

Plasma-derived extracellular vesicles potentially enable prediction and tracking of immunotherapy response.
Abstract
Immune checkpoint inhibitors (ICIs) show promise, but most patients do not respond. We identify and validate biomarkers from extracellular vesicles (EVs), allowing non-invasive monitoring of tumor- intrinsic and host immune status, as well as a prediction of ICI response. We undertook transcriptomic profiling of plasma-derived EVs and tumors from 50 patients with metastatic melanoma receiving ICI, and validated with an independent EV-only cohort of 30 patients. Plasma-derived EV and tumor transcriptomes correlate. EV profiles reveal drivers of ICI resistance and melanoma progression, exhibit differentially expressed genes/pathways, and correlate with clinical response to ICI. We created a Bayesian probabilistic deconvolution model to estimate contributions from tumor and non-tumor sources, enabling interpretation of differentially expressed genes/pathways. EV RNA-seq mutations also segregated ICI response. EVs serve as a non-invasive biomarker to jointly probe tumor-intrinsic and immune changes to ICI, function as predictive markers of ICI responsiveness, and monitor tumor persistence and immune activation.
INTRODUCTION
Historically, blood-based biomarkers for immunotherapy focused on cell-free DNA (cfDNA) or circulating tumor cells (1, 2), which solely reflect tumor-based properties and not changes in the immune system during treatment. To improve prediction and tracking of immune checkpoint inhibitor (ICI) resistance, simultaneous capture of transcriptomic features from both the tumor and immune system (3) is critical.
Extracellular vesicles (EVs) are produced by many cell types including tumor and immune cells, which contain a subtranscriptome of their cell of origin. EVs are involved in oncogenesis and immune modulation and serve as communicators of genetic and epigenetic signals. In cancers, tumor-secreted EVs modulate the tumor microenvironment and elicit antitumoral immune responses (4), and plasma-derived EV transcripts are markers of antitumor immune activity (5). EVs are also secreted by many immune cell types implicated in ICI response including CD4+/CD8+ T cells, dendritic cells, regulatory T cells, and macrophages (6–9). During tumor progression, both overall and tumor-specific EVs are elevated in plasma (10). We hypothesize that bulk, non-enriched plasma-derived EVs capture both tumor-derived EVs and non–tumor-derived EVs, reflecting tumor-intrinsic and nontumor signals. We analyzed pretreatment and on-treatment peripheral blood–derived bulk EV RNA from 50 patients with metastatic melanoma (discovery cohort; n = 33 responders and n = 17 nonresponders) treated with ICI via transcriptome microarray (fig. S1A and table S1). A subset of patients had posttreatment plasma samples (n = 15) and tumors (n = 26). In addition, we profiled four melanoma cell lines and paired EV. We validated results from the discovery cohort using an independent validation cohort of 30 patients (n = 14 responders and n = 16 nonresponders) using EV RNA sequencing (evRNA-seq). To integrate transcriptomic data from two sequencing platforms, we used a multipronged statistical strategy to minimize cross-platform variance (Materials and Methods and fig. S1B). To interpret mixed plasma-derived EV data, we developed a novel Bayesian deconvolution computational model to partition transcripts into tumor and nontumoral sources.
RESULTS
We analyzed melanoma cell lines and cell line–derived EV to correlate EV transcriptomes with tumor transcriptomes. We observed high correlation between cell lines and EV [average coefficient of determination (R2) = 0.87; fig. S3A]. Cell lines shared similar concordance in gene expression (fig. S3B), and the majority of genes had small differences in expression (fig. S3C). As expected, each cell line had the highest correlation with their EV (fig. S3D), suggesting that EV are reasonable proxies for expression in cell lines. To determine whether a patient’s plasma-derived EV profiles correlate with their tumor’s profile, we analyzed paired EV and tumor transcriptomes from n = 9 patients and observed close correlation of expression between the two profiles (average R2 = 0.82; Fig. 1A). Concordance analysis demonstrated that most genes in bulk tumors are detected in corresponding EV (Fig. 1B). By conducting gene set enrichment analysis via GAGE (11), we found enrichment of immune-related signatures exclusively in EV [e.g., T cell activation and natural killer (NK) activation], while tumor-exclusive transcripts were enriched for metabolic and tumor-related pathways (Fig. 1C and table S2). To identify cell populations represented in EV, we used CIBERSORT to infer immune cell type enrichments in patient EV (12, 13). We observed enrichment in five immune subpopulations exclusively in EV (neutrophils, NK cells, and CD4+/CD8+ T cells) and a relative depletion in macrophages/mast cells (Fig. 1D), suggesting that EV is overenriched for signals from immune populations important for anti-programmed cell death 1 (PD1) responses (3). Therefore, we hypothesized that EV transcripts from patients before and during treatment would predict or reflect resistance to ICI.
Fig. 1. Tumor and EV RNA concordance.
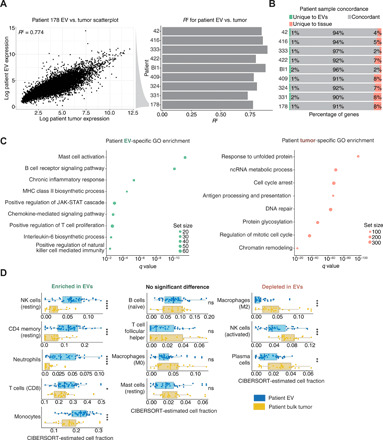
Characterization of transcriptomic similarities between patient melanomas and time-matched plasma-derived EV. (A) Scatter plot displaying the relationship between expression values of tumors and plasma EV in a representative patient (patient 178) and a histogram for R2 between paired tumors and plasma EV across the cohort. If a patient had multiple samples of the same type at the same time point, then the samples were averaged before computing the R2. (B) Concordance was calculated using a low expression threshold cutoff for expressed versus non-expressed status (Materials and Methods). Genes expressed or not expressed in tissue and EV were considered concordant (gray), while a subset of transcripts were unique EV (red) or tumor (green). (C) Selected pathways from gene set enrichment comparison of EV (left) versus patient tumors (right). GO, gene ontology; MHC, major histocompatibility complex; JAK-STAT, Janus kinase–signal transducers and activators of transcription; ncRNA, noncoding RNA. (D) CIBERSORT-inferred deconvolution estimates for all pretreatment patient tumor and pretreatment patient plasma-derived EV samples using LM22 immune reference profiles (12). Technical replicates were averaged, and biological replicates were considered independently. The data are segregated into three categories based on the results of a Mann-Whitney U test between EV- and tumor-inferred CIBERSORT fractions for each deconvolved cell type.
Since tumoral posttreatment signatures are more representative of ICI response than pretreatment values (1, 14), we correlated on-treatment EV transcripts with ICI response. Through differential gene set analysis using the Molecular Signatures Database (MSigDB) (15) via gene set variation analysis (GSVA; Materials and Methods) (16), we observed 258 pathways significantly different between responders and nonresponders in the discovery cohort, of which 25 pathways were significant in the validation cohort. Validated pathways [e.g., T cell receptor, cytotoxic T-lymphocyte-associated protein (CTLA4), transforming growth factor–β (TGF-β), SMAD2/3, Notch, tumor necrosis factor receptor 2, and vascular endothelial growth factor receptor] (Fig. 2A and fig. S4) are implicated in ICI resistance and melanoma progression (17–20). With our longitudinal data, we visualized on-treatment pathway dynamics via single-sample GSVA (16) and observed decreases in T cell receptor pathway activity during treatment in nonresponders (Fig. 2C and fig. S5A) and the CD28 costimulatory pathway (fig. S6A). The CTLA4 pathway diverges over time (fig. S6B), potentially resulting from peripheral tolerance during treatment (21), and similar changes are seen in tumor-related pathways [e.g., P53-hypoxia (fig. S6C) and kinesin activity (fig. S6D) (22)]. At the gene level, there were 1240 nominal and 43 false discovery rate (FDR)–corrected differentially expressed genes (DEGs) in the discovery cohort and 514 nominal and three FDR-corrected DEGs in the validation cohort. Eighty nominal DEGs shared successful P value validation, and 47 nominal DEGs were successfully replicated at P value, minimum expression, and log fold change levels (Materials and Methods, Fig. 2C, and table S3). The replication rate of the 47 DEGs is significantly above that expected by chance (P = 0.00088, hypergeometric test). Many shared DEGs mirrored gene set enrichment findings [e.g., Kruppel like factor 10 (KLF10), a major actor in the TGF-β pathway (23), and WNT8B, affects T effector differentiation (24)]. We detected cancer testis antigens (MAGEA1 and MAGEA3) known to be expressed by melanoma cells (25) in validated DEGs. On-treatment DEGs (MAGEA1, MAGEA2, KLF10, and MIR4519) were plotted to illustrate time dynamics relative to normalized expression changes at first collection (Fig. 2D) and unnormalized expression (fig. S5, B to E).
Fig. 2. Biological pathways and genes that stratify responders and nonresponders in on-treatment EV profiles.
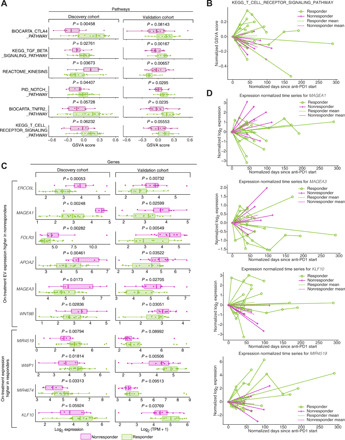
(A) MSigDB canonical (C2) pathway enrichments in on-treatment samples, (purple color, discovery cohort; green color, validation cohort). (B) Time dynamics of T cell receptor Kyoto Encyclopedia of Genes and Genomes (KEGG) signaling in responders versus nonresponders visualized using single-sample gene set enrichment scores derived from GSVA analysis. Individual patient progressions are displayed as connected lines. Both the start time and the relative expression levels were normalized against the time and GSVA score at the first sample collection point. The mean GSVA score changes for responders and nonresponders are plotted in bold green (responders) and purple (nonresponders) lines. (C) Expression comparison between responders (green) and nonresponders (purple) for selected validated DEGs between responders (green) in on-treatment samples across both the discovery and validation cohort. The P values displayed are generated from limma. (D) Time dynamics of several representative validated on-treatment DEGs, including MAGEA1, MAGEA3, KLF10, and MIR4519, in the discovery cohort. Individual patient progressions are displayed as connected lines. Both the start time and the relative expression levels were normalized against the time and expression at the first sample collection point, which were set to 0. The mean expression changes for responders and nonresponders are plotted in bold green (responders) and purple (nonresponders) lines.
We assessed whether pretreatment EV transcriptomes are able to stratify responders from nonresponders by performing gene set enrichment via GSVA (11), showing 101 differentially expressed MSigDB pathways (discovery cohort), of which 26 replicated in the validation cohort (Fig. 3A and fig. S7) (26). Compared to on-treatment pathways, we see differential Notch and TGF-β signaling in the pretreatment cohort and observe differences in mitogen-activated protein kinase (MAPK)–related signaling (ERRB4) and keratinization. Statistical testing revealed 366 nominal and 12 FDR-corrected DEGs in the pretreatment discovery cohort and 1406 nominal and 45 FDR-corrected DEGs in the pretreatment validation cohort. Fifty-four nominal DEGs had replicated P values, while 38 nominal DEGs had replicated P values and log fold changes, representing a replication rate significantly above that of random chance (P = 0.0041, hypergeometric test). DEGs included members of both immune- and tumor-related pathways implicated in ICI resistance or tumor growth [e.g., CD1A, MAPK kinase 4 (MAP2K4), TRBV7-2 (T cell receptor beta variable 7-2), and IGFL1 (insulin growth factor-like family member 1) (17, 27, 28)] (Fig. 3B and table S4), and cancer-associated microRNAs (e.g., miR551A) were enriched in nonresponders. We constructed a pretreatment random forest classifier to predict posttreatment response versus nonresponse status from pretreatment DEGs to quantify their predictive power. To minimize platform-specific differences and demonstrate the robustness of these markers, we pursued a machine learning framework that mixed samples across platforms in both the training and testing sets. To minimize variability from random sampling, we created 100 randomized partitionings (“trial”) of the combined dataset into n = 41 training and n = 30 testing samples. Within each trial, we first conducted K = 5 internal cross-validation to optimize the hyperparameters of a random forest model specific to that trial. We next used the best-performing hyperparameter set to evaluate testing performance on the test set. By summarizing the results from all 100 trials, we are able to generate receiver operating characteristic (ROC) plots displayed for both the training cross-validation performance (top) and the testing set performance (bottom) in Fig. 3C. To evaluate binary classification accuracy, we used the area under the ROC (AUROC); this is the probability that a binary classifier will rank a randomly chosen responder patient higher than a randomly chosen nonresponder one (29). We observed moderate predictive power in both our internal cross-validation (AUROC = 0.784) and our independent testing set (AUROC = 0.737) for our pretreatment DEGs (Materials and Methods). Multiple validated DEGs (IGFL1, TFF2 (trefoil factor 2), and MAP2K4) showed significant stratification in progression-free or overall survival (Fig. 3D and fig. S8).
Fig. 3. Biological pathways and genes stratifying responders/nonresponders in pretreatment EV profiles.
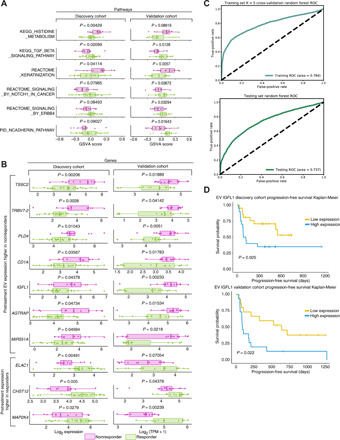
(A) GSVA scores and P values for selected MSigDB pathways differing significantly between responders (green) and nonresponders (purple). (B) Box plots of expression of selected validated pretreatment EV DEGs between responders/nonresponders in the discovery and validation cohorts (purple, nonresponders; green, responders). (C) ROCs generated by a random forest classifier36 using validated pretreatment DEGs to predict response versus nonresponse from EV transcriptomes. To minimize platform-specific differences confounded with response, we used 100 distinct, randomized partitionings of the combined discovery and validation cohorts. Within each 100 trial, we conducted stratified K = 5 internal cross-validation within the training set to determine optimal hyperparameters for a random forest classifier predicting binary response from EV transcriptomes (Materials and Methods): The optimal hyperparameter set and internal cross-validation predictive performance was saved. We used optimal hyperparameters from cross-validation to create a classifier trained on the entire training set and used this to generate predictions for the testing set. We combined predicted results and ground truths across all 100 trials and generated the ROC plot and AUROC values for internal cross-validation within the training set (top) and the held-out testing set (bottom). (D) Kaplan-Meier overall survival plots and log rank test P values for IGFL1 in the discovery and validation cohorts. High- and low-expression classifications were determined for each patient based on expression of IGFL1 that was in the top half (yellow) of their cohort’s IGFL1 expression level.
The bulk EV selection approach raises questions regarding how EVs from nontumor sources affect tumor-derived EV contributions. On the basis of the CIBERSORT results (Fig. 1C), we hypothesized that both tumor-derived EVs and non–tumor-derived EVs are detected. Dissecting the contribution from tumor-derived EVs versus non–tumor-derived sources may reveal whether response-related changes reflect changes in the tumor microenvironment or nontumoral changes (i.e., systemic changes in the immune system). Therefore, we developed a probabilistic deconvolution model to infer (i) a “packaging” coefficient representing depletion/enrichment of transcripts during packaging/export into EVs, (ii) the unobserved non–tumor-derived EV profile (fig. S9), and (iii) a mixing fraction between unobserved tumor-derived EV component and the non–tumor-derived components for each gene (Fig. 4A). To validate our model, we benchmarked deconvolution findings with bulk in silico deconvolution via CIBERSORTx (13) and found concordance between our deconvolution predictions and those generated using an independent algorithm and reference profiles (Supplementary Note). To assess the accuracy of the predictions, we analyzed known genes involved in EV function or immunotherapy response (Fig. 4B) (30, 31) followed by DEGs at pre- and on-treatment time points (Fig. 4C). Our model also probed enrichment across gene sets and calculated a gene set level tumor fraction, demonstrating significant enrichment for tumor-derived transcripts (Fig. 4D). When assessing ICI and melanoma-relevant Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, our results align with expected ranking (e.g., melanoma-related pathways have higher tumor fraction; Fig. 4E). Validated pretreatment DEGs enriched for tumor-derived genes, while on-treatment DEGs had greater nontumor contribution (Fig. 4F). This suggests that on-treatment DEGs preferentially reflect changes induced by ICI, consistent with findings from on-treatment differential pathway analysis. To illustrate the utility of our deconvolution algorithm for interpreting DEGs, we use KLF10, a member of the TGF-β pathway that has both roles as a tumor suppressor (23) and as an inductor of T helper cell 1 (TH1)/TH17 polarity and CD8+ memory T cell formation (32). Our data suggest that KLF10 is derived from the nontumor component, suggesting that differential on-treatment changes may be related to the TH1/TH17 polarity–inducing function of KLF10 as opposed to a tumor suppressor role.
Fig. 4. Deconvolution of EV profiles and analysis of driver mutations in RNA-seq profiles.
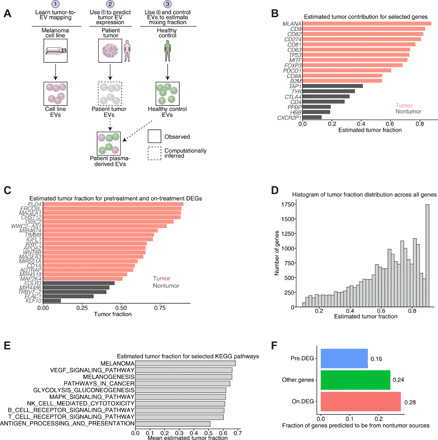
(A) Schematic representation of our deconvolution model (see Supplementary Note). (B) Selected tumor contributions from known tumor and nontumor genes. Red denotes predicted tumor, and gray denotes predicted nontumor tissue of origin for a particular gene. (C) Estimated tumor fraction for pretreatment and on-treatment DEGs demonstrates that a majority of DEGs are predicted to come from tumor sources. (D) Histograms of maximum a posteriori (MAP) estimates of tumor fraction from our model across all genes. (E) Average tumor fraction of all genes involved in several selected KEGG categories. (F) Predicted tumor fraction of validated pretreatment DEGs, on-treatment DEGs, and all other genes predicted to be non–tumor derived (i.e., predicted tumor fractions of <0.5).
Last, we investigated whether mutational information in the evRNA-seq can stratify responder and nonresponder given data relating to tumor mutational burden (TMB) (33). We surveyed the mutational landscape using hg38 genomic reference to call somatic and germline RNA-seq–associated mutations (Materials and Methods). By comparing evRNA-seq mutational calls against patient-matched tumor panel sequencing results [Massachusetts General Hospital (MGH) SNaPshot], we determined that three of our patients had specific driver mutations called by both panel sequencing and evRNA-seq mutational calling (table S1). Since panel data represent a small fraction of somatic tumor-associated mutations, we surveyed the entire somatic mutational landscape to evaluate differences in cancer driver mutational burden. Although patient samples varied in absolute number of mutations detected, we reasoned that differences in the fraction of somatic tumor-related mutations from the Catalogue of Somatic Mutations in Cancer (COSMIC) database relative to the overall mutational pool can be attributed to changes in somatic mutation load and not differences in germline mutations (34). We observed significantly higher COSMIC driver somatic mutational fraction in responders relative to nonresponders (Fig. 5, A and B). This was also reflected in the survival analysis. We observed significant stratification (P = 0.014, log rank test) between patients with high COSMIC mutational fraction (top 50% of cohort) versus those patients with low COSMIC mutational fraction (Fig. 5C), with patients with higher mutational fractions experiencing overall longer survival times. The progression-free survival log rank test was not significant (P = 0.67; fig. S10), suggesting that evRNA-seq cancer driver information may be more effective for predicting long-term effects of ICI treatment rather than short-term effects.
Fig. 5. Comparison of COSMIC mutational driver load between responders and nonresponders.
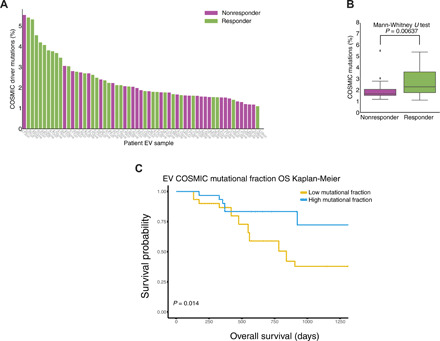
(A) Visualization of cohort-wide percentage of COSMIC driver mutations, as part of total mutations (a combination of somatic and germ line) called for each patient’s evRNA-seq profile in the validation cohort, a higher mutational load in responder patients (purple color, nonresponders; green color, responders). (B) Box plot summarizing the distribution of COSMIC driver mutation fraction between responder and nonresponder profiles. A Mann-Whitney U test was used to test for significant differences between the responder and nonresponder distributions. (C) Kaplan-Meier overall survival (OS) plots for COSMIC survival fraction in the validation evRNA-seq cohort. High- and low-expression classifications were determined for each patient on the basis of whether a particular patient’s COSMIC mutational fraction was in the top half (teal) or bottom half (yellow) of the validation cohort’s COSMIC mutational fraction distribution. A log rank test was used to derive the P value.
DISCUSSION
In this study, we explored the potential usage of plasma-derived EV transcriptomic profiles as a source of biomarkers for predicting and monitoring checkpoint blockade immunotherapy success. Our results show that EVs, in aggregate, correlate with certain aspects of bulk tumoral biology and reflect a number of previously identified differentially regulated biological pathways implicated in ICI resistance or melanoma progression in responders versus nonresponders. The majority of differential pathways and genes at the pretreatment time point primarily reflect differences in metabolic state as opposed to preexisting immune-related differences, suggesting that plasma-derived EVs only start capturing immune-related differences between responders and nonresponders after ICI treatment is administered. This is supported by the enrichment of immune-related pathways in our on-treatment differential pathway analysis and the enrichment for non–tumor-derived DEGs as inferred by our deconvolution model. Although the validated pretreatment DEGs that we discovered are biologically informative and can be used to create predictive models of ICI response with moderate predictive ability, our predictive classifier still lags behind predictive classifiers created from transcriptomic profiles from bulk tumor biopsies in terms of performance; bulk tumor profiles demonstrate performance in the ~0.8 to 0.9 AUROC range (35), compared to our testing AUROC of 0.737. Despite its lower predictive performance, the greater availability and ease of obtaining plasma-derived EV samples relative to bulk tumor biopsies may provide a viable clinical use case for EV profiling in the context of ICI treatment. However, the uncertain tissue of origin of EV transcripts confounds our ability to define a mechanistic basis for differential expression. To make the first steps toward addressing this issue, we developed a novel Bayesian deconvolution model uniquely suited to deconvoluting plasma-derived EV transcriptomic expression to infer tissue of origin. To our knowledge, this is the first computational model that explicitly models preferential packaging of EVs and contribution of nontumor EV components to infer per-gene tumor fractions.
The higher levels of correlation between melanoma cell lines and their EVs as compared to bulk patient tumors and corresponding plasma-derived EVs suggest that bulk plasma-derived EVs reflect a broader repertoire of EV sources. The most robust enrichment in EVs is for immune-related pathways (Fig. 1C). This is reinforced by the relative enrichment of several key immune cell types in our CIBERSORT deconvolution and is validated by our on-treatment DEGs and pathway enrichments. EV transcriptomic biomarkers may complement circulating tumor DNA (ctDNA) to gain transcriptomic information regarding tumor dynamics in addition to genomic information and may give a readout of both tumor-intrinsic and immunologic changes simultaneously.
To address tissue of origin of EV transcripts, we developed a deconvolution model to characterize EV transcripts from tumor versus nontumoral sources that explicitly accounts for differential EV transcript packaging. Currently, our model can only differentiate between tumor and nontumor contributions; however, ongoing experiments using cell-specific EV selection and/or depletion may enable us to differentiate between specific sources. Our deconvolution model is limited by three major factors: (i) the simplifying assumptions regarding the linear nature of the packaging coefficient and how it is shared between in vitro and in vivo samples and (ii) the lack of accounting for both tumor, especially potential immune infiltration in the tumor microenvironment, and patient heterogeneity, which is, in part, due to (iii) the limited number of samples. These limitations could potentially explain the classification of predominantly immune genes (e.g., CD8) into tumor compartments by our model. Thus, quantitative estimates of tumor purity should be interpreted in a qualitative fashion until in-depth in vitro experimental verification. As we continue to analyze data from more patients and perform in vitro EV selection experiments, we anticipate that our current model will serve as the foundation for more sophisticated models that can address these issues. Despite these limitations, our deconvolution model is the first to be able to pinpoint the potential source of EV expression and generate testable hypotheses regarding tissue of origin. Work is ongoing to experimentally validate the predicted source of circulating EVs via both tumor and immune cell–specific EV selection, which will be used to iteratively improve our deconvolution model and establish potential causal roles for EV transcripts in driving ICI resistance.
Our utilization of evRNA-seq technology in the validation cohort brought additional challenges to our analysis when cross-comparing with discovery cohort microarray data. We reasoned that using two separate sequencing technologies on two independent cohorts raises the bar for reproducibility and that findings replicated with distinct methodologies are likely to be robust. In addition, we show that mutational information embedded in the evRNA-seq itself can potentially be exploited to stratify responder and nonresponder populations and to serve as an orthogonal means to determine the tissue of origin of plasma-derived EV transcripts. This approach can be further enhanced through complementary whole-exome sequencing (WES) of EV DNA. Although it is unlikely that the utility of the mutational information from evRNA-seq data will outstrip high-depth cfDNA or WES TMB data in the near future, this mutational information is embedded within a large amount of transcriptomic information provided, which reflects dynamic tumoral changes and can complement other DNA-based sequencing methods (ctDNA and EV DNA sequencing) in ICI monitoring and response prediction tasks.
MATERIALS AND METHODS
Tumor cell lines
Melanoma cell lines (A375, RPMI 7951, SK-MEL-30, SK-MEL-2, and MeWo) were purchased directly from the American Type Culture Collection (ATCC) and maintained in culture as per ATCC recommendations. The A375 cell line was maintained in Dulbecco’s minimal essential medium, whereas RPMI 7951, SK-MEL-30, SK-MEL-2, and MeWo cell lines were cultured in RPMI 1640 media. All growth media consisted of media supplemented with 10% fetal bovine serum (FBS), penicillin (100 IU/ml), streptomycin (100 μg/ml), and l-glutamine (0.292 mg/ml). Cells were grown on plates and incubated at 37°C with a humidified atmosphere of 5% CO2 in air.
Patient samples and plasma isolation
Serial tumor and blood samples were collected from patients with melanoma under protocols approved by the Institutional Review Board at the MGH. Patient samples were linked to clinical data in a retrospective electronic health record database. Blood was collected in sodium citrate cell preparation tubes, with plasma isolated after centrifugation at room temperature for 25 to 30 min at a relative centrifugal force of 1800. Plasma was then frozen and stored at −80°C until use. Peripheral blood mononuclear cells are collected from the same sodium citrate tubes, washed in phosphate-buffered saline (PBS), resuspended in dimethyl sulfoxide with 90% FBS, slow frozen at 1°C/min, and stored at −80°C until use.
Isolation of EVs
Cell lines
When 150-mm plates reached between 50 to 70% confluence, depending on the doubling time of the cell line, the medium was replaced with the appropriate medium containing EV-depleted FBS and was harvested after 48 hours. EVs were isolated from cell-conditioned medium using serial centrifugation to remove cellular debris and filtration with a 0.8 μm or smaller filter (Millipore), followed by ultracentrifugation as previously described (36). Briefly, cell-conditioned medium was collected and centrifuged at 3000 rpm for 10 min at 4°C, after which the supernatant was decanted and filtered using a 0.45- or 0.8-μm filter. The filtered supernatant then underwent ultracentrifugation at 150,000g for 120 min. The pellet was then washed in PBS and underwent a second round of ultracentrifugation for 90 min. The EVs were then resuspended in cold RPMI media on ice and then frozen and stored at −80°C.
Plasma
All EV for RNA transcriptomic analysis from plasma were isolated from 1 ml of plasma using column isolation (QIAGEN exoRNeasy Midi Kit). Column isolation using the QIAGEN exoRNeasy Serum/Plasma Midi Kit resulted in direct isolation of EV RNA (10 to 30 ng of RNA per ml). Approximately 1 in 10 patients had an additional 1 ml of plasma isolated in parallel using ultracentrifugation (for quality control studies only; fig. S2). For ultracentrifugation, plasma was thawed and filtered through a 0.2-μm filter before ultracentrifugation as described for cell lines above.
Nanoparticle tracking analysis, electron microscopy, and Western blot analysis
Nanoparticle tracking analysis
Nanoparticle tracking analysis using the NanoSight LM10 (Malvern) was used to assess size distribution and concentration of particles in cell cultures and selected patient samples. Samples were diluted in PBS either at 1:500 or 1:1000 according to the NanoSight instruction manual.
Transmission electron microscopy
Electron microscopy was used to confirm the presence of EVs in cell culture and selected patient samples by size and morphology. Isolated EV suspensions were diluted 2:1 in 1× PBS, and 8- to 10-μl aliquots of each diluted sample were placed on formvar-carbon–coated Ni mesh grids; samples were allowed to adsorb for 15 min. Following adsorption, grid preparations were placed on drops of primary antibody CD9 rabbit monoclonal (D801A; Cell Signaling Technology, no. 13174), diluted 1:25 (dilutions made in Dako antibody diluent). Samples were allowed to incubate in primary antibody for at least 1 hour at room temperature, then rinsed on drops of PBS, and incubated in drops of a secondary gold conjugate for at least 1 hour at room temperature: goat anti-rabbit 10nm immunoglobulin G (Ted Pella, no. 15726). Grids were then rinsed in drops of 1× PBS and then distilled water, contrast-stained for 10 min in droplets of chilled tylose/uranyl acetate, and air-dried before examining in a JEOL JEM-1011 transmission electron microscope at 80 kV. Images were collected using an Advanced Microscopy Techniques digital camera and imaging system with proprietary image capture software (Advanced Microscopy Techniques, Danvers, MA).
Protein isolation from cell lines or EVs
Protein was isolated using radioimmunoprecipitation assay buffer supplemented with protease inhibitors as described by Wright (37). EVs were lysed directly in a sample buffer containing SDS and dithiothreitol.
Protein quantification
Protein concentration of EV samples was determined using the DC Protein Assay (Bio-Rad) according to the manufacturer’s protocol.
Western blot
Western blot was performed on samples of EV isolated from cell lines (fig. S2) to confirm their presence in the samples. Antibodies for EV markers CD9 (38) were used as a positive marker, given their consistency in expression across EV samples, whereas calnexin, an endoplasmic reticulum protein, is not found in EV and was used as a negative control. Actin was used as a loading control for cell lines using standard protocols.
RNA extraction and sequencing
Cell lines
Cells were trypsinized, washed in media for trypsin deactivation, then washed in PBS, counted, and pelleted. The cell pellet was resuspended in TRIzol at a concentration of 5 × 106 cells per 1 ml and RNA-isolated according to manufacturer’s instructions (Invitrogen). RNA was then quantified using a NanoDrop spectrometer (Thermo Fisher Scientific).
Cell line–derived EV
RNA was extracted from EV using a QIAGEN exoRNeasy kit.
Tumor
Formalin-fixed tissue was analyzed to confirm that viable tumor was present via standard hematoxylin and eosin staining. DNA was extracted from snap-frozen tissue using QIAGEN’s AllPrep DNA/RNA FFPE Kit.
Peripheral blood–derived/plasma EV
Total EV RNA was extracted from thawed patient plasma using the QIAGEN exoRNeasy Serum/Plasma Midi Kit as per the manufacturer’s protocol. Briefly, 1 ml of plasma was thawed and filtered using a 0.8-μm or smaller filter (Millipore). The sample was then mixed with binding buffer and placed on a spin column. After a wash step, the EVs were lysed on the column using QIAzol, and the eluate was then treated with chloroform to achieve phase separation. The aqueous phase was combined with 100% ethanol and then underwent column extraction with wash steps and final elution of EV RNA in ribonuclease-free water.
EV RNA sequencing
Sequencing was performed using an Illumina HiSeq 2000 system in the Ting Lab at MGH. RNA was prepared with the SMARTer Stranded Total RNA-Seq Kit v2 (Pico Input Mammalian) as per supplier’s instructions. For EV sequencing, RNA with a RNA integrity number of 4 or less was not subjected to fragmentation. Libraries were purified using AMPure beads and ZapR v2 for the depletion of ribosomal cDNA.
Discovery cohort microarray processing
We performed microarray analysis (via a subcontract to Thermo Fisher Scientific) using Applied Biosystems GeneChip Human Transcriptome Array 2.0. Raw .cel files provided by Thermo Fisher Scientific were read using the R package “oligo.” We performed background subtraction, quantile normalization, and summarization via the Robust Multichip Average algorithm using the R package oligo (39). We used the R package “pd.hta.2.0” to provide functional annotations for the probes. We filtered all probes that did not map to an annotated gene, as well as duplicate probes. We corrected for batch effects using the “ComBat” algorithm from the R “sva” package (40). For analyses that required validation by evRNA-seq, the resulting matrix was further corrected for platform-specific effects using ComBat and was quantile-normalized (fully described in the RNA-seq processing section). Analyses that only used microarray data (fig. S1B) was performed using the non–platform-corrected data since cross-platform normalization with evRNA-seq log2(TPM + 1) may unduly bias microarray-only analyses.
Validation cohort RNA-seq processing
Raw Illumina .fastq files were first filtered using “fastp” (41) with default settings and then aligned to human hg38 transcriptomic reference using STAR v2.4.1 with default settings. To summarize the count data from the aligned .bam files, we used the function “featurecounts” function from the R package “Rsubread” (42). Next, we derived log2(TPM + 1) values using a custom R function. To make the RNA-seq count matrix coanalyzable with our microarray data, we essentially treated the resulting ComBat-corrected log2(TPM + 1) matrix as an additional microarray batch. To minimize platform-based effects, we concatenated log2(TPM + 1) matrix with the microarray expression matrix to make a joint data matrix. Next, we used the ComBat (40) algorithm from the R sva package (40) to perform a “batch” correction step where the two platforms were treated as two distinct batches. Existing biological treatments were included in the ComBat normalization step as an input argument to preserve true biological effects during the platform correction step. Next, we quantile-normalized the resulting data matrix using R “preprocessCore” library. The resulting platform-normalized matrix was then split back into evRNA-seq and microarray matrices for relevant downstream analyses.
Differential expression analysis
To calculate differential expression in our microarray-based discovery and our evRNA-seq validation cohort, we used the R “limma” package to compute the P values that corresponded to the comparisons (43). For the samples that had multiple replicates, we modeled biological replicates as a random effect. Confounding variables such as age and prior immunotherapy treatment were tested for association against ICI response and did not exhibit significant associations, and as a result, they were not included as covariates. To find the top DEGs, we used limma’s Empirical Bayes linear modeling framework (43). DEGs were defined by 1.5 log fold change between responders and nonresponders and a nominal P value cutoff of P = 0.1 from limma. To be considered validated, a gene has to fulfill both the nominal P value cutoff and log fold change cutoff in both the validation and discovery cohorts. We note that this nominal P value cutoff is ordinarily insufficient to control for false-positive discoveries in a single-cohort study; however, we require explicit confirmation for putative discovery cohort DEGs in our validation cohort; thus, the combined false-positive rate for a gene to be both falsely discovered and falsely validated is substantially lower than what the nominal P value cutoff would suggest.
Concordance and differential pathway analysis
Concordance analysis was performed by first binarizing the mean expression values of either the cell line or patient data based on a 1.5 log expression cutoff using non–platform-corrected microarray data. If a gene is either present or absent in both groups, then it is labeled as concordant. We averaged the expression signal from two pretreatment patients present in the pretreatment time point; the posttreatment replicates were considered separately since they were from separate time points. To find differential pathways that are different between patient tumors and patient EV, we used gene set enrichment between responders and nonresponders with default parameters and Gene Ontology biological processes database (11). To find the canonical (C2) MSigDB pathways (15) that are significantly different between responders and nonresponders in both the discovery and validation cohorts, we used the GSVA program (16) with default settings to generate per-patient GSVA scores (a normalized statistic summarizing enrichment relative to the entire cohort analogous to single-sample gene set enrichment analysis scores) across our platform-corrected discovery and validation cohorts datasets. We then used a Mann-Whitney U test to test for differential GSVA scores between responders and nonresponders. Similar to the rationale used for DEG analysis, we used a nominal P value cutoff of 0.1 to flag differential pathways (Figs. 2A and 3A). A pathway was considered validated if it achieved significance in both the discovery and validation cohorts.
Survival analysis and time series analysis
To compute and plot the Kaplan-Meier curves, we used overall survival data and censoring information as inputs into the Kaplan-Meier computation and plotting functions in the R package “survminer.” To generate the time series plots in Fig. 2B, we used the R GSVA package (16) to generate a normalized enrichment score for each sample for target KEGG pathways. To generate the per-patient time dynamic plots illustrated in Fig. 2C, we normalized discovery cohort expression data by subtracting the expression of a patient’s first sample from all samples from the same patient.
Building a predictive classifier
To build a random forest predictive classifier from our selected pretreatment DEGs, we first merged the post–platform-corrected evRNA-seq and microarray data into a single combined matrix with 71 samples (n = 41 from discovery cohort and n = 30 from validation cohort). To reduce potential bias from platform and batch effects not corrected for by our ComBat correction steps, we randomized the selection of samples in the training and testing cohort by including samples from both sequencing platforms in the training and testing groups. To accomplish this, we randomly selected n = 30 samples to be (reflecting the size of our discovery cohort) the size of the held-out testing cohort and n = 41 samples (reflecting the size of our validation cohort) to be the training set. To minimize variability due to random sampling and potential nonlinear bias between platforms that remains uncorrected for by ComBat, we ran 100 trials for our machine learning pipeline, each with a n = 30 random selection of pretreatment samples as testing set and the remaining n = 41 as random samples. Note that the randomly partitioned training and testing sets will typically contain both evRNA-seq and microarray samples and may contain different proportions of responders and nonresponders depending on the partitioning.
Within each trial, we first conducted K = 5-fold internal cross-validation within the training set using the function “StratifiedKFold” from the Python library “sklearn.model_selection” to optimize the hyperparameters (number of trees) of a random forest model within the training set by optimizing cross-validation AUROC (44). For each trial, we searched T = {10,20,30} as a potential number of trees for our random forest classifier. The optimal hyperparameters within the internal cross-validation and the predicted probabilities for all K = 5 cross-validation folds for each trial were saved. We next used the optimal hyperparameters to train a random forest model on the entire training set and then evaluated the performance of this trained model on the testing set. Both the predicted probabilities for the internal and ground truths for each trial were saved. To generate the plots visualized in Fig. 3C, we concatenated the predicted probabilities and ground truths across 100 trials for both the internal training cross-validation and testing sets. We then used the Python “roc_curve” from the Python library “sklearn.metrics” to compute the ROC and the AUROC for both the results from the internal training set cross-validation (Fig. 3C, top) and the performance of the best-performing model on the held-out testing set (Fig. 3C, bottom) across all 100 trials (44).
EV deconvolution modeling
We formulated a Bayesian probabilistic model to deconvolve for each gene the observed mixed plasma EV expression profiles into two components: tumor and nontumor. We explicitly model the per-gene enrichment/depletion of RNA abundance as a result of export from the tumor to tumor-derived EV by leveraging information from patient-derived EV. To model the nontumor component, we use EV transcript abundances from healthy controls. A full mathematical description of the models and the source code are presented in Supplementary Note. The model was implemented in the probabilistic programming language Stan (45) through its Python interface. Inference for key genes was conducted using the No-U-Turn Hamiltonian Monte Carlo sampler (46). We performed 10,000 iterations with four chains to generate random samples; all other parameters were set using the PyStan defaults. Semi-informative priors for several variables of interest were selected because of the limited availability of the data; we used a Beta(2,2) prior and a Normal(0,2) prior for the scaling coefficient for the mixing fraction to (i) model our prior belief regarding the predicted nature of mixing and scaling and (ii) to constrain the posterior results somewhat in cases where certain genes with low expression but high variability in measurements may yield unreasonable mixing fraction results. We used a weakly informative prior on the variance parameter. Our model is able to return full posteriors for the packaging coefficient, mixing fraction, and each patient’s unobserved tumor profile (fig. S7). Although our full probabilistic algorithm returns the full posteriors for many parameters of interest, there is a high computational cost associated with running Markov chain Monte Carlo on tens of thousands of genes. Thus, we also created a simplified version of our model that is far more computationally efficient but only returns maximum a posteriori estimate of the mixing fraction instead of giving full posterior estimates of several parameters. It does so by using the Sequential Least Squares Programming algorithm from the Python “scipy.optimize.” We used this simplified model to generate the full list of mixing fractions in Fig. 4 (B to F) and table S5.
Mutational calling and analysis from the evRNA-seq data
To derive the mutational information shown in table S1 and Fig. 4 (G and H), we first mapped all reads using “bwa” mem v0.7.17 (with default settings) against human hg38 reference; bwa was chosen to include reads lying outside of the reference transcriptome with potentially useful mutational information. Next, we used GATK “HaplotypeCaller” submodule (with default settings) to call mutations from our patient RNA-seq libraries against hg38 reference and compared the resulting mutational information with summaries of SNaPshot panel sequencing results from clinical records. SNaPshot is a multiplexed polymerase chain reaction assay aimed at identifying somatic variants in 70 different loci from 15 cancer genes; the SNaPshot assay was performed by the MGH’s pathology department (Boston, USA). We used a custom Python script to overlap the resulting .vcf files produced by GATK against the “CosmicCodingMuts.vcf” file downloaded from the COSMIC mutation database v89 (34).
Supplementary Material
Acknowledgments
We thank L. He, Y. Park, Y. Li, and D. Liu for statistical consultations. We also thank D. Ting and members of his laboratory for allowing us to perform evRNA-seq in their laboratory. Funding: This work was supported by funding from the American Surgical Association Foundation, the Association of Women Surgeons, and the Adelson Medical Research Foundation. It was also conducted with support from the Harvard Catalyst and the Harvard Clinical and Translational Science Center (National Center for Research Resources and the National Center for Advancing Translational Sciences, NIH Award UL1 TR001102) and financial contributions from Harvard University and its affiliated academic health care centers. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University, and its affiliated academic health care centers or the NIH. A.S. is supported by an NSF Graduate Research Fellowship (award no. 2016226995). Author contributions: G.M.B., G.G.K., and S.C. designed the experiment. W.A.M., J.C.-G., S.C., D. P., and M.D.-M. carried out the EV experiments. A.S. and I.C. performed the bioinformatic and statistical analyses. A.S. designed and implemented the deconvolution model. D.J. was a material contributor. A.S., A.M., K.T.F., R.J.S., M.K., and G.M.B were involved in data interpretation and integration. J.O. was involved in data analysis and data review. D.T.F. was involved in blood/tissue collection, experimental data creation, and data analysis. A.S., G.M.B., J.C.-G., G.G.K., and M.K. wrote the manuscript. Competing interests: K.T.F. is on the Board of Directors of Loxo Oncology, Clovis Oncology, Strata Oncology, Vivid Biosciences, and Checkmate Pharmaceuticals. K.T.F. is also a member of the scientific advisory board of PIC Therapeutics, Sanofi, Amgen, Asana, Adaptimmune, Fount, Aeglea, Shattuck Labs, Tolero, Apricity, Oncoceutics, FogPharma, Neon, Tvardi, xCures, Monopteros, and Vibliome. K.T.F. has consulted for Eli Lilly and Company, Novartis, Genentech, BMS, Merck, Takeda, Verastem, Boston Biomedical, Pierre Fabre, and Debiopharm. K.T.F. is also a member of the corporate advisory board of X4 Pharmaceuticals. K.T.F. has also received research funding from Novartis and Sanofi. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The raw microarray chip and evRNA-seq files, processed data files, and metadata have been uploaded to NCBI’s GEO database https://github.com/alvinshi20/ExosomeData. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/46/eabb3461/DC1
REFERENCES AND NOTES
- 1.Hong X., Sullivan R. J., Kalinich M., Kwan T. T., Giobbie-Hurder A., Pan S., LiCausi J. A., Milner J. D., Nieman L. T., Wittner B. S., Ho U., Chen T., Kapur R., Lawrence D. P., Flaherty K. T., Sequist L. V., Ramaswamy S., Miyamoto D. T., Lawrence M., Toner M., Isselbacher K. J., Maheswaran S., Haber D. A., Molecular signatures of circulating melanoma cells for monitoring early response to immune checkpoint therapy. Proc. Natl. Acad. Sci. U.S.A. 115, 2467–2472 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashida A., Sakaizawa K., Uhara H., Okuyama R., Circulating tumour DNA for monitoring treatment response to anti-PD-1 immunotherapy in melanoma patients. Acta Derm. Venereol. 97, 1212–1218 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Riaz N., Havel J. J., Makarov V., Desrichard A., Urba W. J., Sims J. S., Hodi F. S., Martín-Algarra S., Mandal R., Sharfman W. H., Bhatia S., Hwu W.-J., Gajewski T. F., Slingluff C. L. Jr., Chowell D., Kendall S. M., Chang H., Shah R., Kuo F., Morris L. G. T., Sidhom J.-W., Schneck J. P., Horak C. E., Weinhold N., Chan T. A., Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 171, 934–949.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alipoor S. D., Mortaz E., Varaham M., Movassaghi M., Kraneveld A. D., Garssen J., Adcock I. M., The potential biomarkers and immunological effects of tumor-derived exosomes in lung cancer. Front. Immunol. 9, 819 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muller L., Muller-Haegele S., Mitsuhashi M., Gooding W., Okada H., Whiteside T. L., Exosomes isolated from plasma of glioma patients enrolled in a vaccination trial reflect antitumor immune activity and might predict survival. Oncoimmunology 4, e1008347 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McDonald M. K., Tian Y., Qureshi R. A., Gormley M., Ertel A., Gao R., Lopez E. A., Alexander G. M., Sacan A., Fortina P., Ajit S. K., Functional significance of macrophage-derived exosomes in inflammation and pain. Pain 155, 1527–1539 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai Z., Yu L., Yu Z., Jiang L., Wang Q., Yang Y., Wang L., Cao X., Wang J., Activated T cell exosomes promote tumor invasion via Fas signaling pathway. J. Immunol. 188, 5954–5961 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Greening D. W., Gopal S. K., Xu R., Simpson R. J., Chen W., Exosomes and their roles in immune regulation and cancer. Semin. Cell Dev. Biol. 40, 72–81 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Chatila T. A., Williams C. B., Regulatory T cells: Exosomes deliver tolerance. Immunity 41, 3–5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang T., Deng C.-X., Current progresses of exosomes as cancer diagnostic and prognostic biomarkers. Int. J. Biol. Sci. 15, 1–11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo W., Friedman M. S., Shedden K., Hankenson K. D., Woolf P. J., GAGE: Generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10, 161 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newman A. M., Liu C. L., Green M. R., Gentles A. J., Feng W., Xu Y., Hoang C. D., Diehn M., Alizadeh A. A., Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newman A. M., Steen C. B., Liu C. L., Gentles A. J., Chaudhuri A. A., Scherer F., Khodadoust M. S., Esfahani M. S., Luca B. A., Steiner D., Diehn M., Alizadeh A. A., Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37, 773–782 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roh W., Chen P.-L., Reuben A., Spencer C. N., Prieto P. A., Miller J. P., Gopalakrishnan V., Wang F., Cooper Z. A., Reddy S. M., Gumbs C., Little L., Chang Q., Chen W.-S., Wani K., De Macedo M. P., Chen E., Austin-Breneman J. L., Jiang H., Roszik J., Tetzlaff M. T., Davies M. A., Gershenwald J. E., Tawbi H., Lazar A. J., Hwu P., Hwu W.-J., Diab A., Glitza I. C., Patel S. P., Woodman S. E., Amaria R. N., Prieto V. G., Hu J., Sharma P., Allison J. P., Chin L., Zhang J., Wargo J. A., Futreal P. A., Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med. 9, eaah3560 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tomayo P., Mesirov J. P., Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hänzelmann S., Castelo R., Guinney J., GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jenkins R. W., Barbie D. A., Flaherty K. T., Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 118, 9–16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janghorban M., Xin L., Rosen J. M., Zhang X. H.-F., Notch signaling as a regulator of the tumor immune response: To target or not to target? Front. Immunol. 9, 1649 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinnberg T., Levesque M. P., Krochmann J., Cheng P. F., Ikenberg K., Meraz-Torres F., Niessner H., Garbe C., Busch C., Wnt-signaling enhances neural crest migration of melanoma cells and induces an invasive phenotype. Mol. Cancer 17, 59 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ni X., Tao J., Barbi J., Chen Q., Park B. V., Li Z., Zhang N., Lebid A., Ramaswamy A., Wei P., Zheng Y., Zhang X., Wu X., Vignali P., Yang C.-P., Li H., Pardoll D., Lu L., Pan D., Pan F., YAP is essential for treg-mediated suppression of antitumor immunity. Cancer Discov. 8, 1026–1043 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buchbinder E. I., Desai A., CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am. J. Clin. Oncol. 39, 98–106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huszar D., Theoclitou M.-E., Skolnik J., Herbst R., Kinesin motor proteins as targets for cancer therapy. Cancer Metastasis Rev. 28, 197–208 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Memon A., Lee W. K., KLF10 as a tumor suppressor gene and its TGF-β signaling. Cancer 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gattinoni L., Zhong X.-S., Palmer D. C., Ji Y., Hinrichs C. S., Yu Z., Wrzesinski C., Boni A., Cassard L., Garvin L. M., Paulos C. M., Muranski P., Restifo N. P., Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 15, 808–813 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Öhman Forslund K., Nordqvist K., The melanoma antigen genes—Any clues to their functions in normal tissues? Exp. Cell Res. 265, 185–194 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Hugo W., Zaretsky J. M., Sun L., Song C., Moreno B. H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G., Seja E., Lomeli S., Kong X., Kelley M. C., Sosman J. A., Johnson D. B., Ribas A., Lo R. S., Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165, 35–44 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng J., Wang E. S., Jenkins R. W., Li S., Dries R., Yates K., Chhabra S., Huang W., Liu H., Aref A. R., Ivanova E., Paweletz C. P., Bowden M., Zhou C. W., Herter-Sprie G. S., Sorrentino J. A., Bisi J. E., Lizotte P. H., Merlino A. A., Quinn M. M., Bufe L. E., Yang A., Zhang Y., Zhang H., Gao P., Chen T., Cavanaugh M. E., Rode A. J., Haines E., Roberts P. J., Strum J. C., Richards W. G., Lorch J. H., Parangi S., Gunda V., Boland G. M., Bueno R., Palakurthi S., Freeman G. J., Ritz J., Haining W. N., Sharpless N. E., Arthanari H., Shapiro G. I., Barbie D. A., Gray N. S., Wong K.-K., CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 8, 216–233 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang L., Zhang Z., Recharacterizing tumor-infiltrating lymphocytes by single-cell RNA sequencing. Cancer Immunol. Res. 7, 1040–1046 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Bradley A. P., The use of the area under the ROC curve in the evaluation of machine learning algorithms. Pattern Recognit. 30, 1145–1159 (1997). [Google Scholar]
- 30.Lu J., Li J., Liu S., Wang T., Ianni A., Bober E., Braun T., Xiang R., Yue S., Exosomal tetraspanins mediate cancer metastasis by altering host microenvironment. Oncotarget 8, 62803–62815 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jenkins R. W., Aref A. R., Lizotte P. H., Ivanova E., Stinson S., Zhou C. W., Bowden M., Deng J., Liu H., Miao D., He M. X., Walker W., Zhang G., Tian T., Cheng C., Wei Z., Palakurthi S., Bittinger M., Vitzthum H., Kim J. W., Merlino A., Quinn M., Venkataramani C., Kaplan J. A., Portell A., Gokhale P. C., Phillips B., Smart A., Rotem A., Jones R. E., Keogh L., Anguiano M., Stapleton L., Jia Z., Barzily-Rokni M., Cañadas I., Thai T. C., Hammond M. R., Vlahos R., Wang E. S., Zhang H., Li S., Hanna G. J., Huang W., Hoang M. P., Piris A., Eliane J.-P., Stemmer-Rachamimov A. O., Cameron L., Su M.-J., Shah P., Izar B., Thakuria M., LeBoeuf N. R., Rabinowits G., Gunda V., Parangi S., Cleary J. M., Miller B. C., Kitajima S., Thummalapalli R., Miao B., Barbie T. U., Sivathanu V., Wong J., Richards W. G., Bueno R., Yoon C. H., Miret J., Herlyn M., Garraway L. A., Van Allen E. M., Freeman G. J., Kirschmeier P. T., Lorch J. H., Ott P. A., Hodi F. S., Flaherty K. T., Kamm R. D., Boland G. M., Wong K.-K., Dornan D., Paweletz C. P., Barbie D. A., Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Discov. 8, 196–215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papadakis K. A., Krempski J., Reiter J., Svingen P., Xiong Y., Sarmento O. F., Huseby A., Johnson A. J., Lomberk G. A., Urrutia R. A., Faubion W. A., Krüppel-like factor KLF10 regulates transforming growth factor receptor II expression and TGF-β signaling in CD8+ T lymphocytes. Am. J. Physiol. Cell Physiol. 308, C362–C371 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goodman A. M., Kato S., Bazhenova L., Patel S. P., Frampton G. M., Miller V., Stephens P. J., Danierls G. A., Kurzrock R., Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 16, 2598–2608 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tate J. G., Bamford S., Jubb H. C., Sondka Z., Beare D. M., Bindal N., Boutselakis H., Cole C. G., Creatore C., Dawson E., Fish P., Harsha B., Hathaway C., Jupe S. C., Kok C. Y., Noble K., Ponting L., Ramshaw C. C., Rye C. E., Speedy H. E., Stefancsik R., Thompson S. L., Wang S., Ward S., Campbell P. J., Forbes S. A., COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Auslander N., Zhang G., Lee J. S., Frederick D. T., Miao B., Moll T., Tian T., Wei Z., Madan S., Sullivan R. J., Boland G., Flaherty K., Herlyn M., Ruppin E., Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat. Med. 24, 1545–1549 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Théry C., Amigorena S., Raposo G., Clayton A., Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. Chapter 3, Unit 3.22 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Wright K., Antibodies a laboratory manual by E. Harlow, and D. Lane. Biochem. Educ. 17, 220 (1989). [Google Scholar]
- 38.Lobb R. J., Becker M., Wen S. W., Wong C. S. F., Wiegmans A. P., Leimgruber A., Möller A., Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracell. Vesicles 4, 27031 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carvalho B. S., Irizarry R. A., A framework for oligonucleotide microarray preprocessing. Bioinformatics 26, 2363–2367 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson W. E., Li C., Rabinovic A., Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Chen S., Zhou Y., Chen Y., Gu J., fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liao Y., Smyth G. K., Shi W., The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.G. K. Smith, limma: Linear models for microarray data, in Bioinformatics and Computational Biology Solutions Using R and Bioconductor (Springer, 2005), pp. 397–420. [Google Scholar]
- 44.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V., Vanderplas J., Passos A., Cournapeau D., Scikit-learn: Machine learning in python. J. Mach. Learn. Res. 12, 2825–2830 (2011). [Google Scholar]
- 45.Gelman A., Lee D., Guo J., Stan: A probabilistic programming language for bayesian inference and optimization. J. Educ. Behav. Stat. 40, 530–543 (2015). [Google Scholar]
- 46.M. D. Hoffman, A. Gelman, The No-U-Turn sampler: Adaptively setting path lengths in Hamiltonian Monte Carlo. arXiv:1111.4246 [stat.CO] (2011).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/46/eabb3461/DC1