

Group B streptococcus (GBS) is a human-pathogenic bacterium inducing a strong inflammatory response that may be detrimental for host tissues if not finely regulated. The inflammatory response can be modulated by different molecular mechanisms, among which growing evidence points toward the crucial role of microRNAs (miRNAs). Regarding innate inflammatory response, studies have reported that miR-223 is essential for the control of granulocyte proliferation and activation. Moreover, a number of investigations on miRNA expression profiling performed in various inflammatory settings have revealed that miR-223 is among the top differentially expressed miRNAs.
KEYWORDS: group B streptococcus, inflammation, innate immunity, miR-223
ABSTRACT
Group B streptococcus (GBS) is a human-pathogenic bacterium inducing a strong inflammatory response that may be detrimental for host tissues if not finely regulated. The inflammatory response can be modulated by different molecular mechanisms, among which growing evidence points toward the crucial role of microRNAs (miRNAs). Regarding innate inflammatory response, studies have reported that miR-223 is essential for the control of granulocyte proliferation and activation. Moreover, a number of investigations on miRNA expression profiling performed in various inflammatory settings have revealed that miR-223 is among the top differentially expressed miRNAs. Yet the dynamic pattern of expression of miR-223 in vivo with respect to the evolution of the inflammatory process, especially in microbial infection, remains elusive. In this study, we analyzed the kinetic expression of miR-223 in an inflammatory model of GBS-induced murine pneumonia and looked for correlates with inflammatory markers, including innate cell infiltrates. We found that miR-223 expression is rapidly induced at very early time points (3 to 6 h postinfection [p.i.]) mainly by lung-infiltrating neutrophils. Interestingly, the level of miR-223 accumulating in the lungs remains higher at later stages of infection (24 h and 48 h p.i.), and this correlates with reduced expression of primary inflammatory cytokines and chemokines and with a shift in infiltrating monocyte and macrophage subtypes toward a regulatory phenotype. Transient inhibition of miR-223 by an antagomir resulted in significant increase of CXCL2 expression and partial enhancement of infiltrating neutrophils in GBS-infected lung tissues. This suggests the potential contribution of miR-223 to the resolution phase of GBS-induced acute inflammation.
INTRODUCTION
Group B streptococcus (GBS), or Streptococcus agalactiae, is a Gram-positive bacterium responsible for severe pneumonia, meningitis, and sepsis in humans (1–3). GBS is endowed with a high inflammatory potential that may be detrimental for host tissues. Animal and human studies have shown that GBS infection is associated with production of high levels of inflammatory cytokines (4–7). The immune inflammatory response can be regarded as beneficial for the host, as it promotes a defense mechanism against the infection; however, the magnitude of the inflammatory response has to be tightly controlled to avoid host tissue damage (7–9).
MicroRNAs (miRNAs) have emerged as important regulators of many biological processes, and their contribution in the modulation of immunity, including the inflammatory response, is now well acknowledged (10–14). miRNAs are small noncoding RNA sequences of more or less 22 nucleotides that regulate gene expression at the posttranscriptional level by repressing translation of specific mRNA targets through complementary pairing with their 3′ untranslated regions (15, 16). Among miRNAs involved in inflammatory response, there has been growing interest in miR-223 given its key role in innate immunity (17, 18). miR-223 is expressed by hematopoietic cells of myeloid lineage and is highly conserved across species, which highlights its important biological function (19, 20). Studies have shown that miR-223 deficiency results in spontaneous inflammation, which is characterized by massive neutrophil proliferation and hyperactivation, hence providing direct evidence for the contribution of this miRNA in granulopoiesis (21–23). Moreover, aberrant miR-223 expression was shown to be associated with pathophysiology of a number of human diseases (17, 24–29). Regarding inflammatory diseases, elevated levels of miR-223 expression were found in the lungs of patients with chronic obstructive pulmonary disease (30). In contrast, low levels of miR-223 expression have been reported for septic patients (31). On the other hand, animal studies have shown that mice challenged with lipopolysaccharide (LPS) exhibited a rapid increase of miR-223 (32). These studies performed in different inflammatory models clearly indicate that miR-223 is among the top differentially expressed miRNAs. However, there is a paucity of data with regard to dynamic expression of miR-223 in relation to the evolution of the inflammatory process in vivo, especially in microbial infection models. In GBS infection, Pu et al. showed that mammary gland tissues from dairy cows challenged locally with S. agalactiae exhibited a high degree of miR-223 upregulation (33). Similarly, in vitro culture studies demonstrated that bovine monocyte-derived macrophages significantly upregulated miR-223 expression upon challenge with GBS (34). In this work, we sought to determine the kinetic of expression of miR-223 within lung tissues upon GBS-induced pneumonia and looked for correlates with local inflammatory parameters, including innate immune cell infiltrates.
RESULTS
Kinetics of expression of miR-223 upon GBS infection.
To investigate the kinetics of miR-223 expression in the context of pathogen-induced innate inflammatory responses, we used an experimental model of GBS-induced murine pneumonia. Mice were intranasally (i.n.) inoculated with a sublethal dose of bacteria (1 × 108 CFU) and were followed up to 48 h postinfection (p.i.). Under this experimental condition, infected mice exhibited acute inflammatory symptoms and weight loss, but no mortality was observed; bacteria were cleared from the lungs within 48 h (see Fig. S1 in the supplemental material). We evaluated the relative expression of miR-223-3p in the lung tissues by real-time PCR. As shown in Fig. 1, miR-223 was induced in the first hours of infection, with a peak at 6 h p.i. The expression decreased slightly at 24 h p.i. and remained constant until 48 h p.i. When contrasted with bacterial burden, miR-223 expression correlated negatively with GBS counts recovered from the lung tissues (Fig. 2). As a comparison, we also evaluated the level of miR-155, a proinflammatory miRNA which is mainly expressed by lymphocytes and involved in adaptive immune response through enhancing inflammatory T cell development (35, 36). The level of miR-155 was found to be significantly elevated in the fetal lung in a nonhuman primate model of choriodecidual GBS infection (37). As expected, the expression of miR-155 was delayed compared to that of miR-223, as it increased significantly only at 24 h p.i. and reached a high level at 48 h p.i. Our finding indicates a rapid expression of the anti-inflammatory miR-223 during the first hours of infection, which may impact the evolution of the innate inflammatory process and bacterial burden.
FIG 1.
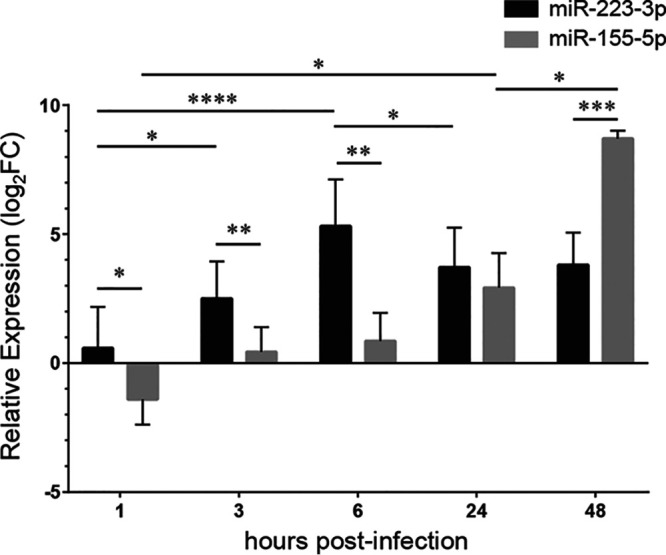
Kinetics of miR-223-3p and miR-155-5p expression in the lungs of mice infected with Streptococcus agalactiae (GBS). Relative levels of miR-223 and miR-155 were measured using real-time PCR (n = 5 to 10 for each time point). Mice injected with PBS were used as a control. The values were normalized with three internal controls: RNU6b, snoRNA-202, and snoRNA-234. The fold change values (2−ΔΔCT) are relative to those with PBS treatment. Data are means ± SD and are representative of those from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (Mann-Whitney U test).
FIG 2.
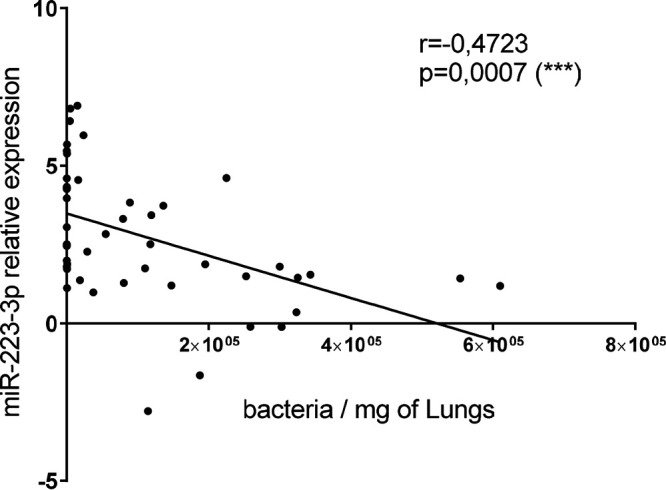
miR-223 expression inversely correlates with GBS counts in the lungs. Mice were infected intranasally with 1 × 108 CFU of GBS (detection limit: 0.75 bacteria/mg of lung). Relative levels of miR-223 expression were measured by RT-PCR using three internal controls: RNU6b, snoRNA-202, and snoRNA-234. The fold change values (2−ΔΔCT) are relative to those with PBS treatment. Significant results are indicated by asterisks. Data are representative of those from three independent experiments.
Dynamic of the innate immune infiltrating cells in the lung upon GBS infection.
To address whether the kinetics of miR-223 expression could be linked to the resolution phase of GBS-induced inflammation, we examined in parallel the dynamic of infiltrating innate immune cells along the infection. Intranasal inoculation of mice with GBS induces a strong inflammatory response characterized by massive infiltration of immune cells in the lungs. We analyzed by flow cytometry the recruitment of different innate immune cells within pulmonary tissues (Fig. 3). The gating strategy used to differentiate the innate cells is shown in Fig. S2 and S3. As expected, there was a progressive increase of leukocytes that reached maximal level at 24 h p.i. We evaluated recruitment of neutrophils, key cell players representing the front line defense against invading pathogens (38). As shown in Fig. 3, neutrophils were recruited as early as 3 h p.i., reached a peak at 6 h p.i., and started to decrease significantly at 48 h p.i. Next, we looked at macrophages, the phagocytic cells that play an important role in the innate immunity and tissue homeostasis (39). Two macrophage subpopulations termed according to their anatomical location in the lung, alveolar macrophages (AM) and interstitial macrophages (IM), can be identified based on their specific markers and function. While the major function of AM is to cure infection in the alveoli by phagocytosis (40), IM are known to play a role in the regulation of the inflammation and antigen presentation (41). As shown in Fig. 3, the level of AM within the lung tissues remained constant, with no significant change along the infection. However, the level of IM went from very low during the first hours of infection to higher at the late phase of infection, indicating the progressive accumulation of cells mainly endowed with regulatory function.
FIG 3.

Innate immune cell recruitment in the lungs of mice infected with Streptococcus agalactiae. Flow cytometry was performed at different time points of infection (n = 5 to 10 for each time point) using specific antibodies. Levels of leucocytes (CD45+), neutrophils (CD45+ CD11b+ MHCII− CD24+ Ly6G+), alveolar macrophages (CD45+ CD64+ CD24− CD11c+ CD11b−), and interstitial macrophages (CD45+ CD64+ CD24− CD11c− CD11b+) are shown. Mice injected with PBS as a control were also monitored at different time points, but no significant change was observed. The PBS bars represent the means of data recorded at different time points. Significant results are indicated (*, P < 0.05; **, P < 0.01; ***, P < 0.001 [Mann-Whitney U test]). Data are means ± SD and are representative of those from 3 independent experiments.
Next, we examined monocytes that are known to have a cardinal role in the immune defense against microbial infection (42). In mice, different monocyte subpopulations with distinct features and functions can be defined according to the expression of Gr-1, referred to as Ly6C (43). We clearly observed three distinct subpopulations expressing high, intermediate, and low Ly6C levels. Ly6C high (Ly6Chi) monocytes are considered proinflammatory, while Ly6C intermediate (Ly6Cint) are thought to be less inflammatory and mainly endowed with antigen-presenting cell capacity. These cells are also believed to differentiate to Ly6C low (Ly6Clo) patrolling monocytes (44, 45). Interestingly, GBS infection induced a sharp increase of Ly6Chi monocytes at 6 h p.i., which tended to decrease at 24 h and 48 h p.i., whereas a gradual increase of Ly6Cint and Ly6Clo monocytes was observed from 6 h p.i. (Fig. 4). These results indicate that monocytes that are likely endowed with anti-inflammatory properties are progressively the predominant cells at the late phase of infection.
FIG 4.

Recruitment of monocyte subpopulations in the lungs of mice infected with Streptococcus agalactiae. Flow cytometry was performed during infection (n = 5 to 10 for each time point) using specific antibody. Monocytes (CD45+ MHCII- CD11b+ CD64+) with different Ly6C phenotypes—Ly6C high (Ly6Chi), Ly6C intermediate (Ly6Cint), and Ly6C low (Ly6Clo)—were quantified. Mice injected with PBS as a control were also monitored at different time points, but no significant changes were observed. The PBS bars represent the means of data recorded at different time points. Significant results are indicated (*, P < 0.05; **, P < 0.01; ***, P < 0.001 [Mann-Whitney U test]). Data are means ± SD and are representative of those from 3 independent experiments.
Taken together, these experiments indicate that the accumulation of miR-223 within infected cells coincided with infiltration of monocytes and macrophage subtypes displaying regulatory phenotypes.
Kinetic evaluation of inflammatory cytokines and chemokines in the lung tissues upon GBS infection.
To further evaluate the inflammatory process in the lung tissues during GBS infection with regard to time course expression of miR-223, we quantified the local expression of primary inflammatory cytokines interleukin 1β (IL-1β) and tumor necrosis factor alpha (TNF-α) by enzyme-linked immunosorbent assay (ELISA) (Fig. 5). As expected, a strong production of these cytokines was seen at early time points of GBS infection, with a peak at 6 h p.i. Both cytokines returned to basal level at 48 h p.i. We also evaluated by reverse transcription-quantitative PCR (qRT-PCR) the mRNA transcripts of CXCL1 and CXCL2, two chemoattractant chemokines for neutrophils and monocytes. The expression of CXCL1 mRNA reached maximal level at 1 h and 3 h p.i. and decreased significantly from 6 h p.i. (Fig. 5). A similar profile was observed for CXCL2 transcript, with a slight peak shift to 6 h p.i. Of note, the decrease in expression levels of these inflammatory cytokines and chemokines was associated with the accumulation of a large amount of miR-223 within infected tissues (Fig. 1).
FIG 5.

Evaluation of cytokines and chemokines in the lungs of mice infected with Streptococcus agalactiae (GBS). TNF-α and IL-1β were measured in suspension fluids of pulmonary tissue by ELISA. Relative mRNA transcripts of CXCL1 and CXCL2 were evaluated in the lungs using real-time RT-PCR (n = 5 to 10 for each time point). Mice injected with PBS as a control were also monitored at different time points, but no significant changes were observed. The PBS bars represents the means of data recorded at different time points. The values were normalized with three internal controls, GAPDH, beta-actin, and RPL-19. Data are means ± SD and are representative of those from 3 independent experiments. Significant results are indicated (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 [Mann-Whitney U test]).
Neutrophils are the major cellular source of miR-223-3p expressed in lung tissues upon GBS infection.
We noticed that the profile of kinetic expression of miR-223 (Fig. 1) matched the dynamic of neutrophil recruitment within lung tissues (Fig. 3), both peaking at 6 h p.i. and remaining significantly higher at the late phase of infection, with a trend toward a decrease at 24 and 48 h p.i. This suggests that neutrophils recruited during the inflammatory process could be the major cellular source of miR-223. To test this, we evaluated the relative expression of miR-223 in lung cell fractions isolated 24 h p.i. from GBS-infected mice before and after immune-magnetic depletion of Ly6G+ neutrophils. As shown in Fig. 6, depletion of neutrophils resulted in a sharp decrease of the expression of miR-223. A similar trend was observed at the basal level in cell fractions from control mice injected with phosphate-buffered saline (PBS), revealing a steady-state expression of miR-223 in neutrophils. These data indicate that neutrophils recruited massively in the context of GBS-induced inflammation are the predominant cellular source of miR-223.
FIG 6.

The neutrophils are the major source of miR-223 in lungs of mice infected with Streptococcus agalactiae. Cell fractions from the lungs of mice injected with GBS or PBS were analyzed by flow cytometry before and after depletion of neutrophils using immunomagnetic Ly6G antibody (A). Percentages of neutrophils in cell fractions isolated from mice injected with GBS and PBS (control) are indicated in panel B. Relative expression of miR-223 (C) in cells derived from mice injected with GBS or PBS was evaluated by real-time RT-PCR before and after Ly6G+ cell depletion. The values were normalized with three internal controls, RNU6b, snoRNA-202, and snoRNA-234. Data are means ± SD and are representative of those from 3 independent experiments. *, P < 0.05; ****, P < 0.0001 (Mann-Whitney U test).
Transient inhibition of miR-223 by an antagomir results in increase of CXCL2 production and partial enhancement of neutrophil count in lung tissues during GBS infection.
A previous study in tuberculosis infection model highlighted the key role of miR-223 as a negative regulator of the innate inflammatory response by targeting CXCL2, a chemokine involved in neutrophil recruitment (46). To evaluate the implication of miR-223 in GBS infection, we performed transient inhibition experiments by intranasal injection of an antagomiR-223 or anti-scrambled RNA as a control 30 min before GBS challenge and we measured changes in the level of CXCL2 mRNA and neutrophil counts within the lungs at 6 h postinfection, the time when both parameters reached their peak under GBS infection. We found that the administration of antagomiR-223 strongly impacts the expression of miR-223. Consequently, antagomiR-223-treated mice exhibited a significant increase in the expression of CXCL2 compared to controls (Fig. 7). We also observed a slight increase, albeit not significant (P = 1), in infiltrating neutrophil counts in lung tissues of antagomiR-223-treated mice compared to those in controls. This transient-inhibition experiment supports previous findings that miR-223 targets CXCL2, one of the chemoattractant molecules implicated in neutrophil recruitment, which may contribute to host mechanisms that aim to control the GBS-induced innate inflammatory response.
FIG 7.

Impact of the transient inhibition of miR-223 by an antagomir on CXCL2 expression and neutrophil counts in lung tissues of mice infected with GBS. Mice (n = 5) received intranasal injection of GBS alone (108 CFU), or antagomiR-223 (50 μg/50 μl of sterile PBS) or scrambled RNA (50 μg/50 μl of sterile PBS) 30 min before GBS infection and checked for changes in mRNA target (CXCL2) and neutrophils counts at 6 h postinfection. (A) The relative expression of miR-223 was evaluated in the lungs by real-time RT-PCR. The values were normalized with three internal controls, RNU6b, snoRNA-202, and snoRNA-234. The fold change values (2−ΔΔCT) are relative to values with PBS treatment. (B) The relative expression of CXCL2 was evaluated in the lungs by real-time RT-PCR. The values were normalized with three internal controls, GAPDH, beta-actin, and RPL-19. (C) Neutrophil count was evaluated by flow cytometry using specific antibody (CD45+ CD11b+ Ly6G+). Data are means ± SD and are representative of those from 3 independent experiments. *, P < 0.05; ***, P < 0.001 (Mann-Whitney U test).
DISCUSSION
The inflammatory response is a beneficial process that mobilizes the immune system to eliminate invading pathogens and protect the host. However, the inflammatory process may cause host tissue damage if not tightly regulated. miRNAs are critical regulators of many biological processes and play an important role among other regulatory mechanisms in the control of the inflammatory response. We specifically focused in this study on miR-223 given the well-documented role of this miRNA in the negative regulation of the innate inflammatory response, through the control of myeloid cell proliferation (17, 18, 21–23). The purpose of this in vivo study was to examine the expression pattern of miR-223 during GBS infection and to evaluate in parallel the evolution of the inflammatory state. Our data show that GBS infection induces rapid miR-223 expression, with a peak preceding a gradual decrease of inflammatory markers. We found that lung-infiltrating neutrophils were the major cellular source of miR-223, in accordance with a previous study performed in an acute lung injury model induced by mitochondrial damage-associated molecular patterns (47). However, the contribution of other innate immune cells in the production of miR-223 cannot be excluded. Of note, a recent report has shown that miR-223 can be transferred from neutrophils to alveolar epithelial cells during coculture, suggesting that this intercellular cross talk may favor the accumulation of miR-223 within lung tissues (48). It is therefore likely that the progressive expression of miR-223 during neutrophil proliferation and its accumulation within the lung tissues may be part of regulatory mechanisms aiming to control the GBS-induced innate inflammatory response. Previous studies showed that miR-223 regulates negatively the recruitment of neutrophils by targeting directly or indirectly their chemoattractant molecules CXCL1 and CXCL2 (46, 49). Our findings are in line with these observations, as the accumulation of miR-223 with lung tissues correlates with the downregulation of both chemokines. In support of this, transient inhibition of miR-223 using specific antagomir resulted in a significant increase of CXCL2 expression and partial enhancement of infiltrating neutrophils in GBS-infected lung tissues. Also, it has been shown that miR-223 has a crucial role in hemostasis of mature neutrophils through targeting of a transcription factor that drives granulocyte-monocyte progenitor proliferation (21). Collectively, these data suggest that miR-223 produced by neutrophils may engender a negative-feedback loop to control granulocyte proliferation and differentiation and to dampen lung injury.
On the other hand, we found that increased levels of miR-223 were associated with a downregulation of the inflammatory cytokines IL-1β and TNF-α. This is in agreement with previous studies showing that miR-223 targeted the NLRP3 inflammasome and STAT3, hence contributing to the downregulation of inflammatory cytokines (47, 49–53).
To our knowledge, this is the first study on kinetic evaluation of monocyte and macrophage subpopulations infiltrating the lung in a GBS infection model. While we found no difference in AM, we noted that IM tended to gradually predominate in late stages, concomitantly with an elevated level of miR-223. IM are characterized by lower phagocytic potential than for AM and are believed to attenuate the inflammation and to maintain lung homeostasis (45). The potential implication of miR-223 in IM differentiation or activation remains unclear. Of note, AM can be subtyped into two main phenotypes, M1 (classical macrophages) and M2 (alternative macrophages). M1 macrophages are considered proinflammatory, as they can produce large amounts of inflammatory mediators and exhibit high phagocytic potential that is crucial for microbial clearance, while M2 macrophages are rather anti-inflammatory and contribute to tissue repair (54, 55). Studies have reported that miR-223 contributes to macrophage differentiation and activation and may help to steer the activation pattern from the M1 to M2 phenotype (56–58). It would be interesting to analyze in a future study these specific AM subsubpopulations and their relation to miR-223 in the context of GBS infection.
Additionally, we observed a distinct kinetic distribution of infiltrating monocyte subpopulations, with Ly6Chi cells being predominant in the early phase of infection while Ly6Cint and Ly6Clo cells gradually predominating at later stages of infection. It has been suggested that proinflammatory Ly6Chi monocytes differentiate to Ly6Cint monocytes, which are thought to be less inflammatory than Ly6Chi and to have high antigen presentation capacity (59). In turn, Ly6Cint cells are believed to give rise to Ly6Clo patrolling monocytes (41, 42, 60). The progressive increase of monocytes with a regulatory phenotype correlates with the increase of expression of miR-223. Whether miR-223 could directly or indirectly influence monocyte differentiation is currently unknown. It would be interesting to investigate in future studies the immune cell populations in lymphoid organs as well in comparison to miR-223 expression.
Overall, based on compelling evidence supporting the key role of miR-223 as a negative regulator of the innate inflammatory response, we believe that our study provides further evidence on the dynamic and the potent role of miR-223 in GBS infection. The rapid accumulation of the anti-inflammatory miR-223 at early infection stages is likely one of the compensatory mechanisms to limit excessive acute inflammation and restore immune homeostasis.
MATERIALS AND METHODS
Mouse infection model.
The clinical Streptococcus agalactiae isolate (COH1; serotype III) was kindly provided by Victor Nizet, University of California San Diego, La Jolla, CA (61). The bacteria were grown to the exponential phase without agitation at 37°C in Todd-Hewitt broth (THB) with nalidixic acid, and the CFU count was determined in THA medium plates with nalidixic acid at 15 mg/ml. A stock of aliquots of known bacterial concentrations was then stored at −80°C with 50% glycerol. Before use, aliquots of bacteria were thawed and centrifuged (5,000 × g for 5 min) and then resuspended in PBS at a concentration of 1 × 108 CFU/100 μl. The CFU count of each aliquot was checked again in THA medium plates with nalidixic acid at 15 mg/ml to verify the exact inoculum dose. C57BL/6 male mice (6 to 8 weeks old) were weighed and then anesthetized with 4% isoflurane gas before intranasal (i.n.) inoculation of 1 × 108 CFU in 100 μl of PBS. The sublethal dose (108 CFU), resulting in a strong inflammatory response, was determined by testing i.n. inoculation of various doses ranging from 107 to 5 × 108 CFU (data not shown). For tested parameters, mice were monitored at different time points (1 h to 48 h) in at least 3 independent experiments, except for the 1-h time point, for which monitoring was performed 2 times. Experiments with mice were approved by the local ethical committee for the humane use of laboratory animals (approval reference number 20180125-01).
RNA expression analysis.
The lungs were collected and crushed in 5 ml of PBS. After centrifugation, cell pellets were treated with 700 μl of QiaZol lysis reagent (Qiagen) and stored at −80°C. Total RNA was isolated using the miRNeasy minikit (Qiagen) following the manufacturer’s instructions. For miRNA expression analysis, reverse transcription was performed with a miScript II RT kit (Qiagen) following the manufacturer’s instructions and qRT-PCR was performed with the miScript SYBR green PCR kit (Qiagen) according to standard protocols. Three miRNA internal controls were used for high-standard miRNA quantification by qRT-PCR: U6 small nuclear ribonucleoprotein RNU6b and two small nucleolar RNAs (snoRNAs): snoRNA-202 and snoRNA-234 (62). For the expression of chemokine transcripts, reverse transcription was performed with the GoScript reverse transcription system kit (Promega). Three housekeeping genes (those for glyceraldehyde-3-phosphate dehydrogenase [GAPDH], beta-actin, and RPL-19) were used as internal controls. A no-template control and no-reverse transcriptase control were performed, and no amplicon was detected. The specificity of each primer pairs was further verified by controlling the melt curve profile. The primers used in this study are listed in Table 1. Relative quantification of miRNA and mRNA expression was calculated using the threshold cycle (2−ΔΔCT) method; the fold change values were relative to findings for PBS-treated mice.
TABLE 1.
Oligonucleotide primers used for miRNA and mRNA qRT-PCR analysis
| Gene | Sequence (5′ → 3′) or name of primer |
|
|---|---|---|
| Forward primer | Reverse primer | |
| miR-223-3p | TGT-CAG-TTT-GTC-AAA-TAC-CCC-A | miScript universal primer (Qiagen) |
| miR-155-5p | TTA-ATG-CTA-ATT-GTG-ATA-GGG-GT | miScript universal primer (Qiagen) |
| snoRNU6b | CTC-GCT-TCG-GCA-GCA-CA | miScript universal primer (Qiagen) |
| snoRNA-202 | AGT-ACT-TTT-GAA-CCC-TTT-TCC-A | miScript universal primer (Qiagen) |
| snoRNA-234 | TTA-ACA-AAA-ATT-CGT-CAC-TAC-CA | miScript universal primer (Qiagen) |
| CXCL1 | CCG-AAG-TCA-TAG-CCA-CAC-TCA-A | GCA-GTC-TGT-CTT-CTT-TCT-CCG-TTA-C |
| CXCL2 | AGA-CAG-AAG-TCA-TAG-CCA-CTC-TCA-AG | CCT-CCT-TTC-CAG-GTC-AGT-TAG-C |
| GAPDH | CTC-GGC-CTT-GAC-TGT-GCC-GTT | TGC-CAG-CCT-CGT-CCC-GTA-GAC-AAA |
| Beta-actin | AAA-TCT-GGC-ACC-ACA-CCT-TC | GGG-GTG-TTG-AAG-GTC-TCA-AA |
| RPL-19 | GAA-GGT-CAA-AGG-GAA-TGT-GTT-CA | CCT-TGT-CTG-CCT-TCA-GCT-TGT |
Detection of inflammatory cytokines.
The quantification of cytokine levels in the lungs was performed by ELISA following a standard protocol as previously described (63). Briefly, the lungs were collected and homogenized in 5 ml of PBS. After centrifugation, the cell-free suspensions were recovered for ELISAs performed with specific kits (Invitrogen) according to the manufacturer’s instructions. Plate reading and quantification were performed using the Infinite F50 absorbance microplate reader, and the results were analyzed using the Magellan program.
Lung cell isolation and flow cytometry.
To analyze the infiltrated cell population after GBS infection, the lungs were harvested and processed for 30 min at 35°C, in digestion buffer (1 mg/ml of collagenase A and 0.1 mg/ml of DNase I in Hanks balanced salt solution [HBSS] with 5% fetal bovine serum). Homogenized lungs were passed through 40-mm nylon mesh to obtain a single-cell suspension. Cells were counted, and 1 × 106 cells per sample were stained with a LIVE/DEAD fixable aqua dead cell stain kit (Invitrogen) according to the manufacturer’s instructions. Innate cell immunophenotyping was performed as previously described (64), with minor adaptations. Cells were incubated with a panel of specific antibodies for 30 min on ice in the dark. Panel 1 included I-A/I-E fluorescein isothiocyanate (FITC), CD45 phycoerythrin (PE), CD11c PE-Cy5.5, CD11b PE-CyTM7, Ly6C allophycocyanin (APC), CD24 APC-eFluor 780 (Thermo Fisher Scientific), and CD64 BV421 (BioLegend). Panel 2 included I-A/I-E FITC, CD45 PE, Ly6G peridinin chlorophyll protein (PerCP)-CyTM5.5, CD11b PE-CyTM7 (Thermo Fisher Scientific), Siglec F APC-CyTM7, and CD24 BV421 (BD Biosciences). After staining, cells were washed and fixed with 4% paraformaldehyde. Data were acquired with a BD FACSVerse flow cytometer and analyzed with BD FACSuite flow cytometry software. The gates used to properly interpret the data were determined by fluorescence minus one (FMO) controls.
Immunomagnetic depletion of Ly6G+ cells.
A lung single-cell suspension was incubated with anti-mouse Ly6G RB6-8C5 biotin (eBioscience) (5 μl/1 × 106 cells) for 30 min at 4°C and washed with PBS and 2 mM bovine serum albumin (BSA). After centrifugation, the cell pellet was incubated with Streptavidin Particles Plus-DM (BD Biosciences) (5 μl/106 cells) for 30 min on ice. The sample volume was then adjusted to 1 ml with PBS, 2 mM EDTA, and 0.5% BSA and placed in a magnetic rack for 7 to 8 min. The supernatant was collected and placed again in the magnetic rack. The procedure was repeated three times to ensure an efficient depletion of Ly6G+ cells. To determine the percentage of depletion, the cell suspensions were analyzed by flow cytometry using anti-mouse Ly6G PerCP-CyTM5.5 and CD11b PE-CyTM7 (Thermo Fisher Scientific).
Supplementary Material
ACKNOWLEDGMENTS
We thank IRIS research for their valuable support. Maud Deny is a Ph.D. fellow of the Belgian Kids Fund.
We are grateful to Giresse Hermann Tima, Pauline Lehebel Percier, Aude Ytebrouck, and Christophe Van Den Poel for their scientific advice and technical help.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Edwards MS, Baker CJ. 2005. Group B streptococcal infections in elderly adults. Clin Infect Dis 41:839–847. doi: 10.1086/432804. [DOI] [PubMed] [Google Scholar]
- 2.Edmond KM, Kortsalioudaki C, Scott S, Schrag SJ, Zaidi AK, Cousens S, Heath PT. 2012. Group B streptococcal disease in infants aged younger than 3 months: systematic review and meta-analysis. Lancet 379:547–556. doi: 10.1016/S0140-6736(11)61651-6. [DOI] [PubMed] [Google Scholar]
- 3.Dauby N, Chamekh M, Melin P, Slogrove AL, Goetghebuer T. 2016. Increased risk of group B Streptococcus invasive infection in HIV-exposed but uninfected infants: a review of the evidence and possible mechanisms. Front Immunol 16:505. doi: 10.3389/fimmu.2016.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams PA, Bohnsack JF, Augustine NH, Drummond WK, Rubens CE, Hill HR. 1993. Production of tumor necrosis factor by human cells in vitro and in vivo, induced by group B streptococci. J Pediatr 123:292–300. doi: 10.1016/S0022-3476(05)81706-8. [DOI] [PubMed] [Google Scholar]
- 5.De Bont ESJM, Martens A, Van Raan J, Samson G, Fetter WPF, Okken A, Leij LHFM. 1993. Tumor necrosis factor-α, interleukin-1β, and interleukin-6 plasma levels in neonatal sepsis. Pediatr Res 33:380–383. doi: 10.1203/00006450-199304000-00013. [DOI] [PubMed] [Google Scholar]
- 6.Mancuso G, Tomasello F, Hunolstein C, Orefici G, Teti G. 1994. Induction of tumor necrosis factor alpha by the group- and type-specific polysaccharides from type III group B Streptococci. Infect Immun 62:2748–2753. doi: 10.1128/IAI.62.7.2748-2753.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teti G, Mancuso G, Tomasello F. 1993. Cytokine appearance and effects of anti-tumor necrosis factor alpha antibodies in a neonatal rat model of group B streptococcal infection. Infect Immun 61:227–235. doi: 10.1128/IAI.61.1.227-235.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cusumano V, Mancuso G, Genovese F, Delfino D, Beninati C, Losi E, Teti G. 1996. Role of gamma interferon in a neonatal mouse model of group B streptococcal disease. Infect Immun 64:2941–2944. doi: 10.1128/IAI.64.8.2941-2944.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wennekamp J, Henneke P. 2008. Induction and termination of inflammatory signaling in group B streptococcal sepsis. Immunol Rev 225:114–127. doi: 10.1111/j.1600-065X.2008.00673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taganov KD, Boldin MP, Baltimore D. 2007. MicroRNAs and immunity: tiny players in a big field. Immunity 26:133–137. doi: 10.1016/j.immuni.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. 2008. Widespread changes in protein synthesis induced by microRNAs. Nature 455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 12.Lodish HF, Zhou B, Liu G, Chen CZ. 2008. Micromanagement of the immune system by microRNAs. Nat Rev Immunol 8:120–130. doi: 10.1038/nri2252. [DOI] [PubMed] [Google Scholar]
- 13.O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. 2010. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol 10:111–122. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- 14.Dai R, Ahmed SA. 2011. microRNA, a new paradigm for understanding immunoregulation, inflammation, and autoimmune diseases. Transl Res 157:163–179. doi: 10.1016/j.trsl.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Filipowicz W, Bhattacharyya SN, Sonenberg N. 2008. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 16.Huntzinger E, Izaurralde E. 2011. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet 12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 17.Haneklaus M, Gerlic M, O’Neill LAJ, Masters SL. 2013. miR-223: infection, inflammation and cancer. J Intern Med 274:215–226. doi: 10.1111/joim.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan X, Berg N, Jing N, Lee JW, Eltzschig H, Le T, Neudecker V. 2018. MicroRNA miR-223 as regulator of innate immunity. J Leukoc Biol 104:515–510. doi: 10.1002/JLB.3MR0218-079R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramkissoon SH, Mainwaring LA, Ogasawara Y, Keyvanfar K, Mccoy JP, Sloand EM, Kajigaya S, Young NS. 2006. Hematopoietic-specific microRNA expression in human cells. Leuk Res 30:643–647. doi: 10.1016/j.leukres.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 20.Fukao T, Fukuda Y, Kiga K, Sharif J, Hino K, Enomoto Y, Kawamura A, Nakamura K, Takeuchi T, Tanabe M. 2007. An evolutionarily conserved mechanism for microRNA-223 expression revealed by microRNA gene profiling. Cell 129:617–631. doi: 10.1016/j.cell.2007.02.048. [DOI] [PubMed] [Google Scholar]
- 21.Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, Brummelkamp TR, Fleming MD, Camargo FD. 2008. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 451:1125–1129. doi: 10.1038/nature06607. [DOI] [PubMed] [Google Scholar]
- 22.Zardo G, Ciolfi A, Vian L, Starnes LM, Billi M, Racanicchi S, Maresca C, Fazi F, Travaglini L, Noguera N, Mancini M, Nanni M, Cimino G, Lo-Coco F, Grignani F, Nervi C. 2012. Polycombs and microRNA-223 regulate human granulopoiesis by transcriptional control of target gene expression. Blood 119:4034–4046. doi: 10.1182/blood-2011-08-371344. [DOI] [PubMed] [Google Scholar]
- 23.Trissal MC, DeMoya RA, Schmidt AP, Link DC. 2015. MicroRNA-223 regulates granulopoiesis but is not required for HSC maintenance in mice. PLoS One 10:e0119304. doi: 10.1371/journal.pone.0119304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stamatopoulos B, Meuleman N, Haibe-Kains B, Saussoy P, Van Den Neste E, Michaux L, Heimann P, Martiat P, Bron D, Lagneaux L. 2009. microRNA-29c and microRNA-223 down-regulation has in vivo significance in chronic lymphocytic leukemia and improves disease risk stratification. Blood 113:5237–5245. doi: 10.1182/blood-2008-11-189407. [DOI] [PubMed] [Google Scholar]
- 25.Fulci V, Scappucci G, Sebastiani GD, Giannitti C, Franceschini D, Meloni F, Colombo T, Citarella F, Barnaba V, Minisola G, Galeazzi M, Macino G. 2010. miR-223 is overexpressed in T-lymphocytes of patients affected by rheumatoid arthritis. Hum Immunol 71:206–211. doi: 10.1016/j.humimm.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 26.Wang H, Peng W, Ouyang X, Li W, Dai Y. 2012. Circulating microRNAs as candidate biomarkers in patients with systemic lupus erythematosus. Transl Res 160:198–206. doi: 10.1016/j.trsl.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 27.Taïbi F, Meuth VM, Massy ZA, Metzinger L. 2014. miR-223: an inflammatory oncomiR enters the cardiovascular field. Biochim Biophys Acta 1842:1001–1009. doi: 10.1016/j.bbadis.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 28.Ye D, Zhang T, Lou G, Liu Y. 2018. Role of miR-223 in the pathophysiology of liver diseases. Exp Mol Med 50:128. doi: 10.1038/s12276-018-0153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong QWL, Lung RWM, Law PTY, Lai PBS, Chan KYY, To KF, Wong N. 2008. MicroRNA-223 is commonly repressed in hepatocellular carcinoma and potentiates expression of stathmin1. Gastroenterology 135:257–269. doi: 10.1053/j.gastro.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 30.Ezzie ME, Crawford M, Cho J, Orellana R, Zhang S, Gelinas R, Batte K, Yu L, Nuovo G, Galas D, Diaz P, Wang K, Nana-Sinkam SP. 2012. Gene expression networks in COPD: microRNA and mRNA regulation. Thorax 67:122–131. doi: 10.1136/thoraxjnl-2011-200089. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Yu M, Yu G, Bian J, Deng X, Wan X, Zhu K. 2010. Serum miR-146a and miR-223 as potential new biomarkers for sepsis. Biochem Biophys Res Commun 394:184–188. doi: 10.1016/j.bbrc.2010.02.145. [DOI] [PubMed] [Google Scholar]
- 32.Moschos SA, Williams AE, Perry MM, Birrell MA, Belvisi MG, Lindsay MA. 2007. Expression profiling in vivo demonstrates rapid changes in lung microRNA levels following lipopolysaccharide-induced inflammation but not in the anti-inflammatory action of glucocorticoids. BMC Genomics 8:240. doi: 10.1186/1471-2164-8-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pu J, Li R, Zhang C, Chen D, Liao X, Zhu Y, Geng X, Ji D, Mao Y, Gong Y, Yang Z. 2017. Expression profiles of miRNAs from bovine mammary glands in response to Streptococcus agalactiae- induced mastitis. J Dairy Res 84:300–308. doi: 10.1017/S0022029917000437. [DOI] [PubMed] [Google Scholar]
- 34.Lewandowska-Sabat AM, Hansen SF, Solberg TR, Østerås O, Heringstad B, Boysen P, Olsaker I. 2018. MicroRNA expression profiles of bovine monocyte-derived macrophages infected in vitro with two strains of Streptococcus agalactiae. BMC Genomics 19:241. doi: 10.1186/s12864-018-4591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR. 2007. Requirement of bic/micro-RNA-155 for normal immune function. Science 316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Connell RM, Kahn D, Gibson WSJ, Round JL, Scholz RL, Chaudhuri AA, Kahn ME, Rao DS, Baltimore D. 2010. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 33:607–619. doi: 10.1016/j.immuni.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McAdams RM, Bierle CJ, Boldenow E, Weed S, Tsai J, Beyer RP, Macdonald JW, Bammler TK, Liggitt HD, Farin FM, Vanderhoeven J, Rajagopal L, Waldorf KMA. 2015. Choriodecidual group B streptococcal infection induces miR-155-5p in the fetal lung in Macaca nemestrina. Infect Immun 83:3909–3917. doi: 10.1128/IAI.00695-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayadas TN, Cullere X, Lowell CA. 2014. The multifaceted functions of neutrophils. Annu Rev Pathol 9:181–218. doi: 10.1146/annurev-pathol-020712-164023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirayama D, Iida T. 2018. The phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. Int J Mol Sci 19:92. doi: 10.3390/ijms19010092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gordon SB, Read RC. 2002. Macrophage defences against respiratory tract infections. Br Med Bull 61:45–61. doi: 10.1093/bmb/61.1.45. [DOI] [PubMed] [Google Scholar]
- 41.Schyns J, Bureau F, Marichal T. 2018. Lung interstitial macrophages: past, present, and future. J Immunol Res 2018:5160794. doi: 10.1155/2018/5160794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Auffray C, Sieweke MH, Geissmann F. 2009. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol 27:669–692. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- 43.Geissmann F, Jung S, Littman DR. 2003. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19:71–82. doi: 10.1016/S1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 44.Gordon S, Taylor PR. 2005. Monocyte and macrophage heterogeneity. Nat Rev Immunol 5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 45.Kratofil RM, Kubes P, Deniset JF. 2017. Monocyte conversion during inflammation and injury. Arterioscler Thromb Vasc Biol 37:35–42. doi: 10.1161/ATVBAHA.116.308198. [DOI] [PubMed] [Google Scholar]
- 46.Dorhoi A, Iannaccone M, Farinacci M, Faé KC, Schreiber J, Moura-Alves P, Nouailles G, Mollenkopf H, Oberbeck-Müller D, Jörg S, Heinemann E, Hahnke K, Löwe D, Del Nonno F, Goletti D, Capparelli R, Kaufmann SHE. 2013. MicroRNA-223 controls susceptibility to tuberculosis by regulating lung neutrophil recruitment. J Clin Invest 123:4836–4848. doi: 10.1172/JCI67604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feng Z, Qi S, Zhang Y, Qi Z, Yan L, Zhou J, He F, Li Q, Yang Y, Chen Q, Xiao S, Li Q, Chen Y, Zhang Y. 2017. Ly6G+ neutrophil-derived miR-223 inhibits the NLRP3 inflammasome in mitochondrial DAMP-induced acute lung injury. Cell Death Dis 8:e3170-14. doi: 10.1038/cddis.2017.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neudecker V, Brodsky KS, Clambey ET, Schmidt EP, Packard TA, Davenport B, Standiford TJ, Weng T, Fletcher AA, Barthel L, Masterson JC, Furuta GT, Cai C, Blackburn MR, Ginde AA, Graner MW, Janssen WJ, Zemans RL, Evans CM, Burnham EL, Homann D, Moss M, Kreth S, Zacharowski K, Henson PM, Eltzschig HK. 2017. Neutrophil transfer of miR-223 to lung epithelial cells dampens acute lung injury in mice. Sci Transl Med 9:eaah5360. doi: 10.1126/scitranslmed.aah5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neudecker V, Haneklaus M, Jensen O, Khailova L, Masterson JC, Tye H, Biette K, Jedlicka P, Brodsky KS, Gerich ME, Mack M, Robertson AAB, Cooper MA, Furuta GT, Dinarello CA, O’Neill LA, Eltzschig HK, Masters SL, McNamee EN. 2017. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J Exp Med 214:1737–1752. doi: 10.1084/jem.20160462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haneklaus M, Gerlic M, Rainey A, Pich D, Mcinnes IB, Hammerschmidt W, O’Neill LAJ, Masters SL. 2012. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1 β production. J Immunol 189:3795–3799. doi: 10.4049/jimmunol.1200312. [DOI] [PubMed] [Google Scholar]
- 51.Bauernfeind F, Rieger A, Schildberg FA, Knolle A, Schmid-Burgk JL, Hornung V. 2012. NLRP3 inflammasome activity is negatively controlled by miR-223. J Immunol 189:4175–4181. doi: 10.4049/jimmunol.1201516. [DOI] [PubMed] [Google Scholar]
- 52.Chen Q, Wang H, Liu Y, Song Y, Lai L, Han Q, Cao X, Wang Q. 2012. Inducible microRNA-223 down-regulation promotes TLR-triggered IL-6 and IL-1β production in macrophages by targeting STAT3. PLoS One 7:e42971. doi: 10.1371/journal.pone.0042971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poon K-S, Palanisamy K, Chang S-S, Sun K-T, Chen K-B, Li P-C, Lin T-C, Li C-Y. 2017. Plasma exosomal miR-223 expression regulates inflammatory responses during cardiac surgery with cardiopulmonary bypass. Sci Rep 7:10807. doi: 10.1038/s41598-017-09709-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gordon S, Martinez FO. 2010. Alternative activation of macrophages: mechanism and functions. Immunity 32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 55.Aggarwal NR, King LS, D’Alessio FR. 2014. Diverse macrophage populations mediate acute lung inflammation and resolution. Am J Physiol Lung Cell Mol Physiol 306:L709–L725. doi: 10.1152/ajplung.00341.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhuang G, Meng C, Guo X, Cheruku PS, Shi L, Xu H, Li H, Wang G, Evans AR, Safe S, Wu C, Zhou B. 2012. A novel regulator of macrophage activation miR-223 in obesity-associated adipose tissue inflammation. Circulation 125:2892–2903. doi: 10.1161/CIRCULATIONAHA.111.087817. [DOI] [PubMed] [Google Scholar]
- 57.Li T, Morgan MJ, Choksi S, Zhang Y, Kim Y-S, Liu Z-G. 2010. MicroRNAs modulate the noncanonical NF-κB pathway by regulating IKKα expression during macrophage differentiation. Nat Immunol 11:799–805. doi: 10.1038/ni.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tran TH, Krishnan S, Amiji MM. 2016. MicroRNA-223 induced repolarization of peritoneal macrophages using CD44 targeting hyaluronic acid nanoparticles for anti-inflammatory effects. PLoS One 11:e0152024. doi: 10.1371/journal.pone.0152024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sprangers S, De Vries TJ, Everts V. 2016. Monocyte Heterogeneity: consequences for monocyte-derived immune cells. J Immunol Res 2016:1475435. doi: 10.1155/2016/1475435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mitchell AJ, Roediger B, Weninger W. 2014. Monocyte homeostasis and the plasticity of inflammatory monocytes. Cell Immunol 291:22–31. doi: 10.1016/j.cellimm.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 61.Chang YC, Olson J, Beasley FC, Tung C, Zhang J, Crocker PR, Varki A, Nizet V. 2014. Group B Streptococcus engages an inhibitory Siglec through sialic acid mimicry to blunt innate immune and inflammatory responses in vivo. PLoS Pathog 10:e1003846. doi: 10.1371/journal.ppat.1003846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bouhaddioui W, Provost PR, Tremblay Y. 2014. Identification of most stable endogenous control genes for microRNA quantification in the developing mouse lung. PLoS One 9:e111855. doi: 10.1371/journal.pone.0111855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chamekh M, Phalipon A, Quertainmont R, Salmon I, Sansonetti P, Allaoui A. 2008. Delivery of biologically active anti-inflammatory cytokines IL-10 and IL-1ra in vivo by the Shigella type III secretion apparatus. J Immunol 180:4292–4298. doi: 10.4049/jimmunol.180.6.4292. [DOI] [PubMed] [Google Scholar]
- 64.Yu Y-RA, O’Koren EG, Hotten DF, Kan MJ, Kopin D, Nelson ER, Que L, Gunn MD. 2016. A protocol for the comprehensive flow cytometric analysis of immune cells in normal and inflamed murine non-lymphoid tissues. PLoS One 11:e0150606. doi: 10.1371/journal.pone.0150606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.