

Abstract
Developing new technologies to study metabolism is increasingly important as metabolic disease prevalence increases. Mitochondria control cellular metabolism and dynamic changes in mitochondrial function are associated with metabolic abnormalities in cardiovascular disease, cancer, and obesity. However, a lack of precise and reversible methods to control mitochondrial function has prevented moving from association to causation. Recent advances in optogenetics have addressed this challenge, and mitochondrial function can now be precisely controlled in vivo using light. A class of genetically-encoded, light-activated membrane channels and pumps has addressed mechanistic questions that promise to provide new insights into how cellular metabolism downstream of mitochondrial function contributes to disease. Here, we highlight emerging reagents – mitochondria-targeted light-activated cation channels or proton pumps – to decrease or increase mitochondrial activity upon light exposure, a technique we refer to as mitochondrial light switches, or mtSWITCH. The mtSWITCH technique is broadly applicable, as energy availability and metabolic signaling are conserved aspects of cellular function and health. Here, we outline the use of these tools in diverse cellular models of disease. We review the molecular details of each optogenetic tool, summarize the results obtained with each, and outline best practices for using optogenetic approaches to control mitochondrial function and downstream metabolism.
Keywords: hypoxia, AMPK, calcium signaling, Parkinson’s, bioenergetics, apoptosis, mitophagy, diabetes
Graphical Abstract
Mitochondrial light switches (mtSWITCH) are novel optogenetic tools to precisely, reversibly and instantaneously decrease or increase mitochondrial function using light. Light-sensitive membrane channels or proton pumps confer this control by altering the electrochemical gradient that drives mitochondrial function, the protonmotive force. This advance allows unprecedented, fundamental control over metabolism and physiology, and will establish causal mechanisms in models of metabolic diseases by providing experimental isolation of mitochondrial function.
INTRODUCTION
An increasingly aged population and the prevalence of metabolic diseases represents a global health care crisis [1–3]. Basic science research focused on understanding metabolism is therefore critical to identify novel molecular targets and mechanisms to develop new therapies. At the core of metabolism are mitochondria, the organelles responsible for making energy available to cells. Mitochondria are metabolically flexible and can adapt to changing cellular conditions and energy needs. Mitochondria affect many cellular decisions, ranging from adaptation and survival to death. Much is still unknown about how mitochondrial function regulates diverse and complicated signaling in vivo.
Mitochondria make ATP [4], regulate calcium (Ca++) signaling [5], coordinate redox signaling and reactive oxygen species (ROS) production [6], modulate cell death through apoptosis [7–9], and integrate metabolic signaling across cells and tissues [10]. These factors underlie cellular signaling programs such as unfolded protein responses [11], autophagy [12], and stress adaptation [9]. In this way, mitochondria are essential cellular signaling hubs that impact all areas of cellular life. Each of these processes and pathways is regulated by the driving force of mitochondrial function, the protonmotive force (PMF).
Mitochondrial protonmotive force (PMF)
The mitochondrial PMF is an electrochemical gradient across the mitochondrial inner membrane (IM) made up of a charge separation, or membrane potential, denoted ΔΨm, and a chemical pH separation denoted ΔpH. The PMF is like a battery, in that potential energy is stored for eventual release to do work. The PMF is created by the electron transport chain (ETC) in the IM when electrons from metabolic substrates from food are passed along the chain and protons are pumped from the mitochondrial matrix to the intermembrane space (IMS) as oxygen is consumed (Figure 1). The battery power of the PMF can then drive ATP synthase to phosphorylate ADP to ATP, the cellular energy source. The PMF also drives other mitochondrial functions such as metabolite and ion transport. In particular, the abundant intracellular cation potassium (K+) is subject to the PMF, as well as Ca++ [5, 13]. These ions can flux across the IM in regulated ways to impact cellular metabolism and physiology.
Figure 1. Endogenous protonmotive force (PMF) regulation.
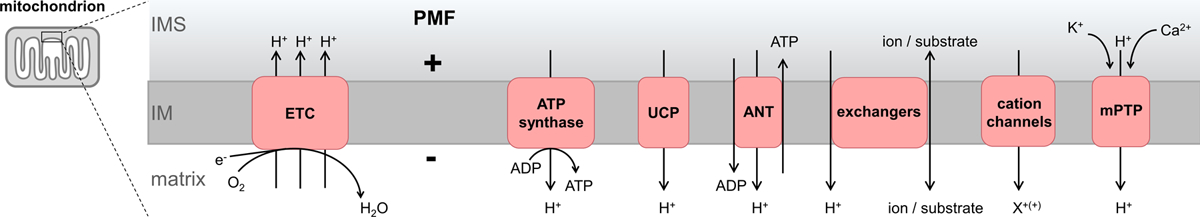
The mitochondrial electron transport chain (ETC) pumps protons from the mitochondrial matrix to the intermembrane space (IMS) to establish the PMF. The PMF drives ATP synthase to phosphorylate ADP to ATP. Other proteins in the inner membrane (IM) regulate mitochondrial function through proton transport which affects the PMF. Uncoupling proteins (UCP) are regulated to dissipate the PMF. Similarly, adenine nucleotide transporters (ANT) can also dissipate the PMF in a regulated manner. Several metabolite and ion exchangers also use the PMF to drive their functions. Cation channels in mitochondria can allow cation (X+(+)) accumulation into the matrix. The mitochondrial permeability transition pore (mPTP) is a large conductance channel that can cause total PMF collapse in different physiologic and pathologic conditions.
The PMF is central to mitochondrial function and is tightly regulated under different metabolic conditions. For example, substrate and oxygen availability [10, 14] or energetic demand from exercise [15–17] can alter the PMF. Metabolic conditions can vary between tissues, and accordingly there are tissue differences in mitochondrial PMF [18, 19]. The PMF can also be differentially regulated within individual mitochondria, with separate matrix cristae compartments able to experience different driving forces [20]. These differences across tissues, metabolic states, and within mitochondria may be responses to adjust metabolism to specific cellular and tissue demands. Despite the clear observations and underlying premise suggesting an important role for PMF in cellular metabolism, specific mechanisms through which changes in PMF cause downstream effects are still not fully understood. Nevertheless, regulation of the PMF is important for mounting physiologic responses to stress and to changes in energy availability for organisms to adapt and to survive. When the PMF is not regulated properly to respond to these changes or stresses, cellular damage can occur [11].
Significant insights have evolved from studying downstream functions of endogenous proteins that dissipate the PMF (Figure 1). A major example of these proteins is the uncoupling proteins (UCP). UCPs are named for their ability to uncouple the reactions of the ETC from ATP synthesis [21, 22]; when protons can leak back into the mitochondrial matrix without driving ATP synthesis through ATP synthase, uncoupled respiration results. To maintain PMF, the ETC increases activity resulting in increased oxygen consumption. The results of uncoupling can range from total energy collapse when using pharmacologic uncouplers, to regulated oxidative stress reduction and energy-sensing signaling to adjust metabolism to meet energy demand through UCPs [23, 24].
Proteins other than UCPs can uncouple mitochondria as well, such as the adenine nucleotide transporter (ANT), which can transport protons into the matrix in a regulated manner [25, 26]. There are also many substrate and ion transporters that use the energy from the PMF to drive their function, such as the mitochondrial phosphate carrier and the glutamate/aspartate exchanger [27, 28]. Cation channels in mitochondria are also subject to the PMF, a driving force for cation accumulation into the matrix. Some examples of cation channels include the ATP-sensitive potassium channel, sodium-activated potassium channels, and the calcium uniporter [29–32]. These channels can regulate responses to different metabolic conditions and to stresses [33, 34]. A large conductance, non-selective channel known as the mitochondrial permeability transition pore (mPTP) is also present in mitochondria and can lead to complete PMF collapse [35, 36]. Though the molecular details of the mPTP are debated, its presence is observed under many regulated metabolic processes and in disease models [37, 38]. While there are many proteins that regulate and respond to the PMF (Figure 1), studying how the PMF controls metabolism and vice versa can be challenging.
Historically, there have been limited experimental approaches to modulate the PMF. Small molecule protonophores, such as FCCP, can be used to shuttle protons across the IM, resulting in PMF dissipation. However, this approach lacks tissue specificity, reversibility, and selectivity. Protonophores can act on cell membrane potential and gradients across other biologic membranes such as those present in the lysosome or in secretory vesicles. Protonophores also lack tissue specificity and reversibility such that FCCP cannot be selectively targeted to one tissue of an organism, and there is no acute means to remove the activity. In addition, until recently there has been no way to specifically increase the PMF in isolation, except by fueling upstream metabolism to provide electrons to the ETC. Because of these difficulties, interpreting physiologic responses to PMF interventions can generate misleading conclusions or opposite conclusions in different models (thoroughly reviewed elsewhere [6]). To circumvent some of these limitations, a mitochondria-targeted light-activated protonophore, mito-photo-DNP, was created. Mito-photo-DNP conferred better spatiotemporal control, allowing specific regions of cells containing mitochondria to be targeted with light for PMF dissipation. [39, 40]. This advance paved the way for optogenetic approaches targeted to mitochondria.
THE MITOCHONDRIAL LIGHT SWITCH TECHNIQUES (mtSWITCH)
Optogenetics
Optogenetics is the expression of light-sensitive proteins to control an aspect of cell physiology, such as ion gradients across the plasma membrane of cells. Optogenetics flourished in neuroscience, where depolarizing or hyperpolarizing the cell membrane potential of neurons in response to specific wavelengths of light was used to functionally dissect neural networks. This led to novel insights regarding physiologic regulation of behavior by the nervous system, with approaches designed to test necessity and sufficiency of precise neuronal inputs [41–44]. There are many widely used optogenetic tools such as the cation-selective Channelrhodopin (ChR2), anion-selective Halorhodopsin, and several specific, directional proton pumps like bacteriorhodopsin (bR), the related delta-rhodopsin (dR) [45], Arch and its derivatives [46], and Mac [47]. While the diversity of optogenetic tools is rapidly expanding, we will focus on two classes that were recently targeted to mitochondria to modulate the PMF: ChR2 and proton pumps.
ChR2 and proton pumps can alter the PMF by fostering ion transport across membranes, but there are important differences between ChR2 and proton pumps that contribute to unique aspects of their function. Proton pumps are unidirectional, and can selectively move protons even against a concentration gradient, with directionality determined by orientation of the pump in the membrane [47–49]. ChR2 is less selective, and allows cations (H+, K+, Ca++) to move in either direction according to that ion’s driving force across a membrane [50, 51]. Notably, proton specific pumps [47] avoid the potential influence of K+ or Ca++ ion flux that may occur with ChR2. Another difference between ChR2 and proton pumps is that ChR2 has an extended desensitization time when the channel remains open [52], whereas proton pumps quickly cease activity in the absence of light [53]. Different proteins in this family can also have different optimal light activation wavelengths, allowing combination of proteins using different irradiation colors [48, 49]. Using proteins with different wavelengths of light would allow differential manipulation of physiology at different times or in different tissues, depending on the color and duration of irradiation. Regardless of their differences, each of these pumps or channels are structurally similar, consisting of 7 transmembrane helices arranged around a covalently bound cofactor all-trans retinal, which is required for photocurrents [48, 51].
Subcellular targeting sequences can be used to direct optogenetic tools toward specific cellular compartments, like vesicles or lysosomes [51, 54]. Orientation in membranes matters for unidirectional proton pumps, and different organelle targeting sequences can be used to orient proton pumping in a selected direction. [54]. Proton pumps translocate protons toward their N terminus, which is natively from the cytoplasm to outside the cell [55, 56]. Based on these principles of targeting and directionality, several pumps and channels were targeted to the mitochondrial IM to increase or decrease the PMF in response to light. These optogenetic tools can dissipate the PMF by moving protons from the IMS to the mitochondrial matrix, resulting in respiratory uncoupling. Conversely, proton pumps can pump protons directionally across the IM from the matrix to the IMS to generate a PMF and energize mitochondria. Collectively, we will refer to these tools as the mitochondrial light switch technique (mtSWITCH), as the reagents are switches that can either selectively “turn off” or “turn on” mitochondrial function through PMF manipulation using light. Targeting optical tools to mitochondria can be challenging. There are many mitochondria-targeting sequences (MTS) that can be used to direct proteins to mitochondria [57], and different targeting strategies can orient proteins differently in the IM [54] (Figure 2). It is important to note that a targeting sequence may work for one approach, but it may not be sufficient to target a different protein to mitochondria. Therefore, proper localization and function should be confirmed.
Figure 2. mtSWITCH constructs increase or decrease the PMF in response to light.
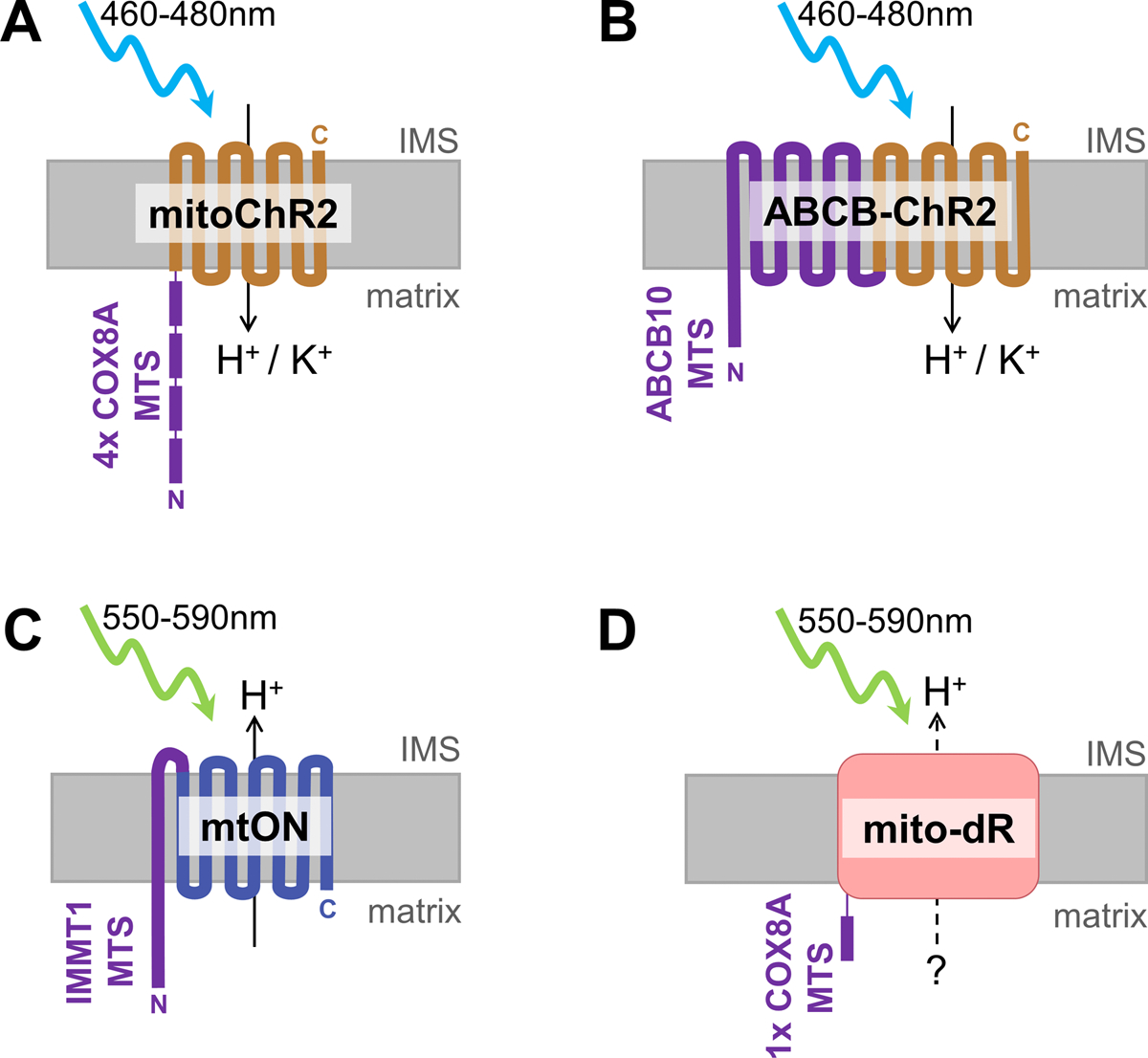
mtSWITCH constructs are shown with their respective mitochondria-targeting sequences (MTS, shown in purple) in the mitochondrial inner membrane. Light-activated proteins are shown in either orange or blue to indicate that they decrease or increase the PMF, respectively. Topology of light-activated proteins are indicated where ions are moved either from the intermembrane space (IMS) to the matrix, or vice versa. N and C termini of peptides are indicated in each construct. A) mitoChR2 uses the COX8A mitochondria-targeting sequence (MTS) repeated in tandem four times to achieve mitochondrial expression. mitoChR2 is activated by 460–480 nm light, and results in decreased PMF. ChR2 is capable of passing protons (H+) as well as other cations, potassium (K+) the most prevalent in the cytosol. B) ABCB-ChR2 uses the ABCB10 MTS to direct ChR2 to mitochondria, and is topologically oriented the same as mitoChR2. ABCB-ChR2 is also activated by 460–480 nm light and results in decreased PMF. C) mtON is targeted to mitochondria by the IMMT1 MTS and some of the transmembrane protein sequence to flip the topology to direct proton-specific translocation from the matrix to the IMS. The native N terminus of the light-activated proton pump is therefore located on the IMS side, opposite of the MTS N terminus. mtON is activated by 550–590 nm light and results in increased PMF. D) mito-dR uses one COX8A MTS, and the topology and direction of proton translocation is unclear in mitochondria. mito-dR is activated by 550–590 nm light and is proposed to increase the PMF.
Once successfully targeted to mitochondria optogenetic tools can be used to study several broad phenomena of cellular metabolism downstream of bioenergetics. Using light allows rapid reversibility and spatial restriction of illumination to confer precise spatiotemporal control of the PMF, a major advance in experimental control. The following sections summarize the mitochondria-targeting details and the findings using individual constructs of the mtSWITCH technique.
mitoChR2
An early example of a mtSWITCH approach targeted ChR2 to mitochondria (mitoChR2) [52], with cation flux across the IM and PMF collapse occurring in response to light. Targeting was through N-terminal fusion to 4 repeats of the well-characterized 29 amino acid COX8A MTS (116 amino acids total) [52, 58, 59]. Our laboratory has found that targeting approaches using a single COX8A targeting sequence are insufficient to direct ChR2 or proton pumps to mitochondria and result instead in plasma membrane targeting, a finding that has been corroborated by others [52, 60]. Mitochondrial targeting using the COX8A repeated sequence was achieved only after deleting a small leading section of the ChR2 sequence (24 amino acids) thought to contain a plasma membrane targeting sequence [52]. Once targeted to mitochondria, topological studies confirmed the orientation of mitoChR2 in the IM (Figure 2A). Confocal fluorescent images of mitoChR2 tagged with YFP combined with protease protection and fluorescence quenching assays confirmed mitoChR2 localization in the IM. mitoChR2 was also functionally characterized in cells to demonstrate PMF dissipation in vivo [52].
In response to 460–480 nm light, mitoChR2 depolarized ΔΨm light-dose dependently in cells [52]. In addition, using a stabilized step function opsin version of this construct allowed closure of the channel using 590 nm light. Using these two colors of light conferred precise, reversible control over PMF dissipation with mitoChR2. This study further showed that ΔΨm changes were not due to mPTP formation, providing extra evidence that mtSWITCH functions independent of endogenous mitochondrial PMF changes.
Mitochondria can modulate cellular Ca++ signaling through uptake of Ca++ into the matrix driven by the PMF. When activated, mitoChR2 modulated Ca++ signaling and respiration in cells. Light activation decreased Ca++ accumulation in the mitochondrial matrix, and increased oxygen consumption rate in respiring cells, which is the expected effect of increased ETC activity following PMF dissipation [61]. Decreases in both ΔΨm and Ca++ uptake mimicked treatment with a protonophore, showing mitoChR2 can completely depolarize mitochondria in cells. These changes in bioenergetics and Ca++ handling have broad implications for many experimental models. Ca++ signaling is intertwined with many cellular processes that also involve mitochondria [5, 62, 63], and optogenetic approaches will help define these molecular mechanisms with better spatial and temporal control.
Next, mitoChR2 was used to control downstream physiologic functions of metabolism that require mitochondrial function. Muscle contraction relies on Ca++ signaling and energy availability, both of which can be altered by mitoChR2. The activation of mitoChR2 almost completely prevented cardiomyocytes from contracting in culture. The loss of cardiomyocyte beating was therefore likely due to dysregulated Ca++ signaling and loss of ATP production downstream of mitoChR2 activation. In another model, mitoChR2 prevented the well-characterized increase in PMF and resultant ATP production in pancreatic β-cells in response to glucose stimulation [64]. These two metabolically distinct effects of muscle contraction and ATP production in β-cells shows the fundamental control mtSWITCH proteins could have in diverse models. Finally, subcellular illumination showed that mitoChR2 functioned only where irradiated, observed through decreased ΔΨm and Ca++ uptake only in the illuminated area. This study demonstrates the precise spatiotemporal control of optical dissipation of PMF using mtSWITCH techniques to alter metabolism [52].
ABCB-ChR2
Another approach to selectively dissipate the PMF again targeted ChR2 to mitochondria using a different strategy. Similar to initial targeting strategies for mitoChR2 [52], ChR2 did not reach mitochondria using one, two or three repeats of the COX8A MTS [60]. ABCB-ChR2 was successfully targeted using the large transmembrane ABCB10 MTS [65]. Again, confocal images of ABCB-ChR2 tagged with YFP and immunolabeling confirmed mitochondrial targeting. The ABCB10 MTS would orient ChR2 with the same topology as mitoChR2 [65] (Figure 2A&B), however, orientation in the membrane should not alter the effect of ChR2 as the channel has no directionality. Indeed, the activation of ABCB-ChR2 also resulted in PMF dissipation in cells [60].
Mitochondrial depolarization can result in apoptotic cell death in many contexts [66, 67]. This optogenetic approach allowed precise control to better understand the link between PMF and cell death. ABCB-ChR2 depolarized mitochondria, and cell viability with increasing light exposure was then measured. Light activation of ABCB-ChR2 caused increased cell death and decreased viability. These effects were reversible using Z-VAD-FMK, a specific caspase-dependent apoptosis inhibitor, indicating cell death was occurring through apoptosis. Observation of mitochondrial cytochrome c release confirmed the involvement of apoptosis in the light-induced cell death, as cytochrome c release is a hallmark of mitochondria-driven apoptosis [66].
Mitochondrial fitness is often signaled through the PMF. Broadly, healthy mitochondria will have a PMF, while mitochondria without a PMF are dysfunctional. Dysfunctional mitochondria are removed through a process called mitochondrial autophagy, or mitophagy. Mitophagy is a mitochondrial quality control process and could be a protective mechanism in different metabolic disorders [68, 69]. Activation of ABCB-ChR2 could then be used to trigger or affect mitophagy through PMF loss. Light activation of ABCB-ChR2 resulted in increased markers of mitophagy, mediated by Parkin and subsequent LC3 accumulation at mitochondria. Consistent with the idea that the activation of mitophagy may protect against stress, ABCB-ChR2 activation exhibited a preconditioning effect; when mitochondrial PMF was dissipated with light, cells were more resistant to subsequent PMF dissipation and cell death. This result indicates that altering the PMF can lead to cellular adaptation for stress resistance. This study highlights the use of mtSWITCH techniques to identify molecular details of the consequences of metabolic shifts in cell fate determination [60].
mtON
While mitochondria-targeted ion channels have been used to dissipate the PMF, PMF generation requires a unidirectional proton-selective pump. The eukaryotic proton pump Mac was expressed in mitochondria and named mtON for its ability to generate a PMF and “turn on” mitochondrial function. Unlike channelrhodopsins, proton pumps like Mac are unidirectional proton transporters, allowing directional control with different targeting sequences. mtON was targeted to the IM by fusion with the MTS and part of the coding region of the IMMT1 protein [70–72]. Importantly, this MTS contains a transmembrane region, required for orientation of mtON to pump protons from the matrix to the IMS to energize the PMF (Figure 2C). Protease protection studies and detailed confocal fluorescent image scans of single mitochondria expressing mtON indicated IM targeting and expected topology oriented to pump protons out of mitochondria. Full characterization of mtON in isolated mitochondria revealed increased PMF in response to light, confirming the direction of proton pumping [71].
The proton pump Mac can generate proton gradients in response to light [47, 49]. Likewise, mtON in isolated mitochondria caused a light dose-dependent energized PMF in response to 550–590 nm light that matched the PMF generated by the ETC [71]. Increased PMF was observed through both ΔΨm and ΔpH components. These results indicated a PMF was generated using only the energy from light, bypassing the ETC, as no source of electrons (metabolic substrates to generate electron donors for respiratory function) were required. To further test this, oxygen consumption was measured in isolated mitochondria fueled with substrates to drive respiration. When activated, mtON decreased reliance on oxygen to make ATP, demonstrating that mtON can supplement ETC activity by using light rather than oxygen and electrons. This showed that mtSWITCH tools can be used to control the PMF independent of oxygen or substrate availability.
In addition to supplementing ETC activity, mtON was able to compensate for lost ETC activity. In a whole-animal C. elegans model, animals exposed to ETC toxins survived more when mtON was active compared to controls, indicating mtON activation can overcome dysfunctional respiration. These results were confirmed using a mitochondrial mutant strain with decreased animal locomotion, a phenotype rescued by mtON activation. Impaired energetic function of mitochondria is signaled throughout cells and organisms, and AMP-activated protein kinsase (AMPK) is a molecular energy sensor responsible for some of this signaling [73]. mtON silenced AMPK activation under starvation conditions and decreased animal locomotion when starved. These acute changes in AMPK signaling could have broad and lasting impacts on metabolism and physiology [74–76].
One example of a lasting impact downstream of AMPK and other signaling pathways is the ability to adapt to stress. mtON was used to study hypoxia-adaptation as a readout of stress resistance. In C. elegans and in mammals a short exposure to hypoxia is protective against a later, more damaging exposure [34, 77]. The protection conferred therein can be suppressed by mtON activation, suggesting that a decreased PMF during the short hypoxia is required for its protective effect. These results are consistent with those using ABCB-ChR2 [60]; PMF dissipation triggers stress resistance prophylactically. The mtON system highlights how temporal control of the PMF can reveal fundamental requirements for stress resistance in models of mitochondrial and metabolic dysfunction [71].
mito-dR
Another attempt to polarize the PMF targeted the proton pump delta-rhodopsin (dR) to mitochondria. mito-dR was expressed in Drosophila and proposed to increase the PMF in response to 550–590 nm light [78]. mito-dR function was assessed in dopaminergic nerve terminals by observing ΔΨm, ATP levels, and Ca++ handling in tissues, each after long-term light exposure, indicating mito-dR can affect energetics in fly nervous tissue. In addition, oxidized lipids and mitochondrial redox state were assayed, and mito-dR decreased signs of oxidative stress. In many contexts, however, decreased rather than increased PMF is associated with alleviated oxidative stress [6, 79, 80], especially in Parkinson’s disease models [81–84].
Mitochondrial dysfunction is involved in models of Parkinson’s disease [85–87]. mito-dR was originally used in a cell model of Parkinson’s disease dysfunction [88]. In a Drosophila knockout model of Parkinson’s disease, mito-dR decreased aggregated α-synuclein levels, a protein causally implicated in Parkinson’s pathology involving mitochondria [85–87]. Light activation of mito-dR also increased flight and movement behavior associated with protection against neurodegeneration, indicating that mito-dR acts to improve dopaminergic neuron function in vivo [78]. This study shows how mtSWITCH techniques can be used in simple models to better understand the fundamental principles of neurodegenerative metabolism.
In comparison to other mtSWITCH approaches, there is limited evidence surrounding the topology and pumping direction of mito-dR. Similar to Mac and bR, dR is a unidirectional proton pump that translocates protons toward its native N terminus [45, 89]. To generate mito-dR, dR was fused to a single COX8A targeting sequence on the N terminus to achieve expression in mitochondria [88]. With this targeting strategy, the dR N terminus is be predicted to be in the matrix. No topology studies or functional measures in isolated mitochondria or cells were carried out, however, so the direction of mito-dR proton pumping is unclear. While proton pump activity of dR was confirmed in bacteria, the activity of mito-dR remains to be fully characterized [78] (Figure 2D). This uncertainty together with observations that decreased PMF (rather than increased PMF) results in decreased oxidative stress is a significant caveat in the interpretation of results from mito-dR.
Other Optical Tools in Mitochondria
While the focus of this article is on new optogenetic approaches to control the PMF, there are other optogenetic tools used in mitochondria to compliment mtSWITCH techniques. For example, mitochondrial position within cells can be reversibly controlled using light-induced recruitment of cytoskeletal motor proteins [90, 91]. Mitochondrial tethering to endoplasmic reticulum can also be controlled using light [92]. Using modified fluorescent proteins, ROS can be generated upon light exposure, modeling mitochondrial ROS production in a tightly controlled manner [93–95]. These ROS generating proteins have been targeted to different compartments in cells, and specifically to different compartments in mitochondria to assess nuances of mitochondrial redox signaling [96]. Similarly, combining optogenetics and pharmacology to generate mitochondrial ROS revealed redox communication between mitochondria and chromosomes in the nucleus [97]. This study highlights the broad reach of mitochondrial signals throughout the cell. Each of these mitochondria-targeted optogenetic tools is likely to confer precise spatiotemporal control over different aspects of mitochondrial biology, and each could be used along with the mtSWITCH techniques to study mitochondria in powerful new ways using light.
PERSPECTIVES
mtSWITCH has been used to control mitochondrial function and resulting metabolism in diverse ways thus far. The technique is poised to become a powerful and efficient means to study metabolism in many new contexts. As is true for all optogenetic approaches, mtSWITCH approaches must include experimental controls for light exposure and, where appropriate, cofactor supplementation. In mammalian cell systems all-trans retinal may be present at sufficient levels, but exogenous supplementation may increase optogenetic effects [52]. Some systems such as C. elegans and Drosophila require exogenous all-trans retinal supplementation for functional optogenetics [41], which must be controlled for due to potential confounding effects, such as antioxidant capabilities of the cofactor [98]. Off-target effects can be controlled for either using non-functional optogenetic protein controls [52, 78], or by combining experiments both with and without supplemented all-trans retinal [71]. Controlling for light exposure is also important, especially when using blue excitation wavelengths that are damaging to biological samples [99]. This is particularly relevant to mitochondria [100] which contain many light-absorbing cytochromes and flavins. Flavins can create ROS when exposed to blue light [101], which can cause damage or alter redox signaling that heavily impacts metabolism [102]. In addition to off-target effects from light or all-trans retinal, protein expression artifacts can be kept to a minimum in living systems by using CRISPR/Cas9 approaches to drive single-copy expression, decreasing the likelihood of overexpression artifacts.
When appropriately controlled, mtSWITCH is a powerful toolkit to investigate how precise changes in mitochondrial function regulate metabolism and downstream physiology. Selective dissipation of the PMF overcomes the significant challenges of using protonophores, especially when used tissue- or cell-specifically. Increasing the PMF with light is a significant advance to understand metabolism, as other means to increase the PMF necessarily fuel the ETC or inhibit ATP synthesis; both interventions have myriad other effects on metabolism. Fueling mitochondria to increase the PMF would result in changed glycolysis and Krebs cycle flux, two pathways that are regulated and that participate in cellular signaling [10]. Inhibiting ATP synthase increases PMF by removing the ability to dissipate the PMF through ATP synthesis. This inhibition could result in non-physiologic responses such as cell damage or death due to lack of energy availability, or conversely adaptive signaling to respond to the energetic crisis [103, 104]. Using mtSWITCH to increase the PMF avoids these confounding variables. Together, mtSWITCH can be combined to take unprecedented control of mitochondrial function and metabolism – decreasing or increasing the PMF in isolation – to determine cause and effect in complicated metabolic models.
mtSWITCH techniques have been used in diverse cell and organism models to lay the foundation of mitochondrial optogenetics going forward (Figure 3). The studies reviewed here show the feasibility of employing mtSWITCH and highlight the broad range of diseases that may be impacted. ATP availability, Ca++ signaling, AMPK signaling, and oxidative stress are each implicated in many diseases and pathologies, and each are cellular outputs affected by mtSWITCH. Using mitoChR2 to learn more about glucose-stimulated ATP production may advance understanding in models of diabetes and metabolic syndromes. mitoChR2 could also inform myopathy research through its ability to control muscle contraction. ABCB-ChR2 control of apoptotic cell death and mitophagy could guide future experimental models to understand aging and cellular senescence. Energizing mitochondria with mtON to modulate AMPK signaling and hypoxia resistance could be translated to models of ischemia reperfusion injury, with outcomes of heart attack and stroke dependent on PMF changes that are not fully understood [105, 106]. mito-dR showed that metabolic control will be important in understanding the fundamentals of neurodegenerative disease progression and protection.
Figure 3. The mtSWITCH technique affects broad metabolic outputs through PMF manipulation.
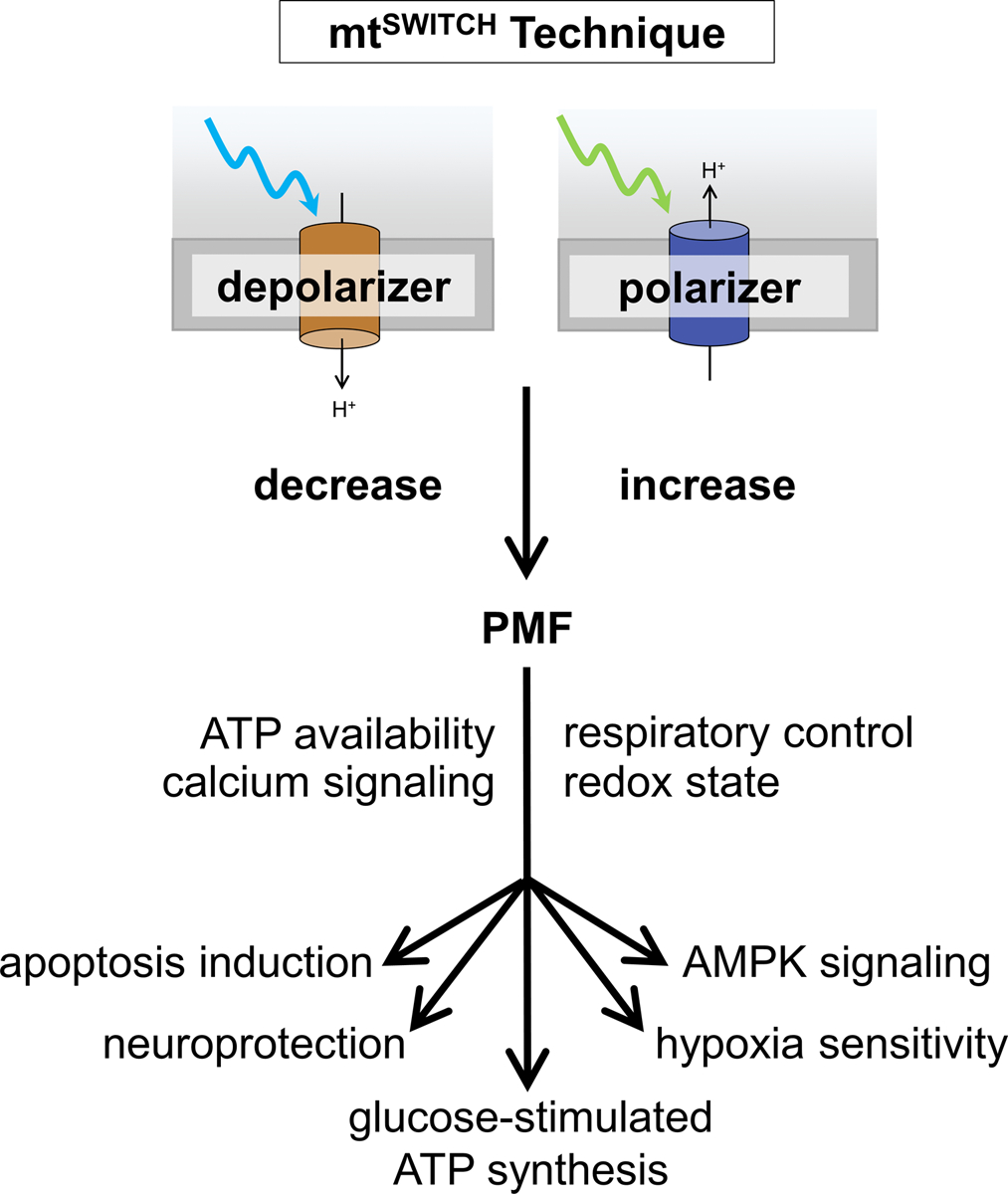
mtSWITCH constructs directly alter mitochondrial functions that impact downstream metabolism. Decreasing or increasing the PMF results in altered ATP production, O2 consumption by the electron transport chain, Ca++ handling and signaling, and cellular redox state. Each of these factors affect cellular signaling. Changing these parameters has impacted physiologic outputs such as apoptosis and mitophagy, glucose-stimulated ATP production in β-cells, AMPK signaling and hypoxia resistance in living animals, and biochemical and behavioral outcomes in neurodegeneration models. These are just a few validated examples of the impact mtSWITCH will have on many fields of research to understand disease and how metabolism impacts it.
Using mtSWITCH to establish causal interactions in these metabolic models of disease will lead to discovery of new pathways to target therapeutically. The most wide-ranging unanswered question mtSWITCH can address is the following: How do mitochondria truly behave in vivo, and how does that behavior impact cellular life? This general question is integral to many models of health and disease, from understanding the benefits of exercise, to understanding how metabolic shifts impact aging tissue, tumor microenvironments, or adaptations to obesity. Light as an experimental tool confers precise control in both space and time that no other reagent can manage in mitochondria, especially in vivo. The tools reviewed here will be essential to advance our understanding of complex cellular functions by increasing the spatial and temporal resolution of metabolic control.
ACKNOWLEDGEMENTS
The laboratory of A.P.W. is supported by grants from the National Institutes of Health (R01 NS092558, R01 NS115906 and R21 CA242843), and B.J.B. is supported by a grant from the American Heart Association (18PRE33990054).
Abbreviations:
- mtSWITCH
mitochondrial light switches
- Ca++
calcium
- ROS
reactive oxygen species
- PMF
protonmotive force
- IM
inner mitochondrial membrane
- ΔΨm
mitochondrial membrane potential
- ΔpH
pH gradient
- ETC
electron transport chain
- IMS
intermembrane space
- K+
potassium
- UCP
uncoupling protein
- ANT
adenine nucleotide transporter
- mPTP
mitochondrial permeability transition pore
- ChR2
channelrhodopsin 2
- bR
bacteriorhodopsin
- dR
delta-rhodopsin
- MTS
mitochondrial targeting sequence
- AMPK
AMP-activated protein kinase
Footnotes
Conflicts of Interest: The authors declare there are no conflicts of interest.
REFERENCES
- 1.Lozano, Naghavi R, Foreman M, Lim K, Shibuya S, Aboyans K, Abraham V, Adair J, Aggarwal T, Ahn R, Alvarado SY, Anderson M, Anderson HR, Andrews LM, Atkinson KG, Baddour C, Barker-Collo LM, Bartels S, Bell DH, Benjamin ML, Bennett EJ, Bhalla D, Bikbov K, Bin Abdulhak B, Birbeck A, Blyth G, Bolliger F, Boufous I, Bucello S, Burch C, Burney M, Carapetis P, Chen J, Chou H, Chugh D, Coffeng SS, Colan LE, Colquhoun SD, Colson S, Condon KE, Connor J, Cooper MD, Corriere LT, Cortinovis M, de Vaccaro M, Couser KC, Cowie W, Criqui BC, Cross MH, Dabhadkar M, Dahodwala KC, De Leo N, Degenhardt D, Delossantos L, Denenberg A, Des Jarlais J, Dharmaratne DC, Dorsey SD, Driscoll ER, Duber T, Ebel H, Erwin B, Espindola PJ, Ezzati P, Feigin M, Flaxman V, Forouzanfar AD, Fowkes MH, Franklin FG, Fransen R, Freeman M, Gabriel MK, Gakidou SE, Gaspari E, Gillum F, Gonzalez-Medina RF, Halasa D, Haring YA, Harrison D, Havmoeller JE, Hay R, Hoen RJ, Hotez B, Hoy PJ, Jacobsen D, James KH, Jasrasaria SL, Jayaraman R, Johns S, Karthikeyan N, Kassebaum G, Keren N, Khoo A, Knowlton JP, Kobusingye LM, Koranteng O, Krishnamurthi A, Lipnick R, Lipshultz M, Ohno SE, S. L., et al. (2012) Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010, Lancet 380, 2095–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ng, Fleming M, Robinson T, Thomson M, Graetz B, Margono N, Mullany C, Biryukov EC, Abbafati S, Abera C, Abraham SF, Abu-Rmeileh JP, Achoki NM, AlBuhairan T, Alemu FS, Alfonso ZA, Ali R, Ali MK, Guzman R, Ammar NA, Anwari W, Banerjee P, Barquera A, Basu S, Bennett S, Bhutta DA, Blore Z, Cabral J, Nonato N, Chang IC, Chowdhury JC, Courville R, Criqui KJ, Cundiff MH, Dabhadkar DK, Dandona KC, Davis L, Dayama A, Dharmaratne A, Ding SD, Durrani EL, Esteghamati AM, Farzadfar A, Fay F, Feigin DF, Flaxman VL, Forouzanfar A, Goto MH, Green A, Gupta MA, Hafezi-Nejad R, Hankey N, Harewood GJ, Havmoeller HC, Hay R, Hernandez S, Husseini L, Idrisov A, Ikeda BT, Islami N, Jahangir F, Jassal E, Jee SK, Jeffreys SH, Jonas M, Kabagambe JB, Khalifa EK, Kengne SE, Khader AP, Khang YS, Kim YH, Kimokoti D, Kinge RW, Kokubo JM, Kosen Y, Kwan S, Lai G, Leinsalu T, Li M, Liang Y, Liu X, Logroscino S, Lotufo G, Lu PA, Ma Y, Mainoo J, Mensah NK, Merriman GA, Mokdad TR, Moschandreas AH, Naghavi J, Naheed M, Nand A, Narayan D, Nelson KM, Neuhouser EL, Nisar ML, Ohkubo MI, Oti T, Pedroza SO, A., et al. (2014) Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013, Lancet 384, 766–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ, Lin JK, Farzadfar F, Khang YH, Stevens GA, Rao M, Ali MK, Riley LM, Robinson CA, Ezzati M & Global Burden of Metabolic Risk Factors of Chronic Diseases Collaborating, G. (2011) National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants, Lancet 378, 31–40. [DOI] [PubMed] [Google Scholar]
- 4.Mitchell P (1961) Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism, Nature 191, 144–8. [DOI] [PubMed] [Google Scholar]
- 5.Bartok A, Weaver D, Golenar T, Nichtova Z, Katona M, Bansaghi S, Alzayady KJ, Thomas VK, Ando H, Mikoshiba K, Joseph SK, Yule DI, Csordas G & Hajnoczky G (2019) IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer, Nat Commun 10, 3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berry BJ, Trewin AJ, Amitrano AM, Kim M & Wojtovich AP (2018) Use the Protonmotive Force: Mitochondrial Uncoupling and Reactive Oxygen Species, J Mol Biol 430, 3873–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rasola A & Bernardi P (2011) Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis, Cell Calcium 50, 222–33. [DOI] [PubMed] [Google Scholar]
- 8.Ly JD, Grubb DR & Lawen A (2003) The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update, Apoptosis 8, 115–28. [DOI] [PubMed] [Google Scholar]
- 9.Ortega SP, Chouchani ET & Boudina S (2017) Stress turns on the heat: Regulation of mitochondrial biogenesis and UCP1 by ROS in adipocytes, Adipocyte 6, 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez-Reyes I & Chandel NS (2020) Mitochondrial TCA cycle metabolites control physiology and disease, Nat Commun 11, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rolland SG, Schneid S, Schwarz M, Rackles E, Fischer C, Haeussler S, Regmi SG, Yeroslaviz A, Habermann B, Mokranjac D, Lambie E & Conradt B (2019) Compromised Mitochondrial Protein Import Acts as a Signal for UPR(mt), Cell Rep 28, 1659–1669 e5. [DOI] [PubMed] [Google Scholar]
- 12.Haeussler S, Kohler F, Witting M, Premm MF, Rolland SG, Fischer C, Chauve L, Casanueva O & Conradt B (2020) Autophagy compensates for defects in mitochondrial dynamics, PLoS Genet 16, e1008638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aldakkak M, Stowe DF, Cheng Q, Kwok WM & Camara AK (2010) Mitochondrial matrix K+ flux independent of large-conductance Ca2+-activated K+ channel opening, Am J Physiol Cell Physiol 298, C530–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aon MA, Cortassa S & O’Rourke B (2010) Redox-optimized ROS balance: a unifying hypothesis, Biochim Biophys Acta 1797, 865–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mendham AE, Larsen S, George C, Adams K, Hauksson J, Olsson T, Fortuin-de Smidt MC, Nono Nankam PA, Hakim O, Goff LM, Pheiffer C & Goedecke JH (2020) Exercise training results in depot-specific adaptations to adipose tissue mitochondrial function, Sci Rep 10, 3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leckey JJ, Hoffman NJ, Parr EB, Devlin BL, Trewin AJ, Stepto NK, Morton JP, Burke LM & Hawley JA (2018) High dietary fat intake increases fat oxidation and reduces skeletal muscle mitochondrial respiration in trained humans, FASEB J 32, 2979–2991. [DOI] [PubMed] [Google Scholar]
- 17.Korzeniewski B (2017) Contribution of proton leak to oxygen consumption in skeletal muscle during intense exercise is very low despite large contribution at rest, PLoS One 12, e0185991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willingham TB, Zhang Y, Andreoni A, Knutson JR, Lee DY & Glancy B (2019) MitoRACE: evaluating mitochondrial function in vivo and in single cells with subcellular resolution using multiphoton NADH autofluorescence, J Physiol 597, 5411–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim Y, Yang DS, Katti P & Glancy B (2019) Protein composition of the muscle mitochondrial reticulum during postnatal development, J Physiol 597, 2707–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolf DM, Segawa M, Kondadi AK, Anand R, Bailey ST, Reichert AS, van der Bliek AM, Shackelford DB, Liesa M & Shirihai OS (2019) Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent, EMBO J 38, e101056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicholls DG (1977) The effective proton conductance of the inner membrane of mitochondria from brown adipose tissue. Dependency on proton electrochemical potential gradient, Eur J Biochem 77, 349–56. [DOI] [PubMed] [Google Scholar]
- 22.Parker N, Crichton PG, Vidal-Puig AJ & Brand MD (2009) Uncoupling protein-1 (UCP1) contributes to the basal proton conductance of brown adipose tissue mitochondria, J Bioenerg Biomembr 41, 335–42. [DOI] [PubMed] [Google Scholar]
- 23.Chouchani ET, Kazak L & Spiegelman BM (2019) New Advances in Adaptive Thermogenesis: UCP1 and Beyond, Cell Metab 29, 27–37. [DOI] [PubMed] [Google Scholar]
- 24.Trewin AJ, Berry BJ & Wojtovich AP (2018) Exercise and Mitochondrial Dynamics: Keeping in Shape with ROS and AMPK, Antioxidants (Basel) 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brand MD, Pakay JL, Ocloo A, Kokoszka J, Wallace DC, Brookes PS & Cornwall EJ (2005) The basal proton conductance of mitochondria depends on adenine nucleotide translocase content, Biochem J 392, 353–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bertholet AM, Chouchani ET, Kazak L, Angelin A, Fedorenko A, Long JZ, Vidoni S, Garrity R, Cho J, Terada N, Wallace DC, Spiegelman BM & Kirichok Y (2019) H(+) transport is an integral function of the mitochondrial ADP/ATP carrier, Nature 571, 515–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kramer R (1996) Structural and functional aspects of the phosphate carrier from mitochondria, Kidney Int 49, 947–52. [DOI] [PubMed] [Google Scholar]
- 28.Hoek JB & Njogu RM (1980) The role of glutamate transport in the regulation of the pathway of proline oxidation in rat liver mitochondria, J Biol Chem 255, 8711–8. [PubMed] [Google Scholar]
- 29.Smith CO, Wang YT, Nadtochiy SM, Miller JH, Jonas EA, Dirksen RT, Nehrke K & Brookes PS (2018) Cardiac metabolic effects of KNa1.2 channel deletion and evidence for its mitochondrial localization, FASEB J, fj201800139R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paggio A, Checchetto V, Campo A, Menabo R, Di Marco G, Di Lisa F, Szabo I, Rizzuto R & De Stefani D (2019) Identification of an ATP-sensitive potassium channel in mitochondria, Nature 572, 609–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davidson SM, Szabadkai G & Duchen MR (2019) Fantastic beasts and how to find them-Molecular identification of the mitochondrial ATP-sensitive potassium channel, Cell Calcium 84, 102100. [DOI] [PubMed] [Google Scholar]
- 32.Wescott AP, Kao JPY, Lederer WJ & Boyman L (2019) Voltage-energized Calcium-sensitive ATP Production by Mitochondria, Nat Metab 1, 975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wojtovich AP, Smith CO, Haynes CM, Nehrke KW & Brookes PS (2013) Physiological consequences of complex II inhibition for aging, disease, and the mKATP channel, Biochim Biophys Acta 1827, 598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wojtovich AP, Nadtochiy SM, Brookes PS & Nehrke K (2012) Ischemic preconditioning: the role of mitochondria and aging, Exp Gerontol 47, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bernardi P & Di Lisa F (2015) The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection, J Mol Cell Cardiol 78, 100–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He J, Carroll J, Ding S, Fearnley IM & Walker JE (2017) Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase, Proc Natl Acad Sci U S A 114, 9086–9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly-Dyson E, Di Lisa F & Forte MA (2006) The mitochondrial permeability transition from in vitro artifact to disease target, FEBS J 273, 2077–99. [DOI] [PubMed] [Google Scholar]
- 38.Mnatsakanyan N, Beutner G, Porter GA, Alavian KN & Jonas EA (2017) Physiological roles of the mitochondrial permeability transition pore, J Bioenerg Biomembr 49, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chalmers S, Caldwell ST, Quin C, Prime TA, James AM, Cairns AG, Murphy MP, McCarron JG & Hartley RC (2012) Selective uncoupling of individual mitochondria within a cell using a mitochondria-targeted photoactivated protonophore, J Am Chem Soc 134, 758–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glancy B, Hartnell LM, Malide D, Yu ZX, Combs CA, Connelly PS, Subramaniam S & Balaban RS (2015) Mitochondrial reticulum for cellular energy distribution in muscle, Nature 523, 617–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Husson SJ, Liewald JF, Schultheis C, Stirman JN, Lu H & Gottschalk A (2012) Microbial light-activatable proton pumps as neuronal inhibitors to functionally dissect neuronal networks in C. elegans, PLoS One 7, e40937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boyden ES, Zhang F, Bamberg E, Nagel G & Deisseroth K (2005) Millisecond-timescale, genetically targeted optical control of neural activity, Nat Neurosci 8, 1263–8. [DOI] [PubMed] [Google Scholar]
- 43.Deisseroth K (2011) Optogenetics, Nat Methods 8, 26–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Azimi Hashemi N, Bergs ACF, Schuler C, Scheiwe AR, Steuer Costa W, Bach M, Liewald JF & Gottschalk A (2019) Rhodopsin-based voltage imaging tools for use in muscles and neurons of Caenorhabditis elegans, Proc Natl Acad Sci U S A 116, 17051–17060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kamo N, Hashiba T, Kikukawa T, Araiso T, Ihara K & Nara T (2006) A light-driven proton pump from Haloterrigena turkmenica: functional expression in Escherichia coli membrane and coupling with a H+ co-transporter, Biochem Biophys Res Commun 341, 285–90. [DOI] [PubMed] [Google Scholar]
- 46.Chow BY, Han X & Boyden ES (2012) Genetically encoded molecular tools for light-driven silencing of targeted neurons, Prog Brain Res 196, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Waschuk SA, Bezerra AG Jr., Shi L & Brown LS (2005) Leptosphaeria rhodopsin: bacteriorhodopsin-like proton pump from a eukaryote, Proc Natl Acad Sci U S A 102, 6879–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kandori H (2015) Ion-pumping microbial rhodopsins, Front Mol Biosci 2, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chow BY, Han X, Dobry AS, Qian X, Chuong AS, Li M, Henninger MA, Belfort GM, Lin Y, Monahan PE & Boyden ES (2010) High-performance genetically targetable optical neural silencing by light-driven proton pumps, Nature 463, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schneider F, Gradmann D & Hegemann P (2013) Ion selectivity and competition in channelrhodopsins, Biophys J 105, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P & Bamberg E (2003) Channelrhodopsin-2, a directly light-gated cation-selective membrane channel, Proc Natl Acad Sci U S A 100, 13940–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tkatch T, Greotti E, Baranauskas G, Pendin D, Roy S, Nita LI, Wettmarshausen J, Prigge M, Yizhar O, Shirihai OS, Fishman D, Hershfinkel M, Fleidervish IA, Perocchi F, Pozzan T & Sekler I (2017) Optogenetic control of mitochondrial metabolism and Ca(2+) signaling by mitochondria-targeted opsins, Proc Natl Acad Sci U S A 114, E5167–E5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Althaus T & Stockburger M (1998) Time and pH dependence of the L-to-M transition in the photocycle of bacteriorhodopsin and its correlation with proton release, Biochemistry 37, 2807–17. [DOI] [PubMed] [Google Scholar]
- 54.Rost BR, Schneider F, Grauel MK, Wozny C, Bentz C, Blessing A, Rosenmund T, Jentsch TJ, Schmitz D, Hegemann P & Rosenmund C (2015) Optogenetic acidification of synaptic vesicles and lysosomes, Nat Neurosci 18, 1845–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gerber GE, Gray CP, Wildenauer D & Khorana HG (1977) Orientation of bacteriorhodopsin in Halobacterium halobium as studied by selective proteolysis, Proc Natl Acad Sci U S A 74, 5426–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sumii M, Furutani Y, Waschuk SA, Brown LS & Kandori H (2005) Strongly hydrogen-bonded water molecule present near the retinal chromophore of Leptosphaeria rhodopsin, the bacteriorhodopsin-like proton pump from a eukaryote, Biochemistry 44, 15159–66. [DOI] [PubMed] [Google Scholar]
- 57.Pfanner N & Geissler A (2001) Versatility of the mitochondrial protein import machinery, Nat Rev Mol Cell Biol 2, 339–49. [DOI] [PubMed] [Google Scholar]
- 58.Zong S, Wu M, Gu J, Liu T, Guo R & Yang M (2018) Structure of the intact 14-subunit human cytochrome c oxidase, Cell Res 28, 1026–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pendin D, Greotti E, Lefkimmiatis K & Pozzan T (2017) Exploring cells with targeted biosensors, J Gen Physiol 149, 1–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ernst P, Xu N, Qu J, Chen H, Goldberg MS, Darley-Usmar V, Zhang JJ, O’Rourke B, Liu X & Zhou L (2019) Precisely Control Mitochondria with Light to Manipulate Cell Fate Decision, Biophys J 117, 631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brand MD & Nicholls DG (2011) Assessing mitochondrial dysfunction in cells, Biochem J 435, 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nicholls DG (2005) Mitochondria and calcium signaling, Cell Calcium 38, 311–7. [DOI] [PubMed] [Google Scholar]
- 63.Szibor M, Gizatullina Z, Gainutdinov T, Endres T, Debska-Vielhaber G, Kunz M, Karavasili N, Hallmann K, Schreiber F, Bamberger A, Schwarzer M, Doenst T, Heinze HJ, Lessmann V, Vielhaber S, Kunz WS & Gellerich FN (2020) Cytosolic, but not matrix, calcium is essential for adjustment of mitochondrial pyruvate supply, J Biol Chem [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kennedy ED, Maechler P & Wollheim CB (1998) Effects of depletion of mitochondrial DNA in metabolism secretion coupling in INS-1 cells, Diabetes 47, 374–80. [DOI] [PubMed] [Google Scholar]
- 65.Graf SA, Haigh SE, Corson ED & Shirihai OS (2004) Targeting, import, and dimerization of a mammalian mitochondrial ATP binding cassette (ABC) transporter, ABCB10 (ABC-me), J Biol Chem 279, 42954–63. [DOI] [PubMed] [Google Scholar]
- 66.Heiskanen KM, Bhat MB, Wang HW, Ma J & Nieminen AL (1999) Mitochondrial depolarization accompanies cytochrome c release during apoptosis in PC6 cells, J Biol Chem 274, 5654–8. [DOI] [PubMed] [Google Scholar]
- 67.Bhola PD & Letai A (2016) Mitochondria-Judges and Executioners of Cell Death Sentences, Mol Cell 61, 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schofield JH & Schafer ZT (2020) Mitochondrial ROS and Mitophagy: A Complex and Nuanced Relationship, Antioxid Redox Signal [DOI] [PubMed] [Google Scholar]
- 69.Palikaras K, Lionaki E & Tavernarakis N (2018) Mechanisms of mitophagy in cellular homeostasis, physiology and pathology, Nat Cell Biol 20, 1013–1022. [DOI] [PubMed] [Google Scholar]
- 70.John GB, Shang Y, Li L, Renken C, Mannella CA, Selker JM, Rangell L, Bennett MJ & Zha J (2005) The mitochondrial inner membrane protein mitofilin controls cristae morphology, Mol Biol Cell 16, 1543–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Berry BJ, Trewin AJ, Milliken AS, Baldzizhar A, Amitrano AM, Lim Y, Kim M & Wojtovich AP (2020) Optogenetic control of mitochondrial protonmotive force to impact cellular stress resistance, EMBO Rep, e49113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fischer LR, Igoudjil A, Magrane J, Li Y, Hansen JM, Manfredi G & Glass JD (2011) SOD1 targeted to the mitochondrial intermembrane space prevents motor neuropathy in the Sod1 knockout mouse, Brain 134, 196–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ahmadi M & Roy R (2016) AMPK acts as a molecular trigger to coordinate glutamatergic signals and adaptive behaviours during acute starvation, Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mihaylova MM & Shaw RJ (2011) The AMPK signalling pathway coordinates cell growth, autophagy and metabolism, Nat Cell Biol 13, 1016–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cunningham KA, Hua Z, Srinivasan S, Liu J, Lee BH, Edwards RH & Ashrafi K (2012) AMP-activated kinase links serotonergic signaling to glutamate release for regulation of feeding behavior in C. elegans, Cell Metab 16, 113–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang Y, Lanjuin A, Chowdhury SR, Mistry M, Silva-Garcia CG, Weir HJ, Lee CL, Escoubas CC, Tabakovic E & Mair WB (2019) Neuronal TORC1 modulates longevity via AMPK and cell nonautonomous regulation of mitochondrial dynamics in C. elegans, Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dasgupta N, Patel AM, Scott BA & Crowder CM (2007) Hypoxic preconditioning requires the apoptosis protein CED-4 in C. elegans, Curr Biol 17, 1954–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Imai Y, Inoshita T, Meng H, Shiba-Fukushima K, Hara KY, Sawamura N & Hattori N (2019) Light-driven activation of mitochondrial proton-motive force improves motor behaviors in a Drosophila model of Parkinson’s disease, Commun Biol 2, 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Onukwufor JO, Berry BJ & Wojtovich AP (2019) Physiologic Implications of Reactive Oxygen Species Production by Mitochondrial Complex I Reverse Electron Transport, Antioxidants (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Komlodi T, Geibl FF, Sassani M, Ambrus A & Tretter L (2018) Membrane potential and delta pH dependency of reverse electron transport-associated hydrogen peroxide production in brain and heart mitochondria, J Bioenerg Biomembr 50, 355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT & Surmeier DJ (2010) Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1, Nature 468, 696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mao W, Yu XX, Zhong A, Li W, Brush J, Sherwood SW, Adams SH & Pan G (1999) UCP4, a novel brain-specific mitochondrial protein that reduces membrane potential in mammalian cells, FEBS Lett 443, 326–30. [DOI] [PubMed] [Google Scholar]
- 83.Ramsden DB, Ho PW, Ho JW, Liu HF, So DH, Tse HM, Chan KH & Ho SL (2012) Human neuronal uncoupling proteins 4 and 5 (UCP4 and UCP5): structural properties, regulation, and physiological role in protection against oxidative stress and mitochondrial dysfunction, Brain Behav 2, 468–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu K, Liu J, Zhuang N & Wang T (2014) UCP4A protects against mitochondrial dysfunction and degeneration in pink1/parkin models of Parkinson’s disease, FASEB J 28, 5111–21. [DOI] [PubMed] [Google Scholar]
- 85.Grassi D, Howard S, Zhou M, Diaz-Perez N, Urban NT, Guerrero-Given D, Kamasawa N, Volpicelli-Daley LA, LoGrasso P & Lasmezas CI (2018) Identification of a highly neurotoxic alpha-synuclein species inducing mitochondrial damage and mitophagy in Parkinson’s disease, Proc Natl Acad Sci U S A 115, E2634–E2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Imaizumi Y, Okada Y, Akamatsu W, Koike M, Kuzumaki N, Hayakawa H, Nihira T, Kobayashi T, Ohyama M, Sato S, Takanashi M, Funayama M, Hirayama A, Soga T, Hishiki T, Suematsu M, Yagi T, Ito D, Kosakai A, Hayashi K, Shouji M, Nakanishi A, Suzuki N, Mizuno Y, Mizushima N, Amagai M, Uchiyama Y, Mochizuki H, Hattori N & Okano H (2012) Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue, Mol Brain 5, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kumar A, Ganini D & Mason RP (2016) Role of cytochrome c in alpha-synuclein radical formation: implications of alpha-synuclein in neuronal death in Maneb- and paraquat-induced model of Parkinson’s disease, Mol Neurodegener 11, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hara KY, Wada T, Kino K, Asahi T & Sawamura N (2013) Construction of photoenergetic mitochondria in cultured mammalian cells, Sci Rep 3, 1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Khorana HG (1988) Bacteriorhodopsin, a membrane protein that uses light to translocate protons, J Biol Chem 263, 7439–42. [PubMed] [Google Scholar]
- 90.Duan L, Che D, Zhang K, Ong Q, Guo S & Cui B (2015) Optogenetic control of molecular motors and organelle distributions in cells, Chem Biol 22, 671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.van Bergeijk P, Adrian M, Hoogenraad CC & Kapitein LC (2015) Optogenetic control of organelle transport and positioning, Nature 518, 111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shi F, Kawano F, Park SE, Komazaki S, Hirabayashi Y, Polleux F & Yazawa M (2018) Optogenetic Control of Endoplasmic Reticulum-Mitochondria Tethering, ACS Synth Biol 7, 2–9. [DOI] [PubMed] [Google Scholar]
- 93.Bulina ME, Chudakov DM, Britanova OV, Yanushevich YG, Staroverov DB, Chepurnykh TV, Merzlyak EM, Shkrob MA, Lukyanov S & Lukyanov KA (2006) A genetically encoded photosensitizer, Nat Biotechnol 24, 95–9. [DOI] [PubMed] [Google Scholar]
- 94.Onukwufor JO, Trewin AJ, Baran TM, Almast A, Foster TH & Wojtovich AP (2020) Quantification of reactive oxygen species production by the red fluorescent proteins KillerRed, SuperNova and mCherry, Free Radic Biol Med 147, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Trewin AJ, Berry BJ, Wei AY, Bahr LL, Foster TH & Wojtovich AP (2018) Light-induced oxidant production by fluorescent proteins, Free Radic Biol Med 128, 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Trewin AJ, Bahr LL, Almast A, Berry BJ, Wei AY, Foster TH & Wojtovich AP (2019) Mitochondrial Reactive Oxygen Species Generated at the Complex-II Matrix or Intermembrane Space Microdomain Have Distinct Effects on Redox Signaling and Stress Sensitivity in Caenorhabditis elegans, Antioxid Redox Signal 31, 594–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Qian W, Kumar N, Roginskaya V, Fouquerel E, Opresko PL, Shiva S, Watkins SC, Kolodieznyi D, Bruchez MP & Van Houten B (2019) Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction, Proc Natl Acad Sci U S A 116, 18435–18444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Siddikuzzaman & Grace VM (2013) Antioxidant potential of all-trans retinoic acid (ATRA) and enhanced activity of liposome encapsulated ATRA against inflammation and tumor-directed angiogenesis, Immunopharmacol Immunotoxicol 35, 164–73. [DOI] [PubMed] [Google Scholar]
- 99.De Magalhaes Filho CD, Henriquez B, Seah NE, Evans RM, Lapierre LR & Dillin A (2018) Visible light reduces C. elegans longevity, Nat Commun 9, 927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Godley BF, Shamsi FA, Liang FQ, Jarrett SG, Davies S & Boulton M (2005) Blue light induces mitochondrial DNA damage and free radical production in epithelial cells, J Biol Chem 280, 21061–6. [DOI] [PubMed] [Google Scholar]
- 101.Yang MY, Chang CJ & Chen LY (2017) Blue light induced reactive oxygen species from flavin mononucleotide and flavin adenine dinucleotide on lethality of HeLa cells, J Photochem Photobiol B 173, 325–332. [DOI] [PubMed] [Google Scholar]
- 102.Pryde KR & Hirst J (2011) Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer, J Biol Chem 286, 18056–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yi HS, Chang JY & Shong M (2018) The mitochondrial unfolded protein response and mitohormesis: a perspective on metabolic diseases, J Mol Endocrinol 61, R91–R105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang YT, Lim Y, McCall MN, Huang KT, Haynes CM, Nehrke K & Brookes PS (2019) Cardioprotection by the mitochondrial unfolded protein response requires ATF5, Am J Physiol Heart Circ Physiol 317, H472–H478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Murphy MP & Hartley RC (2018) Mitochondria as a therapeutic target for common pathologies, Nat Rev Drug Discov 17, 865–886. [DOI] [PubMed] [Google Scholar]
- 106.Yang JL, Mukda S & Chen SD (2018) Diverse roles of mitochondria in ischemic stroke, Redox Biol 16, 263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]