

Abstract
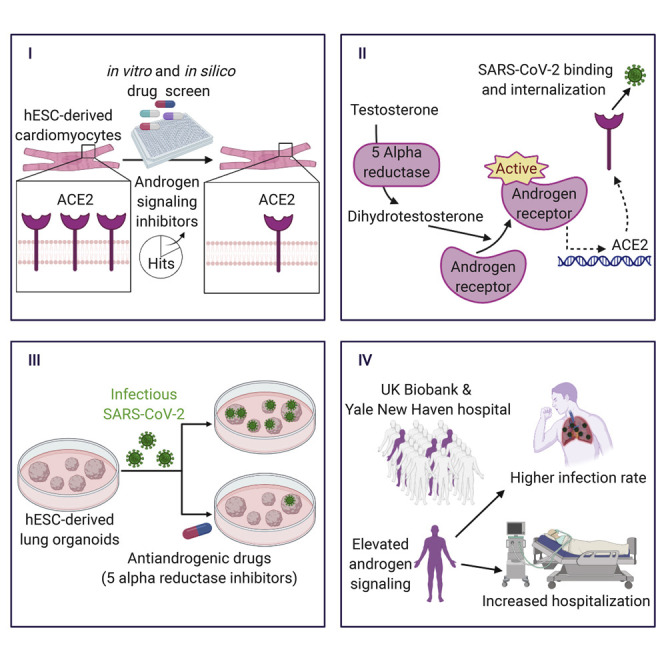
SARS-CoV-2 infection has led to a global health crisis, and yet our understanding of the disease and potential treatment options remains limited. The infection occurs through binding of the virus with angiotensin converting enzyme 2 (ACE2) on the cell membrane. Here, we established a screening strategy to identify drugs that reduce ACE2 levels in human embryonic stem cell (hESC)-derived cardiac cells and lung organoids. Target analysis of hit compounds revealed androgen signaling as a key modulator of ACE2 levels. Treatment with antiandrogenic drugs reduced ACE2 expression and protected hESC-derived lung organoids against SARS-CoV-2 infection. Finally, clinical data on COVID-19 patients demonstrated that prostate diseases, which are linked to elevated androgen, are significant risk factors and that genetic variants that increase androgen levels are associated with higher disease severity. These findings offer insights on the mechanism of disproportionate disease susceptibility in men and identify antiandrogenic drugs as candidate therapeutics for COVID-19.
Keywords: hPSC-based disease modeling, COVID-19 sex bias, drug re-purposing, high content screening, deep learning, virtual drug screen, 5-alpha reductase inhibitors, COVID-19 risk factors, ACE2 regulation, SARS-CoV-2 infection model
Graphical Abstract
Highlights
-
•
Drug screens on hESC cardiac cells identify modulators of SARS-CoV-2 receptor ACE2
-
•
Targets of drugs that reduce ACE2 converge on androgen signaling pathway
-
•
Androgen signaling inhibition reduces SARS-CoV-2 infection in hESC lung organoids
-
•
Elevated androgen increases COVID-19 susceptibility and severity in men
Men are more susceptibility to severe COVID-19 complications. Fattahi and colleagues leverage stem cell models, high-throughput drug screens, and patient records to demonstrate that male sex hormones regulate SARS-CoV-2 receptors and increase disease severity in men. They identify drug candidates that reduce viral infection by inhibiting these hormones.
Introduction
Coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has become a pandemic affecting millions of people worldwide. Limited understanding of the disease pathophysiology has impeded our ability to develop effective preventative and therapeutic strategies.
Multi-organ failure is the most lethal complication of SARS-CoV-2 infection (Guan et al., 2020). It has been well documented that organ involvement and disease manifestation are correlated with the expression of SARS-CoV-2 receptor and co-receptors on the membrane of target cells (Hamming et al., 2004). The spike (S) protein, which is responsible for the characteristic crown-like shape of coronaviruses, facilitates the binding of the virus to its receptors on the cell membrane (Hoffmann et al., 2020; Vabret et al., 2020). ACE2 has been identified as the main receptor utilized by SARS-CoV-2 and SARS-CoV-1 to enter cells (Lan et al., 2020). Additionally, recent findings using human and animal cell lines have demonstrated that SARS-CoV-2 relies on the serine protease TMPRSS2 for S protein priming prior to ACE2-facilitated internalization (Hoffmann et al., 2020). Research after the SARS-CoV-1 epidemic shows that knocking-out ACE2 results in markedly decreased viral entry in lungs of mice infected with the virus (Kuba et al., 2005). In this study, we identified pharmacological strategies to reduce the levels of ACE2 in human embryonic stem cell (hESC)-derived lung organoids and cardiac cells in order to facilitate therapeutic interventions aimed at reducing viral entry, thereby mitigating the multi-organ complications induced by SARS-CoV-2 infection.
To identify candidate drugs capable of modulating ACE2 protein levels, we took advantage of our previously established methods to generate cardiac cells from hESCs at large scale (Ghazizadeh et al., 2018, 2020; Tsai et al., 2020) and performed a high-throughput screen with a library of 1,443 FDA-approved drugs and a subsequent in silico screen with the ZINC15 library of more than 9 million drug-like compounds. We discovered that drugs most effective in reducing ACE2 protein levels converge on androgen receptor (AR) signaling inhibition as a common mechanism of action. These drugs were effective in reducing ACE2 and TMPRSS2 levels in both lung epithelial cells and cardiac cells and resulted in reduced infectivity of SARS-CoV-2 in hESC-derived lung organoids.
Clinical case studies have identified male sex as a major risk factor for SARS-CoV-2 complications. In fact, 70% of the patients on ventilators in the ICU were found to be males (Guan et al., 2020). The sex gap is closed in prepubescent patients; children of both sexes are relatively protected from adverse effects of COVID-19 compared to adults (Toubiana et al., 2020; Wu et al., 2020). To explore the possible role of sex hormones on poor disease outcomes in adults and male patients in particular, we conducted a study on two independent cohorts of patients tested for SARS-CoV-2. Among males, we found a significant positive association between prostatic diseases and genetic factors that elevate androgen levels and the risk of COVID-19 susceptibility and severity. Our data provide a potential mechanistic link between clinical observations and pathways involved in COVID-19 pathogenesis. The results identify AR signaling inhibition as a potential therapeutic strategy to reduce SARS-CoV-2 viral entry and mitigate severe manifestations in COVID-19 patients.
Results
High-Throughput Drug Screen Identifies ACE2 Modulators in Human Cardiac Cells
Our analysis of previously published single-cell RNA sequencing datasets (Madissoon et al., 2019; Smillie et al., 2019; Wang et al., 2020) showed abundant expression of SARS-CoV-2 receptor ACE2 and of co-receptors TMPRSS2 and FURIN in adult cardiac cells, lung alveolar and ciliated epithelial cells, and esophageal and colon tissues (Figure S1A). Given the highly significant association of poor outcomes in COVID-19 patients with cardiovascular complications (Guo et al., 2020; Shi et al., 2020) and the significant role of ACE2 in cardiac physiology (Guzik et al., 2020), we chose to initially focus on the regulation of ACE2 levels in cardiac cells. Due to limitations associated with isolation and maintenance of human cells from primary tissue, we used our previously established hESC differentiation method as an alternative strategy to generate cardiac cells (Figure S1B; Tsai et al., 2020). Previously published transcriptomics data on hESC-derived cells generated using this method (Tsai et al., 2020) confirmed the expression of SARS-CoV-2 receptor and co-receptors mRNAs in cardiomyocytes and non-cardiomyocytes (Figure S1C). The differentiated cells also stained positive for ACE2 as assessed by immunofluorescence imaging (Figure S1D).
Searching for modulators of ACE2 levels in hESC-derived cardiac cells, we screened a Selleckchem small molecule library composed of 1,443 FDA-approved drugs (Figure 1 A). ACE2 levels were measured in drug-treated cells using high-throughput imaging and a list of drugs that significantly downregulate or upregulate ACE2 were identified based on their normalized ACE2 expression z-scores (Figure 1B, Table S1). We confirmed the effect of a compound with a high positive z-score (vincristine) and a compound with a low negative z-score (dronedarone) on ACE2 fluorescence intensity (Figure 1C), and subsequently selected several hit compounds with low and high z-scores (Figures S1E and S1F) for further analysis and validation at 1 μM and 2 μM concentrations (Figures 1D and 1E). Interestingly, Vero cells, which are commonly used in COVID-19 drug discovery, did not show any significant changes in ACE2 expression in response to treatment with the hit compounds, highlighting that the underlying regulatory mechanisms are cell-type and species specific (Figures S1G and S1H). The high-quality cell-based measurements and the inherent diversity of the FDA library provided a unique opportunity to develop a virtual high-throughput screening (vHTS) approach that allowed for rapid in silico screening and cost-effective identification of compounds that can elicit the desired biological response. Combined analysis of these in vitro measurements and in silico predictions allows us to nominate molecular entities that can effectively modulate the signaling pathways responsible for ACE2 regulation.
Figure 1.

High-Throughput In Vitro and In Silico Screenings Identify Drugs that Modulate ACE2 Expression in hESC-Derived Cardiomyocytes
(A and B) High-throughput screening of Selleckchem FDA-approved drug library identifies drugs that increase and decrease ACE2 expression in hESC-derived cardiomyocytes.
(C) Representative immunofluorescent images of cells treated with vehicle, vincristine, and dronedarone at 1 μM. Scale bar: 200 μm.
(D and E) Dose response of hits that (D) decreased and (E) increased ACE2 expression in hESC-derived cardiomyocytes culture.
(F) Two-dimensional visualization of molecular features (Morgan fingerprints) for the in vitro and in silico tested compounds using UMAP. For the ZINC15 library, the points are sub-sampled by a factor of 103.
(G) UMAP visualization of the in vitro (labeled) and in silico (unlabeled) hit compounds. Also shown are the K-means cluster memberships based on their Morgan fingerprints.
(H) Dose response analysis of selected in silico hit compounds in hESC-derived cardiac cells. Data are represented as mean ± SEM.
(I) Effect of selected in silico hit compounds on ACE2 expression in human primary alveolar epithelial cells. Data are represented as mean ± SEM.
∗p value < 0.05, ∗∗p value < 0.01, ∗∗∗p value < 0.001. See also Figure S1 and Table S1.
To achieve this goal, we first randomly split our in vitro screening results into three datasets as training, validation, and test inputs (Figures S1I–S1K). We used the model trained on the in vitro screening data to predict changes in ACE2 expression in response to treatments with the in silico library. We then variance normalized the model predictions to determine their associated z-scores. We selected the compounds with a z-scores smaller than −4 as “in silico hit compounds.” To visualize these hits, we used Morgan fingerprints to generate molecular features for all in vitro and in silico tested compounds and visualized their relationship in a 2-dimensional space using Uniform Manifold Approximation and Projection (UMAP) (Figure 1F). We then used K-means clustering to categorize the in silico and in vitro hits and grouped them into 15 coarse-grained clusters based on their molecular features (Figure 1G). We observed a consistent co-clustering of the FDA-approved drugs that are known to have structural similarities, such as the cluster containing finasteride and dutasteride, confirming the utility of these clustering representations. We next tested a subset of in silico hit compounds (Figure S1L) selected from different clusters on hESC-derived cardiomyocytes and human primary alveolar epithelial cells and confirmed their ability to significantly reduce ACE2 expression in both cell types (Figures 1H and 1I). Taken together, this integrative cell-based and in silico screening strategy enabled the identification and nomination of highly efficacious drug-like compounds with similar structures to the FDA-approved drugs utilized in the in vitro screen.
Drugs that Reduce ACE-2 Regulate Steroid Signaling and Peptidase Activities
We explored whether a potential shared pathway exists among the candidates that decrease ACE2 levels on the membrane of cardiac cells. We acquired isometric simplified molecular-input line-entry system (SMILES) for each drug in the FDA-approved library from Selleckchem and used them to predict drug-protein interactions via the similarity ensemble approach (SEA) computational tool (Keiser et al., 2007). The SEA-predicted drug-protein pairs were filtered, selecting human proteins and predicted interaction p values < 0.05, which yielded 2,150 predicted proteins targeted by the drug library.
Weighted combined z-scores were then calculated by adding normalized z-scores across all compounds that target each target protein (Zaykin, 2011). The p values were then calculated based on the combined z-scores and adjusted using p.adjust (method = false discovery rate [FDR]). As an orthogonal approach, for each protein, we recorded the number of treatments with negative normalized z-scores as well as the total number of compounds predicted to target that protein. Using the sum of counts for all other targets and drugs, we performed a Fisher’s exact test to evaluate the degree to which negative z-scores were enriched among the drugs likely to target a protein of interest. As expected, the two p values, i.e., combined z-score and Fisher’s, were generally correlated (R = 0.6, p < 1e−200). In Figure 2 A, we have specifically compared these p values across the genes with negative average z-scores. Finally, we selected the likely target genes using the following criteria: average z-score < 0, FDR < 0.25 based on combined z-score analysis, and Fisher’s p < 0.05. This selection process resulted in 30 proteins nominated as significant drug targets (Figures 2B and 2C, Table S2).
Figure 2.

Target Prediction Analysis Identified Shared Pathways among ACE2 Regulators
(A) We employed two independent tests for identifying the genes that are most likely targeted by the effective treatments: (1) a combined z-score approach, where normalized z-scores from all the treatments associated with a gene are integrated, and (2) a Fisher’s exact test to assess the enrichment of a gene among those that are targets of treatments with negative z-scores. Here we have shown the correlation between the p values reported by these two independent approaches.
(B) A one-sided volcano plot showing the average z-score versus −log of p value for all genes with negative z-scores. The genes that pass our statistical thresholds are marked in gold (combined z-score FDR < 0.25 and Fisher’s p value < 0.05).
(C) The identified target genes along with their combined z-score, associated p value, and FDRs. Also included are the total number of compounds each gene is likely targeted by, and the number of those that result in lower ACE2 expression (z-score < 0).
(D) Gene-set enrichment analysis using iPAGE for the target genes identified from FDA-approved library with negative z-score. Genes were ordered based on their combined z-score from left to right and divided into nine equally populated bins. The enrichment and depletion pattern of various gene-sets is then assessed across this spectrum using mutual information. Red boxes show enrichment and blue boxes show depletion.
(E) Gene-set enrichment analysis for the in silico hits. Similar to (D), genes were grouped into those that are likely targeted by the identified compounds and those that are not (i.e., background). We then assessed the enrichment of each pre-compiled gene-set among the targets using iPAGE.
The previously published bulk transcriptomics data (Tsai et al., 2020) confirmed that 28 of these 30 proteins were expressed by the in vitro generated cardiomyocytes and/or non-cardiomyocytes supporting their potential as drug targets (Figure S2A). Analysis of the collective expression of all 30 predicted targets using module scoring revealed that some ACE2-expressing cell types, such as cardiac fibroblasts and ciliated cells in the lung, express high levels of multiple predicted drug targets (Figure S2B). Additionally, expression of the predicted targets was detected in ACE2-expressing cell types of the adult human heart, and the majority were also detected in ACE2-expressing cell types of other commonly affected organs (Figure S2C). This analysis also provides insight into non-ACE2-expressing cell types that may be affected by treatment with the hit compounds. In particular, tissue-resident immune populations of the myeloid lineage in the heart, esophagus, and lung also collectively express many of the predicted target genes (Figure S2C). This is particularly interesting given the recent reports that myeloid lineage and epithelial cells are affected in severe SARS-CoV-2 infection cases (Bost et al., 2020).
To identify biological pathways associated with changes in ACE2 expression, we used the combined z-scores across all proteins using iPAGE gene ontology (GO) analysis (Figure S2D). Interestingly, target proteins associated with compounds that reduce ACE2 expression were associated with various GO terms related to peptidase activity and steroid metabolic processes (Figure 2D). These pathways were then validated with the SEA-predicted protein targets from the in silico library screen. Interestingly, hit compounds identified in the in silico screen also showed enrichment for targets involved in steroid hormone activity, steroid metabolic processes, and peptidase activity (Figure 2E). Given the strong clinical evidence on COVID-19 disproportionately affecting men, the well-established role of peptidase in modulating ACE2 and SARS-Co-V-2 co-receptors, and the possible link between ACE2 expression and sex hormones, we sought to uncover the specific drug-protein interactions driving enrichment of steroid and peptidase pathways. We first mapped the drug-protein interactions for the full list of predicted targets and compounds with z-score < −1.5 (Figure 3 A). This revealed a list of 48 compounds with significant interactions with target proteins (Figure S3A). Next, we created drug-protein matrices on proteins in “steroid metabolic process” and “serine-type peptidase” activity GO terms to map the interaction of drugs with corresponding targets (Figures S3B and S3C). This analysis highlighted the interaction of several drugs including ketoconazole, spironolactone, finasteride, and dutasteride with androgen-signaling modulators such as SRD5A1 and SHBG (Figures 3A, S3B, and S3C), whereas other drugs from the in vitro screen, such as camostat mesilate, sotagliflozin, and guanabenz acetate, were specific to the serine-type peptidase activity pathway (Figure S3C).
Figure 3.

Androgen Signaling Regulates Peptidase Expression
(A) The drug-gene interaction matrix for the 30 significantly enriched drug target genes from Figure 2C that are deemed functional in their respective analyses. Shading represents the significance of the predicted interaction.
(B) STRING protein-protein interaction network was used to identify interactions between our list of significantly enriched genes from Figure 2C (depicted as significantly predicted targets and yellow circles), androgen signaling pathway components (AR and SRD5A2), and proteins implicated in ACE2 regulation (ACE, ADAM10, ADAM17, FURIN, REN, TMPRSS2). Minimum required interaction score was set to 0.7 corresponding to high confidence and edge thickness indicates the degree of data support.
(C) SEA predicted drug-protein target interactions (blue lines and boxes) in the androgen signaling pathway. Yellow ovals represent significantly enriched genes from Figure 2C. Dashed lines represent MaxTC < 1.
(D) The expression of ACE2-related peptidases is regulated by AR and other transcription factors that are targets of our candidate drugs. MaxTC, maximum tanimoto similarity between compounds from ref_target to compounds from query_target in [0,1] with 1 being identical up to the resolution of the fingerprint.
(E and F) Dose response analysis of the effects of antiandrogenic drug candidates on ACE2 expression in cardiac cells generated from hECS lines WA09 (E) and WA01 (F). Data are represented as mean ± SEM.
(G) Representative images of immunofluorescence staining for ACE2 and TMPRSS2 in hESC-derived cardiomyocytes treated with antiandrogenic drugs. Scale bar = 50 μm.
∗p value < 0.05, ∗∗p value < 0.01, ∗∗∗p value < 0.001. See also Figure S3 and Table S3.
Androgen Receptor Signaling Modulates SARS-CoV-2 Receptors
To build a working model of the drug-protein interactions in ACE2 regulation, we conducted a protein-protein interaction (PPI) network analysis to identify interactions between our list of significant predicted targets (Figure 2C), androgen signaling pathway components (AR and SRD5A2), and proteins implicated in ACE2 function regulation (ACE, ADAM10, ADAM17, FURIN, REN, TMPRSS2). We used the STRING physical interaction database to draw a high confidence network of associating proteins (Figure 3B). In the resulting network, AR and IL6 had the highest degree centrality, connecting to seven other nodes in the network. Furthermore, AR and IL6 share high betweenness centrality, connecting the androgen pathway module to the peptidases. The observed link between AR and IL6 is clinically important given the elevated IL6 response in severe cases of COVID-19 infection (Liu et al., 2020).
The remarkable convergence of the gene set enrichment analysis and the PPI network on steroid hormone-related genes and pathways prompted us to hypothesize that the drug candidates may be reducing ACE2 expression via inhibition of AR signaling and peptidase pathways (Figures 3C and 3D). Seven of the predicted drug targets are upstream regulators of AR signaling and are targeted by multiple drug candidates that reduce ACE2 (Figure 3C). Furthermore, peptidases such as FURIN and TMPRSS2, which are important players in ACE2 regulation and thereby SARS-CoV-2 viral entry (Hasan et al., 2020; Heurich et al., 2014), are among the downstream targets of AR. These receptors and their upstream regulators are also predicted to be targeted by the drug candidates (Figure 3D).
Although most highly expressed in male reproductive organs, AR mediates hormone signaling in many male and female tissues (Matsumoto et al., 2013). In agreement, expression of AR- and testosterone-converting enzymes SRD5A1 and SRD5A2 are detected in ACE2-expressing cell types in the adult heart, lung, esophagus, and colon (Figures S3D–S3G). Additionally, collective expression of genes involved in AR signaling (GO:0030521), common receptors upstream of AR activation (Azevedo et al., 2011; Cao and Kyprianou, 2015; Girling et al., 2007; Wang et al., 2020), and common gene targets of AR transcription factor activity (Jin et al., 2013), reveals potential organ and cell-type-specific differences in AR signaling regulation (Figures S3D–S3G). A list of genes included in each module is provided in Table S3.
To evaluate the effect of AR inhibition on ACE2 levels, we treated cardiac cells derived from two different hESC lines (WA09 and WA01) with the drug candidates that are known to inhibit AR signaling and observed significant reductions of ACE2 in a dose-dependent manner (Figures 3E–3G). Finasteride and dutasteride reduce AR signaling by inhibiting 5 alpha reductases, which are the enzymes that convert testosterone to an AR ligand and agonist, 5a-dihydrotestosterone (DHT). Treatment with these drugs resulted in a significant decrease in TMPRSS2 in addition to ACE2 (Figures S4A and S4B).
To determine whether AR signaling regulates the expression of SARS-CoV-2 receptors directly, we used an existing AR ChIP-seq dataset generated in LNCaP cells (Tran et al., 2020) to identify direct transcriptional targets. The genes with AR binding to this 5 kb downstream or upstream of transcription start sites (TSS) were selected as direct AR-bound targets. We next used a transcriptomics dataset generated using RNAi-mediated knockdown of AR (Zhang et al., 2020) and compared gene expression changes in response to AR knockdown. We divided log-fold expression changes into nine equally populated bins, which were also shown along with the patterns of AR enrichment and depletion at the corresponding TSS across the data (Figure 4 A). This analysis identified ACE2 and other SARS-CoV-2 co-receptors (TMPRSS2 and FURIN) as direct transcriptional targets that are downregulated in response to AR knockdown (Figure 4A).
Figure 4.

Androgen Receptor Signaling Modulates ACE2 and TMPRSS2 Levels in Heart and Lung Cells
(A) Volcano plot visualizing gene expression changes in response to AR knockdown in LNCaP cells. Genes of interest are labeled and shown in red. Also shown are the enrichment and depletion pattern of AR target genes (i.e., genes with promoter AR binding) as a heatmap along with the mutual information value and its associated z-score. The log-fold change values were divided into equally populated bins and the enrichment of AR-bound genes in each bin was assessed using hypergeometric p values and colored accordingly (gold for enrichment and blue for depletion; red and blue borders mark bins that are statistically significant.
(B and C) Effect of CRISPR-Cas9 ribonucleoproteins containing AR and SRD5A2 sgRNAs on ACE2 expression in hESC-derived cardiomyocyes and human primary alveolar epithelial cells. Dots represent fluorescence intensity values in individual cells.
(D–G) Differential effect of dutasteride (potent inhibitor of testosterone to DHT conversion) and DHT on the membrane ACE2 levels and spike-RBD protein entry to cardiomyocytes (D and E) and alveolar epithelial cells (F and G) with their corresponding immunofluorescence images.
(H) Immunofluorescence staining of bronchial epithelial cells isolated from human lung tissue for epithelial marker ECAD and ciliated cell marker TUBA.
(I and J) Effect of antiandrogenic drug candidates on ACE2 expression in human primary bronchial epithelial cells isolated from three independent donors. Individual values represent normalized fluorescence intensity in independent imaging fields across different Transwell inserts.
Scale bar = 100 μm in (E), (G), and (H). ∗p value < 0.05, ∗∗p value < 0.01, ∗∗∗p value < 0.001. Data are represented as mean ± SEM. See also Figure S4.
To further interrogate the role of AR signaling in regulation of ACE2, we used CRISPR-Cas9 ribonucleoproteins (RNPs) delivery to knock out AR and SRD5A2 in hESC-derived cardiac cells and human primary alveolar epithelial cells. Treatment with AR and SRD5A2 RNPs resulted in significant reductions in ACE2 levels in both cell types (Figures 4B and 4C). To perform the gain-of-function experiment, we treated the hESC-derived cardiac cells and human primary alveolar epithelial cells with DHT. Remarkably, DHT treatment significantly increased ACE2 levels and internalization of recombinant spike-RBD protein in both cell types, while the 5 alpha reductase inhibitor dutasteride had the opposite effect (Figures 4D–4G). Furthermore, finasteride, which is another well-known 5 alpha reductase inhibitor, was able to significantly reduce the internalization of SARS-CoV-2 pseudotyped virus in human primary alveolar epithelial cells (Figure S4C).
We next evaluated the ability of our candidate antiandrogenic drugs in reducing SARS-CoV-2 receptors in bronchial epithelial cells. Human bronchial epithelial cell cultures were generated from donated tissue samples from lung transplant recipients as described previously (Bonser et al., 2020; Fulcher et al., 2005) and are enriched in ciliated cells that express ECAD and TUBA (Figure 4H). Ciliated cells belong to another COVID-19-relevant subtype of lung epithelial cells that shows high levels of ACE2 expression (Figure S1A). Treatment with the 5 alpha reductase inhibitors, finasteride and dutasteride, resulted in significant reductions in ACE2 and TMPRSS2 in bronchial epithelial cells from two out of three donors (Figures 4I, 4J, and S4D–S4F). Together, these results indicate that AR signaling regulates the expression of SARS-CoV-2 receptors in COVID-19 target tissues.
Inhibition of Androgen Receptor Signaling Protects hESC-Derived Human Lung Organoids against SARS-CoV-2 Infection
Our results indicate that antiandrogenic drugs can lower the levels of SARS-CoV-2 receptors in target cell types. To build an in vitro model of viral infection in human lung tissue, we set out to generate lung organoids from hESC using a slightly modified combination of previously established differentiation methods (Figure S5A) (de Carvalho et al., 2019; Jacob et al., 2017; Miller et al., 2019). These human lung organoids (HLOs) expressed key lineage markers of alveolar epithelial precursors such as SOX9, NKX2.1, and ECAD, and high levels of ACE2 (Figure 5 A). Bulk RNA sequencing of differentiating cells at various time point showed the upregulation of subtype-specific transcript panels confirming the emergence of different lung epithelial lineages in HLOs (Figure 5B). Single-cell RNA sequencing of fully differentiated organoids demonstrated that they were enriched in lung epithelial cells (Figures S5B and S5C) that contain a diverse array of subtypes (Figures 5C, 5D, and S5D). The most abundant subtype was the alveolar type 2 cells which are considered to be the primary targets of SARS-CoV-2. The bulk and single-cell RNA sequencing data confirmed the expression of SARS-CoV-2 receptors ACE2 and TMPRSS2 and androgen signaling genes AR, SRD5A1, and SRD5A2 in the differentiated HLOs (Figures 5B and 5E).
Figure 5.

Antiandrogenic Drugs Reduce ACE2 and SARS-CoV-2 Infection in hESC-Derived Lung Organoids
(A) Immunofluorescence staining of hESC-derived lung organoids (HLOs) for epithelial lineage markers SOX9, NKX2.1, and ECAD and SARS-CoV-2 receptor ACE2.
(B) Expression analysis of lineage-specific markers during HLO differentiation measured using bulk RNA sequencing at days 0, 5, 9, 15, 25, 35, and 50.
(C) UMAP visualization of single-cell RNA sequencing data showing different epithelial cell clusters in differentiated HLOs.
(D) Dot plot visualization of single-cell expression of epithelial subtype-specific lineage markers in different cell clusters.
(E) Violin plot visualization of single-cell expression of SARS-CoV-2 receptors and androgen signaling genes in different lung epithelial cell clusters.
(F) Effect of antiandrogenic drugs on ACE2 levels in HLOs.
(G and H) Quantification (G) and representative images of immunofluorescence staining (H) of SARS-CoV-2 N-protein in HLOs treated with antiandrogenic drugs prior to infection with SARS-CoV-2 isolate USA/CA-UCSF-0001C/2020.
(I) Plaque assay quantification of viral titers in supernatants of infected HLOs treated with antiandrogenic drugs.
(J and K) Quantification (J) and representative images of immunofluorescence staining (K) of viral double-strand RNA (dsRNA) and SARS-CoV-2 N-protein in infected HLOs treated with dutasteride prior to infection with SARS-CoV-2 isolate USA-WA1/2020, NR-52281.
Scale bar = 100 μm in (A) and 50 μm in (H) and (K). ∗p value < 0.05, ∗∗p value < 0.01, ∗∗∗p value < 0.001. Data are represented as mean ± SEM. See also Figure S5.
We next used these hESC-derived HLOs to test the ability of our drug candidates in reducing SARS-CoV-2 infection. Similar to primary lung epithelial cells, treatment with the antiandrogenic drugs led to a significant reduction in ACE2 levels in HLOs (Figures 5F, S5E, and S5F). To determine whether drug treatment can protect the HLOs against SARS-CoV-2 infection, we subjected the HLOs to the virus and quantified the number of infected cells based on the detection of double-strand RNA (dsRNA) or SARS-CoV-2 N-protein in the cytoplasm. Remarkably, drug-treated HLOs showed a dramatic reduction in the number of N-protein+ infected cells compared to untreated controls (Figures 5G and 5H). Plaque assay of the supernatants showed a substantial reduction in viral titers produced by drug-treated HLOs (Figure 5I). To assess reproducibility, we tested an independent SARS-CoV-2 isolate on dutastetride-treated HLOs and observed a similar reduction in the number of cells that contain viral dsRNA or N-protein. These results provide crucial evidence for the efficacy of antiandrogenic drugs in attenuating SARS-CoV-2 infections in the target cells.
Androgen Imbalance States Are Associated with COVID-19 Complications in Male Patients
Our results suggest that androgen can increase viral receptor and co-receptor expression which could lead to increased SARS-CoV-2 infectivity in target tissues. To determine whether androgen plays a role in COVID-19 disease manifestation, we explored the effect of disorders related to androgen imbalance on COVID-19-induced cardiac injury, measured by elevated troponin T levels (Figure 6 A). We also included the previously described risk factors associated with organ failure (Goyal et al., 2020) such as age, BMI, diabetes, and hypertension in our data collection. In the de-identified aggregate data from Yale New Haven Hospital, 1,577 individuals tested positive for COVID-19 and had serum troponin T measured during the same encounter. There was a larger number of males with abnormal serum troponin T levels in both selected age groups (Figure S6A). Association analysis in the COVID-19 patients showed that most risk factors were correlated with abnormal levels of serum troponin T, but the risk factors were also correlated with each other (Figure S6B). To account for associations between individual risk factors, we tested multiple multi-variate models (as described in the STAR Methods section). In our final model, prostatic diseases (hyperplasia of prostate or neoplasm of prostate) increased the odds of having abnormal troponin T by 50.5% (OR = 1.505, 95% CI, p value 0.046), independent of the other risk factors (Figure 6B).
Figure 6.

Effects of Androgen Signaling on Outcomes Associated with COVID-19
(A) Schematic representation of the patients’ outcome analysis with COVID-19 at Yale New Haven Hospital.
(B) The effects of BMI, prostatic disease, hypertension, and diabetes on the odds of having abnormal troponin T in male patients with COVID-19 in Yale patients. Troponin T and BMI were dichotomized during data collection. BMI, <30 versus ≥30; troponin T, normal (<0.01 ng/mL) versus abnormal (≥0.01 ng/mL). For the primary outcome, the odds ratio were calculated for the pre-specified subgroups.
(C) Schematic representation of the outcome studied in the UK Biobank (UKBB) cohort.
(D) Association of BPH with COVID-19 hospitalization, in multivariate logistic models adjusted for age, hypertension, type 2 diabetes, normalized BMI, Townsend deprivation index, and principal components of genetic ancestry.
(E) Gene set enrichment analysis of androgen signaling genes on COVID-19 hospitalization using the COVID-19 host genetics initiative GWAS results.
(F) Mendelian randomization between bioavailable testosterone and COVID-19 hospitalization. MR-Egger (visualized through the blue fitted line) was performed between 6 independent variants near the androgen signaling genes from the drug screen that are genome-wide significantly associated with bioavailable testosterone and their respective associations from the COVID-19 host genetics initiative release GWAS. OR = odds ratio, CI = confidence interval.
To further explore the association between androgen and COVID-19 disease severity, we analyzed an independent cohort of patient records in the UK Biobank (UKBB) (Figure 6C). A total of 190,150 men in the UKBB passed quality-control criteria and were from the UKBB English recruitment centers with COVID-19 tests reported back to UKBB. Among these individuals, median age at enrolment was 57 (SD 8) years and mean BMI was 27.8 (SD 4.2), 19,794 (10.4%) had type 2 diabetes mellitus and 83,510 (43.9%) had hypertension. A total of 8,146 men were tested for COVID-19, of which 831 (10.2%) had at least one positive COVID-19 test. Of all the men who tested positive for COVID-19, 534 individuals (64.3%) were hospitalized (Figure S6C).
Our analysis showed that benign prostatic hypertrophy (BPH) was independently associated with both COVID-19 susceptibility (OR 1.4, 95% CI 1.2–1.8, p = 0.00087) and COVID-19 hospitalization (OR 1.6, 95% CI 1.2–2.1, p = 0.00022) in multivariate models adjusted for age, hypertension, type 2 diabetes, BMI, Townsend deprivation index, and principal components 1–10 of genetic ancestry (Figures 6D and S6D). In particular, only 12.0% of controls also had BPH while 17.9% of COVID-19-positive men and 21.2% of COVID-19 hospitalized men had BPH (Figure S6E).
To identify potential associations between androgen signaling genes (Table S3) and the 8 androgen signaling genes identified from our drug screen (Figure S6F) with COVID-19 severity, we performed gene set enrichment analysis and identified a significant enrichment of both gene sets in the hospitalized patients versus population in the COVID-19 host genetics initiative (Figure S6G).
To investigate the relationship between androgen signaling and disease severity, we used Mendelian randomization which leverages randomization of genetic variants during meiosis at conception, using genetic instruments for an exposure to mitigate risks for confounding, thereby facilitating more robust causal inference (Davies et al., 2018). Here, we performed two-sample Mendelian randomization between variants within 1 MB of the 8 androgen signaling genes that were genome-wide significantly associated with bioavailable testosterone (Ruth et al., 2020) and the GWAS summary statistics for those same genes in the COVID-19 host genetics initiative. Significant robust MR-Egger association was identified across 6 genetic variants (COVID-19 hospitalization OR 5.2 per SD sex-specific bioavailable testosterone, 95% CI: 2.48–10.97, p = 1.68 × 10−5), with significant intercept term suggesting pleiotropy also present (p = 7.5 × 10−5) (Figures 6E and S6H). The top genetic variant in the 2-sample Mendelian randomization was rs545206972, a non-coding variant located at 25 kb downstream of SHBG which was strongly associated with bioavailable testosterone (beta 0.43 SD, p = 1.8 × 10−265; COVID-19 hospitalization OR = 1.62, p = 0.095). In further sensitivity analysis, removing this top variant still maintained significant Robust MR-Egger association (p = 0.016), with a significant intercept term suggestive of significant pleiotropy (p = 0.009) (Figure S6I). These observations provide strong clinical evidence for the role of androgen signaling in COVID-19 susceptibility and severity.
Discussion
There are two important observations in the COVID-19 pandemic: the higher prevalence of severe complications in male individuals and the relative immunity in children. Our study identifies a link between male sex hormone signaling and regulation of the SARS-CoV-2 receptor ACE2 and co-receptor TMPRSS2, providing a potential explanation for these observations. Our results demonstrate that inhibitors of 5 alpha reductases, which dampen androgen signaling, can reduce ACE2 levels in the target cells and thereby decrease SARS-CoV-2 infectivity. These drugs, commonly prescribed for prostatic disorders, have good safety profiles and show repurposing potential for the treatment of COVID-19.
The most lethal complication of COVID-19 is multi-organ failure affecting the lungs, the kidneys, and the heart. Although there has been tremendous effort toward understanding the biology of SARS-CoV-2 infection at the molecular level, the use of relevant experimental models has been limited. Modeling the infection in commonly used cell lines that do not adequately recapitulate human pathophysiology could potentially delay the identification of effective therapeutic targets. Taking advantage of directed hESC differentiation, we generated scalable cultures of disease-relevant human cardiac cells and lung organoids and performed high-throughput screening to identify drugs that regulate ACE2 expression in these cell types. These experiments underscore the potential of hESC-derived cells for in-depth investigation of cell-type-specific processes and offer a framework for rapid identification and validation of therapeutically relevant compounds. Other reports on the use of hESC-derived cell or organoid models highlight the utility of hESC-derived cells in modeling SARS-CoV-2 infection and COVID-19 pathophysiology (Han et al., 2020).
Results from our in vitro high-throughput screening of the FDA-approved drugs enabled us to develop a deep learning strategy to screen millions of drug-like compounds in silico and identify candidates predicted to show superior potency and efficacy. The diverse pharmacokinetic properties in these candidates may enable the development of drugs with improved distribution to disease-relevant tissues. Further experiments are needed to validate these compounds and characterize their pharmacokinetic and pharmacodynamic properties in vitro and in vivo.
A common characteristic among our validated hit compounds is their ability to target androgen signaling. Analysis of disease outcomes in COVID-19 patients in two independent cohorts revealed a significant association between elevated free androgen and COVID-19 complications, pointing to a possible link between androgen-mediated ACE2 regulation and disease severity. Pathway and gene target analysis on compounds that reduce ACE2 levels also highlighted the regulatory roles of peptidase pathways. Interestingly, protein interaction maps suggested a possible crosstalk between AR signaling pathways, inflammatory markers, and peptidases relevant to the viral receptor and co-receptors, offering insights into alternative pathways involved in ACE2 regulation.
Our FDA drug screen data revealed that many commonly used medications modulate ACE2 levels and could affect disease severity in COVID-19 patients. Further studies evaluating the relationships between these drugs and disease outcomes will be necessary to assess potential clinical impact and the need to substitute medications that might pose a heightened risk for COVID-19 patients.
In conclusion, our results provide key insights into ACE2 regulatory mechanisms, present strong molecular and clinical evidence for the role of androgen signaling in SARS-CoV-2 infection, and identify therapeutic candidates for the treatment of COVID-19.
Limitations of the Study
There are a number of challenges and limitations in this study. The performance of our deep learning model used for the in silico screen could be improved with additional in vitro datasets containing larger number of experimentally tested compounds. Genetic validation of AR signaling components could be further validated using alternative CRISPR-Cas9 strategies with higher efficiency of RNP delivery and gene editing in target cell types. Finally, we were not able to perform observational studies to determine the effect of antiandrogenic drugs on mitigating COVID-19 outcomes due to limitations in power for such analyses even in large biobanks. Future investigations will be necessary to define the effects of androgen signaling and its inhibition on COVID-19 severity both in men and women.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC Mouse IgG1, κ isotype control (FC) antibody | Biolegend | 400121 |
| APC Mouse IgG2a, kappa isotype control antibody | Biolegend | 400219 |
| donkey α-goat AF488 | Invitrogen | A11055 |
| donkey α-goat AF568 | Invitrogen | A11057 |
| donkey α-goat AF647 | Invitrogen | A21447 |
| donkey α-mouse AF488 | Invitrogen | A21202 |
| donkey α-mouse AF568 | Invitrogen | A10037 |
| donkey α-mouse AF647 | Invitrogen | A31571 |
| donkey α-rabbit AF488 | Invitrogen | A11008 |
| donkey α-rabbit AF568 | Invitrogen | A10042 |
| donkey α-rabbit AF647 | Invitrogen | A31573 |
| goat α-chicken AF488 | Invitrogen | A11039 |
| Mouse monoclonal anti-CD324(E-CAD) | BD Biosciences | 610181 |
| Mouse monoclonal anti-acetylated Tubulin (TUBA) | Sigma | T7451 |
| Mouse monoclonal anti-ACE2 | Proteintech | 66699-1-AP |
| Mouse monoclonal anti-dsRNA | Absolute Antibodies | Ab01299-2.0 |
| Mouse monoclonal anti-SARS/SARS-CoV-2 Nucleocapsid | Invitrogen | MA1-7404 |
| Rabbit monoclonal anti-Sox9 | Cell Signaling Technologies | 82630T |
| Mouse monoclonal APC anti-human CD184 (CXCR4) | Biolegend | 306509 |
| Mouse Monoclonal PE anti-human CD117 (c-kit) | Biolegend | 313203 |
| PE Mouse IgG1, kappa isotype control antibody | Biolegend | 400111 |
| PE Mouse IgG2a, κ isotype control antibody | Biolegend | 400211 |
| Goat polyclonal anti-ACE2 | R&D Systems | AF933 |
| Rabbit polyclonal anti-ACE2 | Proteintech | #21115-1-AP |
| Chicken polyclonal anti-GFP | abcam | ab13970 |
| Rabbit polyclonal anti-NKX2.1 | Seven Hills Bioreagents | WMAB-1231 |
| Goat polyclonal anti-TMPRSS2 | Novus Biologicals | #NBP1-20984 |
| Bacterial and Virus Strains | ||
| SARS-CoV-2/human/USA/CA-UCSF-0001C/2020 | UCSF Clinical Specimen | N/A |
| USA-WA1/2020, NR-52281 isolate | BEI Resources | N/A |
| G-complemented G∗ΔG-GFP rVSV | Kerafast | EH1024-PM |
| Biological Samples | ||
| Donated lung tissue from transplant recipients | Laboratory of David J. Erle | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 8-Br-cAMP | Sigma-Aldrich Inc | B5386 |
| Accutase | Stemcell Tech | 7922 |
| Activin A | R&D Systems | 338AC010 |
| All-trans retinoic acid | Stemcell Tech | 72264 |
| Ascorbic acid | Sigma-Aldrich Inc | A4034 |
| BMP4 | Stemcell Tech | 78211.2 |
| Camostat mesilate | Selleck Chem (FDA-Approved Library) | N/A |
| cAMP | Sigma-Aldrich Inc | D0627 |
| Cas9-NLS purified protein | QB3 MacroLab | N/A |
| CHIR99021 | Stem-RD | CHIR |
| Collagen, Type IV, Human Placenta | Sigma-Aldrich Inc | 234154 |
| DAPI | Thermo Fisher Scientific | D1306 |
| Dexamethasone | Sigma-Aldrich Inc | D4902 |
| DHT | Sigma-Aldrich Inc | D-073 |
| Dispase | Corning | 354235 |
| Dutasteride | Selleck Chemicals LLC | S1202 |
| 0.5M EDTA, pH 8.0 | Thermo Fisher Scientific | 15575020 |
| FGF10 | Stemcell Tech | 78037.2 |
| FGF4 | Stemcell Tech | 78103.2 |
| FGF7 | Stemcell Tech | 78046.1 |
| Fibronectin | Corning | DLW354008 |
| Finasteride | Selleck Chemicals LLC | S1197 |
| Guanabenz acetate | Selleck Chem (FDA-Approved Library) | N/A |
| IBMX | Sigma-Aldrich Inc | I5879 |
| Ketoconazole | Selleck Chemicals LLC | S1353 |
| Laminin | Corning | 354232 |
| LDN193189 | Stemgent | #04-0074-02 |
| Library of FDa-approved drugs (1443 compounds) | Selleck Chem | L1300 |
| Monothioglycerol | Sigma-Aldrich Inc | M6145 |
| Paraformaldehyde | Thermo Fisher Scientific | AAJ19943K2 |
| Paraformaldehyde, 4% solution | SCBT | sc-281692 |
| Poly-L-ornithine hydrobromide | Sigma | P3655 |
| SAG | Stemcell Tech | 73412 |
| SARS-CoV-2 (2019-nCoV) Spike RBD-Fc Recombinant Protein | Sino Biological | 40592-V02H |
| SB431542 | PeproTech | 3014193 |
| Sotagliflozin | Selleck Chem (FDA-Approved Library) | N/A |
| 0.05% Trypsin/0.53mM EDTA | Corning | 25-051-Cl |
| XAV939 | R&D | 3746 |
| Critical Commercial Assays | ||
| PowerUp™ SYBR™ Green Master Mix | Thermo Fisher Scientific | A25743 |
| Cell Staining Buffer | Biolegend | 420201 |
| Chromium Next GEM Single Cell 3¢ Kit v3.1 | 10X Genomics | 1000121 |
| eBioscience™ Permeabilization Buffer (10X) | Invitrogen | 00-8333-56 |
| Gene Knockout Kit v2 - AR | Synthego | N/A |
| Gene Knockout Kit v2 - SRD5A1 | Synthego | N/A |
| Lipofectamine 3000 Transfection Kit (0.1 mL) | Fisher Scientific | L3000001 |
| Lipofectamine Cas9 Plus Reagent (ThermoFisher Scientific, USA) | Fisher Scientific | CMAX00008 |
| Lipofectamine CRISPRMAX transfection reagent | Fisher Scientific | CMAX00008 |
| P3 Primary Cell 96-Well Kit | Lonza | V4SP-3096 |
| QuantSeq FWD Kit (with UMI module) | Lexogen | #015 |
| Quick-RNA 96 Kit (Zymo) | Zymo Research | R1052 |
| RNA/DNA/Protein Purification Plus Kit (Norgen Biotek, Thorold, ON, Canada) | Norgen Biotek | 47700 |
| SuperScript III First-Strand Synthesis System | ThermoFisher Scientific | 18080051 |
| TotalSeq HTO antibodies | Biolegend | #394601-394629 |
| Deposited Data | ||
| Deidentified aggregate patient data from Yale New Haven Hospital | available upon request | N/A |
| Bulk and single cell RNA sequencing data on hESC-derived lung organoids | GEO | GEO: GSE161264 |
| UK Biobank patient data | available upon request | N/A |
| Experimental Models: Cell Lines | ||
| Human embryonic stem cells, H1 | WiCell | WA01 |
| Human embryonic stem cells, H9 | WiCell | WA09 |
| Human Pulmonary Alveolar Epithelial Cells | Sciencell | #3200 |
| Calu-3 (ATCC® HTB-55™) | ATCC | ATCC® HTB-55™ |
| VERO C1008 [Vero 76, clone E6, Vero E6] | ATCC | CRL-1586 |
| Recombinant DNA | ||
| Plasmid: PLVX H2B Cherry | Laboratory of Deepak Lamba | N/A |
| Mammalian expression plasmid for SARS-CoV-2 surface glycoprotein (Spike) for VSV pseudotyping (Ginkgo Bioworks) | addgene | pGBW-m4137382 |
| Software and Algorithms | ||
| GE Developer Toolbox v1.9.1 | GE Healthcare | https://www.gehealthcare.com/ |
| ImageJ (FIJI) | NIH | https://imagej.net/Fiji |
| iPAGE gene ontology analysis | Goodarzi et al. (2009) | https://tavazoielab.c2b2.columbia.edu/iPAGE/ |
| R package: cutadapt v2.10 | CRAN package repository | https://cran.r-project.org/web/packages/ |
| R package: DESeq2 v1.30.0 | CRAN package repository | https://cran.r-project.org/web/packages/ |
| R package: DropletUtils v1.10.0 | CRAN package repository | https://cran.r-project.org/web/packages/ |
| R package: HTSeq v0.12.4 | CRAN package repository | https://cran.r-project.org/web/packages/ |
| R package: kb-python v0.24.4 | CRAN package repository | https://cran.r-project.org/web/packages/ |
| R package: Seurat v3.2.2 | CRAN package repository | https://cran.r-project.org/web/packages/ |
| R package: STAR v2.7.6a | CRAN package repository | https://cran.r-project.org/web/packages/ |
| R package: umi_tools v1.1.0 | CRAN package repository | https://cran.r-project.org/web/packages/ |
| R v3.6.3 | The R Project for Statistical Computing | https://www.r-project.org/ |
| SEA computational tool | Keiser et al., 2007 | http://sea.bkslab.org/ |
| STRING v11 protein-protein interaction (PPI) network analysis | Szklarczyk et al. Nucleic acids research 47.D1 (2018): D607-D613.2 | https://string-db.org/ |
| Other | ||
| AR ChIP-seq dataset generated in LNCaP cells | Tran et al., 2020 | N/A |
| Previously published single cell RNA sequencing datasets | Madissoon et al., 2019; Smillie et al., 2019; Wang et al., 2020 | N/A |
| Previously published transcriptomics data on hESC-derived cells | Tsai et al., 2020 | N/A |
| SMILES for each drug in FDA-approved library | Selleckchem | https://www.selleckchem.com/screening/fda-approved-drug-library.html |
| ZINC15 library | Sterling and Irwin, 2015 | https://zinc15.docking.org/ |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Faranak Fattahi (Faranak.Fattahi@ucsf.edu).
Materials Availability
DNA constructs and other research reagents generated by the authors will be distributed upon request to other researchers.
Data and Code Availability
Original source data for previously published scRNA-seq of human organ tissues used in this study are available by: heart data from Wang et al. (2020) from the Gene Expression Omnibus (GEO) at GEO: GSE109816, lung and esophagus data from Madissoon et al. (2019) at https://www.tissuestabilitycellatlas.org, colon data from Smillie et al. (2019) at https://singlecell.broadinstitute.org/single_cell/study/SCP259/intra-and-inter-cellular-rewiring-of-the-human-colon-during-ulcerative-colitis.
Original source data for the AR ChIP-seq experiment is available from GEO at GEO: GSM3148987.
Original source data for AR knockdown RNA-seq are available from GEO under accession code GEO: GSE114052, GEO: GSE128515, and GEO: GSE139962.
Training datasets and code used for in silico drug screening are available upon request to other researchers.
Code used for SEA drug target prediction is available upon request to other researchers.
The raw dataset from bulk and scRNA-seq of hESC-derived lung organoid are available on GEO under accession number GEO: GSE161264.
Yale New Haven Hospital and UK Biobank patient datasets and code used for analysis are available upon request to other researchers.
Experimental Models and Subject Details
Female H9 (WA09) and male H1 (WA01) human embryonic stem cells (hESCs) were obtained from WiCell, cultured in mTeSR (Stem Cell Technologies) and grown on Geltrex™ Growth Factor Reduced (GFR) Basement Membrane Matrix (GIBCO) at 37°C with 5% CO2. hESCs were fed daily and passaged every 5-6 days using 0.5mM EDTA (Thermo Fisher).
Human bronchial epithelial cells (HBECs) were isolated from explanted tissue from lung transplant donor recipients (n = 3), as previously described by Fulcher et al. (2005) and cryopreserved until culture. Briefly, explanted bronchial tissue was dissected and the epithelial layer was isolated and dissociated. HBECs (passage 0) were thawed and seeded on human placental collagen (HPC; MilliporeSigma)-coated 12-mm Transwell inserts (Corning, Corning, NY) at a density of ∼0.5x106 cells/cm2 and, once confluent, maintained at air-liquid interface (ALI).
Human alveolar epithelial cells (AECs) were purchased from Sciencell (cat#3200) and maintained in HLO media (described below in “Differentiation of HLOs”) and grown on poly-ornithine (Sigma), fibronectin (Corning) and laminin (Corning) plate coating at 37°C with 5% CO2. AECS were fed every other day and re-plated into final assay plates with Accutase (StemCell Tech).
VERO C1008 were purchased from ATCC (CRL-1586) and maintained in EMEM+10%FBS on tissue culture treated plates at 37°C with 5% CO2. Cells were fed every other day and passaged once plates reached 80% confluency with 0.05% Trypsin (Corning).
Method Details
Analysis of published scRNA-seq datasets
Quality control. Cells were identified as poor quality and subsequently removed based on the criteria implemented by the original authors of each dataset.
Lung and esophagus datasets: RData objects deposited to Tissue Stability Cell Atlas were pre-filtered by the authors. Filtering criteria can be found in the methods of Madissoon et al. (2019). Briefly, cells were excluded if they did not meet the following criteria: more than 300 and fewer than 5,000 genes detected, fewer than 20,000 UMI, and less than 10% mitochondrial reads. Genes were removed if they were detected in fewer than three cells per tissue.
Heart dataset: The gene by cell counts matrix deposited to GEO was pre-filtered by the authors. Filtering criteria can be found in the methods of Wang et al. (2020). Briefly, cells were removed if they did not meet the following criteria: detected at least 500 genes, UMI’s within 2 standard deviations of the mean of log10UMI of all cells, unique read alignment rate at least 50%, and fewer than 72% mitochondrial reads. Further, cardiomyocytes (CM) from CM-enriched datasets were included if UMIs detected were greater than 10,000.
Colon dataset: Based on the methods of Smillie et al. (2019), cells were removed if they did not meet one of the following criteria: minimal expression of 500 genes per cell; nUMIs within two standard deviations from the mean of log10 nUMIs of all cells; unique read alignment rate (# of assigned reads/ # total aligned reads) greater that 50%; and a mitochondrial read percentage of less than 72%. Mitochondrial genes were subsequently removed from the dataset prior to dimensionality reduction.
Data integration, dimensionality reduction and cell clustering. Different methods available in Seurat (Butler et al., 2018) were implemented respective to individual datasets.
Lung and esophagus datasets: Batch correction, data normalization, variable gene identification, data scaling, principal component analysis (PCA), and uniform manifold approximation and projection (UMAP) dimensionality reduction was performed by the original authors and included in the RData objects deposited to Tissue Stability Cell Atlas. The original UMAP coordinates generated by the authors were used for visualizations.
Colon Dataset: Batch correction by patient sample was performed using the Seurat v3 integration functions. The dataset was split by “Subject,” individual gene matrices were log normalized using a scaling factor of 10,000 and 2,000 variable features were identified per individual gene matrix using variance stabilizing transformation. 2,000 integration anchors were found using the first 30 dimensions of the canonical correlation analysis and individual datasets were integrated using the same number of dimensions. PCA was performed and UMAP coordinates were found based on the first 20 principal components.
Heart Dataset: Batch correction by patient sample was performed using mutual nearest neighbor (MNN) (Haghverdi et al., 2018) matching through the “RunFastMNN” function from the SeuratWrappers package. The dataset was log normalized using a scaling factor of 10,000 and 2,000 variable features were identified per individual gene matrix using variance stabilizing transformation. “RunFastMNN” was performed on the dataset split by “Individual” and UMAP coordinates were found based on the first 30 MNN dimensions. A shared nearest neighbor graph was constructed with the same number of MNN dimensions and clusters were identified using clustering resolution of 0.2.
Cell type identification and gene expression analysis. Lung, esophagus and colon datasets: Cell type cluster annotations determined by the original authors were available through the metadata downloaded with each dataset. These original cluster annotations were used for any visualizations.
Heart Dataset: Clusters were annotated based on cluster specific expression of marker genes identified in the original publication. Cluster markers were identified using a Wilcoxon Rank Sum test.
Following cell type annotation, gene dropout values were imputed using adaptively-thresholded low rank approximation (ALRA) (Linderman et al., 2018). The rank-k approximation was automatically chosen for each dataset and all other parameters were set as the default values. The imputed gene expression in shown in all plots and used in all downstream analysis.
Gene signature scoring. The Seurat “AddModuleScore” function was used to score each cell in the datasets for their expression of high confidence drug target genes (Figure 2C) and genes associated with AR signaling (Table S3). 100 control genes selected from the same bin per analyzed gene were used to calculate each module score. “Upstream AR Activators” were identified through literature search (Azevedo et al., 2011; Cao and Kyprianou, 2015; Girling et al., 2007; Wang et al., 2012), “AR Signaling” genes were taken from the androgen receptor signaling pathway gene ontology term (GO:0030521), and “Common AR Target Genes” are the common “core” target genes transcribed by AR identified by Jin et al. (2013) through comparison of multiple microarray studies.
Differentiation and characterization of cardiac cells
Differentiation of cardiac cells. hESCs were re-plated 72 h prior to initiating differentiation. The cardiac differentiation was started with a mesoderm induction cocktail comprised of 1.5 μM CHIR99021 (CHIR, Stem-RD), 20 ng/mL BMP4 and 20 ng/mL Activin A in RPMI (Cellgro) supplemented with B27 minus insulin, 2 mM GlutaMAX, 1x NEAA and 1x Normocin (InvivoGen) for 3 days (RPMI+B27 w/o insulin). Next, cells were treated with 5 μM XAV939 from days 3-6 in RB27-INS. From day 6 onward, differentiation of cells was carried out in RPMI supplemented with B27, 2 mM GlutaMAX, 1x NEAA and 1x Normocin (RPMI+complete B27). The protocol is outlined in Figure S1B.
Analysis of published RNA-seq data. The bulk RNA-seq dataset analyzed here was previously reported (Tsai et al., 2020). Briefly, human pluripotent stem cell (H9 with knock-in MYH6:mCherry reporter)-derived cardiomyocytes were prepared as described previously (Ghazizadeh et al., 2020; Tsai et al., 2020). Reporter tagged cardiomyocytes were isolated from the negative fraction of the culture by FACS. Both the purified cardiomyocytes and negative fractions were prepared and sent for bulk RNA-seq. The resulting datasets included two negative fraction biological replicates and two positive fraction biological replicates. Gene expression between the cardiomyocytes and non-cardiomyocytes was compared by averaging the read count per gene and normalizing by the average read count of GAPDH.
Immunofluorescence staining of cardiac cells. Cells were washed 3 times with PBS and fixed with 4% paraformaldehyde for 30 min at 4°C. Non-specific antigen binding was blocked by incubation with PBS+0.5% BSA for 30 min at room temperature (RT) prior to adding primary antibodies. The following primary antibodies were used: rabbit anti-ACE2 (1:500, ProteinTech, 21115-1-AP) and goat anti-TMPRSS2 (1:200, Novus Biologicals, NBP1-20984). Cells were incubated with the primary antibody solution overnight at 4°C then washed 3x5 min with PBS. Secondary antibodies and fluorophore conjugated anti-human Fc antibody were incubated for 30 min at RT. Finally, cells were washed 3x5 min with PBS and stained with DAPI for nuclear counterstaining. Cells were imaged on an EVOS™ FL digital inverted fluorescence microscope (Invitrogen) and images were processed using NIH ImageJ software.
High-throughput drug screening
In vitro high-throughput drug screening. hESC-derived cardiac cells were replated at day 25 of differentiation in 384 well plates at 2,000 cells per well. 72 h after re-plating, the cells were treated with compounds from an FDA-approved chemical library (Selleckchem) at 1 μM. 24 h after treatment, cells were fixed and stained with ACE2 antibody, as described previously. High-throughput imaging was carried out using the In Cell Analyzer 2000 (GE Healthcare, USA). ACE2 signal intensity was normalized to the total cell number within each well as measured by DAPI staining with GE Developer Toolbox v1.9.1.
Candidate drug selection and validation. The z-score was calculated and normalized for each plate. Hit compounds were determined by a normalized z-score of ± 1.5. A handful of hits that both increased and decreased ACE2 levels were selected to be validated by dose response. hESC-derived cardiac cells were replated at day 25 of differentiation in 96 well plates at 10,000 cells per well. 72 h after re-plating, the cells were treated with selected compounds (See Figure 1D-E) at 1uM and 2uM for 24 h. After treatment, cells were fixed and stained with ACE2 antibody, as described previously. Representative images for select compounds (See Figure 1C) were taken on an EVOS™ FL digital inverted fluorescence microscope (Invitrogen) and images were processed using NIH ImageJ software. High-throughput imaging and quantification was performed using the In Cell Analyzer 2000 and GE Developer Toolbox, as previously described. The above drug treatment, immunofluorescence staining, imaging and image analysis protocols were repeated on VERO cells at 70% confluency in 96 well format.
In silico high-throughput screening. To train the vHTS platform, we first split the data into training (n = 1048), validation (n = 131), and test datasets (n = 131) (Figure S1I-K), and used the validation set to evaluate increasingly complex models. We used Morgan fingerprints to represent the chemical features of each compound in the screened FDA-approved library (CircularFingerprint function available in DeepChem with SMILES as input). We tested a random forest regressor (scikit-learn) which failed to perform adequately (validation R2 score ∼0). We then used an XGBoost model (XGBRegressor) with the following parameters: colsample_bytree = 0.3, learning_rate = 0.1, max_depth = 5, alpha = 10, n_estimators = 10. While the model performance improved (correlation coefficient of 0.1), it was largely driven by positive z-score values and the prediction of the negative side was substantially less reliable. We then tested graph convolutional neural networks after featurizing the molecules using ConvMolFeaturizer (DeepChem) and testing a default model architecture with dropout set to 0.2. This initial model was promising with an R of 0.05; therefore, we performed a systematic hyperparameter tuning using a grad search on the size of the graph convolutional, dense layers and dropout rates. The best-performing model contained two graph convolutional (size 64) and dropout of 0.5 (and uncertainty set to true) and achieved an R of 0.1. We also tested message-passing models (MPNN) in a similar fashion; however, the performance was not improved. Therefore, to further boost the performance of the models, we first focused on improving the dataset itself. In addition to the ACE2 in vitro screening data, we included data from five additional screens with the same library, added an inverse normal transformation when appropriate, and performed a multi-task learning. These modifications allowed the model to generalize better and learn faster with better R scores (this allowed us to reduce the dropout to 0.25). Second, instead of using a single model, we utilized a bagging ensemble model where five models were trained on random samplings (with replacement) of training data. This ensemble model achieved an R of > 0.2 on the validation dataset. We then continued training while the Pearson coefficient for validation set remained above 0.2 (early stopping). We tested this final ensemble model on the test dataset, which achieved a similar R of 0.22 (p-value 0.01).
The final model described above was used to evaluate 9.2 million compounds from the ZINC15 database. We downloaded SMILES from this database and featurized them (same as above). The resulting predictions were variance normalized and the molecules with z-scores below −4 for ACE2 expression were selected as hits. This resulted in 169 potential small molecules. To visualize the relationship between the identified hits in a lower dimension, we used Morgan fingerprints to represent all compounds and used UMAPs to project each compound down to 2 dimensions. For the screened ZINC15 database, we sampled 1 from 1000 compounds for visualization purposes. We also used the Morgan fingerprints to cluster in silico and in vitro hits using K-means clustering (number of clusters set to 15).
In silico screen hit validation. 6 of the 169 hit compounds identified in the in silico screen were purchased from mcule.com (A01: MCULE-3280389582, A02: MCULE-3932122770, A03: MCULE-8963404982, A04: MCULE-8989818798, A05: MCULE-5352701202, A06: MCULE-5411082744).
hESC-derived cardiac cells were replated in 96 well plates, as previously described, and were treated with each compound at 1, 3, or 5μM. 24 h after treatment, cells were fixed and stained with ACE2 antibody, as described previously, followed by high-throughput imaging and quantification using the In Cell Analyzer 2000 (GE Healthcare, USA). ACE2 signal intensity was normalized to the total cell number within each well as measured by DAPI staining. Each well was then normalized to the average normalized fluorescence intensity of the no treatment condition.
Primary alveolar epithelial cells were replated in 96 well plates, as previously described, and were treated with each compound at 5μM. 72 h after treatment, cells were fixed and stained with ACE2 antibody, as described previously, followed by high-throughput imaging and quantification using the In Cell Analyzer 2000 (GE Healthcare, USA). ACE2 signal intensity was normalized to the total cell number within each well as measured by DAPI staining. Each well was then normalized to the average normalized fluorescence intensity of the no treatment condition.
Drug-protein-pathway interaction analysis
Identification of drug targets in FDA-approved library. Isomeric SMILES for each drug in the library were acquired from Selleckchem and used to run a similarity ensemble approach (SEA) library search. The SEA predicted targets were filtered, selecting human targets and predicted interaction p-values < 0.05, which yielded 2150 predicted proteins targeted by the drug library.
Target drug selection. The normalized z-score values reported for all the compounds were first transformed to N(0,1) using the bestNormalize package (v1.4.0) in R (v3.5.1). The treatments with transformed z-scores smaller than −1.5 were selected, which resulted in 41 compounds.
Target gene selection. For every compound, possible target genes were identified as above. Weighted combined z-scores were then calculated for each gene by combining normalized z-scores across all treatments. The p-values were then calculated based on the combined z-scores and adjusted using p.adjust (method = FDR). As an orthogonal approach for each gene, we recorded the number of treatments with negative normalized z-scores as well as the total number of compounds predicted to target that gene. Using the sum of counts for all other genes and drugs, we performed a Fisher’s exact test to evaluate the degree to which negative z-scores were enriched among the treatments likely to affect a gene of interest. As expected, the two p-values, i.e., combined z-score and Fisher’s, are generally correlated (R = 0.6, p < 1e-200).
Gene-set enrichment analysis. We used the combined z-scores across all genes to identify pathways and gene-sets that are associated with changes in ACE2 expression. For this analysis, we used our iPAGE toolkit (Goodarzi et al., 2009), in conjunction with annotations from MSigDB and Gene Ontology (GO). The following parameters were set:–ebins = 9–nodups = 1–independence = 0.
Drug-protein-pathway analysis. For drugs with a normalized z-score < -1, drug-protein interactions per biological pathway were analyzed. The SEA predicted drug-protein interaction dataset was filtered to exclude drugs with a normalized z-score 3-1. Protein pathway gene lists were generated per gene set, comprised of the genes present in the pre-ranked gene list. For each protein pathway gene list, drug-protein matrices were plotted by the SEA p-value for the interaction. The reported interaction significance scores represent the interaction z-scores calculated by SEA.
Protein-protein interaction network analysis
Protein-protein interaction network analysis was performed using the Search Tool for the Retrieval of Interacting Genes (STRING) database. The minimum required interaction score was set to 0.7, corresponding to high confidence with the edge thickness, indicating the degree of data support from the following active interaction sources: textmining, experiments, databases, co-expression, neighborhood, gene fusion, and co-occurrence.
SEA predicted drug-protein interactions were connected by lines. Lines were dashed if the MaxTC score was below 1. Proteins corresponding with the 30 significantly enriched genes from 2C were highlighted in yellow. To identify pathways involving our candidate proteins, we combined data from the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000) database and previous reports in the literature. Adobe Illustrator 24.1 was used for visualization.
AR ChIP-seq analysis
We used an existing AR ChIP-seq dataset generated in LNCaP cells to identify direct transcriptional targets of AR. We downloaded processed peaks from the Gene Expression Omnibus (GSM3148987) and lifted the peaks from hg19 over to hg38. We then used annotatePeak function (package ChIPseeker v1.8) to identify peaks 5kb downstream or upstream of transcription start sites (TSS). The genes with AR binding to this 10kb window around their TSS were selected as direct AR-bound targets.
AR knockdown RNA-seq analysis
We used an existing RNA-seq dataset generated using RNAi-mediated knockdown of AR (SRR7120653, SRR7120649, SRR7120646, and SRR7120642). We downloaded raw fastq files and aligned them to the transcriptome (gencode.v28) using Salmon (v0.14.1 using–validateMappings and -l ISR flags). We then used DESeq2 (1.22.2) to compare gene expression changes in response to AR knockdown. We then used the AR-bound gene-set from the previous step to assess how the AR targets respond to its downregulation. For this, we used our iPAGE tool (Goodarzi et al., 2009) which uses mutual information (MI) and an associated z-score to assess the enrichment/depletion patterns of a gene-set across gene expression modulations. To visualize this data, we included a volcano plot, with genes of interest shown in red. For this analysis, we divided log-fold changes into nine equally populated bins, which were also included along with the patterns of enrichment and depletion across the data.
Analysis of anti-androgenic drug treatment
hESC cardiac treatment and immunofluorescence analysis. Cardiac cells derived from both WA09 and WA01 hESCs were re-plated at day 25 of differentiation in 96 well plates at 10,000 cells per well. 72 h after re-plating, the cells were treated with ketoconazole, finasteride, dutasteride or spironolactone at 1uM, 2uM or 5uM for 24 h. After treatment, cells were fixed and stained with ACE2 antibody, as described previously. Representative images for select compounds were taken on an EVOS™ FL digital inverted fluorescence microscope (Invitrogen) and images were processed using NIH ImageJ software. High-throughput imaging and quantification were performed using the In Cell Analyzer 2000 and GE Developer Toolbox, as previously described.
HBECs treatment and immunofluorescence analysis. HBECs were isolated and cultured as mentioned above. For treatment with selected anti-androgenic drugs, media was supplemented with 5 uM finasteride, dutasteride or ketoconazole for 72 h. Prior to fixation, the apical surface was washed with PBS for 10 min to remove accumulated mucus. Basolateral media was aspirated, and the basolateral membrane rinsed briefly with 750 μL PBS. HBECs were fixed for 1 h at RT in 4% PFA; 0.2 mL and 0.7 mL PFA were added to the apical and basolateral compartments, respectively. PFA was removed and the cells were stored in PBS until staining.
For all staining incubations, solutions were only changed in the apical membrane side of the Transwell insert and all incubations occurred on an orbital shaker at RT. HBECs were permeabilized and blocked in 0.5% Triton-X with 5% Donkey Serum (Jackson Laboratories) in PBS for 1 h. HBECs were incubated overnight in primary antibody in 0.1% Triton-X with 5% Donkey Serum in PBS. The following primary antibodies were used: rabbit anti-ACE2 (1:200, ProteinTech, 21115-1-AP), mouse anti-ACE2 (1:200, ProteinTech, 66699-1-AP), goat anti-TMPRSS2 (1:200, Novus Biologicals, NBP1-20984), mouse anti-CD324 (E-CAD) (1:500, BD Biosciences, 610191), and mouse anti-TUBA (1:500, Sigma, T7451). Antibodies were detected using AF488-, AF568-, and AF647-conjugated donkey antibodies raised against mouse, rabbit, or goat IgG (all 1:1000, all Invitrogen) in 0.1% Triton-X with 5% Donkey Serum in PBS for 1 h at RT. DNA was labeled with DAPI in 0.05% PBS-Tween-20 for 5 min at RT. Organoids were washed 3x10 min with 0.05% PBS-Tween-20 at RT after primary and secondary antibody incubations. For imaging, the Transwell membranes were removed from the inserts with a razor and slide-mounted in a drop of Fluoromount-G (SouthernBiotech). Slides were imaged using a Leica Sp8 confocal microscope and processed using NIH ImageJ software.
All images compared were from paralleled staining rounds and imaged using equal laser intensity settings during image capture. All quantifications were done using NIH ImageJ software. For comparison of ACE2 and TMPRSS2 levels in drug treated HBECs, z-projections of each image stack were generated using the sum slices method. The integrated density of each channel was then measured and normalized to the DAPI channel integrated density of the same field of view. At least three images were taken per transwell insert for quantification.
HBECs qRT-PCR gene expression analysis. mRNA was quantified as previously described (Koh et al., 2020). Briefly, HBECs were lysed in Buffer SKP and homogenized by vortexing. Total RNA was isolated using the RNA/DNA/Protein Purification Plus Kit (Norgen Biotek, Thorold, ON, Canada) according to manufacturers’ protocols. RNA was reverse-transcribed using SuperScript III First-Strand Synthesis System (ThermoFisher Scientific) and the resulting cDNA was analyzed by quantitative real-time PCR (qRT-PCR) using PowerUp SYBR Green (ThermoFisher Scientific). mRNA levels were normalized to housekeeping gene levels and comparisons in ACE2 expression were made using the deltaCt method.
CRISPR knockout of androgen signaling
Targeting and analysis in hESC derived cardiac cells. AR and SRD5A2 ribonucleoprotein (RNP) complexes were assembled by mixing 180 pmol of multi-guide sgRNA (Synthego, USA) and 20 pmol of Cas9 2NLS (Berkeley QB3) in Lonza electroporation buffer P3 (Lonza, Switzerland) per reaction. hESC-derived cardiac cells were dissociated using accutase, washed with PBS, and passed through a cell strainer before resuspension in Lonza electroporation buffer P3 immediately before electroporation. Cells were mixed with the RNPs and were electroporated using a Lonza 4D 96 well electroporation system with pulse code CA137. Cells were then diluted in warm medium and plated in 96 well plates. Medium was changed on the following day and the cells were fixed 3 days after transfection. Cells were fixed and stained as described previously with the following primary antibodies: rabbit anti-ACE2 (1:500, ProteinTech, 21115-1-AP), mouse anti-AR (1:300, ProteinTech, 66747-1-IG), and goat anti-SRD5A2 (1:100, Abcam, ab27469). Plates were imaged by high-throughput imaging using the In Cell Analyzer 2000 (GE Healthcare, USA). For quantification, images were analyzed with NIH ImageJ software by measuring the fluorescence intensity of individual cells by manual region of interest selection.
Targeting and analysis in alveolar epithelial cells. For the human primary alveolar epithelial cells, AR and SRD5A2 RNP complexes were assembled by mixing 0.7 pmol of multi-guide sgRNA (Synthego, USA), 0.5 pmol of Cas9 2NLS (Berkeley QB3) and Lipofectamine Cas9 Plus Reagent (ThermoFisher Scientific, USA) in Opti-MEM I reduced serum medium (ThermoFisher Scientific, USA) per reaction (one well of a 96 well plate). In a separate tube, Lipofectamine CRISPRMAX transfection reagent was first diluted in Opti-MEM I reduced serum medium at a 1.5:25 ratio and was then immediately added to the RNPs (1:1 ratio). The mixture was incubated for 10 min at RT before addition to the alveolar epithelial monolayer cultures. Medium was changed on the following day and the cells were fixed 3 days after transfection. Cells were fixed and stained as described previously with the following primary antibodies: rabbit anti-ACE2 (1:500, ProteinTech, 21115-1-AP), mouse anti-AR (1:300, ProteinTech, 66747-1-IG), and goat anti-SRD5A2 (1:100, Abcam, ab27469). Plates were imaged by high-throughput imaging using the In Cell Analyzer 2000 (GE Healthcare, USA). For quantification, images were analyzed with NIH ImageJ software by measuring the fluorescence intensity of individual cells by manual region of interest selection.
Spike-RBD cell internalization assay
hESC-derived cardiac cells and primary alveolar epithelial cells were re-plated in 96 well plates, as previously described, and were treated with 300nM DHT or 10uM dutasteride. After 24 h, 0.5 μM/mL human Fc-tagged recombinant spike-RBD protein (Sino Biological Inc. 40592-V02H) was added to the medium and incubated for 30 min. The cells were subsequently fixed, permeabilized, and stained with antibodies against ACE2 and human Fc receptor, as described previously, followed by high-throughput imaging and quantification using the In Cell Analyzer 2000 (GE Healthcare, USA). ACE2 and spike-RBD signal intensity were normalized to the total cell number within each well as measured by DAPI staining.
Generation of pseudotyped SARS-CoV-2 VSVΔG-GFP
Chimeric vesicular stomatitis virus (VSV) was produced based on existing protocols (Hoffmann et al., 2020; Whitt, 2010). In brief, HEK293T cells were grown to 70% confluency in DMEM hi-glucose with 10% FBS in a 6 well tissue culture plate (Fisher 0877233). HEK293T cells were transfected with 2.5 μg of SARS-CoV-2 S protein expression plasmid optimized for mammalian expression, 2.5 μg mCherry plasmid (generous gift of Lamba Lab, UCSF), 7.5 μL Lipofectamine 3000 (ThermoFisher Scientific L3000001), and 10 μL P3000 reagent (ThermoFisher Scientific L3000001). For bald virus control, a separate well was transfected with mCherry plasmid, but no S protein expression construct. After 20 h, HEK293T cells were given fresh media. 24-48 h after transfection, when 80% of cells were mCherry positive, both S protein expressing and bald control cells were inoculated with replication-deficient, G-complemented G∗ΔG-GFP rVSV (Kerafast EH1024-PM) at an MOI of 5 and incubated at 37°C. Inoculum was removed after 1 h and replaced with fresh media. Supernatant was collected 16 h after inoculation and clarified by centrifugation at 1320xg for 10 min at RT. Aliquots were immediately frozen at −80°C.
Functional titration of the pseudotyped virus was performed on Vero E6 cells (ATCC CRL-1586). Vero E6 cells were grown to 70% confluency in 96 well tissue culture-treated black optical plates (Fisher 1256670) in EMEM + 10% FBS. Cells were infected with serial 2-fold dilutions of either S-complemented chimeric VSV or bald VSV control. GFP expression of infected cells was measured 24 h post-inoculation.
SARS-CoV-2 VSVΔG-GFP inhibition assays
Human alveolar epithelial cells (AECs) were cultured to 70% confluency in 96 well tissue culture-treated black optical plates (Fisher 1256670) in HLO media. AECs were treated with finasteride, dutasteride, or ketoconazole at 1 μM in HLO media for 72 h. AECs were then inoculated with SARS-CoV-2 S-complemented VSVΔG-GFP pseudovirus in fresh HLO media containing 1 μM finasteride, dutasteride, or ketoconazole. 24 h post-inoculation, cells were fixed for 30 min in 4% PFA (SCBT sc-281692) at RT then rinsed twice with PBS. Cells were permeabilized and blocked with perm block buffer (Invitrogen 00-8333-56) for 1 h at RT, incubated with αGFP (ab13970) 1:2000 in perm block buffer for 1 h at RT, and washed three times in perm block buffer. Cells were stained with α-chicken AF488 secondary antibody at 1:1000 in perm block buffer for 1 h followed by three 10 min washes in perm block buffer. Images were taken with GE inCell 2000 imager. Image segmentation and cell counting were performed with GE Developer Toolbox v1.9.1.
Differentiation and characterization of human lung organoids (HLOs)
Differentiation of HLOs. Human lung organoids (HLOs) were derived from hESCs as previously described (de Carvalho et al., 2019; Jacob et al., 2017; Miller et al., 2019), with minor modifications. Briefly, H9 hESCs were maintained in mTeSR Plus media and were seeded on Day 0 onto matrigel (Corning) coated surface. On days 1-4, cells were treated with 100 ng/mL Activin A and increasing concentrations of defined FBS (dFBS, HyClone) in RPMI 1640 medium (ThermoFisher Scientific) to induce definitive endoderm formation (day 1: 3 μM CHIR99021 and no dFBS; day 2: 0.2% dFBS; days 3-4: 2% dFBS). On day 4, cells were > 95% double positive for CXCR4/CD184 and c-Kit/CD117 as determined by flow cytometry. On days 5-9, cells were treated with anterior foregut induction medium containing 10 μM SB431542, 100 nM LDN193189, 2 μM CHIR99021, 1 μM SAG, 500 ng/mL FGF4 in foregut basal medium (Advanced DMEM, 1x B27, 1x N2, 10 mM HEPES, 1x Glutagro (all ThermoFisher Scientific), 50 μg/mL ascorbic acid (Sigma), 0.4 μM monothioglycerol (Sigma)). On day 9, anterior foregut spheroids were harvested by gentle pipetting and transferred into an ultra-low attachment plate, in lung organoid medium I (3 μM CHIR99021, 10 ng/mL BMP4, 10 ng/mL FGF7, 10 ng/mL FGF10, 50 nM all-trans retinoic acid, in foregut basal medium). The medium was changed every two days until day 15. On day 15, the medium was changed to lung organoid medium II (3 μM CHIR99021, 10 ng/mL FGF7, 10 ng/mL FGF10, in foregut basal medium). On day 25, the organoids were embedded in matrigel, in transwell inserts, and grown in lung organoid medium III (3 μM CHIR99021, 10 ng/mL FGF7, 10 ng/mL FGF10, 50 nM dexamethasone (Sigma), 100 μM 8-bromo-cAMP (Sigma), 100 μM IBMX (Sigma), in foregut basal medium). The medium was changed every 3 days. The CHIR99021 was withdrawn on days 35-42. All media components were from Stem Cell Technologies unless noted otherwise.
Immunofluorescence staining of HLO sections. Matrigel embedded organoids were extracted in 1 mL Organoid Recovery Solution (Cultrex) 1 h 30 at 4°C with end-to-end rotation and then fixed 1 h at RT in 4% PFA. Organoids were incubated in increasing concentrations of sucrose (15%–30%) and embedded in OCT (Tissue-Tek). Organoids were sectioned on a cryostat and tissue sections kept at −80°C until immunofluorescent analysis. OCT tissue blocks were kept at −80°C. Organoid sections were equilibrated to RT for 10 min and OCT was removed by washing with PBS. Organoid sections were permeabilized and blocked in 0.1% Triton X-100 with 5% Donkey Serum (Jackson Laboratories) in PBS for 1 h at RT. Organoid sections were incubated overnight at 4°C in primary antibody in 0.1% Triton X-100 with 5% Donkey Serum in PBS. The following primary antibodies were used: mouse anti-CD324 (E-CAD) (1:500, BD Biosciences, 610191), mouse anti-NKX2-1 (1:1000, Seven Hills Bioreagents, WMAB-1231), mouse anti-SOX9 (1:500, Cell Signaling, 82630T), and mouse anti-ACE2 (1:200, ProteinTech, 66699-1-AP). Antibodies were detected using AF488-, AF568-, and AF647-conjugated donkey antibodies raised against mouse, rabbit, or goat IgG (all 1:1000, Invitrogen) in 0.1% Triton X-100 with 5% Donkey Serum in PBS for 1 h at RT. DNA was labeled with DAPI (1:1000, Invitrogen, D1306) in 0.05% PBS-Tween-20 for 1 min at RT. Sections were washed 3x5 min with 0.05% PBS-Tween-20 at RT after primary and secondary antibody incubations. Slides were mounted using Fluoromount-G (SouthernBiotech) and tissue was imaged using a Leica Sp8 confocal microscope and processed using NIH ImageJ software.
Bulk RNA sequencing. Cells during HLO differentiation were harvested on days 0, 5, 9, 15, 25, 35, 50, and RNA was extracted using Quick-RNA 96 Kit (Zymo) with on-column DNaseI treatment. RNA-seq libraries were constructed with QuantSeq FWD Kit (with UMI module, Lexogen) and sequenced on Illumina HiSeq sequencer in the Center for Advanced Technology (UCSF). UMIs were extracted from the fastq files with umi_tools and cutadapt was used to remove short and low-quality reads. The reads were aligned to human GENCODE v.34 reference genome using STAR aligner and the duplicate reads were collapsed using umi_tools. Gene level counts were measured using HTSeq and compared using DESeq2.
Single cell RNA sequencing sample preparation and data collection. All tubes and pipet tips used for cell harvesting were pre-treated with 1% BSA in 1X PBS. The HLOs were transferred with a wide-end pipet tip from matrigel to 1 mL Organoid Harvesting solution (Cultrex) and incubated 1 h at 4°C with end-to-end rotation. Then the cells were dissociated in Accutase (Stem Cell) with DNaseI (200 U/mL, Worthington) and Dispase (100 U/mL, Corning) at 37°C, in 10 min increments with end-to-end rotation, until a single cell suspension was obtained. The cells were washed in Cell Staining Buffer (Biolegend) and stained with TotalSeq HTO antibodies for 30 min on ice. The cells were washed twice in Cell Staining Buffer and filtered through a 40 μm pipette tip strainer (BelArt). The cells were counted using Trypan Blue dye and a hemocytometer and pooled for sequencing. scRNA-seq libraries were prepared with Chromium Next GEM Single Cell 3¢ Kit v3.1 (10x Genomics), with custom amplification of TotalSeq HTO sequences (Biolegend). The libraries were sequenced on an Illumina NovaSeq sequencer in the Center for Advanced Technologies (UCSF). The cell feature matrices were extracted using kallisto/bustools and demultiplexed using Seurat.
Single-cell RNA sequencing data analysis. All further downstream analysis was carried out with Seurat and SeuratWrapper packages. Cells were identified as poor quality and subsequently removed if they did not meet one of the following criteria: greater than 200 unique features detected; unique read alignment rate (log10(nFeatures)/log10(nCounts)) greater that 80%; and a mitochondrial read percentage of less than 30%. Cell-by-gene count matrices from independent experiments were merged, log normalized using a scaling factor of 10,000, and 2,000 variable features were identified using variance stabilizing transformation. Batch correction by experiment was performed using the “RunFastMNN” function from the SeuratWrappers package (Haghverdi et al., 2018) and UMAP coordinates were found based on the first 20 MNN dimensions. A shared nearest neighbor graph was constructed using the same number of MNN dimensions and clusters were identified using clustering resolution of 0.1. Broad cell type clusters were annotated based on cluster specific expression of marker genes identified using a Wilcoxon Rank Sum test. Following cell type annotation, gene dropout values were imputed using adaptively-thresholded low rank approximation (ALRA) (Linderman et al., 2018). The rank-k approximation was automatically chosen for each dataset and all other parameters were set as the default values. The imputed gene expression is shown in all plots and used in all downstream analysis. Epithelial cells were subsetted based on expression of EPCAM and CDH1 for subtype clustering and analysis. The subsetted epithelial cell-by-gene count matrix was re-analyzed using the same workflow as the full dataset. UMAP coordinates were found based on the first 20 MNN dimensions. A shared nearest neighbor graph was constructed using the same number of MNN dimensions and clusters were identified using clustering resolution of 0.2. Epithelial cell type clusters were annotated based on cluster specific expression of marker genes identified using a Wilcoxon Rank Sum test in combination with module scoring of cell type marker gene lists using the “AddModuleScore” function. 100 control genes selected from the same bin per analyzed gene were used to calculate each module score.
Wild-type SARS-CoV-2 HLO infection
SARS-CoV-2 Isolation and Amplification. SARS-CoV-2/human/USA/CA-UCSF-0001C/2020 was isolated from a UCSF clinical specimen (nasopharyngeal swab collected in viral holding medium) that was RT-qPCR positive. The clinical specimen was serially diluted and isolated on Calu-3 cells grown in EMEM in the presence of penicillin, streptomycin, amphotericin B and 5% heat-inactivated fetal bovine serum (FBS). Virus was subsequently amplified by passage on Calu-3 cells and sequence verified using next-generation sequencing.
Infection of HLOs with SARS-CoV-2. Infections using the live USA/CA-UCSF-0001C/2020 isolate and USA-WA1/2020, NR-52281 isolate (BEI resources) of SARS-CoV-2 were performed in two independent Biosafety Level 3 labs (Andino lab and Ott lab respectively). SARS-CoV-2 stocks were passaged in Vero cells (ATCC) and titer was determined via plaque assay on Vero cells. Organoids were plated in suspension in fresh media or fresh media supplemented with 5 uM ketoconazole, finasteride and dutasteride for 72 h prior to infection. For infection, organoids were plated in suspension in fresh media plus virus (MOI 0.1) for 2 h, washed with PBS and then replated in suspension with fresh media for 72 h. Organoids were next fixed in 4% PFA for a minimum of 30 min before removal from the BSL-3 lab.
HLO Whole Mount Immunofluorescence staining and quantification. Organoids grown in suspension were fixed overnight at 4°C in 4% PFA and then kept at 4°C in PBS until immunofluorescent analysis. All incubations were performed in Eppendorf tubes on an orbital rocker at RT. Organoids were permeabilized and blocked in 0.5% Triton-X with 5% Donkey Serum (Jackson Laboratories) in PBS for 1 h. Organoids were incubated overnight in primary antibody in 0.1% Triton-X with 5% Donkey Serum in PBS. The following primary antibodies were used: rabbit anti-ACE2 (1:200, ProteinTech, 21115-1-AP), goat anti-ACE2 (1:20, R&D Systems, af933), mouse anti-dsRNA (1:200, Absolute Antibodies, ab0129.2.0), and mouse anti-N-protein (1:100, Invitrogen, MA1-7404). Antibodies were detected using AF488-, AF568-, and AF647-conjugated Donkey antibodies raised against mouse, rabbit, or goat IgG (all 1:1000, Invitrogen) in 0.1% Triton-X with 5% Donkey Serum in PBS for 3 h at RT. DNA was labeled with DAPI in 0.05% PBS-Tween-20 for 10 min at RT. Organoids were washed 3x30 min with 0.05% PBS-Tween-20 at RT after primary and secondary antibody incubations. Organoids were suspended in PBS in Ibidi imaging dishes imaged using a Leica Sp8 confocal microscope and processed using NIH ImageJ software.
All images compared were from paralleled staining rounds and imaged using equal laser intensity settings during image capture. All quantifications were done using NIH ImageJ software. For comparison of ACE2 levels in drug treated lung organoids, z-projections of each image stack were generated using the sum slices method. Next, an automatic threshold was applied to the ACE2 channel of each image to determine the areas of positive ACE2 staining. The raw integrated density of the threshold area was measured and normalized to the area of positive staining. For comparison of SARS-CoV-2 infection by dsRNA and nucleocapsid protein staining in drug treated organoids, the number of cells positive for either antigen was determined by manual counting using z-projections of each image stack generated using the sum slices method in combination with orthogonal views. The number of infected cells was normalized to the area the organoid, found by drawing and measuring an ROI around the organoid in the z-projection window.
Plaque assay. Vero E6 cells were maintained in MEM supplemented with 10% fetal calf serum (FCS), penicillin-streptomycin (100 IU/mL) and glutamine (292 μg/mL; pen-strep-glutamine). 6 well plates were seeded with cells a day prior to the assay to be fully confluent on the day of infection. Samples were serially diluted 10-fold in MEM containing 2% FCS and pen-strep-glutamine. After removing the culture media, 250 uL of sample dilution was added to the corresponding cell monolayer and incubated for 1 h at 37°C at 5% CO2. To make the overlay, boiled 2% agar made in cell-grade water was mixed 1:1 with 2X MEM media (powdered MEM in cell grade water, 2.2g/L NaHCO3 and pen-strep-glutamine) kept at 4°C until mixing. Cells with 3 mL of overlay were incubated for 72 h at 37°C in a 5% CO2 incubator and fixed in 4% PFA for 2 h. Agar plugs were flipped and plaques were counted following staining with 0.1% crystal violet solution and presented as plaque forming units (pfu)/mL.
Yale New Haven Hospital patient data analysis
EPIC’s SlicerDicer feature was used to collect the Yale New Haven Hospital (YNHH) electronic health record anonymous aggregate-level data. Using this feature, a total of 8657 patients had a COVID-19 ICD-10 diagnosis code at YNHH during November 28, 2019-April 28 2020 time period. Among them, 1577 patients had a Troponin T checked during the same encounter and were selected for subsequent analysis. Out of 1577 patients, 787 were female and 790 were male.
Next, we focused on males above age 40 for subsequent risk factor analysis. Risk factors associated with disease outcome, including age, BMI, status of hypertensive disorder, status of diabetes mellitus disorder and prostate disease, were collected as dichotomized variables during data download. Variable cutoffs were set as follows: age, 40-65y versus 65+ y, BMI, < 30 versus > = 30, troponin T < 0.01 ng/mL versus > = 0.01 ng/mL. Diabetes, hypertension and prostatic disease (hyperplasia of prostate or neoplasm of prostate) were binary originally and were collected as a visit diagnosis. Appropriate SNOMED-CT (systematized nomenclature of medicine–clinical terms) were used to include or exclude patients according to their pre-existing conditions. The following terms were used to identify patients with pre-existing conditions in the database: “disorders of glucose metabolism” to select the patients with any form of the Diabetes Mellitus, “Hypertensive disorder” to select the patients with any form of hypertension and “hyperplasia of prostate or Neoplasm of Prostate” to select patients with disorders of prostate gland. The study was in accordance with Institutional Review Board policy.
A total of 681 subjects exist in this dataset with 239 high troponin and 442 low troponin cases. The interdependence of all variables was explored through their correlation matrix (Figure S6B) and pairwise Fisher’s exact tests. To find the optimal model, we used a backward selection approach. We started with a logistic regression model with troponin as response and all the other variables and their pairwise interaction terms as covariates. In successive steps, least significant terms were eliminated one at a time and the model was refitted until all remaining terms were significant at p < 0.05. Odds of having abnormal troponin T in the baseline group (40-65y, no prostate disease, low BMI, no diabetes, no hypertension, n = 58) was 0.038.
UK Biobank data analysis
Individual-level longitudinal phenotypic data from the UK Biobank, a large-scale population-based cohort with genotype and phenotype data in approximately 500,000 volunteer participants recruited from 2006-2010, was used (Bycroft et al., 2018). Baseline assessments were conducted at 22 assessment centers across the UK using touch screen questionnaire, computer assisted verbal interview, physical tests, and sample collection including for DNA. Additional details regarding the study protocol are described online (https://www.ukbiobank.ac.uk).
Of 488,377 individuals genotyped in the UK Biobank, we used data for 190,150 men from English recruiting centers where COVID-19 testing was reported in the UK Biobank, as done previously (Armstrong et al., 2020). Additionally, we further filtered to individuals with white British ancestry consenting to genetic analyses, with genotypic-phenotypic sex concordance, without sex aneuploidy, and removed one individual from each pair of 1st or 2nd degree relatives selected randomly. Participants provided informed consent as previously described. Secondary use of the data was approved by the Massachusetts General Hospital institutional review board (protocol 2013P001840) and facilitated through UK Biobank Application 7089.
UK Biobank phenotypes. Definitions for phenotypes are included in Table S4; all phenotypes were compiled using the most updated phenotypes from the July 2020 release. In brief, benign prostatic hyperplasia was defined by combining self-reported enlarged prostate and ICD-9 and ICD-10 billing codes for prostatic hyperplasia. Hypertension was defined by self-reported hypertension and billing codes for essential hypertension, hypertensive disease with and without heart failure, hypertensive heart and renal diseases, and secondary hypertension. Type 2 diabetes was defined by billing codes for non-insulin-dependent diabetes mellitus or self-reported type 2 diabetes. Body mass index was from enrollment.
UK Biobank COVID-19 Case/Control Models. COVID-19 phenotypes used in the present analysis are from UK Biobank downloaded on Sept 22, 2020 and include combined nose/throat swabs and lower respiratory samples analyzed using PCR. Two case/control definitions were used in the analysis to iterate over COVID-19 susceptibility and severity across all individuals with COVID-19 testing reported from the UK Biobank English recruitment centers (Armstrong et al., 2020). COVID-19 positive cases were defined as any individual with at least one positive test. COVID-19 hospitalized cases were defined as any individual with at least one positive test who also had evidence that they were a hospitalized inpatient. Individuals with COVID-19 of unknown severity were excluded from the COVID-19 hospitalization analyses. Controls included all white British men from the UK Biobank English recruitment centers who either tested negative for COVID-19 or did not have a COVID-19 test.
UK Biobank Association with Benign Prostatic Hypertrophy. Association of benign prostatic hypertrophy (BPH) with COVID-19 severity and susceptibility was performed among men using logistic regression in R-3.5. Adjustment for age, hypertension, type 2 diabetes, normalized BMI at enrollment, normalized Townsend deprivation index (a marker of socioeconomic status), and the first ten principal components of genetic ancestry were performed.
Gene set enrichment analysis of androgen signaling genes in the COVID-19 human genetics consortium GWAS. Gene set enrichment analysis was performed using MAGMA (de Leeuw et al., 2015) on a set of 8 genes identified from our FDA drug screen as well as all 76 androgen signaling genes listed in Table S3 on the COVID-19 Host Genetics Initiative GWAS release 3 results (https://www.covid19hg.org/results/) across 3,199 COVID-19 hospitalized cases and 897,488 controls.
Mendelian randomization of bioavailable testosterone with COVID-19 hospitalization. Two-sample Mendelian randomization analysis of bioavailable testosterone on COVID-19 hospitalization was performed. The exposure was identified using genome-wide significant independent variants in the bioavailable testosterone GWAS (Ruth et al., 2020) performed using sex-specific inverse rank normal transformation of the phenotype) within 1 MB of the 8 androgen signaling genes from the drug screen across 425,097 individuals in the UK Biobank (Table S8 of Ruth et al., 2020). The outcome was using GWAS summary stats from the COVID-19 hospitalized versus population GWAS across 3199 cases and 897,488 controls from the COVID-19 Host Genetics Initiative release 3 GWAS (https://www.covid19hg.org/results/). The robust MR-Egger method was utilized from the MendelianRandomization R-package to perform Mendelian randomization as it adjusts for directional pleiotropy (Bowden et al., 2015).
Quanitification and Statistical Analysis
Statistical methods relevant to each figure are outlined in the accompanying figure legend or described in the Results section. Unless otherwise indicated, experiments were carried out in a minimum of 3 biological replicates. Two-tailed Student’s t test was performed to compare treatment groups. Error bars in figures represent standard error of the mean.
Acknowledgments
We are grateful to the UCSF Program for Breakthrough Biomedical Research and Sandler Foundation and the UCSF QBI Coronavirus Research Group (QCRG) for the grants to F.F. that supported this work. F.F. is also supported by the NIH Director’s New Innovator Award (DP2NS116769) and the National Institute of Diabetes and Digestive and Kidney Diseases (R01DK121169). H.M. is supported by Larry L. Hillblom Foundation postdoctoral fellowship. S.M.Z. is supported by the National Institutes of Health’s NHLBI under award number 1F30HL149180-01 and the National Institutes of Health’s Medical Scientist Training Program at the Yale School of Medicine. A.N. was supported by a DoD PRCRP Horizon Award (DoD: W81XWH-19-1-0594). H.A. is supported by the NIH postdoctoral fellowship F32GM133118 and the postdoctoral grant from the Program for Breakthrough Biomedical Research and the Sandler Foundation at UCSF. H.G. is supported by the National Cancer Institute (R01 CA240984), the National Institute of General Medical Sciences (R01 GM123977), and an LGR-ERA Award. P.N. is supported by a Hassenfeld Scholar Award from the Massachusetts General Hospital, and grants from the National Heart, Lung, and Blood Institute (R01HL1427, R01HL148565, and R01HL148050) and Fondation Leducq (TNE-18CVD04). L.R.B. and D.J.E. are supported by NIH grants U19 AI077439 and R35 HL145235. Special thanks to the Center for Advanced Technology (UCSF) for their help with RNA sequencing experiments and the sectioning in Histology and Light Microscopy Core (Gladstone Institutes). M.O acknowledges support through a gift from the Roddenberry Foundation and by NIH 5DP1DA038043. We are grateful to Sohit Miglani, Abolfazl Arab, and Aidan Winters for their help with RNA sequencing data analysis. The following reagent was obtained through BEI Resources, NIAID, NIH: SARS-Related Coronavirus 2, Isolate USA-WA1/2020, NR-52281, deposited by the Centers for Disease Control. Graphical abstract created with BioRender.com.
Author Contributions
R.M.S., H.M., M.N.R., Z.G., A.N., J.T.R., C.R.S., L.R.B., K.D.K., M.G.-K., M.T., S.S., S.F., and A.K. performed the experiments, H.A. performed statistical analysis, S.M.Z., W.L., H.Z., and P.N. performed the UKBB study analysis, D.J.E. supervised experiments with primary pulmonary epithelial cells, R.A. and M.O. supervised SARS-CoV-2 infection experiments; H.G. performed pathway analysis and in silico library screen; F.F. designed, conceptualized, performed, and supervised the experiments; and Z.G., H.M., M.N.R., R.M.S., H.G., and F.F. wrote the manuscript.
Declaration of Interests
P.N. receives grants from Apple, Amgen, and Boston Scientific, and personal fees from Apple and Blackstone Life Sciences, all outside the submitted work. F.F. receives grants from Takeda Pharmaceuticals on projects outside of the submitted work.
Published: November 17, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stem.2020.11.009.
Supplemental Information
References
- Armstrong J., Rudkin J.K., Allen N., Crook D.W., Wilson D.J., Wyllie D.H., O’Connell A.-M. Dynamic linkage of COVID-19 test results between Public Health England’s Second Generation Surveillance System and UK Biobank. Microb. Genom. 2020;6:6. doi: 10.1099/mgen.0.000397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo A., Cunha V., Teixeira A.L., Medeiros R. IL-6/IL-6R as a potential key signaling pathway in prostate cancer development. World J. Clin. Oncol. 2011;2:384–396. doi: 10.5306/wjco.v2.i12.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonser L.R., Koh K.D., Johansson K., Choksi S.P., Cheng D., Liu L., Sun D.I., Zlock L.T., Eckalbar W.L., Finkbeiner W.E. Flow cytometric analysis and purification of airway epithelial cell subsets. bioRxiv. 2020 doi: 10.1101/2020.04.20.051383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bost P., Giladi A., Liu Y., Bendjelal Y., Xu G., David E., Blecher-Gonen R., Cohen M., Medaglia C., Li H. Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell. 2020;181:1475–1488.e12. doi: 10.1016/j.cell.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden J., Davey Smith G., Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015;44:512–525. doi: 10.1093/ije/dyv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z., Kyprianou N. Mechanisms navigating the TGF-β pathway in prostate cancer. Asian J. Urol. 2015;2:11–18. doi: 10.1016/j.ajur.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies N.M., Holmes M.V., Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601. doi: 10.1136/bmj.k601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho A.L.R.T., Strikoudis A., Liu H.-Y., Chen Y.-W., Dantas T.J., Vallee R.B., Correia-Pinto J., Snoeck H.-W. Glycogen synthase kinase 3 induces multilineage maturation of human pluripotent stem cell-derived lung progenitors in 3D culture. Development. 2019;146:146. doi: 10.1242/dev.171652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw C.A., Mooij J.M., Heskes T., Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015;11:e1004219. doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulcher M.L., Gabriel S., Burns K.A., Yankaskas J.R., Randell S.H. Well-differentiated human airway epithelial cell cultures. Methods Mol. Med. 2005;107:183–206. doi: 10.1385/1-59259-861-7:183. [DOI] [PubMed] [Google Scholar]
- Ghazizadeh Z., Fattahi F., Mirzaei M., Bayersaikhan D., Lee J., Chae S., Hwang D., Byun K., Tabar M.S., Taleahmad S. Prospective Isolation of ISL1+ Cardiac Progenitors from Human ESCs for Myocardial Infarction Therapy. Stem Cell Reports. 2018;10:848–859. doi: 10.1016/j.stemcr.2018.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazizadeh Z., Kiviniemi T., Olafsson S., Plotnick D., Beerens M.E., Zhang K., Gillon L., Steinbaugh M.J., Barrera V., Sui S.H. Metastable Atrial State Underlies the Primary Genetic Substrate for MYL4 Mutation-Associated Atrial Fibrillation. Circulation. 2020;141:301–312. doi: 10.1161/CIRCULATIONAHA.119.044268. [DOI] [PubMed] [Google Scholar]
- Girling J.S., Whitaker H.C., Mills I.G., Neal D.E. Pathogenesis of prostate cancer and hormone refractory prostate cancer. Indian J. Urol. 2007;23:35–42. doi: 10.4103/0970-1591.30265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi H., Elemento O., Tavazoie S. Revealing global regulatory perturbations across human cancers. Mol. Cell. 2009;36:900–911. doi: 10.1016/j.molcel.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal P., Choi J.J., Pinheiro L.C., Schenck E.J., Chen R., Jabri A., Satlin M.J., Campion T.R., Jr., Nahid M., Ringel J.B. Clinical Characteristics of Covid-19 in New York City. N. Engl. J. Med. 2020;382:2372–2374. doi: 10.1056/NEJMc2010419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan W.-J., Ni Z.-Y., Hu Y., Liang W.-H., Ou C.-Q., He J.-X., Liu L., Shan H., Lei C.-L., Hui D.S.C., China Medical Treatment Expert Group for Covid-19 Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T., Fan Y., Chen M., Wu X., Zhang L., He T., Wang H., Wan J., Wang X., Lu Z. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19) JAMA Cardiol. 2020;5:811–818. doi: 10.1001/jamacardio.2020.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik T.J., Mohiddin S.A., Dimarco A., Patel V., Savvatis K., Marelli-Berg F.M., Madhur M.S., Tomaszewski M., Maffia P., D’Acquisto F. COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc. Res. 2020;116:1666–1687. doi: 10.1093/cvr/cvaa106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L., Lun A.T.L., Morgan M.D., Marioni J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018;36:421–427. doi: 10.1038/nbt.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamming I., Timens W., Bulthuis M.L.C., Lely A.T., Navis G., van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004;203:631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y., Yang L., Duan X., Duan F., Nilsson-Payant B.E., Yaron T.M., Wang P., Tang X., Zhang T., Zhao Z. Identification of Candidate COVID-19 Therapeutics using hPSC-derived Lung Organoids. bioRxiv. 2020 doi: 10.1101/2020.05.05.079095. [DOI] [Google Scholar]
- Hasan A., Paray B.A., Hussain A., Qadir F.A., Attar F., Aziz F.M., Sharifi M., Derakhshankhah H., Rasti B., Mehrabi M. A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1754293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurich A., Hofmann-Winkler H., Gierer S., Liepold T., Jahn O., Pöhlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014;88:1293–1307. doi: 10.1128/JVI.02202-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob A., Morley M., Hawkins F., McCauley K.B., Jean J.C., Heins H., Na C.-L., Weaver T.E., Vedaie M., Hurley K. Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell. 2017;21:472–488.e10. doi: 10.1016/j.stem.2017.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H.-J., Kim J., Yu J. Androgen receptor genomic regulation. Transl. Androl. Urol. 2013;2:157–177. doi: 10.3978/j.issn.2223-4683.2013.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiser M.J., Roth B.L., Armbruster B.N., Ernsberger P., Irwin J.J., Shoichet B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007;25:197–206. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- Koh K.D., Siddiqui S., Cheng D., Bonser L.R., Sun D.I., Zlock L.T., Finkbeiner W.E., Woodruff P.G., Erle D.J. Efficient RNP-directed Human Gene Targeting Reveals SPDEF Is Required for IL-13-induced Mucostasis. Am. J. Respir. Cell Mol. Biol. 2020;62:373–381. doi: 10.1165/rcmb.2019-0266OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba K., Imai Y., Rao S., Gao H., Guo F., Guan B., Huan Y., Yang P., Zhang Y., Deng W. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Linderman G.C., Zhao J., Kluger Y. Zero-preserving imputation of scRNA-seq data using low-rank approximation. bioRxiv. 2018 doi: 10.1101/397588. [DOI] [Google Scholar]
- Liu B., Li M., Zhou Z., Guan X., Xiang Y. Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS)? J. Autoimmun. 2020;111:102452. doi: 10.1016/j.jaut.2020.102452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madissoon E., Wilbrey-Clark A., Miragaia R.J., Saeb-Parsy K., Mahbubani K.T., Georgakopoulos N., Harding P., Polanski K., Huang N., Nowicki-Osuch K. scRNA-seq assessment of the human lung, spleen, and esophagus tissue stability after cold preservation. Genome Biol. 2019;21:1. doi: 10.1186/s13059-019-1906-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T., Sakari M., Okada M., Yokoyama A., Takahashi S., Kouzmenko A., Kato S. The androgen receptor in health and disease. Annu. Rev. Physiol. 2013;75:201–224. doi: 10.1146/annurev-physiol-030212-183656. [DOI] [PubMed] [Google Scholar]
- Miller A.J., Dye B.R., Ferrer-Torres D., Hill D.R., Overeem A.W., Shea L.D., Spence J.R. Generation of lung organoids from human pluripotent stem cells in vitro. Nat. Protoc. 2019;14:518–540. doi: 10.1038/s41596-018-0104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruth K.S., Day F.R., Tyrrell J., Thompson D.J., Wood A.R., Mahajan A., Beaumont R.N., Wittemans L., Martin S., Busch A.S., Endometrial Cancer Association Consortium Using human genetics to understand the disease impacts of testosterone in men and women. Nat. Med. 2020;26:252–258. doi: 10.1038/s41591-020-0751-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S., Qin M., Shen B., Cai Y., Liu T., Yang F., Gong W., Liu X., Liang J., Zhao Q. Association of Cardiac Injury With Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol. 2020;5:802–810. doi: 10.1001/jamacardio.2020.0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smillie C.S., Biton M., Ordovas-Montanes J., Sullivan K.M., Burgin G., Graham D.B., Herbst R.H., Rogel N., Slyper M., Waldman J. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell. 2019;178:714–730.e22. doi: 10.1016/j.cell.2019.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling T., Irwin J.J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015;55:2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toubiana J., Poirault C., Corsia A., Bajolle F., Fourgeaud J., Angoulvant F., Debray A., Basmaci R., Salvador E., Biscardi S. Kawasaki-like multisystem inflammatory syndrome in children during the covid-19 pandemic in Paris, France: prospective observational study. BMJ. 2020;369:m2094. doi: 10.1136/bmj.m2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran M.G.B., Bibby B.A.S., Yang L., Lo F., Warren A.Y., Shukla D., Osborne M., Hadfield J., Carroll T., Stark R. Independence of HIF1a and androgen signaling pathways in prostate cancer. BMC Cancer. 2020;20:469. doi: 10.1186/s12885-020-06890-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai S.-Y., Ghazizadeh Z., Wang H.-J., Amin S., Ortega F.A., Badieyan Z.S., Hsu Z.-T., Gordillo M., Kumar R., Christini D.J. A human embryonic stem cell reporter line for monitoring chemical-induced cardiotoxicity. Cardiovasc. Res. 2020;116:658–670. doi: 10.1093/cvr/cvz148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabret N., Britton G.J., Gruber C., Hegde S., Kim J., Kuksin M., Levantovsky R., Malle L., Moreira A., Park M.D., Sinai Immunology Review Project Immunology of COVID-19: current state of the science. Immunity. 2020;52:910–941. doi: 10.1016/j.immuni.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Gu H., Brewster B.D., Huang C. Role of endogenous testosterone in TNF-induced myocardial injury in males. Int. J. Clin. Exp. Med. 2012;5:96–104. [PMC free article] [PubMed] [Google Scholar]
- Wang L., Yu P., Zhou B., Song J., Li Z., Zhang M., Guo G., Wang Y., Chen X., Han L., Hu S. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat. Cell Biol. 2020;22:108–119. doi: 10.1038/s41556-019-0446-7. [DOI] [PubMed] [Google Scholar]
- Whitt M.A. Generation of VSV pseudotypes using recombinant ΔG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J. Virol. Methods. 2010;169:365–374. doi: 10.1016/j.jviromet.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Zhu H., Yuan C., Yao C., Luo W., Shen X., Wang J., Shao J., Xiang Y. Clinical and Immune Features of Hospitalized Pediatric Patients With Coronavirus Disease 2019 (COVID-19) in Wuhan, China. JAMA Netw. Open. 2020;3:e2010895. doi: 10.1001/jamanetworkopen.2020.10895. e2010895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaykin D.V. Optimally weighted Z-test is a powerful method for combining probabilities in meta-analysis. J. Evol. Biol. 2011;24:1836–1841. doi: 10.1111/j.1420-9101.2011.02297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D., Hu Q., Liu X., Ji Y., Chao H.-P., Liu Y., Tracz A., Kirk J., Buonamici S., Zhu P. Intron retention is a hallmark and spliceosome represents a therapeutic vulnerability in aggressive prostate cancer. Nat. Commun. 2020;11:2089. doi: 10.1038/s41467-020-15815-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original source data for previously published scRNA-seq of human organ tissues used in this study are available by: heart data from Wang et al. (2020) from the Gene Expression Omnibus (GEO) at GEO: GSE109816, lung and esophagus data from Madissoon et al. (2019) at https://www.tissuestabilitycellatlas.org, colon data from Smillie et al. (2019) at https://singlecell.broadinstitute.org/single_cell/study/SCP259/intra-and-inter-cellular-rewiring-of-the-human-colon-during-ulcerative-colitis.
Original source data for the AR ChIP-seq experiment is available from GEO at GEO: GSM3148987.
Original source data for AR knockdown RNA-seq are available from GEO under accession code GEO: GSE114052, GEO: GSE128515, and GEO: GSE139962.
Training datasets and code used for in silico drug screening are available upon request to other researchers.
Code used for SEA drug target prediction is available upon request to other researchers.
The raw dataset from bulk and scRNA-seq of hESC-derived lung organoid are available on GEO under accession number GEO: GSE161264.
Yale New Haven Hospital and UK Biobank patient datasets and code used for analysis are available upon request to other researchers.