

Abstract
Tuberous sclerosis complex (TSC) is an autosomal dominant, multisystem disorder that is characterized by cellular and tissue dysplasia in several organs. With the advent of genetic and molecular techniques, mutations in the TSC1 or TSC2 genes were discovered to be responsible for mTOR overactivation, which is the underlying mechanism of pathogenesis. TSC is a highly heterogenous clinical entity with variable presentations and severity of disease. The brain, heart, skin, eyes, kidneys, and lungs are commonly involved in this syndrome, with neurologic symptoms comprising a significant source of morbidity and mortality. In 2012, the diagnostic criteria for TSC were revised by the International Tuberous Sclerosis Complex Consensus panel, and genetic testing was incorporated into the guidelines. Early detection of cardiac rhabdomyomas or TSC-associated skin lesions can suggest the diagnosis and underlie the importance of clinical vigilance. Animal studies have demonstrated the benefit of using mTOR inhibitors for various symptoms of TSC, and they have been successfully translated into clinical trials with significant improvement in symptom burden. Subependymal giant cell astrocytomas, renal angiomyolipomas, and epilepsy are the three FDA-approved indications in relation to TSC for the use of everolimus, which is a first generation mTOR inhibitor. Rapamycin has been FDA approved for lymphangioleiomyomatosis. Other TSC symptoms that could potentially benefit from this class of medication are currently under investigation. TSC constitutes a unique combination of protean physical symptoms and neurobehavioral abnormalities. TSC associated neuropsychiatric disorders (TAND), including intellectual disability, mood disorders, and autism spectrum disorder, represent significant challenges but remain underdiagnosed and undertreated. The TAND checklist is a useful tool for routine use in the clinical evaluation of TSC patients. A multidisciplinary treatment plan, based on the specific problems and needs of individuals, is the key to management of this genetic condition. Ongoing research studies have been providing promising leads for developing novel mechanistic strategies to address the pathophysiology of TSC.
Keywords: Tuberous sclerosis, mTOR, autism, epilepsy, rapamycin
1. Introduction
Tuberous sclerosis complex (TSC) is a rare, multisystem, autosomal dominant syndrome characterized by tumorigenesis and is associated with neurologic and behavioral abnormalities. The pathogenesis is driven by hyperactivation in the mTOR pathway due to de novo or inherited mutations in the TSC1 or TSC2 genes. TSC was first identified by German pathologist Friedrich Daniel von Recklinghausen, in 1862, in a baby with cardiac myotomas and sclerotic brain lesions who died shortly after birth [1].
It was better defined in 1880 by French neurologist Désiré-Magloire Bourneville as “tuberous sclerosis of the cerebral convolutions”; hence, the disease was named “Bourneville’s disease” after him. His patient reportedly had seizures, intellectual disability, and renal angiomyolipomas (AMLs) in the form of “hard masses, one the size of a walnut” [2]. In 1908, another German neurologist, Heinrich Vogt, established the Vogt triad for TSC, comprising intellectual disability, intractable seizures, and facial angiofibromas [3]. The first use of the term tuberous sclerosis complex was by Sylvan Moolten in 1942 [4]. In 1972, Spanish-American neurologist Manuel Rodriguez Gomez established the first diagnostic criteria for TSC, and he has since been viewed as the father of TSC in the USA [5].
Even though there are no pathognomonic signs for TSC, various clinical stigmata are commonly seen as part of the syndrome, which raise suspicion for the diagnosis. Common manifestations include cortical tubers, subependymal nodules, subependymal giant cell astrocytomas (Figure 1), seizures, cardiac rhabdomyomas, renal AMLs, retinal hamartomas, pulmonary lymphangioleiomyomatosis (LAM), facial angiofibromas (Figure 2), ash-leaf spots, shagreen patches, intellectual disability, and autism spectrum disorder [1,3]. Once the diagnosis is suspected, genetic testing can be performed to look for mutations in the TSC1 or TSC2 genes and guide genetic counseling. As a matter of fact, improvements in the realm of genetics opened a new era for TSC in the 1990s, when the TSC1 and TSC2 genes were identified, which led to the exploration of the molecular pathways involved [6].
Figure 1.
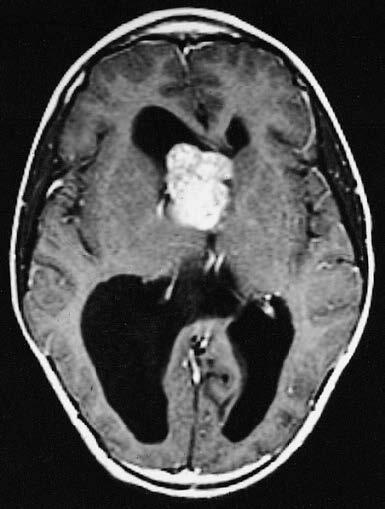
MRI of a SEGA (Image courtesy of Wikimedia Commons [online]. Website https://commons.wikimedia.org/wiki/File:MRI_of_brain_with_subependymal_giant_cell_astrocytoma.jpg [accessed 05 January 2020].).
Figure 2.
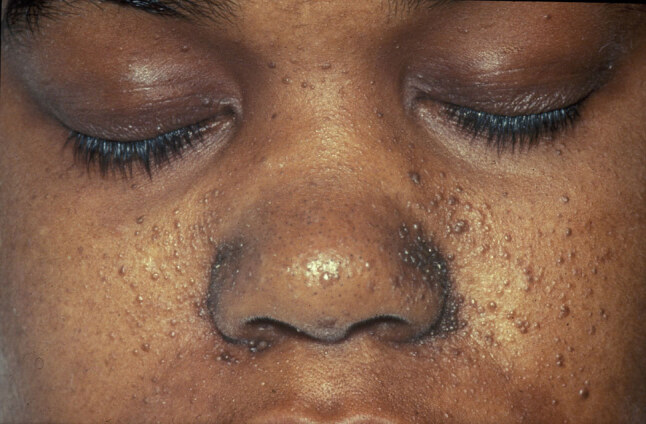
Facial angiofibromas (Image courtesy of Wikimedia Commons [online]. Website https://commons.wikimedia.org/wiki/File:Patient_with_facial_angiofibromas_caused_by_tuberous_sclerosis.jpg [accessed 05 January 2020].).
TSC1 and TSC2 genes encode the proteins hamartin and tuberin, respectively. These proteins compose the TSC complex, which acts as a brake on the mTOR signaling cascade [7,8]. In agreement with this, mTOR inhibitors, such as rapamycin and its analogs (rapalogs), are commonly used in a clinical setting for various symptoms of this condition [9]. The current management of TSC is mostly symptomatic, with pharmacologic, surgical, or behavioral intervention options. Due to the vast phenotypic heterogeneity encountered, not all therapeutic approaches benefit the entirety of symptoms or patients to the same extent; hence, a personalized treatment strategy is critical [10].
2. Epidemiology
The incidence of TSC has been estimated as occurring in 1/6000–10,000 newborns annually, and therefore, it is categorized as a rare disease. It affects approximately 2 million people globally [11]. The prevalence in Europe is approximately 11,500–25,000 [12]. There is no sex or ethnicity predilection [13]. However, differences in sex predominance have been observed in numerous symptoms (see the Clinical presentation section). In 2012, a conference was held in Washington, DC, USA, by the International Tuberous Sclerosis Complex Consensus group to revise the diagnostic criteria for TSC, providing a comprehensive list consisting of major and minor criteria for use by physicians in a clinical setting [14,15] (Table).
Table.
TSC diagnostic criteria.
|
Diagnostic criteria according to the 2012 International Tuberous Sclerosis Complex Consensus Conference
Definite diagnosis: Two major features, or 1 major feature with greater than 2 minor features, or the presence of a TSC1 or TSC2 mutation of confirmed pathogenicity Possible diagnosis: Either 1 major feature or greater than 2 minor features Major criteria: Skin/oral cavity • Hypomelanotic macules (n > 3, at least 5 mm in diameter) • Angiofibromas (n > 3) or fibrous cephalic plaque • Ungual fibromas (n > 2) • Shagreen patch Central nervous system • Cortical dysplasias (includes tubers and cerebral white matter radial migration lines) • Subependymal nodules • Subependymal giant cell astrocytoma Heart • Cardiac rhabdomyoma Lungs • Lymphangioleiomyomatosis Kidney • Angiomyolipoma (n > 2) Eyes • Multiple retinal hamartomas Minor criteria: Skin/oral cavity • Confetti skin lesions • Dental enamel pits (n > 3) • Intraoral fibromas (n > 2) Kidney • Multiple renal cysts Eyes • Retinal achromic patch Other organs • Nonrenal hamartomas Genetics: Identification of either a TSC1 or a TSC2 pathogenic mutation in DNA from normal tissue is sufficient to make a definite diagnosis |
3. Molecular genetics
TSC is caused by mutations in either of the TSC1 or TSC2 genes, which were discovered through the use of Drosophila models in the 1990s [16]. TSC1 is found on chromosome 9 (9q34) and TSC2 is found on chromosome 16 (16p13.3), encoding the proteins hamartin and tuberin, respectively [7,8]. TSC is also genetically linked to autosomal dominant polycystic kidney disease, as the PKD1 and TSC2 genes are closely located (48 base pairs apart) on chromosome 16. When both genes are affected due to a contiguous gene deletion, it may lead to a clinical picture called PKD-TSC (MIM #600273) with severe renal symptoms [17].
TSC can arise due to de novo or inherited mutations. De novo mutations constitute two-thirds of all TSC diagnoses. The remaining one-third is inherited in an autosomal dominant fashion [18]. The genetic pattern of TSC can be described by Knudson’s 2-hit hypothesis, where the acquisition of a somatic mutation in a previously functional allele of TSC1 or TSC2 , in addition to the existing germline mutation in the other allele, leads to the disease state [19]. Most TSC2 cases are sporadic and have more severe manifestations, while the ratio of TSC1 to TSC2 mutations in familial cases is close to one [20]. TSC shows almost complete penetrance with wide phenotypic variability. This means that any individual carrying a TSC mutation would be afflicted with the disease, but to varying degrees. The large number of possible mutations in the TSC genes also contributes to the heterogeneity within the patient population [21].
The TSC1 and TSC2 genes differ from each other in that TSC1 mutations are mostly nonsense or frameshift mutations, leading to protein truncation, whereas missense mutations, large deletions, or rearrangements are seen more with TSC2 [22]. Additionally, TSC1 mutations have been identified in ~10–20% of patients clinically diagnosed with TSC, while TSC2 mutations have been identified in ~70–90% [23,24]. Several thousand small mutations have been shown to cause TSC, which can be found in the online Leiden Open Variation Database http://chromium.lovd.nl/LOVD2/TSC/home.php. However, genetic testing may be negative in 10–25% of inherited cases due to reasons such as somatic mosaicism, or mutations in intron or promoter regions [25].
The protein products of the TSC genes, hamartin and tuber, work together within the same intracellular pathway, which explains why a mutation in either gene can give rise to the same disease. The downstream target of these proteins is the mammalian target of rapamycin complex 1 (mTORC1), which is a protein serine/threonine kinase complex involved in many important anabolic and catabolic processes, such as translation, cellular growth, proliferation, stress response, and autophagy [9,26,27].
4. Pathophysiology
mTORC1 is a protein complex that contains mTOR, a rapamycin-associated protein of TOR (raptor), and mLST8 [28] (Figure 3). The major driver of the cellular hyperplasia and tissue dysplasia seen in TSC is the overactivation of the mTORC1 signaling pathway. Hamartin and tuberin bind to a third protein, TBC1D7, to form the TSC protein complex as part of this cascade [29]. The heterotrimeric TSC complex acts as a GTPase-activating protein for RAS homologue enriched in brain (Rheb), which is the functional mediator between the TSC complex and mTORC1 [30]. Under normal circumstances, the TSC complex functions to keep it in an inactive GDP-bound state, thus rendering Rheb unable to stimulate mTORC1.
Figure 3.

mTOR signaling pathway.
When second hit mutations affect either TSC1 or TSC2 , the brake on Rheb by the TSC complex is released as the complex can no longer be formed [9]. Therefore, mTORC1 is constitutively activated by Rheb, regardless of the upstream signals. The mechanism by which Rheb regulates mTORC1 still needs exploration. Rheb-induced mTOR activation results in the stimulation of S6 kinase and inhibition of 4EBP1, the eukaryotic translation initiation factor 4E-binding protein 1, leading to unrestricted protein synthesis and proliferation [31].
The mTOR pathway can be pharmacologically manipulated by rapamycin (Sirolimus), which binds to FKPB12 and causes the mTORC1 complex component raptor to dissociate, rendering mTORC1 unable to stimulate the downstream targets of anabolism [32,33]. Rapamycin was discovered in the 1970s on Easter Island and was originally used as an antifungal compound [34]. Later, it drew further attention due to its immunosuppressant and antiproliferative properties, causing it to become an important agent in oncology. In the early 2000s, rapamycin was demonstrated to be effective for symptom control in TSC mouse models by several labs, and the bench-to-bedside studies that followed [35,36].
The activity of mTORC1 is also influenced by the upstream components of several intersecting pathways. mTORC1 is negatively regulated by PRAS40, the proline-rich Akt substrate of 40 kDa and DEPTOR, the DEP domain containing mTOR-interacting protein, which works as a component of the GATOR complex. [37]. In addition to growth-inducing states in the presence of sufficient energy and nutrients, mTORC1 is also turned on by tyrosine kinase growth factor receptors, such as insulin, insulin-like growth factor 1, brain-derived neurotrophic factor, and epidermal growth factor receptor [38]. This results in the activation of the Ras/ERK and PI3K/Akt signaling pathways, which converge on the TSC complex and cause its activation favoring a progrowth state [39].
mTORC1 has been demonstrated to regulate both anabolic and catabolic processes. When activated, it stimulates protein synthesis, nucleotide synthesis, gluconeogenesis, lipogenesis, and ATP production. When it is turned off, it inhibits cell growth through several mechanisms including autophagy [40,41]. mTORC2 complex is less well understood, especially in terms of its role in the pathogenesis of TSC [42]. It is involved in the regulation of cytoskeletal dynamics. Additionally, it is not sensitive to the effects of acute rapamycin treatment because the core component, RICTOR, does not bind FKBP12 [43]. Further studies could help to characterize its functions and explore the possibility of interactions with the mTORC1 pathway.
Both mTOR and Rheb are ubiquitous in the body, causing multisystem involvement in the presence of TSC mutations [43]. Thanks to the successful implementation of genetic and molecular discoveries into clinical settings, TSC serves as model for many other diseases involving similar pathways or cellular processes [44].
5. Clinical presentation
The clinical manifestations of TSC are protean in terms of severity and the range of tissues it can involve. Despite the potential of TSC to involve any organ system in the body, some organs are more affected than others. Neurologic manifestations, including seizures and cognitive impairment, are the primary source of patient and caretaker burden, followed by renal abnormalities [45,46].
Although there is no single symptom specific to TSC, a constellation of findings on physical examination and imaging raises the suspicion for diagnosis [47]. The revision of the diagnostic criteria for TSC by the International Tuberous Sclerosis Complex Consensus group incorporated genetic testing into the clinical framework of TSC [14]. Even before genetic testing is undertaken, the presence of a first-degree relative with TSC puts the patient at 50% risk for having the disease [48].
The 2012 diagnostic criteria list includes major and minor features, determining a TSC diagnosis to be categorized as definite or possible (Table). A definite diagnosis can be made when 2 major or 1 major and 2 minor criteria are fulfilled. Patients with 1 major or 2 and more minor criteria meet the criteria for having a possible TSC diagnosis. A thorough clinical evaluation should be followed by genetic testing for confirmation of the disease and prognostication [45].
5.1. Neurologic manifestations
The neurologic issues of TSC comprise the most important cause of impairment in the majority of patients, owing to their prevalence and the severity of symptoms [46]. The array of manifestations include epilepsy, cortical tubers, subependymal nodules and giant cell astrocytomas, intellectual disability, autism spectrum disorder, and behavioral problems [49,50]. The foremost neurologic symptom in TSC is epilepsy, afflicting 90% of patients [51].
The onset of epilepsy is variable, but most patients present before the age of 1 year [52]. All seizure types can be seen with TSC. The most common seizure type in early life is infantile spasms, which affects nearly 40% of patients with TSC-associated epilepsy [53]. Although earlier studies supported the hypothesis of seizures originating from cortical tubers, the exact origin and mechanism of epileptogenesis are still debated [54]. Several studies have demonstrated a lack of correlation between tuber burden and epilepsy severity [55]. Up to 38–50% of seizure patients are refractory to the point of necessitating surgical intervention [56]. The age of onset and severity of seizures are most predictive of long-term cognitive and behavioral outcomes [57,58].
Cortical tubers (80–90%) are one type of brain malformation that present as part of TSC and give the disease its name. They are thought to be caused by a failure of cellular differentiation and neuronal migration during neurodevelopment [59]. Radial migration lines occur due to a similar process and can be observed with the tubers [45]. The cortical tubers contain giant dysplastic neurons and astrocytes, and they tend to stay stable in size [60]. Microtubers may also be found in normal appearing white matter [61].
Subependymal nodules (SENs) are formations that arise mostly along the lateral and third ventricle walls, and are seen in >80% of patients. Approximately 5–15% of these growths transform into subependymal giant cell astrocytomas (SEGAs) [47]. SEGAs are composed of ganglion-like giant cells expressing both neuronal and astrocytic markers [62]. SEN/SEGAs may remain asymptomatic [51]. However, if they are located at the foramen of Monro, they can potentially cause obstructive hydrocephalus and increased intracranial pressure. Both SENs and SEGAs can be detected prenatally or at birth, and it is rare for SEGAs to grow after the age of 20 [14]. Most of these lesions tend to progressively calcify [63].
5.2. Renal manifestations
Renal abnormalities can also lead to significant morbidity and mortality, as they are commonly encountered in TSC patients, and can lead to complications if left untreated [64]. The most common renal lesion is angiomyolipoma, a hamartoma composed of blood vessels, smooth muscle, and adipose tissue [65]. They are often bilateral and multiple. [66]. Although benign in nature, these lesions have a tendency to bleed, and therefore should be watched closely for timely intervention. In severe cases, renal AMLs may lead to renal failure [67].
The second most frequent lesion in the kidney is single or multiple simple cysts. They are seen in 45% of patients and can result in hypertension or kidney failure [68]. The combination of AMLs and cysts in the same patient is highly suspicious for a TSC diagnosis [69]. In early-onset severe cases, they may constitute stigmata of PKD-TSC syndrome (see also the Genetic background section).
5.3. Dermatologic manifestations
The dermatologic lesions seen with TSC are of paramount value, as their presence heralds the diagnosis in a considerable number of cases [70]. Many of the major features listed in the 2012 diagnostic criteria for TSC included cutaneous manifestations. Among these, hypomelanotic macules or ash-leaf spots, which were named after the European Mountain Ash Tree, can be seen in up to 90% of patients [71]. They are detected at birth or during early infancy. They may be difficult to visualize especially in fair-skinned individuals or small babies; therefore, the use of ultraviolet light (Wood’s lamp) can be helpful [51].
Another important dermatologic finding is facial angiofibromas or adenoma sebaceum. These lesions are comprised of connective and adipose tissue and are found in 75% patients over 9 years of age [71,72]. They appear in the central face and increase in number with age, which is a common source of concern [73]. TSC patients can also present with fibrous plaques in the forehead (20%), shagreen patches in the lumbosacral region (20%), and ungual or gingival fibromas (20%) [74].
5.4. Cardiac manifestations
Intracardiac rhabdomyomas are seen in nearly 50% of patients [75]. They are one of the earliest TSC lesions to emerge as the cardiac rhabdomyomas can be detected on prenatal ultrasound as early as 22 weeks of gestation [11]. On average, the lesions tend to cluster as a few lesions, grow to a size of 3–25 mm, and are mostly found in the ventricles of the heart along the septum. They tend to regress within 3 years of life [76]. Although rare, some rhabdomyomas may lead to arrhythmia, valvular defects, or cardiac failure, so prenatal and postnatal surveillance are critical until regression [77].
5.5. Pulmonary manifestations
Pulmonary involvement is much less common than the previously mentioned manifestations of TSC. The most frequent pulmonary lesion is LAM, which is almost exclusively seen in adult females [78]. This suggests that the pathogenesis may be driven by estrogen, which has also been evidenced by animal models of LAM [79]. Infiltration of the lung tissue by smooth muscle cells is characteristic of LAM, leading to cystic lung changes and potential complications of pneumothorax, pleural effusion and hemoptysis [52]. Multifocal micronodular pneumocyte hyperplasia (MMPH) is another type of pulmonary lesion associated with TSC [80].
5.6. Ophthalmic manifestations
The eye is another organ that can be commonly affected in TSC, as they arise from ectoderm like the central nervous system. Retinal astrocytic hamartomas are seen in 35–50% of patients and are typically benign unless they compress the optic disc [81]. Additionally, areas of hypopigmentation around the retina called retinal achromic patch can be observed in 39% of TSC patients [82].
5.7. Other manifestations
TSC can involve the gastrointestinal system (liver AML, colon polyps, or hamartomas), thyroid, pituitary gland, pancreas, and gonads (AML, fibroadenoma) [83]. However, the number of cases reported in the literature is limited.
5.8. Tuberous sclerosis associated neuropsychiatric disorders (TAND)
The group of cognitive and behavioral problems associated with TSC causes a great burden, both for TSC patients and their caretakers. Most neuropsychiatric symptoms of TSC present a challenge for treatment and ironically do not receive as much attention as the physical stigmata of the disease [84]. The gap between burden and treatment exhibited for the tuberous sclerosis associated neuropsychiatric disorders (TAND) was found to be similar to those seen in the approach to HIV, where the major focus is also on the physical symptoms [85]. The umbrella term TAND was coined through the inspiration by HAND, which defines the HIV-associated neurocognitive disorders [86].
Among the neuropsychiatric manifestations of TSC, ASD has a special place, both in terms of clinical approach and research focus. Approximately 26–50% of TSC patients fulfill the criteria for autism spectrum disorder (ASD) [87]. ASD in TSC has many overlaps with idiopathic ASD, which is another factor that makes TSC an important genetic model to study this condition [88]. It has been observed that TSC patients with ASD tend to have a more severe epilepsy phenotype, in line with several studies demonstrating that poorly controlled seizures contribute to worse cognitive outcomes [89]. Overall in TSC, there is a wide range of severity for intellectual disability and patients are affected by the neurologic comorbidities at varying degrees [90]. The exact nature of the relationship between autism, intellectual disability, and epilepsy needs further investigation.
TAND is a broad category of symptoms encompassing multiple dimensions of cognitive, psychological, and social issues encountered within the context of TSC. The 2012 International Tuberous Sclerosis Complex Consensus group established the guidelines for evaluation of neuropsychiatric manifestations of TSC, by providing the practitioners with a TAND checklist. This proved to be a practical clinical tool for practitioners to address TAND, which are often missed and therefore undertreated. Properly addressing TAND can dramatically improve the quality of life of TSC patients and their families [91].
The multiple levels covered in the TAND checklist included the following: behavioral level (mood swings, self-injury, obsessions, aggression, impulsivity, eating, and sleeping difficulties), psychiatric level (autism spectrum disorder, attention deficit hyperactivity disorder, anxiety, and depression), intellectual level (IQ assessments and adaptive behaviors; e.g., daily living skills), academic level (reading, writing, spelling, and mathematics) and psychosocial level (quality of life, self-esteem, parental stress, and relationship difficulties) [84]. The different levels provide a common ground between patients and physicians to have a conversation and come up with a personalized management plan. This is of great importance, as up to 90% of TSC patients are observed to have TAND features during at least one period of their life [92].
One should bear in mind that the TAND checklist was not designed to be used as a rating scale, but rather a tool to make an individual action plan based on the TAND profile [84]. Neuropsychiatric reevaluations are recommended by the 2012 TSC guidelines, as the profile of the individual may change over time. Sudden changes in behavior should prompt evaluation for underlying medical causes, such as new brain lesions or seizures [93].
6. Treatment options
As a consequence of the multisystem involvement in TSC, each symptom demands evaluation and management within the relevant clinical context. mTOR inhibitors have been groundbreaking in the TSC world due to their ability to target the molecular defect in the disorder. However, animal models and clinical studies have shown that not all TSC-related symptoms benefit from mTOR inhibitors to the same extent [94]. Research is still ongoing for the optimization of mTOR inhibition for each symptom and what other pharmacologic and nonpharmacologic approaches could be employed for tackling the challenges in TSC. In particular, the timing of treatment may be crucial for the neurocognitive symptoms, and treatment during early critical windows may be necessary [95]. Regardless of the choice of intervention, genetic counseling should be included in the discussion with families.
6.1. Neurologic management
Once a diagnosis of TSC is reached, a baseline MRI is recommended to look for the presence of any cortical malformations as tubers, SENs, or SEGAs [68]. Surgery and mTOR inhibitors are the current treatment options for asymptomatic SEGAs; however, surgical intervention is recommended in acute cases [91]. After the success of the EXIST-1 clinical trial, the FDA approved the use of everolimus for individuals with tuberous sclerosis who present with SEGAs that are not amenable to surgery [96].
As mentioned previously, epilepsy remains one of the primary sources of morbidity for TSC patients. Early intervention is key for both better control of seizures and improved neurocognitive outcomes. For infantile spasms, vigabatrin is the best choice of medication, and ACTH is an alternative in cases with insufficient response [97]. Vigabatrin is an irreversible inhibitor of GABA transaminase. It helps to increase the GABA concentration and potentially reset the imbalance between GABA and glutamate neurotransmitters, which is a proposed mechanism of epileptogenesis [98]. Patients should be counseled about the possible side effects of retinal toxicity and evaluated for vision changes.
Most antiseizure medications can be used for epilepsy in TSC. Alternative or complementary therapeutic options include surgery, ketogenic low glycemic index diet, and vagal nerve stimulation. Despite the presence of these modalities, refractory epilepsy is still a big concern, with seizures persisting in more than 60% of patients [53]. The EXIST-3 trial demonstrated the benefit of everolimus in patients with treatment-resistant focal seizures [99].
Neuropsychiatric conditions associated with TSC should be managed with a multidisciplinary team, focusing on the individual’s level of psychosocial and neurocognitive functioning. Despite contributing greatly to the burden of care, TAND has received little clinical attention, with less than 20% of cognitive and behavioral symptoms being treated [84]. During clinical visits, the TAND checklist is a useful diagnostic tool to determine which neuropsychiatric symptoms require special attention. An individualized educational plan should be established for school-aged patients [14].
Animal studies have shown correlations between seizure frequency and the extent of social deficits, which are improved by the early treatment of epilepsy [100]. Several clinical trials have aimed to investigate the relationship between mTOR inhibition and neurocognition. In a 6-month clinical trial of an mTOR inhibitor (everolimus) in children and adolescents with TSC (6–21 years of age), no improvement was detected in the active drug group when compared to those taking a placebo [101]. Another trial in Europe with a similar age group also failed to show an effect on cognitive abilities or neuropsychological functioning [102].
While there are several possible explanations for these results, one important aspect may be the age of treatment. It should be kept in mind that infancy is a critical course of the disease, where pharmacologic interventions may also lead to long-lasting unfavorable changes [103]. Therefore, determining the optimum therapeutic window is crucial [88]. A randomized clinical trial of early intervention with vigabatrin to prevent seizure development in TSC (EPISTOP) recently concluded in Europe, and an National Institutes of Health-funded trial to prevent epilepsy and improve neurocognitive outcomes in infants with TSC (PREVeNT) is currently ongoing in the USA.
6.2. Nonneurologic management
The EXIST-2 trial demonstrated the benefit of mTOR inhibitors for renal AMLs, and everolimus was approved by the FDA for asymptomatic and growing renal AMLs larger than 3 cm [104]. Second-line treatment options in cases with unsatisfactory response or a lack of access to everolimus include selective arterial embolization or radiofrequency ablation, especially for hemorrhaging or compressive lesions [105]. Sirolimus was approved by the FDA for use in patients with LAM in 2015, after obtaining successful results in the MILES trial [106]. Pulmonary function and capacity need to be regularly monitored for signs of clinical improvement. Last, but not least, topical rapamycin may be used for facial angiofibromas in cases of significant disfigurement or psychological stress [107].
For most TSC-related hamartomas, lifelong treatment will continue to be necessary, as many lesions tend to regrow and seizures may recur upon the discontinuation of medication [15,108]. Reported side effects of mTOR inhibitors include stomatitis, menstrual irregularities, acne, hyperlipidemia, infections, and poor wound healing, which are related to suppression of the immune system and changes in cellular metabolism [109–111].
In TSC, an interdisciplinary approach with consultations from neurologists, cardiologists, nephrologists, pulmonologists, psychiatrists, psychologists, social workers, educational specialists, genetic counselors and additional practitioners, based on the needs of the specific individual, is essential. To achieve this goal, TSC clinics have been established in numerous hospitals across the USA. TSC has been one of the great examples in medicine where the bench research has been successfully translated to improve the diagnostic and therapeutic yield in a clinical setting.
More recent studies have started to focus on developing alternative strategies for the treatment of TSC. One notable approach is the elimination of TSC-deficient cells through the induction of autophagy or oxidative stress by exploiting the inadaptability of the overactivated mTOR pathway [112–115]. Such research questions will continue to be explored and can be promising for the development of preemptive therapy for TSC.
7. Conclusion
TSC is a rare genetic disorder characterized by hamartoma formation in multiple organs. The pathogenesis is driven by uncontrolled mTOR activation, which is the target of rapamycin and rapalogs to control the disease symptomatology with varying success. TSC serves as a model for epilepsy, autism, and tumorigenesis and many other diseases involving the mTOR pathway.
Despite the morbidity and mortality that TSC symptoms are associated with, the medical world is fortunate for the discoveries into the genetic and molecular aspects of this disease. Owing to the immense endeavors of TSC researchers, new therapeutic options targeting vulnerabilities in the TSC-related pathways will continue to be developed that maximize efficacy and minimize toxicity. Biomarkers to demonstrate disease progression and treatment efficacy need to be identified for maximizing the benefit for all patient profiles. An individually tailored multidisciplinary approach, with special attention to cognitive and psychosocial comorbidities, is the key to success in the management of this disease.
Disclosures
Mustafa Sahin reports grant support from Novartis, Roche, Pfizer, Ipsen, LAM Therapeutics, and Quadrant Biosciences. He has served on Scientific Advisory Boards for Sage, Roche, Celgene, Aeovian, and Takeda.
Acknowledgments
We thank Dr. Kellen Winden for critical reading of the manuscript.
References
- Roach ES Applying the lessons of tuberous sclerosis: The 2015 Hower Award Lecture. Pediatric Neurology . 2016;63:6–22. doi: 10.1016/j.pediatrneurol.2016.07.003. [DOI] [PubMed] [Google Scholar]
- Bourneville D. Sclerose tubereuse des circonvolutions cerebrales: idioties et epilepsie hemiplegique. Archives of Neurology . 1880;1:81–91. [Google Scholar]
- Tuberous sclerosis complex: from basic science to clinical phenotypes 2003.
- Peron A Northrup H Part C Tuberous sclerosis complex. American Journal of Medical Genetics . 2018;178:274–277. doi: 10.1002/ajmg.c.31657. [DOI] [PubMed] [Google Scholar]
- Huang J Manning BD The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochemical Journal . 2018;412:179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapon N Ito N Dickson BJ Treisman JE Hariharan IK The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell . 2001;105:345–355. doi: 10.1016/s0092-8674(01)00332-4. [DOI] [PubMed] [Google Scholar]
- Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell . 1993;75:1305–1315. doi: 10.1016/0092-8674(93)90618-z. [DOI] [PubMed] [Google Scholar]
- van Slegtenhorst M de Hoogt R Hermans C Nellist M Janssen B Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science . 1997;277:805–08. doi: 10.1126/science.277.5327.805. [DOI] [PubMed] [Google Scholar]
- Tee AR Fingar DC Manning BD Kwiatkowski DJ Cantley LC Blenis J Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. u2ef6 2002 Oct . 15:13571–6. doi: 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au KS Ward CH Northrup H Tuberous sclerosis complex: disease modifiers and treatments. 20:628–33. doi: 10.1097/MOP.0b013e328318c529. [DOI] [PubMed] [Google Scholar]
- Hyman MH Whittemore VH National Institutes of Health consensus conference: tuberous sclerosis complex. 57:662–5. doi: 10.1001/archneur.57.5.662. [DOI] [PubMed] [Google Scholar]
- Devlin LA Shepherd CH Crawford H Morrison PJ Tuberous sclerosis complex: clinical features, diagnosis, and prevalence within Northern Ireland. Developmental Medicine & Child Neurology . 2006;48:495–99. doi: 10.1017/S0012162206001058. [DOI] [PubMed] [Google Scholar]
- ’Callaghan O Shiell FJ Osborne AW Martyn JP CN Prevalence of tuberous sclerosis estimated by capture-recapture analysis. Lancet . 1998;351:1490–1490. doi: 10.1016/S0140-6736(05)78872-3. [DOI] [PubMed] [Google Scholar]
- Northrup H Krueger DA on behalf of the International Tuberous Sclerosis Complex Consensus Group. Pediatric Neurology . 2013;49:243–54. doi: 10.1016/j.pediatrneurol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henske EP Jozwiak S Kingswood JC Sampson JR Thiele EA Tuberous sclerosis complex. Nature Reviews Disease Primers . 2016;2:16035–16035. doi: 10.1038/nrdp.2016.35. [DOI] [PubMed] [Google Scholar]
- Zoncu R Bar-Peled L Efeyan A Wang S Sancak Y Sabatini DM mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science . 2011;34:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon BP Hulbert JC Bissler JJ Tuberous sclerosis complex renal disease. u2ef8 . 2011;118:e15–20. doi: 10.1159/000320891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AC Daniells CE Snell RG Tachataki M Idziaszczyk SA Molecular genetic and phenotypic analysis reveals differences between TSC1 and TSC2 associated familial and sporadic tuberous sclerosis. Human Molecular Genetics . 1997;6:2155–2161. doi: 10.1093/hmg/6.12.2155. [DOI] [PubMed] [Google Scholar]
- Kobayashi T Hirayama Y Kobayashi E Kubo Y Hino O A germline insertion in the tuberous sclerosis (Tsc2) gene gives rise to the Eker rat model of dominantly inherited cancer. Nature Genetics . 1995;9:70–74. doi: 10.1038/ng0195-70. [DOI] [PubMed] [Google Scholar]
- Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. u2ef3 2001 Jan; 68 . pp. 64–80. [DOI] [PMC free article] [PubMed]
- Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. u2ef4 2007 Feb; 9 . pp. 88–100. [DOI] [PubMed]
- Mayer K Ballhausen W Rott HD Mutation screening of the entire coding regions of the TSC1 and the TSC2 gene with the protein truncation test (PTT) identifies frequent splicing defects Human Mutation. 1999;14:401–411. doi: 10.1002/(SICI)1098-1004(199911)14:5<401::AID-HUMU6>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Sancak O Nellist M Goedbloed M Elfferich P Wouters C Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype– phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. European Journal of Human Genetics . 2005;13:731–41. doi: 10.1038/sj.ejhg.5201402. [DOI] [PubMed] [Google Scholar]
- Cheadle JP Reeve MP Sampson JR Kwiatkowski DJ Human Genetics . 2000;107:97–114. doi: 10.1007/s004390000348. [DOI] [PubMed] [Google Scholar]
- Tyburczy ME Dies KA Camposano S Chekaluk Y Thorner AR Mosaic and intronic mutations in TSC1/TSC2 explain the majority of TSC patients with no mutation identified by conventional testing. Public Library of Science Genetics . 2015;11:e1005637–e1005637. doi: 10.1371/journal.pgen.1005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino PB mTOR: a pathogenic signaling pathway in developmental brain malformations. Trends in Molecular Medicine . 2011;17:734–42. doi: 10.1016/j.molmed.2011.07.008. [DOI] [PubMed] [Google Scholar]
- Crino PB Nathanson KL Henske EP The tuberous sclerosis complex. New England Journal of Medicine . 2006;355:1345e56–1345e56. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- Winden KD Ebrahimi-Fakhari D Sahin M Abnormal mTOR Activation in Autism. u2ef1 2018 Jul . 8:1–23. doi: 10.1146/annurev-neuro-080317-061747. [DOI] [PubMed] [Google Scholar]
- Dibble CC Elis W Menon S Qin W Klekota J TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. u2ef2 2012 Aug . 24:535–46. doi: 10.1016/j.molcel.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y Gao X Saucedo LJ Ru B Edgar BA Pan D Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nature Cell Biology. 2003;5:578–581. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
- Guertin DA Sabatini DM Defining the role of mTOR in cancer. Cancer Cell . 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Sampson JR Therapeutic targeting of mTOR in tuberous sclerosis. Biochemical Society Transactions . 2009;37:259e64–259e64. doi: 10.1042/BST0370259. [DOI] [PubMed] [Google Scholar]
- Kim DH Sarbassov DD Ali SM Latek RR Guntur KV GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Molecular Cell . 2003;11:895–904. doi: 10.1016/s1097-2765(03)00114-x. [DOI] [PubMed] [Google Scholar]
- Arriola Apelo SI Lamming DW Rapamycin: An Inhibitor of Aging Emerges From the Soil of Easter Island. u2eef;71:841–9. doi: 10.1093/gerona/glw090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L Pollizzi K Egnor A Kramvis I Lane H Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. The Journal of neuroscience: the official journal of the Society for Neuroscience . 2008;28:5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler W Sedrani R Cottens S Schulz M Schuurman HJ Zenke G Zerwes HG Schreier MH SDZ RAD, a new rapamycin derivative: pharmacological properties in vitro and in vivo. Transplantation . 1997;64:36–42. doi: 10.1097/00007890-199707150-00008. [DOI] [PubMed] [Google Scholar]
- Yang H Rudge DG Koos JD mTOR kinase structure, mechanism and regulation. u2ef0 2013 May . 9:217–23. doi: 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPiccolo J Blumenthal GM Bernstein WB Dennis PA Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist Update . 2008;11:32e50–32e50. doi: 10.1016/j.drup.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J Alberts I Li X Dysregulation of the IGF-I/PI3K/AKT/mTOR signaling pathway in autism spectrum disorders. International Journal of Developmental Neuroscience . 2014;35:35–41. doi: 10.1016/j.ijdevneu.2014.03.006. [DOI] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D Wahlster L Hoffmann GF Kölker S Emerging role of autophagy in pediatric neurodegenerative and neurometabolic diseases. Pediatric Research . 2014;75:217–26. doi: 10.1038/pr.2013.185. [DOI] [PubMed] [Google Scholar]
- Dodd KM Dunlop EA Tuberous sclerosis--A model for tumour growth. Seminars in Cell and Developmental Biology . 2016;52:3–11. doi: 10.1016/j.semcdb.2016.01.025. [DOI] [PubMed] [Google Scholar]
- Oh WJ Jacinto E. mTOR complex 2 signaling and functions. Cell Cycle . 2011;10:2305–2316. doi: 10.4161/cc.10.14.16586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEneaney LJ Tee AR Finding a cure for tuberous sclerosis complex: From genetics through to targeted drug therapies. Advances in Genetics . 2019;103:91–118. doi: 10.1016/bs.adgen.2018.11.003. [DOI] [PubMed] [Google Scholar]
- Switon K Kotulska K Janusz-Kaminska A Zmorzynska J Jaworski J Molecular neurobiology of mTOR. Neuroscience . 2017;341:112–153. doi: 10.1016/j.neuroscience.2016.11.017. [DOI] [PubMed] [Google Scholar]
- Lu DS Karas PJ Krueger DA Weiner HL Part C Central nervous system manifestations of tuberous sclerosis complex. American Journal of Medical Genetics . 2018;178:291–298. doi: 10.1002/ajmg.c.31647. [DOI] [PubMed] [Google Scholar]
- Curatolo P Moavero R Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurology . 2015;14:733–745. doi: 10.1016/S1474-4422(15)00069-1. [DOI] [PubMed] [Google Scholar]
- Kohrman MH Emerging treatments in the management of tuberous sclerosis complex. Pediatric Neurology . 2012;46:267–275. doi: 10.1016/j.pediatrneurol.2012.02.015. [DOI] [PubMed] [Google Scholar]
- Tuberous sclerosis. In: Child Neurology . 2000. pp. 865–872.
- FJ Brain abnormalities in tuberous sclerosis complex. Journal of Child Neurology . 2004;19:650–57. doi: 10.1177/08830738040190090401. [DOI] [PubMed] [Google Scholar]
- Luat AF Makki M Chugani HT Neuroimaging in tuberous sclerosis complex. Current Opinion in Neurology . 2007;20:142–50. doi: 10.1097/WCO.0b013e3280895d93. [DOI] [PubMed] [Google Scholar]
- Curatolo P Bombardieri R Tuberous sclerosis. Lancet . 2008;372:657–668. doi: 10.1016/S0140-6736(08)61279-9. [DOI] [PubMed] [Google Scholar]
- Islam MP Roach ES Tuberous sclerosis complex. Handbook of Clinical Neurology . 2015;132:97–109. doi: 10.1016/B978-0-444-62702-5.00006-8. [DOI] [PubMed] [Google Scholar]
- Chu-Shore CJ Major P Camposano S Muzykewicz D. Thiele EA The natural history of epilepsy in tuberous sclerosis complex. Epilepsia . 2010;51:1236–1241. doi: 10.1111/j.1528-1167.2009.02474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowska M Jurkiewicz E Domanska-Pakiela D Syczewska M Lojszczyk B Cerebral tuber count and its impact on mental outcome of patients with tuberous sclerosis complex. Epilepsia . 2011;52:22–21. doi: 10.1111/j.1528-1167.2010.02892.x. [DOI] [PubMed] [Google Scholar]
- Tuberous sclerosis. In: Neurology in Clinical Practice . 2004. pp. 1867–1873.
- Zhang K Hu WH Zhang C Meng FG Chen N Zhang JG Predictors of seizure freedom after surgical management of tuberous sclerosis complex: a systematic review and meta-analysis. Epilepsy Research . 2013;105:377–83. doi: 10.1016/j.eplepsyres.2013.02.016. [DOI] [PubMed] [Google Scholar]
- Capal JK Bernardino-Cuesta B Horn PS Murray D Byars AW Pt A Influence of seizures on early development in tuberous sclerosis complex. Epilepsy & Behavior . 2017;70:245–252. doi: 10.1016/j.yebeh.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen FE Vincken KL Algra A Anbeek P Braams O Cognitive impairment in tuberous sclerosis complex is a multifactorial condition. Neurology . 2008;70:916–923. doi: 10.1212/01.wnl.0000280579.04974.c0. [DOI] [PubMed] [Google Scholar]
- Ehninger D Sano Y de Vries PJ Dies K Franz D Gestational immune activation and Tsc2 haploinsufficiency cooperate to disrupt fetal survival and may perturb social behavior in adult mice. Molecular Psychiatry . 2012;17:62–70. doi: 10.1038/mp.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz DN Bissler JJ McCormack FX Tuberous sclerosis complex: Neurological, renal, and pulmonary manifestations. Neuropediatrics . 2010;41:199e208–199e208. doi: 10.1055/s-0030-1269906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte L Aronica E Baybis M Crino PB Cytoarchitectural alterations are widespread in cerebral cortex in tuberous sclerosis complex. Acta Neuropathologica . 2012;123:685–693. doi: 10.1007/s00401-012-0950-3. [DOI] [PubMed] [Google Scholar]
- Sharma MC Ralte AM Gaekwad S Santosh V Shankar SK Sarkar C Subependymal giant cell astrocytoma—a clinicopathological study of 23 cases with special emphasis on histogenesis. Pathology & Oncology Research . 2004;10:219–224. doi: 10.1007/BF03033764. [DOI] [PubMed] [Google Scholar]
- Grajkowska W Kotulska K Jurkiewicz E Matyja E Brain lesions in tuberous sclerosis complex. Review. Folia Neuropathologica . 2010;48:139–49. [PubMed] [Google Scholar]
- Shepherd C Gomez M Lie J Crowson C Causes of death in patients with tuberous sclerosis. Mayo Clinic Proceedings . 1991;66:792–796. doi: 10.1016/s0025-6196(12)61196-3. [DOI] [PubMed] [Google Scholar]
- DiMario FJ Sahin M Ebrahimi-Fakhari D Tuberous sclerosis complex. Pediatric Clinics of North America . 2015;62:633–648. doi: 10.1016/j.pcl.2015.03.005. [DOI] [PubMed] [Google Scholar]
- Renal angiomyolipoma. In: Pediatric Nephrology . 2004. pp. 1120–1121.
- Franz DN Non-neurologic manifestations of tuberous sclerosis complex. Journal of Child Neurology . 2004;19:690–698. doi: 10.1177/08830738040190091001. [DOI] [PubMed] [Google Scholar]
- Randle SC Tuberous Sclerosis Complex: A Review. Pediatric Annals . 2017;46:e166–e171. doi: 10.3928/19382359-20170320-01. [DOI] [PubMed] [Google Scholar]
- Brook-Carter P Peral B Ward C Thompson P Hughes J Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease—a contiguous gene syndrome. Nature Genetics . 1994;8:328–332. doi: 10.1038/ng1294-328. [DOI] [PubMed] [Google Scholar]
- Teng JM Cowen EW Wataya-Kaneda M Gosnell ES Witman PM Dermatologic and dental aspects of the 2012 International Tuberous Sclerosis Complex Consensus Statements. JAMA Dermatology . 2014;150:1095–1101. doi: 10.1001/jamadermatol.2014.938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozwiak S Schwartz RA Janniger CK Michalowicz R Chmielik J Skin lesions in children with tuberous sclerosis complex: their prevalence, natural course, and diagnostic significance. Internation Journal of Dermatology . 1998;37:911–917. doi: 10.1046/j.1365-4362.1998.00495.x. [DOI] [PubMed] [Google Scholar]
- Webb D Clarke A Fryer A Osborne J. The cutaneous features of tuberous sclerosis: a population study. British Journal of Dermatology . 1996;135:1–5. [PubMed] [Google Scholar]
- McEneaney LJ Tee AR Finding a cure for tuberous sclerosis complex: From genetics through to targeted drug therapies. Advances in Genetics . 2019;103:91–118. doi: 10.1016/bs.adgen.2018.11.003. [DOI] [PubMed] [Google Scholar]
- Roach ES Sparagana SP Diagnosis of tuberous sclerosis complex. Journal of Child Neurology . 2004;19:643–649. doi: 10.1177/08830738040190090301. [DOI] [PubMed] [Google Scholar]
- Hinton RB Prakash A Romp RL Krueger DA Knilans TK International Cardiovascular manifestations of tuberous sclerosis complex and summary of the revised diagnostic criteria and surveillance and management recommendations from the International Tuberous Sclerosis Consensus Group. 2014;3:e001493–e001493. doi: 10.1161/JAHA.114.001493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joźwiak S Kotulska K Kasprzyk-Obara J Domanska-Pakiela D Tomyn-Drabik M Clinical and genotype studies of cardiac tumors in 154 patients with tuberous sclerosis complex. Pediatrics . 2006;118:e1146–51. doi: 10.1542/peds.2006-0504. [DOI] [PubMed] [Google Scholar]
- Staley BA Vail EA Thiele EA Tuberous sclerosis complex: diagnostic challenges, presenting symptoms, and commonly missed signs. Pediatrics . 2011;127:e117–25. doi: 10.1542/peds.2010-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudzilo CJ Szczesniak RD Brody AS Rattan MS Krueger DA Lymphangioleiomyomatosis screening in women with tuberous sclerosis. Chest . 2013;144:578–85. doi: 10.1378/chest.12-2813. [DOI] [PubMed] [Google Scholar]
- Yu JJ Robb VA Morrison TA Ariazi EA Magdalena K Estrogen promotes the survival and pulmonary metastasis of tuberin-null cells. Proceedings of the National Academy of Sciences of the USA . 2009;106:2635e40–2635e40. doi: 10.1073/pnas.0810790106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz D Brody A Meyer C Leonard J Chuck G Mutational and radiographic analysis of pulmonary disease consistent with lymphangioleiomyomatosis and micronodular pneumocyte hyperplasia in women with tuberous sclerosis. American Journal of the Respiratory and Critical Care Medicine . 2001;164:661–668. doi: 10.1164/ajrccm.164.4.2011025. [DOI] [PubMed] [Google Scholar]
- Hodgson N Kinori M Goldbaum MH Robbins SL Ophthalmic manifestations of tuberous sclerosis: a review. Journal of Clinical and Experimental Ophthalmology . 2017;45:81–86. doi: 10.1111/ceo.12806. [DOI] [PubMed] [Google Scholar]
- Roach ES Gomez MR Northrup H Tuberous sclerosis complex consensus conference: Revised clinical diagnostic criteria. Journal of Child Neurology . 1998;13:624–628. doi: 10.1177/088307389801301206. [DOI] [PubMed] [Google Scholar]
- Tuberous sclerosis. 2003. pp. 858–867.
- de Vries PJ Whittemore VH Leclezio L Byars AW Dunn D Tuberous sclerosis associated neuropsychiatric disorders (TAND) and the TAND Checklist. Pediatric Neurology . 2015;52:25–35. doi: 10.1016/j.pediatrneurol.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund C Tomlinson M De Silva M Fekadu A Shidhaye R PRIME: a programme to reduce the treatment gap for mental disorders in five low- and middle-income countries. Public Library of Science Medicine . 2012;9:e1001359–e1001359. doi: 10.1371/journal.pmed.1001359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antinori A Arendt G Becker JT Brew BJ Byrd DA Updated research nosology for HIV-associated neurocognitive disorders. Neurology . 2007;69:1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeste SS Sahin M Bolton P Ploubidis GB Humphrey A Characterization of autism in young children with tuberous sclerosis complex. Journal of Child Neurology . 2008;23:520–525. doi: 10.1177/0883073807309788. [DOI] [PubMed] [Google Scholar]
- Davis PE Peters JM Krueger DA Sahin M. Tuberous Sclerosis: A New Frontier in Targeted Treatment of Autism. Neurotherapeutics . 2015;12:572–583. doi: 10.1007/s13311-015-0359-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eeghen AM Black ME Pulsifer MB Kwiatkowski DJ Thiele EA Genotype and cognitive phenotype of patients with tuberous sclerosis complex. European Journal of Human Genetics . 2012;20:510–515. doi: 10.1038/ejhg.2011.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numis AL Major P Montenegro MA Muzykewicz DA Pulsifer MB Thiele EA Identification of risk factors for autism spectrum disorders in tuberous sclerosis complex. Neurology . 2011;76:981–987. doi: 10.1212/WNL.0b013e3182104347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger DA Northrup H Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatric Neurology . 2013;49:255–265. doi: 10.1016/j.pediatrneurol.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclezio L Jansen A Whittemore VH Pilot validation of the Tuberous Sclerosis Associated Neuropsychiatric Disorders (TAND) Checklist. Pediatric Neurology . 2015;52:16–24. doi: 10.1016/j.pediatrneurol.2014.10.006. [DOI] [PubMed] [Google Scholar]
- de Vries PJ Humphrey A McCartney D Prather P Bolton P Consensus clinical guidelines for the assessment of cognitive and behavioural problems in tuberous sclerosis. European Child & Adolescent Psychiatry . 2005;14:183–190. doi: 10.1007/s00787-005-0443-1. [DOI] [PubMed] [Google Scholar]
- Li M Zhou Y Chen C Yang T Zhou S Efficacy and safety of mTOR inhibitors (rapamycin and its analogues) for tuberous sclerosis complex: a meta-analysis. Orphanet Journal of Rare Diseases . 2019;14:39–39. doi: 10.1186/s13023-019-1012-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PT Rudolph S Guo C Ellegood J Gibson M Sensitive Periods for Cerebellar-Mediated Autistic-like Behaviors. Cell Reports . 2018;25:357–367. doi: 10.1016/j.celrep.2018.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz DN Belousova E Sparagana S Bebin EM Frost M Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet . 2013;381:116–116. doi: 10.1016/S0140-6736(12)61134-9. [DOI] [PubMed] [Google Scholar]
- Curatolo P Joźwiak S Nabbout R on behalf of the participants of the TSC Consensus Meeting for SEGA and Epilepsy Management. Management of epilepsy associated with tuberous sclerosis complex (TSC): clinical recommendations. European Journal of Paediatric Neurology . 2012;16:582–86. doi: 10.1016/j.ejpn.2012.12.008. [DOI] [PubMed] [Google Scholar]
- Zhang B McDaniel SS Rensing NR Wong M. Vigabatrin inhibits seizures and mTOR pathway activation in a mouse model of tuberous sclerosis complex. PLoS One . 2013;8:e57445–e57445. doi: 10.1371/journal.pone.0057445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French JA Lawson JA Yapici Z Ikeda H Polster T Adjunctive everolimus therapy for treatment resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): A phase 3, randomised, double-blind, placebo-controlled study. Lancet . 2016;388:2153–2163. doi: 10.1016/S0140-6736(16)31419-2. [DOI] [PubMed] [Google Scholar]
- Julich K Sahin M Mechanism-based treatment in tuberous sclerosis complex. Pediatric Neurology . 2014;50:290–296. doi: 10.1016/j.pediatrneurol.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger DA Sadhwani A Byars AW de Vries PJ Franz DN Whittemore VH Filip-Dhima R Murray D Kapur K Sahin M Everolimus for treatment of tuberous sclerosis complex-associated neuropsychiatric disorders. Annals of Clinical and Translational Neurology . 2017;4:877–887. doi: 10.1002/acn3.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overwater IE Rietman AB Mous SE Bindels-de Heus K Rizopoulos D Ten Hoopen LW van der Vaart T Jansen FE Elgersma Y Moll HA de Wit MY ENCORE Expertise Centre for Neurodevelopmental Disorders. A randomized controlled trial with everolimus for IQ and autism in tuberous sclerosis complex. Neurology . 2019;9:e200–e209. doi: 10.1212/WNL.0000000000007749. [DOI] [PubMed] [Google Scholar]
- Way SW Rozas NS Wu HC McKenna J Reith RM The differential effects of prenatal and/or postnatal rapamycin on neurodevelopmental defects and cognition in a neuroglial mouse model of tuberous sclerosis complex. Human Molecular Genetics . 2012;21:3226–3236. doi: 10.1093/hmg/dds156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissler JJ Kingswood JC Radzikowska E Zonnenberg BA Frost M Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet . 2013;381:817–824. doi: 10.1016/S0140-6736(12)61767-X. [DOI] [PubMed] [Google Scholar]
- Sooriakumaran P Gibbs P Coughlin G Attard V Elmslie F Angiomyolipomata: Challenges, solutions, and future prospects based on over 100 cases treated. British Journal of Urology International . 2010;105:101e6–101e6. doi: 10.1111/j.1464-410X.2009.08649.x. [DOI] [PubMed] [Google Scholar]
- McCormack FX Inoue Y Moss J Singer LG Strange C Efficacy and safety of sirolimus in lymphangioleiomyomatosis. 2011;364:1595–606. doi: 10.1056/NEJMoa1100391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J Yun SK Cho YS Song KH Kim HU Treatment of angiofibromas in tuberous sclerosis complex: the effect of topical rapamycin and concomitant laser therapy. Dermatology . 2014;228:37–41. doi: 10.1159/000357033. [DOI] [PubMed] [Google Scholar]
- Sparagana SP Delgado MR Batchelor LL Roach ES Seizure remission and antiepileptic drug discontinuation in children with tuberous sclerosis complex. Archives of Neurology . 2003;60:1286–1289. doi: 10.1001/archneur.60.9.1286. [DOI] [PubMed] [Google Scholar]
- Nashan B Citterio F Wound healing complications and the use of mammalian target of rapamycin inhibitors in kidney transplantation: a critical review of the literature. Transplantation . 2012;94:547–561. doi: 10.1097/TP.0b013e3182551021. [DOI] [PubMed] [Google Scholar]
- Houde VP Brule S Festuccia WT Blanchard PG Bellmann K Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes . 2010;59:1338–1348. doi: 10.2337/db09-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J Anglicheau D Elie C Bererhi L Timsit MO Sirolimus may reduce fertility in male renal transplant recipients. American Journal of Transplant . 2008;8:1471–1479. doi: 10.1111/j.1600-6143.2008.02267.x. [DOI] [PubMed] [Google Scholar]
- Parkhitko AA Priolo C Coloff JL Yun J Wu JJ Autophagy-dependent metabolic reprogramming sensitizes TSC2-deficient cells to the antimetabolite 6-aminonicotinamide. Molecular Cancer Research . 2014;12:48–57. doi: 10.1158/1541-7786.MCR-13-0258-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alayev A Berger SM Holz MK Resveratrol as a novel treatment for diseases with mTOR pathway hyperactivation. Annals of the New York Academy of Sciences . 2015;1348:116–123. doi: 10.1111/nyas.12829. [DOI] [PubMed] [Google Scholar]
- Johnson CE Hunt DK Wiltshire M Herbert TP Sampson JR Endoplasmic reticulum stress and cell death in mTORC1-overactive cells is induced by nelfinavir and enhanced by chloroquine. Molecular Oncology . 2015;9:675–688. doi: 10.1016/j.molonc.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siroky BJ Yin H Babcock JT Lu L Hellmann AR Human TSC-associated renal angiomyolipoma cells are hypersensitive to ER stress. American Journal of Renal Physiology . 2012;303:F831–F844. doi: 10.1152/ajprenal.00441.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]