

HSF1 neutralizes soluble amyloid oligomers to safeguard the mitochondria, thereby enabling uncontrolled growth.
Abstract
The role of proteomic instability in cancer, particularly amyloidogenesis, remains obscure. Heat shock factor 1 (HSF1) transcriptionally governs the proteotoxic stress response to suppress proteomic instability and enhance survival. Paradoxically, HSF1 promotes oncogenesis. Here, we report that AKT activates HSF1 via Ser230 phosphorylation. In vivo, HSF1 enables megalencephaly and hepatomegaly, which are driven by hyperactive phosphatidylinositol 3-kinase/AKT signaling. Hsf1 deficiency exacerbates amyloidogenesis and elicits apoptosis, thereby countering tissue overgrowth. Unexpectedly, HSF1 physically neutralizes soluble amyloid oligomers (AOs). Beyond impeding amyloidogenesis, HSF1 shields HSP60 from direct assault by AOs, averting HSP60 destabilization, collapse of the mitochondrial proteome, and, ultimately, mitophagy and apoptosis. The very same mechanism occurs in Alzheimer’s disease. These findings suggest that amyloidogenesis may be a checkpoint mechanism that constrains uncontrolled growth and safeguards tissue homeostasis, congruent with its emerging tumor-suppressive function. HSF1, by acting as an anti-amyloid factor, promotes overgrowth syndromes and cancer but may suppress neurodegenerative disorders.
INTRODUCTION
Phosphatidylinositol 3-kinase (PI3K)/AKT signaling governs a wide array of cellular functions including survival, cell cycle progression, metabolism, and growth (1). Its hyperactivation, frequently caused by genetic mutations, drives oncogenesis (1). While both PI3K and AKT are oncoproteins, PTEN, a phosphatase with opposite functions from PI3K, is a tumor suppressor (1). Aberrant activation of PI3K/AKT signaling has been implicated in overgrowth syndromes in humans, including the PIK3CA-related overgrowth spectrum (PROS) and autism spectrum disorder (ASD), a developmental disability associated with brain overgrowth (2, 3).
As a serine/threonine kinase, AKT mediates most of the effects of PI3K signaling by phosphorylating numerous cellular targets. Among them are TSC2 and PRAS40, both of which are key regulators of mTORC1 signaling and translational control (1). Congruently, AKT/mTORC1 signaling plays a key role in stimulating protein biosynthesis and thereby controlling cell size (4).
Protein synthesis, which contributes to cellular protein quantity, is an integral part of the dynamic network that maintains proteomic balance. However, another equally important aspect is protein quality. Disruptions to this quantity-quality equilibrium destabilize the cellular proteome, eliciting system-wide protein misfolding, aggregation, and, ultimately, amyloidogenesis. Proteomic imbalance has been associated with aging and a number of pathological conditions in humans. Amyloidogenesis is, of course, a hallmark of neurodegenerative disorders, particularly Alzheimer’s disease (AD), but emerging evidence has also revealed its tumor-suppressive nature (5, 6).
In living cells, several mechanisms sustain the delicate proteomic balance, especially in the face of environmental insults. Among them is the evolutionarily conserved heat shock, or heat shock factor 1 (HSF1)–mediated proteotoxic stress, response (HSR/PSR). Through the induction of heat shock proteins (HSPs) or molecular chaperones, this powerful transcriptional program ensures the quality of the cellular proteome by preventing protein misfolding, facilitating proteasomal degradation of misfolded proteins, and antagonizing protein aggregation (7). In contrast to HSF1’s broadly acclaimed role in countering protein-misfolding diseases, including neurodegenerative disorders, a growing body of evidence has pinpointed an unexpected prooncogenic effect as well. Moreover, cancerous cells, unlike their normal counterparts, become dependent on HSF1 for their growth and survival (8).
Here, we report HSF1 as a previously unidentified physiological substrate for AKT. Through Ser230 phosphorylation, AKT activates HSF1 and its mediated HSR/PSR. In mice, constitutive activation of PI3K/AKT signaling in the brain and liver drives megalencephaly and hepatomegaly, respectively, leading to rapid postnatal death. Concurrent Hsf1 deficiency mitigates tissue overgrowth and prolongs animal survival. Excessive protein translation driven by PI3K/AKT hyperactivation is sufficient to evoke amyloidogenesis; however, apoptosis occurs only in Hsf1-deficient overgrown tissues. HSF1 unexpectedly neutralizes soluble amyloid oligomers (AOs) via physical interactions, which means that, beyond impeding amyloidogenesis, HSF1 shields the essential mitochondrial chaperone HSP60 against degradation and aggregation caused by the direct assault of AOs. In the face of amyloids accrued during overgrowth, HSF1 defends the stability of the mitochondrial proteome to avert apoptosis. HSP60 is a bona fide target of AOs in human AD, and this transcription-independent anti-amyloid action of HSF1 also protects human neurons against Aβ1–42–induced toxicity in vitro. These findings illuminate HSP60 and its governed mitochondrial proteome as a prime target of AOs and pinpoint HSF1 as an intrinsic anti-amyloid factor. Contrary to its neurodegenerative effect, amyloidogenesis serves as a safeguard to limit uncontrolled growth and preserve tissue homeostasis.
RESULTS
AKT activates HSF1 via Ser230 phosphorylation
In mammalian cells, phosphorylation plays an important part in activating HSF1 during heat shock (9). Our previous studies showed that the oncoprotein mitogen-activated protein kinase kinase (MEK) activates HSF1 via Ser326 phosphorylation (5). We were curious whether other oncogenic pathways also activate HSF1. To this end, we turned our attention to PI3K/AKT signaling, which is often hyperactivated in human cancers (1).
Heat shock activates AKT, as evidenced by its elevated phosphorylation, particularly at Thr308 in human embryonic kidney (HEK) 293T cells (fig. S1A), which is a PDK1-mediated modification (1). Furthermore, pharmacological inhibition of PI3K by LY294002 or AKT by MK2206 and RG7440 blocked the induction of Hsp mRNAs and proteins by heat shock in NIH 3T3 cells (fig. S1, B and C), which demonstrates AKT’s causative role in activating the HSR/PSR. Contrary to its canonical stress-inducible activation, HSF1 remains constitutively active in malignant cells (10, 11). In HEK293T cells under basal conditions, pharmacological AKT inhibition impaired HSF1 binding to the HSP72 promoter, a classic HSF1 target gene (fig. S1D) (12). This demonstrates a causal effect of AKT on constitutive HSF1 activation. Conversely, AKT1Myr, a constitutively active mutant with an src myristoylation sequence (13), heightened its HSP promoter binding and induced HSP mRNAs (fig. S1, E and F). AKT2Myr and AKT3Myr, similar to AKT1Myr, activated the HSF1 reporter (fig. S1G), demonstrating that AKT is sufficient for HSF1 activation. To exclude the potential confounding effects of protein overexpression, we activated AKT by knocking down the tumor suppressor PTEN in HEK293T cells. Just like AKT1Myr overexpression, PTEN deficiency activated the HSF1 reporter and enhanced its DNA binding; importantly, both effects were blocked by pharmacological AKT inhibition (fig. S1, H and I), thus pinpointing the causality of AKT activation. Notably, endogenous AKT and HSF1 coprecipitated in HEK293T cells under basal conditions, indicating constitutive activation of AKT and HSF1, which was markedly enhanced by heat shock (fig. S1J). This physical AKT-HSF1 interaction was confirmed and visualized in situ by the proximity ligation assay (PLA) in mouse embryonic fibroblasts (MEFs). Despite sparse interaction under basal conditions, which reflects the lack of HSF1 activation, heat shock drastically induced this interaction (fig. S1K).
To demonstrate HSF1 phosphorylation by AKT, we searched the human HSF1 protein sequence for the consensus AKT phosphorylation motif “RXRXXS/T” (1). Ser230 (KYSRQFS) partially conforms to this criterion (fig. S1L). Our in vitro kinase assays revealed that all three AKT isoforms phosphorylated HSF1 at Ser230, which were fully blocked by AKT inhibitors (fig. S1M). Thus, the oncoproteins AKT and MEK phosphorylate HSF1 at distinct sites (fig. S1N). Beyond in vitro assays, heat shock induced HSF1 Ser230 phosphorylation in HEK293T cells, which was blocked by either PI3K or AKT inhibitors (fig. S1O). Like heat stress, PTEN deficiency was sufficient to induce Ser230 phosphorylation in a dose-dependent manner (fig. S1P). To address the necessity of Ser230 phosphorylation for HSF1 activation, we generated HSF1S230A mutants. In contrast to HSF1WT, HSF1S230A was refractory to activation by AKT1Myr and displayed impaired transcriptional activities under both basal and heat-shock conditions (fig. S1, Q and R). AKT inhibitors diminished the transcriptional activity of HSF1WT. By contrast, they did not affect HSF1S230A (fig. S1R), highlighting the importance of Ser230 phosphorylation to AKT-mediated HSF1 activation. Abolishing Ser230 phosphorylation did not impede HSF1 nuclear translocation (fig. S1S); instead, HSF1S230A displayed diminished occupancy on HSP gene promoters (fig. S1T), indicating the importance of Ser230 phosphorylation to HSF1 DNA binding. The residual DNA binding is likely due to constitutively activated MEK in HEK293T cells. Notably, death-associated protein kinase and calcium/calmodulin-dependent kinase II reportedly phosphorylate HSF1 at Ser230 as well (14, 15). All three kinases can recognize the minimal phosphorylation motif “RXXS/T.” Thus, they likely share the same phosphorylation site on HSF1. These results collectively identify HSF1 as a previously unidentified physiological substrate for AKT and demonstrate that AKT activates HSF1 via Ser230 phosphorylation in response to both heat stress and oncogenic stimuli (fig. S1U).
HSF1 supports megalencephaly driven by constitutively active PI3K
We were interested in the biological significance of this AKT-mediated HSF1 activation beyond the canonical HSR/PSR. One of the fundamental questions in biology is the control of body and organ size. Organ size is dictated by both cell size, driven by cellular growth, and cell number, controlled by proliferation and survival (4). By stimulating mTORC1 and its mediated protein translation, PI3K/AKT signaling controls cellular growth and cell size (4). Activating mutations of PI3KCA or AKT have unsurprisingly been associated with human overgrowth syndromes including megalencephaly, a growth development disorder characterized by abnormally enlarged brains (16, 17). Moreover, hyperactivation of PI3K/AKT signaling drives malignant growth (1).
To avoid a proteomic imbalance, protein synthesis must orchestrate with the quality-control machineries that comprise the HSF1-governed chaperone system. Thus, we reasoned that HSF1 activation counterbalanced the heightened protein synthesis stimulated by PI3K/AKT signaling. To address the need for HSF1 in PI3K/AKT-driven growth in vivo, we used a genetically engineered mouse strain, R26StopFLp110*. In this strain, the expression of a constitutively active p110 catalytic subunit of PI3K (p110*) from the Rosa26 locus is silenced by a floxed transcriptional STOP cassette (18). With a Cre deleter strain, PI3K signaling can be constitutively activated in a tissue-specific manner. We first induced p110* expression in the brain by crossing with hGFAP-Cre transgenic mice, wherein the Cre recombinase expression is driven by the human GFAP (glial fibrillary acidic protein) promoter (19). Cre-mediated recombination is widespread in both neurons and astrocytes because of its expression in radial glia, a key population of neural stem/progenitor cells (19). These hGFAP-Cre; R26StopFLp110* mice displayed brain enlargement in full penetrance, requiring euthanasia within 16 days postnatal and thereby demonstrating a causative role of PI3K in human megalencephaly (Fig. 1, A and B). Of note, concurrent Hsf1 deletion, mediated by the same hGFAP-Cre expression, reduced brain size and prolonged animal median survival by 21%. This shows that HSF1 is required for brain overgrowth.
Fig. 1. HSF1 is required for megalencephaly driven by constitutively active PI3K.
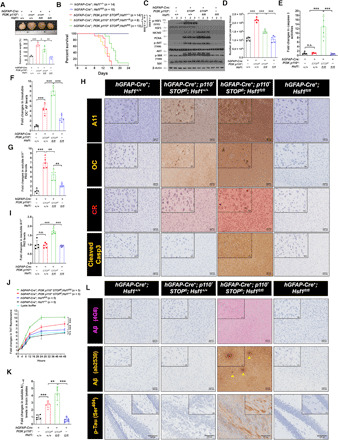
(A) Brains were collected at 15 days postnatal (n = 3 mice). Photo credit: Zijian Tang, NCI. (B) Kaplan-Meier survival curves (n = 8 to 15 mice). (C) Immunoblotting of brain lysates (n = 3 mice). (D) Quantitation of the numbers of nuclei in frozen brain tissues (n = 5 mice). (E) Quantitation of caspase 3 activities in brain lysates (n = 5 mice). (F and G) Enzyme-linked immunosorbent assay (ELISA) quantitation of detergent-insoluble amyloid fibrils (AFs) and detergent-soluble prefibrillar AOs (PAOs) in brain lysates (n = 5 mice). (H) Representative staining images of paraffin brain sections (from three brains). (I) ELISA quantitation of PAOs levels in detergent-insoluble fractions of brain lysates (n = 5 mice). (J) Aβ1–42 (2 μM) peptides were incubated with 100-μg detergent-soluble fractions of brain lysates and the Aβ1–42 fibrillation was monitored by thioflavin T (ThT) binding (n = 5 mice). (K) ELISA quantitation of endogenous mouse Aβ1–42 levels in the soluble fractions of brain lysates (n = 5 mice). (L) Representative immunohistochemistry images of paraffin brain sections (from three brains). Arrowheads denote plaque-like Aβ deposits. Scale bars, 100 μm for main images; 20 μm for insets. Statistical analyses: Log-rank test for (B); two-way analysis of variance (ANOVA) for (J); and one-way ANOVA for the rest. (A) to (D) and (J) were done once; (E) to (G) and (I) were repeated twice; (K) was repeated thrice; (H) and (L) were repeated thrice with different sets of brains. n.s., not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
Constitutive activation of PI3K stimulated AKT/mTORC1 signaling and cell proliferation in the brain, as evidenced by increased AKT/S6K phosphorylation and proliferation markers including MCM2 and proliferating cell nuclear antigen (PCNA). Hsf1 deficiency did not diminish them (Fig. 1C). MCM2 and PCNA were further elevated by Hsf1 deficiency, suggesting an enhanced proliferation of certain cell populations in these brains. Consistent with elevated S6K phosphorylation, p110* expression stimulated the global protein translation rate, measured by puromycin labeling (20), in primary astrocytes derived from these mice (fig. S2A). Although Hsf1 deficiency reduced basal protein translation in cells without p110* expression, as reported in our previous study (21), it did not alter the heightened translation rate in p110*-expressing cells. Thus, these findings exclude impaired cell proliferation or protein translation as the cause of reduced brain size in p110*-expressing, Hsf1-deficient (P*H−) mice. Despite elevated proliferation markers, the overall DNA contents were not increased in these P*H− brains (fig. S2B), suggesting a concurrent loss of other cell populations. Quantitation of nuclei extracted from these brains revealed that while PI3K activation doubled the cell number, Hsf1 deficiency mitigated this increase (Fig. 1D). Given the lack of proliferation inhibition, we reasoned that this reduction in cell number must be due to impaired cell survival. We detected markedly elevated apoptosis in P*H− brains (Fig. 1E). Consistent with widespread apoptosis in neurons and astrocytes, where p110* was expressed, there was a global reduction in neuronal and synaptic markers, including βIII-tubulin, synaptophysin, AMPA, and N-methyl-d-aspartate (NMDA) receptors, in the whole-brain lysates of P*H− mice and the astroglial marker GFAP. By contrast, the microglial marker Iba-1 was increased (fig. S2C). In the brain, microglia become activated upon apoptosis and play a prime role in clearing apoptotic cells via phagocytosis (22). Thus, these findings strongly suggest a proliferation and activation of microglia in response to apoptosis in P*H− brains. Congruently, activated microglia closely surrounded neurons in these brains (fig. S2D). This pathology is known as neuronophagia. While resting microglia showing ramified morphology were prevalent in the other three brain groups, the microglia in P*H− brains displayed an amoeboid morphology with enlarged cell bodies and retracted branches (illustrated by the Iba-1 staining), indicative of the active phagocytic state (23). Of note, staining of P*H+ brains revealed increased microglia, contrasting with the unaltered Iba-1 expression detected by immunoblotting (fig. S2C). This discrepancy is likely due to the proportional increase of most cell populations in P*H+ brains, a scenario under which the percentage of microglia would remain unchanged, reflected by immunoblotting using equal protein normalization. This contrasts with a disproportionate loss of neurons and astrocytes in P*H− brains. Despite unimpaired AKT/mTORC1 signaling and protein translation, we cannot exclude a reduction in cell size in these P*H− brains, as other factors may also control cell size. Nonetheless, apoptotic cell death, by diminishing cell numbers, at least partially underlies the reduced brain size and prolonged survival of P*H− mice.
How does Hsf1 deficiency cause apoptosis? As our findings exclude impaired AKT signaling, we focused on the impacts of Hsf1 deficiency on the proteome, with a particular interest in amyloidogenesis, an indicator of severe proteomic instability. Several reagents have been commonly applied to detect amyloids. Aside from the amyloid dyes thioflavin T (ThT) and Congo red (CR) (24), two conformation-specific antibodies (Abs), A11 and OC, detect distinct amyloid species. While A11 recognizes soluble prefibrillar AOs (PAOs), OC recognizes both soluble fibrillar AOs (FAOs) and insoluble amyloid fibrils (AFs) (25, 26). We detected markedly elevated detergent-insoluble OC+ AFs and membrane-associated A11+ PAOs in p110*-expressing brains. Hsf1 deficiency further elevated their levels (Fig. 1F and fig. S2, E and F). Although p110* expression also elevated free, detergent-soluble A11+ PAOs in the brains, Hsf1 deficiency unexpectedly lowered their levels (Fig. 1G). The presence of amyloids in p110*-expressing brains was confirmed by immunostaining, except that P*H− brains displayed higher levels of PAOs than P*H+ brains (Fig. 1H). We reasoned that this discrepancy might be due to the precipitation of some PAOs during centrifugation to prepare detergent-soluble brain fractions. Increased PAOs were detected in the insoluble fractions of P*H− brains (Fig. 1I), suggesting the formation of high-molecular-weight (HMW) PAO-containing complexes that cannot remain soluble during centrifugation. Consistent with the soluble nature of AOs, A11 and OC Abs produced largely diffuse staining, both intracellular and extracellular (Fig. 1H). The amyloidogenesis accompanied by brain overgrowth was further confirmed by in situ CR staining as well as in vitro amyloid fibrillation assays, wherein soluble brain lysates supplied amyloid seeds preformed in vivo to nucleate the assembly of human Aβ1–42 fibrils in vitro (Fig. 1, H and J). p110*-expressing brains contained elevated endogenous Aβ1–42 (Fig. 1K and fig. S2, G and H), a hallmark of human AD (27). This finding was further confirmed by immunohistochemistry using either the widely used mouse monoclonal 4G8 Ab, which recognizes an epitope located within Aβ17–24, or the rabbit polyclonal ab2539 Ab, which recognizes an epitope located within Aβ1–14. The specificity of ab2539 toward mouse Aβ (β-amyloid) was validated (fig. S2I). Compared to the relatively homogeneous 4G8 staining, ab2539 produced some focally intense staining, reminiscent of extracellular Aβ plaques (Fig. 1L). These distinct staining patterns probably reflect the differential accessibility of the corresponding epitopes within discrete Aβ conformers. Plaque-like Aβ deposits in P*H− brains were either spherical and diffuse or small and compact (fig. S2J). Also, intracellular, even intranuclear, Aβ accumulation and amyloid angiopathy were observed. Of note, intracellular Aβ has been detected in patients with AD and animal models (28, 29). In contrast to the enzyme-linked immunosorbent assay (ELISA) and staining with oligomer-specific Abs (Fig. 1K and fig. S2G), immunohistochemistry of both 4G8 and ab2539 failed to detect Aβ in P*H+ brains (Fig. 1L). Given the apparent detection of Aβ by immunoblotting (fig. S2E), we reasoned that this discrepancy might be due to the inaccessibility of the epitopes located within Aβ1–24 under nondenaturing conditions in P*H+ brains. Apparently, the epitopes recognized by the ELISA Abs were not obscured in P*H+ brains. Besides the production of Aβ, P*H− brains also displayed Tau hyperphosphorylation, particularly in the hippocampus (Fig. 1L), which is another characteristic of human AD (30). Tau Ser404 phosphorylation, an epitope recognized by the widely used PHF-1 Abs, has been closely associated with the neurofibrillary tangles in AD brains (31). Apart from these resemblances to AD, widespread p110* expression caused neurodevelopmental defects as well, most notably in the hippocampus and cerebellum (fig. S2K). Moreover, P*H− brains displayed pale, uneven Nissl staining, congruent with neuronal loss. Together, these results reveal that both amyloidogenesis and Tau hyperphosphorylation, two pathological features suppressed by HSF1, are associated with brain overgrowth.
The prevalent apoptosis in P*H− brains was further confirmed by TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) (fig. S2G). To establish a causative role of amyloids in apoptosis, we used the primary astrocytes derived from these mice. Similar to the whole brains, p110* expression in astrocytes elevated amyloids but induced apoptosis only in the context of Hsf1 deficiency (fig. S2, L and M). In addition to staining amyloids, CR can impede amyloidogenesis (5, 32). As expected, CR treatment diminished amyloids in these p110*-expressing astrocytes (fig. S2, N and O). CR largely reduced caspase 3 activities in P*H− astrocytes; by contrast, it exerted no effects on P*H+ cells (Fig. 2A), concurrent with the lack of apoptosis in these cells.
Fig. 2. AOs are causally related to apoptosis.
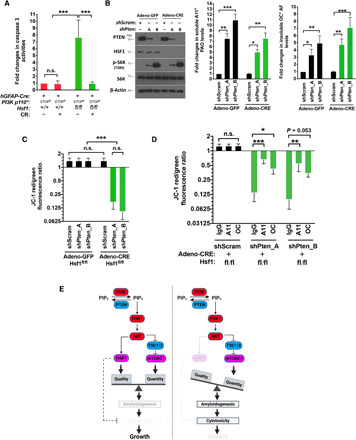
(A) Quantitation of caspase 3 activities in p110*-expressing astrocytes treated with and without 10 μM CR for 2 days (n = 3 lines of astrocytes). (B) Primary Hsf1fl/fl astrocytes were first stably transduced with lentiviral short hairpin RNAs. To delete Hsf1, these astrocytes were transiently transduced with adenoviral green fluorescent protein (GFP) or Cre. Both PAOs and AFs were quantitated by ELISA (n = 3 lines of astrocytes). (C) Quantitation of the mitochondrial membrane potentials in Pten-deficient astrocytes with and without Hsf1 deletion by fluorescence-activated cell sorting (FACS) using JC-1 dyes. The red/green (FL2-H/FL1-H) fluorescence ratios were calculated using geometric means (n = 3 lines of astrocytes). (D) Rescue of the mitochondrial membrane potentials in Pten-deficient astrocytes by neutralizing AOs. Following transfection of 100-ng antibodies for 2 days, Pten-deficient astrocytes with Hsf1 deletion were stained with JC-1 dyes (n = 3 lines of astrocytes). (E) Schematic depiction of the roles of HSF1 in supporting the tissue overgrowth. In addition to maintaining the balance between protein quantity and quality, which suppresses the initiation of amyloidogenesis, HSF1 also plays a critical role in antagonizing the amyloid-induced cytotoxicity, once amyloidogensis becomes inevitable. Thereby, HSF1 empowers overgrowth. Statistical analyses: one-way ANOVA. (A) to (D) were repeated thrice with different sets of astrocytes. n.s., not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
To exclude the possibility that exogenous p110* expression from the Rosa26 locus intrinsically creates proteotoxic stress that leads to amyloidogenesis, we activated AKT signaling in mouse brains by deleting Pten using the same hGFAP-Cre strain. For reasons unknown, these mice did not develop megalencephaly. Alternatively, we stably knocked down Pten in primary Hsf1fl/fl astrocytes with and without concurrent Hsf1 deletion by adenoviral Cre transduction. Similar to p110* expression, Pten knockdown induced amyloidogenesis (Fig. 2B). Again, Hsf1 deletion prompted apoptosis only in Pten-deficient cells, evidenced by the loss of mitochondrial membrane potential (Fig. 2C). Accumulating evidence has strongly implicated soluble AOs rather than insoluble AFs as the highly toxic amyloid species in neurodegenerative disorders (33–35). To determine whether soluble AOs are causally related to apoptosis in our models, we transfected A11 or OC Abs into these Pten-deficient astrocytes to neutralize PAOs and FAOs, respectively. Both Abs markedly rescued the mitochondrial membrane potential individually, although A11 seemed more effective (Fig. 2D), which supports the idea that both PAOs and FAOs are responsible for toxicity.
Owing to heightened mTORC1 signaling and protein translation, Pten loss enlarged cell size, as expected. Unexpectedly, concurrent Hsf1 deletion mitigated this enlargement without impairing mTORC1 signaling and protein translation (Fig. 2B and fig. S2, P and Q). This is likely due to proteomic instability caused by Hsf1 deficiency, which provokes extensive protein misfolding and aggregation, thereby rendering heightened protein synthesis “unproductive.” These results together indicate that aberrant activation of PI3K/AKT signaling is sufficient to trigger amyloidogenesis. HSF1 prevents amyloid-induced apoptosis beyond impeding amyloidogenesis, thereby supporting robust growth (Fig. 2E).
HSF1 enables hepatomegaly driven by hyperactivation of PI3K or PTEN deficiency
To test the generality of our findings, we established another in vivo model in which p110* expression in hepatocytes—achieved by crossing with Albumin-Cre (Alb-Cre) mice—led to hepatomegaly. Compared to the megalencephaly model, these mice lived longer and required euthanasia by 30 days postnatal (Fig. 3A). Concurrent deletion of Hsf1 in hepatocytes prolonged animal median survival by 64%. Even Hsf1 haplo-deficiency extended the life span (Fig. 3A). Similarly, p110* expression elicited amyloidogenesis in livers but only induced apoptosis in the absence of Hsf1 (Fig. 3, B to D, and fig. S3, A and B).
Fig. 3. HSF1 enables hepatomegaly.
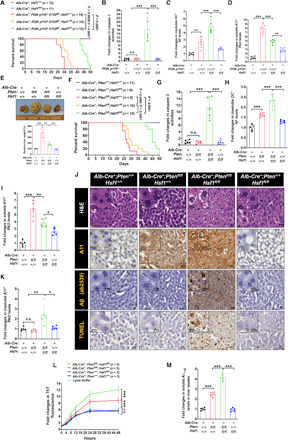
(A) Kaplan-Meier survival curves (n = 10 to 12 mice). (B) Quantitation of caspase 3 activities in liver lysates (n = 5 mice). (C and D) ELISA quantitation of AFs and soluble PAOs in liver lysates (n = 5 mice). (E) Representative photographs of livers. As one set of livers was harvested at a later time point, the weights of each set of livers are normalized (n = 3 mice). Photo credit: Zijian Tang, NCI. (F) Kaplan-Meier survival curves (n = 8 to 11 mice). (G) Quantitation of caspase 3 activities in liver lysates (n = 5 mice). (H and I) ELISA quantitation of AFs and soluble PAOs in liver lysates (n = 5 mice). (J) Representative staining images of frozen liver sections (from three livers, and the TUNEL staining was performed by two individuals). Scale bars, 20 μm for main images; 10 μm for insets. (K) ELISA quantitation of insoluble PAOs in liver lysates (n = 5 mice). (L) Aβ1–42 peptides (2 μM) were incubated with 100-μg soluble liver lysates, and the Aβ1–42 fibrillation was monitored (n = 5 mice). (M) ELISA quantitation of endogenous Aβ1–42 levels in the soluble fractions of liver lysates (n = 5 mice). Statistical analyses: log-rank test for (A) and (F); two-way ANOVA for (L); and one-way ANOVA for the rest. (A), (E), (F), (L), and (M) were done once; (B) to (D), (G) to (I), and (K) were repeated twice; and (J) was repeated thrice with different sets of livers. H&E, hematoxylin and eosin. n.s., not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
Whereas we could not induce megalencephaly by deleting Pten in the brain, we did generate a hepatomegaly model (Fig. 3E). The results are very similar to those obtained in the p110*-expressing brains and livers (Fig. 3, F to I, and fig. S3C), thus pinpointing that the overgrowth and amyloidogenic phenotypes are due to AKT activation rather than p110* transgene expression per se. Livers deficient for both Pten and Hsf1 displayed the highest levels of amyloids, accompanied by widespread apoptosis and cytoplasmic vacuolization/fragmentation in hepatocytes (Fig. 3, J to L, and fig. S3D). Endogenous Aβ1–42 was also detected in these Pten-deficient livers (Fig. 3, J and M, and fig. S3D). Of note, immunohistochemistry showed compact Aβ deposits in livers deficient for both Pten and Hsf1 (Fig. 3J). Together, these findings suggest that amyloidogenesis is a common consequence of uncontrolled growth.
Excessive protein synthesis causes amyloidogenesis
Amyloids were elevated even in overgrown Hsf1+/+ tissues, indicating that HSF1 cannot preclude amyloidogenesis. Considering protein translation’s prominent role in driving cellular growth, we asked whether uncontrolled translation might underlie amyloidogenesis.
We elected to inhibit translation in p110*-expressing astrocytes, which displayed a heightened protein translation rate. To avoid inducing autophagy through mTORC1 inhibition, we blocked translation using either 4EGI-1, which disrupts eIF4F complexes (36), or the selective p70S6K inhibitor LY2584702 (37). To accurately quantitate amyloids, these cultured astrocytes were cotreated with Q-VD-OPH, a pan-caspase inhibitor (CI), to prevent the loss of apoptotic cells containing high levels of AOs. Both 4EGI-1 and LY2584702 mitigated the translation rate in p110*-expressing astrocytes, as expected (fig. S4A). This mitigation diminished amyloidogenesis (Fig. 4, A to D). Corroborating the loss of soluble AOs owing to apoptotic cell death in culture, CI treatments elevated PAOs in P*H− astrocytes (Fig. 4, A and C). Moreover, translation inhibition diminished amyloids in astrocytes deficient for both Pten and Hsf1, rescuing the mitochondrial membrane potential (fig. S4, B and C). These findings indicate that excessive protein synthesis is causally related to amyloidogenesis.
Fig. 4. Hsf1 deficiency leads to HSP60 loss and mitochondrial damage.
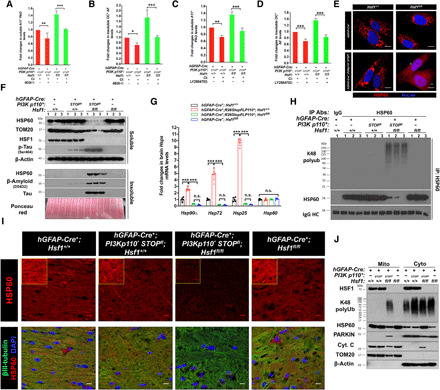
(A to D) Quantitation of PAOs and AFs in p110*-expressing astrocytes treated with and without 50 μM 4EGI-1 or 20 μM LY2584702 overnight. All cells were cotreated with 20 μM Q-VD-OPH to block apoptosis (n = 3 lines of astrocytes). (E) Representative images of HSP60 immunofluorescence in two lines of astrocytes. Scale bars, 10 μm. (F) Immunoblotting of HSP60 and TOM20 in detergent-soluble and detergent-insoluble fractions of brain lysates (n = 3 mice). Equal amounts of detergent-insoluble fractions, following re-solubilization by sonication, were loaded, shown by ponceau red staining. (G) Quantitation of Hsp mRNAs in brains by quantitative reverse transcription polymerase chain reaction (n = 5 mice). (H) Detection of HSP60 polyubiquitination in brain lysates by immunoprecipitation (IP) with the EasyBlot reagents (n = 3 mice). (I) Representative images of HSP60 immunofluorescence in three frozen brains. βIII-tubulin served as a neuronal marker. Scale bars, 10 μm. (J) Immunoblotting of the mitochondrial and cytosolic fractions of astrocytes (representative images of two lines of astrocytes). Equal amounts of the same fractions were loaded. TOM20 and β-actin were used as the mitochondrial and cytosolic markers, respectively. Statistical analyses: one-way ANOVA. (F) to (H) were done once; (E) and (J) were repeated twice with different sets of astrocytes; and (A) to (D) and (I) were repeated thrice with different sets of astrocytes or brains. DAPI, 4′,6-diamidino-2-phenylindole. n.s., not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
HSF1 safeguards HSP60 and the mitochondrial proteome to avert apoptosis
How did amyloids induce apoptosis? Serendipitously, in our independent efforts to identify tumor-associated amyloids by mass spectrometry, we discovered that A11 Abs precipitated HSP60 in human melanoma cells challenged with proteotoxic stressors. HSP60, by forming a chaperonin complex with HSP10, is an essential mitochondrial chaperone in charge of protein importation, folding, and anti-aggregation (38). Thus, we posited that PAOs might target HSP60 to undermine the mitochondrial proteome, thereby causing mitochondrial dysfunction and apoptosis.
Revealed by immunofluorescence, HSP60 became dispersed throughout the cytoplasm and nucleus in P*H− astrocytes, a pattern vastly different from its typical reticulated mitochondrial distribution (Fig. 4E). Apart from this mislocalization, there were numerous small puncta, suggesting HSP60 aggregation. In vivo, HSP60 protein was drastically depleted from the detergent-soluble fractions of P*H− brains; by contrast, its levels remained comparable among the other three groups (Fig. 4F). This depletion is not due to reduced transcription, as the levels of brain Hsp60 mRNA remained unchanged (Fig. 4G). On the other hand, Hsf1 deficiency fully blocked the induction of Hsp90α, Hsp72, and Hsp25 mRNAs, all of which are well-known stress-inducible Hsps, in p110*-expressing brains. These findings not only confirm HSF1 activation by PI3K but also reveal a dispensable role of HSF1 in Hsp60 transcription in these brains. Consistent with aggregation, HSP60—along with Aβ and Tau proteins, both of which are closely implicated in human AD (30)—was detected in the detergent-insoluble fractions (Fig. 4F). Moreover, Tau was hyperphosphorylated in P*H− brains. Filter-trap assays using equal amounts of brain tissues, which also revealed the most severe protein aggregation in P*H− brains, further confirmed HSP60 aggregation (fig. S4D). Besides aggregation, Hsf1 deficiency also caused Lys48 polyubiquitination of HSP60 (Fig. 4H), which also contributed to its depletion via proteasomal degradation. Immunofluorescence also demonstrated HSP60 depletion and aggregation in P*H− brains (Fig. 4I), which together support the misfolding and destabilization of HSP60. Accompanying this depletion of soluble HSP60 was a reduction in soluble TOM20 (Fig. 4F), which is a mitochondrial import receptor subunit serving as a mitochondrial marker (39). However, TOM20 did not become detergent-insoluble, suggesting mitochondrial damage and loss of mitochondrial mass owing to HSP60 depletion, which is expected to elicit apoptosis. To test this, we separated the cytosolic and mitochondrial fractions of cultured astrocytes. In line with proteomic imbalance due to excessive protein synthesis, p110* expression elevated global protein polyubiquitination (Fig. 4J). Whereas increased polyubiquitination occurred mainly in the cytosol in P*H+ astrocytes, a notable increase occurred in the mitochondria in P*H− astrocytes (Fig. 4J). HSP60, which is typically localized within the mitochondria, re-distributed to the cytosol in P*H− astrocytes (Fig. 4J). A similar mitochondria-to-cytosol re-distribution of cytochrome C, a key inducer of apoptosis (40), was observed in these astrocytes, confirming apoptosis. Of note, some HSF1 was localized within the mitochondria. Despite the evident mislocalization, these cultured astrocytes did not display marked depletion of soluble HSP60, suggesting mild misfolding and aggregation. Likely, this is due to the continuous loss of apoptotic cells with abundant AOs and severe HSP60 destabilization during culture.
Contrasting with HSP60 and cytochrome C, PARKIN, an E3 ubiquitin ligase associated with Parkinson’s disease (41), re-distributed from the cytosol to the mitochondria (Fig. 4J). The PARKIN-mediated ubiquitination of mitochondrial proteins plays a key part in the lysosomal degradation of damaged mitochondria, a process referred to as mitophagy (41). Thus, this increased PARKIN association aligns with elevated polyubiquitination of mitochondrial proteins, supporting mitochondrial damage and mitophagy. In accordance with reduced TOM20, P*H− astrocytes displayed diminished mitochondrial mass; importantly, chloroquine (CQ), an autophagy inhibitor (42), partially rescued this defect (Fig. 5A and fig. S4E), confirming mitophagy. Also, CR rescued mitochondrial mass partially, indicating a causative role of amyloids in mitochondrial defects.
Fig. 5. Loss of HSP60 causes mitochondrial damage, mitophagy, and apoptosis.
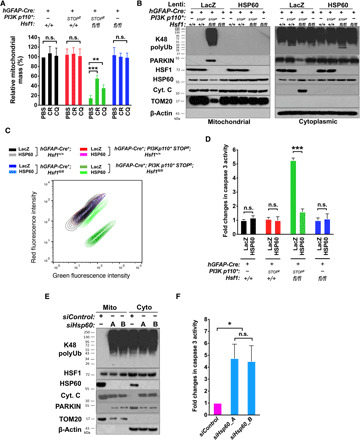
(A) Quantitation of mitochondrial mass in astrocytes treated with 10 μM CR or 20 μM CQ for 6 days by FACS (FL1-H, geometric means) using MitoView Green (n = 3 lines of astrocytes). (B) Immunoblotting of astrocytes transduced with lentiviral LacZ or HSP60 (multiplicity of infection, MOI = 10) for 6 days (representative images of two lines of astrocytes). Equal amounts of the same fraction were loaded. (C) JC-1 staining of astrocytes described in (B). The contour plot represents one line of astrocytes. The red/green (FL2-H/FL1-H) fluorescence ratios from three lines of astrocytes are quantitated in fig. S4F. (D) Quantitation of caspase 3 activities in astrocytes described in (B) (n = 3 lines of astrocytes). (E) Detection of molecular changes in hGFAP-Cre+; Hsf1+/+ astrocytes transfected with control or Hsp60-targeting siRNAs for 4 days (representative images of three lines of astrocytes). Equal amounts of the same fraction were loaded. (F) Quantitation of caspase 3 activities in astrocytes described in (E) (n = 3 lines of astrocytes). Statistical analyses: one-way ANOVA. (B) was repeated twice with different sets of astrocytes; (A) and (C) to (F) were repeated thrice with different sets of astrocytes. n.s., not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
Given the apparent HSP60 defects, we reasoned that loss of HSP60 function might be a prime culprit for the mitochondrial damage. In P*H− astrocytes, lentiviral HSP60 overexpression largely reversed all the molecular mitochondrial changes (Fig. 5B). Accordingly, HSP60 overexpression restored the mitochondrial membrane potential and blocked apoptosis in both p110*-expressing and Pten-deficient astrocytes (Fig. 5, C and D, and fig. S4, F and G). Further supporting the key causality of HSP60 deficiency, Hsp60 knockdown alone recapitulated all the major mitochondrial defects and induced apoptosis in Hsf1+/+ astrocytes without p110* expression (Fig. 5, E and F, and fig. S4H). Similar to overgrown brains, enlarged livers deficient for both Pten and Hsf1 also displayed HSP60 insolubility and TOM20 loss (fig. S4, I and J). In aggregate, these results pinpoint HSP60 destabilization as a key causal event underpinning the cytotoxicity in Hsf1-deficient cells with hyperactive PI3K/AKT signaling.
Soluble AOs directly attack HSP60
Given the causative role of amyloids in damaging mitochondria, we postulated that soluble AOs might directly cause HSP60 degradation and aggregation in the absence of HSF1. In p110*-expressing brains, A11 Abs readily precipitated both Aβ and Tau (fig. S5A), indicating the formation of PAOs from both proteins in vivo. Whereas PAOs physically interacted with HSF1 in P*H+ brains, they interacted with HSP60 in P*H− brains instead, which was revealed by co-immunoprecipitation (co-IP) experiments (Fig. 6A). Besides PAOs, there are other subtypes of soluble AOs, such as OC+ FAOs, which share a common conformation with insoluble AFs (34). Similar to PAOs, FAOs also interacted with either HSF1 or HSP60 (Fig. 6B). By contrast, AOs did not interact with other major chaperones, including HSP90, HSP72, and HSP25, highlighting the specificity of AO-HSP60 interactions. Concurrent with the fact that PAOs comprised Aβ oligomers, 4G8 Abs also precipitated HSP60 from P*H− brain lysates. Aβ was not precipitated from P*H+ brain lysates by 4G8 (fig. S5B). This is not due to the absence of Aβ, as 4G8 Abs clearly detected Aβ in P*H+ brain lysates by immunoblotting, a denaturing condition. Contrary to the 4G8 Ab, HSF1 Abs precipitated both Aβ and Tau, but not HSP60, from P*H+ brain lysates (fig. S5C), confirming the AO-HSF1 interaction and supporting the masking of the 4G8 epitope in Hsf1+/+ tissues under nondenaturing conditions. Given the HSF1-Aβ interaction, we asked whether HSF1 could mask the 4G8 epitope. To test this, we incubated HSF1 proteins with the highly amyloidogenic Aβ1–42 in vitro and analyzed them using nondenaturing dot blotting. We found that HSF1 blocked the recognition of Aβ1–42 by both 4G8 and ab2539 Abs in a dose-dependent fashion but did not block recognition by another Aβ Ab, D54D2 (fig. S5D). This demonstrates the masking effect of HSF1 on Aβ epitopes.
Fig. 6. HSF1 shields HSP60 from the attack of AOs.
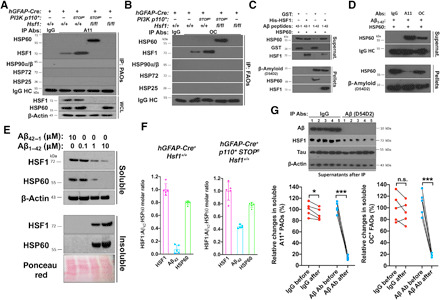
(A and B) Representative images of AO-HSF1 and AO-HSP60 co-IP from three (A) and two (B) brains with the EasyBlot reagents (performed by two individuals). (C) In vitro reconstitution of interactions among Aβ1–42, HSP60, and HSF1 using recombinant proteins at a 1:1:1 molar ratio. After incubation, reconstituted mixtures were centrifuged to collect the supernatants and pellets for immunoblotting (representative images of three experiments). (D) Aβ1–42, HSP60, and A11 or OC antibodies were reconstituted in vitro at a 1:1:1 molar ratio at RT for 4 hours (representative images of three experiments). (E) Immunoblotting of HSF1 and HSP60 in immortalized Rosa26-CreERT2; Hsf1fl/fl MEFs transfected with Aβ42–1 or Aβ1–42 for 2 days (representative images of three experiments). (F) HSF1:Aβ42:HSP60 molar ratios in soluble Hsf1+/+ brain lysates (n = 5 mice). (G) ELISA quantitation of soluble AOs in P*H+ brain lysates before and after IP of Aβ (n = 5 mice, two-tailed paired Student’s t test). Biotinylated A11 and OC Abs were used to detect AOs. (G) was done once; (F) was repeated twice; (B) was repeated twice with different sets of brains; (A) was repeated thrice with different sets of brains; and (C) to (E) were repeated thrice with the same cell line or reagents. n.s., not significant, P > 0.05; *P < 0.05; ***P < 0.001.
Similar to p110*-expressing brains, AOs also interacted with either HSF1 or HSP60 in Pten-deficient livers (fig. S5, E and F). Of note, even in normal Hsf1+/+ tissues, trace amounts of AOs were present and interacted with HSF1 (Fig. 6, A and B, and fig. S5, E and F). Furthermore, despite slightly elevated amyloids in Hsf1fl/fl tissues without p110* expression or Pten deletion, no evident AO-HSP60 interactions were detected. This may be due to either additional defensive mechanisms or simply the inability of low-level AOs to access mitochondrial HSP60. The PAO-HSP60 interaction was further visualized by PLA in cultured P*H− astrocytes (fig. S5, G and H). Collectively, these findings strongly suggest that HSF1 precludes the binding between AOs and HSP60.
To validate this model, we reconstituted the interactions between Aβ and HSP60 or HSF1 in vitro. The control non-amyloidogenic Aβ42–1, as expected, did not exert any detectable effects on recombinant HSP60 or HSF1 proteins at the equal molar ratio; by contrast, Aβ1–42 caused HSP60 to precipitate, confirming the physical attack on HSP60 by amyloids (Fig. 6C and fig. S5I). Demonstrating the specificity of this attack, Aβ1–42 did not cause precipitation of a panel of major chaperones, including HSP90β, HSP72, HSP27, and HSP10 (fig. S5J). Coincubation with HSF1 proteins largely blocked this HSP60 precipitation (Fig. 6C and fig. S5I), corroborating that HSF1 protects HSP60 against amyloid attacks. Of note, HSF1 did not evidently improve the solubility of Aβ1–42 at this equal molar ratio and even became precipitated itself (Fig. 6C and fig. S5I), suggesting HSF1 destabilization by amyloids. Just like HSF1, A11 and OC Abs both rescued the HSP60 solubility in vitro (Fig. 6D) by neutralizing PAOs and FAOs, respectively. Although neither fully rescued HSP60 solubility as HSF1 did, A11 demonstrated a higher potency than OC. Of note, Aβ1–42 did not markedly destabilize A11 or OC Abs, suggesting a distinction between Ab-AOs interactions and HSF1-AOs interactions, which is supported by the fact that HSF1 did not mask the AO epitopes recognized by A11 and OC in P*H+ brains (Fig. 1H). These results confirm the causative role of AOs in destabilizing HSP60 and support both PAOs and FAOs as toxic species.
To demonstrate that amyloids destabilize HSP60 in live cells, we transfected Aβ1–42 into Hsf1-proficient MEFs without p110* expression. Concurrent with our in vitro findings, we found that high doses of Aβ1–42 diminished the solubility of both endogenous HSP60 and HSF1 proteins. On the other hand, low doses exerted no detectable effects (Fig. 6E), resembling the scenario in P*H+ brains (Fig. 4F). These findings show that abundant endogenous HSF1 forms a critical barrier against attacks on HSP60 by amyloids. This action is not limited to specific cell types. To shed light on this barrier, we quantitated the absolute levels of soluble HSF1, Aβ42, and HSP60 in mouse brain lysates by ELISA (fig. S5K). The results show that HSF1 is more abundant than Aβ42 and HSP60 (Fig. 6F). IP of Aβ depleted ~80% of soluble AOs from p110*-expressing brain lysates (Fig. 6G and fig. S5L), revealing that Aβ is the major constituent of soluble AOs. Together, these results pinpoint the essential mitochondrial chaperone HSP60 as an immediate cellular target of soluble AOs that is shielded from the assault of AOs by HSF1.
HSF1 physically neutralizes AOs to defend the mitochondria
It has been broadly recognized that HSF1, as a canonical transcription factor, up-regulates HSPs under proteotoxic stress, thereby stabilizing the proteome. Thus, HSF1 is presumed to suppress the emergence of amyloids in this indirect manner. Unusually, though, our studies indicate that HSF1 is capable of antagonizing amyloids directly.
To demonstrate the transcription-independent rescue of HSP60 stability and mitochondrial function by HSF1 in living cells, we expressed two FLAG-tagged mutants, HSF11–323 lacking the C-terminal transactivation domain (TAD) and HSF1324–529 lacking the N-terminal DNA binding domain (DBD), both of which are devoid of transcriptional activity (Fig. 7A), in p110*-expressing astrocytes through lentiviral transduction. Moreover, HSF11–323, retaining the ability to trimerize via the HR-A/B domain and bind DNAs, functioned as a dominant negative mutant to impair the activity of endogenous HSF1 compared to the LacZ control (Fig. 7A). HSF11–323, but not HSF1324–529, largely rescued the mitochondrial membrane potential and blocked apoptosis in P*H− astrocytes, just like the full-length HSF11–529 (Fig. 7, B and C). This also held true in Pten-deficient astrocytes (fig. S5M). Mechanistically, lentiviral transduction of HSF11–529 and HSF11–323, but not HSF1324–529, abolished the PAO-HSP60 interaction, which was accompanied by physical interactions between PAOs and exogenously expressed FLAG-HSF1 (Fig. 7D and fig. S5N). This confirms the causative, transcription-independent action of HSF1 and strongly suggests that HSF1, by physically neutralizing AOs, antagonizes amyloid-induced cytotoxicity. Nonetheless, this capacity could be overwhelmed by excessive AOs (Fig. 7E).
Fig. 7. HSF1 protects HSP60 independently of transcription.
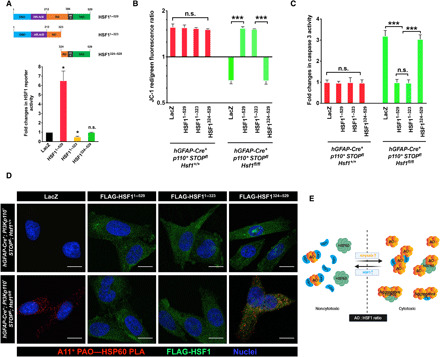
(A) The dual HSF1 reporter plasmids were cotransfected with HSF1 mutants into HEK293T cells (n = 3 experiments). (B and C) Quantitation of the mitochondrial membrane potential (B) and caspase activities (C) in p110*-expressing astrocytes transduced with lentiviral LacZ, HSF11–529, HSF11–323, or HSF1324–529 (MOI = 10) for 6 days (n = 3 lines of astrocytes). (D) PLA visualization of PAO-HSP60 interactions in astrocytes described in (B) and (C) (representative images of two lines of astrocytes). Scale bars, 10 μm. (E) Schematic depiction of the model wherein the AO:HSF1 molar ratio determines the fates of both HSP60 and HSF1. Excessive HSF1 fully neutralizes AOs and protects HSP60. However, the AO-HSF1 complexes may either remain soluble or become nonamyloid aggregates, depending on their interaction stoichiometry (AO << HSF1 or AO < HSF1). When AOs rise and become excessive, on the one hand, AOs that are not neutralized by HSF1 start attacking HSP60, leading to HSP60 misfolding and aggregation. On the other hand, the stoichiometry of AO-HSF1 interactions may be reversed (AO > HSF1), which would cause HSF1 misfolding and aggregation. Statistical analyses: one-way ANOVA. (D) was repeated twice with different sets of astrocytes; (B) and (C) were repeated thrice with different sets of astrocytes; and (A) was repeated thrice with the same cell line. n.s., not significant, P > 0.05; *P < 0.05; ***P < 0.001.
HSF1 directly impedes amyloidogenesis
In vitro fluorescence resonance energy transfer assays further supported the physical interaction between HSF1 and amyloids, wherein Dabcyl-labeled HSF1 quenched the fluorescence of Aβ1–42 labeled with fluorophores (Fig. 8A). Beyond stopping the attack on HSP60 by AOs, this interaction also dose-dependently impeded the amyloidogenesis of Aβ1–42 in vitro, evidenced by diminished ThT binding and reduced formation of PAOs, FAOs, and AFs (Fig. 8, B to E). Glutathione S-transferase (GST) and non-amyloidogenic Aβ42–1 served as the negative controls (fig. S6, A to C). At a 1:4 molar ratio, HSF1 abolished Aβ1–42 fibrillation (Fig. 8, B and E). By contrast, increased HSF1 did not further diminish PAOs and FAOs (Fig. 8, C and D), suggesting their stabilization, rather than abolition, by HSF1. Since both AOs can self-propagate (43, 44), this finding also suggests a halt of their self-propagation by HSF1, even at a 1:1 molar ratio. However, the complete blockade of AF assembly required a 1:4 molar ratio, indicating that HSF1 impedes amyloid fibrillation with a lower efficiency compared to its impact on AOs. OC Abs, but not A11 Abs, blocked Aβ1–42 fibrillation in a dose-dependent manner (Fig. 8F and fig. S6D). These findings both contradict the participation of PAOs in the fibrillation process and suggest FAOs as the primary intermediate of AF assembly. Despite the disappearance of AFs at a 1:4 molar ratio, HSF1 did not eradicate Aβ1–42 aggregation, revealed by the nephelometric turbidity measurement and filter-trap assay (Fig. 8G and fig. S6E). We reasoned that following the blockade of fibril assembly, HSF1 might transform FAOs into non-amyloid aggregates. This blockade of amyloid fibrillation and diversion to “off-pathway” aggregation was visualized by transmission electron microscopy (TEM). After 48-hour incubation with GST, Aβ1–42 formed mature fibrils, as expected; by contrast, coincubation with HSF1 at a 1:4 molar ratio resulted in amorphous aggregates (Fig. 8H). The fact that HSF1 dose-dependently diminished the turbidity of Aβ1–42 prompted us to test whether excessive HSF1 could fully stabilize Aβ1–42 at the soluble state. Excitingly, the incubation of Aβ1–42 with HSF1 at a 1:32 molar ratio resulted in no detectable aggregation and turbidity increase (Fig. 8I and fig. S6F).
Fig. 8. HSF1 blocks in vitro amyloidogenesis.
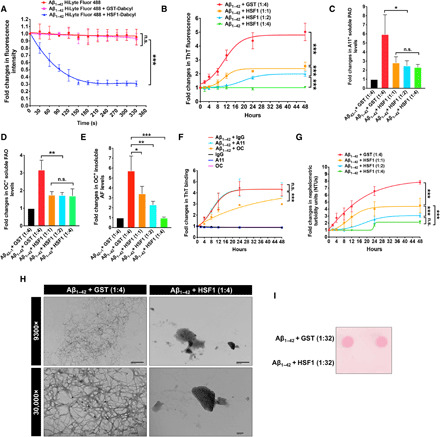
(A) HiLyte Fluor 488–labeled Aβ1–42 was incubated with Dabcyl-labeled HSF1 or GST at a 1:1 molar ratio (n = 3 experiments). (B) Quantitation of the fibrillation of 0.8 μM Aβ1–42 incubated with recombinant HSF1 proteins in vitro at increased molar ratios (n = 3 experiments). (C to E) ELISA quantitation of amyloids after incubation of Aβ1–42 with GST or HSF1 for 48 hours (n = 3 experiments). Aβ42–1 served as the negative control. (F) Quantitation of the fibrillation of 0.8 μM Aβ1–42 incubated with normal rabbit IgG, A11, or OC Abs in vitro at a 1:1 molar ratio (n = 3 experiments). (G) Quantitation of the nephelometric turbidities of Aβ1–42 described in (B) (n = 3 experiments). (H) Visualization of in vitro fibrillation of Aβ1–42 coincubated with either GST or HSF1 at 37°C for 48 hours by transmission electron microscopy (representative images of three experiments). Scale bars, 600 nm for 9300×; 100 nm for 30,000×. (I) Detection of the aggregation of 0.2 μM Aβ1–42 incubated with HSF1 at a 1:32 molar ratio for 48 hours by filter trap assays. The aggregates were stained with ponceau red (representative images of two experiments). The curves in (B), (F), and (G) are fitted with the Boltzmann sigmoid equation. Statistical analyses: two-way ANOVA for (A), (B), (F), and (G); and one-way ANOVA for (C) to (E). (I) was repeated twice; and (A) to (H) were repeated thrice. n.s., not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
While HSF1 can block in vitro amyloidogenesis, it remains unclear how it affects amyloids naturally formed in vivo. The turbidity of detergent-soluble fractions of Hsf1-deficient brains, irrespective of p110* expression, continued to increase during a 48-hour incubation period; by contrast, no changes were detected in Hsf1-proficient brain lysates (Fig. 9A). This increase is not due to the formation of large, insoluble aggregates, as filter-trap assays did not detect them after incubation. Instead, accompanying this turbidity increase was an elevation in PAOs (Fig. 9B), due to either the primary assembly from misfolded monomers or rapid self-propagation. Although not tested, a similar increase in FAOs is expected. A simple addition of recombinant HSF1 proteins into these brain lysates blocked the increases in turbidity and PAOs completely (Fig. 9, C and D), indicating the specific, transcription-independent action of HSF1. This turbidity increase is unlikely to be caused by promiscuous interactions among misfolded cellular proteins, as HSF1 would not be expected to prevent these stochastic, nonspecific interactions. Moreover, despite their correlated increases, the turbidities did not reflect the amount of PAOs before incubation (fig. S6G). This suggests that the turbidity measurement likely detected the formation of HMW complexes between AOs and cellular proteins, such as the AO-HSP60/chaperonin complexes, rather than a mere increase in AO quantity. Contrasting with the absence of in vivo AO-HSP60/chaperonin complexes in hGFAP-Cre+; Hsf1fl/fl brains, in vitro AOs, in the absence of neutralizing Abs, may freely access HSP60 released from mitochondria during brain tissue lysis.
Fig. 9. HSF1 contains amyloids formed in vivo.
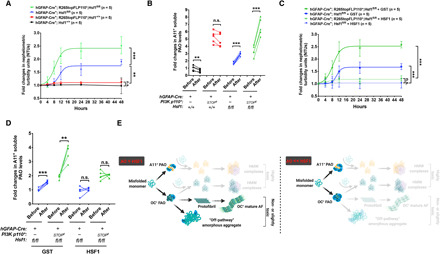
(A and B) Quantitation of the changes in nephelometric turbidities (A) and soluble PAOs (B) of 100-μg detergent-soluble brain lysates incubated at 37°C with shaking for 48 hours (n = 5 mice). (C and D) Quantitation of the changes in nephelometric turbidities (C) and soluble PAOs (D) of 100-μg detergent-soluble fractions of Hsf1-deficient brain lysates supplemented with 200-ng GST or HSF1 proteins (n = 5 mice). (E) Schematic depiction of the multilayered regulations of amyloidogenesis by HSF1. When HSF1 is slightly or moderately excessive (AOs < HSF1), the low interaction stoichiometry fully blocks the self-propagation of both PAOs and FAOs and, importantly, stops their attack on HSP60. This low stoichiometry, however, is inadequate to block the assembly of FAOs into mature AFs, only achieving partial impairments. Under this scenario, FAOs either continue to mature into fibrils (very low stoichiometry) or are transformed into amorphous aggregates (intermediately low stoichiometry), owing to the increased antagonizing force of HSF1. When HSF1 is considerably excessive (AOs << HSF1), the high interaction stoichiometry stabilizes FAOs at the soluble, nontoxic state, preventing the formation of both AFs and amorphous aggregates. The curves are fitted with the Boltzmann sigmoid equation. Statistical analyses: two-way ANOVA for (A) and (C); two-tailed paired Student’s t test for (B) and (D). (A) to (D) were done once. n.s., not significant, P > 0.05; **P < 0.01; ***P < 0.001.
Despite its interaction with AOs, HSP60 did not evidently impede amyloidogenesis in vitro (fig. S6, H to J). Neither did other major chaperones, including HSP90β, HSP72, HSP27, and HSP10 (fig. S6K), underscoring the specific, transcription-independent anti-amyloid action of HSF1. It remains possible that HSF1 may interact with amyloid monomers to block the genesis of AOs. However, in this case, HSF1 would block the initial steps of AO assembly, which predicts zero or very few AOs in Hsf1+/+ overgrown tissues and a dose-dependent elimination of AOs in the in vitro Aβ1–42 fibrillation assays. Apparently, our findings contradict these predictions (Figs. 1G and 8, C, and D), thus supporting a blockade of AO self-propagation by HSF1. Collectively, these results reveal HSF1 as a versatile anti-amyloid factor. Beyond blocking the self-propagation of AOs and preventing their assaults on cellular targets with high efficiency, HSF1 impedes the assembly of AFs from FAOs and further remodels them into non-amyloid conformers, albeit less efficiently (Fig. 9E).
Implications of the anti-amyloid action of HSF1 in human AD
We were intrigued by the central question of whether the same mechanism underlies both overgrowth syndromes and AD. Thus, we asked whether this direct anti-amyloid effect of HSF1 could confer neuroprotection.
First, we used a cell culture AD model, wherein primary human neurons were transfected with Aβ1–42. To visualize the interactions between Aβ1–42 and HSP60 in neurons by PLA, we used biotinylated Aβ peptides and a mouse monoclonal anti-biotin Ab (Fig. 10A). All Abs were validated by immunofluorescence (fig. S7A). Consistent with our findings in murine astrocytes, biotinylated Aβ1–42, but not the control Aβ42–1, interacted with neuronal HSP60. Lentiviral transduction of HSF11–529 or HSF11–323, but not HSF1324–529, abolished this interaction (Fig. 10A). Accompanying the disappearance of HSP60–Aβ1–42 interactions was the advent of interactions between transduced HSF11–529 or HSF11–323 and Aβ1–42 (Fig. 10B). In line with their attack on HSP60, Aβ1–42 impaired the viability of cultured human neurons. Lentiviral transduction of either HSF11–529 or HSF11–323, but not HSF1324–529, largely rescued neuronal viability (Fig. 10C). These results suggest that HSF1 may neutralize Aβ oligomers in human neurons to protect HSP60 and the mitochondria, thereby guarding against neurotoxicity.
Fig. 10. HSF1 protects human neurons against Aβ.
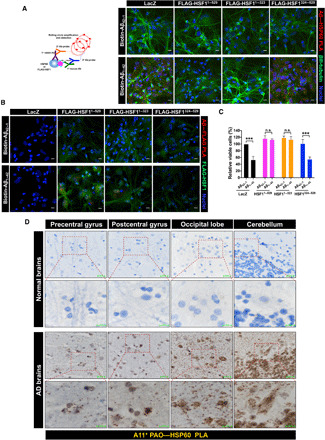
(A and B) PLA visualization of HSP60–Aβ1–42 interactions (A, representative images of three experiments performed by two individuals) or HSF1-Aβ1–42 interactions (B, a single experiment) in cultured primary human neurons. Following transduction with lentiviral LacZ or HSF1 mutants (MOI = 20) for 4 days, primary human neurons were transfected with 1 μM biotinylated Aβ42–1 or Aβ1–42 overnight. βIII-tubulin (green) was costained as a neuronal marker. Scale bars, 10 μm. (C) Measurement of the viabilities of primary human neurons transfected with 10 μM Aβ42–1 or Aβ1–42 for 24 hours (n = 3 experiments, one-way ANOVA). The neurons were transduced with lentivirus as described in (A) before transfection with Aβ42–1 or Aβ1–42. (D) Visualization of HSP60-PAOs interactions in the brains of patients with AD by bright-field PLA using a mouse anti-HSP60 Ab (LK1) and the rabbit anti-PAOs (A11) Ab (representative images of five experiments performed by two individuals). The brain sections on tissue arrays are from three patients with late-onset AD and three aged normal controls. Scale bars, 20 μm for low magnification; 10 μm for high magnification. (B) was done once; (A) and (C) were repeated thrice; and (D) was repeated five times. n.s., not significant, P > 0.05; ***P < 0.001.
This attack on HSP60 by AOs was confirmed in the brains of patients with AD. Compared to aged normal control brains, brain tissues from patients with late-onset AD displayed evident A11 and OC immunoreactivities accompanied by elevated apoptosis (figs. S7, B to D), as expected. In several regions of AD brains, including the precentral and postcentral gyrus, occipital lobe, and cerebellum, PAO-HSP60 interactions, both cytoplasmic and nuclear, were readily detected by PLA (Fig. 10D). In stark contrast, no such interactions were detected in the counterparts of normal control brains. The mouse monoclonal anti-HSP60 Ab used for PLA was validated as well (fig. S7E). On the other hand, no neuritic or senile plaques were detected in these AD brain regions, which are known to be less afflicted by amyloid plaques (30), despite intense, homogeneous Aβ (4G8) staining (fig. S7F). Confirming the absence of plaques in these brain areas, 4G8 did stain many Aβ plaques in positive control AD brain sections (fig. S7F). This PAO-HSP60 interaction was further confirmed by co-IP using AD brain lysates from different patients. PAO-HSF1 interactions were also detected in AD brains (Fig. 11A). Similarly, FAOs also interacted with both HSP60 and HSF1 in AD brains (Fig. 11B and fig. S7G), strongly suggesting that endogenous HSF1 has already been overwhelmed by AOs. The simple addition of recombinant HSF1 proteins into AD brain lysates was sufficient to obliterate these preexisting AO-HSP60 interactions (Fig. 11, A and B). Again, AOs did not attack endogenous HSP90, HSP72, and HSP27 in AD brains. Furthermore, analogous to the MEFs transfected with high doses of Aβ1–42 (Fig. 6E), AD brain lysates displayed diminished soluble HSF1 and HSP60 proteins (Fig. 11A). These findings were validated by immunostaining of AD brain sections from different patients. Compared to normal counterparts, varying degrees of HSF1 depletion were observed in different AD brain regions, which may be partly ascribed to the differential HSF1 expression between individuals. By contrast, all tested AD brain regions displayed consistent HSP60 depletion (fig. S7, H and I). Because of the unavailability of insoluble fractions of AD brain lysates, we could not examine HSP60 aggregation; nonetheless, similar to the P*H− mouse brains, human AD brains displayed elevated HSP60 polyubiquitination (Fig. 11C), supporting its destabilization by AOs and subsequent proteasomal degradation. Congruent with its depletion, HSF1 was also polyubiquitinated in AD brains (Fig. 11C), a decisive event expected to exacerbate the attack on HSP60 by AOs. HSF1 polyubiquitination is, likely, the consequence of destabilization by AOs, supported by its aggregation provoked by Aβ1–42 in vitro (Fig. 6C). Supporting hyperactivation of AKT/mTORC1 signaling, AD brains displayed markedly elevated phosphorylation of AKT and S6K (Fig. 11D). Given the primary focus of this manuscript on overgrowth, we only tested a limited number of human AD samples randomly (table S1). Despite this caveat, all AD samples, but not control samples, displayed AO-HSP60 interactions. These findings do suggest that at least in some cases of AD, HSF1 shields HSP60 from the assault of AOs, thereby preserving the stability of mitochondrial proteome and bestowing neuroprotection (Fig. 11E), the very same mechanism underpinning tissue overgrowth.
Fig. 11. Implications of HSF1 in human AD.
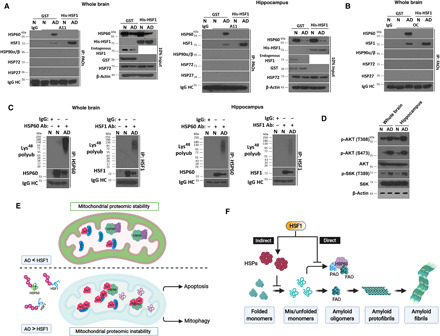
(A and B) Detection of AO-HSP60 and AO-HSF1 interactions by co-IP with the EasyBlot reagents in AD whole brain or hippocampus lysates (500 μg) with addition of 200 ng of recombinant GST or HSF1. The lysates are from two patients with late-onset AD and two aged normal controls, distinct from the ones on the tissue arrays. N, normal controls. (C) Detection of HSP60 and HSF1 polyubiquitination in AD brain lysates by IP with the EasyBlot reagents. (D) Immunoblotting of AKT/mTOCR1 signaling in AD brain lysates. (E) Schematic depiction of the model wherein HSF1 safeguards the mitochondria against amyloids. With sufficient HSF1, the attack on HSP60 by AOs is blocked. Accordingly, HSP60 preserves the stability of the mitochondrial proteome. Continuous rise of AOs overwhelms and destabilizes HSF1, causing its deficiency. Thus, those nonneutralized AOs start attacking HSP60, a decisive event devastating the entire mitochondrial proteome. The ensuing mitochondrial damage instigates both apoptosis and mitophagy. (F) Schematic depiction of the dual roles of HSF1 in antagonizing amyloidogenesis. Through transcriptional up-regulation of HSPs, HSF1 ensures appropriate protein quality to sustain proteomic stability, thereby preventing the emergence of amyloids, which represents an indirect mechanism. Nonetheless, once amyloidogenesis becomes inevitable, HSF1, by physically sequestering AOs, both alleviates the buildup of amyloids and safeguards the mitochondria to avert toxicity, which represents a direct mechanism. (A) to (D) were done once.
DISCUSSION
Amyloid deposits in the nervous system are a hallmark of neurodegenerative disorders. Serendipitously, our studies of tissue overgrowth driven by oncogenic signaling, a seemingly unrelated pathological condition, have yielded insights into both the genesis and mechanism of action of amyloids, which may have broad implications in overgrowth syndromes, cancer, and neurodegenerative disorders in humans.
HSF1 dictates growth capacity
Our studies uncovered HSF1 as a previously unidentified physiological substrate for AKT, a key controller of protein translation. It appears logical for AKT to activate HSF1, thereby providing a necessary chaperoning capability. Thus, the coregulation of mTORC1 and HSF1 by AKT promotes the balance between protein quantity and quality, a prerequisite for robust growth. Without adequate quality, increased protein quantity would be infertile or even deleterious, diminishing cell size and triggering cell death, as demonstrated in our mouse models. Apart from tissue overgrowth, constitutive activation of HSF1 by oncogenic AKT in the absence of environmental stress is expected to promote malignant growth as well. Moreover, our previous study revealed that HSF1, by sustaining mTORC1 activity, supports normal growth (21). Thus, HSF1 is an indispensable element of growth signaling that ensures proteomic stability, thereby dictating growth capacity. This superfluous growth capacity empowered by HSF1, ironically, becomes crucial to its generic prooncogenic effects.
Amyloidogenesis may be a mechanism to preserve tissue homeostasis
Our studies strongly suggest that an imbalance between protein quantity and quality, prompted by heightened protein synthesis and/or Hsf1 deficiency, elicits amyloidogenesis. Of note, even orchestrated protein quantity- and quality-control machineries cannot preclude amyloidogenesis entirely, exemplified by elevated amyloids in Hsf1-proficient tissues with hyperactive PI3K/AKT signaling. Thus, amyloidogenesis is likely an inevitable sequel of uncontrolled cell growth. Nonetheless, it remains unclear whether amyloids are indeed induced in human PROS, ASD, and fragile X syndrome, all of which display hyperactivation of PI3K signaling and brain overgrowth (2, 3).
Mechanistically, our findings indicate that excessive protein synthesis is causally related to amyloidogenesis. The manifestation of both Aβ and Tau aggregates, the two pathological hallmarks of human AD, in our mouse models suggests a general defect caused by excessive protein synthesis, which disturbs both the amyloid precursor protein (APP) metabolism and Tau protein stability. Likely, a global surge in protein quantity overwhelms the cellular protein quality-control machineries, resulting in prevalent protein misfolding and aggregation. This proteomic instability may ultimately instigate amyloidogenesis. Nonetheless, it remains to be elucidated how the APP metabolism and Tau protein stability are affected by heightened protein synthesis specifically.
Whereas our models mimic tissue overgrowth, an analogous scenario is malignancy, which is characterized in part by uncontrolled clonal cell growth. Amyloidogenesis is expected to occur in malignancy, as shown in our previous study (5). Numerous studies in neurodegenerative disorders have demonstrated that amyloids are neurotoxic and have a devastating effect. However, our previous study revealed that amyloidogenesis is tumor-suppressive (5). In line with this, our current study illuminates the underlying mechanism through which uncontained amyloidogenesis, owing to Hsf1 deficiency, potently induces apoptosis. Despite the apparent involvement of apoptosis, however, we cannot exclude the contributions of other types of cell death, such as necroptosis. It remains possible that different cell death mechanisms coexist. Together, our findings suggest that amyloidogenesis serves to restrain aberrant growth at least in part via apoptotic cell elimination, thereby preserving tissue homeostasis and suppressing tumorigenesis, which is, paradoxically, beneficial.
HSP60, guardian of the mitochondrial proteome, is a prime target of soluble AOs
The impacts of amyloids on neurons are broad. A number of direct neuronal targets of Aβ have been reported, including the Ephrin-type B2 receptor, NMDA receptors, and α7 nicotinic acetylcholine receptors (45–47). Likely, these interactions underlie the amyloid-induced neuronal dysfunction. Mitochondrial dysfunction, an expected cause of neurotoxicity, has also been reported in human neurodegenerative disorders (48).
Our studies now pinpoint HSP60 as a direct target of amyloids. Physical assault by AOs causes mislocalization, polyubiquitination, and aggregation of HSP60, resulting in loss of HSP60 functions and subsequent destabilization of the whole mitochondrial proteome. In turn, mitochondrial damage, apoptosis, and mitophagy ensue. The substantial rescue of mitochondrial defects and blockade of apoptosis by HSP60 overexpression authenticate it as a pivotal cellular target of amyloids.
HSF1 is a genuine anti-amyloid factor
Despite evident amyloidogenesis, the lack of apoptosis in Hsf1+/+ overgrown tissues is unusual. Apparently, amyloidogenesis alone is insufficient to provoke toxicity, underlining the importance of intrinsic antagonizing mechanisms.
Our previous studies have demonstrated that HSF1 suppresses amyloidogenesis in cancer cells (5), a mechanism promoting oncogenesis. As the master regulator of the HSR/PSR, HSF1 is widely assumed to exert this amyloid-suppressing effect via its transcriptional regulation of HSPs. Our results unexpectedly showed that HSF1 physically interacts with soluble AOs, both impeding the amyloidogenic process and, more importantly, stopping their assaults on HSP60. Our findings do not support an evident role of HSPs in countering amyloids directly; nonetheless, the transcriptional induction of HSPs by HSF1, which warrants appropriate protein quality and thereby maintains a healthy proteome, could prevent the origination of amyloids from mis/unfolded proteins, thus representing an indirect action of HSF1 and HSPs. Once amyloidogenesis begins, the direct action of HSF1 then becomes crucial to containment and neutralization of amyloids (Fig. 11F).
In aggregate, our findings support a molecular model wherein HSF1, via physical interactions with soluble AOs, potently suppresses the amyloidogenic process and its induced toxicity. At a low interaction stoichiometry (AOs < HSF1), HSF1 efficiently blocks the self-propagation of AOs and stops their attack on cellular targets, thereby averting toxicity. At a high stoichiometry (AOs << HSF1), HSF1 can also impede the assembly of FAOs into mature fibrils and further convert them into amorphous aggregates or stabilize them at the soluble, nontoxic state. This model concurs with our finding that insoluble OC+ AFs still formed in Hsf1+/+ overgrown tissues in the absence of apoptosis. Whereas the abundance of HSF1 completely blocks the attack by AOs on HSP60, it could only partially impede the AF assembly from FAOs in these tissues. This was recapitulated in vitro. At a 1:1 molar ratio, HSF1 fully rescued the solubility of HSP60 but only partially blocked Aβ1–42 fibrillation (Figs. 6C and 8B). Thus, HSF1 preferentially antagonizes the highly toxic pathway during amyloidogenesis.
The absence of HSP60 depletion and apoptosis in Hsf1+/+ overgrown tissues, albeit with elevated amyloids, is indeed notable. This finding not only underscores the pivotal role of HSF1 in inhibiting amyloid-induced cytotoxicity but also agrees with previous reports that Aβ loads are not well correlated with cognitive impairment or neurotoxicity (49). However, excessive amyloids can overcome the neutralizing capacity of endogenous HSF1, leading to HSP60 destabilization and apoptosis. Of note, amyloids could also destabilize HSF1, which is an undesirable price for shielding HSP60, as evidenced by HSF1 aggregation induced by Aβ1–42 in vitro and HSF1 polyubiquitination in AD brains (Figs. 6C and 11C). Likely, the HSF1:AO interaction stoichiometry decides the fate of HSF1, a notion supported by the dose-dependent effect of Aβ1–42 on HSF1 depletion and aggregation (Fig. 6E). In Hsf1+/+ overgrown tissues, there was no depletion of soluble HSF1, suggesting a relatively high HSF1:AO stoichiometry and absence of HSF1 destabilization. However, the existence of additional in vivo factor(s) that facilitate HSF1 to stabilize AOs cannot be excluded. Our finding that HSF1 is depleted in AD brains, likely a consequence of interactions with amyloids, is congruent with a reduction in plasma exosomal HSF1 proteins in patients with AD (50). Depletion of soluble HSF1 would not only aggravate the assault on HSP60 by AOs but also impair the cytoprotective HSR/PSR, greatly accelerating the progression of AD. Hence, HSF1 constitutes a critical line of defense that buffers amyloid-induced toxicity. Our findings suggest that the intracellular AO:HSF1 molar ratio may be a determinant of neurotoxicity and a prognostic marker for AD progression. Whereas the precise threshold of AO:HSF1 ratios deciding the neurotoxicity in patients with AD remains to be determined, the 4G8 immunoreactivity under nondenaturing conditions and AO-HSP60 interactions may signify the excessiveness of AOs and a breach of this threshold.
While HSF1 antagonizes the cytotoxicity associated with amyloidogenesis by neutralizing both PAOs and FAOs, we cannot exclude the possibility that HSF1 may interact with other amyloid conformers as well. Its interactions with AOs suggest a conformation-dependent recognition of amyloids by HSF1, a notion supported by its binding to both Aβ and Tau (fig. S5C). Thus, it remains elusive whether HSF1 is a generic anti-amyloid factor. Whereas Aβ is the major constituent of AOs in overgrown brains, other non-Aβ AOs can also attack HSP60 and interact with HSF1. HSF1 either binds to a common conformation shared by PAOs and FAOs via a single interface or recognizes PAOs and FAOs via discrete interfaces. Our current study has already pinpointed the anti-amyloid interfaces within the N-terminal 323 amino acids of HSF1, encompassing the DBD but excluding the TAD. The detailed characterization of these interactions, a critical step toward harnessing this powerful anti-amyloid action, is underway and will be reported in a separate manuscript. While both HSF1 and HSP60 bind FAOs, only HSF1 can block the AF assembly, suggesting that the HSP60-binding site is not engaged in the AF assembly. Aside from the HSP60-binding site, HSF1 likely interacts with FAOs at other sites that are crucial to the AF assembly, thereby both neutralizing the attack on HSP60 and impeding the fibril assembly. Binding of AOs at a high stoichiometry is predicted to sequestrate HSF1 apart from its DNA binding. This may represent a means for amyloids to incapacitate the HSR/PSR and destabilize the proteome, thereby exerting tumor suppression. Moreover, despite its importance to HSF1 DNA binding, it remains unclear whether Ser230 phosphorylation affects the AO-HSF1 interaction.
Neurodegeneration, overgrowth, and malignancy converge upon amyloidogenesis
On the one hand, the anti-amyloid action of HSF1 confers neuroprotection; on the other, it promotes overgrowth and malignancy. The two seemingly opposite human pathologies, cancer and neurodegeneration, converge upon proteomic instability and, in particular, amyloidogenesis. Of note, both cancerous cells and neurons are characterized by high rates of protein synthesis. Moreover, PI3K/AKT/mTORC1 signaling, closely implicated in aging and age-related diseases (51), is hyperactivated in AD brains (Fig. 11D) (52, 53). Although it remains possible that this hyperactivation may be secondary to amyloidogenesis, it is tantalizing to postulate that dysregulated protein synthesis during aging may instigate or at least aggravate amyloidogenesis in sporadic AD. Our findings indicate that extensive activation of PI3K/AKT signaling in neural cells suffices to induce pathologies bearing some similarities to AD, including Aβ deposits, amyloid angiopathy, and Tau hyperphosphorylation and aggregation. Nonetheless, the characteristic senile plaques are largely absent, which may be in part due to the very short animal life span. In support of this notion, P*H− brains also display evident intranuclear Aβ accumulation, which has been proposed as an early event leading to senile plaques in AD (54). It is noteworthy that in our overgrowth models, PI3K signaling is hyperactivated in most neurons and astrocytes, leading to ubiquitous amyloid accumulation throughout the brain. Moreover, the rapid demise of P*H+ mice, owing to severe brain enlargement, precludes the investigation of how the amyloidogenesis evolves in vivo until endogenous HSF1 is overwhelmed. Hence, it remains interesting whether heightening protein translation in certain subsets of neurons could recapitulate AD phenotypes more closely.
Whereas, in neurodegenerative disorders, the compromised proteostatic mechanisms, including depleted HSF1, fail to contain amyloidogenesis, cancerous cells succeed in doing so, in part through up-regulation of HSF1 (55). Thus, HSF1 may play a role in balancing cancer and neurodegenerative disorders, two major age-related diseases in humans whose incidences demonstrate an inverse correlation (56, 57).
MATERIALS AND METHODS
The detailed information of all cell lines, animals, reagents, and assay kits is listed in table S2.
Animal studies
The generation of Hsf1fl/fl mice on the inbred C57BL/6J background was described previously (21). All other mice were purchased from the Jackson Laboratory. R26StopFLP110*, Alb-Cre, and Ptenfl/fl mice were all on the C57BL/6J background. hGFAP-Cre mice on the FVB background were first backcrossed onto the C57BL/6J background for five generations. Both males and females were included in the experiments. All mouse experiments were approved by the Institutional Animal Care and Use Committees.
Quantitation of AOs, AFs, and Aβ1–42 by ELISA
To quantitate soluble PAOs and FAOs, 100 μg of detergent-soluble fractions or membrane-associated fractions was diluted in phosphate-buffered saline (PBS) and coated in a 96-well ELISA plate at 4°C overnight. Following blocking with 5% nonfat milk in PBS-T at room temperature (RT) for 1 hour, each well was incubated with 100 μl of A11 Abs or OC Abs (1:1000) at RT for 2 hours. After washing with PBS-T, each well was incubated with 100 μl of goat anti-rabbit immunoglobulin G (IgG) horseradish peroxidase (HRP) conjugates (1:5000) at RT for 1 hour. Following washing, 100 μl of 1-Step Ultra TMB-ELISA substrates was added to each well.
To quantitate AFs, detergent-insoluble fractions were first re-solubilized by sonication with 2% SDS in PBS for 10 min. Following protein quantitation using a Pierce BCA protein assay kit, 100 μg of solubilized aggregates diluted in PBS was added to each well and incubated at 37°C without cover overnight to dry the wells. Following blocking, each well was incubated with 100 μl of OC Abs (1:1000) at RT for 2 hours. Mouse endogenous Aβ1–42 in tissues was quantitated by a sandwich Amyloid beta 42 Mouse ELISA kit.
Proximity ligation assay
The PLA procedure was described in detail previously (21). Duolink In Situ PLA anti-rabbit Plus probes; anti-mouse Minus probes; and Detection Reagents Red, Green, or Brightfield were all purchased from Sigma-Aldrich. All PLA primary Abs were incubated at 4°C overnight.
For detection of endogenous AKT-HSF1 interactions in MEFs, rabbit anti-AKT (pan) Abs (C67E7) (1:100) and mouse anti-HSF1 Ab (E-4) (1:100) were combined. For detection of PAO-HSP60 interactions in cultured astrocytes, rabbit anti-AOs A11 Abs (1:100) and mouse anti-HSP60 Abs clone LK1 (1:100) were combined. To detect interactions between biotinylated Aβ1–42 and HSP60 or FLAG-HSF1 in neurons, cultured primary human neurons were fixed in 1× Hanks’ balanced salt solution (prewarmed to 37°C) containing 4% formaldehyde for 20 min. For staining, mouse anti-biotin Ab-2 BTN.4 Abs (1:100) and rabbit anti-HSP60 (D6F1) Abs (1:100) or rabbit anti-DYKDDDDK Tag (D6W5B) Abs Alexa Fluor 488 conjugates (1:100) were combined. For the biotinylated Aβ1–42–HSP60 PLA, following the coincubation of anti-rabbit Plus and anti-mouse Minus PLA probes, neurons were incubated with mouse anti–βIII-tubulin Abs clone 2G10-TB3 Alexa Fluor 488 conjugates (1:100) at RT for 2 hours. The PLA procedure was continued after the βIII-tubulin staining. Nuclei were counterstained with Hoechst 33342. PLA signals were documented using a Zeiss LSM 780 confocal microscope.
For detection of AO-HSP60 interactions in human AD brains, the tissue arrays were first immersed in sodium citrate buffer (pH 6.0) for heat-induced antigen retrieval using a vegetable steamer for 30 min. After quenching endogenous peroxidase and alkaline phosphatase activities using the BLOXALL blocking solution and blocking with 2.5% normal horse serum, the tissue arrays were incubated with rabbit A11 or OC Abs (1:100) and mouse anti-HSP60 Abs clone LK1 (1:100) at 4°C overnight. Following incubation with anti-rabbit Plus and anti-mouse Minus probes at 37°C for 1 hour and subsequent ligation at 37°C for 30 min, amplification was performed at 37°C for 4 hours or overnight. Following washing with buffer A, the arrays were incubated with Detection Brightfield reagents at RT for 60 min. The substrate development was performed using an ImmPACT DAB Peroxidase (HRP) substrate kit. A set of arrays incubated without the primary Abs served as negative controls. Counterstained tissue arrays were imaged using an Aperio AT2 Digital Whole Slide Scanner (Leica Biosystems).
Immunohistochemistry
When staining frozen sections, tissues were first air-dried at 37°C overnight. Antigen retrieval was performed by treating sections with 1% SDS at RT for 5 min (A11 and Aβ1–42 oligomer staining) or 20 min (cleaved caspase 3 staining). When staining paraffin-embedded mouse brain tissues and human AD brain tissue arrays, antigen retrieval was performed using sodium citrate buffer (pH 6.0) in a vegetable steamer for 30 min. When staining Aβ (4G8 and ab2539), antigen retrieval was performed by treating frozen sections for 5 min or paraffin-embedded sections for 20 min with 70% formic acids at RT. Following blocking with BLOXALL blocking solution and 2.5% normal horse serum, tissue sections were incubated with primary Abs, including rabbit anti-AOs (A11) or anti-AFs (OC) Abs (1:100), rabbit anti-Aβ1–42 Abs (oligomer-specific) (1:100), rabbit anti-Aβ Ab ab2539 (1:100) or mouse anti-Aβ17–24 (4G8) Abs (1:100), rabbit anti-phospho-Tau (Ser404) (D2Z4G) Abs (1:100), rabbit anti-cleaved caspase 3 (Asp175) (5A1E) (1:100), rabbit anti-Iba1/AIF1 Abs (1:100), mouse anti–βIII-tubulin (AA10) Abs (1:100), rabbit anti-HSF1 (EP1710Y) Abs (1:100), or rabbit anti-HSP60 (D6F1) Abs (1:100). Except for 4G8, ab2539, Iba-1, and βIII-tubulin Abs, which were incubated at RT for either 2 or 4 hours, all other primary Abs were incubated at 4°C overnight. Following washing with PBS-T, slides were incubated with either ImmPRESS HRP anti-rabbit IgG or AP anti-mouse IgG polymers at RT for 30 min. Subsequently, the ImmPACT DAB or ImmPACT Vector Red Alkaline Phosphatase substrate (for 4G8 staining in Fig. 1L) was added. For the OC, ab2539, phospho-Tau, and cleaved Caspase 3 staining, an ImmPRESS Excel Amplified HRP Polymer Staining Kit (anti-rabbit IgG) was applied to amplify the weak staining signals following the incubation with primary Abs. For staining with primary mouse Abs, mouse on mouse (M.O.M.) blocking reagents were used, and the primary Abs were incubated at RT for 2 or 4 hours. For double staining of microglia and neurons, slides were coincubated with rabbit anti-Iba1 and mouse anti–βIII-tubulin (AA10) Abs at RT for 2 hours. To visualize the double staining, ImmPACT DAB HRP and Vector Blue Alkaline Phosphatase substrates were used without counterstaining. All counterstained slides were imaged using an Aperio AT2 Digital Whole Slide Scanner (Leica Biosystems).
In vitro amyloid seeding and Aβ1–42 fibrillation assays
For amyloid fibrillation assays, each reaction was carried out in PBS containing 2 μM synthetic human Aβ1–42 or Aβ42–1 peptides (200 μM stock in 0.01 M NaOH), various doses of recombinant proteins that were predialyzed against PBS, and 10 μM ThT. For amyloid seeding experiments, each reaction contained 100-μg detergent-soluble fractions of tissue lysates diluted in PBS, 2 μM synthetic Aβ1–42 peptides, and 10 μM ThT. All reactions were incubated at 37°C with gentle shaking at 600 rpm in an Eppendorf ThermoMixer C (Eppendorf North America). Fluorescence was measured at Ex 450 nm/Em 482 nm using a CLARIOstar microplate reader (BMG LABTECH).
Filter-trap assay
A total of 1 × 106 cells or 10-mg pulverized frozen tissues were re-suspended in 100 μl of 1× PBS containing 1× Halt phosphatase inhibitor cocktail and processed by a Q125 sonicator (Qsonica LLC) using 20% amplitude for 3-min processing time with 5S pulsing cycle to prepare lysates. After sonication, 900 μl of PBS containing 2% SDS was added to the samples. Immobilon polyvinylidene difluoride membranes with 0.45-μm pore size were presoaked in PBS containing 2% SDS, and the slots of a 96-well Bio-Dot Microfiltration Apparatus (Bio-Rad Laboratories) were washed twice with PBS containing 2% SDS. Two hundred microliters of samples was applied to each slot, and the samples were allowed to filter through the membranes by gravity flow for 30 to 40 min. A vacuum was then applied to drain any residual liquids. Following staining with 0.1% ponceau red solution to visualize protein aggregates, the membranes were blocked with 5% nonfat dry milk in PBS. Primary and secondary Ab-HRP conjugates were subsequently applied to detect specific protein aggregates.
In vitro reconstitution of Aβ1–42, HSP60, and HSF1 interactions
Recombinant proteins and Abs were all predialyzed against PBS. Each reaction was carried out in PBS containing 2 μM Aβ1–42 or Aβ42–1 peptides, HSP60, and HSF1 or Abs at a 1:1:1 molar ratio. Reactions were incubated at RT for 4 hours, followed by centrifugation at 15,000 rpm for 10 min at 4°C in an Eppendorf Benchtop 5424 Microcentrifuge. The pellets were then re-solubilized by sonication for 10 min in 100 μl of PBS containing 2% SDS for SDS–polyacrylamide gel electrophoresis.
Fluorescence resonance energy transfer
HiLyte Fluor 488–labeled human Aβ1–42 peptides were purchased from AnaSpec EGT Corporate. Recombinant GST and HSF1 proteins, which predialyzed against PBS, were labeled with DABCYL acid N-hydroxysucciminide ester using a Molecular Probes Alexa Fluor 594 Microscale Protein Labeling kit. HiLyte Fluor 488–labeled Aβ1–42 and DABCYL-labeled GST or HSF1 were mixed at a 1:1 molar ratio in a 96-well black-walled, clear-bottom microplate and the fluorescence was immediately measured at Ex/Em = 503/528 nm.
Turbidity measurement
Aβ1–42 peptides (2 μM) were mixed with either recombinant GST or HSF1 proteins, which were predialyzed against PBS, at different molar ratios in a clear 96-well microplate. For monitoring the turbidity of mouse brain lysates, detergent-soluble fractions were loaded onto a clear 96-well microplate (100 μg per well diluted in PBS). The plates were incubated at 37°C with 600 rpm shaking for 48 hours, and the turbidity was measured at different time points using a NEPHELOstar Plus microplate nephelometer (BMG Labtech).
Transmission electron microscopy
Following incubation, AFs or aggregates were pelleted and re-suspended in distilled H2O. One drop of fibril solution was placed on a 200-mesh carbon-coated nickel grid. After 1 min, the remaining liquid was wicked. Immediately, a drop of 2% uranyl acetate solution was placed on the grid for 1 min. After wicking, the grids were air-dried and examined under a Tecnai T12 transmission electron microscope (FEI Company, USA) operating at 80 kV.
Neuronal viability measurement
Primary human neurons (1 × 104 cells per well) were plated in 96-well culture plates coated with poly-l-lysine (50 μg/ml). Following transduction with lentiviral particles overnight, neurons were further cultured for 4 days, followed by transfection of 10 μM Aβ42–1 or Aβ1–42 for 24 hours. Neuronal viabilities were measured by CellTiter-Blue Cell Viability Assay.
Statistical methods
All error bars represent means ± SD. Statistical analyses were performed using Prism 7.0 or 8.0 (GraphPad software). Detailed statistical methods were described in figure legends. Statistical significance: n.s., not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
Code availability
No codes were generated during this study.
Supplementary Material
Acknowledgments
We thank H. Morris for technical support for the animal care and studies, the Electron Microscopy Laboratory for assistance with the TEM studies, and BioRender.com for the creation of illustrations. Funding: This work was supported by a grant from NIH to C.D. (1DP2OD007070) and by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government. Author contributions: Z.T., K.-H.S., and M.X. designed and conducted the experiments. C.D. conceptualized and supervised this study, analyzed the results, and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/46/eabc6871/DC1
REFERENCES AND NOTES
- 1.Manning B. D., Cantley L. C., AKT/PKB signaling: Navigating downstream. Cell 129, 1261–1274 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keppler-Noreuil K. M., Parker V. E., Darling T. N., Martinez-Agosto J. A., Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & therapeutic strategies. Am. J. Med. Genet. C Semin. Med. Genet. 172, 402–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen J., Alberts I., Li X., Dysregulation of the IGF-I/PI3K/AKT/mTOR signaling pathway in autism spectrum disorders. Int. J. Dev. Neurosci. 35, 35–41 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Tumaneng K., Russell R. C., Guan K.-L., Organ size control by Hippo and TOR pathways. Curr. Biol. 22, R368–R379 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang Z., Dai S., He Y., Doty R. A., Shultz L. D., Sampson S. B., Dai C., MEK guards proteome stability and inhibits tumor-suppressive amyloidogenesis via HSF1. Cell 160, 729–744 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selkoe D. J., Hardy J., The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morimoto R. I., The heat shock response: Systems biology of proteotoxic stress in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 76, 91–99 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Dai C., The heat-shock, or HSF1-mediated proteotoxic stress, response in cancer: From proteomic stability to oncogenesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 373, 20160525 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gomez-Pastor R., Burchfiel E. T., Thiele D. J., Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 19, 4–19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dai C., Santagata S., Tang Z., Shi J., Cao J., Kwon H., Bronson R. T., Whitesell L., Lindquist S., Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J. Clin. Invest. 122, 3742–3754 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mendillo M. L., Santagata S., Koeva M., Bell G. W., Hu R., Tamimi R. M., Fraenkel E., Ince T. A., Whitesell L., Lindquist S., HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150, 549–562 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McMillan D. R., Xiao X., Shao L., Graves K., Benjamin I. J., Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J. Biol. Chem. 273, 7523–7528 (1998). [DOI] [PubMed] [Google Scholar]
- 13.Kohn A. D., Summers S. A., Birnbaum M. J., Roth R. A., Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J. Biol. Chem. 271, 31372–31378 (1996). [DOI] [PubMed] [Google Scholar]
- 14.Benderska N., Ivanovska J., Rau T. T., Schulze-Luehrmann J., Mohan S., Chakilam S., Gandesiri M., Ziesché E., Fischer T., Söder S., Agaimy A., Distel L., Sticht H., Mahadevan V., Schneider-Stock R., DAPK–HSF1 interaction as a positive-feedback mechanism stimulating TNF-induced apoptosis in colorectal cancer cells. J. Cell Sci. 127, 5273–5287 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Holmberg C. I., Hietakangas V., Mikhailov A., Rantanen J. O., Kallio M., Meinander A., Hellman J., Morrice N., MacKintosh C., Morimoto R. I., Eriksson J. E., Sistonen L., Phosphorylation of serine 230 promotes inducible transcriptional activity of heat shock factor 1. EMBO J. 20, 3800–3810 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jansen L. A., Mirzaa G. M., Ishak G. E., O’Roak B. J., Hiatt J. B., Roden W. H., Gunter S. A., Christian S. L., Collins S., Adams C., Rivière J.-B., St-Onge J., Ojemann J. G., Shendure J., Hevner R. F., Dobyns W. B., PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain 138, 1613–1628 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mirzaa G. M., Rivière J.-B., Dobyns W. B., Megalencephaly syndromes and activating mutations in the PI3K-AKT pathway: MPPH and MCAP. Am. J. Med. Genet. C Semin. Med. Genet. 163C, 122–130 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Srinivasan L., Sasaki Y., Calado D. P., Zhang B., Paik J. H., DePinho R. A., Kutok J. L., Kearney J. F., Otipoby K. L., Rajewsky K., PI3 kinase signals BCR-dependent mature B cell survival. Cell 139, 573–586 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhuo L., Theis M., Alvarez-Maya I., Brenner M., Willecke K., Messing A., hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis 31, 85–94 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Schmidt E. K., Clavarino G., Ceppi M., Pierre P., SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 6, 275–277 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Su K.-H., Cao J., Tang Z., Dai S., He Y., Sampson S. B., Benjamin I. J., Dai C., HSF1 critically attunes proteotoxic stress sensing by mTORC1 to combat stress and promote growth. Nat. Cell Biol. 18, 527–539 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elliott M. R., Ravichandran K. S., Clearance of apoptotic cells: Implications in health and disease. J. Cell Biol. 189, 1059–1070 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu R., Shen Q., Xu P., Luo J. J., Tang Y., Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 49, 1422–1434 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buell A. K., Dobson C. M., Knowles T. P., Welland M. E., Interactions between amyloidophilic dyes and their relevance to studies of amyloid inhibitors. Biophys. J. 99, 3492–3497 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., Glabe C. G., Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Kayed R., Head E., Sarsoza F., Saing T., Cotman C. W., Necula M., Margol L., Wu J., Breydo L., Thompson J. L., Rasool S., Gurlo T., Butler P., Glabe C. G., Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2, 18 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Brien R. J., Wong P. C., Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 34, 185–204 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gouras G. K., Tsai J., Naslund J., Vincent B., Edgar M., Checler F., Greenfield J. P., Haroutunian V., Buxbaum J. D., Xu H., Greengard P., Relkin N. R., Intraneuronal Aβ42 accumulation in human brain. Am. J. Pathol. 156, 15–20 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LaFerla F. M., Green K. N., Oddo S., Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 8, 499–509 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Serrano-Pozo A., Frosch M. P., Masliah E., Hyman B. T., Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1, a006189 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otvos L. Jr., Feiner L., Lang E., Szendrei G. I., Goedert M., Lee V. M.-Y., Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J. Neurosci. Res. 39, 669–673 (1994). [DOI] [PubMed] [Google Scholar]
- 32.Lorenzo A., Yankner B. A., Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc. Natl. Acad. Sci. U.S.A. 91, 12243–12247 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eisenberg D., Jucker M., The amyloid state of proteins in human diseases. Cell 148, 1188–1203 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glabe C. G., Structural classification of toxic amyloid oligomers. J. Biol. Chem. 283, 29639–29643 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haass C., Selkoe D. J., Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Moerke N. J., Aktas H., Chen H., Cantel S., Reibarkh M. Y., Fahmy A., Gross J. D., Degterev A., Yuan J., Chorev M., Halperin J. A., Wagner G., Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell 128, 257–267 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Tolcher A., Goldman J., Patnaik A., Papadopoulos K. P., Westwood P., Kelly C. S., Bumgardner W., Sams L., Geeganage S., Wang T., Capen A. R., Huang J., Joseph S., Miller J., Benhadji K. A., Brail L. H., Rosen L. S., A phase I trial of LY2584702 tosylate, a p70 S6 kinase inhibitor, in patients with advanced solid tumours. Eur. J. Cancer 50, 867–875 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Hayer-Hartl M., Bracher A., Hartl F. U., The GroEL–GroES chaperonin machine: A nano-cage for protein folding. Trends Biochem. Sci. 41, 62–76 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Wiedemann N., Pfanner N., Mitochondrial machineries for protein import and assembly. Annu. Rev. Biochem. 86, 685–714 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Jiang X., Wang X., Cytochrome C-mediated apoptosis. Annu. Rev. Biochem. 73, 87–106 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Youle R. J., Narendra D. P., Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mizushima N., Yoshimori T., Levine B., Methods in mammalian autophagy research. Cell 140, 313–326 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kayed R., Canto I., Breydo L., Rasool S., Lukacsovich T., Wu J., Albay R. III, Pensalfini A., Yeung S., Head E., Marsh J. L., Glabe C., Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Aβ oligomers. Mol. Neurodegener. 5, 57 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu J. W., Breydo L., Isas J. M., Lee J., Kuznetsov Y. G., Langen R., Glabe C., Fibrillar oligomers nucleate the oligomerization of monomeric amyloid β but do not seed fibril formation. J. Biol. Chem. 285, 6071–6079 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cissé M., Halabisky B., Harris J., Devidze N., Dubal D. B., Sun B., Orr A., Lotz G., Kim D. H., Hamto P., Ho K., Yu G.-Q., Mucke L., Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 469, 47–52 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Felice F. G., Velasco P. T., Lambert M. P., Viola K., Fernandez S. J., Ferreira S. T., Klein W. L., Aβ oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 282, 11590–11601 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Wang H. Y., Lee D. H., D’Andrea M. R., Peterson P. A., Shank R. P., Reitz A. B., β-Amyloid1–42 binds to α7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J. Biol. Chem. 275, 5626–5632 (2000). [DOI] [PubMed] [Google Scholar]
- 48.Moreira P. I., Carvalho C., Zhu X., Smith M. A., Perry G., Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 1802, 2–10 (2010). [DOI] [PubMed] [Google Scholar]
- 49.Morris G. P., Clark I. A., Vissel B., Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2, 135 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goetzl E. J., Boxer A., Schwartz J. B., Abner E. L., Petersen R. C., Miller B. L., Carlson O. D., Mustapic M., Kapogiannis D., Low neural exosomal levels of cellular survival factors in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2, 769–773 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnson S. C., Rabinovitch P. S., Kaeberlein M., mTOR is a key modulator of ageing and age-related disease. Nature 493, 338–345 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Griffin R. J., Moloney A., Kelliher M., Johnston J. A., Ravid R., Dockery P., O’Connor R., O’Neill C., Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J. Neurochem. 93, 105–117 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Tramutola A., Triplett J. C., Di Domenico F., Niedowicz D. M., Murphy M. P., Coccia R., Perluigi M., Butterfield D. A., Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): Analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J. Neurochem. 133, 739–749 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Pensalfini A., Albay R. III, Rasool S., Wu J. W., Hatami A., Arai H., Margol L., Milton S., Poon W. W., Corrada M. M., Kawas C. H., Glabe C. G., Intracellular amyloid and the neuronal origin of Alzheimer neuritic plaques. Neurobiol. Dis. 71, 53–61 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ciocca D. R., Arrigo A. P., Calderwood S. K., Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: An update. Arch. Toxicol. 87, 19–48 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li J.-M., Liu C., Hu X., Cai Y., Ma C., Luo X.-G., Yan X.-X., Inverse correlation between Alzheimer’s disease and cancer: Implication for a strong impact of regenerative propensity on neurodegeneration? BMC Neurol. 14, 211 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Plun-Favreau H., Lewis P. A., Hardy J., Martins L. M., Wood N. W., Cancer and neurodegeneration: Between the devil and the deep blue sea. PLOS Genet. 6, e1001257 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dai C., Celestino J. C., Okada Y., Louis D. N., Fuller G. N., Holland E. C., PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 15, 1913–1925 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/46/eabc6871/DC1