

The obligate intracellular pathogen Chlamydia trachomatis is the leading cause of noncongenital blindness and causative agent of the most common sexually transmitted infection of bacterial origin. With a reduced genome, C. trachomatis is dependent on its host for survival, in part due to a need for the host cell to compensate for incomplete bacterial metabolic pathways. However, relatively little is known regarding how C. trachomatis is able to hijack host cell metabolism.
KEYWORDS: aldolase A, Chlamydia, glycolysis, inclusion membrane, metabolism
ABSTRACT
The obligate intracellular pathogen Chlamydia trachomatis is the leading cause of noncongenital blindness and causative agent of the most common sexually transmitted infection of bacterial origin. With a reduced genome, C. trachomatis is dependent on its host for survival, in part due to a need for the host cell to compensate for incomplete bacterial metabolic pathways. However, relatively little is known regarding how C. trachomatis is able to hijack host cell metabolism. In this study, we show that two host glycolytic enzymes, aldolase A and pyruvate kinase, as well as lactate dehydrogenase, are enriched at the C. trachomatis inclusion membrane during infection. Inclusion localization was not species specific, since a similar phenotype was observed with C. muridarum. Time course experiments showed that the number of positive inclusions increased throughout the developmental cycle. In addition, these host enzymes colocalized to the same inclusion, and their localization did not appear to be dependent on sustained bacterial protein synthesis or on intact host actin, vesicular trafficking, or microtubules. Depletion of the host glycolytic enzyme aldolase A resulted in decreased inclusion size and infectious progeny production, indicating a role for host glycolysis in bacterial growth. Finally, quantitative PCR analysis showed that expression of C. trachomatis glycolytic enzymes inversely correlated with host enzyme localization at the inclusion. We discuss potential mechanisms leading to inclusion localization of host glycolytic enzymes and how it could benefit the bacteria. Altogether, our findings provide further insight into the intricate relationship between host and bacterial metabolism during Chlamydia infection.
INTRODUCTION
The pathogen Chlamydia trachomatis is the leading cause of noncongenital blindness and causative agent of the most common sexually transmitted infection of bacterial origin (1). As an obligate intracellular pathogen with a small genome (1 Mb), C. trachomatis is strictly dependent on the host cell to complete its developmental cycle. Upon entering epithelial cells, C. trachomatis resides within a membranous vacuole, the inclusion. In the lumen of the inclusion, the bacteria undergo a biphasic developmental cycle, alternating between the infectious elementary body (EB) form and the replicative reticulate body (RB) form (2, 3). After entry, the EBs differentiate into RBs, and the RBs continue to replicate within the inclusion until the asynchronous differentiation of RBs back to EBs begins approximately 24 h postinfection (3). The EBs are then released from the host cell through extrusion or host cell lysis, allowing for infection of neighboring cells (4–6).
It is still unclear how much C. trachomatis relies on the host cell for energy production throughout development. For almost 40 years, C. trachomatis was thought to be an energy parasite (7–10). However, sequencing of the C. trachomatis genome revealed that the bacteria possess components of the electron transport chain and ATP synthase complex, suggesting that C. trachomatis can drive a minimal electron transport chain to produce ATP through oxidative phosphorylation (11). In addition, C. trachomatis was found to have an intact pentose phosphate pathway and a partial citric acid (tricarboxylic acid [TCA]) cycle (11, 12). Although these findings indicate that C. trachomatis is not merely an energy parasite, there is evidence to suggest that C. trachomatis is in part dependent on the host cell for energy production. For example, the nucleotide transporters Npt1 and Npt2 are highly expressed in Chlamydia RBs (13, 14), indicating that Chlamydia is reliant on scavenging ATP and NAD+ from the host (15, 16).
In addition to oxidative phosphorylation, glycolysis is another major source of cellular energy. Glycolysis relies on the function of 10 different enzymes to sequentially convert glucose into pyruvate and NAD+ to NADH, producing ATP in the process (17). Sequencing of the C. trachomatis genome identified a nearly full set of glycolytic enzymes, lacking only the gene for hexokinase, the first enzyme of the pathway responsible for converting glucose to glucose-6-phosphate (11). Instead, glucose-6-phosphate is taken directly from the host cell via the UhpC antiporter produced by Chlamydia (18). Heterologous expression of C. trachomatis glycolytic enzymes in Escherichia coli confirmed their functionality (19). However, the C. trachomatis glycolytic enzymes were proposed to be expendable, as saturated ethyl methanesulfonate mutagenesis resulted in a loss-of-function mutation in bacterial glucose-6-phosphate isomerase (pgi), the enzyme responsible for shuttling glucose-6-phosphate into the glycolytic pathway (20).
More recently, a genome-wide RNA interference screen performed by Rother et al. suggested that two host glycolytic enzymes, glucose-6-phosphate isomerase and 6-phosphofructokinase, were potentially involved in Chlamydia progeny production (21). However, these results were not validated. The same study also profiled metabolites of central carbon metabolism after Chlamydia infection and observed elevated levels of pyruvate, lactate, and glutamate. An increase in these metabolites is indicative of Warburg metabolism, a metabolic state commonly observed in cancer cells that is characterized by the increased utilization of glycolysis rather than oxidative phosphorylation, resulting in the increased production of lactate (22–24). In this metabolic state, the upregulation of glycolysis leads to an accumulation of glycolytic intermediates that can be shuttled into the pentose phosphate pathway and used for ribonucleotide synthesis. Thus, Rother et al. (21) concluded that, much like what occurs in cancer cells, Chlamydia is able to shift the host cell into a hypermetabolic state in order to meet the high energetic demand of bacterial replication. However, whether this upregulation of host glycolysis is influenced by the localization of host glycolytic enzymes in relation to the C. trachomatis inclusion remains unknown.
In the present study, we show that several of the host glycolytic enzymes localized at the inclusion membrane and that the number of positive inclusions increased as the developmental cycle progressed. Moreover, knockdown of the host glycolytic enzyme aldolase A (AldoA) resulted in reduced inclusion size and decreased infectious progeny production, suggesting a role for host glycolysis in bacterial development. Lastly, quantitative PCR (qPCR) analysis of C. trachomatis glycolytic enzymes showed their downregulation throughout the developmental cycle. These novel findings further shed light on host and bacterial metabolism throughout Chlamydia development.
RESULTS
Host glycolytic enzymes localize at the Chlamydia inclusion membrane.
In order to investigate how Chlamydia hijacks host metabolism, specifically glycolysis, we generated 3×FLAG-tagged constructs of two host enzymes involved in glycolysis (aldolase A and pyruvate kinase) and one acting directly on the end product of glycolysis (lactate dehydrogenase). In HeLa cells transfected with these constructs, the enzymes exhibited diffuse expression throughout the cytosol with enrichment at the leading edge of the cell (see Fig. S1 in the supplemental material). However, when HeLa cells expressing these constructs were infected with a strain of C. trachomatis serovar L2 expressing mCherry (mCherry CtL2) for 24 h, the signal corresponding to the enzymes appeared enriched as a ring around the inclusion (Fig. 1A). Quantification of this enrichment revealed that the enzymes were enriched by 40% at the inclusion compared to the surrounding cytosol (Fig. 1B). Moreover, quantification of the number of positive inclusions revealed that, for all three enzymes, 60% of the inclusions were positive (Fig. 1C).
FIG 1.

Host glycolytic enzymes are enriched at the Chlamydia inclusion membrane. (A) Confocal micrographs of HeLa cells expressing 3×FLAG-tagged aldolase A (3×F-AldoA), pyruvate kinase (3×F-PKM2), or lactate dehydrogenase (3×F-LDHA), infected for 24 h with the mCherry CtL2 strain (red) and immunostained with anti-FLAG antibodies (green). The merge is shown on the right. Scale bar, 10 μm. (B and C) For each of the indicated 3×FLAG-tagged host enzymes, quantification of the percent enrichment at the inclusion (B) and the percentage of positive inclusions (C) was performed. The quantification methods are described in Materials and Methods. Data show means and SD of a combination of three independent experiments. (D) Confocal micrographs of HeLa cells expressing the indicated 3×FLAG-tagged enzyme, infected for 24 h with the mCherry CtL2 strain (red) and coimmunostained with anti-FLAG (yellow) and anti-IncA (blue) antibodies. The merge is shown on the right. Scale bar, 5 μm. (E) Line intensity scan analyses of the micrographs depicted in panel D, indicating the coincidence of 3×FLAG-tagged host enzyme constructs (yellow lines) and the inclusion membrane protein IncA (blue lines). (F) Confocal micrographs of HeLa cells expressing the indicated 3×FLAG-tagged enzyme, infected for 24 h with a C. muridarum strain expressing mCherry (red) and stained with anti-FLAG (yellow) antibodies. The merge is on the right. Scale bar, 10 μm.
To determine whether the enzymes were localized specifically at the inclusion membrane or simply to the periphery of the inclusion, HeLa cells expressing the 3×FLAG-tagged enzymes were infected with the mCherry CtL2 strain for 24 h and stained with the IncA antibody to denote the inclusion membrane (25, 26). Confocal imaging and line intensity scan analysis showed that enrichment of the host glycolytic enzymes overlap with the IncA staining, indicating that the host glycolytic enzymes are localizing at the inclusion membrane (Fig. 1D and E). In addition, localization of host glycolytic enzymes extends to other Chlamydia species as the 3×FLAG-tagged enzymes were localized at Chlamydia muridarum inclusions at 24 h postinfection (Fig. 1F). Altogether, these results indicate that host glycolytic enzymes are localized at the Chlamydia inclusion membrane and that this localization is not species specific.
Host glycolytic enzymes localize together at the C. trachomatis inclusion membrane.
We next sought to determine whether the host glycolytic enzymes were localized together at the same inclusions. HeLa cells were cotransfected to express both HA-tagged aldolase A and 3×FLAG-tagged pyruvate kinase, or lactate dehydrogenase (Fig. 2A). Approximately 60% of inclusions were positive for both the HA- and 3×FLAG-tagged enzymes, matching the level of localization observed for the individual enzymes in Fig. 1C. Fewer than 10% of inclusions were positive for only one enzyme, while approximately 30% of inclusions were negative (Fig. 2B). These results indicate that host glycolytic enzymes are localized together at the same inclusions.
FIG 2.

Host glycolytic enzymes localize together at the inclusion membrane. (A) Confocal micrographs of HeLa cells coexpressing HA-tagged AldoA (yellow) and 3×FLAG-tagged, PKM2, or LDHA (blue) and infected with the mCherry CtL2 strain (red). The infected cells were fixed at 24 h postinfection and coimmunostained with anti-HA and anti-FLAG antibodies. The merge is shown on the right. Scale bar, 5 μm. (B) Quantification of the percentage of inclusions positive for both the HA- and 3×FLAG-tagged enzyme (green bars), neither enzyme (white bars), the 3×FLAG-tagged enzyme alone (cyan bars), or the HA-tagged enzyme alone (yellow bars). Data show the means and SD of a combination of three independent experiments.
The percentage of aldolase A positive inclusions increases over time and is not due to nutrient deprivation or sustained de novo bacterial protein synthesis.
We next characterized the inclusion localization of the host glycolytic enzymes over time. Since our data showed that all three enzymes behaved similarly (Fig. 1), we chose to focus on Aldolase A for further experiments. HeLa cells expressing 3×FLAG-Aldolase A were infected with the mCherry CtL2 strain and, at the indicated time point (8, 18, 24, or 32 h) postinfection, the cells were fixed, immunostained with anti-Flag antibodies, and processed for confocal microscopy. Inclusions were negative for aldolase A at 8 h postinfection (see Fig. S2 in the supplemental material). However, the number of positive inclusions steadily increased over time, with 48, 66, and 81% of inclusions exhibiting inclusion localization at 18, 24, and 32 h postinfection, respectively (Fig. 3A). An increase in the number of positive inclusions over time was still observed when infection was performed in the presence of cycloheximide to inhibit eukaryotic protein synthesis (Fig. 3A), ruling out that the phenotype was an artifact of increased overexpression of the aldolase A over time. Moreover, overexpression of two metabolic enzymes involved in the purine biosynthesis pathway, and unrelated to glycolysis, (i.e., green fluorescent protein-tagged glycinamide ribonucleotide transformylase [GART] and Phosphoribosylaminoimidazole carboxylase [PAICS]) did not result in inclusion localization of these enzymes (see Fig. S3 in the supplemental material).
FIG 3.

The percentage of aldolase A-positive inclusions increases over time and is not due to nutrient deprivation or sustained de novo bacterial protein synthesis. HeLa cells expressing 3×FLAG-tagged AldoA were infected with the mCherry CtL2 strain then fixed and stained with anti-FLAG antibodies at the indicated time postinfection. (A) Quantification of the percentage of inclusions positive for 3×FLAG-AldoA at 8, 18, 24, and 32 h postinfection in the absence or presence of 1 μg/ml cycloheximide (CHX). (B) Quantification of the percentage of 3×FLAG-AldoA-positive inclusions at 18, 24, or 24 h postinfection with medium replacement at 18 h postinfection (24 h*). (C) Quantification of the percentage of 3×FLAG-AldoA positive inclusions at 24 h postinfection (–CHL) or after an additional 8 h of incubation with 40 μg/ml chloramphenicol and fixed at 32 h postinfection (+CHL). Data show the means and SD of a combination of three independent experiments. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not statistically significant (Student t test or one-way ANOVA with multiple comparisons).
Since we observed an increase in the number of positive inclusions over time, we wondered whether this increase could be due to depletion of nutrients as the bacteria and cells continued to grow and divide. To answer this question, HeLa cells expressing 3×FLAG-aldolase A were infected with the mCherry CtL2 strain for 18 h, at which point nutrients were replenished, or not, by the addition of fresh media. The infected cells were fixed at 24 h postinfection, and the number of aldolase A-positive inclusions was determined (Fig. 3B). If nutrient deprivation played a significant role in host glycolytic enzyme inclusion localization, we expected that at 24 h postinfection, the percentage of positive inclusions in the media replacement condition would be that of an 18h infection. However, the number of positive inclusions did not significantly differ from conditions in which the medium was not replaced (Fig. 3B, 24 h versus 24 h*).
We next wanted to determine whether continuous de novo bacterial protein synthesis was required to maintain aldolase A localization at the inclusion. HeLa cells expressing 3×FLAG-aldolase A were infected with the mCherry CtL2 strain for 24 h, followed by 8 h of treatment with 40 μg/ml chloramphenicol to halt bacterial protein synthesis. At 32 h postinfection, the cells were fixed and processed for confocal microscopy. Untreated cells, fixed at 24 h postinfection, served as a control. Treatment with chloramphenicol did not result in a decrease in the number of positive inclusions compared to the untreated control (Fig. 3C).
Together, these results indicate that the number of aldolase A-positive inclusions increases throughout the development cycle and that this increase is not due to the depletion of nutrients. Moreover, continuous de novo bacterial protein synthesis is not required for sustained localization of aldolase A at the inclusion.
F-actin, host cell microtubule dynamics, and vesicular trafficking do not play a role in aldolase A localization at the inclusion.
Since sustained active bacterial protein synthesis did not appear to play a role in aldolase A localization at the inclusion, we tested whether host cell factors were responsible. Aldolase A has been previously shown to bind F-actin (27, 28). It has also been shown that Chlamydia inclusions are encased within a cage of F-actin filaments (29). To determine whether host cell actin played a role in aldolase A localization at the inclusion, HeLa cells expressing 3×FLAG-aldolase A were infected with the mCherry CtL2 strain. At 23.5 h postinfection, infected cells were incubated with or without cytochalasin D to inhibit actin polymerization (see Fig. S4A in the supplemental material). The cells were fixed at 24 h postinfection and immunostained with anti-Flag antibodies, and the percentage of aldolase A-positive inclusions was determined. Treatment with cytochalasin D did not significantly decrease the percentage of positive inclusions (Fig. 4A). However, cytochalasin D can affect cell morphology and have a confounding effect on quantification. Thus, we addressed a possible role for actin with two additional approaches. First, we utilized the R42A and R148A mutants of aldolase A that have been previously shown to have minimal binding to F-actin (28). HeLa cells expressing either wild-type (WT), R42A, or R148A aldolase A were infected with the mCherry CtL2 strain. At 24 h postinfection, the cells were fixed, immunostained with anti-Flag antibodies, and imaged by confocal microscopy (see Fig. S5A in the supplemental material). The R42A and R148A mutants did not result in a significant decrease in the percentage of positive inclusions compared to the WT enzyme (Fig. 4B). Second, we tested whether actin cage formation is required for localization of aldolase A to the inclusion by using an inaC::aadA mutant strain of C. trachomatis. It was previously shown that the bacterial inclusion membrane protein InaC modulates F-actin assembly around the inclusion and that inaC mutant inclusions lack actin cages (20, 30). Aldolase A still localized to inaC::aadA mutant inclusions (see Fig. S5B in the supplemental material), and no significant difference in the percentage of positive inclusions was observed between WT and inaC::aadA mutant inclusions (Fig. 4C). Next, we addressed whether other major host cell factors could be mediating the localization of aldolase A at the inclusion. Short treatment with nocodazole to inhibit microtubule polymerization (see Fig. S4B in the supplemental material) or overnight treatment with brefeldin A to inhibit vesicular trafficking (see Fig. S4C in the supplemental material) had no observable effect on the percentage of inclusions positive for aldolase A (Fig. 4D and E). Altogether, these results indicate that the actin cytoskeleton or microtubules are not required for sustained localization of aldolase A at the inclusion and that vesicular trafficking or the Chlamydia actin cage do not mediate the inclusion localization of aldolase A.
FIG 4.
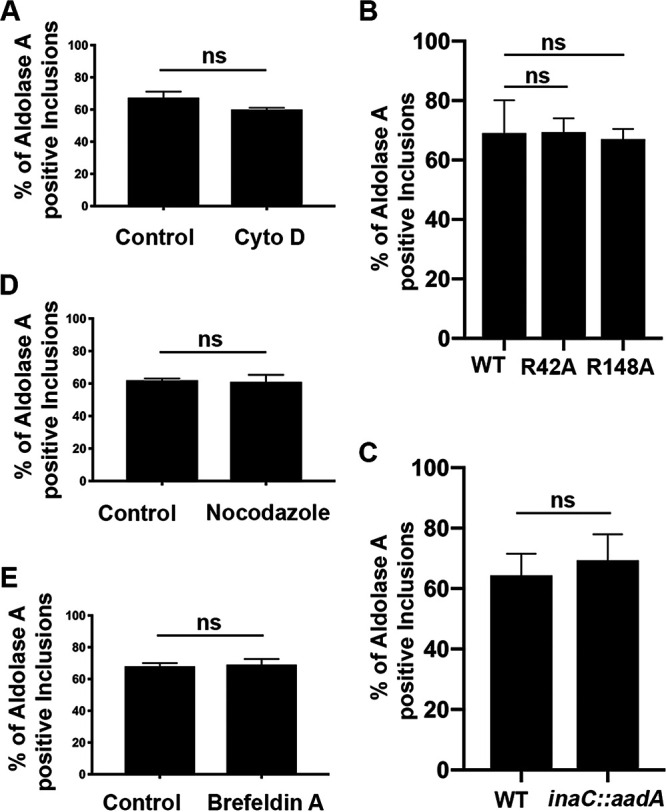
F-actin, host cell microtubule dynamics, and vesicular trafficking do not play a role in the localization of aldolase A at the inclusion. HeLa cells expressing 3×FLAG-AldoA and infected with the mCherry CtL2 strain were fixed at 24 h postinfection and immunostained with anti-FLAG antibodies. (A, D, and E) Quantification of the percentage of inclusions positive for 3×FLAG-AldoA following treatment with 1 μM cytochalasin D (Cyto D) at 23.5 h postinfection (A), 33 μM nocodazole at 23.5 h postinfection (D), or 1 μg/ml brefeldin A at 6 h postinfection (E). (B) Quantification of the percentage of inclusions positive for 3×FLAG- AldoA in HeLa cells expressing wild-type 3×FLAG-AldoA (WT), or point mutants no longer able to bind actin 3×FLAG-R42A AldoA (R42A) and 3×FLAG-R148A AldoA (R148A). (C) Quantification of the percentage of inclusions positive for 3×FLAG-AldoA in HeLa cells infected with wild-type C. trachomatis (WT) or an inaC mutant strain (inaC::aadA). Data show the means and the SD of a combination of three independent experiments. ns, not statistically significant (Student t test).
Aldolase A confers a developmental advantage to C. trachomatis.
To determine whether aldolase A played a role in C. trachomatis intracellular growth and development, we first assessed whether aldolase A inclusion localization was associated with larger inclusion size. HeLa cells expressing 3×FLAG-aldolase A were infected with the mCherry CtL2 strain. At 24 h postinfection, the cells were fixed and immunostained with anti-Flag antibodies, and the size of aldolase A-positive and -negative inclusions was determined. Aldolase A-positive inclusions were significantly larger than aldolase A-negative inclusions (Fig. 5A).
FIG 5.

Aldolase A confers a developmental advantage to C. trachomatis. (A) HeLa cells expressing 3×FLAG-tagged AldoA were infected with the mCherry CtL2 strain and the area of 3×FLAG-AldoA positive and negative inclusions (arbitrary units; each circle represents data from a single inclusion) was quantified at 24 h postinfection. (B to D) HeLa cells were transfected with individual aldolase A siRNA duplexes or siRNA buffer alone (Mock) for 3 days. (B) Immunoblots of the corresponding lysates were probed using antibodies against endogenous aldolase A (top blot) and actin (bottom blot). The knockdown efficacy of each duplex targeting aldolase A is indicated. (C and D) HeLa cells treated with the indicated siRNA duplexes targeting aldolase A were infected with C. trachomatis. For each condition, the total area of the inclusions (arbitrary units; each circle represents data from a single inclusion) at 32 h postinfection (C) and the number of infectious bacteria (IFUs/ml) at 48 h postinfection (D) was determined. The cell density of the mock-treated conditions was matched to the cell density of the siRNA-treated conditions. The data in panel A show the mean and the standard errors of the mean (SEM) of a representative experiment. The data in panel C show the means and the SEM of a combination of three independent experiments. The data shown in panel D is from a representative experiment. Error bars indicate the SD. ****, P < 0.0001 (Student t test).
We next assessed whether depletion of host aldolase A had an impact on C. trachomatis intracellular growth or infectious progeny production. Aldolase A was depleted by four independent small interfering RNA (siRNA) duplexes, each of which exhibited >90% efficacy of aldolase knockdown (Fig. 5B). Although duplex 2 exhibited over 90% efficacy, it was consistently the least efficient of the duplexes. In order to determine the effect of host aldolase A depletion on inclusion establishment and bacterial replication, we assessed inclusion size. HeLa cells were treated with independent siRNA duplexes for 3 days prior to infection with the mCherry CtL2 strain. At 32 h postinfection, the cells were fixed and imaged, and the inclusion size was determined by computer-assisted image analysis. We noted that aldolase A depletion affected the cell number, and since cell density affects the multiplicity of infection and inclusion size, each siRNA duplex condition was matched to a control with equal cell number. Compared to mock-treated cells, aldolase A depletion led to a significant, albeit modest, decrease in inclusion size for three of the four independent aldolase A siRNA duplexes (duplexes 1, 3, and 4) (Fig. 5C). Next, we wanted to determine whether depletion of host aldolase A affected infectious progeny production. HeLa cells depleted of host aldolase A were infected with the mCherry CtL2 strain for 48 h. At 48 h postinfection, lysates from infected cells were used to infect a fresh monolayer of HeLa cells to determine the number of infectious bacteria. The number of infectious bacteria recovered at 48 h postinfection was significantly reduced following aldolase A depletion compared to mock-treated cells (Fig. 5D). We note that aldolase A siRNA duplex 2 did not significantly decrease inclusion size and had the smallest impact on infectious progeny production. We attribute this to the lower efficacy of this duplex compared to the other three. Nonetheless, we observed significant decrease in inclusion size and infectious progeny production with three independent siRNA duplexes, indicating that the observed phenotypes are specifically due to aldolase A depletion. Altogether, our data show that host aldolase A plays a role in inclusion development and infectious progeny production, suggesting that host glycolysis is important for C. trachomatis intracellular growth.
Bacterial glycolytic enzyme expression is downregulated during development.
Having shown that host glycolytic enzymes play a role in C. trachomatis intracellular growth, and knowing that C. trachomatis possesses its own glycolytic enzymes, we wanted to determine the pattern of bacterial glycolytic enzyme expression throughout infection. Three bacterial glycolytic enzymes—glucose-6-phosphate isomerase (pgi), fructose-bisphosphate aldolase (dhnA), and pyruvate kinase (pykF)—were selected based on their respective positions at the beginning, middle, and end of the glycolytic pathway. The mid- to late-cycle gene omcA served as a control. Gene expression of the bacterial enzymes was measured at 8, 12, 24, 36, and 48 h postinfection by quantitative PCR (qPCR). The expression of each gene was normalized to Chlamydia 16S rRNA and relative to expression at 8 h postinfection. As expected, omcA expression was strongly upregulated at 36 and 48 h postinfection (Fig. 6, omcA). Although expression of the bacterial glycolytic enzymes was detected throughout the time course of infection, all three enzymes were significantly downregulated over time (Fig. 6, pgi, dhnA, and pykF). Interestingly, the downregulation of bacterial glycolytic enzymes over time correlated with the increase in the number of inclusions that were positive for the host glycolytic enzyme aldolase A (Fig. 3A).
FIG 6.
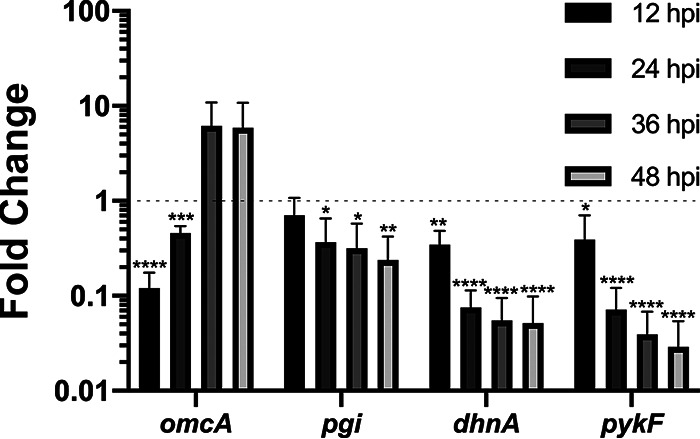
Chlamydia glycolytic enzymes are downregulated during development. The relative gene expression of a subset of C. trachomatis glycolytic enzymes (glucose-6-phosphate isomerase, pgi; aldolase A, dhnA; pyruvate kinase, pykF) at 12, 24, 36, and 48 h postinfection of HeLa cells was determined by RT-qPCR. Gene expression was normalized to 16S rRNA levels and the expression at 8 h postinfection (dotted line). The late-expressed gene encoding the outer membrane protein OmcA (omcA) was included as a control. The y axis is a log scale. The data show the means and the SD of a combination of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (Student t test).
DISCUSSION
Host glycolytic enzymes support C. trachomatis intracellular growth and replication.
In the present study, we showed that aldolase A-positive inclusions were significantly larger than aldolase-negative inclusions and that depletion of host aldolase A resulted in smaller inclusions and decreased infectious progeny production (Fig. 5). These results suggest that the specific inclusion localization of aldolase A is beneficial for C. trachomatis, however, we cannot rule out that aldolase A function in general (i.e., glycolysis) and regardless of its cellular localization is responsible for the phenotype observed. Determining whether an siRNA-resistant form of aldolase A targeted to specific organelles or the plasma membrane can rescue the growth defect could address this question.
In line with our results, a recent study described a genome-wide RNA interference screen to identify host factors essential for C. trachomatis growth and identified the host glycolytic enzymes glucose-6-phosphate isomerase and phosphofructokinase as high confidence hits in the primary screen (21). Although not validated, these results further support the reliance of C. trachomatis on host glycolysis for propagation and growth. Moreover, systematic depletion of glycolytic enzymes will be required to unequivocally determine that host glycolysis is essential for Chlamydia intracellular development. The same study also profiled metabolites of central carbon metabolism and observed upregulation of pyruvate, lactate, and glutamate, features indicative of Warburg metabolism (22–24). During Warburg metabolism, pyruvate is converted into lactate, and glycolytic intermediates are shuttled into the pentose phosphate pathway, where they are converted to ribonucleotides. Thus, Rother et al. (21) suggested that by shifting the cell into this hypermetabolic state, C. trachomatis is effectively forcing the host cell to generate anabolic substrates that the bacteria can then scavenge from the host. This was supported by the observed upregulation of carbon flux through the pentose phosphate pathway upon C. trachomatis infection. These authors concluded that C. trachomatis is able to modulate host cell metabolism in order to meet the high energetic demand of bacterial replication.
Localization of host glycolytic enzymes at the inclusion membrane.
We show here that at least three different glycolytic enzymes localized at the Chlamydia inclusion (Fig. 1). One of the limitations of our study is the overexpression of tagged constructs. While antibodies against eukaryotic glycolytic enzymes are widely available and successfully detect their targets via Western blotting, we were unable to determine the conditions that would allow for detection by immunofluorescence. This technical limitation precluded us from performing an important control and validating that endogenous enzymes localize at the Chlamydia inclusion. However, our results are strengthened by the fact that inclusion localization did not extend to other enzymes outside of the glycolytic pathway (see Fig. S3 in the supplemental material) and was not a result of increased expression of the construct over time (Fig. 3A).
Our data also indicate that enzymes localized together at the same inclusions (Fig. 2). The colocalization of multiple glycolytic enzymes is in line with the hypothesis that glycolytic enzymes form localized, multienzyme complexes. Although this hypothesis has existed for many years (31), it has lacked direct experimental evidence (32, 33). More recently, a model in which three separate glycolytic subcomplexes come together to form an active glycolytic complex has been proposed (34) and supports a dynamic complex formation.
Further supporting glycolytic enzyme complex formation, kinetoplastid parasites, such as Trypanosoma brucei, possess a glycosome, a membrane-bound organelle that specifically contains the majority of the enzymes of the glycolytic pathway (35–37). Glycosomes, and hence the compartmentalization of glycolytic enzymes, have also been shown to be essential for trypanosomatid metabolic regulation and viability (38). These glycosomes provide evidence that the compartmentalization of glycolytic enzymes can be advantageous to survival within a host. Although the localization of host glycolytic enzymes at the inclusion likely differs from that of the glycosomes, in that the glycolytic enzymes are not clustered within small membrane bound organelles, it is possible that the host enzymes may become bound to the surface of the inclusion membrane or associate with inclusion-bound factors.
In addition, work by Jang et al. demonstrated that under conditions of energy depletion or high energetic demand, the glycolytic enzymes of C. elegans neurons redistribute from a diffuse cytosolic localization to puncta localized to the presynaptic sites of neurons (39). Moreover, disrupting the localization of the glycolytic enzyme phosphofructokinase to the presynaptic sites fails to restore a phosphofructokinase mutant phenotype, indicating that localization of glycolytic enzymes to the presynaptic sites of neuron is necessary to meet the energy demands at C. elegans synapses. Similarly, the localization, and thus compartmentalization, of glycolytic enzymes at the C. trachomatis inclusion membrane could facilitate efficient channeling of substrates through the sequential enzymes of the pathway.
Mechanism of host glycolytic enzyme localization at the inclusion.
In the present study, we inhibited three major host cells factors that could be reasonable contributors to host glycolytic enzyme localization at the inclusion, the actin cytoskeleton, microtubules, and vesicular trafficking. Based on our results, we ruled out a role for the Chlamydia actin cage and for vesicular trafficking (Fig. 4B, C, and E). Moreover, host cell actin and microtubules were not necessary for sustained localization at the inclusion membrane (Fig. 4A and D).
The results presented in Fig. 3C indicate that sustained bacterial protein synthesis is not necessary to maintain inclusion localization of aldolase A. However, it is possible that treatment with chloramphenicol at 24 h postinfection may not affect the involvement of stable bacterial factors synthesized prior to treatment. Unfortunately, because aldolase A did not localize to the inclusion at early time points (Fig. 3A; see also Fig. S2 in the supplemental material) and because inhibition of bacterial protein synthesis would halt the developmental cycle, this hypothesis could not be tested. Of particular interest would be the Inclusion membrane proteins (Incs), a family of C. trachomatis type III translocated effector proteins that are inserted into the inclusion membrane to allow for the interaction of the inclusion with host molecules and organelles (6, 20, 40–55).
A pulldown of 3×FLAG-tagged pyruvate kinase or aldolase A to identify interacting partners that could play a role in inclusion localization of the host glycolytic enzymes did not identify any candidates (data not shown). However, we have only tested two of the glycolytic enzymes, and given that the enzymes could form individual subcomplexes that come together through interaction of these subcomplexes (34), it is difficult to predict which of the host enzymes could be interacting with bacterial and/or host factors at the inclusion. Therefore, testing each of the host glycolytic enzymes systematically would be necessary to completely rule out a role for bacterial and/or host factors in the localization of host glycolytic enzymes at the inclusion.
Finally, it is also possible that inclusion localization of the enzymes is not actively mediated by inclusion localized bacterial or host factors. This hypothesis is harder to test because the cell biology of the glycolytic pathway has not been investigated and the tools are underdeveloped. However, one could envision a host response, where sensing of the depletion of glycolytic substrates (discussed below) in the proximity of the inclusion brings the host glycolytic enzymes together.
Potential role for host glycolytic enzyme localization at the Chlamydia inclusion.
The expression of C. trachomatis glycolytic enzymes was significantly downregulated beyond 24 h postinfection (Fig. 6), when RBs are known to asynchronously transition into EBs (56, 57). The overall decrease in expression of bacterial glycolytic enzymes at mid-developmental cycle is consistent with previous RT- and quantitative RT-PCR data (19, 58). However, we do note that our data differ from previous publications in that we saw the highest level of enzyme expression at 8 h postinfection rather than at 24 h postinfection. These differences could be due to differences in cell lines and normalization conditions used. Moreover, the transcriptional downregulation is in line with the quantitative measurements of C. trachomatis encoded glycolytic enzymes indicating that they are more abundant in RBs than EBs (14). If the abundance of bacterial glycolytic enzymes is indicative of the pathway’s activity, the results presented above would suggest that RBs rely more on bacterial glycolytic pathway than EBs. However, this hypothesis would have to be reconciled with the energy source requirements of purified RBs and EBs in axenic media, where RBs exclusively used ATP and EBs preferentially required G6P, suggesting that RB are not reliant on substrate level phosphorylation and scavenge ATP from the host (59). One possibility is that energy source requirements differ in axenic media and in vivo and that during infection RBs utilize both G6P and cellular ATP. The latter could be provided and made more readily accessible by the localization of host glycolytic enzymes at the inclusion. As bacteria transition into EBs, their metabolic demand may be met by importing and using host G6P for substrate level phosphorylation by producing glycolytic enzymes, although at a lower level than RBs, or by using enzymes previously transcribed and translated by the RB form of the bacteria.
If Chlamydia can utilize its own glycolytic pathway, why would host glycolytic enzymes localize to the inclusion? One possibility mentioned above is that an increased local concentration of ATP around the inclusion could facilitate ATP import across the inclusion membrane for utilization in the lumen of the inclusion and in the cytosol of the bacteria. It is also possible that the ATP generated by host glycolysis is used at the surface of the inclusion to fuel the myriad of reactions that mediate host-inclusion interactions (60). The glycolytic pathway also generates glycolytic intermediates which are shuttled into the pentose phosphate pathway and converted to ribonucleotides. If these reactions were to occur in the close proximity of the inclusion, it would also increase the local pool of host-synthesized nucleotides that Chlamydia relies on for replication. Alternatively, Chlamydia may directly use intermediate glycolytic substrates. While it is not clear whether these specific host metabolic intermediates can be taken up by the bacteria, there are examples that demonstrate Chlamydia’s ability to acquire metabolites from the host cell. As discussed above, the glycolytic intermediate G6P must be obtained from the host and is imported via the bacterial UhpC antiporter (18). In addition, work by Mehlitz et al. demonstrated that the TCA-derived dicarboxylate malate is also taken up and metabolized by C. trachomatis (12). Moreover, while the inclusion membrane was originally thought to be impermeable to low-molecular-weight compounds, such as microinjected fluorophores greater than 520 Da (61), further studies showed that the inclusion membrane is permeable to ions (62). Thus, host metabolites such as glycolytic substrates, which are smaller than 520 Da, may also be able to passively enter the inclusion. Further experiments would be required to validate that it is indeed the case and whether and how these metabolites can be used to support bacterial growth.
Altogether, our study further highlights the complex and elaborate metabolic relationship between Chlamydia and its host and reveal that the spatial and temporal distribution of host glycolytic enzymes around the inclusion may facilitate the intracellular development of this obligate intracellular pathogen and therefore identify potential routes for future therapeutic drug development.
MATERIALS AND METHODS
Ethics statement.
All genetic manipulations and containment work were approved by the University of Virginia Biosafety Committee and are in compliance with the section III-D-1-a of the NIH guidelines for research involving recombinant DNA molecules.
Cell lines and bacterial strains.
HeLa cells were obtained from the ATCC (CCL-2) and cultured at 37°C with 5% CO2 in high-glucose Dulbecco modified Eagle medium (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen). C. trachomatis lymphogranuloma venereum (LGV) type II was obtained from the American Type Culture Collection (L2/434/Bu VR-902B). C. trachomatis propagation and infection were performed as previously described (63). C. muridarum was obtained from Michael Starnbach (Harvard Medical School, Boston, MA). mCherry-expressing C. trachomatis (mCherry CtL2) and C. muridarum strains were described previously (57).
Plasmid construction.
Restriction enzymes and T4 DNA ligase were obtained from New England BioLabs (Ipswich, MA). PCR was performed using Herculase DNA polymerase (Stratagene). PCR primers were obtained from Integrated DNA Technologies.
Vectors for expression in mammalian cells.
DNA fragments corresponding to pyruvate kinase, aldolase A, and the R42A and R148A mutants of aldolase A were amplified by PCR and cloned into the BamHI and XhoI restriction sites of pCDNA 4/TO 3×FLAG. Lactate dehydrogenase was cloned into the EcoRI and XhoI restriction sites of pCDNA 4/TO 3×FLAG. HA-aldolase A was cloned into the KpnI and NotI restriction sites of pCDNA3.1+. Pyruvate kinase, aldolase A and lactate dehydrogenase DNA fragments were amplified using uninfected HeLa cell cDNA as the template, and the primers are listed in Table S1 in the supplemental material.
DNA transfection.
DNA transfection was performed using X-tremeGENE 9 DNA transfection reagent (Roche), according to the manufacturer’s recommendations.
Immunofluorescence and confocal microscopy.
All steps were performed at room temperature. At the indicated times, HeLa cells seeded on glass coverslips were fixed with 4% paraformaldehyde in 1× phosphate-buffered saline (PBS) for 30 min. Coverslips were sequentially incubated with primary and secondary antibodies diluted in 0.1% Triton X-100 in 1× PBS for 1 h. Coverslips were washed with 1× PBS and mounted with DABCO antifade-containing mounting media. Imaging was performed using the Leica DMi8 microscope equipped with an Andor iXon ULTRA 888BV EMCCD camera and a CSU-W1 confocal scanner unit and driven by the IQ software. Images were processed using the Imaris software (Bitplane, Belfast, United Kingdom). Line intensity scan analyses presented in Fig. 1E were performed using ImageJ (NIH).
Antibodies.
The following primary antibodies were used for immunofluorescence microscopy (IF) and immunoblotting (Western blotting): mouse monoclonal anti-FLAG (1:1,000 for IF; Sigma), rabbit polyclonal anti-C. trachomatis IncA (1:200 for IF; kindly provided by T. Hackstadt, Rocky Mountain Laboratories), rabbit polyclonal anti-HA (1:100 for IF; Sigma), mouse monoclonal anti-aldolase A (1:1,000 for Western blotting; Santa Cruz), rabbit polyclonal anti-actin (1:10,000 for Western blotting; Sigma), mouse monoclonal anti-α-tubulin (1:2000 for IF; Sigma), and Alexa Fluor 514-conjugated phalloidin (1:200 for IF; Invitrogen). The following secondary antibodies were used: Alexa Fluor 488-, Alexa Fluor 514-, or Pacific Blue-conjugated goat anti-mouse antibody (1:500 for IF; Molecular Probes), Pacific Blue- or Alexa Fluor 514-conjugated goat anti-rabbit antibody (1:500 for IF; Molecular Probes), peroxidase-conjugated goat anti-mouse IgG (1:10,000 for Western blotting; Jackson ImmunoResearch), and peroxidase-conjugated goat anti-rabbit IgG (1:10,000 for Western blotting; Jackson ImmunoResearch).
Quantification of host glycolytic enzyme enrichment at the inclusion.
HeLa cells were transfected with the indicated 3×FLAG-tagged enzyme construct 18 h before infection with the mCherry CtL2 strain. The samples were processed for confocal microscopy and analyzed using Imaris imaging software. For each inclusion, the quantification was performed on a 1-μm slice located in the middle of the inclusion. Three-dimensional reconstructions of the raw signal corresponding to the enzyme signal at the inclusion and to the enzyme signal in the surrounding cytosol were generated using the Imaris imaging software. The average intensity of these three-dimensional objects was calculated using Imaris imaging software. The average intensity of the enzyme signal at the inclusion divided by the average intensity of the surrounding cytosol was used to determine the percent enrichment of the enzyme at the inclusion. Each experiment was performed in triplicate. Fifteen to thirty inclusions were analyzed per condition. The graphs were generated using GraphPad Prism. Averages and standard deviations (SD) are shown. A Student t test was performed, and statistical significance was set to P < 0.05.
Quantification of the percentage of inclusions positive for a given enzyme.
HeLa cells were transfected with the indicated 3×FLAG-tagged enzyme construct 18 h before infection with the mCherry CtL2 strain. The samples were processed for immunofluorescence microscopy and analyzed using an epifluorescence microscope. Transfected cells were scored for the presence (positive) or absence (negative) of a ring of enzyme enrichment at the inclusion (as seen in Fig. 1A). One hundred inclusions were analyzed per condition. The average percent positive inclusions and SD from three replicate experiments are presented. A Student t test was performed. and statistical significance was set to P < 0.05.
Quantification of the graph presented in Fig. 2B was performed as described above, except that HeLa cells were cotransfected with HA-aldolase A and the indicated 3×FLAG-tagged enzyme construct 18 h before infection with the mCherry CtL2 strain. Inclusions in cotransfected cells were scored for the presence or absence the HA- and 3×FLAG-tagged constructs. Quantification of the graph presented in Fig. 4B was performed as described above, except that the cells were transfected with the R42A, R148A, or WT aldolase A. Quantification of the graph presented in Fig. 4C was performed as described above, except that the cells were infected the inaC::aadA mutant or corresponding WT C. trachomatis strain.
Inhibition of eukaryotic protein synthesis.
HeLa cells were transfected with the 3×FLAG-tagged aldolase A construct 18 h before infection with the mCherry CtL2 strain. At the time of infection, cells were incubated in the presence or absence of 1 μg/ml cycloheximide for 18, 24, or 32 h. At the indicated time points, the samples were processed for confocal microscopy, and the percentage of positive inclusions was determined. One hundred inclusions were analyzed per condition. The average percentage of positive inclusions and SD from three replicate experiments are presented. A one-way analysis of variance (ANOVA) with multiple comparisons was performed, and statistical significance was set to P < 0.05.
Inhibition of C. trachomatis protein synthesis.
HeLa cells were transfected with the 3×FLAG-tagged aldolase A construct 18 h before infection with the mCherry CtL2 strain. At 24 h postinfection, infected cells were incubated in the presence of 40 μg/ml of chloramphenicol for 8 h. At 32 h postinfection, the samples were processed for confocal microscopy, and the percentage of positive inclusions was determined. One hundred inclusions were analyzed per condition. The average percentage of positive inclusions and SD from three replicate experiments are presented. A Student t test was performed, and statistical significance was set to P < 0.05.
Drug treatments.
HeLa cells were transfected with the indicated 3×FLAG-tagged enzyme construct 18 h before infection with the mCherry CtL2 strain. For cytochalasin D and nocodazole treatment, at 23.5 h postinfection, infected cells were incubated in the presence of 1 μM cytochalasin D or 33 μM nocodazole for 30 min. For brefeldin A treatment, at 6 h postinfection, infected cells were incubated in the presence of 1 μg/ml brefeldin A overnight. At 24 h postinfection, the samples for all drug treatments were processed for confocal microscopy, and the percentage of positive inclusions was determined. One hundred inclusions were analyzed per condition. The average percentage of positive inclusions and SD from three replicate experiments are presented. A Student t test was performed, and statistical significance was set to P < 0.05.
inaC mutant generation.
An inaC::aadA mutant was generated in our lab C. trachomatis LGV L2 strain background using TargeTron, as described by previous studies (20, 30). Using PCR, the GrpII intron was retargeted for C. trachomatis 434/Bu inaC using the primers CTL0184 129 130 IBS1/2, CTL0184 129 130 EBS1/delta, and CTL0184 129 130 EBS2 designed by the TargeTron computer algorithm (TargeTronics) (the primer sequences are listed in Table S1 in the supplemental material). The resulting PCR product was digested with BsrGI and HindIII and cloned into the BsrGI/HindIII site of the pDFTT3-aadA suicide vector (64). C. trachomatis serovar L2 was transformed with pDFTT3-aadA-inaC according to our calcium-based transformation protocol as previously described (57). After three passages, transformants were plaque purified and amplified. Genomic DNA was prepared using an illustra bacteria genomicPrep Mini Spin kit (GE Healthcare), according to the manufacturer’s recommendations. The inaC open reading frame was amplified from WT or inaC::aadA mutant genomic DNA by PCR using the primers CTL0184 Up and CTL0184 Dwn (see Table S1). The resulting PCR products were analyzed by DNA gel electrophoresis and sequenced to verify proper insertion of the group II intron (see Fig. S6).
Aldolase A depletion.
The protocol for siRNA transfection has been described previously (63). Aldolase A depletion was performed by transfection of four independent siRNA duplexes. The sequences of the Aldolase A siRNA duplexes were: duplex 1 (GGACAAAUGGCGAGACUAC), duplex 2 (UUGAAGCGCUGCCAGUAUG), duplex 3 (GGCGUUGUGUGCUGAAGAU), and duplex 4 (UGACAUCGCUCACCGCAUC).
Immunoblotting.
Protein samples were separated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked for 1 h at room temperature in 1× PBS containing 0.05% Tween and 5% fat-free milk. Primary and horseradish peroxidase-conjugated secondary antibodies were diluted in 1× PBS containing 0.05% Tween and 5% fat-free milk and incubated overnight at 4°C and 1 h at room temperature, respectively. Proteins were detected using the Amersham ECL immunoblotting detection reagent, according to the manufacturer’s recommendation, and a Bio-Rad ChemiDoc imaging system. Immunoblots were quantified using ImageJ (NIH) software.
Inclusion size quantification and computer-assisted image analysis.
HeLa cells were transfected with the indicated 3×FLAG-tagged enzyme construct 18 to 24 h before infection with the mCherry CtL2 strain. The samples were processed for immunofluorescence microscopy and imaged using an epifluorescence microscope. Computer-assisted image analysis, using the analytical tools of the MetaExpress software, was used to determine the area of each aldolase A-positive and -negative inclusion. One hundred inclusions were analyzed per condition.
For quantification of the graph presented in Fig. 5C, siRNA-treated cells were infected with the mCherry CtL2 strain and fixed at 32 h postinfection. The nuclei were labeled with the DNA dye Hoechst. The cells were subjected to automated fluorescence microscopy using an ImageXpress automated system to capture images corresponding to the cell nuclei and the inclusion. Computer-assisted image analysis, using the analytical tools of the MetaExpress software, was used to determine the number of nuclei and the total area of each inclusion.
Infectious progeny production.
HeLa cells incubated with the indicated siRNA duplexes for 3 days were collected 48 h postinfection and lysed with water, and dilutions of the lysate were used to infect fresh HeLa cells. The cells were fixed 24 h postinfection and the number of inclusion-forming units (IFU) was determined after assessment of the number of infected cells by automated imaging using an ImageXpress automated system.
Real-time PCR analysis of bacterial glycolytic gene expression.
At the indicated time point, infected cells were homogenized with TRIzol (Thermo Fisher Scientific) to extract RNA from infected cells. Each RNA sample was treated with DNase according to the Turbo DNA-free kit protocol (Thermo Fisher Scientific). Complementary DNA (cDNA) was synthesized using SuperScript II reverse transcriptase (Thermo Fisher Scientific) according to the manufacturer’s protocol. Samples were primed using random primers (Thermo Fisher Scientific). mRNA levels were determined by quantitative real-time PCR using the Universal Probe Library (Roche Biochemicals, Indianapolis, IN) and Luna Universal qPCR master mix (New England Biolabs). Thermal cycling was carried out using a LightCycler 96 instrument (Roche Diagnostics) under the following conditions: 95°C for 5 min, followed by 45 cycles of 95°C for 10 s and 56°C for 25 s. Quantification cycle (Cq) values were derived using the LightCycler 96 software, and fold changes were calculated using threshold cycle (CT) 16S rRNA for normalization (65, 66). Statistical analysis was performed by using the Student t test, and statistical significance was set to P < 0.05. PCR primers used for quantitative real-time PCR were obtained from Integrated DNA Technologies. Primer sequences and corresponding probes are listed in Table S2 in the supplemental material.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ted Hackstadt (NIH-Rocky Mountain Laboratories) for the anti-IncA antibodies and María Eugenia Cortina, Erin Weddle, Clayton Bishop, and Hervé Agaisse (University of Virginia) for critical reading of the manuscript.
This study was supported by NIH grants T32AI007046 to R.J.E. and R01AI101441 to I.D. and by the generosity of a Wagner Fellowship to R.J.E.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Malhotra M, Sood S, Mukherjee A, Muralidhar S, Bala M. 2013. Genital Chlamydia trachomatis: an update. Indian J Med Res 138:303–316. [PMC free article] [PubMed] [Google Scholar]
- 2.Moulder JW. 1991. Interaction of chlamydiae and host cells in vitro. Microbiol Rev 55:143–190. doi: 10.1128/MMBR.55.1.143-190.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abdelrahman YM, Belland RJ. 2005. The chlamydial developmental cycle. FEMS Microbiol Rev 29:949–959. doi: 10.1016/j.femsre.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Todd WJ, Caldwell HD. 1985. The interaction of Chlamydia trachomatis with host cells: ultrastructural studies of the mechanism of release of a biovar II strain from HeLa 229 cells. J Infect Dis 151:1037–1044. doi: 10.1093/infdis/151.6.1037. [DOI] [PubMed] [Google Scholar]
- 5.Hybiske K, Stephens RS. 2007. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc Natl Acad Sci U S A 104:11430–11435. doi: 10.1073/pnas.0703218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lutter EI, Barger AC, Nair V, Hackstadt T. 2013. Chlamydia trachomatis inclusion membrane protein CT228 recruits elements of the myosin phosphatase pathway to regulate release mechanisms. Cell Rep 3:1921–1931. doi: 10.1016/j.celrep.2013.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moulder JW. 1970. Glucose metabolism of L cells before and after infection with Chlamydia psittaci. J Bacteriol 104:1189–1196. doi: 10.1128/JB.104.3.1189-1196.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gill SD, Stewart RB. 1970. Effect of metabolic inhibitors on the production of Chlamydia psittaci by infected L cells. Can J Microbiol 16:1079–1085. doi: 10.1139/m70-182. [DOI] [PubMed] [Google Scholar]
- 9.Becker Y, Asher Y. 1972. Obligate parasitism of trachoma agent: lack of trachoma development in ethidium bromide-treated cells. Antimicrob Agents Chemother 1:171–173. doi: 10.1128/aac.1.2.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hatch TP, Al-Hossainy E, Silverman JA. 1982. Adenine nucleotide and lysine transport in Chlamydia psittaci. J Bacteriol 150:662–670. doi: 10.1128/JB.150.2.662-670.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, Koonin EV, Davis RW. 1998. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282:754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- 12.Mehlitz A, Eylert E, Huber C, Lindner B, Vollmuth N, Karunakaran K, Goebel W, Eisenreich W, Rudel T. 2017. Metabolic adaptation of Chlamydia trachomatis to mammalian host cells. Mol Microbiol 103:1004–1019. doi: 10.1111/mmi.13603. [DOI] [PubMed] [Google Scholar]
- 13.Saka HA, Thompson JW, Chen YS, Kumar Y, Dubois LG, Moseley MA, Valdivia RH. 2011. Quantitative proteomics reveals metabolic and pathogenic properties of Chlamydia trachomatis developmental forms. Mol Microbiol 82:1185–1203. doi: 10.1111/j.1365-2958.2011.07877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skipp PJ, Hughes C, McKenna T, Edwards R, Langridge J, Thomson NR, Clarke IN. 2016. Quantitative proteomics of the infectious and replicative forms of Chlamydia trachomatis. PLoS One 11:e0149011. doi: 10.1371/journal.pone.0149011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tjaden J, Winkler HH, Schwoppe C, Van Der Laan M, Mohlmann T, Neuhaus HE. 1999. Two nucleotide transport proteins in Chlamydia trachomatis, one for net nucleoside triphosphate uptake and the other for transport of energy. J Bacteriol 181:1196–1202. doi: 10.1128/JB.181.4.1196-1202.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fisher DJ, Fernandez RE, Maurelli AT. 2013. Chlamydia trachomatis transports NAD via the Npt1 ATP/ADP translocase. J Bacteriol 195:3381–3386. doi: 10.1128/JB.00433-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson DL, Cox MM. 2008. Glycolysis, gluconeogenesis, and the pentose phosphate pathway, p 527–568. In Lehninger principles of biochemistry, 5th ed. WH Freeman, New York, NY. [Google Scholar]
- 18.Schwoppe C, Winkler HH, Neuhaus HE. 2002. Properties of the glucose-6-phosphate transporter from Chlamydia pneumoniae (HPTcp) and the glucose-6-phosphate sensor from Escherichia coli (UhpC). J Bacteriol 184:2108–2115. doi: 10.1128/jb.184.8.2108-2115.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iliffe-Lee ER, McClarty G. 1999. Glucose metabolism in Chlamydia trachomatis: the ‘energy parasite’ hypothesis revisited. Mol Microbiol 33:177–187. doi: 10.1046/j.1365-2958.1999.01464.x. [DOI] [PubMed] [Google Scholar]
- 20.Kokes M, Dunn JD, Granek JA, Nguyen BD, Barker JR, Valdivia RH, Bastidas RJ. 2015. Integrating chemical mutagenesis and whole-genome sequencing as a platform for forward and reverse genetic analysis of Chlamydia. Cell Host Microbe 17:716–725. doi: 10.1016/j.chom.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rother M, Gonzalez E, Teixeira da Costa AR, Wask L, Gravenstein I, Pardo M, Pietzke M, Gurumurthy RK, Angermann J, Laudeley R, Glage S, Meyer M, Chumduri C, Kempa S, Dinkel K, Unger A, Klebl B, Klos A, Meyer TF. 2018. Combined human genome-wide RNAi and metabolite analyses identify IMPDH as a host-directed target against Chlamydia infection. Cell Host Microbe 23:661–671. doi: 10.1016/j.chom.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 22.Warburg O. 1956. On the origin of cancer cells. Science 123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 23.Vander Heiden MG, Cantley LC, Thompson CB. 2009. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asgari Y, Zabihinpour Z, Salehzadeh-Yazdi A, Schreiber F, Masoudi-Nejad A. 2015. Alterations in cancer cell metabolism: the Warburg effect and metabolic adaptation. Genomics 105:275–281. doi: 10.1016/j.ygeno.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 25.Rockey DD, Heinzen RA, Hackstadt T. 1995. Cloning and characterization of a Chlamydia psittaci gene coding for a protein localized in the inclusion membrane of infected cells. Mol Microbiol 15:617–626. doi: 10.1111/j.1365-2958.1995.tb02371.x. [DOI] [PubMed] [Google Scholar]
- 26.Bannantine JP, Stamm WE, Suchland RJ, Rockey DD. 1998. Chlamydia trachomatis IncA is localized to the inclusion membrane and is recognized by antisera from infected humans and primates. Infect Immun 66:6017–6021. doi: 10.1128/IAI.66.12.6017-6021.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arnold H, Pette D. 1968. Binding of glycolytic enzymes to structure proteins of the muscle. Eur J Biochem 6:163–171. doi: 10.1111/j.1432-1033.1968.tb00434.x. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Morris AJ, Tolan DR, Pagliaro L. 1996. The molecular nature of the F-actin binding activity of aldolase revealed with site-directed mutants. J Biol Chem 271:6861–6865. doi: 10.1074/jbc.271.12.6861. [DOI] [PubMed] [Google Scholar]
- 29.Kumar Y, Valdivia RH. 2008. Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell Host Microbe 4:159–169. doi: 10.1016/j.chom.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wesolowski J, Weber MM, Nawrotek A, Dooley CA, Calderon M, St Croix CM, Hackstadt T, Cherfils J, Paumet F. 2017. Chlamydia hijacks ARF GTPases to coordinate microtubule posttranslational modifications and Golgi complex positioning. mBio 8:e02280-16. doi: 10.1128/mBio.02280-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clarke FM, Masters CJ. 1975. On the association of glycolytic enzymes with structural proteins of skeletal muscle. Biochim Biophys Acta 381:37–46. doi: 10.1016/0304-4165(75)90187-7. [DOI] [PubMed] [Google Scholar]
- 32.Boiteux A, Hess B. 1981. Design of glycolysis. Philos Trans R Soc Lond B Biol Sci 293:5–22. doi: 10.1098/rstb.1981.0056. [DOI] [PubMed] [Google Scholar]
- 33.Brooks SP, Storey KB. 1991. Where is the glycolytic complex? A critical evaluation of present data from muscle tissue. FEBS Lett 278:135–138. doi: 10.1016/0014-5793(91)80101-8. [DOI] [PubMed] [Google Scholar]
- 34.Menard L, Maughan D, Vigoreaux J. 2014. The structural and functional coordination of glycolytic enzymes in muscle: evidence of a metabolon? Biology (Basel) 3:623–644. doi: 10.3390/biology3030623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Opperdoes FR, Borst P. 1977. Localization of nine glycolytic enzymes in a microbody-like organelle in Trypanosoma brucei: the glycosome. FEBS Lett 80:360–364. doi: 10.1016/0014-5793(77)80476-6. [DOI] [PubMed] [Google Scholar]
- 36.Michels PA, Bringaud F, Herman M, Hannaert V. 2006. Metabolic functions of glycosomes in trypanosomatids. Biochim Biophys Acta 1763:1463–1477. doi: 10.1016/j.bbamcr.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 37.Crowe LP, Wilkinson CL, Nicholson KR, Morris MT. 2020. Trypanosoma brucei Pex13.2 is an accessory peroxin that functions in the import of peroxisome targeting sequence type 2 proteins and localizes to subdomains of the glycosome. mSphere 5:e00744-19. doi: 10.1128/mSphere.00744-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haanstra JR, Gonzalez-Marcano EB, Gualdron-Lopez M, Michels PA. 2016. Biogenesis, maintenance and dynamics of glycosomes in trypanosomatid parasites. Biochim Biophys Acta 1863:1038–1048. doi: 10.1016/j.bbamcr.2015.09.015. [DOI] [PubMed] [Google Scholar]
- 39.Jang S, Nelson JC, Bend EG, Rodriguez-Laureano L, Tueros FG, Cartagenova L, Underwood K, Jorgensen EM, Colon-Ramos DA. 2016. Glycolytic enzymes localize to synapses under energy stress to support synaptic function. Neuron 90:278–291. doi: 10.1016/j.neuron.2016.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scidmore MA, Hackstadt T. 2001. Mammalian 14-3-3β associates with the Chlamydia trachomatis inclusion membrane via its interaction with IncG. Mol Microbiol 39:1638–1650. doi: 10.1046/j.1365-2958.2001.02355.x. [DOI] [PubMed] [Google Scholar]
- 41.Rzomp KA, Moorhead AR, Scidmore MA. 2006. The GTPase Rab4 interacts with Chlamydia trachomatis inclusion membrane protein CT229. Infect Immun 74:5362–5373. doi: 10.1128/IAI.00539-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Derré I, Swiss R, Agaisse H. 2011. The lipid transfer protein CERT interacts with the Chlamydia inclusion protein IncD and participates to ER-Chlamydia inclusion membrane contact sites. PLoS Pathog 7:e1002092. doi: 10.1371/journal.ppat.1002092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aeberhard L, Banhart S, Fischer M, Jehmlich N, Rose L, Koch S, Laue M, Renard BY, Schmidt F, Heuer D. 2015. The proteome of the isolated Chlamydia trachomatis containing vacuole reveals a complex trafficking platform enriched for retromer components. PLoS Pathog 11:e1004883. doi: 10.1371/journal.ppat.1004883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mirrashidi KM, Elwell CA, Verschueren E, Johnson JR, Frando A, Von Dollen J, Rosenberg O, Gulbahce N, Jang G, Johnson T, Jager S, Gopalakrishnan AM, Sherry J, Dunn JD, Olive A, Penn B, Shales M, Cox JS, Starnbach MN, Derré I, Valdivia R, Krogan NJ, Engel J. 2015. Global mapping of the Inc-human interactome reveals that retromer restricts Chlamydia infection. Cell Host Microbe 18:109–121. doi: 10.1016/j.chom.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mital J, Lutter EI, Barger AC, Dooley CA, Hackstadt T. 2015. Chlamydia trachomatis inclusion membrane protein CT850 interacts with the dynein light chain DYNLT1 (Tctex1). Biochem Biophys Res Commun 462:165–170. doi: 10.1016/j.bbrc.2015.04.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elwell CA, Czudnochowski N, von Dollen J, Johnson JR, Nakagawa R, Mirrashidi K, Krogan NJ, Engel JN, Rosenberg OS. 2017. Chlamydia interfere with an interaction between the mannose-6-phosphate receptor and sorting nexins to counteract host restriction. Elife 6:e22709. doi: 10.7554/eLife.22709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paul B, Kim HS, Kerr MC, Huston WM, Teasdale RD, Collins BM. 2017. Structural basis for the hijacking of endosomal sorting nexin proteins by Chlamydia trachomatis. Elife 6:e22311. doi: 10.7554/eLife.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sixt BS, Bastidas RJ, Finethy R, Baxter RM, Carpenter VK, Kroemer G, Coers J, Valdivia RH. 2017. The Chlamydia trachomatis inclusion membrane protein CpoS counteracts STING-mediated cellular surveillance and suicide programs. Cell Host Microbe 21:113–121. doi: 10.1016/j.chom.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weber MM, Lam JL, Dooley CA, Noriea NF, Hansen BT, Hoyt FH, Carmody AB, Sturdevant GL, Hackstadt T. 2017. Absence of specific Chlamydia trachomatis inclusion membrane proteins triggers premature inclusion membrane lysis and host cell death. Cell Rep 19:1406–1417. doi: 10.1016/j.celrep.2017.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stanhope R, Flora E, Bayne C, Derré I. 2017. IncV, a FFAT motif-containing Chlamydia protein, tethers the endoplasmic reticulum to the pathogen-containing vacuole. Proc Natl Acad Sci U S A 114:12039–12044. doi: 10.1073/pnas.1709060114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nguyen PH, Lutter EI, Hackstadt T. 2018. Chlamydia trachomatis inclusion membrane protein MrcA interacts with the inositol 1,4,5-trisphosphate receptor type 3 (ITPR3) to regulate extrusion formation. PLoS Pathog 14:e1006911. doi: 10.1371/journal.ppat.1006911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Almeida F, Luis MP, Pereira IS, Pais SV, Mota LJ. 2018. The human centrosomal protein CCDC146 binds Chlamydia trachomatis inclusion membrane protein CT288 and is recruited to the periphery of the chlamydia-containing vacuole. Front Cell Infect Microbiol 8:254. doi: 10.3389/fcimb.2018.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shaw JH, Key CE, Snider TA, Sah P, Shaw EI, Fisher DJ, Lutter EI. 2018. Genetic inactivation of Chlamydia trachomatis inclusion membrane protein CT228 alters MYPT1 recruitment, extrusion production, and longevity of infection. Front Cell Infect Microbiol 8:415. doi: 10.3389/fcimb.2018.00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Faris R, Merling M, Andersen SE, Dooley CA, Hackstadt T, Weber MM. 2019. Chlamydia trachomatis CT229 subverts Rab GTPase-dependent CCV trafficking pathways to promote chlamydial infection. Cell Rep 26:3380–3390. doi: 10.1016/j.celrep.2019.02.079. [DOI] [PubMed] [Google Scholar]
- 55.Agaisse H, Derré I. 2014. Expression of the effector protein IncD in Chlamydia trachomatis mediates recruitment of the lipid transfer protein CERT and the endoplasmic reticulum-resident protein VAPB to the inclusion membrane. Infect Immun 82:2037–2047. doi: 10.1128/IAI.01530-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee JK, Enciso GA, Boassa D, Chander CN, Lou TH, Pairawan SS, Guo MC, Wan FYM, Ellisman MH, Sutterlin C, Tan M. 2018. Replication-dependent size reduction precedes differentiation in Chlamydia trachomatis. Nat Commun 9:45. doi: 10.1038/s41467-017-02432-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cortina ME, Ende RJ, Bishop RC, Bayne C, Derré I. 2019. Chlamydia trachomatis and Chlamydia muridarum spectinomycin-resistant vectors and a transcriptional fluorescent reporter to monitor conversion from replicative to infectious bacteria. PLoS One 14:e0217753. doi: 10.1371/journal.pone.0217753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang M, Rajeeve K, Rudel T, Dandekar T. 2019. Comprehensive flux modeling of Chlamydia trachomatis proteome and qRT-PCR data indicate biphasic metabolic differences between elementary bodies and reticulate bodies during infection. Front Microbiol 10:2350. doi: 10.3389/fmicb.2019.02350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Omsland A, Sager J, Nair V, Sturdevant DE, Hackstadt T. 2012. Developmental stage-specific metabolic and transcriptional activity of Chlamydia trachomatis in an axenic medium. Proc Natl Acad Sci U S A 109:19781–19785. doi: 10.1073/pnas.1212831109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Elwell C, Mirrashidi K, Engel J. 2016. Chlamydia cell biology and pathogenesis. Nat Rev Microbiol 14:385–400. doi: 10.1038/nrmicro.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heinzen RA, Hackstadt T. 1997. The Chlamydia trachomatis parasitophorous vacuolar membrane is not passively permeable to low-molecular-weight compounds. Infect Immun 65:1088–1094. doi: 10.1128/IAI.65.3.1088-1094.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grieshaber S, Swanson JA, Hackstadt T. 2002. Determination of the physical environment within the Chlamydia trachomatis inclusion using ion-selective ratiometric probes. Cell Microbiol 4:273–283. doi: 10.1046/j.1462-5822.2002.00191.x. [DOI] [PubMed] [Google Scholar]
- 63.Derré I, Pypaert M, Dautry-Varsat A, Agaisse H. 2007. RNAi screen in Drosophila cells reveals the involvement of the Tom complex in Chlamydia infection. PLoS Pathog 3:1446–1458. doi: 10.1371/journal.ppat.0030155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lowden NM, Yeruva L, Johnson CM, Bowlin AK, Fisher DJ. 2015. Use of aminoglycoside 3′ adenyltransferase as a selection marker for Chlamydia trachomatis intron-mutagenesis and in vivo intron stability. BMC Res Notes 8:570. doi: 10.1186/s13104-015-1542-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Borges V, Ferreira R, Nunes A, Nogueira P, Borrego MJ, Gomes JP. 2010. Normalization strategies for real-time expression data in Chlamydia trachomatis. J Microbiol Methods 82:256–264. doi: 10.1016/j.mimet.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 66.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.