

Abstract
Among the plethora of catalytic methods developed for hydrocarbofunctionalization of olefins to date, reactions that regioselectively install a functionalized alkyl unit at the 2-position of a terminal unactivated C=C bond to afford branched products are scarce. Here, we show that a Ni-based catalyst in conjunction with a stoichiometric reducing agent promote Markovnikov-selective hydroalkylation of unactivated alkenes tethered to a recyclable 8-aminoquinaldine directing auxiliary. These mild reductive processes employ readily available primary and secondary haloalkanes as both the hydride and alkyl donor. Reactions of alkenyl amides with ≥ five-carbon chain length regioselectively afforded β-alkylated products through remote hydroalkylation, underscoring the fidelity of the catalytic process and the directing group’s capability in stabilizing five-membered nickelacycle intermediates. The operationally simple protocol exhibits exceptional functional group tolerance and is amenable to the synthesis of bioactive molecules as well as regioconvergent transformations.
Subject terms: Homogeneous catalysis, Reaction mechanisms, Synthetic chemistry methodology
Methods that regioselectively install a functionalized alkyl unit at the 2-position of a terminal unactivated C=C bond are scarce. Here, the authors report a Markovnikov-selective hydroalkylation of unactivated amide-tethered alkenes catalyzed by nickel in conjunction with a stoichiometric reductant.
Introduction
The abundance, low cost and distinct reactivity profiles of alkenes have enabled these feedstock molecules to be widely utilized in olefin functionalization reactions for various chemical synthesis applications1–3. In this respect, the installation of a hydrogen and carbon-based moiety across π-systems represents an effective strategy for C–C bond construction4–6. Numerous hydrocarbofunctionalization protocols rely on conjugation (i.e., 1,3-dienes7–11, olefins such as styrenes12–14, alkenyl boronates15,16, and Michael acceptors17) to deliver high regioselectivity. In contrast, reactions with unactivated aliphatic C=C bonds are typically plagued by lower substrate reactivity and/or poorer regiochemical differentiation leading to unsatisfactory levels of site selectivity. Notwithstanding these difficulties, remarkable advances for both anti-Markovnikov18–25 (linear)- and Markovnikov26–30 (branched)-selective hydroarylations of alkyl-substituted alkenes have been made. A single report on Pd-catalyzed olefin hydroalkynylation/hydroalkenylation to afford linear products was also recently disclosed31. On the other hand, there is growing demand for methods that furnish C–C bonds between two sp3-hybridized motifs, which are crucial for assembling the skeletal backbone of organic entities en route to bioactive compounds32,33. Accordingly, hydroalkylations across aliphatic olefins have been devised, although the vast majority involved linear-selective additions (Fig. 1a). Of these cases, either a limited range of ureas34, organometallic reagents35,36 or carbonyl compounds37–39 were employed as nucleophiles, or an exogenous protic source40 or hydrosilane/base reagent41–45 is needed to (i) promote protodemetallation or (ii) generate the requisite metal-hydride species.
Fig. 1. The significance of developing branched- and β-selective hydroalkylation of unactivated alkenes.

a Reported methods that involve anti-Markovnikov-selective hydroalkylation of aliphatic olefins. b Reported methods that involve Markovnikov-selective hydroalkylation of aliphatic olefins. c An attractive catalytic approach for Markovnikov- and β-selective olefin hydroalkylation takes advantage of haloalkanes to transfer the hydride and alkyl motif under mild reductive conditions without additional hydrosilane, acidic or basic additives. d The resulting hydroalkylation products are versatile building blocks that may be used to access biologically active molecules. R, functional group; Phth, phthaloyl; pyr., pyridinium; cat., catalyst.
To date, highly Markovnikov-selective hydroalkylations of aliphatic π-systems have been largely achieved with 1,1-disubstituted/trisubstituted alkenes and primary haloalkanes through a Mn/Ni dual catalytic metal-hydride hydrogen atom transfer approach46, or with olefins linked to a tridendate directing group and 1,3-dicarbonyl nucleophiles using a Pd-based catalyst40 (Fig. 1b). Therefore, there is compelling motivation to develop a complementary catalytic regime that accomplishes efficient and branched-selective hydroalkylation of unactivated acyclic olefins with exceptional control of regioselectivity in the presence of commonly occurring functionalities. For operational simplicity, we speculated that aliphatic halides could serve as mild donors of both the hydride (by facile in situ β-H elimination47,48) and alkyl component without external acidic or basic additives, which might otherwise compromise functional group compatibility (Fig. 1c). The products resulting from successful implementation of this strategy can be readily elaborated to a variety of important biologically active compounds (e.g., 1–3, Fig. 1d). Herein, we report a directed Ni-catalyzed reductive protocol that achieves these goals.
Results
Reaction design and optimization
A hallmark of catalytic reductive transformations49–57 is the use of stoichiometric amounts of an inexpensive reducing agent to drive single-electron transfer processes mediated by an appropriate (e.g., Ni-based) catalyst. This led us to conceive a reductive strategy for alkene hydroalkylation that takes advantage of the characteristic mild reaction conditions. Specifically, we aimed to avoid the use of hydrosilanes, acidic and basic reagents that could engender undesired side reactions with certain sensitive functional units (see below for further discussion). However, the question remains how we could effectively deliver hydride to the C=C bond in this scenario.
To this end, we envisioned a catalytic approach that merges aliphatic alkenes tethered to a suitable directing unit 458 with alkyl halides directly in the presence of a Ni-based catalyst and a reductant (Fig. 2). This strategy relies on the propensity of haloalkanes to undergo facile β-H elimination, an observation that was previously exploited in Ni catalysis to generate catalytic amounts of nickel-hydride species47,48. As illustrated in the putative catalytic pathway, a Ni(0) species i could first associate with 4 and react with an equivalent of the alkyl halide through a halogen atom abstraction/radical recombination59,60 process to give ii. At this juncture, a directing auxiliary with appropriate steric and/or electronic properties could induce alkene dissociation from the metal center in ii, providing an opportunity for the Ni–alkyl moiety to preferentially undergo β-H elimination47,48 leading to nickel-hydride iii with concomitant discharge of an alkene by-product (vs. olefin alkylnickelation to form viii). Regioselective β-hydride insertion across the associated C=C bond in iii then affords quinoline-chelated Ni(II) species iv. Following single-electron reduction with a reducing agent, Ni(I) complex v would be generated that could subsequently react with a second equivalent of alkyl halide to furnish vi. Reductive elimination of vi furnishes the desired hydroalkylation product 5 and vii, which eventually gets reduced back to i to turn over the catalytic cycle. The key to efficient transformation of 4 to 5 entails faster conversion of intermediate ii to iv via iii (vs. ii to viii) in order to suppress adventitious formation of the undesired dialkylation adduct 6.
Fig. 2. The challenges involved in developing site-selective reductive hydroalkylation reactions.

As shown in the Ni catalytic cycle, the key to efficient transformation of 4 to 5 requires faster conversion of intermediate ii to iv in order to suppress adventitious formation of the undesired dialkylation adduct 6. Under the reductive conditions, both the hydrogen and alkyl unit are derived from the haloalkane reagent. R, G, functional group; X, halide; L, ligand; cat., catalyst.
We first examined conditions that facilitate hydroalkylation of β,γ-unsaturated amides with 1-iodobutane 8 (Table 1). After an extensive survey, we found that the reaction between 8-aminoquinaldine-tethered alkene 7a and 8 (2 equiv.) using NiCl2(PPh3)2 (15 mol%), manganese (1.5 equiv.) as reductant and NMP as solvent gave the best results, affording 9a in 90% GC (88% isolated) yield and complete Markovnikov selectivity at ambient temperature (Table 1, entry 1). There was no appreciable diminution in yield or selectivity when the reaction was carried out on larger scale (2 mmol). Intriguingly, changing the directing group to other variants gave poorer yields and site selectivities, demonstrating the unexpected beneficial role of the ortho-methyl appendage. We reasoned that the effect might arise from an elevated steric strain inherent within complex ii (cf. Fig. 2), forcing Ni-olefin dissociation and allowing the sterically less demanding Cβ–H bond to coordinate and trigger β-H elimination47,48 prior to the ensuing β-H insertion to iv. Further studies to rationalize this effect will be reported in due course.
Table 1.
Evaluation of reaction conditions for reductive hydroalkylation.
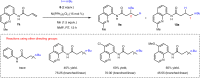 | |||
|---|---|---|---|
| Entry | Deviation from standard conditions | Yield (%) | 9a:10aa |
| 1 | None | 90(88)(81b) | >95:5 |
| 2 | NiCl2, NiCl2·DME, NiCl2(Py)4, NiI2 or Ni(COD)2 instead of NiCl2(PPh3)2 | 50–74 | 92:8–>95:5 |
| 3 | DMF, DMA, DMPU or MeCN instead of NMP | 70–92 | 92:8–94:6 |
| 4 | DMSO, THF or toluene instead of NMP | Trace–10 | ND |
| 5 | No Mn | Trace | ND |
| 6 | Mn (1 equiv.) instead of Mn (1.5 equiv.) | 70 | >95:5 |
| 7 | Mn (2 equiv.) instead of Mn (1.5 equiv.) | 90 | >95:5 |
| 8 | Zn instead of Mn | 44 | 80:20 |
| 9 | 8 (1.5 equiv.) instead of 8 (2 equiv.) | 70 | >95:5 |
| 10 | 8 (2.5 equiv.) instead of 8 (2 equiv.) | 91 | >95:5 |
| 11 | 40 °C instead of RT | 90 | >95:5 |
| 12 | 10 mol % NiCl2(PPh3)2 instead of 15 mol % | 92 | 92:8 |
Reactions were carried out on 0.1 mmol scale.
DMA N,N-dimethylacetamide, DMF N,N-dimethylformamide, NMP N-methyl-2-pyrrolidone, DMPU N,N′-dimethylpropyleneurea, DMSO dimethyl sulfoxide, THF tetrahydrofuran, DME 1,2-dimethoxyethane, Py pyridine, COD 1,5-cyclooctadiene, RT room temperature, ND not determined.
aYields and regioisomeric ratios were determined by GC analysis with n-tridecane as internal standard. Values in parentheses denote yields for isolated and purified products.
bThe reaction was conducted on 2 mmol scale.
Changing the Ni-based catalyst afforded lower yields across the board (Table 1 entry 2), while replacing NMP with other solvents did not improve results (Table 1, entries 3–4). Unsurprisingly, the reaction did not proceed in the absence of Mn, with 1.5 equiv. of Mn being optimal (Table 1, entries 5–7). Switching Mn to Zn as the reductant led to a drastic reduction in yield and regioselectivity (Table 1 entry 8). Decreasing the equivalents of 8 resulted in diminished reaction efficiency, whereas no appreciable improvement was detected at higher loadings (Table 1 entries 9–10). Carrying out the reaction at 40 °C gave similar results as RT (Table 1, entry 11). A slight drop in site selectivity was observed with 10 mol% loading of NiCl2(PPh3)2 (Table 1, entry 12). Performing the hydroalkylation by replacing Mn with hydrosilane/base as the hydride source41–45 led to lower yields of 9a (see Supplementary Methods 2.6 for details), highlighting the importance of the reductive conditions.
Substrate scope
With the established conditions in hand, we proceeded to evaluate the scope by examining various functionalized aliphatic halides and alkenyl amides (Fig. 3). Primary alkyl iodides and bromides containing different functional groups underwent efficient hydroalkyl additions to 7a, furnishing the desired branched compounds 9b–w in 32–94% yield and high regioselectivities. Transformations with less activated organobromides were performed at 60 °C for optimal efficiency. These include products that contain an ester (9d), an enoate (9e), an aldehyde (9h), an alkene (9n) as well as unprotected protic units (problematic with basic organometallic reagents)57,61 such as carboxylic acid (9k), alcohol (9p–q) and phenol (9r). It merits mention that previous methods which rely on hydrosilane/base to generate the hydride source may cause undesired silylation side reactions with hydroxyl groups46. Both acid-labile (acetal 9o) and base-labile (9k, 9r and boronate 9t) functionalities, which could be vulnerable under conditions that require acid/base additives40–46, as well as Lewis basic heterocyclic motifs (9f, 9u–v) are tolerated. Among the products bearing a derivatizable halogen appendage (9i–j, 9s), the reaction that afforded 9j highlights the inherent chemoselectivity of an iodoalkane over an aryl iodide, although arene hydrodeiodination was detected as a side reaction. The reductive protocol is also compatible with substrates derived from complex multifunctional bioactive molecules such as base-sensitive sulbactam62 (9l) and indometacin (9m).
Fig. 3. The range of products accessible by reductive olefin hydroalkylation.

The protocol is compatible with both primary and secondary alkyl halides bearing Brønsted/Lewis acidic and basic functionalities, including those derived from complex bioactive molecules. Both mono- and 1,2-disubstituted alkenyl amides are tolerated in the catalytic system. For 9w, the reaction was conducted with neopentyl bromide (3 equiv.) and isopropyl bromide (1.2 equiv.) using NiI2 as the catalyst. For 9ac and 9af, reactions were conducted using iodides (X = I). For 9ad, 9ae, and 9ag, reactions were conducted using bromides (X = Br). For 9j, ~20% of an inseparable hydrodeiodination side product was detected. For 9m, ~3% of an inseparable self-coupling side product of iodide substrate was detected. 9g, 9i, 9u, 9ab, and 9af were obtained as 88:12, 93:7, 92:8, 90:10, and 91:9 regioisomeric mixtures, respectively. 9l and 9z were obtained as 5:1 and 1:1 diastereomeric mixtures, respectively. Regioisomeric and diastereomeric ratios were determined by 1H NMR analysis. Yields are for isolated and purified products. R, functional group; X, halide; NMP, N-methyl-2-pyrrolidone; Boc, tert-butoxycarbonyl.
Secondary alkyl iodides and bromides also served as effective reagents under the standard conditions, delivering the expected products 9x–ab in 51–73% yield and offering complementary scope to a previous report in which secondary haloalkanes were low-yielding46. However, organohalides that lack a Cβ–H bond and therefore incapable of generating the requisite nickel-hydride species by β-H elimination pose a challenge in our system. Attempts to carry out reductive hydroalkylation using neopentyl bromide (3 equiv.) as alkyl donor and isopropyl bromide (1.2 equiv.) as hydride donor47,48 gave the desired hydroalkylation adduct 9w in 32% yield, along with side products derived from dialkylation with neopentyl bromide as well as reductive hydroalkylation with isopropyl bromide (see Supplementary Methods 2.7 for details). β,γ-Unsaturated amides with 1,2-disubstitiuted C=C bonds underwent reaction to form the corresponding β-alkylated products (9ac–ag), but those with 1,1-disubstituted and trisubstituted olefins were ineffective substrates (<5% conv. to desired product).
Synthetic applications
The first application that demonstrates utility of our Markovnikov-selective reductive hydroalkylation protocol involves the synthesis of a family of therapeutic compounds 1 for the treatment of metabolic disorders63 (Fig. 4a). Chemoselective removal of the amide directing group in 9y (recovered 8-aminoquinaldine in 95% yield) afforded acid 11, a key intermediate employed in the preparation of 1, in 83% yield.
Fig. 4. Application to synthesis of biologically active molecules and remote hydroalkylation.

a The products resulting from reductive hydroalkylation can be conveniently transformed to a variety of medicinal compounds of interest. b Alkenyl amides bearing ≥ five-carbon chain length undergo remote hydroalkylation through in situ isomerization of alkylnickel intermediates, providing reliable access to β-alkylated molecules. Regioisomeric olefin mixtures can be regioconvergently converted to a single value-added product. Regioisomeric ratios were determined by 1H NMR analysis. Yields are for isolated and purified products. 9ah was obtained as a 96:4 regioisomeric mixture. For 18a, 18d, 18e, and 18g, reactions were conducted using iodides (X = I). For 18b, 18c, and 18f, reactions were conducted using bromides (X = Br). For 18g, 5 equiv. of 1-iodobutane was used. R, functional group; X, halide; NMP, N-methyl-2-pyrrolidone; DMA, N,N-dimethylacetamide; NHPI, N-hydroxyphthalimide; DIC, N,N′-diisopropylcarbodiimide; DMAP, 4-dimethylaminopyridine; dtbbpy, 4,4′-di-tert-butyl-2,2′-bipyridine; Boc, tert-butoxycarbonyl; Phth, phthaloyl; Cp, cyclopentadienyl; RT, room temperature.
In a second instance, chemoselective reduction of the 8-aminoquinaldine amide in 9ah to aldehyde 12 followed by reductive amination with benzylamine 13 and N-Boc protection delivered 14, a precursor for the synthesis of an anti-cancer compound 264, in 51% overall yield. The entire sequence is more concise compared to a previous report64. The preparation of 16 highlights yet another compelling example of the versatility of the branched hydroalkylation products. Facile conversion of 9ai to its redox-active ester derivative 15 (70% overall yield) set the stage for a catalytic decarboxylative cross-coupling53 with 1-(benzyloxy)-3-iodobenzene to furnish 16, which has been further elaborated to another anti-cancer agent 365.
A corollary to the present reductive approach is the implementation of remote olefin hydroalkylation66–70, specifically with alkenyl amides 17 containing an extended hydrocarbon backbone (≥ five-carbon chain length). As showcased in Fig.4b, we postulated that the organonickel intermediate ix generated from nickel-hydride addition (cf. Fig. 2) could potentially isomerize to the relatively more stabilized five-membered nickelacycle x66,67,71 through consecutive β-H elimination/olefin insertion steps. Following a similar mechanism in Fig. 2, x could be converted to 18 with net remote alkylation at the β position. Although nickel-hydride-promoted remote hydrocarbonfunctionalizations have been disclosed, most instances involve functionalizations at either the sterically exposed terminus or the α-carbon site adjacent to an electron-stabilizing moiety68,69.
Gratifyingly, γ,δ-unsaturated amides bearing terminal and internal C=C bonds were found to participate in remote hydroalkylation with primary and secondary haloalkanes, affording products 18a–e in 47–82% yield as single β regioisomers through single double-bond migrations (Fig. 4b). Cyclic olefins also underwent reaction as exemplified by 18e, which was obtained as a single syn diastereomer71. Using the regioisomeric β,γ-unsaturated amide substrate leading to 18e would be, however, less practical since the corresponding β,γ-unsaturated carboxylic acid is much more expensive. Transformations with δ,ε-unsaturated amides (involving two C=C bond isomerizations) were similarly efficient and site-selective (18d and 18f). Remarkably, subjecting the 6-heptenoic acid-derived alkenyl amide to established conditions gave β-alkylated 18g in 60% yield, underscoring the fidelity of the catalytic process that features an alkene transposition over three positions67,68. Furthermore, a streamlined synthesis of β-alkylated 18d could be attained in 62% yield through regioconvergent hydroalkylation of an isomeric mixture of olefin substrates (Fig. 4b, gray inset).
Mechanistic studies
Subjecting the α,β-unsaturated isomer of 7a to the standard conditions only gave the fully hydrogenated product 19, thereby ruling out the likelihood of olefin isomerization prior to the hydroalkylation event (Fig. 5a). To shed light on the importance of the alkyl halide partner in our catalytic system, we carried out deuterium labeling studies with 7a in the presence of two equivalents of d-20 under the standard reaction conditions (Fig. 5b). Accordingly, the hydroalkylation product d-9aj and olefin by-product d-21 were isolated. Deuterium incorporation on C1 of d-9aj and deuterium scrambling in d-21 suggest that a nickel-deuteride species was likely generated from reaction with d-20 (i.e., ii→iii in Fig. 2). The reversible nature of β-H(D) elimination and re-insertion means that adventitious deuterium scrambling in d-21 and formation of Ni–H cannot be avoided. Competitive addition of Ni–H and Ni–D across the olefin in 7a eventually gave rise to d-9aj with 40% D incorporation at C1. Overall, these results support the haloalkane’s key role as a donor of both the hydride and alkyl group.
Fig. 5. Mechanistic studies.

a Control experiment ruling out the intermediacy of 7a′. b Deuterium labeling experiment. c Radical clock experiment. d Complete stereochemical erosion with an enantioenriched alkyl halide. Diastereomeric ratios were determined by 1H NMR analysis. Enantiomeric ratios were determined by chiral HPLC analysis. Yields are for isolated and purified products. NMP, N-methyl-2-pyrrolidone; RT, room temperature.
The radical nature of the reductive hydroalkylation reaction was substantiated through a radical clock experiment using (bromomethyl)cyclopropane 22 as an electrophile (Fig. 5c). A 20:80 mixture of 9ak and the ring-ruptured product 9n in 84% yield was detected, intimating that 22 likely reacts with the catalytic organonickel species (cf. i→ii and v→vi in Fig. 2) through a bromine abstraction/radical recombination process59,60 via a cyclopropylmethyl radical that is prone to ring opening. Further support for the intermediacy of radicals could be obtained from the corresponding reactions using enantioenriched 23, in which the desired product 9al was generated in 51–73% yield and ~70:30 d.r. as a racemic mixture (Fig. 5d).
In summary, we demonstrated that Ni-catalyzed branched-selective hydroalkyl additions to unactivated alkenyl amides can be achieved by using haloalkanes and manganese as reductant. With long-chain olefins, remote and regioconvergent hydroalkylations proceed to furnish products with reliable β selectivities. Mechanistic experiments corroborate the aliphatic halide’s dual role as a hydride and alkyl donor. Equally crucial is the 8-aminoquinaldine tether that effectively suppresses any adventitious dialkylation side reaction. The robust conditions are compatible with diverse functional groups, including those that are sensitive to hydrosilane, acidic or basic additives. In conjunction with existing methods, we expect our catalytic strategy to find significant utility in chemical synthesis.
Methods
General reductive hydroalkylation procedure
In a N2-filled glovebox, to an oven-dried 5 mL vial equipped with a magnetic stir bar were added alkene substrate (0.1 mmol), alkyl iodide or bromide (if solid, added at this time) (0.2 mmol), Ni(PPh3)2Cl2 (9.8 mg, 0.015 mmol) and Mn powder (0.15 mmol). The mixture was then dissolved in 0.3 mL dry NMP. The vial was tightly capped and removed from the glovebox. The alkyl iodide or bromide (if liquid, added at this time) was added by a micro-syringe. The mixture was allowed to vigorously stir at ambient temperature (for alkyl iodide) or 60 °C (for alkyl bromide) for 12–20 h. When alkene was almost fully consumed (monitored by TLC), the mixture was directly subjected to flash silica gel column chromatography to afford the pure product.
Supplementary information
Acknowledgements
This research was supported by the National University of Singapore Academic Research Fund Tier 1: R-143-000-A77-114 (M.J.K.). X.C. acknowledges support from Project Funded by the National First-class Disciplines (PNFD). W.R. acknowledges support from Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Author contributions
X.C., W.R., and T.Y. developed the catalytic method. M.J.K. and T.Y. directed the investigations. M.J.K. wrote the paper with revisions provided by the other authors.
Data availability
All data are available from the corresponding authors upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks Christian D.-T. Nielsen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Tao Yang, Email: chmyt@nus.edu.sg.
Ming Joo Koh, Email: chmkmj@nus.edu.sg.
Supplementary information
Supplementary information is available for this paper at 10.1038/s41467-020-19717-6.
References
- 1.Saini V, Stokes BJ, Sigman MS. Transition-metal-catalyzed laboratory-scale carbon-carbon bond-forming reactions of ethylene. Angew. Chem. Int. Ed. 2013;52:11206–11220. doi: 10.1002/anie.201303916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coombs JR, Morken JP. Catalytic enantioselective functionalization of unactivated terminal alkenes. Angew. Chem. Int. Ed. 2016;55:2636–2649. doi: 10.1002/anie.201507151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McDonald RI, Liu GS, Stahl SS. Palladium(II)-catalyzed alkene functionalization via nucleopalladation: stereochemical pathways and enantioselective catalytic applications. Chem. Rev. 2011;111:2981–3019. doi: 10.1021/cr100371y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beller M, Seayad J, Tillack A, Jiao H. Catalytic markovnikov and anti-markovnikov functionalization of alkenes and alkynes: recent developments and trends. Angew. Chem. Int. Ed. 2004;43:3368–3398. doi: 10.1002/anie.200300616. [DOI] [PubMed] [Google Scholar]
- 5.Crossley SWM, Obradors C, Martinez RM, Shenvi RA. Mn-, Fe-, and Co-Catalyzed radical hydrofunctionalizations of olefins. Chem. Rev. 2011;116:8910–9000. doi: 10.1021/acs.chemrev.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yadav JS, Antony A, Rao TS, Reddy BVS. Recent progress in transition metal catalyzed hydrofunctionalisation of less activated olefins. J. Organmet. Chem. 2011;696:16–36. doi: 10.1016/j.jorganchem.2010.09.052. [DOI] [Google Scholar]
- 7.Baker R, Popplestone RJ. Reactions of active methylene and carbonyl compounds with myrcene catalyzed by palladium and nickel complexes. Tetrahedron Lett. 1978;19:3575–3578. doi: 10.1016/S0040-4039(01)94998-6. [DOI] [Google Scholar]
- 8.Yang XH, Dong VM. Rhodium-catalyzed hydrofunctionalization: enantioselective coupling of indolines and 1,3-dienes. J. Am. Chem. Soc. 2017;139:1774–1777. doi: 10.1021/jacs.6b12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng L, Li MM, Xiao LJ, Xie JH, Zhou QL. Nickel(0)-catalyzed hydroalkylation of 1,3-dienes with simple ketones. J. Am. Chem. Soc. 2018;140:11627–11630. doi: 10.1021/jacs.8b09346. [DOI] [PubMed] [Google Scholar]
- 10.Chen XW, et al. Highly selective and catalytic generation of acyclic quaternary carbon stereocenters via functionalization of 1,3-dienes with CO2. J. Am. Chem. Soc. 2019;141:18825–18835. doi: 10.1021/jacs.9b09721. [DOI] [PubMed] [Google Scholar]
- 11.Lv L, Zhu D, Qiu Z, Li J, Li C-J. Nickel-catalyzed regioselective hydrobenzylation of 1,3-dienes with hydrazones. ACS Catal. 2019;9:9199–9205. doi: 10.1021/acscatal.9b02483. [DOI] [Google Scholar]
- 12.Gligorich KM, Cummings, Sarah A, Sigman MS. Palladium-catalyzed reductive coupling of styrenes and organostannanes under aerobic conditions. J. Am. Chem. Soc. 2007;129:14193–14195. doi: 10.1021/ja076746f. [DOI] [PubMed] [Google Scholar]
- 13.Xiao L-J, et al. Nickel(0)-catalyzed hydroarylation of styrenes and 1,3-dienes with organoboron compounds. Angew. Chem. Int. Ed. 2018;57:461–464. doi: 10.1002/anie.201710735. [DOI] [PubMed] [Google Scholar]
- 14.Kumar GS, et al. Nickel-catalyzed chain-walking cross-electrophile coupling of alkyl and aryl halides and olefin hydroarylation enabled by electrochemical reduction. Angew. Chem. Int. Ed. 2020;59:6513–6519. doi: 10.1002/anie.201915418. [DOI] [PubMed] [Google Scholar]
- 15.Bera S, Hu X. Nickel-catalyzed regioselective hydroalkylation and hydroarylation of alkenyl boronic esters. Angew. Chem. Int. Ed. 2019;58:13854–13859. doi: 10.1002/anie.201907045. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Han B, Zhu SL. Rapid access to highly functionalized alkyl boronates by NiH-catalyzed remote hydroarylation of boron-containing alkenes. Angew. Chem. Int. Ed. 2019;58:13860–13864. doi: 10.1002/anie.201907185. [DOI] [PubMed] [Google Scholar]
- 17.Lan Y, et al. An iron-catalyzed hydroalkylation reaction of α,ß-unsaturated ketones with ethers. Chem. Commun. 2017;53:12353–12356. doi: 10.1039/C7CC07235J. [DOI] [PubMed] [Google Scholar]
- 18.Dong Z, Re Z, Thompson SJ, Xu Y, Dong G. Transition-metal-catalyzed C–H alkylation using alkenes. Chem. Rev. 2017;117:9333–9403. doi: 10.1021/acs.chemrev.6b00574. [DOI] [PubMed] [Google Scholar]
- 19.Friis SD, Pirnot MT, Dupuis LN, Buchwald SL. A dual palladium and copper hydride catalyzed approach for alkyl–aryl cross-coupling of aryl halides and olefins. Angew. Chem. Int. Ed. 2017;56:7242–7246. doi: 10.1002/anie.201703400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boyington AJ, Riu M-LY, Jui NT. Anti-markovnikov hydroarylation of unactivated olefins via pyridyl radical intermediates. J. Am. Chem. Soc. 2017;139:6582–6585. doi: 10.1021/jacs.7b03262. [DOI] [PubMed] [Google Scholar]
- 21.Bair JS, et al. Linear-selective hydroarylation of unactivated terminal and internal olefins with trifluoromethyl-substituted arenes. J. Am. Chem. Soc. 2014;136:13098–13101. doi: 10.1021/ja505579f. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen J, Chong A, Lalic G. Nickel-catalyzed anti-Markovnikov hydroarylation of alkenes. Chem. Sci. 2019;10:3231–3236. doi: 10.1039/C8SC05445B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gurak JA, Jr., Engle KM. Practical intermolecular hydroarylation of diverse alkenes via reductive heck coupling. ACS Catal. 2018;8:8987–8992. doi: 10.1021/acscatal.8b02717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang C, et al. Palladium-catalyzed regiocontrollable reductive heck reaction of unactivated aliphatic alkenes. J. Am. Chem. Soc. 2018;140:9332–9336. doi: 10.1021/jacs.8b03619. [DOI] [PubMed] [Google Scholar]
- 25.Saper NI, et al. Nickel-catalysed anti-Markovnikov hydroarylation of unactivated alkenes with unactivated arenes facilitated by non-covalent interactions. Nat. Chem. 2020;12:276–283. doi: 10.1038/s41557-019-0409-4. [DOI] [PubMed] [Google Scholar]
- 26.Ma X, Herzon SB. Intermolecular hydropyridylation of unactivated alkenes. J. Am. Chem. Soc. 2016;138:8718–8721. doi: 10.1021/jacs.6b05271. [DOI] [PubMed] [Google Scholar]
- 27.Green SA, Matos JLM, Yagi A, Shenvi RA. Branch-selective hydroarylation: iodoarene–olefin cross-coupling. J. Am. Chem. Soc. 2016;138:12779–12782. doi: 10.1021/jacs.6b08507. [DOI] [PubMed] [Google Scholar]
- 28.Shevick SL, Obradors C, Shenvi RA. Mechanistic interrogation of Co/Ni-dual catalyzed hydroarylation. J. Am. Chem. Soc. 2018;140:12056–12068. doi: 10.1021/jacs.8b06458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen F, et al. Remote migratory cross-electrophile coupling and olefin hydroarylation reactions enabled by in situ generation of NiH. J. Am. Chem. Soc. 2017;139:13929–13935. doi: 10.1021/jacs.7b08064. [DOI] [PubMed] [Google Scholar]
- 30.Green SA, Vasquez-Céspedes S, Shenvi RA. Iron–nickel dual-catalysis: a new engine for olefin functionalization and the formation of quaternary centers. J. Am. Chem. Soc. 2018;140:11317–11324. doi: 10.1021/jacs.8b05868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bai Z, et al. Palladium-catalyzed amide directed hydrocarbon-functionalization of 3-alkenamides with alkynes. ACS Catal. 2020;10:933–940. doi: 10.1021/acscatal.9b04285. [DOI] [Google Scholar]
- 32.Geist E, Kirschning A, Schmidt T. sp3-sp3 Coupling reactions in the synthesis of natural products and biologically active molecules. Nat. Prod. Rep. 2014;31:441–448. doi: 10.1039/c3np70108e. [DOI] [PubMed] [Google Scholar]
- 33.Lovering F, Bikker J, Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 34.Yamauchi D, Nishimura T, Yorimitsu H. Hydroxoiridium-catalyzed hydroalkylation of terminal alkenes with ureas by C(sp3)-H bond activation. Angew. Chem. Int. Ed. 2017;56:7200–7204. doi: 10.1002/anie.201702169. [DOI] [PubMed] [Google Scholar]
- 35.DeLuca RJ, Sigman MS. Anti-markovnikov hydroalkylation of allylic amine derivatives via a palladium-catalyzed reductive cross-coupling reaction. J. Am. Chem. Soc. 2011;133:11454–11457. doi: 10.1021/ja204080s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeLuca RJ, Sigman MS. The palladium-catalyzed anti-markovnikov hydroalkylation of allylic alcohol derivatives. Org. Lett. 2013;15:92–95. doi: 10.1021/ol303129p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang KS, Gurak JA, Jr., Liu Z, Engle KM. Catalytic, regioselective hydrocarbofunctionalization of unactivated alkenes with diverse C–H nucleophiles. J. Am. Chem. Soc. 2016;138:14705–14712. doi: 10.1021/jacs.6b08850. [DOI] [PubMed] [Google Scholar]
- 38.Shen H-C, et al. Enantioselective addition of cyclic ketones to unactivated alkenes enabled by amine/Pd(II) cooperative catalysis. ACS Catal. 2019;9:791–797. doi: 10.1021/acscatal.8b04654. [DOI] [Google Scholar]
- 39.Sayed M, Shen H-C, Zhang L, Gong L-Z. Hydroalkylation of unactivated alkenes with ketones and 5-benzylfurfurals enabled by amine/Pd(II) cooperative catalysis. Synthesis. 2020;52:703–710. doi: 10.1055/s-0039-1690245. [DOI] [Google Scholar]
- 40.O’Duill ML, et al. Tridentate directing groups stabilize 6-membered palladacycles in catalytic alkene hydrofunctionalization. J. Am. Chem. Soc. 2017;139:15576–15579. doi: 10.1021/jacs.7b08383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu X, et al. Practical carbon–carbon bond formation from olefins through nickel-catalyzed reductive olefin hydrocarbonation. Nat. Commun. 2016;7:11129. doi: 10.1038/ncomms11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Z, Yin H, Fu GC. Catalytic enantioconvergent coupling of secondary and tertiary electrophiles with olefins. Nature. 2018;563:379–383. doi: 10.1038/s41586-018-0669-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu X, Xiao Bin, Liu L, Fu Y. Formation of C(sp3)–C(sp3) bonds through nickel-catalyzed decarboxylative olefin hydroalkylation reactions. Chem. Eur. J. 2016;22:11161–11164. doi: 10.1002/chem.201602486. [DOI] [PubMed] [Google Scholar]
- 44.Sun S-Z, Romano C, Martin R. Site-selective catalytic deaminative alkylation of unactivated olefins. J. Am. Chem. Soc. 2019;141:16197–16201. doi: 10.1021/jacs.9b07489. [DOI] [PubMed] [Google Scholar]
- 45.Qian D, Hu X. Ligand-controlled regiodivergent hydroalkylation of pyrrolines. Angew. Chem. Int. Ed. 2019;58:18519–18523. doi: 10.1002/anie.201912629. [DOI] [PubMed] [Google Scholar]
- 46.Green SA, Huffman TR, McCourt RO, Puyl V, Shenvi RA. Hydroalkylation of olefins to form quaternary carbons. J. Am. Chem. Soc. 2019;141:7709–7714. doi: 10.1021/jacs.9b02844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Nakajima M, Serrano E, Martin R. Alkyl bromides as mild hydride sources in Ni-catalyzed hydroamidation of alkynes with isocyanates. J. Am. Chem. Soc. 2016;138:15531–15534. doi: 10.1021/jacs.6b10351. [DOI] [PubMed] [Google Scholar]
- 48.Juliá-Hernández F, Moragas T, Cornella J, Martin R. Remote carboxylation of halogenated aliphatic hydrocarbons with carbon dioxide. Nature. 2017;545:84–88. doi: 10.1038/nature22316. [DOI] [PubMed] [Google Scholar]
- 49.Weix DJ. Methods and mechanisms for cross-electrophile coupling of Csp2 halides with alkyl electrophiles. Acc. Chem. Res. 2015;48:1767–1775. doi: 10.1021/acs.accounts.5b00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gu J, Wang X, Xue W, Gong H. Nickel-catalyzed reductive coupling of alkyl halides with other electrophiles: concept and mechanistic considerations. Org. Chem. Front. 2015;2:1411–1421. doi: 10.1039/C5QO00224A. [DOI] [Google Scholar]
- 51.Yu X, Yang T, Wang S, Xu H, Gong H. Nickel-catalyzed reductive cross-coupling of unactivated alkyl halides. Org. Lett. 2011;13:2138–2141. doi: 10.1021/ol200617f. [DOI] [PubMed] [Google Scholar]
- 52.Everson DA, Jones BA, Weix DJ. Replacing conventional carbon nucleophiles with electrophiles: nickel-catalyzed reductive alkylation of aryl bromides and chlorides. J. Am. Chem. Soc. 2012;134:6146–6159. doi: 10.1021/ja301769r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huihui KMM, et al. Decarboxylative cross-electrophile coupling of N-hydroxyphthalimide esters with aryl iodides. J. Am. Chem. Soc. 2016;138:5016–5019. doi: 10.1021/jacs.6b01533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Konev MO, Hanna LE, Jarvo ER. Intra- and intermolecular nickel-catalyzed reductive cross-electrophile coupling reactions of benzylic esters with aryl halides. Angew. Chem. Int. Ed. 2016;55:6730–6733. doi: 10.1002/anie.201601206. [DOI] [PubMed] [Google Scholar]
- 55.Biswas S, Weix DJ. Mechanism and selectivity in nickel-catalyzed cross electrophile coupling of aryl halides with alkyl halides. J. Am. Chem. Soc. 2013;135:16192–16197. doi: 10.1021/ja407589e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.García-Domínguez A, Li Z, Nevado C. Nickel-catalyzed reductive dicarbofunctionalization of alkenes. J. Am. Chem. Soc. 2017;139:6835–6838. doi: 10.1021/jacs.7b03195. [DOI] [PubMed] [Google Scholar]
- 57.Yang T, Chen X, Rao W, Koh MJ. Broadly applicable directed catalytic reductive difunctionalization of alkenyl carbonyl compounds. Chem. 2020;6:738–751. doi: 10.1016/j.chempr.2019.12.026. [DOI] [Google Scholar]
- 58.Zaitsev VG, Shabashov D, Daugulis O. Highly regioselective arylation of sp3 C–H bonds catalyzed by palladium acetate. J. Am. Chem. Soc. 2005;127:13154–13155. doi: 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]
- 59.Derosa J, van der Puyl VA, Tran VT, Liu M, Engle KM. Directed nickel-catalyzed 1,2-dialkylation of alkenyl carbonyl compounds. Chem. Sci. 2018;9:5278–5283. doi: 10.1039/C8SC01735B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Y, Zou L, Bai R, Lan Y. Ni(I)–Ni(III) vs. Ni(II)–Ni(IV): mechanistic study of Ni-catalyzed alkylation of benzamides with alkyl halides. Org. Chem. Front. 2018;5:615–622. doi: 10.1039/C7QO00850C. [DOI] [Google Scholar]
- 61.Manolikakes G, Dong ZB, Mayr H, Li JS, Knochel P. Negishi cross-couplings compatible with unprotected amide functions. Chem. Eur. J. 2009;15:1324–1328. doi: 10.1002/chem.200802349. [DOI] [PubMed] [Google Scholar]
- 62.Haginaka J, Yasuda H, Uno T, Nakagawa T. Alkaline degradation and determination by high-performance liquid chromatography. Chem. Pharm. Bull. 1984;32:2752–2758. doi: 10.1248/cpb.32.2752. [DOI] [PubMed] [Google Scholar]
- 63.Barba, O. et al. Compounds for the treatment of metabolic disorders. Patent: WO 034388 A1 (2009).
- 64.Shouksmith AE, et al. Synthesis and activity of putative small-molecule inhibitors of the F-box protein SKP2. Aust. J. Chem. 2015;68:660–679. doi: 10.1071/CH14586. [DOI] [Google Scholar]
- 65.Yamauchi S, et al. Design of 92 new 9-norlignan derivatives and their effect on cell viabilities of cancer and insect cells. J. Agric. Food Chem. 2019;67:7880–7885. doi: 10.1021/acs.jafc.9b03171. [DOI] [PubMed] [Google Scholar]
- 66.Basnet P, et al. Ni-Catalyzed regioselective β,δ-diarylation of unactivated olefins in ketimines via ligand-enabled contraction of transient nickellacycles: rapid access to remotely diarylated ketones. J. Am. Chem. Soc. 2018;140:7782–7786. doi: 10.1021/jacs.8b03163. [DOI] [PubMed] [Google Scholar]
- 67.Li W, Boon JK, Zhao Y. Nickel-catalyzed difunctionalization of allyl moieties using organo boronic acids and halides with divergent regioselectivities. Chem. Sci. 2018;9:600–607. doi: 10.1039/C7SC03149A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.He Y, Cai Y, Zhu S. Mild and regioselective benzylic C–H functionalization: Ni-catalyzed reductive arylation of remote and proximal olefins. J. Am. Chem. Soc. 2017;139:1061–1064. doi: 10.1021/jacs.6b11962. [DOI] [PubMed] [Google Scholar]
- 69.Xiao J, He Y, Ye F, Zhu S. Remote sp3 C–H amination of alkenes with nitroarenes. Chem. 2018;4:1645–1657. doi: 10.1016/j.chempr.2018.04.008. [DOI] [Google Scholar]
- 70.Zhou F, Zhu J, Zhang Y, Zhu S. NiH-catalyzed reductive relay hydroalkylation: a strategy for the remote C(sp3)–H alkylation of alkenes. Angew. Chem. Int. Ed. 2018;57:4058–4062. doi: 10.1002/anie.201712731. [DOI] [PubMed] [Google Scholar]
- 71.Derosa J, Tran VT, Boulous MN, Chen JS, Engle KM. Nickel-catalyzed β,γ-dicarbofunctionalization of alkenyl carbonyl compounds via conjunctive cross-coupling. J. Am. Chem. Soc. 2017;139:10657–10660. doi: 10.1021/jacs.7b06567. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the corresponding authors upon reasonable request.