


Abstract
The six major mammalian DNA repair pathways were discovered as independent processes, each dedicated to remove specific types of lesions, but the past two decades have brought into focus the significant interplay between these pathways. In particular, several studies have demonstrated that certain proteins of the nucleotide excision repair (NER) and base excision repair (BER) pathways work in a cooperative manner in the removal of oxidative lesions. This review focuses on recent data showing how the NER proteins, XPA, XPC, XPG, CSA, CSB and UV-DDB, work to stimulate known glycosylases involved in the removal of certain forms of base damage resulting from oxidative processes, and also discusses how some oxidative lesions are probably directly repaired through NER. Finally, since many glycosylases are inhibited from working on damage in the context of chromatin, we detail how we believe UV-DDB may be the first responder in altering the structure of damage containing-nucleosomes, allowing access to BER enzymes.
INTRODUCTION
Reactive oxygen and nitrogen species (ROS/RNS), such as singlet oxygen, superoxide, hydrogen peroxide, hydroxyl radical, nitric oxide and peroxynitrite, can be generated endogenously by normal cellular metabolism or inflammation, or by exogenous sources such as ultraviolet (UV) or ionizing radiation (IR) (1–3). Oxidation can either directly or indirectly introduce a wide spectrum of base lesions in the DNA (4). Due to the extensive DNA damage caused by oxidation, these lesions have been associated with a large number of human maladies including neurodegeneration, cancer and aging (5).
Some of the most widely studied DNA lesions resulting from oxidation are shown in Figure 1. One of the best characterized oxidative lesions is 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxoG), the major product produced from the oxidation of guanine. Further oxidation of 8-oxoG results in the formation of spiroiminodihydantoin (Sp) and 5-guanidinohydantoin (Gh). Purine oxidation can also result in the formation of 5′,8-cyclo-purine adducts. An important product of thymine oxidation is thymine glycol (Tg). Cytosine is subject to methylation, resulting in the formation of 5-methylcytosine (5mC). Oxidative removal of 5-methylcytosine (5mC) occurs through an active enzymatic process in which 5mC is oxidized in three steps by a family of TET dioxygenases to form 5-hydroxymethyl-C (5hmC), 5-formylC (5fC) and 5-carboxyC (5caC).
Figure 1.

Chemical structures of oxidative lesions formed in DNA. (A) Various oxidation products of guanine. (B) Formation of cyclic guanosine by oxidation. (C) Formation of cyclic adenosine by oxidation. (D) Enzymatic oxidative demethylation of 5-methylcytosine. (E) Oxidation of thymine to thymine glycol. ROS produces over 100 different types of lesions in DNA, and this figure displays the structures of those damages that are discussed in this review. ROS: Reactive oxygen species; DNMT: DNA methyltransferase; TET: Ten-eleven translocation enzymes.
Oxidative base lesions are commonly repaired via base excision repair (BER) pathway (6). BER is initiated after a lesion-specific DNA glycosylase cleaves the glycosidic bond, which frees the lesion, and creates an abasic site (1) (Figure 2). Currently, there are 11 known mammalian DNA glycosylases that can be categorized as monofunctional or bifunctional. Monofunctional glycosylases only possess the ability to break the glycosidic bond between the damaged base and the sugar moiety, resulting in an abasic site, which is processed by AP endonuclease 1 (APE1) to form a 3′OH and a deoxyribose-5′-phosphate (dRP). This dRP is removed by the lyase activity of DNA polymerase β (pol β). Bifunctional glycosylases have an additional AP lyase activity which allows for cleavage of the phosphate backbone, creating a single strand break, leaving a free 5′ phosphate and either a 3′-phospho-α, β-unsaturated aldehyde (3′-PUA) (β-elimination) or 3′ phosphate (β,δ-elimination). APE1 acts on the β-elimination product, while polynucleotide kinase phosphate (PNKP) is required to process the 3′phosphate after β,δ-elimination. The resulting 3′OH is bound by PARP1 which recruits the BER complex consisting of pol β, XRCC1 and DNA ligase. The one base gap is then filled by pol β and the nick in the DNA is sealed by DNA ligase (7). The human 8-oxoG glycosylase (OGG1) is a bifunctional glycosylase responsible for the recognition and removal of 8-oxoG. Like several glycosylases, OGG1 is product inhibited, binding avidly to abasic sites, and turns over slowly in the absence of other proteins such as APE1 (7). Sp and Gh are removed by the actions of the bifunctional glycosylases Endonuclease VIII-like 1–3 (NEIL1–3), which will be discussed in detail in a later section. Tg is removed by the bifunctional glycosylase Endonuclease III-like 1 (NTHL1). 5fC and 5caC are removed by the action of thymine DNA glycosylase (TDG), which is a monofunctional glycosylase. The structures of these glycosylases and their substrates are given in Figure 3.
Figure 2.

Mammalian base excision repair (BER) pathway. The base lesion is excised by a lesion-specific DNA glycosylase. Monofunctional glycosylases break the glycosidic bond between the damaged base and the sugar moiety, resulting in an abasic site. AP endonuclease 1 (APE1) processes the abasic site to form a 3′OH and a deoxyribose-5′-phosphate (dRP), which is removed by the lyase activity of DNA polymerase β (pol β). Bifunctional glycosylases utilize their AP lyase activity to cleave the phosphate backbone, creating a single strand break, leaving a free 5′ phosphate and either a 3′-phospho-α, β-unsaturated aldehyde (3′-PUA) (β-elimination) or a 3′ phosphate (β,δ-elimination). APE1 acts on the β-elimination product while polynucleotide kinase phosphate (PNKP) is required to process the 3′phosphate after β,δ-elimination. The resulting 3′OH is bound by PARP1 which recruits the BER complex consisting of pol β, XRCC1 and DNA ligase. The one base gap is then filled by pol β and the nick in the DNA is sealed by DNA ligase. Adapted from Kumar et al. (8) with permission.
Figure 3.

BER glycosylases, their structures and respective substrates. The glycosylases (green) are bound to DNA (purple) containing a lesion (purple, space-filled). All structures are human except SMUG1 (Xenopus laevis), MUTYH (Geobacillus stearothermophilus), NEIL2 (Monodelphis domestica), NEIL3 (Mus musculus), NTHL1 (EndoIII, Geobacillus stearothermophilus). PDB: SMUG1 (1OE4), MBD4 (5CHZ), UNG (1EMH), TDG (3UFJ), MPG (1BNK), MUTYH (4YOQ), OGG1 (1EBM), NEIL1 (5ITY), NEIL2 (6VJI), NEIL3 (3W0F), NTHL1 (1ORN). Abbreviations: U, uracil; A, adenine; T, thymine; C, cytosine; G, guanine; 5-FU, 5-fluorouracil; 5-hmU, 5-hydroxymethyluracil; ϵ, etheno; FaPy, 2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine; 8-oxoG, 8-oxoguanine; Gh, Guanidonohydantoin; Sp, Spiroiminodihydantonin; Im, iminoallantoin; 5fC, 5-formylcytosine; 5caC, 5-carboxycytosine; 5-BrU, 5-Bromouracil; Tg, Thymine Glycol; meA, 3-methyladenine; meG, 3-methylguanine; 5-hC, 5-hydroxycytosine; 5-hU, 5-hydroxyuracil; 2-hA, 2-hydroxyadenine
For more than a decade, studies have provided evidence suggesting a role for NER proteins in the repair of oxidative damage through interactions with BER proteins, reviewed (8–11). NER is the major pathway for the repair of bulky adducts and other helix-distorting lesions, such as UV-induced photoproducts, such as 6–4 photoproducts (6–4 PP) and cyclobutane pyrimidine dimers (CPD) (12). Unlike BER that has a set of glycosylases each tuned to find and process specific altered bases (Figure 3), damage recognition proteins of NER are remarkable in that they have a broad ability to dynamically detect many different structurally and chemically diverse lesions (13). There are two sub-pathways in NER: global-genome NER (GG-NER) and transcription-coupled NER (TC-NER), reviewed in (12,14). These sub-pathways differ in the manner of lesion recognition. In GG-NER, damage recognition proteins scan the entire genome, including heterochromatic, transcriptionally inactive regions, or the non-transcribed strand for damage-induced structural distortions. In contrast, TC-NER is initiated when RNA polymerase (RNAP) stalls at damaged site on the transcribed strand of active genes, in euchromatic regions of the genome. Defects in NER are associated with two important human diseases, xeroderma pigmentosum (XP) and Cockayne syndrome (CS). Damage recognition in GG-NER is initiated by two proteins, UV-DDB and XPC-RAD23B. In response to UV-induced DNA damage, UV-DDB in complex with CUL4 and RBX forms a ubiquitin E3 ligase complex and binds to the chromatin to ubiquitinate histones, making the lesion more accessible to downstream repair proteins in the NER pathway, including XPC-RAD23B. XPC-RAD23B binds with high affinity to the strand opposite to the distorted lesion, which begins the damage verification step of NER. XPC-RAD23B facilitates recruitment of the transcription factor TFIIH. TFIIH is a multi-subunit protein, consisting of 10 proteins, including XPB and XPD, proteins that have DNA helicase folds. When XPD encounters the lesion, its strand opening activity stalls and facilitates the recruitment of XPA, RPA and XPG, collectively known as the pre-incision complex. XPB is believed to act as a translocase to help reel the DNA into the pre-incision complex. XPA and RPA recruit the heterodimeric endonuclease, XPF-ERCC1. Recruitment of XPF-ERCC1 produces an endonucleolytic incision 5′ to the lesion. DNA polymerases δ, ϵ or κ begin to fill in the repair patch, which stimulates the 3′ endonucleolytic activity of XPG, leading to release of an oligonucleotide of 22–27 nucleotides containing both the lesion and TFIIH. DNA ligase I seals the remaining nick in the repair patch. TC-NER damage recognition is initiated by the presence of a stalled RNAP at a lesion site, which facilitates recruitment of Cockayne syndrome proteins (CSB and CSA), and the accessory proteins (UVSSA, XAB2, and HMGN1) to the lesion site on the transcribed strand (15). XAB2 facilitates recruitment of XPA and subsequently TFIIH which intertwines the two NER sub-pathways at the damage verification step. CS patients and a subset of XP patients display signs of neurological degeneration and have been shown to accumulate unrepaired oxidative DNA lesions (16,17). Therefore, it is important to understand any crosstalk which exists between the two repair pathways to gain a better understanding of disease progression.
COULD CYCLOPURINE DEOXYNUCLEOSIDES EXPLAIN NEURODEGENRATION IN XERODERMA PIGMENTOSUM PATIENTS?
5′,8-Cyclopurine deoxynucleotides (cyPu) are endogenous oxidative DNA lesions formed by the reaction of hydroxyl radicals with DNA, and first identified after ionizing radiation (18). These lesions contain damage to both the purine base and the 2′-deoxyribose sugar moiety by forming a covalent bond between the C8 of the base and C5 of 2′-deoxyribose (19). Both cyclo-deoxyadenosine (cdA) and cyclo-deoxyguanosine (cdG) lesions exist as 5′R- and 5′S-diaestereomers (20) (Figure 1) and cause significant distortions to DNA and therefore, can act as good substrates for nucleotide excision repair (NER) (11). Estimates of cyPu lesions vary, but are generally considered to be less frequent than 8-oxoG, with cdG lesions about an order of magnitude more prevalent that cdA lesions (Table 1).
Table 1.
Oxidative lesions and NER proteins
| Lesion | Frequency/106 bases * | Reference for lesion frequency | Technique used | Source of DNA | Repaired by | Evidence for NER proteins involved (reference) |
|---|---|---|---|---|---|---|
| 8-oxoG | 1 | (113) | COMET assay | HeLa cells | BER/ TC-NER | XPA (67,69,73,75,76), XPC (29,38,67,69,72,73), XPG (67–69), UV-DDB (56), CSA (75), CSB (75) |
| 1.2 | (29) | LC/MS | Human primary keratinocytes | |||
| 4.6 | (40) | HPLC-ESI-MS | Rat Liver | |||
| 25** | (114) | LC-MS/MS | Calf Thymus | |||
| 0.35 | (115) | NaI extraction; HPLC-EC | Mice Liver | |||
| Gh | 0.01–0.07 | (116) | LC/MS | Mice Liver and Colon | BER/NER | XPA (83), XPC (83) |
| Sp | 0.01–0.07 | (116) | LC/MSfC | Mice Liver and Colon | BER/NER | XPA (83), XPC (83) |
| 8-cyclo-2′dG | 2.8 (5′S); 0.7 (5′R) | (29) | GC/MS*** | Human primary keratinocytes | NER | XPC (29) |
| 8-cyclo-2′dA | 0.1–0.15 | (20) | LC/MS; GC/MS*** | Calf Thymus | NER | XPA (23), XPC (29), CSA (30), CSB (30) |
| 0.015–0.03 | (117) | 32P-Postlabeling Assay | Fetal and postnatal rat liver | |||
| 0.2 | (29) | LC/MS | Human primary keratinocytes | |||
| 5hmC | 247 | (118) | Tet-assisted bisulfite sequencing (TAB-Seq) | Human embryonic stem cells | BER | XPC (60) |
| 1300 | (119) | 2D-TLC; LC/MS/MS | Mouse embryonic stem cells | |||
| 5fC | 20 | (119) | 2D-TLC; LC/MS/MS | Mouse embryonic stem cells | BER | XPC (60) |
| 5caC | 3 | (119) | 2D-TLC; LC/MS/MS | Mouse embryonic stem cells | BER | XPC (60) |
| Tg | 0.01 | (120) | Capillary electrophoresis and laser-induced fluorescence detection | human lung carcinoma cells | BER | XPC (60) |
| Oxazolone | 0.02–0.41 | (40,114) | HPLC-ESI-MS | Rat Liver | BER | XPC (80) |
Abbreviations: 8-oxoG, 8-oxoguanine; Gh, Guanidonohydantoin; Sp, Spiroiminodihydantonin; 8-cyclo-2′dG, 8-cyclo-2′deoxyguanosine; 8-cyclo-2′dA, 8-cyclo-2′deoxyadenosine; 5hmC, 5-hydroxymethylcytosine; 5fC, 5-formylcytosine; 5caC, 5-carboxycytosine; Tg, Thymine Glycol
*Steady state levels of damage found in purified DNA from various sources.
**Commercially available Calf thymus DNA probably contains higher than normal levels of 8-oxoG due to oxidation in purification and processing (114,121)
***GC/MS – method of preparation contributes to high lesion frequency.
As mentioned earlier, defects in NER genes can cause the rare disorder xeroderma pigmentosum (XP). About 20% of XP patients exhibit neurological symptoms, which have been posited to be caused by endogenously accumulated oxidative DNA damage (16,17). Work by Lindahl's group supported this hypothesis by demonstrating that a subclass of base lesions formed by γ-irradiation were repaired by normal cell extracts, but not by XP cell extracts (21). The first direct evidence of NER involvement in the repair of cyPu lesions was provided by Kuraoka et al. Primer extension assays using mammalian polymerase δ or T7 DNA polymerase were performed to show that both 5′S- and 5′R-cdA can block DNA synthesis by terminating product extension at the cdA site in a 5′-32P-labeled DNA template-primer substrate containing a site-specific 5′S- or 5′R-cdA lesion. Therefore, if left unrepaired, cdA lesions can be highly cytotoxic by blocking DNA replication (22). Moreover, when HeLa cell extracts were incubated with 20 bp cdA substrates, no DNA glycosylase-mediated cleavage was observed suggesting that human DNA glycosylases cannot act on cyPu residues. Finally, the authors used a dual-incision assay with cell extracts and closed circular DNA substrates harboring a 5′S- or 5′R-cdA lesion to show that the cyPu residues are repaired by excision of a 26–28 bp DNA product. The excision was significantly suppressed in the presence of an XPA antibody, indicating the dependence of repair on the NER pathway. Interestingly, the R form of cdA was repaired more efficiently (∼2-fold) than the S form, however, both diastereoisomers were relatively poor (∼40–150-fold) NER substrates as compared to the 1–3-intrastrand d(GpTpG)-cisplatin crosslink substrate. This study clearly shows that cyPu lesions are removed by NER in vitro, and have the ability to cause local helix distortions and block polymerases. Brooks et al performed a host reactivation assay (HCR) in Chinese hamster ovary (CHO) cells using a plasmid expressing the luciferase (Luc) gene that contained a cdA lesion on the transcribed strand (23). They showed that a single cdA lesion dramatically decreased the Luc gene expression, suggesting that cdA can act as a strong block to transcription. They also found that repair of cdA lesion on the plasmid was significantly reduced in XPG and ERCC1 mutant cells as compared to wildtype. They further confirmed this result by employing the HCR assay in SV-40 transformed normal (GM00637) and XP-A (XP12BE) cell lines, providing evidence for defective repair of cdA in the XP-A cells. The XP12BE cell line was derived from a XP-A patient (XP2OS) who exhibited severe neurological symptoms (24). Therefore, these studies provide a strong correlation between defective NER and neurodegeneration in XP patients. In this scenario, an obvious prediction would be that NER deficient XP patients would have elevated levels of cyPu lesions in their DNA. Indeed, cyPu lesions have been shown to accumulate over age in the brain tissue of Xpa−/− and Csb−/− mice (25,26), although unlike humans, Xpa−/− mice do not display any neurological abnormalities (27,28). In another study, D’Errico et al used X-rays to introduce base lesions in the DNA of normal and XP-C keratinocytes and measured the accumulation of cyPu by HPLC/MS. While both normal and XP-C cells accumulated equal numbers of cyPu lesions after 5Gy of X-rays, XP-C cells were inefficient in the removal of damage over time (29). Similar accumulation of cdA was also observed in CSA deficient (CS-A) fibroblasts treated with X-rays (30). This is consistent with it being a strong transcription blocking lesion as CSA and CSB both recognize DNA damage in the context of transcription. Further studies are required in more XP and CS patients with neurological disorders to establish a direct link between cyPu accumulation and XP neurodegeneration. Despite the low prevalence of XP throughout the world, it would be of interest to set-up a rapid autopsy program to measure cyPu lesions in brain tissues from deceased XP patients.
Whether cdG lesions could be processed by DNA glycosylases was investigated by Pande et al. (31). In this study, seven purified glycosylases (bacterial: Fpg, EndoIII, EndoV, EndoVIII; human: NEIL1, NEIL2, and OGG1) failed to incise a 12 or 36 bp duplex containing a S-cdA or a S-cdG lesion. Even at high concentrations of the enzymes (200 fmol), no cleavage activity was observed. They also performed binding assays with the glycosylases and found that at very high concentrations (10–20 pmol), NEIL1 bound to both cdA and cdG substrates as well as undamaged DNA, suggesting that the binding was non-specific. Strikingly, an earlier study had shown that both S- and R-cdA lesions accumulate in Neil1−/− mice (32), suggesting the role of NEIL1 in cyPu repair, although the exact mechanism is still unclear. Furthermore, this study went on to demonstrate that S-cdG was repaired slightly better than S-cdA by NER in human HeLa cell extracts and that the base complementary to the lesion affected the efficiency of repair. They speculated that both base pairing and base stacking are important for the recognition of cyPu lesions in the DNA and that an abnormal Watson-Crick base pairing (e.g. S-cdG:dT) acts as a better substrate for NER. NMR combined with molecular dynamics have proven highly successful in understanding the alterations in the conformation of the DNA helix induced by DNA lesions. It would be of interest to use these techniques to determine the distortions formed on the DNA by the cyPu lesions and investigate the interactions of NEIL1 and other NER recognition proteins with cyPu lesions (33,34).
Recently, for the first time, all four cyPu lesions (5′R-cdA, 5′S-cdA, 5′R-cdG, 5′S-cdG) were examined in the same sequence context by Shafirovich's group and the NER efficiencies were measured by excision assays using HeLa cell extracts (35). In agreement with the study mentioned earlier (22), 5′R-cyPu were repaired more efficiently than 5′S-cyPu. Molecular dynamics (100ns) revealed greater DNA backbone distortions and diminished base stacking in the R form of cyPu as compared to the S form. In cells, DNA lesions are embedded in the nucleosome which can hinder the accessibility of some repair proteins (36–46). Therefore, to explore the effect of CyPu on histone-DNA interactions, Shafirovich et al. embedded cyPu lesions in an ‘In’ (fourth nucleotide on the 5′-side of dyad) and ‘Out’ (eighth nucleotide facing the aqueous solution environment) rotational setting near the dyad axis in nucleosomes reconstituted with either recombinant histones or histones extracted from HeLa cells (47). In both cases, they made the surprising discovery that cyPu lesions were completely resistant to excision by NER proteins in human cell extracts. This suggests that even though cyPu lesions cause significant distortions to naked DNA duplex, they either do not significantly disturb the DNA-histone interactions at these specific positions or these lesions when embedded in a nucleosome escape detection by NER proteins. It also remains unknown whether these results are relevant in physiological conditions in vivo. It would be, therefore, of great interest to extend these studies to living cells, although there are no tools available yet that could be used to specifically introduce cyPu lesions in cells.
As mentioned earlier, NER proteins recognize and repair UV-induced photoproducts, 6–4 PP and CPD. While a 6–4 PP causes a significant distortion in nucleosomal DNA (48), a CPD causes less of a distortion (49). It is well-established that while XPC-RAD23B can recognize 6–4PP, this heterodimer has limited ability to detect CPDs, (50) and in cells recruitment of XPC to sites of CPD in chromatin requires UV-DDB. Furthermore, it is interesting to note that XPC does not efficiently bind DNA configured on a nucleosome (51). On the other hand, UV-DDB has been shown to bind lesions directly in the nucleosome and even shift the nucleosomal register to provide access to more occluded sites (49,52). Moreover, the binding of UV-DDB seems to precede the activity of ATP-mediated chromatin remodelers (53,54). More recently, as discussed in the last section, our group has demonstrated that the oxidative base damage 8-oxoG, which only causes a mild helix distortion, is recognized by UV-DDB in naked duplex DNA, as well as in living cells (55,56). The relatively mild nucleosome distortion caused by CPD and 8-oxoG is analogous to cyPu. Therefore, it would be interesting to determine the full substrate repertoire of UV-DDB and if UV-DDB is capable of recognizing CyPu and other lesions in the context of nucleosomes.
OXIDATIVE REMOVAL OF 5mC MOIETIES IS STIMULATED BY XPC
5-methylcytosine (5mC) is formed by the addition of a methyl group to carbon 5 of cytosine through the action of a DNA methyltransferase (57). As previously mentioned, during an active enzymatic demethylation process, 5mC is oxidized by TET dioxygenases to 5hmC, 5fC, and 5caC. The latter two lesions are removed by TDG. 5fC, and 5caC are some of the more common oxidative lesions with steady state levels at 20 and 3 lesions per million bases, respectively (Table 1). TDG has been shown to also remove deaminated 5mC (T:G) moieties from DNA (58).
TDG is a monofunctional glycosylase, which as previously mentioned, binds avidly to abasic sites and thus becomes product inhibited. Studies have demonstrated roles for other BER proteins, including NEIL1 and APE1 in facilitating TDG turnover. However, the mechanism of DNA demethylation by TDG in cells remains unclear (59). To this end, Ho et al investigated the role of XPC in epigenetic gene regulation through stimulation of TDG. Using an ELISA specific to 5mC, they were able to show XPC-dependent DNA demethylation (60). While these authors reported that XPC plays a role in DNA demethylation, they argued based on a W690S variant of XPC that this activity was independent of XPC’s role in NER. This W690S XPC variant, discovered in an XP-C patient, XP13PV, was previously shown to have reduced stability in cells, and cells expressing this variant showed reduced rates of removal of UV-induced photoproducts (61). However, careful analysis of data presented in the Ho et al. study indicates that the W609S XPC variant showed reduced stimulation, ∼20% increase of TDG excision compared to TDG alone, of either 5fC:G or 5caC:G. In comparison, WT XPC fully doubled the activity of TDG. These data suggest that reduced damage recognition and/or DNA binding of this W690S XPC variant prevented its stimulation of TDG. To provide further support for the role of XPC in TDG stimulation, the authors performed a ChIP-seq analysis and determined co-enrichment of TDG and the XPC subunit, RAD23B at the promoter region of embryonic stem cells. Additionally, using MeDIP-seq to measure 5mC levels globally in the genome, the authors were able to show reduced DNA methylation in cells overexpressing WT XPC. Single-particle tracking experiments utilizing Halo-tagged TDG and SNAP-tagged XPC, revealed that overexpression of XPC led to a reduction in the length of time Halo-tagged TDG remained bound to DNA. Using shRNA to knockdown XPC expression the authors demonstrated longer retention of TDG on the DNA. These data support the role of XPC in stimulating TDG activity by facilitating turnover of TDG from the abasic DNA product, (Figure 4). Lastly, the authors determined that TDG stimulation by XPC occurs through interactions between the N-terminus of TDG and the C-terminus of XPC. This study only looked at the role of XPC in TDG stimulation, it would be interesting to determine if any other NER or BER proteins are recruited to the 5mC moiety in response to XPC stimulation. Finally, it should be pointed out that XP-C patients and Xpc-/- mice develop normally and do not appear to have large defect in epigenetic programing during cellular differentiation. Thus, further work is needed on the role of XPC, and NER proteins in the oxidative removal of 5mC.
Figure 4.
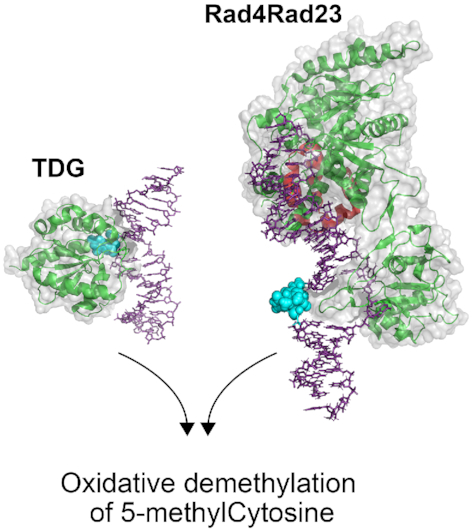
XPC and TDG in oxidative demethylation of 5-methylcytosine. Based on the work by Ho et al. (60), XPC works to help turnover TDG, which like other glycosylases, is product inhibited binding tightly to abasic sites. Shown here is the structure of the yeast, XPC homolog, Rad4 (green) Rad23 (red) bound to a DNA duplex (purple) containing a 6–4 photoproduct (blue space-filled), PDB: 6CFI; Human TDG (green) bound to a DNA duplex (purple) containing 2′-fluoro-2′-deoxyuridine (blue space-filled), PDB:3UFJ (122).
NER PROTEINS HELP MEDIATE THE REMOVAL OF THYMINE GLYCOL AND 8-OXOG
Guanine oxidation is a well-characterized DNA lesion. ROS acting on guanine results in the formation 8-oxoG, through two subtle modifications on guanine (Figure 1): the addition of an oxo group on carbon 8 and the addition of hydrogen to the seventh position nitrogen (62). These modifications causes the base to rotate from the anti- to the syn-conformation with respect to the deoxyribose moiety around the glycosidic bond causing 8-oxoG to pair with A during replication creating T:A transversions, if left unrepaired. The formation of 8-oxoG lesions in cells is estimated to occur up to 10 000 times per cell per day in humans, with the estimated steady-state levels of about 1–2 8-oxoG lesions/106 guanines (63,64) (Table 1). While older literature has referred to this lesion as 8-hydroxy-guanine, this tautomeric form at physiological pH (7.4) is a minor product (65). This relatively high lesion frequency of 8-oxoG (Table 1) coupled with the implications in genome instability emphasize the need for repair pathways dedicated to the removal of the 8-oxoG lesion. As mentioned earlier, 8-oxoG is commonly removed through base excision repair (BER), through the actions of the DNA glycosylase OGG1. The work by the Mitra laboratory in showing OGG1 is product inhibited and needs the actions of APE1 to facilitate OGG1 turnover, imply the potential for other co-factors outside of BER to stimulate either OGG1 activity or processing of 8-oxoG (7).
The first implications of NER protein involvement in oxidative DNA damage repair was shown using the Escherichia coli NER system consisting of the UvrABC complex (66). The authors used a DNA substrate containing a thymine glycol (Tg) lesion to show that UvrABC efficiently recognizes and incises the lesion. This finding was recapitulated in the mammalian system by Sancar and coworkers (67). It has been estimated that the steady-state levels of Tg in mammalian cells are about two orders of magnitude lower than 8-oxoG adducts (Table 1). Using human cell free extracts lacking any one of the XPA-XPG proteins, they were able to show reduced excision of two common oxidative lesions, 8-oxoG and Tg, from damaged DNA substrate (67). The basis of this assay is that a DNA substrate is created by ligating a 5′ 32-P end-labeled oligonucleotide containing an 8-oxoG or Tg moiety into a 139 bp DNA duplex. Dual incisions by XPF/ERRC1 on the 5′ side and XPG on the 3′ side liberates excision products of ∼22–26 bases containing the label. Additionally, they showed using a system of purified proteins the need for a complete NER system containing XPC-RAD23B, XPA, RPA, TFIIH (XPB and XPD), XPG and XPF-ERCC1, in order for proper excision of 8-oxoG or Tg. This work from the Sancar laboratory clearly demonstrates the ability of purified NER proteins to remove oxidative lesions, but did not assess whether there was any interaction between NER and BER proteins or whether NER is an important pathway for their removal in cells. The authors hypothesize the role of NER is to act as a slower alternative pathway for oxidative damage removal by BER, and the loss of this activity in XP patients, contributes to the accumulation of oxidative damage and subsequent neurodegeneration. It is important to point out the ability of the NER machinery to excise 8-oxoG had not been confirmed in any other laboratory or through any other approaches, and it remains to be determined whether NER is a back-up system for the removal of 8-oxoG in the absence of BER.
Following this pioneering work by the Sancar laboratory, which established an in vitro role of NER proteins in oxidative DNA damage repair, attention shifted to understanding the roles of specific NER proteins in the repair of 8-oxoG and other lesions induced by oxidative DNA damage. Klungland et al began by characterizing the role of XPG in BER of the oxidative lesions, thymine glycol and dihydrouracil. These lesions are excised by the bifunctional DNA glycosylase, NTH1. Using a reconstituted BER system containing hNTH1, APE1, pol β, and XRCC1-DNA ligase III, the Lindahl laboratory was able to show stimulation of NTH1 by XPG (68). Specifically, they were able to show enhanced binding of NTH1 to damaged DNA in the presence of XPG. The authors also looked at the ability of XPG to stimulate OGG1 excision of 8-oxoG but were unable to detect any enhanced OGG1 activity in the presence of XPG. In a later study of oxidative damage repair in melanocytes by Wang et al. cells deficient in XPG protein were shown to have decreased repair of hydrogen peroxide (H2O2)-mediated oxidative damage, when measured using a luciferase-based host cell reactivation assay (HCR) (69). They were also able to show that cells with defective XPA or XPC proteins showed reduced repair of oxidative damage. This study primarily focused on understanding oxidative DNA damage repair capacity of melanocytes, as melanoma incidence is increased ∼1000-fold in XP patients (70). Wang et al. went on to further investigate the role of XPA in oxidative DNA damage repair and showed XPA deficient cells had approximately a 4-fold reduction in oxidative damage repair capacity. While this study showed an apparent involvement of NER proteins in the repair of oxidative DNA damage, it remained unclear the specific roles of XPA, XPC and XPG in this process. The authors hypothesized in the case of melanocytes, the increased presence of melanin which binds to DNA may inhibit the recognition of the damage by both BER and NER proteins. It would be interesting to further investigate whether the levels of melanin prevent recruitment of damage recognition proteins and inhibit subsequent DNA repair.
The Dogliotti lab provided the first evidence of an NER protein, XPC, having a protective role against oxidative stress in human skin cells. Looking at keratinocytes and fibroblasts with a nonsense mutation in the XPC protein, they were able to demonstrate an increased sensitivity to oxidizing damage, such as that from X-rays or potassium bromate, through a colony formation assay (29). While X-rays produce a wide spectrum of DNA lesions including various forms of base damage, single-strand and double-strand breaks, potassium bromide produces primarily 8-oxoG and to a lesser extent other base damages (71). Additionally, D’Errico et al demonstrated using HPLC/MS an accumulation and subsequent delayed removal of 8-oxoG from XPC-deficient skin cells. To strengthen support for the role of XPC in oxidative DNA damage repair, they also showed XPC-RAD23B-mediated stimulation of OGG1, the DNA glycosylase responsible for 8-oxoG removal. They were unable to demonstrate XPA stimulation of OGG1, even at high protein concentrations, even though previous studies alluded to a role for XPA in oxidative damage repair (67,69). Furthermore, through far western blot analysis the authors demonstrated a direct binding between OGG1 and XPC-RAD23B, showing XPC enhances the ability of OGG1 to recognize 8-oxoG lesions. The authors did not show a direct interaction between XPC and damaged DNA, indicating its role is possibly to facilitate the turnover of OGG1. In a later study by the Rainbow lab, XPC deficient fibroblasts were shown to have reduced removal of 8-oxoG (72). 8-oxoG was introduced onto the β-galactosidase reporter gene via generation of a singlet oxygen by methylene blue plus visible light. Using this reporter gene, they conducted a host cell reactivation assay (HCR) to investigate the effect of XPC on DNA damage repair. Additionally, while the authors demonstrated pre-treatment of cells with UVC does not change the relative repair rate of 8-oxoG in XPC-deficient cells, pre-treatment with UVC resulted in an approximate 1.5-fold increase in overall repair of 8-oxoG. These data imply a potential role of other proteins induced by UV damage mediated through p53 stabilization induced gene expression, in the repair of oxidative damage. One such protein may be DDB2.
Taken together these data suggested BER may not work in isolation to remove oxidative damage. However, the specific molecular roles NER proteins may play in the repair of oxidative damage remained unclear. To this end, Parlanti et al provided significant insights on the roles of NER proteins, specifically XPA, XPC, CSA, and CSB, on 8-oxoG repair (73). First, using fibroblasts derived from NER deficient mice, they showed reduced repair and increased accumulation of 8-oxoG in XPA−/−, XPC−/−, CSA−/−, CSB−/− and OGG1−/−, knockout (KO) cells. Unsurprisingly, the OGG1−/− KO cells showed the greatest reduction in repair and subsequent accumulation of 8-oxoG. However, all of the NER protein MEF KO cell lines also showed a noticeable reduction in the rates of 8-oxoG repair compared to the WT cells with sufficient NER proteins. They also compared repair capacity in both the single and double KO cell lines and were able to demonstrate a more pronounced reduction in repair in the XPA−/−/ CSB−/−, XPC−/−/CSB−/−, double KO cell lines, which resembled the repair capacity of the OGG1 −/− cell line. Alternatively, the repair capacities of the XPA−/− and XPC−/− double KO cells resembled that of the single KO cells. These data suggest CSB and OGG1 are involved in the same repair pathway, one that may be different from that of XPC and XPA. However, the exact molecular details of these pathways remain unresolved. This group were also able to recapitulate the results from the mouse experiments in human XPA fibroblasts, by showing decreased repair of 8-oxoG in addition to increased sensitivity to oxidizing agents. Using siRNA targeting OGG1 in the XP12SV40 cell line, an XP-A deficient cell line they were able to show impaired repair of 8-oxoG, when compared to XP-A cells or cells with deficient XPC. It remains unclear how the authors were able to demonstrate impaired repair of 8-oxoG in XPA deficient cells in one study, but failed to show XPA-mediated stimulation of OGG1, the glycosylase mediating the repair of the lesion, in another study (29). These conflicting results further reiterates the ambiguity of the role of XPA in 8-oxoG removal, and other published studies show contrasting results (74). The Spivak and Hanawalt laboratory developed a cutting-edge strategy to investigate the role of XPA in repair of relatively low levels of oxidative damage. Using a comet-FISH (combining single-cell gel electrophoresis with a fluorescence in situ hybridization assay), they were able to demonstrate roles for XPA and CSB in the transcription-coupled repair of 8-oxoG (75). Using the ATM gene, they fluorescently labeled the 5′ and 3′ ends using different probes and tracked the increasing distance between the probes after treatment with potassium bromate, as a measure of BER-mediated single-strand break formation. In this way the authors were able to show the repair rates of CSB and XPA deficient cells were comparable to the repair rates of the wild-type non-transcribed strand showing the repair of 8-oxoG is coupled to transcription. To further support the idea of 8-oxoG processing to be coupled to transcription, the authors performed the comet-FISH assay on cells deficient in UVSSA and RNAPII, key proteins in TC-NER, and were able to show that these cells displayed a repair rate similar to that of the wild-type non-transcribed strand. Finally, they showed that in the absence of OGG1, this transcriptional effect of XPA and CSB was lost, suggesting that RNAP is not inhibited by 8-oxoG, but instead by the resulting SSB created by the actions of OGG1 and APE1. The protective role of XPA was further investigated by the Yasui lab, which utilized the TATAM (tracing DNA adducts in the targeted mutagenesis) system to study 8-oxoG lesions into XPA knockout cells (76). Introduction of a single 8-oxoG lesion in cells deficient of XPA had no effect on mutagenesis. Tracks of ionizing radiation can create closely spaced multiple lesions and it was interesting to note that this group was able to show increased mutagenesis in XPA deficient cells with the introduction of multiple 8-oxoG lesions, specifically when the lesions were introduced on the actively transcribed strand.
The Vermeulen lab developed an imaging system to study the roles of NER proteins in 8-oxoG repair. They locally induced 8-oxoG lesions via singlet oxygen by using a photosensitizer and a 405nm laser (38). Using this highly innovative approach, the authors were able to show a difference in the recruitment kinetics of CSB and XPC to damaged sites, with CSB showing slightly enhanced recruitment over XPC (t1/2 = ∼9 s for CSB versus ∼13 s for XPC). CSB and XPC are involved in different sub-pathways of NER, offering an explanation for their different rates of recruitment to damage sites. In support of a role of CSB in transcription-coupled repair of 8-oxoG, they were able to show enhanced CSB recruitment to the transcriptionally active nucleolus, while XPC was seen more in the heterochromatic nucleoplasm, supporting its role in GG-NER. This study while demonstrating the recruitment of CSB and XPC to sites of oxidative damage, did not address whether these proteins are directly involved in the removal of 8-oxoG, either through DNA glycosylase stimulation or in some other step in BER. In a later study, these same authors looked at the role of CSB in 8-oxoG repair by looking at OGG1 recruitment (77). Using the previously described photosensitizer and laser strategy, they induced 8-oxoG lesions and monitored recruitment of CSB and OGG1 to the damage site and demonstrated that CSB recruitment to damage sites is independent of OGG1. These data seem at odds with the previous work by Spivak and Hanawalt who suggested TCR of 8-oxoG can only occur after processing of the lesion to a strand break. Furthermore, the Menoni et al. study (77) demonstrated CSB is able to stimulate XRCC1 recruitment to 8-oxoG, in transcriptionally active regions. The authors hypothesize the role of CSB is to aid in the recruitment of XRCC1 and other BER proteins by facilitating chromatin remodeling to aid accessing lesions, especially single-strand breaks. Additionally, it is important to note that APE1 has been shown to interact directly with CSB and is stimulated up to 6-fold in an ATP-independent manner (78). Thus, CSB may provide an important role during transcription-coupled BER by orchestrating both backtracking of stalled RNAP, as well as coordinating downstream BER proteins. Finally, it was shown that CSB deficient cells are hypersensitive to killing by hydroxymethyl-deoxyuridine (hmdU) treatment, suggesting a direct role of CSB in SMUG1-mediated removal of this oxidized base from DNA (78).
NER AND BER WORK COOPERATIVELY TO REPAIR OXIDIZED 8-OXOGUANINE LESIONS
The oxidation of guanine can create 2,2-diamino-4-[(2-deoxy-β-d-ery-pentofuranosyl) amino)-5(2H)-oxazolone (Oz), and 8-oxoG. The Oz lesion is normally processed by BER in mammalian cells by the activities of NEIL1 and NTH1 (79). However, using a defined system, Hanaoka and coworkers showed that Oz is also a poor substrate for NER, and that the overall affinity of XPC-RAD23B to a DNA duplex containing Oz was significantly lower than a 6–4 PP containing duplex (80). The 8-oxoG lesion is several orders of magnitude more sensitive to oxidation than the parent guanine moiety resulting in two oxidation products, Sp and Gh, which are less common oxidative lesions, having steady state levels in the range of 0.01–0.07 lesions per million bases (Table 1). These lesions can be removed by BER proteins, however work from several laboratories suggest that NER can also process these lesions. Early studies with the bacterial NER UvrABC system indicated that this enzyme incised a duplex containing several lesions to different extents, 8-oxoG:A the worst (10% completion) with Gh (23%) and Sp (32%) lesions being moderate and amine modified Sp to 62% completion (81). These results suggest that a larger and more distorting lesion is recognized and incised the most efficiently by the bacterial NER proteins.
Work from the Shavirovich and Geacintov laboratories indicated that human NER proteins from cell extracts can perform dual incisions on Sp or Gh lesions embedded in a 135 bp duplex. XPA depletion studies with antibodies to XPA or extracts from XPC−/− fibroblasts failed to produce dual incisions (82). Adding back purified XPC to the latter extract was able to restore NER activity. The same study showed that NEIL1 can also process these lesions in an extract and it was not clear if these two pathways work synergistically or in an antagonistic manner. This question was elegantly explored in living cells by the same group in which they transfected internally 32P-labeled DNA hairpin in which the label was placed near the lesion (83). By transfecting this DNA duplex into human cells, they were able to follow the excision of the oligonucleotide containing the lesion via a NER pathway or direct incision by the activity of NEIL1 initiating BER. They found that compared to a substrate containing a benzo[a]pyrene-dG lesion, Sp or Gh lesions were processed by NER 8- or 6-fold, respectively, less efficiently. Transfection of the Gh substrate into XPA−/− cells failed to produce the characteristic excision product by NER, whereas transfection into NEIL1−/− cells reduced but did not eliminate the BER incision product. Taken together these data suggested to the authors that the amount of XPC and XPA and perhaps their relatively low affinity for Sp and Gh substrates versus 5–10-fold higher levels of NEIL1 with high catalytic efficiency in the cell, probably pushes repair of these substrates into a BER pathway. However, it is not clear whether these transfected DNA hairpins were associated with nucleosomes and what role chromatin structure may play on the processing of Sp and Gh lesions by NER and BER pathways. Finally, to better explore the question of relative affinities of XPC-RAD23B for Sp and Gh lesions and its ability to compete with NEIL1 for these lesions, the same group studied relative binding affinities of XPC-RAD23 and NEIL for 147 bp duplexes containing Sp or Gh lesions using EMSA analysis (84). They found that XPC-RAD23 bound to Sp containing substrate with high affinity (low nM range) and that XPC could effectively compete away NEIL1 binding to these substrates at equal molar concentrations. They then followed NEIL1 incision burst kinetics under single turnover conditions on both substrates in the absence and presence of XPC-RAD23 and found that equal molar concentrations of added XPC-RAD23 greatly reduced the amplitude of the burst phase of NEIL1 cleavage. Together these data strongly suggest that XPC-RAD23 and NEIL1 can directly compete for Sp and Gh lesions. Additionally, it has been demonstrated NEIL2 and NEIL3 can act to remove Sp and Gh lesions (85,86). Further cellular work such as transfecting plasmids carrying these defined lesions into cells and using immunofluorescence to measure the binding kinetics of these proteins will be necessary to better understand if XPC or other NER proteins can bind to these lesions in the cell nucleus.
A NEW ROLE OF UV-DDB IN THE REMOVAL OF 8-OXOG
As previously mentioned, OGG1 is product-inhibited and needs the activity of APE1 to turnover and work on other 8-oxoG lesions (7). Another factor which can impair the activity of BER proteins is the inaccessibility of oxidative lesions in the context of chromatin. DNA glycosylases have been shown to have impaired activity on damage when the lesion is contained within a nucleosome (42–46,87–90). It is important to note, while certain glycosylases such as SMUG1 are completely inhibited, others such as OGG1, AAG or UDG can recognize outward facing lesions and can readily initiate BER (88,90–92). In addition, both NEIL1 and NTH1 have been shown to show reduced activity on Tg substrates embedded in nucleosomes (91,93,94). The issue of lesion accessibility in the context of chromatin is an important factor in NER (36,37). UV-damaged DNA binding protein (UV-DDB), a heterodimeric protein consisting of DDB1 and DDB2, has a demonstrated role in NER for damage recognition in the context of chromatin (95). In response to UV-induced damage, UV-DDB as part of a ubiquitin E3 ligase complex with Cul4A and RBX, ubiquitinates histone H2A to facilitate chromatin remodeling to increase lesion accessibility (55). UV-DDB is known to recognize and bind sites of UV damage, but has also been shown to bind more strongly to a short DNA duplex containing an abasic site, as compared to a DNA substrate containing a CPD, suggesting a possible role for UV-DDB in BER (96,97). In order to directly test this hypothesis, we recently investigated whether UV-DDB plays an important role in initiating the repair of 8-oxoG lesions (56). It should be noted that previous studies missed that UV-DDB was able to discriminate between non-damaged DNA and DNA containing 8-oxoG (96). We have found that the presence of magnesium helps increase damage specificity by greatly decreasing binding to non-damaged DNA duplexes (55,56). EMSA assays conducted in the presence of 5 mM Mg2+ showed that UV-DDB preferentially bound abasic sites, CPD and 8-oxoG, with equilibrium dissociation constants, Kd, of 3.9, 30 and 160 nM, respectively, with high specificity as compared to undamaged DNA (Kd = 1108 nM) (55,56). Using an incision assay, with 8-oxoG and THF as substrates for OGG1 and APE1 specifically, we were able to show that UV-DDB increased the incision activity of both OGG1 and APE1 by 3- and 8-fold, respectively. We then utilized single-molecule fluorescence microscopy of quantum dot (Qdot) labeled proteins and a unique DNA tightrope optical platform to elucidate that UV-DDB increases the turnover and thus the incision activity of both OGG1 and APE1. In this assay DNA containing an abasic site every 2 kb was suspended from poly-Lysine coated 5 micron beads (34) and Qdot labeled OGG1 or APE1 binding to DNA was followed over time in the absence and presence of UV-DDB. We demonstrated that UV-DDB helped dissociate these proteins from the DNA in a concentration dependent manner. Furthermore, by orthogonally labeling UV-DDB and OGG1 or APE1 with different colored Qdots, we were able to show UV-DDB can form transient complexes with OGG1 and APE1 to facilitate their removal from the lesion site. Using an in vitro BER reaction, the activity of pol β was measured though the incorporation of radiolabeled dCTP into the gap created by the dual action of APE1 incision and pol β dRPase activity. This assay revealed that addition of UV-DDB increased BER product formation by 30-fold. Additionally, we were able to show that the newly incorporated dCTP could be ligated into full length product, indicating that UV-DDB does not have an inhibitory effect on downstream steps in BER. Lastly, we used a highly innovative chemoptogenetic system to generate site specific 8-oxoG lesions at telomeres (Figure 5). This system consists of a fluorogen activating peptide (FAP) that can bind to a malachite green dye (MG-2I) with high affinity and be excited at a far-red light wavelength (660 nm). When excited, the FAP-MG-2I combination generates singlet oxygen, which is highly reactive and short-lived and can oxidize nearby macromolecules including proteins and DNA. When fused to a telomere binding protein (TRF1), the FAP-MG-2I plus light system generates targeted singlet oxygen and oxidizes telomeric DNA to form almost exclusively 8-oxoG lesions. Using this FAP-TRF1 targeting system, we found that UV-DDB was recruited to telomeric 8-oxoG lesions prior to OGG1, indicating a direct role for UV-DDB as an early damage sensor in chromatin, acting before the initiation of BER (Figure 5).
Figure 5.

Role of UV-DDB in 8-oxoguanine (8-oxoG) repair. (A) A chemoptogenetic approach to introduce 8-oxoG at telomeres. Fluorogen-activating peptide (FAP) is fused to a telomere binding protein (TRF1). In the presence of a malachite green dye (MG2I), the FAP-TRF1-MG2I combination is excitable at far red wavelength (660nm) and generates singlet oxygen. Singlet oxygen reacts with telomeric DNA to form 8-oxoG lesions. (B) Immunofluorescence images of mCherry-DDB2 (red) and OGG1-GFP (green) recruitment to and departure from 8-oxoG lesions at telomeres (blue). (C) Quantitative analysis of immunofluorescence images in (B). (D) Working model of the potential role of UV-DDB in BER of 8-oxoG. Figure adapted from Jang et al. (56) with permission. Model of human UV-DDB-CUL4A-RBX bound to a 6–4 photoproduct in the context of a nucleosome, built from PDB codes: 4A0K and 6R8Y (52,123). Human UV-DDB bound to a THF lesion, PDB: 4E54 (124). Human OGG1 bound to 8-oxoG, PDB: 1EBM (125). Human APE1 bound to a 3′ deoxyribose phosphate moiety, PDB: 5DFF (126). DNA polymerase beta bound to gapped and nicked DNA, PDB: 1BPX (127)
Two independent studies have highlighted the role of the DDB2 subunit in chromatin decompaction (53,54) and may give insight into how DDB2 could help facilitate BER. In these studies, the authors used cell lines containing heterochromatic lac operator (lacO) regions and found that the lacO array size significantly increased when bound with lac repressor (lacR) tethered to DDB2 (DDB2-LacR) as compared to being bound with lacR alone. The increase in lacO array size is consistent with significant chromatin unfolding. They further showed a similar change in chromatin state triggered by DDB2 at sites of UV damage. Additionally, recruitment of DDB2 was independent of its E3 ligase activity and ATP-driven chromatin remodeling. Based on these data and the inefficiency of OGG1 to act on lesions embedded in nucleosomes (98), we postulate UV-DDB facilitates the recognition of 8-oxoG by BER proteins by first modifying the chromatin architecture at sites of damage, particularly in inaccessible heterochromatic regions, and then stimulates the down-stream processing of the lesion. Future structural and real-time imaging studies should focus on the mechanism of 8-oxoG recognition by DDB2 in reconstituted nucleosomes and in mammalian cells. Future experiments also need to address whether the DDB1-Cul4A E3 ligase is involved in 8-oxoG recognition.
CONCLUSION AND OUTLOOK
Here we have illustrated the cooperativity, and in the case of Sp and Gh lesions, antagonism, between NER and BER proteins during oxidative DNA damage repair, as well as instances where the oxidative lesions are repaired by NER proteins directly. The structure of the oxidative lesion influences the role of NER proteins in the oxidative damage repair response. A comprehensive summary of the involvement of UV-DDB, XPC, CSA, CSB and XPG in the removal of oxidative lesions, specifically 8-oxoG, is presented in Figure 6. Our lab was recently able to demonstrate early recruitment of UV-DDB to sites of 8-oxoG damage, preceding OGG1 recruitment (56), strengthening the argument for interaction between NER and BER pathways. There are 11 mammalian glycosylases which each recognize a range of lesions induced by oxidative damage (6). To this end, current work in our lab is focused on understanding the role of UV-DDB in stimulation of the other mammalian glycosylases beyond OGG1. We have preliminary data suggesting MUTYH activity is stimulated by UV-DDB (56). Additionally, we are examining the role of UV-DDB in the recruitment of downstream NER or BER proteins at oxidative lesion sites to elucidate a role for UV-DDB in facilitating the repair process. The evidence of interplay between the two repair pathways could suggest direct interactions between BER and NER proteins, both on and off DNA, which needs further study by yeast-2-hybrid screens, co-IP and/or biochemical/biophysical methods, such as single-molecule in vitro techniques with a purified protein system or cell extracts to examine protein-protein and protein–DNA interactions at a defined lesion site (99). Moreover, these techniques can be used to examine, in real-time, how proteins act in unison to faithfully process DNA lesions. With the advancement of single-particle tracking in living cells and in situ labeling of proteins (39–41), and introduction of site-specific lesions (56), we are currently developing methods to monitor the behavior of DNA repair proteins at damage sites in different chromatin states in real time.
Figure 6.

Oxidative DNA damage lesions repaired by BER, NER or both. NER proteins involved in the removal of these oxidative lesions are highlighted. Also depicted above is stimulation of BER proteins, especially glycosylases, by NER proteins. In this schematic, NER refers to both global and transcription-coupled repair. cdG: cyclo-deoxyguanosine; cdA: cyclo-deoxyadenosine; 8-oxoG: 8-oxoguanine; Oz: Oxazolone; Gh: Guanidinohydantoin; Sp: Spiroiminodihydantoin; Tg: Thymine glycol; 5mC: 5-methylcytosine; 5fC: 5-formylcytosine; 5caC: 5-carboxycytosine.
Most glycosylases cannot efficiently process lesions embedded in a nucleosome (42–46,87–90). Therefore, additional studies need to be conducted to further understand the molecular mechanisms of lesion recognition, specifically in the context of nucleosomes, chromatin remodeling, and glycosylase activity. Work from the Thoma laboratory, strengthened the argument for a role for UV-DDB in nucleosome remodeling by demonstrating the ability of UV-DDB to modify the register of the DNA on the nucleosome, increasing lesion accessibility (100). The direct role of DDB2 in chromatin decompaction also provides a mechanism of how DDB2 could help facilitate BER (53,54). Thus, DDB2 either acting alone or in conjunction with its associated E3 ligase activity as part of the DDB1-Cul4A-RBX complex to direct the BER of oxidative damage. Furthermore, it is possible UV-DDB is working cooperatively with other chromatin remodelers, such as PARP1 to increase lesion accessibility (101). As we and other groups have demonstrated, UV-DDB has a high affinity for undamaged DNA, and perhaps the binding of UV-DDB to undamaged DNA alters the chromatin structure and changes gene expression, leading to a modification in DNA repair. Moreover, it would be of note to determine if the rates of 8-oxoG repair are similar throughout the genome. Several groups are currently examining this question and it would appear that the human genome contains hotspots for 8-oxoG formation and possibly repair (102–109). Studying oxidative DNA damage repair in vivo has been challenging due to the lack of tools capable of introducing oxidative lesions at defined regions in the nucleus. Several groups including our own have developed tools to overcome this challenge (38,110,111), which can be used to understand differences in repair kinetics in euchromatin and heterochromatin. Perhaps the chromatin state may help dictate whether NER or BER proteins are necessary and sufficient to remove 8-oxoG (112). One hypothesis that needs to be tested is if 8-oxoG occurring in heterochromatin is refractory to BER and needs the additional factors of UV-DDB and XPC to help facilitate chromatin opening. Harnessing our new innovative FAP-dye plus light technology, through direct fusion of FAP to other DNA binding proteins will allow new insights into the repair of 8-oxoG throughout the genome as we attempt to watch DNA repair proteins at high spatial and temporal resolution in living cells. Gaining an understanding of the dynamic process of oxidative DNA damage repair in the context of different chromatin states, should help aid in the development of improved therapies for diseases associated with defects in DNA repair proteins.
ACKNOWLEDGEMENTS
We would like to thank Wim Vermeulen for useful discussions. All data from our laboratory cited in this article is available upon request
Contributor Information
Namrata Kumar, Molecular Genetics and Developmental Biology Graduate Program, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15213 USA; UPMC Hillman Cancer Center, University of Pittsburgh, PA 15213, USA.
Sripriya Raja, UPMC Hillman Cancer Center, University of Pittsburgh, PA 15213, USA; Molecular Pharmacology Graduate Program, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15213 USA.
Bennett Van Houten, Molecular Genetics and Developmental Biology Graduate Program, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15213 USA; UPMC Hillman Cancer Center, University of Pittsburgh, PA 15213, USA; Molecular Pharmacology Graduate Program, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15213 USA; Department of Pharmacology and Chemical Biology, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15213, USA.
FUNDING
NIH [R01ES019566, R35ES031638 to B.V.H.]. Funding for open access charge: NIEHS [R35ES031638].
Conflict of interest statement. None declared.
REFERENCES
- 1. Markkanen E. Not breathing is not an option: how to deal with oxidative DNA damage. DNA Repair (Amst.). 2017; 59:82–105. [DOI] [PubMed] [Google Scholar]
- 2. Reuter S., Gupta S.C., Chaturvedi M.M., Aggarwal B.B.. Oxidative stress, inflammation, and cancer: how are they linked. Free Radic. Biol. Med. 2010; 49:1603–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tubbs A., Nussenzweig A.. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017; 168:644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Poetsch A.R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 2020; 18:207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sedelnikova O.A., Redon C.E., Dickey J.S., Nakamura A.J., Georgakilas A.G., Bonnera W.M.. Role of oxidatively induced DNA lesions in human pathogenesis. Mutat. Res. 2010; 704:152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Whitaker A.M., Schaich M.A., Smith M.R., Flynn T.S., Freudenthal B.D.. Base excision repair of oxidative DNA damage: from mechanism to disease. Front. Biosci. (Landmark Ed.). 2017; 22:1493–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hill J.W., Hazra T.K., Izumi T., Mitra S.. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001; 29:430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kumar N., Moreno N.C., Feltes B.C., Menck C.F.M., Van Houten B.. Cooperation and interplay between base and nucleotide excision repair pathways: From DNA lesions to proteins. Genet. Mol. Biol. 2020; 43:e20190104–e20190117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Limpose K.L., Corbett A.H., Doetsch P.W.. BERing the burden of damage: pathway crosstalk and posttranslational modification of base excision repair proteins regulate DNA damage management. DNA Repair (Amst.). 2017; 56:51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Melis J.P.M., van Steeg H., Luijten M.. Oxidative DNA damage and nucleotide excision repair. Antioxid. Redox Signal. 2013; 18:2409–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shafirovich V., Geacintov N.E.. Removal of oxidatively generated DNA damage by overlapping repair pathways. Free Radic. Biol. Med. 2017; 107:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Scharer O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013; 5:a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mu H., Geacintov N.E., Broyde S., Yeo J.-.E., Schärer O.D.. Molecular basis for damage recognition and verification by XPC-RAD23B and TFIIH in nucleotide excision repair. DNA Repair (Amst.). 2018; 71:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marteijn J.A., Lans H., Vermeulen W., Hoeijmakers J.H.J.. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014; 15:465–481. [DOI] [PubMed] [Google Scholar]
- 15. Fousteri M., Mullenders L.H.. Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res. 2008; 18:73–84. [DOI] [PubMed] [Google Scholar]
- 16. Rapin I., Lindenbaum Y., Dickson D.W., Kraemer K.H., Robbins J.H.. Cockayne syndrome and xeroderma pigmentosum. Neurology. 2000; 55:1442–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Robbins J.H., Brumback R.A., Mendiones M., Barrett S.F., Carl J.R., Cho S., Denckla M.B., Ganges M.B., Gerber L.H., Guthrie R.A. et al.. Neurological disease in xeroderma pigmentosum. Documentation of a late onset type of the juvenile onset form. Brain. 1991; 114:1335–1361. [DOI] [PubMed] [Google Scholar]
- 18. Keck K. [Formation of cyclonucleotides during irradiation of aqueous solutions of purine nucleotides]. Z. Naturforsch. B. 1968; 23:1034–1043. [PubMed] [Google Scholar]
- 19. Raleigh J.A., Kremers W., Whitehouse R.. Radiation chemistry of nucleotides: 8,5′-cyclonucleotide formation and phosphate release initiated by hydroxyl radical attack on adenosine monophosphates. Radiat. Res. 1976; 65:414–422. [PubMed] [Google Scholar]
- 20. Dizdaroglu M., Dirksen M.L., Jiang H.X., Robbins J.H.. Ionizing-radiation-induced damage in the DNA of cultured human cells. Identification of 8,5-cyclo-2-deoxyguanosine. Biochem. J. 1987; 241:929–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Satoh M.S., Jones C.J., Wood R.D., Lindahl T.. DNA excision-repair defect of xeroderma pigmentosum prevents removal of a class of oxygen free radical-induced base lesions. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:6335–6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuraoka I., Bender C., Romieu A., Cadet J., Wood R.D., Lindahl T.. Removal of oxygen free-radical-induced 5′,8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:3832–3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brooks P.J., Wise D.S., Berry D.A., Kosmoski J.V., Smerdon M.J., Somers R.L., Mackie H., Spoonde A.Y., Ackerman E.J., Coleman K. et al.. The oxidative DNA lesion 8,5′-(S)-cyclo-2′-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J. Biol. Chem. 2000; 275:22355–22362. [DOI] [PubMed] [Google Scholar]
- 24. Ramkumar H.L., Brooks B.P., Cao X., Tamura D., Digiovanna J.J., Kraemer K.H., Chan C.-.C.. Ophthalmic manifestations and histopathology of xeroderma pigmentosum: two clinicopathological cases and a review of the literature. Surv. Ophthalmol. 2011; 56:348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mori T., Nakane H., Iwamoto T., Krokidis M.G., Chatgilialoglu C., Tanaka K., Kaidoh T., Hasegawa M., Sugiurag S.. High levels of oxidatively generated DNA damage 8,5′-cyclo-2′-deoxyadenosine accumulate in the brain tissues of xeroderma pigmentosum group A gene-knockout mice. DNA Repair (Amst.). 2019; 80:52–58. [DOI] [PubMed] [Google Scholar]
- 26. Kirkali G., de Souza-Pinto N.C., Jaruga P., Bohr V.A., Dizdaroglu M.. Accumulation of (5′S)-8,5′-cyclo-2′-deoxyadenosine in organs of Cockayne syndrome complementation group B gene knockout mice. DNA Repair (Amst.). 2009; 8:274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. de Vries A., van Oostrom C.T., Hofhuis F.M., Dortant P.M., Berg R.J., de Gruijl F.R., Wester P.W., van Kreijl C.F., Capel P.J., van Steeg H. et al.. Increased susceptibility to ultraviolet-B and carcinogens of mice lacking the DNA excision repair gene XPA. Nature. 1995; 377:169–173. [DOI] [PubMed] [Google Scholar]
- 28. Nakane H., Takeuchi S., Yuba S., Saijo M., Nakatsu Y., Murai H., Nakatsuru Y., Ishikawa T., Hirota S., Kitamura Y. et al.. High incidence of ultraviolet-B-or chemical-carcinogen-induced skin tumours in mice lacking the xeroderma pigmentosum group A gene. Nature. 1995; 377:165–168. [DOI] [PubMed] [Google Scholar]
- 29. D’Errico M., Parlanti E., Teson M., Bernardes de Jesus B.M., Degan P., Calcagnile A., Jaruga P., Bjørås M., Crescenzi M., Pedrini A.M. et al.. New functions of XPC in the protection of human skin cells from oxidative damage. EMBO J. 2006; 25:4305–4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. D’Errico M., Parlanti E., Teson M., Degan P., Lemma T., Calcagnile A., Iavarone I., Jaruga P., Ropolo M., Pedrini A.M. et al.. The role of CSA in the response to oxidative DNA damage in human cells. Oncogene. 2007; 26:4336–4343. [DOI] [PubMed] [Google Scholar]
- 31. Pande P., Das R.S., Sheppar C., Kow Y.W., Basua A.K.. Repair efficiency of (5′S)-8,5′-cyclo-2′-deoxyguanosine and (5′S)-8,5′-cyclo-2′-deoxyadenosine depends on the complementary base. DNA Repair (Amst.). 2012; 11:926–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jaruga P., Xiao Y., Vartanian V., Stephen Lloyd R., Dizdaroglu M.. Evidence for the involvement of DNA repair enzyme NEIL1 in nucleotide excision repair of (5′R)- and (5′S)-8,5′-cyclo-2′-deoxyadenosines. Biochemistry. 2010; 49:1053–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beckwitt E.C., Kong M., Van Houten B.. Studying protein-DNA interactions using atomic force microscopy. Semin. Cell Dev. Biol. 2018; 73:220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kong M., Beckwitt E.C., Springall L., Kad N.M., Houten B.V.. Single-molecule methods for nucleotide excision repair: building a system to watch repair in real time. Methods Enzymol. 2017; 592:213–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kropachev K., Ding S., Terzidis M.A., Masi A., Liu Z., Cai Y., Kolbanovskiy M., Chatgilialoglu C., Broyde S., Geacintov N.E. et al.. Structural basis for the recognition of diastereomeric 5′,8-cyclo-2′-deoxypurine lesions by the human nucleotide excision repair system. Nucleic Acids Res. 2014; 42:5020–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wellinger R.E., Thoma F.. Nucleosome structure and positioning modulate nucleotide excision repair in the non-transcribed strand of an active gene. EMBO J. 1997; 16:5046–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Z., Wu X., Friedberg E.C.. Nucleotide-excision repair of DNA in cell-free extracts of the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:4907–4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Menoni H., Hoeijmakers J.H., Vermeulen W.. Nucleotide excision repair-initiating proteins bind to oxidative DNA lesions in vivo. J. Cell Biol. 2012; 199:1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mazza D., Abernathy A., Golob N., Morisaki T., McNally J.G.. A benchmark for chromatin binding measurements in live cells. Nucleic Acids Res. 2012; 40:e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matter B., Malejka-Giganti D., Saari Csallany A., Tretyakova N.. Quantitative analysis of the oxidative DNA lesion, 2,2-diamino-4-(2-deoxy-beta-D-erythro-pentofuranosyl)amino)-5(2H)-oxazolone (oxazolone), in vitro and in vivo by isotope dilution-capillary HPLC-ESI-MS/MS. Nucleic Acids Res. 2006; 34:5449–5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lord S.J., Lee H.L., Moerner W.E.. Single-molecule spectroscopy and imaging of biomolecules in living cells. Anal. Chem. 2010; 82:2192–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hinz J.M., Rodriguez Y., Smerdon M.J.. Rotational dynamics of DNA on the nucleosome surface markedly impact accessibility to a DNA repair enzyme. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:4646–4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hinz J.M., Mao P., McNeill D.R., Wilson D.M. III. Reduced nuclease activity of apurinic/apyrimidinic endonuclease (APE1) variants on nucleosomes: identification of access residues. J. Biol. Chem. 2015; 290:21067–21075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hinz J.M. Impact of abasic site orientation within nucleosomes on human APE1 endonuclease activity. Mutat. Res. 2014; 766–767:19–24. [DOI] [PubMed] [Google Scholar]
- 45. Bilotti K., Tarantino M.E., Delaney S.. Human axoguanine glycosylase 1 removes solution accessible 8-oxo-7,8-dihydroguanine lesions from globally substituted nucleosomes except in the dyad region. Biochemistry. 2018; 57:1436–1439. [DOI] [PubMed] [Google Scholar]
- 46. Beard B.C., Wilson S.H., Smerdon M.J.. Suppressed catalytic activity of base excision repair enzymes on rotationally positioned uracil in nucleosomes. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:7465–7470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shafirovich V., Kolbanovskiy M., Kropachev K., Liu Z., Cai Y., Terzidis M.A., Masi A., Chatgilialoglu C., Amin S., Dadali A. et al.. Nucleotide excision repair and impact of site-specific 5′,8-cyclopurine and bulky DNA lesions on the physical properties of nucleosomes. Biochemistry. 2019; 58:561–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Osakabe A., Tachiwana H., Kagawa W., Horikoshi N., Matsumoto S., Hasegawa M., Matsumoto N., Toga T., Yamamoto J., Hanaoka F. et al.. Structural basis of pyrimidine-pyrimidone (6-4) photoproduct recognition by UV-DDB in the nucleosome. Sci. Rep. 2015; 5:16330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Horikoshi N., Tachiwana H., Kagawa W., Osakabe A., Matsumoto S., Iwai S., Sugasawa K., Kurumizaka H.. Crystal structure of the nucleosome containing ultraviolet light-induced cyclobutane pyrimidine dimer. Biochem. Biophys. Res. Commun. 2016; 471:117–122. [DOI] [PubMed] [Google Scholar]
- 50. Fitch M.E., Nakajima S., Yasui A., Ford J.M.. In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product. J. Biol. Chem. 2003; 278:46906–46910. [DOI] [PubMed] [Google Scholar]
- 51. Yasuda T., Sugasawa K., Shimizu Y., Iwai S., Shiomi T., Hanaoka F.. Nucleosomal structure of undamaged DNA regions suppresses the non-specific DNA binding of the XPC complex. DNA Repair (Amst.). 2005; 4:389–395. [DOI] [PubMed] [Google Scholar]
- 52. Matsumoto S., Cavadini S., Bunker R.D., Grand R.S., Potenza A., Rabl J., Yamamoto J., Schenk A.D., Schübeler D., Iwai S. et al.. DNA damage detection in nucleosomes involves DNA register shifting. Nature. 2019; 571:79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Adam S., Dabin J., Chevallier O., Leroy O., Baldeyron C., Corpet A., Lomonte P., Renaud O., Almouzni G., Polo S.E.. Real-time tracking of parental histones reveals their contribution to chromatin integrity following DNA damage. Mol. Cell. 2016; 64:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Luijsterburg M.S., Lindh M., Acs K., Vrouwe M.G., Pines A., van Attikum H., Mullenders L.H., Dantuma N.P.. DDB2 promotes chromatin decondensation at UV-induced DNA damage. J. Cell Biol. 2012; 197:267–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Beecher M., Kumar N., Jang S., Rapić-Otrin V., Houten B.V.. Expanding molecular roles of UV-DDB: shining light on genome stability and cancer. DNA Repair. 2020; 94:102860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jang S., Kumar N., Beckwitt E.C., Kong M., Fouquerel E., Rapic-Otrin V., Prasad R., Watkins S.C., Khuu C., Majumdar C. et al.. Damage sensor role of UV-DDB during base excision repair. Nat. Struct. Mol. Biol. 2019; 26:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Moore L.D., Le T., Fan G.. DNA methylation and its basic function. Neuropsychopharmacology. 2013; 38:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shimizu Y., Iwai S., Hanaoka F., Sugasawa K.. Xeroderma pigmentosum group C protein interacts physically and functionally with thymine DNA glycosylase. EMBO J. 2003; 22:164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schomacher L., Han D., Musheev M.U., Arab K., Kienhöfer S., von Seggern A., Niehrs C.. Neil DNA glycosylases promote substrate turnover by Tdg during DNA demethylation. Nat. Struct. Mol. Biol. 2016; 23:116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ho J.J., Cattoglio C., McSwiggen D.T., Tjian R., Fong Y.W.. Regulation of DNA demethylation by the XPC DNA repair complex in somatic and pluripotent stem cells. Genes Dev. 2017; 31:830–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yasuda G., Nishi R., Watanabe E., Mori T., Iwai S., Orioli D., Stefanini M., Hanaoka F., Sugasawa K.. In vivo destabilization and functional defects of the xeroderma pigmentosum C protein caused by a pathogenic missense mutation. Mol. Cell. Biol. 2007; 27:6606–6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. David S.S., O'Shea V.L., Kundu S.. Base-excision repair of oxidative DNA damage. Nature. 2007; 447:941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993; 362:709–715. [DOI] [PubMed] [Google Scholar]
- 64. Nakabeppu Y. Cellular levels of 8-oxoguanine in either DNA or the nucleotide pool play pivotal roles in carcinogenesis and survival of cancer cells. Int. J. Mol. Sci. 2014; 15:12543–12557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cooke M.S., Loft S., Olinski R., Evans M.D., Bialkowski K., Richard Wagner J., Dedon P.C., Møller P., Greenberg M.M., Cadet J.. Recommendations for standardized description of and nomenclature concerning oxidatively damaged nucleobases in DNA. Chem. Res. Toxicol. 2010; 23:705–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kow Y.W., Wallace S.S., Van Houten B.. UvrABC nuclease complex repairs thymine glycol, an oxidative DNA base damage. Mutat. Res. 1990; 235:147–156. [DOI] [PubMed] [Google Scholar]
- 67. Reardon J.T., Bessho T., Kung H.C., Bolton P.H., Sancar A.. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:9463–9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Klungland A., Höss M., Gunz D., Constantinou A., Clarkson S.G., Doetsch P.W., Bolton P.H., Wood R.D., Lindahl T.. Base excision repair of oxidative DNA damage activated by XPG protein. Mol. Cell. 1999; 3:33–42. [DOI] [PubMed] [Google Scholar]
- 69. Wang H.T., Choi B., Tang M.S.. Melanocytes are deficient in repair of oxidative DNA damage and UV-induced photoproducts. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:12180–12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kraemer K.H., Lee M.M., Andrews A.D., Lambert W.C.. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch. Dermatol. 1994; 130:1018–1021. [PubMed] [Google Scholar]
- 71. Cadet J., Sage E., Douki T.. Ultraviolet radiation-mediated damage to cellular DNA. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2005; 571:3–17. [DOI] [PubMed] [Google Scholar]
- 72. Kassam S.N., Rainbow A.J.. Deficient base excision repair of oxidative DNA damage induced by methylene blue plus visible light in xeroderma pigmentosum group C fibroblasts. Biochem. Biophys. Res. Commun. 2007; 359:1004–1009. [DOI] [PubMed] [Google Scholar]
- 73. Parlanti E., D’Errico M., Degan P., Calcagnile A., Zijno A., van der Pluijm I., van der Horst G.T.J., Biard D.S.F., Dogliotti E.. The cross talk between pathways in the repair of 8-oxo-7,8-dihydroguanine in mouse and human cells. Free Radic. Biol. Med. 2012; 53:2171–2177. [DOI] [PubMed] [Google Scholar]
- 74. Sassa A., Tada H., Takeishi A., Harada K., Suzuki M., Tsuda M., Sasanuma H., Takeda S., Sugasawa K., Yasui M. et al.. Processing of a single ribonucleotide embedded into DNA by human nucleotide excision repair and DNA polymerase η. Sci. Rep. 2019; 9:13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Guo J., Hanawalt P.C., Spivak G.. Comet-FISH with strand-specific probes reveals transcription-coupled repair of 8-oxoGuanine in human cells. Nucleic Acids Res. 2013; 41:7700–7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sassa A., Kamoshita N., Kanemaru Y., Honma M., Yasui M.. Xeroderma pigmentosum group A suppresses mutagenesis caused by clustered oxidative dna adducts in the human genome. PLoS One. 2015; 10:e0142218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Menoni H., Wienholz F., Theil A.F., Janssens R.C., Lans H., Campalans A., Radicella J.P., Marteijn J.A., Vermeulen W.. The transcription-coupled DNA repair-initiating protein CSB promotes XRCC1 recruitment to oxidative DNA damage. Nucleic Acids Res. 2018; 46:7747–7756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wong H.K., Muftuoglu M., Beck G., Imam S.Z., Bohr V.A., Wilson D.M. 3rd. Cockayne syndrome B protein stimulates apurinic endonuclease 1 activity and protects against agents that introduce base excision repair intermediates. Nucleic Acids Res. 2007; 35:4103–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kino K., Takao M., Miyazawa H., Hanaoka F.. A DNA oligomer containing 2,2,4-triamino-5(2H)-oxazolone is incised by human NEIL1 and NTH1. Mutat. Res. 2012; 734:73–77. [DOI] [PubMed] [Google Scholar]
- 80. Kino K., Sugasawa K., Miyazawa H., Hanaoka F.. 2,2,4-Triamino-5(2H)-oxazolone is a weak substrate for nucleotide excision repair. J. Pharm. Negative Results. 2016; 7:42–45. [Google Scholar]
- 81. McKibbin P.L., Fleming A.M., Towheed M.A., Houten B.V., Burrows C.J., David S.S.. Repair of hydantoin lesions and their amine adducts in DNA by base and nucleotide excision repair. J. Am. Chem. Soc. 2013; 135:13851–13861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shafirovich V., Kropachev K., Anderson T., Liu Z., Kolbanovskiy M., Martin B.D., Sugden K., Shim Y., Chen X., Min J.-.H. et al.. Base and nucleotide excision repair of oxidatively generated guanine lesions in DNA. J. Biol. Chem. 2016; 291:5309–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shafirovich V., Kropachev K., Kolbanovskiy M., Geacintov N.E.. Excision of oxidatively generated guanine lesions by competing base and nucleotide excision repair mechanisms in human cells. Chem. Res. Toxicol. 2019; 32:753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kolbanovskiy M., Shim Y., Min J.-.H., Geacintov N.E., Shafirovich V.. Inhibition of Excision of Oxidatively Generated Hydantoin DNA Lesions by NEIL1 by the Competitive Binding of the Nucleotide Excision Repair Factor XPC-RAD23B. Biochemistry. 2020; 59:1728–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Liu M., Bandaru V., Bond J.P., Jaruga P., Zhao X., Christov P.P., Burrows C.J., Rizzo C.J., Dizdaroglu M., Wallace S.S.. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:4925–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhou J., Fleming A.M., Averill A.M., Burrows C.J., Wallace S.S.. The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res. 2015; 43:4039–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rodriguez Y., Hinz J.M., Smerdon M.J.. Accessing DNA damage in chromatin: preparing the chromatin landscape for base excision repair. DNA Repair (Amst.). 2015; 32:113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Odell I.D., Wallace S.S., Pederson D.S.. Rules of engagement for base excision repair in chromatin. J. Cell. Physiol. 2013; 228:258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Menoni H., Gasparutto D., Hamiche A., Cadet J., Dimitrov S., Bouvet P., Angelov D.. ATP-dependent chromatin remodeling is required for base excision repair in conventional but not in variant H2A.Bbd nucleosomes. Mol. Cell. Biol. 2007; 27:5949–5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Maher R.L, Marsden C.G, Averill A.M., Wallace S.S., Sweasy J.B., Pederson D.S.. Human cells contain a factor that facilitates the DNA glycosylase-mediated excision of oxidized bases from occluded sites in nucleosomes. DNA Repair (Amst.). 2017; 57:91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Prasad A., Wallace S.S., Pederson D.S.. Initiation of base excision repair of oxidative lesions in nucleosomes by the human, bifunctional DNA glycosylase NTH1. Mol. Cell. Biol. 2007; 27:8442–8453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Tarantino M.E., Dowb B.J., Drohatbc A.C., Delaneyd S.. Nucleosomes and the three glycosylases: High, medium, and low levels of excision by the uracil DNA glycosylase superfamily. DNA Repair (Amst.). 2018; 72:56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Maher R.L., Prasad A., Rizvanova O., Wallace S.S., Pederson D.S.. Contribution of DNA unwrapping from histone octamers to the repair of oxidatively damaged DNA in nucleosomes. DNA Repair (Amst.). 2013; 12:964–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Odell I.D., Newick K., Heintz N.H., Wallace S.S., Pederson D.S.. Non-specific DNA binding interferes with the efficient excision of oxidative lesions from chromatin by the human DNA glycosylase, NEIL1. DNA Repair (Amst.). 2010; 9:134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sugasawa K. UV-DDB: a molecular machine linking DNA repair with ubiquitination. DNA Repair (Amst.). 2009; 8:969–972. [DOI] [PubMed] [Google Scholar]
- 96. Fujiwara Y., Masutani C., Mizukoshi T., Kondo J., Hanaoka F., Iwai S.. Characterization of DNA recognition by the human UV-damaged DNA-binding protein. J. Biol. Chem. 1999; 274:20027–20033. [DOI] [PubMed] [Google Scholar]
- 97. Wittschieben B., Iwai S., Wood R.D.. DDB1-DDB2 (xeroderma pigmentosum group E) protein complex recognizes a cyclobutane pyrimidine dimer, mismatches, apurinic/apyrimidinic sites, and compound lesions in DNA. J. Biol. Chem. 2005; 280:39982–39989. [DOI] [PubMed] [Google Scholar]
- 98. Olmon E.D., Delaney S.. Differential ability of five DNA glycosylases to recognize and repair damage on nucleosomal DNA. ACS Chem. Biol. 2017; 12:692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hughes C.D., Simons M., Mackenzie C.E., Houten B.V., Kad N.M.. Single molecule techniques in DNA repair: a primer. DNA Repair (Amst.). 2014; 20:2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Matsumoto S., Cavadini S., Bunker R.D., Grand R.S., Potenza A., Rabl J., Yamamoto J., Schenk A.D., Schübeler D., Iwai S. et al.. DNA damage detection in nucleosomes involves DNA register shifting. Nature. 2019; 571:79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Pines A., Mischa, Vrouwe G., Jurgen, Marteijn A., Typas D., Luijsterburg M.S., Cansoy M., Hensbergen P., Deelder A., de Groot A., Matsumoto S. et al.. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J. Cell Biol. 2012; 199:235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Akatsuka S., Toyokuni S.. Genome-wide assessment of oxidatively generated DNA damage. Free Radic. Res. 2012; 46:523–530. [DOI] [PubMed] [Google Scholar]
- 103. Amente S., Palo G.D.i., Scala G., Castrignanò T., Gorini F., Cocozza S., Moresano A., Pucci P., Ma B., Stepanov I. et al.. Genome-wide mapping of 8-oxo-7,8-dihydro-2′-deoxyguanosine reveals accumulation of oxidatively-generated damage at DNA replication origins within transcribed long genes of mammalian cells. Nucleic Acids Res. 2019; 47:221–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ding Y., Fleming A.M., Burrows C.J.. Sequencing the mouse genome for the oxidatively modified base 8-oxo-7,8-dihydroguanine by OG-Seq. J. Am. Chem. Soc. 2017; 139:2569–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Fleming A.M., Ding Y., Burrows C.J.. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:2604–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Fleming A.M., Ding Y., Burrows C.J.. Sequencing DNA for the oxidatively modified base 8-oxo-7,8-dihydroguanine. Methods Enzymol. 2017; 591:187–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ohno M., Miura T., Furuichi M., Tominaga Y., Tsuchimoto D., Sakumi K., Nakabeppu Y.. A genome-wide distribution of 8-oxoguanine correlates with the preferred regions for recombination and single nucleotide polymorphism in the human genome. Genome Res. 2006; 16:567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Poetsch A.R., Boulton S.J., Luscombe N.M.. Genomic landscape of oxidative DNA damage and repair reveals regioselective protection from mutagenesis. Genome Biol. 2018; 19:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Yoshihara M., Jiang L.i., Akatsuka S., Suyama M., Toyokuni S.. Genome-wide profiling of 8-oxoguanine reveals its association with spatial positioning in nucleus. DNA Res. 2014; 21:603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. He J., Wang Y.i., Missinato M.A., Onuoha E., Perkins L.A., Watkins S.C., St Croix C.M., Tsang M., Bruchez M.P.. A genetically targetable near-infrared photosensitizer. Nat. Methods. 2016; 13:263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Vegh R.B., Solntsev K.M., Kuimova M.K., Cho S., Liang Y., Loo B.L.W., Tolbert L.M., Bommarius A.S.. Reactive oxygen species in photochemistry of the red fluorescent protein “Killer Red”. Chem. Commun. (Camb.). 2011; 47:4887–4889. [DOI] [PubMed] [Google Scholar]
- 112. Lan L., Nakajima S., Wei L., Sun L., Hsieh C.-.L., Sobol R.W., Bruchez M., Houten B.V., Yasui A., Levine A.S.. Novel method for site-specific induction of oxidative DNA damage reveals differences in recruitment of repair proteins to heterochromatin and euchromatin. Nucleic Acids Res. 2014; 42:2330–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Collins A.R. The comet assay for DNA damage and repair: principles, applications, and limitations. Mol. Biotechnol. 2004; 26:249–261. [DOI] [PubMed] [Google Scholar]
- 114. Cui L., Ye W., Prestwich E.G., Wishnok J.S., Taghizadeh K., Dedon P.C., Tannenbaum S.R.. Comparative analysis of four oxidized guanine lesions from reactions of DNA with peroxynitrite, singlet oxygen, and γ-radiation. Chem. Res. Toxicol. 2013; 26:195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Hamilton M.L., Guo Z., Fuller C.D., Van Remmen H., Ward W.F., Austad S.N., Troyer D.A., Thompson I., Richardson A.. A reliable assessment of 8-oxo-2-deoxyguanosine levels in nuclear and mitochondrial DNA using the sodium iodide method to isolate DNA. Nucleic Acids Res. 2001; 29:2117–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Mangerich A., Knutson C.G., Parry N.M., Muthupalani S., Ye W., Prestwich E., Cui L., McFaline J.L., Mobley M., Ge Z.. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E1820–E1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Randerath K., Zhou G.D., Somers R.L., Robbins J.H., Brooks P.J.. A 32P-postlabeling assay for the oxidative DNA lesion 8,5′-cyclo-2′-deoxyadenosine in mammalian tissues: evidence that four type II I-compounds are dinucleotides containing the lesion in the 3′ nucleotide. J. Biol. Chem. 2001; 276:36051–36057. [DOI] [PubMed] [Google Scholar]
- 118. Yu M., Hon G.C., Szulwach K.E., Song C-.X., Zhang L., Kim A., Li X., Dai Q., Shen Y., Park B. et al.. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012; 149:1368–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ito S., Shen L.i., Dai Q., Wu S.C., Collins L.B., Swenberg J.A., He C., Zhang Y.i.. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011; 333:1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Le X.C., Xing J.Z., Lee J., Leadon S.A., Weinfeld M.. Inducible repair of thymine glycol detected by an ultrasensitive assay for DNA damage. Science. 1998; 280:1066–1069. [DOI] [PubMed] [Google Scholar]
- 121. Ravanat J.L., Douki T., Duez P., Gremaud E., Herbert K., Hofer T., Lasserre L., Saint-Pierre C., Favier A., Cadet J.. Cellular background level of 8-oxo-7,8-dihydro-2′-deoxyguanosine: an isotope based method to evaluate artefactual oxidation of DNA during its extraction and subsequent work-up. Carcinogenesis. 2002; 23:1911–1918. [DOI] [PubMed] [Google Scholar]
- 122. Maiti A., Noon M.S., MacKerell A.D. Jr., Pozharski E., Drohat A.C.. Lesion processing by a repair enzyme is severely curtailed by residues needed to prevent aberrant activity on undamaged DNA. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:8091–8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Fischer E.S., Scrima A., Böhm K., Matsumoto S., Lingaraju .M., Faty M., Yasuda T., Cavadini S., Wakasugi M., Hanaoka F. et al.. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell. 2011; 147:1024–1039. [DOI] [PubMed] [Google Scholar]
- 124. Yeh J.I., Levine A.S., Du S.. Damaged DNA induced UV-damaged DNA-binding protein (UV-DDB) dimerization and its roles in chromatinized DNA repair. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E2737–E2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Bruner S.D., Norman D.P., Verdine G.L.. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature. 2000; 403:859–866. [DOI] [PubMed] [Google Scholar]
- 126. Freudenthal B.D., Beard W.A., Cuneo M.J., Dyrkheeva N.S., Wilson S.H.. Capturing snapshots of APE1 processing DNA damage. Nat. Struct. Mol. Biol. 2015; 22:924–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sawaya M.R., Prasad R., Wilson S.H., Kraut J., Pelletier H.. Crystal structures of human DNA polymerase beta complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry. 1997; 36:11205–11215. [DOI] [PubMed] [Google Scholar]