lifetimes of 1–0T–1–2T were similar (66–68 ns), as were those measured for 2–0T–2–3T (75–76 ns). Although less than 100 ns, these lifetimes were still somewhat longer than those for the tris homoleptic parent complexes, suggesting at least some contribution by the proximal phenanthroline portion of the IP-nT ligands to the 3MLCT emissive state. The 3MLCT lifetime of 1–3T was even longer, around 100 ns, suggesting that its decay may be influenced by other excited states. This was further corroborated by the observation of a biexponential decay of the emissive 3MLCT state in 1–4T, whereby the short and long components were 76–110 and 420–520 ns, respectively. The short component was assigned to prompt emission from the 3MLCT state, and the long component was attributed to delayed emission from the same state. Notably, the decay of 2–4T exhibited only the delayed emission component with a lifetime of 650 ns. The observation of delayed emission from the 3MLCT state only for complexes with more π-expansive ligands suggests that the 3MLCT state may be populated from a longer-lived ligand-based triplet state, acting as an excited state reservoir,45 when their excited states are in energetic proximity. This is consistent with behavior we have encountered previously in other oligothiophene-based complexes.41,66 In the case of 1–3T, the decay from the 3MLCT state was monoexponential with a lifetime longer than that for 1–0T–1–2T and 2–3T but not as long as the delayed emission time constant measured for 1–4T and 2–4T. This prompt but prolonged emission for 1–3T (τem=100 ns) was attributed to an 3MLCT state that is in equilibrium with a ligand-based triplet (3ILCT) that is very close in energy.
3.4.3. Transient absorption
The triplet excited states were further probed by nanosecond transient absorption spectroscopy at room temperature using degassed (5× freeze-pump-thaw) dilute (typically 10–20 μM) solutions in MeCN, excited by a 355 nm laser pulse (width ≈5 ns). The differential excited state absorption (ESA) spectra were collected at 10 nm intervals. The spectral profiles immediately following the excitation pulse are compared in Figure 6, the full sets of time-resolved ESA spectra are shown in Figure S54 and S55, and the peak and lifetime data are compiled in Table 4. The ESA spectra and TA lifetimes, along with the emission characteristics, were used to construct the Jablonski diagrams in Figure 7.
Figure 6:
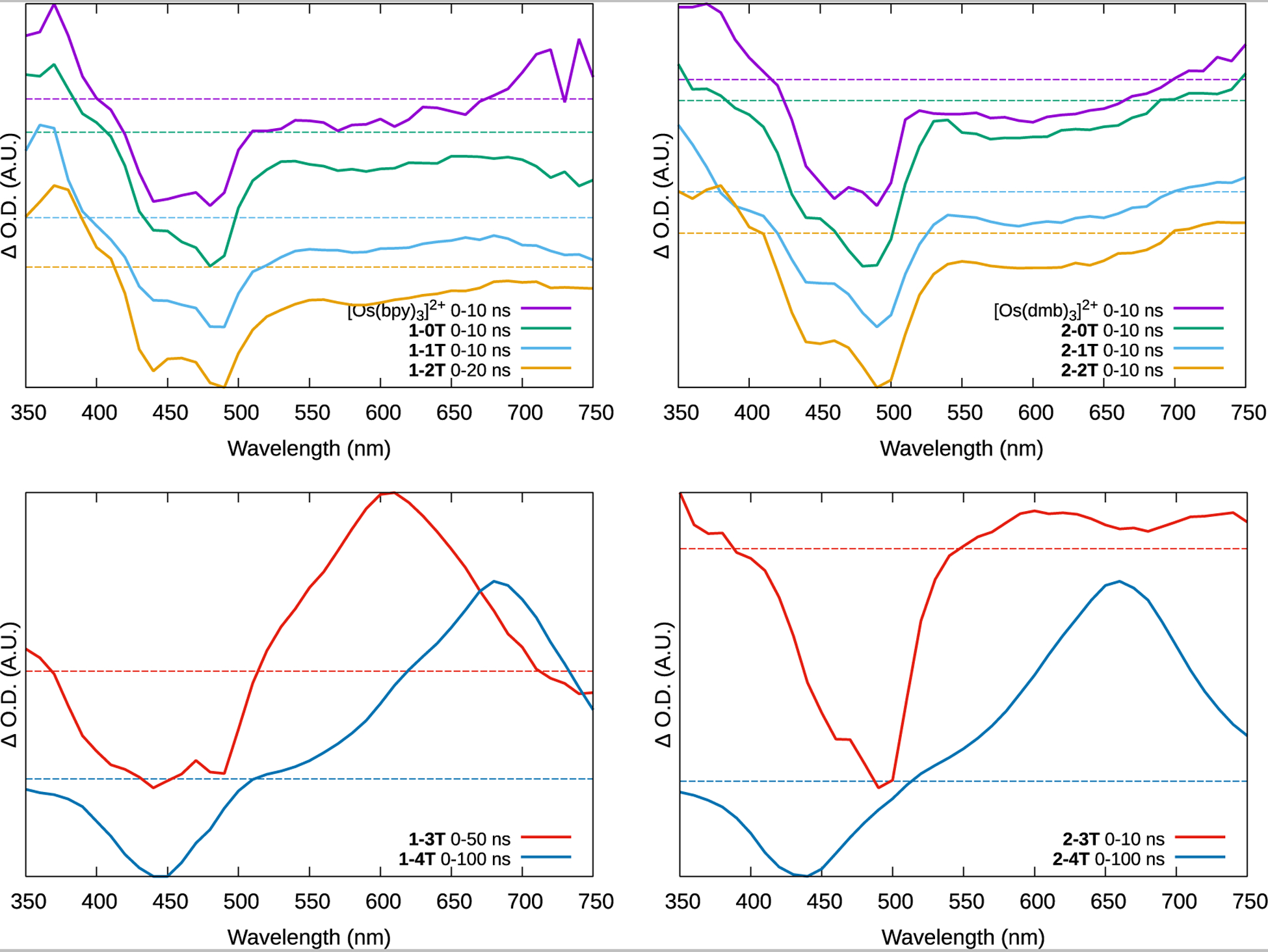
Normalized ESA spectra of the reference compounds [Os(bpy)3]2+ and [Os(dmb)3]2+ and complexes 1-0T–1-4T and 2-0T–2-4T as dilute (≈20 μM) PF6− solutions in deaerated acetonitrile at room temperature, and immediately after the excitation pulse. ΔO.D.=0 is denoted by a color-coded dashed line.
Figure 7:
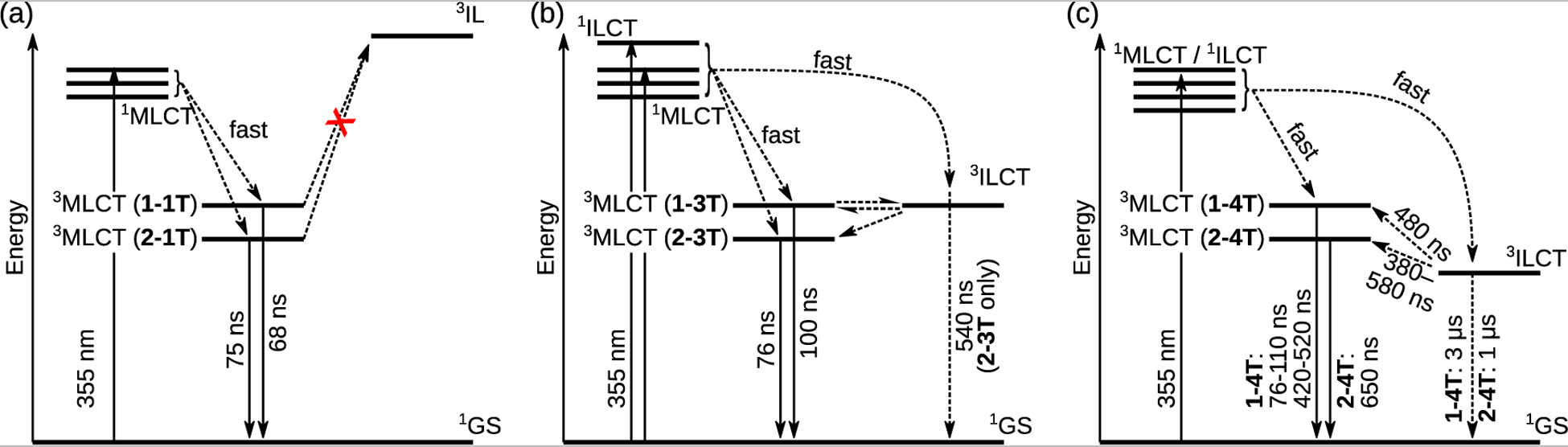
Jablonski diagrams of (a) 1–1T and 2–1T (to represent all x-nT for n≤2), (b) 1–3T and 2–3T, and (c) 1–4T and 2–4T. The energy levels are not drawn to scale.
Generally, the ESA spectra display signatures that are characteristic of either 3MLCT or 3ππ* (3ILCT/3IL) states, where complexes of shorter thienyl chains showed the 3MLCT signature and those with longer chains showed the 3ILCT signature. The 3MLCT signature is characterized by a strong bleach in the 400–500 nm region plus a weaker one in the 550–700 nm region, corresponding to the loss of the ground state 1MLCT absorption seen in the UV-Vis spectra. A moderately strong new absorption around 350–400 nm arose from new excited state transitions originating from the reduced ligand in the 3MLCT state. The tris homoleptic complexes [Os(bpy)3]2+ and [Os(dmb)3]2+, and the shorter-chained 1–0T–1–2T and 2–0T–2–2T all produced this profile. The TA lifetimes were monoexponential, similar within a series, and corresponded well with the emission lifetimes (Table 4). This indicates that the emissive 3MLCT state is the dominant pathway for relaxation in complexes with less π-conjugated ligands.
The signature of the 3ILCT state was observed for almost all of the complexes with n≥3, which qualitatively resembles the spectra of the free IP-3T and IP-4T ligands (Figure S56) and is consistent with long-lived ligand-based 3ππ* states. The strong bleach below 500 nm corresponds to the loss of the overlapping ground-state 1MLCT and IP-nT 1ππ* absorption. The new broad and intense absorptions starting around 500 nm and extending out to the near infrared correspond to transitions associated with 3ππ* states, which are actually mixed 3ILCT/3LLCT character according to the computational study but referred to here as 3ILCT for simplicity. The lack of these characteristics in complexes with less π-expansive ligands indicates that the long-lived 3ILCT state cannot be populated when its energy is sufficiently higher than that of the 3MLCT.
The more π-expansive bpy complexes 1–3T and 1–4T and dmb complex 2–4T exhibited the characteristic ESA signature for the 3ILCT state (Figure 6). Both x-4T complexes had biexponential lifetimes, with the shorter component being similar to the emission lifetime and consistent with the delayed emission from the 3MLCT state. The longer component was around 3 μs in 1–4T and around 1 μs in 2–4T and can be ascribed to a spin-forbidden non-emissive decay. However, the TA lifetime of 1–3T was monoexponential and equal to the emission lifetime, further supporting that the 3MLCT and 3ILCT states in this complex are in equilibrium.
The spectrum of 2–3T mainly exhibited the 3MLCT ESA signature with a strong MLCT bleach in the 450–500 nm region, but there was also a weak 3ILCT absorption spanning around 550–650 nm. The short TA lifetime matches the emission lifetime, and the longer (5.4 × 102 ns) lifetime component was found only at the longer wavelength absorption (although its corresponding bleach may well have been obscured by the much more intense MLCT bleach). The shape of the ESA spectrum suggests that, unlike 1–3T, the contribution of the 3ILCT state to the overall excited state dynamics of 2–3T is small compared to the influence of the 3MLCT state. This could be a reflection of the distribution of populated 1MLCT and 1ILCT states immediately after excitation or the fact that that the computed 3MLCT-3ILCT energy gap is smaller for 2–3T.
The influence of the initial singlet state is also evident in the photophysics of 2–4T. There was no evidence of a prompt 3MLCT emission (τem≈10–100 ns) process that typifies Os(bpy)3-type complexes. Rather, the 3MLCT emission appeared only to be a consequence of thermal equilibration with the non-emissive 3ILCT state. Presumably 1ILCT dominates as the initial excited singlet in these conditions, which undergoes ISC to 3ILCT or fluoresces with τf≈5 ns (Table 4).
3.4.4. Excited state model
The emission and TA data for x-nT were used to construct the Jablonski diagrams in Figure 7. The energy levels are not drawn to scale, however the emission wavelengths (Table 4) show that the 3MLCT energy of 2-nT is consistently lower than 1-nT. The excitation maxima do not vary much in either series, nor do the corresponding UV-Vis absorptions, so the 1MLCT energy can be assumed to be constant. Being centered on the thiophene rings, the 3IL or 3ILCT should vary with n but not x. This concurs with the calculated model presented earlier that estimates when n>2, the state is more ILCT in character.
Following excitation to an initial 1MLCT state, complexes x-0T–x-2T (Figure 7a) undergo rapid ISC to the emissive 3MLCT state. This ISC pathway is common in polypyridyl-type Os2+ complexes and occurs with near unity efficiency.82 The IP-nT 3IL/3ILCT energy is too high to be populated by the 3MLCT, and so the latter relaxes through a simple phosphorescence process. While the computational study suggested some contribution of the ILCT state in x-2T, the experimental results demonstrate that the MLCT state is dominant for complexes with n=2. It should be noted that the computations are estimates of excited state energies and also do not consider the influence of the initially populated excited states on the relaxation dynamics, where population of different singlet excited state configurations may result in different decay pathways.
The 3ILCT energies in x-3T are nearer to the 3MLCT (Figure 7b) so there is opportunity for the states to interact. 1–3T has a strong ESA profile consistent with population of the 3ILCT state. However, its lifetime is unusually short for such a state, and equals the lifetime of the 3MLCT emission, which in turn is longer than the lifetime of the less conjugated members of the series. The common lifetime points to an equilibrium between 3ILCT and 3MLCT in 1–3T, where the forward and reverse rates must be similar for the two states to decay with a common rate constant.45 In contrast, the ESA profile of 2–3T exhibits only a weak 3ILCT ESA. Its TA lifetime is much longer, around 540 ns, and the 3MLCT emission lifetime shows only the prompt emission lifetime consistent with the less π-conjugated members of the series. Evidently the small shift of the 3MLCT to lower energy due to the methyl groups on the coligand was enough to displace its equilibrium with the 3ILCT state.
The TA lifetime of 1–4T had two components: τ1≈480 ns and τ2≈3 μs. The emission also had two components: a prompt emission τ1≈76–110 ns and a delayed emission τ2≈420–520 ns. The short TA lifetime and the long emission lifetime are in agreement, suggesting that the 3MLCT can be populated from the 3ILCT state, as indicated in Figure 7c. Similarly, the shorter TA lifetime of 2–4T matches the emission, but this complex was the only one not to exhibit the prompt 3MLCT emission. It was also the only one to fluoresce noticeably, τf=5 ns (not shown in the diagram). This fluorescence was assigned to the 1ILCT state, and points to a possible difference in the initial excited states and their ensuing decay pathways. In 2–4T the 1ILCT state could be preferentially populated over the 1MLCT state and either populate 3ILCT exclusively or fluoresce. In this case, access to the 3MLCT state only occurs through the 3ILCT state and not the 1MLCT state, which is why prompt 3MLCT emission with a time constant ≤100 ns is not observed. The difference with 1–4T could again be a consequence of the subtle influence the added methyl groups exert on the MLCT energy level.
3.4.5. Singlet oxygen sensitization
The quantum yields for producing singlet oxygen (ΦΔ, Table 5) were calculated from the intensity of the O21Δg ⟶3Σg phosphorescence around 1276 nm, measured at room temperature in air-saturated MeCN with [Ru(bpy)3]2+ as the reference standard (ΦΔ=0.5667), in accordance with Equation 1. The values for ΦΔ were similar for the x-0T to x-3T complexes, as well as the corresponding homoleptic archetypes, around 0.16–0.24 for bpy and 0.13–0.26 for dmb, pointing to sensitization arising from the same quantum state, i.e., 3MLCT based on the ancillary ligand. The quaterthiophene-based complexes were much more efficient at 1O2 sensitization, 0.70 and 0.56 for 1–4T and 2–4T, respectively. This is consistent with sensitization arising from the longer-lived and distally-located 3ILCT state, and reflects the enhanced 1O2 quantum yields we have previously encountered with Ru and Os complexes containing quaterthiophene ligands.36,66 The larger 1O2 quantum yield for 1–4T may be indicative of the larger contribution of its 3ILCT state to the overall excited state dynamics due to a slightly higher-lying 3MLCT state and/or its longer lifetime.
Table 5:
Singlet oxygen quantum yields in acetonitrile at room temperature. The excitation wavelength (in nm) is indicated in parentheses.
| cmpd | ΦΔ | |
|---|---|---|
| LL=bpy; x=1 | LL=dmb; x=2 | |
| [Os(LL)3]2+ | 0.22 (479)a | 0.13 (470) |
| x-0T | 0.24 (475) | 0.26 (470) |
| x-1T | 0.16 (476) | 0.20 (476) |
| x-2T | 0.18 (475) | 0.20 (472) |
| x-3T | 0.21 (489) | 0.15 (417) |
| x-4T | 0.70 (460) | 0.56 (461) |
Literature value83
Notably, most of the complexes exhibited weak but non-zero 1O2 sensitization at long wavelengths where the UV-visible spectrum indicated very little absorption, e.g., 753 nm. While the weak emissions could be integrated, ΦΔ could not be determined because in all cases the absorption at the excitation wavelength was below the detection limit of the instrument.
3.5. Biological Results
3.5.1. Cytotoxicity and photocytotoxicity in normoxia
The dark and light cytotoxicities for the x-nT complexes and their corresponding tris-homoleptic complexes (chloride salts) were measured in the amelanotic84 human melanoma line SK-MEL-28 as 2D adherent monolayers. Briefly, cells growing in log phase were seeded in microplates, dosed with compound (1 nM–300 μM), incubated for 16–21 h, and then given either a sham (dark) or a light treatment. After 48 h, the relative cell viability was assessed using the resazurin assay for cytotoxicity. Dose-response curves (logistic) were prepared from both sets of treatments and used to derive dark and light EC50 values, the effective concentration to reduce relative cell viability by 50%. Phototherapeutic indices (PIs), a measure of the amplification of cytotoxic effects by light, were calculated as the ratios of dark EC50 to light EC50 values. Any deviations from these measurements (hypoxia, specialized light protocols) are described as they are introduced.
3.5.1.1. Dark cytotoxicity
Overall, the complexes were relatively nontoxic in the absence of a light treatment (Figure 8 and Tables S10–S11). The tris homoleptic reference compounds [Os(bpy)3]Cl2 and [Os(dmb)3]Cl2 along with x-0T were the least cytotoxic with dark EC50 values >100 μM. The dark EC50 values were lower for thienyl containing compounds, ranging from 42.2–112 μM for the Os-bpy complexes and 63.8–85.7 μM for the Os-dmb complexes. The greater dark cytotoxicity roughly paralleled increasing lipophilic character (Figure S57, Table S9).
Figure 8:
Summary resazurin-based (photo)cytotoxicity of (a) 1-nT and (c) 2-nT (where n =0–4) with reference tris homoleptics [Os(bpy)3]Cl2 and [Os(dmb)3]Cl2 against SK-MEL-28 in hypoxia (1% O2; open symbols) and normoxia (18.5–21% O2; filled symbols) are plotted as log (EC50±SEM). The phototherapeutic indexes (PI), as the ratio of light to dark cytotoxicity, are plotted in (b) for 1-nT and (d) for 2-nT. Figures are labelled as dark (no light; black circles) and 100 J cm−2 treatments at ~20 mW cm−2 as 633 nm (red, inverted triangle), 523 nm (green, triangle), and cool white visible (blue, square). Results are tabulated in Tables S10–S11.
3.5.1.2. Photocytotoxicity
The photocytotoxicities of the Os complexes were determined with visible (400–700 nm, maxima≈450 nm), green (523 nm), and red (633 nm) light using a fluence of 100 J cm−2 and an irradiance of approximately 20 mW cm−2. The spectral output of the light sources is shown in Figure S45, the EC50 values and PIs are listed in Tables S10–S11, and activity comparisons are highlighted in Figure 8.
Complexes lacking thiophenes were not only the least dark toxic but also the least phototoxic, with light EC50 values >200 μM for the bpy series and ≥50 μM for the dmb series. The cytotoxicities of the thiophene-containing complexes were potentiated by light, with compounds in the bpy family (for a given n) being more potent than those in the dmb family (with the exception of n=1). The EC50 values with visible light ranged from 1.31 nM to 10.6 μM in the bpy series and from 71 nM to 4.78 μM in the dmb series. The photocytotoxicities increased with n, and the increase in potency per thiophene was greatest in the bpy series, where photocytotoxicity increased more than 100-fold on going from n=3 to 4. The most potent compounds, 1–4T and 2–4T, exhibited nanomolar visible light EC50 values (1.31 nM and 71 nM, respectively). These trends were similar for green and red light but attenuated by up to 10-fold at the longer wavelengths. The wavelength dependence was greatest for 1–4T, with light EC50 values near 10 nM for both green and red.
3.5.1.3. Phototherapeutic indices (PIs)
The ratio of dark to light cytotoxicity, or PI, is a better descriptor of the light amplified cytotoxicity given that the light EC50 value necessarily encompasses contributions from the dark cytotoxicity. Light had very little influence on the cytotoxicities of compounds without thienyl groups, resulting in PIs close to 1. For x-1T–x-4T, the trends in PI mirrored their EC50 values, where visible PIs were as large as 55,000 for 1–4T in the bpy series and almost 900 for 2–4T in the dmb series. With regard to the other thienyl-based compounds (n=1–3), the visible PI range was 11–280 for the bpy family and 18–130 for the dmb family. These PI values were attenuated with the longer-wavelength green and red treatments. Generally, the PIs were reduced by less than 5-fold with the exception of the most potent 1–4T, which was attenuated by up to 10-fold. Despite this greater attenuation compared to the PIvis, the PIgreen and PIred of 1–4T were still very large (6700 and 5500, respectively). These longer-wavelength PIs for 1–4T were 6- to 7-fold larger than the PIvis for 2–4T, which was the second most potent compound across the two families. Clearly the coligand (bpy versus dmb) has a very strong influence on the overall light-induced potency of the compound.
The light EC50 values and PIs followed the same trend observed in cell-free photophysical studies, where the compounds with accessible 3ILCT states were the most phototoxic. 1–4T was the most potent of all complexes, with low nanomolar light EC50 values (1–13 nM) and PIs of 5500–55000, and had the longest-lived (3.6 μs) 3ILCT state. 2–4T was the most potent of the dmb series but second overall with submicromolar EC50 values (71–284 nM) that were larger than those for 1–4T and PIs that were significantly lower. Its lower photocytotoxicity compared to 1–4T agrees with its shorter triplet state lifetime (0.98–1.2 μs) and lower singlet oxygen quantum yield (56% vs. 70% in 1–4T). Indeed, the least phototoxic complexes with micromolar light EC50 values had the shortest lifetimes (τ<100 ns) and lowest singlet oxygen quantum yields (ΦΔ≤26%).
3.5.2. Cytotoxicity and photocytotoxicity in hypoxia
The Os complexes were also screened for their dark and light cytotoxic effects in hypoxia (1% O2) to emulate a more challenging environment present in solid or large-volume tumors. While a hypoxic 2D in vitro model lacks certain pathophysiological features present in an actual tumor, it does accurately reflect the oxygen-dependence of a compound’s activity and recapitulates hypoxia-induced drug resistance. In order to avoid false positives, maintenance of low oxygen in solution and headspace is of the utmost importance for drug screening with typically oxygen-dependent compounds. For phototoxic compounds in particular, it is paramount that low oxygen tension be maintained during the entire illumination period. Low gas-permeable and fully transparent films alongside an internal positive control for oxygen-dependence (i.e., [Ru(bpy)2(dppn)]Cl2) made it possible to perform light treatments for up to 1.5 h outside the hypoxic chamber. Briefly, cells were screened in parallel with normoxic experiments, pre-incubated at low oxygen (1% O2) before compound addition, and maintained in hypoxia until completion of light treatment (~20 h DLI) as reported previously.31
3.5.2.1. Hypoxic dark cytotoxicity
The dark cytotoxicities of the complexes in hypoxia were similar to those in normoxia (±10%), with the greatest changes in the bpy series. Compounds without thienyl groups or only one thienyl group were nontoxic (≥100 μM), while those with two thiophenes or more (n=2–4) exhibited dark EC50 values ranging from 55.3 to 76.9 μM for the Os-bpy family and from 62.9 to 74.4 μM for the Os-dmb family. Hypoxia-induced resistance relative to the normoxic cell experiment was observed for two compounds, 1–2T (42.2→76.9 μM) and 1–3T (45.8→71.9 μM), and increased susceptibility in one, 1–4T (71.9→55.3 μM).
3.5.2.2. Hypoxic photocytotoxicity and PIs
Upon light activation in hypoxia, a significant contrast with normoxic activities was observed. For any compound with a PI<100 in normoxia, no appreciable phototoxicity (PI=1) was observed under low oxygen (1% O2). Compounds in the Os-bpy series by comparison were much more active than those in the Os-dmb family. The most potent compound in normoxia, 1–4T, maintained its position as the top performer in hypoxia but with a loss of several orders of magnitude in activity. The wavelength dependence observed in normoxia was not apparent in hypoxia. Light EC50 values for 1–4T in hypoxia averaged 770 nM and PIs ranged from 68 to 76, indicative of a similar phototoxic mechanism accessible even with low energy red (633 nm) light. The close analog 2–4T displayed some phototoxic effects in hypoxia but was at least five-fold less active compared to 1–4T, with a mean EC50 of 6.83 μM and PI=6–15 across all three light conditions.
To our knowledge, the largest PIs for photoactive compounds in hypoxia up to now have been <20, including our own [Ru(6,6′-dmb)2(1-NIP)]Cl2 (reported with Glazer and coworkers) with PIvis=15 and another Ru(II) complex reported by the Bonnet group (PIgreen=16).30,31 2–4T with its PIgreen of 15 is similar. With PI values near 70 in hypoxia, 1–4T stands out as being one of the most hypoxia active photosensitizers reported to date and comparable to its phen-based analog [Os(phen)2(IP-4T)]Cl2 (phen=1,10-phenanthroline) with a PIvis=90 that we just reported.66
The exceptional activities by the Os-based quaterthiophene complexes are some of the first examples of truly hypoxia active photosensitizers. While spectroscopy has afforded key characteristics for 1–4T and 2–4T, such as long lived excited states (τTA>1 μs) and high singlet oxygen quantum yields (ΦΔ=56–70%), the mode of action for hypoxic activity has not been definitively established. Our parallel work with [Os(phen)2(IP-4T)]Cl2 also demonstrated that the longest-lived excited states were 3ILCT in character and that these states were also the most hypoxia active.66 By comparison, the phen analog was the top performer with several unique properties but the bpy and dmb analogs reported here demonstrate unequivocally that the extremely high potency in normoxia that leads to hypoxia activity is a general property of this limited Os(II) IP-4T compound class. While still early to conclude, long-lived excited states with a high degree of charge transfer character (i.e., ILCT) are preliminary leads for installing hypoxic photoactivity and the subject of future mechanistic work.
3.5.3. Fluence dependence
Complexes with three or four thiophenes (n=3,4) were further evaluated for their fluence dependence since examples from Figure 8 only used a single light dose. The effect of fluence (10–100 J cm−2) on light EC50 values for complexes was determined in normoxia with the monochromatic light sources 523 (Figure 9b) and 633 nm (Figure 9a). Fluence dependent EC50 values and PIs are included in Tables S13 with PDT dose product EC50 values in Table S12. The maximum photocytotoxicity for these Os complexes occurred in the 50–100 J cm−2 range with similar activities for 523 and 633 nm. While the terthiophene (n=3) complexes were not highly potent (EC50>1 μM) across the tested fluences, they maintained moderate PDT dose product EC50 values >100 μM·J·cm−2. Addition of just one thiophene (n=4) had a significant impact, resulting in a 15 to 34-fold improvement over the n=3 complexes. Submicromolar activity was achieved for the most potent complex 1–4T at both wavelengths with as little as 10 J cm−2, which was the lowest tested fluence. 1–4T had the lowest PDT dose product EC50 values at 3.06 and 6.12 μM·J·cm−2 for green and red treatments, respectively. This greater potency in 1–4T translated to a minimum 5-fold improvement over 2–4T, further demonstrating the impact of the coligand on potency. While greater photocytotoxicity with higher PS dose or higher fluences is expected, the PDT dose product EC50 values for 1–4T are low (single-digit) and considered quite good. For reference in the same model, our Ru-terthiophene drug in clinical trials, TLD1433, has PDT dose product EC50 values at 19.0 and 266 μM·J·cm−2 for green and red treatments in the same model, respectively. Lower PDT dose products allow greater flexibility since lower PS dosing and fluences can be used when translating to in vivo applications, thereby improving probability for treatment tolerance and (potential) clinical success.
Figure 9:
Fluence dependence of most photoactive compounds 1-nT and 2-nT (n=3,4) against amelanotic SK-MEL-28 human cells. Fluence ranged from 0 (dark) to 100 J cm−2 at 18 mW cm−2 for light treatments using (a) red 633 nm and (b) green 523 nm. Reported log (EC50 ± SEM) values were taken from log-logistic fits. SEM = standard error of the mean. * Values from separate experiment as reported in Figure 8.
3.5.4. MTD results
Since our in vitro models indicated low dark toxicity, intraperitoneal MTD studies were carried out in female C57BL/6J mice (25–200 mg kg−1) to gauge whether in vivo phototoxicity experiments were a logical next step for the most promising complexes 1–4T and 2–4T.
The complexes were well tolerated in the animals at the doses used in this study. Mild toxicity was observed with 2–4T at the higher concentrations of 100, 125, and 200 mg kg−1. In contrast, 1–4T only had mild toxicity at the highest concentration tested (200 mg kg−1). Since no serious toxicity was observed, the acutely mild symptoms quickly resolved after 2 h, and no other symptoms manifested over the 2-week study period, both 1–4T and 2–4T were assigned an MTD ≥ 200 mg kg−1.
4. CONCLUSIONS
Osmium complexes are relatively unexplored in medicinal chemistry compared to their analogous group 8 counterparts, ruthenium and iron. Despite this, osmium complexes have an advantage for photobiological applications due to their extended absorption windows. In our previous work with Ru complexes, we have highlighted the pivotal role of 3ILCT states for installing potent photobiological anticancer activity.20,36 Our photosensitizer in clinical trials, TLD1433, provided preliminary evidence that such excited states could maintain activity even at low oxygen tension.85 This study explores the structural requirements (in terms of the number of thienyl rings n) for accessing low-energy 3ILCT states in the related osmium complexes (which are actually mixed 3ILCT/3LLCT character according to computation but referred to here as 3ILCT for simplicity) and for maintaining photocytotoxic effects in hypoxia. Our hypothesis was that 1–4T and 2–4T would be the most potent compounds based on their 3ILCT energies and lifetimes as well as the inherent photosensitizing power of this state. Further, we hypothesized that the x-4T complexes would maintain some activity in hypoxia. We also probed the influence of the ancillary ligands, which differ only by the presence or absence of methyl substituents on the bpy rings. We expected these ancillary ligands to play a relatively minor role in the overall photodynamics but we found that seemingly trivial changes to the ancillary ligands can lead to meaningful effects on the excited state dynamics and thus on the photobiology of the compounds.
The structure activity relationships based on the number of thienyl groups in both series corroborated the photophysical model derived from spectroscopic measurements. Each of the complexes were characterized by an emissive 3MLCT state lying between 1.60–1.66 eV. The excited state dynamics were governed by this emissive 3MLCT state for n=0–2, while the characteristic 3ILCT signature dominated in the TA of complexes with 3 or 4 thiophenes for the bpy series but only in the TA of 2–4T from the dmb series. The conclusion is that the coligand and the IP-nT ligand affect the 3MLCT–3ILCT energy gap and small changes to this gap can have a profound influence of the excited state dynamics. Furthermore, a similar minor influence on the initial 1MLCT and/or 1ILCT states has important downstream effects.
The excited state lifetimes were also affected by both the number of thiophene rings and the coligand. Only the quaterthiophene complexes 1–4T and 2–4T exhibited the microsecond lifetime characteristic of an 3ILCT state sufficiently lower in energy than the 3MLCT state. While the spectroscopic signature of the 3ILCT state was detected in the TA of 1–3T, the 3MLCT–3ILCT energy difference was not large enough for the 3ILCT state to dominate the photophysics. This assertion was supported by singlet oxygen quantum yields, where all of the complexes with n=0–3 had singlet oxygen quantum yields near 20% or less. The quaterthiophene complexes 1–4T and 2–4T had singlet oxygen quantum yields of 70 and 56%, respectively, that paralleled their microsecond 3ILCT lifetimes. The conclusion is that an 3ILCT of sufficiently low energy relative to the 3MLCT state is required for the most effective singlet oxygen sensitization.
The dominant and highly photosensitizing 3ILCT state in 1–4T and 2–4T led to notable activity against SK-MEL-28 cells under both normoxic and hypoxic conditions. The most potent complex in vitro, 1–4T, had the longest lifetime by TA (τ≈3 μs) and highest singlet oxygen quantum yield (ΦΔ=70%). The methylated 2–4T exhibited lesser potency by several-fold in normoxia (PI=225–900) and hypoxia (PI=6–15) compared to 1–4T. Notably in both hypoxia-active complexes, 1–4T and 2–4T, any wavelength dependence observed under normoxic conditions disappeared in hypoxia. The more active 1–4T exhibited up to a 10-fold difference between red (PIred=550) and visible potencies (PIvis=55,000) in normoxia; these values fell to within ~10% of each other in hypoxia with a mean EC50=770 nM and PI=68–76. The loss of wavelength dependence in hypoxia suggests similar excited state pathways accessible with even low energy red (633 nm) light. While a mechanism of action was not identified in this work, the increased charge transfer character present in the 3ILCT state (and long lifetime) could be responsible for the retention of photocytotoxicity even under hypoxic conditions.
In light of the low dark toxicity in vitro and excellent photobiological properties of 1–4T and 2–4T, MTD studies were carried out to determine whether the compounds were viable candidates for additional in vivo studies. Both compounds were well tolerated with MTD values ≥200 mg kg−1 by intraperitoneal injection. As shown with previous Os(II) complexes,39,66 1–4T and 2–4T dispel the stigma of osmium complexes being inherently toxic and thus not amenable to clinical translation. The lead complexes demonstrated low toxicity in vitro with high tolerance in mice. Studies are currently underway to probe the scope of this demonstrated hypoxic activity in more sophisticated biological models and to delineate those phototoxic mechanism(s) at play.
Supplementary Material
6.2. Acknowledgements
S.A.M., C.G.C., S.G., R.P.T., and G.D. thank the National Cancer Institute (NCI) of the National Institutes of Health (NIH) (Award R01CA222227) for partial support of this work. The content in this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. N.R. and M.E.A. are grateful to the Università della Calabria. M.E.A. would also like to thank Dr. Antonio Francés Monerris for the valuable and constructive discussion. S.A.M. also thanks Dr. Daniel Todd as UNCG’s Triad Mass Spectrometry Facility manager and his assistants Jennifer Simpson and Diane Wallace. S.A.M. likewise thanks Dr. Franklin Moy for his experimental support and instrument maintenance as UNCG’s NMR facility manager. S.A.M. notes that the graphical (TOC) abstract was created with BioRender.com.
5. ABBREVIATIONS
- η
Refractive index
- λ
Wavelength
- τ
Lifetime
- Φ
Quantum Yield
- bpy
2,2′-bipyridine
- COSY
Correlated spectroscopy
- Do/w
Distribution coefficient of 1-octanol and buffered water
- DFT
Density functional theory
- DLI
Drug to Light Interval
- DMEM
Dulbecco’s Modified Eagle Medium
- DMSO
Dimethyl sulfoxide
- dmb
4,4′-dimethyl-2,2′-bipyridine
- EC50
Effective Concentration to reduce relative cell viability by 50%
- EMEM
Eagle’s Minimum Essential Medium
- ESA
Excited State Absorption
- ESI
Electrospray Ionization
- HMBC
Heteronuclear Multiple Bond Correlation
- HPLC
High Performance Liquid Chromatography
- HRMS
High Resolution Mass Spectrometry
- HSQC
Heteronuclear Single Quantum Correlation
- IL
Intraligand
- ILCT
Intraligand Charge Transfer
- IP
1-H-imidazo[4,5-f][1,10]phenanthroline
- IP-nT
imidazo[4,5-f][1,10]phenanthroline with n appended thiophene rings
- ISC
Intersystem crossing
- MeCN
Acetonitrile
- MLCT
Metal – Ligand Charge Transfer
- MTD
Maximum Tolerated Dose
- NIR
Near Infrared
- NMR
Nuclear Magnetic Resonance
- PCT
Photochemotherapy
- PDT
Photodynamic Therapy
- phen
phenanthroline
- ppm
parts per million
- PI
Phototherapeutic Index
- PMT
Photomultiplier Tube
- PS
Photosensitizer
- ROS
Reactive oxygen species
- SD
Standard Deviation
- SEM
Standard Error of the Mean
- TA
Transient Absorption
- TDDFT
Time-dependent density functional theory
- UV
Ultraviolet
- UV-Vis
Ultraviolet – Visible
- XC
Exchange correlation functional
Footnotes
ASSOCIATED CONTENT
Synthetic characterization (1D and 2D NMR, HPLC, HRMS), computational characterization (ground and excited states), spectroscopic characterization (emission, TA), and (photo)biological data are included in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
S.A.M. has a potential research conflict of interest due to a financial interest with Theralase Technologies, Inc. and PhotoDynamic, Inc. A management plan has been created to preserve objectivity in research in accordance with UTA policy.
7. REFERENCES
- (1).Global Cancer Facts & Figures 4th Edition American Cancer Society; 2018. [Google Scholar]
- (2).Falzone L; Salomone S; Libra M Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol 2018, 9, 1300 10.3389/fphar.2018.01300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Loeb LA A Mutator Phenotype in Cancer. Cancer Res. 2001, 61 (8), 3230–3239. [PubMed] [Google Scholar]
- (4).Azmi AS; Bao B; Sarkar FH Exosomes in Cancer Development, Metastasis, and Drug Resistance: A Comprehensive Review. Cancer Metastasis Rev. 2013, 32 (3–4), 623–642. 10.1007/s10555-013-9441-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Rankin EB; Giaccia AJ Hypoxic Control of Metastasis. Science 2016, 352 (6282), 175–180. 10.1126/science.aaf4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Vera-Ramirez L; Hunter KW Tumor Cell Dormancy as an Adaptive Cell Stress Response Mechanism. F1000Research 2017, 6, 2134 10.12688/f1000research.12174.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Fiedler EC; Hemann MT Aiding and Abetting: How the Tumor Microenvironment Protects Cancer from Chemotherapy. Annu. Rev. Cancer Biol 2019, 3 (1), 409–428. 10.1146/annurev-cancerbio-030518-055524. [DOI] [Google Scholar]
- (8).Photodynamic Therapy: Basic Principles and Clinical Applications; Henderson BW, Dougherty TJ, Eds.; Dekker: New York, 1992. [Google Scholar]
- (9).Dougherty TJ; Gomer CJ; Henderson BW; Jori G; Kessel D; Korbelik M; Moan J; Peng Q Photodynamic Therapy. JNCI J. Natl. Cancer Inst 1998, 90 (12), 889–905. 10.1093/jnci/90.12.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bonnett R Chemical Aspects of Photodynamic Therapy; Advanced chemistry texts; Gordon and Breach Science Publishers: Amsterdam, The Netherlands, 2000. [Google Scholar]
- (11).Advances in Photodynamic Therapy: Basic, Translational, and Clinical; Hamblin MR, Mróz P, Eds.; Artech House engineering in medicine & biology series; Artech House: Boston, Mass, 2008. [Google Scholar]
- (12).Plaetzer K; Krammer B; Berlanda J; Berr F; Kiesslich T Photophysics and Photochemistry of Photodynamic Therapy: Fundamental Aspects. Lasers Med. Sci 2009, 24 (2), 259–268. 10.1007/s10103-008-0539-1. [DOI] [PubMed] [Google Scholar]
- (13).Agostinis P; Berg K; Cengel KA; Foster TH; Girotti AW; Gollnick SO; Hahn SM; Hamblin MR; Juzeniene A; Kessel D; Korbelik M; Moan J; Mroz P; Nowis D; Piette J; Wilson BC; Golab J Photodynamic Therapy of Cancer: An Update. CA. Cancer J. Clin 2011, 61 (4), 250–281. 10.3322/caac.20114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Handbook of Photomedicine; Hamblin MR, Huang Y-Y, Eds.; CRC Press: Boca Raton, FL, 2014. [Google Scholar]
- (15).Allison RR Photodynamic Therapy: Oncologic Horizons. Future Oncol. 2014, 10 (1), 123–124. 10.2217/fon.13.176. [DOI] [PubMed] [Google Scholar]
- (16).Benov L Photodynamic Therapy: Current Status and Future Directions. Med. Princ. Pract 2015, 24 (s1), 14–28. 10.1159/000362416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Photodynamic Medicine: From Bench to Clinic; Kostron H, Hasan T, Royal Society of Chemistry (Great Britain), Eds.; Comprehensive series in photochmistry and photobiology; Royal Society of Chemistry: Cambridge, 2016. [Google Scholar]
- (18).Abrahamse H; Hamblin MR New Photosensitizers for Photodynamic Therapy. Biochem. J 2016, 473 (4), 347–364. 10.1042/BJ20150942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).van Straten D; Mashayekhi V; de Bruijn H; Oliveira S; Robinson D Oncologic Photodynamic Therapy: Basic Principles, Current Clinical Status and Future Directions. Cancers 2017, 9 (12), 19 10.3390/cancers9020019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Monro S; Colón KL; Yin H; Roque J; Konda P; Gujar S; Thummel RP; Lilge L; Cameron CG; McFarland SA Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev 2019, 119 (2), 797–828. 10.1021/acs.chemrev.8b00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Spring BQ; Rizvi I; Xu N; Hasan T The Role of Photodynamic Therapy in Overcoming Cancer Drug Resistance. Photochem. Photobiol. Sci 2015, 14 (8), 1476–1491. 10.1039/C4PP00495G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Muz B; de la Puente P; Azab F; Azab AK The Role of Hypoxia in Cancer Progression, Angiogenesis, Metastasis, and Resistance to Therapy. Hypoxia Auckl. NZ 2015, 3, 83–92. 10.2147/HP.S93413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Glazer EC Light-Activated Metal Complexes That Covalently Modify DNA. Isr. J. Chem 2013, 53 (6–7), 391–400. 10.1002/ijch.201300019. [DOI] [Google Scholar]
- (24).Bonnet S Why Develop Photoactivated Chemotherapy? Dalton Trans. 2018, 47 (31), 10330–10343. 10.1039/C8DT01585F. [DOI] [PubMed] [Google Scholar]
- (25).White JK; Schmehl RH; Turro C An Overview of Photosubstitution Reactions of Ru(II) Imine Complexes and Their Application in Photobiology and Photodynamic Therapy. Inorganica Chim. Acta 2017, 454, 7–20. 10.1016/j.ica.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Feng W; Gao C; Liu W; Ren H; Wang C; Ge K; Li S; Zhou G; Li H; Wang S; Jia G; Li Z; Zhang J A Novel Anticancer Theranostic Pro-Prodrug Based on Hypoxia and Photo Sequential Control. Chem Commun 2016, 52 (60), 9434–9437. 10.1039/C6CC02932A. [DOI] [PubMed] [Google Scholar]
- (27).Lameijer LN; Ernst D; Hopkins SL; Meijer MS; Askes SHC; Le Dévédec SE; Bonnet S A Red-Light-Activated Ruthenium-Caged NAMPT Inhibitor Remains Phototoxic in Hypoxic Cancer Cells. Angew. Chem. Int. Ed 2017, 56 (38), 11549–11553. 10.1002/anie.201703890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Al-Afyouni MH; Rohrabaugh TN; Al-Afyouni KF; Turro C New Ru(ii) Photocages Operative with near-IR Light: New Platform for Drug Delivery in the PDT Window. Chem. Sci 2018, 9 (32), 6711–6720. 10.1039/C8SC02094A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Li M-D; Wong N-K; Xiao J; Zhu R; Wu L; Dai S-Y; Chen F; Huang G; Xu L; Bai X; Geraskina MR; Winter AH; Chen X; Liu Y; Fang W; Yang D; Phillips DL Dynamics of Oxygen-Independent Photocleavage of Blebbistatin as a One-Photon Blue or Two-Photon Near-Infrared Light-Gated Hydroxyl Radical Photocage. J. Am. Chem. Soc 2018, 140 (46), 15957–15968. 10.1021/jacs.8b10235. [DOI] [PubMed] [Google Scholar]
- (30).van Rixel VHS; Ramu V; Auyeung AB; Beztsinna N; Leger DY; Lameijer LN; Hilt ST; Le Dévédec SE; Yildiz T; Betancourt T; Gildner MB; Hudnall TW; Sol V; Liagre B; Kornienko A; Bonnet S Photo-Uncaging of a Microtubule-Targeted Rigidin Analogue in Hypoxic Cancer Cells and in a Xenograft Mouse Model. J. Am. Chem. Soc 2019, 141 (46), 18444–18454. 10.1021/jacs.9b07225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Roque J; Havrylyuk D; Barrett PC; Sainuddin T; McCain J; Colón K; Sparks WT; Bradner E; Monro S; Heidary D; Cameron CG; Glazer EC; McFarland SA Strained, Photoejecting Ru(II) Complexes That Are Cytotoxic Under Hypoxic Conditions. Photochem. Photobiol 2020, 96 (2), 327–339. 10.1111/php.13174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Howerton BS; Heidary DK; Glazer EC Strained Ruthenium Complexes Are Potent Light-Activated Anticancer Agents. J. Am. Chem. Soc 2012, 134 (20), 8324–8327. 10.1021/ja3009677. [DOI] [PubMed] [Google Scholar]
- (33).Wachter E; Glazer EC Mechanistic Study on the Photochemical “Light Switch” Behavior of [Ru(Bpy) 2 Dmdppz] 2+. J. Phys. Chem. A 2014, 118 (45), 10474–10486. 10.1021/jp504249a. [DOI] [PubMed] [Google Scholar]
- (34).Yin H; Stephenson M; Gibson J; Sampson E; Shi G; Sainuddin T; Monro S; McFarland SA In Vitro Multiwavelength PDT with 3IL States: Teaching Old Molecules New Tricks. Inorg. Chem 2014, 53, 4548–4559. 10.1021/ic5002368. [DOI] [PubMed] [Google Scholar]
- (35).Stephenson M; Reichardt C; Pinto M; Wächtler M; Sainuddin T; Shi G; Yin H; Monro S; Sampson E; Dietzek B; McFarland SA Ru(II) Dyads Derived from 2-(1-Pyrenyl)-1H-Imidazo[4,5-f][1,10]Phenanthroline: Versatile Photosensitizers for Photodynamic Applications. J Phys Chem A 2014, 118 (45), 10507–10521. 10.1021/jp504330s. [DOI] [PubMed] [Google Scholar]
- (36).Shi G; Monro S; Hennigar R; Colpitts J; Fong J; Kasimova K; Yin H; DeCoste R; Spencer C; Chamberlain L; Mandel A; Lilge L; McFarland SA Ru(II) Dyads Derived from α -Oligothiophenes: A New Class of Potent and Versatile Photosensitizers for PDT. Coord. Chem. Rev 2015, 282–283, 127–138. 10.1016/j.ccr.2014.04.012. [DOI] [Google Scholar]
- (37).Sainuddin T; McCain J; Pinto M; Yin H; Gibson J; Hetu M; McFarland SA Organometallic Ru(II) Photosensitizers Derived from π-Expansive Cyclometalating Ligands: Surprising Theranostic PDT Effects. Inorg. Chem 2016, 55, 83–95. 10.1021/acs.inorgchem.5b01838. [DOI] [PubMed] [Google Scholar]
- (38).Sainuddin T; Pinto M; Yin H; Hetu M; Colpitts J; McFarland SA Strained Ruthenium Metal–Organic Dyads as Photocisplatin Agents with Dual Action. J. Inorg. Biochem 2016, 158, 45–54. 10.1016/j.jinorgbio.2016.01.009. [DOI] [PubMed] [Google Scholar]
- (39).Lazic S; Kaspler P; Shi G; Monro S; Sainuddin T; Forward S; Kasimova K; Hennigar R; Mandel A; McFarland S; Lilge L Novel Osmium-Based Coordination Complexes as Photosensitizers for Panchromatic Photodynamic Therapy. Photochem. Photobiol 2017, 93 (5), 1248–1258. 10.1111/php.12767. [DOI] [PubMed] [Google Scholar]
- (40).Ghosh G; Colón KL; Fuller A; Sainuddin T; Bradner E; McCain J; Monro SMA; Yin H; Hetu MW; Cameron CG; McFarland SA Cyclometalated Ruthenium(II) Complexes Derived from α-Oligothiophenes as Highly Selective Cytotoxic or Photocytotoxic Agents. Inorg. Chem 2018, 57 (13), 7694–7712. 10.1021/acs.inorgchem.8b00689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).McCain J; Colón KL; Barrett PC; Monro SMA; Sainuddin T; Roque III J; Pinto M; Yin H; Cameron CG; McFarland SA Photophysical Properties and Photobiological Activities of Ruthenium(II) Complexes Bearing π-Expansive Cyclometalating Ligands with Thienyl Groups. Inorg. Chem 2019, 58 (16), 10778–10790. 10.1021/acs.inorgchem.9b01044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Monro S; Cameron CG; Zhu X; Colón KL; Yin H; Sainuddin T; Hetu M; Pinto M; Fuller A; Bennett L; Roque J; Sun W; McFarland SA Synthesis, Characterization and Photobiological Studies of Ru(II) Dyads Derived from α -Oligothiophene Derivatives of 1,10-Phenanthroline. Photochem. Photobiol 2019, 95 (1), 267–279. 10.1111/php.13012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Arenas Y; Monro S; Shi G; Mandel A; McFarland S; Lilge L Photodynamic Inactivation of Staphylococcus Aureus and Methicillin-Resistant Staphylococcus Aureus with Ru(II)-Based Type I/Type II Photosensitizers. Photodiag Photodyn Ther 2013, 10, 615–625. 10.1016/j.pdpdt.2013.07.001. [DOI] [PubMed] [Google Scholar]
- (44).Chen Q; Ramu V; Aydar Y; Groenewoud A; Zhou X-Q; Jager MJ; Cole H; Cameron CG; McFarland SA; Bonnet S; Snaar-Jagalska BE TLD1433 Photosensitizer Inhibits Conjunctival Melanoma Cells in Zebrafish Ectopic and Orthotopic Tumour Models. Cancers 2020, 12 (3), 587 10.3390/cancers12030587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).McClenaghan ND; Leydet Y; Maubert B; Indelli MT; Campagna S Excited-State Equilibration: A Process Leading to Long-Lived Metal-to-Ligand Charge Transfer Luminescence in Supramolecular Systems. Coord Chem Rev 2005, 249 (13–14), 1336–1350. 10.1016/j.ccr.2004.12.017. [DOI] [Google Scholar]
- (46).Alberto ME; Pirillo J; Russo N; Adamo C Theoretical Exploration of Type I/Type II Dual Photoreactivity of Promising Ru(II) Dyads for PDT Approach. Inorg. Chem 2016, 55 (21), 11185–11192. 10.1021/acs.inorgchem.6b01782. [DOI] [PubMed] [Google Scholar]
- (47).Kober EM; Caspar JV; Sullivan BP; Meyer TJ Synthetic Routes to New Polypyridyl Complexes of Osmium(II). Inorg Chem 1988, 27 (25), 4587–4598. 10.1021/ic00298a017. [DOI] [Google Scholar]
- (48).Wang Z Radziszewski Reaction In Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons, Inc., Hoboken, NJ, 2010; Vol. 3 10.1002/9780470638859.conrr518. [DOI] [Google Scholar]
- (49).Pazderski L; Pawlak T; Sitkowski J; Kozerski L; Szłyk E 1 H NMR Assignment Corrections and 1 H, 13 C, 15 N NMR Coordination Shifts Structural Correlations in Fe(II), Ru(II) and Os(II) Cationic Complexes with 2,2′-Bipyridine and 1,10-Phenanthroline. Magn. Reson. Chem 2010, 48 (6), 450–457. 10.1002/mrc.2600. [DOI] [PubMed] [Google Scholar]
- (50).Ye B-H; Chen X-M; Zeng T-X; Ji L-N Syntheses, Spectra and Crystal Structures of Ruthenium(II) Complexes with Polypyridyl: [Ru(Bipy)2(Phen)](ClO4)2 · H2O and [Ru(Bipy)2(Me-Phen)](ClO4)2. Inorg. Chim. Acta 1995, 240, 5–11. [Google Scholar]
- (51).Recent Developments and Applications of Modern Density Functional Theory; Seminario JM, Ed.; Theoretical and computational chemistry; Elsevier: Amsterdam ; New York, 1996. [Google Scholar]
- (52).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian09; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- (53).Francés-Monerris A; Magra K; Darari M; Cebrián C; Beley M; Domenichini E; Haacke S; Pastore M; Assfeld X; Gros PC; Monari A Synthesis and Computational Study of a Pyridylcarbene Fe(II) Complex: Unexpected Effects of Fac / Mer Isomerism in Metal-to-Ligand Triplet Potential Energy Surfaces. Inorg. Chem 2018, 57 (16), 10431–10441. 10.1021/acs.inorgchem.8b01695. [DOI] [PubMed] [Google Scholar]
- (54).Francés-Monerris A; Gattuso H; Roca-Sanjuán D; Tuñón I; Marazzi M; Dumont E; Monari A Dynamics of the Excited-State Hydrogen Transfer in a (DG)·(DC) Homopolymer: Intrinsic Photostability of DNA. Chem. Sci 2018, 9 (41), 7902–7911. 10.1039/C8SC03252A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Bertini L; Alberto ME; Arrigoni F; Vertemara J; Fantucci P; Bruschi M; Zampella G; De Gioia L On the Photochemistry of Fe 2 (Edt)(CO) 4 (PMe 3) 2, a [FeFe]-Hydrogenase Model: A DFT/TDDFT Investigation. Int. J. Quantum Chem 2018, 118 (9), e25537 10.1002/qua.25537. [DOI] [Google Scholar]
- (56).Ji S; Ge J; Escudero D; Wang Z; Zhao J; Jacquemin D Molecular Structure–Intersystem Crossing Relationship of Heavy-Atom-Free BODIPY Triplet Photosensitizers. J. Org. Chem 2015, 80 (11), 5958–5963. 10.1021/acs.joc.5b00691. [DOI] [PubMed] [Google Scholar]
- (57).Adamo C; Barone V Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys 1999, 110 (13), 6158–6170. 10.1063/1.478522. [DOI] [Google Scholar]
- (58).Andrae D; Häußermann U; Dolg M; Stoll H; Preuß H Energy-Adjusted Ab Initio Pseudopotentials for the Second and Third Row Transition Elements. Theor. Chim. Acta 1990, 77 (2), 123–141. 10.1007/BF01114537. [DOI] [Google Scholar]
- (59).Cossi M; Barone V Solvent Effect on Vertical Electronic Transitions by the Polarizable Continuum Model. J. Chem. Phys 2000, 112 (5), 2427–2435. 10.1063/1.480808. [DOI] [Google Scholar]
- (60).Tomasi J; Mennucci B; Cammi R Quantum Mechanical Continuum Solvation Models. Chem. Rev 2005, 105 (8), 2999–3094. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- (61).Zhao Y; Truhlar DG The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc 2008, 120 (1–3), 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- (62).Latouche C; Skouteris D; Palazzetti F; Barone V TD-DFT Benchmark on Inorganic Pt(II) and Ir(III) Complexes. J. Chem. Theory Comput 2015, 11 (7), 3281–3289. 10.1021/acs.jctc.5b00257. [DOI] [PubMed] [Google Scholar]
- (63).Alberto ME; Adamo C Synergistic Effects in Pt II -Porphyrinoid Dyes as Candidates for a Dual-Action Anticancer Therapy: A Theoretical Exploration. Chem. - Eur. J 2017, 23 (60), 15124–15132. 10.1002/chem.201702876. [DOI] [PubMed] [Google Scholar]
- (64).Alberto ME; Russo N; Adamo C Synergistic Effects of Metals in a Promising Ru II −Pt II Assembly for a Combined Anticancer Approach: Theoretical Exploration of the Photophysical Properties. Chem. - Eur. J 2016, 22 (27), 9162–9168. 10.1002/chem.201601089. [DOI] [PubMed] [Google Scholar]
- (65).Ponte F; Alberto ME; De Simone BC; Russo N; Sicilia E Photophysical Exploration of Dual-Approach Pt II –BODIPY Conjugates: Theoretical Insights. Inorg. Chem 2019, 58 (15), 9882–9889. 10.1021/acs.inorgchem.9b01002. [DOI] [PubMed] [Google Scholar]
- (66).Roque III JA; Barrett PC; Cole HD; Lifshits LM; Shi G; Monro S; von Dohlen D; Kim S; Russo N; Deep G; Cameron CG; Alberto ME; McFarland SA Breaking the Barrier: An Osmium Photosensitizer with Unprecedented Hypoxic Phototoxicity for Real World Photodynamic Therapy. Chem. Sci 2020, 11, 9784–9806. 10.1039/D0SC03008B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).DeRosa MC; Crutchley RJ Photosensitized Singlet Oxygen and Its Applications. Coord. Chem. Rev 2002, 233–234, 351–371. 10.1016/S0010-8545(02)00034-6. [DOI] [Google Scholar]
- (68).Wenger R; Kurtcuoglu V; Scholz C; Marti H; Hoogewijs D Frequently Asked Questions in Hypoxia Research. Hypoxia 2015, 35 10.2147/HP.S92198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).McFarland SA Metal-Based Thiophene Photodynamic Compounds and Their Use. US 9,345,769 B2, May 24, 2016. [Google Scholar]
- (70).McFarland SA Metal-Based Thiophene Photodynamic Compounds and Their Use. 9,676,806, June 13, 2017. [Google Scholar]
- (71).Tan L-F; Wang F; Chao H; Zhou Y-F; Weng C Ruthenium(II) Mixed-Ligand Complex Containing 2-(4′-Benzyloxy-Phenyl)Imidazo[4,5-f][1,10]Phenanthroline: Synthesis, DNA-Binding and Photocleavage Studies. J. Inorg. Biochem 2007, 101 (4), 700–708. 10.1016/j.jinorgbio.2006.12.016. [DOI] [PubMed] [Google Scholar]
- (72).Tan L-F; Chao H; Li H; Liu Y-J; Sun B; Wei W; Ji L-N Synthesis, Characterization, DNA-Binding and Photocleavage Studies of [Ru(Bpy)2(PPIP)]2+ and [Ru(Phen)2(PPIP)]2+. J Inorg Biochem 2005, 99 (2), 513–520. 10.1016/j.jinorgbio.2004.10.028. [DOI] [PubMed] [Google Scholar]
- (73).Sun B; Chu J; Gao F; Ji L-N; Chao H Synthesis, Characterization, Electrochemical and Photophysical Properties of Ruthenium(II) Complexes Containing 3-Amino-1,2,4-Triazino[5,6-f]-1,10-Phenanthroline. J. Mol. Struct 2008, 890, 203–208. [Google Scholar]
- (74).Xu H; Zheng K-C; Lin L-J; Li H; Gao Y; Ji L-N Effects of the Substitution Positions of Br Group in Intercalative Ligand on the DNA-Binding Behaviors of Ru(II) Polypyridyl Complexes. J. Inorg. Biochem 2004, 98 (1), 87–97. 10.1016/j.jinorgbio.2003.09.002. [DOI] [PubMed] [Google Scholar]
- (75).Jiang C-W; Chao H; Li R-H; Li H; Ji L-N Syntheses, Characterization and Third-Order Nonlinear Optical Properties of Ruthenium(II) Complexes Containing 2-Phenylimidazo-[4,5-f][1,10]Phenanthroline and Extended Diimine Ligands. Polyhedron 2001, 20 (17), 2187–2193. 10.1016/S0277-5387(01)00813-0. [DOI] [Google Scholar]
- (76).Lanzani G; Nisoli M; De Silvestri S; Tubino R Femtosecond Vibrational and Torsional Energy Redistribution in Photoexcited Oligothiophenes. Chem. Phys. Lett 1996, 251 (5–6), 339–345. 10.1016/0009-2614(96)00107-8. [DOI] [Google Scholar]
- (77).Yang J-P; Paa W; Rentsch S Femtosecond Investigations of Photophysics of Ultrafast Intersystem Crossing in Terthiophene by Wavelength Dependent Excitation. Chem. Phys. Lett 2000, 320 (5–6), 665–672. 10.1016/S0009-2614(00)00264-5. [DOI] [Google Scholar]
- (78).Decurtins S; Felix F; Ferguson J; Guedel HU; Ludi A The Electronic Spectrum of Tris(2,2’-Bipyridine)Iron(2+) and Tris (2,2’-Bipyridine)Osmium(2+). J. Am. Chem. Soc 1980, 102 (12), 4102–4106. 10.1021/ja00532a020. [DOI] [Google Scholar]
- (79).Pankuch BJ; Lacky DE; Crosby GA Charge-Transfer Excited States of Osmium(II) Complexes. 1. Assignment of the Visible Absorption Bands. J. Phys. Chem 1980, 84 (16), 2061–2067. 10.1021/j100453a013. [DOI] [Google Scholar]
- (80).Kober EM; Meyer TJ An Electronic Structural Model for the Emitting MLCT Excited States of Ru(Bpy)32+ and Os(Bpy)32+. Inorg Chem 1984, 23 (24), 3877–3886. 10.1021/ic00192a009. [DOI] [Google Scholar]
- (81).Grosshenny V; Harriman A; Romero FM; Ziessel R Electron Delocalization in Ruthenium(II) and Osmium(II) 2,2’-Bipyridyl Complexes Formed from Ethynyl-Bridged Ditopic Ligands. J. Phys. Chem 1996, 100 (44), 17472–17484. 10.1021/jp9616268. [DOI] [Google Scholar]
- (82).Juris A; Balzani V; Barigelletti F; Campagna S; Belser P; von Zelewsky A Ru(II) Polypyridine Complexes: Photophysics, Photochemistry, Eletrochemistry, and Chemiluminescence. Coord. Chem. Rev 1988, 84, 85–277. 10.1016/0010-8545(88)80032-8. [DOI] [Google Scholar]
- (83).Abdel-Shafi AA; Worrall DR; Ershov AY Photosensitized Generation of Singlet Oxygen from Ruthenium(II) and Osmium(II) Bipyridyl Complexes. Dalton Trans 2004, No. 1, 30–36. 10.1039/B310238F. [DOI] [PubMed] [Google Scholar]
- (84).Chen KG; Leapman RD; Zhang G; Lai B; Valencia JC; Cardarelli CO; Vieira WD; Hearing VJ; Gottesman MM Influence of Melanosome Dynamics on Melanoma Drug Sensitivity. JNCI J. Natl. Cancer Inst 2009, 101 (18), 1259–1271. 10.1093/jnci/djp259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Fong J; Kasimova K; Arenas Y; Kaspler P; Lazic S; Mandel A; Lilge L A Novel Class of Ruthenium-Based Photosensitizers Effectively Kills in Vitro Cancer Cells and in Vivo Tumors. Photochem. Photobiol. Sci 2015, 14 (11), 2014–2023. 10.1039/C4PP00438H. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.