

Abstract
RNA sequencing (RNA-seq) is currently the standard method for genome-wide expression profiling. RNA-seq reads often need to be mapped to a reference genome before read counts can be produced for genes. Read trimming methods have been developed to assist read mapping by removing adapter sequences and low-sequencing-quality bases. It is however unclear what is the impact of read trimming on the quantification of RNA-seq data, an important task in RNA-seq data analysis. In this study, we used a benchmark RNA-seq dataset and simulation data to assess the impact of read trimming on mapping and quantification of RNA-seq reads. We found that adapter sequences can be effectively removed by read aligner via ’soft-clipping’ and that many low-sequencing-quality bases, which would be removed by read trimming tools, were rescued by the aligner. Accuracy of gene expression quantification from using untrimmed reads was found to be comparable to or slightly better than that from using trimmed reads, based on Pearson correlation with reverse transcriptase-polymerase chain reaction data and simulation truth. Total data analysis time was reduced by up to an order of magnitude when read trimming was not performed. Our study suggests that read trimming is a redundant process in the quantification of RNA-seq expression data.
INTRODUCTION
RNA-seq technology is a powerful tool for rapid and comprehensive profiling of gene expression at a genome scale (1). The bioinformatic analysis of data generated from this technology however requires significant amount of CPU time and disk storage, due to vast amount of sequence reads generated for even a small RNA-seq experiment. An RNA-seq data analysis includes a number of steps, making it a non-trivial task. There are continuous efforts in the field to try to reduce the data analysis complexity and improve its efficiency (2–5).
Read trimming tools have been developed to remove adapter sequences and bases with low sequencing quality from sequencing reads such as RNA-seq reads, in order to help read aligners to achieve a better read mapping result (6,7). Read trimming is the first operation in a sequencing data analysis pipeline that modifies the read sequences produced by a sequencer. The changes it makes to the raw read sequences may impact all the subsequent steps in the analysis pipeline.
An important step in analyzing RNA-seq data is the quantification of RNA-seq reads, which assigns reads to genes and counts the number of reads assigned to each gene (8,9). RNA-seq quantification is required by many statistical methods that were developed to discover genes with significant expression changes (3,10–11). The accuracy of RNA-seq quantification certainly affects the performance of these methods and other downstream tools. However, it is unclear how read trimming affects RNA-seq quantification and if it can improve the accuracy of RNA-seq quantification. Del Fabbro et al. reported that the total number of reads mapping to annotated genes was reduced when read trimming was performed (12). Didion et al. performed a similar study but found that read trimming led to more reads mapping to annotated genes (13). Williams et al. found that read trimming resulted in a reduced correlation of RNA-seq data to the microarray data (14). To resolve this long-standing issue, more rigorous investigations performed directly on individual genes using data with ground truth are required.
On the other hand, modern read aligners are known to be able to ‘soft-clip’ read bases that cannot be mapped along with the majority of bases in a read (15–17). Soft-clipped bases are still included in the mapping results of the reads but are marked as ‘soft-clipped’. Both soft-clipping and read trimming remove bases from the ends of the reads, but soft-clipping is performed within the read mapping procedure whereas read trimming is performed prior to mapping as a standalone procedure. When performing read trimming, a lot of parameters need to be specified such as adapter sequences and threshold for quality filtering. In contrast, soft-clipping is performed solely based on the matching of read bases with reference sequences and it does not require users to provide any parameters. It would be really interesting to compare soft-clipping with read trimming, however surprisingly no studies have been carried out to do so to the best of our knowledge.
In this study, we first compared read trimming tools to the soft-clipping implemented in the Subread aligner (2,16). Then we assessed if read trimming can improve mapping and quantification of RNA-seq reads. We used a benchmark RNA-seq dataset generated in the SEQC project (18) in this evaluation. The SEQC project also produced real-time polymerase chain reaction (PCR) data for >900 genes, which were used as the truth in our evaluation of quantification accuracy of gene expression.
MATERIALS AND METHODS
SEQC RNA-seq data and TaqMan RT-PCR data
A benchmark RNA-seq dataset generated in the SEQC project was used in this study. The SEQC project is the third stage of the MAQC (MicroArray Quality Control) project. Two reference RNA samples were sequenced in the SEQC project: Universal Human Reference RNA (UHRR) and Human Brain Reference RNA (HBRR). This study includes RNA-seq data generated from a UHRR library and a HBRR library. A total of 15 million pairs of 100 bp reads was generated from the sequencing of each library. These data are part of the large SEQC dataset deposited into the Gene Expression Omnibus database (GSE47774).
In the SEQC project, expression levels of >1000 genes were validated by the TaqMan RT-PCR technique and 949 of these genes have matched symbols with genes in the RNA-seq data. RT-PCR expression levels of these 949 genes were used as the ‘truth’ of gene expression in the evaluation. The TaqMan RT-PCR data are available from the seqc Bioconductor package (19).
Simulation data
Three simulation datasets were generated to simulate different levels of adapter contamination in RNA-seq data. A total of 15 million pairs of 100 bp reads were created for each dataset based on human genome GRCh38/hg38. Reads were extracted from RefSeq gene regions in the genome. RPKM (reads per kilobases per million mapped reads) values were generated from an exponential distribution and randomly assigned to genes. Number of read pairs that need to extracted from each gene was then calculated based on RPKM value of the gene, gene length and library size. Fragment lengths were randomly generated according to a normal distribution with mean 200 and variance 30. The extracted read pairs may fall within exons or span exons.
Illumina TruSeq adapter sequences (version 3) were added to the datasets at base percentages of 0.1, 0.5 and 1%, corresponding to 1.1, 5.5 and 11.0% of adapter-containing reads, respectively. Length of adapter sequences inserted to the reads follows an exponential distribution (λ = 0.1) to simulate variable adapter lengths observed in the real data.
To make the simulation data as close to the real data as possible, we also added biological variants and sequencing errors to the data. Biological SNPs (single nucleotide polymorphism) and short indels (insertions and deletions) were randomly introduced to the genome at the rates of 0.0009 and 0.0001, respectively, before RNA-seq read sequences were extracted. Sequencing errors were simulated by altering read bases based on the Phred scores in each read in the SEQC 100 bp paired-end data.
Software
Two read trimming tools, Trimmomatic and TrimGalore, were included in this study. TrimGalore performs adapter removal and quality filtering via calling the Cutadapt tool (7). Trimmomatic has two trimming modes: ‘adapters and SW’ mode and ‘adapters and MI’ mode. In ‘adapters and SW’ mode, a sliding window approach is used to remove read bases that have a low sequencing quality. In ‘adapters and MI’ mode, a maximum information quality filtering approach is applied for removing low quality bases. Adapter sequences are detected and removed in both modes.
When running read trimmers, we tried to keep their default settings where possible. TrimGalore was run with parameters –illumina -j 8 –paired. The ‘adapters and SW’ mode of Trimmomatic was run with parameters PE -threads 8 -phred33 ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36. The ‘adapters and MI’ mode of Trimmomatic was run with parameters PE -threads 8 -phred33 ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 MAXINFO:50:0.5 MINLEN:36.
Trimmed reads and untrimmed reads were mapped to the human reference genome GRCh38/hg38 using the ‘align’ function in Rsubread package (2). ‘align’ is an R wrapper function for the Subread aligner (16). Soft-clipping is automatically performed by Subread. Reads counts were generated for genes using the featureCounts tool (9). The Rsubread inbuilt annotation for human genes (2), which is an modified version of NCBI RefSeq gene annotation, was used in the quantification of gene expression. Read counts for each gene were converted to log2-RPKM expression values and then compared against the RT-PCR gene expression data which is also at log2 scale.
All software tools were executed with eight CPU threads on a CentOS 6 Linux server with 24 Intel Xeon 2.60 GHz CPU cores and 512 GB of memory. Versions of these software tools are: Trimmomatic v0.39, TrimGalore v0.6.2 and Rsubread v2.0.0.
RESULTS
Using RNA-seq data generated in the SEQC project, we compared read trimming performed prior to read mapping to soft-clipping that was carried out during read mapping at base level, read level and gene level. We used RT-PCR data generated for 949 genes to assess the impact of read trimming on the accuracy of gene expression quantification.
Base-level comparison
We found that 2.3–4.6% of all read bases included in each library were trimmed off, and Trimmomatic removed twice as many bases as TrimGalore (Supplementary Table S1). Total number of successfully mapped bases was reduced by 1.3–4.0% when trimming was applied (Table 1). Subread was found to soft-clip 18–29% of bases that were trimmed off by read trimmers, indicating that a large number of trimmed bases were rescued during read mapping. Out of those commonly removed bases by Subread and a trimmer, 10–27% were found to be adapter sequences and the rest were low-quality bases (Supplementary Table S1). Subread was able to soft-clip almost all adapter sequences (94%) reported and removed by Trimmomatic (Supplementary Table S2). TrimGalore reported about six times more adapter sequences than Trimmomatic, however TrimGalore is likely to have a high false positive rate in adapter calling because a lot of adapter sequences it called are very short. Nonetheless, ∼30% of adapter sequences reported by TrimGalore were soft-clipped by Subread. Put together, Subread was found to be able to effectively remove adapter sequences from the raw reads and rescue a lot of bases with relatively low sequencing qualities which would otherwise be removed by read trimmers. This has led to a non-trivial increase in the number of successfully mapped read bases.
Table 1.
Percentages of mapped read bases with or without read trimming prior to mapping
| Method | UHRR (%) | HBRR (%) |
|---|---|---|
| No trimming + Subread | 86.4 | 85.5 |
| Trimmomatic–adapters and SW + Subread | 82.4 | 81.7 |
| Trimmomatic–adapters and MI + Subread | 83.2 | 82.3 |
| TrimGalore + Subread | 85.1 | 84.2 |
Subread was used for mapping of untrimmed or trimmed reads.
Read-level comparison
We then examined the impact of read trimming on read mapping results. Read trimming may cause a slight change to the mapping location of a read or cause a read to map to a different exon of the same gene, but this normally would not change the quantification of expression of the gene because the read still overlaps the same gene. We therefore call a read as a concordantly mapped read if read trimming only results in a <100 bp change in its mapping location or results in the read mapping to an alternative exon from the same gene. We found that >98% of reads were concordantly mapped when comparing mapping of TrimGalore trimmed reads and untrimmed reads (Supplementary Table S3). Mapping concordance between Trimmomatic trimmed reads and untrimmed reads was ∼97%. Mapping concordances between reads trimmed by different trimmers were also found to be ∼97%. The mapping analysis shows that read trimming only affects the mapping of a very small fraction of reads in a library and that the mapping difference between trimmed and untrimmed reads is similar to the mapping difference between reads that were trimmed by different trimmers.
Gene-level comparison
Finally we investigated if read trimming will affect the quantification of gene expression in RNA-seq data. For both trimmed and untrimmed data, we counted the number of mapped reads assigned to each gene using the featureCounts program (9). Read counts were then converted to log2-RPKM expression values for each gene. The SEQC RNA-seq benchmarking study validated the expression of ∼1000 genes using TaqMan RT-PCR technique (18). 949 of these genes matched the RefSeq genes and were included in this evaluation. We used RT-PCR expression values of these 949 genes as the truth to assess if read trimming is beneficial to the quantification of gene expression in RNA-seq data. In addition to the original 100 bp paired-end SEQC data, we also generated a 50 bp single-end SEQC data by extracting the first reads (R1 reads) from the 100bp paired-end SEQC data and then truncating them to 50 bp long. The first 50 bases were removed from each R1 read so that adapter bases and low-quality bases (usually more abundant at the 3’ end of the Illumina reads) can be kept allowing us to evaluate the impact of trimming these bases on gene expression quantification.
Table 2 shows that performing read trimming before read mapping does not improve the correlation of gene expression values with true values. In fact, the correlation has a slight decrease when the reads were trimmed by TrimGalore or Trimmomatic ‘adapters and SW’ mode.
Table 2.
Correlation of trimmed and untrimmed RNA-seq data with the TaqMan RT-PCR data
| 100 bp PE | 50 bp SE | |||
|---|---|---|---|---|
| Method | UHRR | HBRR | UHRR | HBRR |
| No trimming + Subread | 0.851 | 0.870 | 0.848 | 0.870 |
| Trimmomatic–adapters and SW + Subread | 0.850 | 0.870 | 0.848 | 0.869 |
| Trimmomatic–adapters and MI + Subread | 0.850 | 0.871 | 0.849 | 0.869 |
| TrimGalore + Subread | 0.850 | 0.870 | 0.849 | 0.869 |
Shown are the coefficients of Pearson correlation between log2 expression values of 949 genes measured by the TaqMan RT-PCR technique and their RNA-seq expression levels generated from using each method (log2-RPKM). ‘100 bp PE’ in the table denotes the 100 bp paired-end SEQC dataset. First reads (R1 reads) in this dataset were extracted and truncated to 50 bp long to generate the 50bp single-end dataset used here (‘50 bp SE’).
We have also generated simulation data to assess if read trimming is helpful for RNA-seq expression quantification. We generated three simulation RNA-seq datasets with different levels of adapter contamination. Sequencing errors were introduced to the simulation data based on the error profiles of the 100 bp paired-end SEQC data to make the simulation data as close to the real data as possible (see ‘Materials and Methods’ section for more details). We ran all the methods on the simulation data and computed the coefficients of Pearson correlation between log2-RPKM expression values of genes calculated from each method and the true log2-RPKM expression values of genes we generated in the simulation. In line with the evaluation results from the SEQC data, read trimming was also found to make no discernible difference in the quantification accuracy in the simulation (Supplementary Table S4).
Taken together, our evaluation results from using both real data and simulation data clearly showed that using untrimmed reads to quantify expression levels of genes yielded comparable or slightly better quantification accuracy than using trimmed reads.
Running time and disk usage
Performing read trimming was found to result in a significant increase in data analysis time (Figure 1). The total running time for producing mapped reads was increased by more than an order of magnitude when using TrimGalore for trimming, compared to no trimming performed. Trimming by Trimmomatic increased the running time by nearly five times. Furthermore, the amount of disk storage required increased by ∼40% due to the need to store trimmed read data (Supplementary Table S5). Read trimming has become a significant computational burden in the analysis of RNA-seq expression data.
Figure 1.
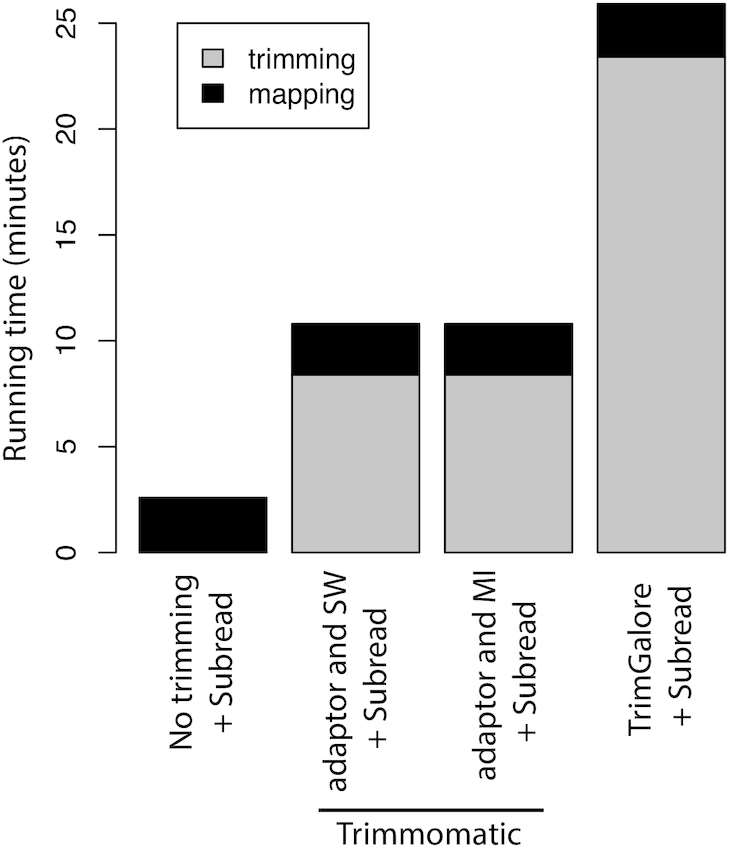
Time cost of different methods running on a UHRR RNA-seq dataset that includes 15 million 100 bp read pairs. All software tools were run with eight CPU threads. Input data to trimming and mapping tools are in gzipped FASTQ format which is the standard format of data generated by Illumina sequencers.
DISCUSSION
In this study we demonstrated that the reference-based soft-clipping implemented in the Subread aligner can effectively remove adapter sequences introduced by sequencers, a major goal that read trimming tools try to achieve. Subread’s soft-clipping was also found to successfully rescue a lot of low-quality bases. Subread makes use of both sequencing quality scores and reference sequences to make a informed decision on whether low-quality read bases should be removed from the read sequence, whereas read trimming tools only rely on the sequencing quality scores for the trimming of such bases. Read bases can be over-trimmed or under-trimmed by trimming tools when different quality thresholds are used. Over-trimming is particularly problematic because read bases lost during trimming cannot be recovered by read aligner. It is very difficult to determine the best threshold for quality trimming just based on the sequencing quality scores alone.
Although we expect that most state-of-the-art RNA-seq aligners are capable of identifying and removing adapter sequences and low-quality bases, Subread has an unique advantage in doing so thanks to its highly flexible and powerful ‘seed-and-vote’ mapping paradigm. Under this paradigm, a large number of seed sequences (16 bp mers) are extracted from each read and then mapped to the reference genome concurrently to determine the candidate mapping locations of the read. Because the extracted seed sequences cover the entire read sequence and the number of seeds is large, the presence of adapter bases and/or low-quality bases is unlikely to cause the read to fail to map as long as there are enough mappable bases existing in the read. Seed-and-vote is therefore more tolerant of adapter and low-quality bases for read mapping than the conventional seed-and-extend paradigm.
The Subread mapping comprises of two passes. In its first pass, it utilizes the consensus mapping of seed sequences extracted from each read to determine the initial mapping locations of all the reads including exon-spanning reads. It also detects short indels (insertions and deletions) and associated breakpoints in this pass. In the second pass, Subread finalizes the alignment of each read by maximizing the largest mapping region in each read and by utilizing the breakpoint data collected in the first pass. The read bases that cannot be mapped along with the rest of the read are also soft-clipped in the second pass. This two-pass procedure allows a reliable detection of adapter sequences and sequencing errors in the reads. It also maximizes the opportunity to confidently include more bases in the final mapping results, enabling more useful data to be provided to downstream analyses such as differential expression analysis and variant analysis (e.g. detection of single nucleotide variant and gene fusion). Subread is the first aligner that implemented this two-pass mapping strategy, which was later adopted in many other aligners.
We compared Subread to the popular RNA-seq aligner STAR (15) on the accuracy of gene expression quantification, using untrimmed SEQC 100 bp paired-end data. We ran STAR with default setting or with the end-to-end mapping setting (‘–alignEndsType EndToEnd’) to disable soft-clipping. Subread was found to yield better Pearson correlation between RNA-seq data and RT-PCR data than STAR in both of its two settings (Supplementary Table S6). Also as expected, running STAR with end-to-end mapping setting resulted in a reduced correlation of RNA-seq data with RT-PCR data due to the failure to soft-clip adapter sequences and low-quality bases, compared to using its default setting (Supplementary Table S6). These results demonstrate that soft-clipping improves the quality of read mapping, which leads to the improvement of gene expression quantification and that the soft-clipping approach implemented in Subread should contribute to its better quantification results.
In conclusion, we found that read trimming performed prior to Subread mapping did not improve read mapping results and consequently the accuracy of gene expression quantification was not improved either. The quantification accuracy was actually found to be slightly higher when read trimming was not performed. Total RNA-seq quantification time was also found to be reduced by up to an order of magnitude for the datasets used in this study, without read trimming being performed. Our study suggested that RNA-seq reads do not need to be trimmed prior to mapping for the purpose of quantifying expression levels of genes.
DATA AVAILABILITY
All the data and analysis code used in this study can be accessed at the following URL: https://github.com/ShiLab-Bioinformatics/ReadTrimming.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Prof. Gordon K Smyth for suggesting this study.
Contributor Information
Yang Liao, Olivia Newton-John Cancer Research Institute, Heidelberg, Victoria 3084, Australia; School of Cancer Medicine, La Trobe University, Heidelberg, Victoria 3084, Australia; Department of Medical Biology, The University of Melbourne, Parkville, Victoria 3010, Australia; The Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, Victoria 3052, Australia.
Wei Shi, Olivia Newton-John Cancer Research Institute, Heidelberg, Victoria 3084, Australia; School of Cancer Medicine, La Trobe University, Heidelberg, Victoria 3084, Australia; The Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, Victoria 3052, Australia; School of Computing and Information Systems, The University of Melbourne, Parkville, Victoria 3010, Australia.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
Australian National Health and Medical Research Council, Project grants [1023454, 1128609 to W.S.]; Walter and Eliza Hall Institute Centenary Fellowship sponsored by CSL (to W.S.); Victorian State Government, Operational Infrastructure Support; Australian Government NHMRC IRIISS.
Conflict of interest statement. None declared.
REFERENCES
- 1. Wang Z., Gerstein M., Snyder M.. Rna-seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009; 10:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liao Y., Smyth G.K., Shi W.. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019; 47:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K.. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Law C.W., Chen Y., Shi W., Smyth G.K.. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014; 15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen Y., Lun A.T., Smyth G.K.. From reads to genes to pathways: differential expression analysis of rna-seq experiments using rsubread and the edger quasi-likelihood pipeline. F1000Res. 2016; 5:1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011; 17:10–12. [Google Scholar]
- 8. Anders S., Pyl P.T., Huber W.. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liao Y., Smyth G.K., Shi W.. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014; 30:923–930. [DOI] [PubMed] [Google Scholar]
- 10. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Del Fabbro C., Scalabrin S., Morgante M., Giorgi F.M.. An extensive evaluation of read trimming effects on Illumina NGS data analysis. PLoS One. 2013; 8:e85024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Didion J.P., Martin M., Collins F.S.. Atropos: specific, sensitive, and speedy trimming of sequencing reads. PeerJ. 2017; 5:e3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Williams C.R., Baccarella A., Parrish J.Z., Kim C.C.. Trimming of sequence reads alters RNA-Seq gene expression estimates. BMC Bioinformatics. 2016; 17:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. Star: ultrafast universal rna-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liao Y., Smyth G.K., Shi W.. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013; 41:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Langmead B., Salzberg S.L.. Fast gapped-read alignment with bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Consortium S.M.I. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat. Biotechnol. 2014; 32:903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liao Y., Shi W.. seqc: RNA-seq data generated from SEQC (MAQC-III) study. R Package Version 1.20.0. 2019; http://bioconductor.org/packages/release/data/experiment/html/seqc.html. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data and analysis code used in this study can be accessed at the following URL: https://github.com/ShiLab-Bioinformatics/ReadTrimming.