

A nanoparticle delivered ENaC-inhibitory mRNA for the treatment of cystic fibrosis.
Abstract
Cystic fibrosis (CF) results from mutations in the chloride-conducting CF transmembrane conductance regulator (CFTR) gene. Airway dehydration and impaired mucociliary clearance in CF is proposed to result in tonic epithelial sodium channel (ENaC) activity, which drives amiloride-sensitive electrogenic sodium absorption. Decreasing sodium absorption by inhibiting ENaC can reverse airway surface liquid dehydration. Here, we inhibit endogenous heterotrimeric ENaC channels by introducing inactivating mutant ENaC α mRNA (αmutENaC). Lipid nanoparticles carrying αmutENaC were transfected in CF-based airway cells in vitro and in vivo. We observed a significant decrease in macroscopic as well as amiloride-sensitive ENaC currents and an increase in airway surface liquid height in CF airway cells. Similarly, intranasal transfection of αmutENaC mRNA decreased amiloride-sensitive nasal potential difference in CFTRKO mice. These data suggest that mRNA-based ENaC inhibition is a powerful strategy for reducing mucus dehydration and has therapeutic potential for treating CF in all patients, independent of genotype.
INTRODUCTION
Cystic fibrosis (CF) is an autosomal recessive disorder that affects 70,000 patients worldwide. Mutations in the CF transmembrane conductance regulator (CFTR) gene leads to defects in CFTR protein that cause impaired Cl− ion transport at the luminal airway surface along with tonic Na+ and water reabsorption (1). The result is thick inspissated mucus that is retained in the airways, leading to permanent bacterial colonization and progressive pulmonary deterioration (2). Current strategies to restore airway hydration include CFTR protein modulator therapy that partially corrects defective membrane trafficking and function of CFTR, restoring chloride transport and mucus hydration in the airways (3). However, partial restoration of CFTR protein function by gene therapy does not appear to alter dysregulation of Na+ absorption (4). Thus, continued Na+ dysregulation and water reabsorption may account for the variability in clinical endpoints (e.g., lung function and sweat chloride) with the otherwise highly effective CFTR modulator therapy (3, 5).
In the airway, epithelial sodium channel (ENaC) performs electrogenic absorption of Na+ from the mucosal surface that results in water reabsorption (6). The drug amiloride specifically inhibits ENaC while being a poor inhibitor of other channels and transporters at lower concentrations. Thus, amiloride-sensitive sodium current is used to measure ENaC activity (7). ENaC activity is proposed to be regulated by CFTR (8). Dysregulation of ENaC activity is a common feature across many CFTR gene mutations (9). In CF, excessive water absorption due to unopposed ENaC activity results in dehydrated airway surface liquid (ASL), which, in turn, impairs ciliary motion (10). Restoring ASL height enhances mucociliary clearance and leads to improvement of pulmonary function (11, 12). One proposed approach to increase ASL height and restore mucus hydration is to reduce mucosal absorption of Na+ and water by inhibiting ENaC. Unlike CFTR modulator therapy that is only effective for specific CFTR mutations, this strategy would benefit all CF patients (3, 5). Previous and current approaches of ENaC inhibition have included drugs acting on ENaC directly, agents targeting ENaC-specific proteases, and RNA interference (RNAi) or antisense-based oligonucleotides against specific ENaC subunits (10). ENaC as a target in CF has not yet translated to a clinical therapeutic because of concerns for efficacy and duration of action as well as off-target or systemic effects of the various molecules tested so far (10).
ENaC is a heterotrimeric channel composed of three homologous α, β, and γ subunits, with the α-ENaC subunit being essential in forming a functional channel (13). Channels lacking α subunit are completely nonfunctional, whereas channels lacking β or γ subunits are hypofunctional (13). Human airways express a lesser studied ENaC δ subunit that is phylogenetically close to the ENaC α subunit (14). The α and δ subunits can be substituted for each other to express an atypical δβγ channels along with the more common αβγ channels, which have differing bioelectric properties (15). The ENaC inhibitor amiloride has an IC50 (median inhibitory concentration) in the concentration range of 0.1 to 0.5 μM for human αβγ ENaC and approximately 2.5 μM for human δβγ ENaC (16). Previous attempts at blocking ENaC activity with amiloride failed because of its rapid clearance from the lung (10). We hypothesized that ENaC activity can be reduced by deploying mRNA that encodes for a channel-inactivating ENaC α (αmutENaC) subunit packaged inside lipid-based nanoparticles (LNPs) for delivery. LNPs are clinically approved for small interfering RNA (siRNA) delivery and are in clinical trials for delivery of mRNA-based therapeutics and vaccines (17). We postulated that exogenous αmutENaC would compete with endogenous α and δ subunits to form inactivated channels resulting in reduced amiloride-sensitive currents in target cells. We also propose that reduction in ENaC activity would increase ASL height in CF airway cells (CFBE41o-). In this study, we used an in vitro model to determine the effect of αmutENaC mRNA transfection on ENaC activity as well as its effect on regulating ASL height in polarized CFBE41o- cells. For in vivo experiments, we used intranasal transfection of αmutENaC mRNA containing LNP and measured amiloride-sensitive nasal potential difference (NPD) in CFTR knockout mice. Together, our experiments show that intracellular delivery of mutant ENaC subunits can be a novel and promising strategy to inhibit sodium and water reabsorption activity in vitro and in vivo and should be pursued as a CF therapeutic.
RESULTS
Formulated LNPs effectively transfect mRNA into CFBE41o- cells
Our LNPs consisted of four types of lipids: (i) ionizable lipid [O-(Z,Z,Z,Z-heptatriaconta-6,9,26,29-tetraen-19-yl)-4-(N,N-dimethylamino) (DLin-MC3-DMA)]; (ii) cholesterol; (iii) helper lipid, distearolyphosphatidycholine; and (iv) dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG2K). They were formulated by microfluidic mixing of mRNA and lipids. Further characterization for size and encapsulation by dynamic light scattering analysis showed a hydrodynamic diameter of 73 nm with a polydispersity index of less than 0.1 (Fig. 1A). LNPs had high mRNA encapsulation (>99%) and a spherical shape (Fig. 1, A and B). LNPs were used to transfect either enhanced green fluorescent protein (EGFP) or firefly luciferase (Luc) mRNA in CFBE41o- cells, as reporter genes. Fluorescent signal was detected in CFBE41o- cells treated with the LNPs encapsulating EGFP mRNA by confocal microscopy 24 hours after transfection (Fig. 1C). Moreover, a dose-dependent response was observed with LNPs used to deliver Luc mRNA transfected at 24 hours after transfection (Fig. 1D). No significant change in cell viability was observed after luciferase transfection, whereas 10% dimethyl sulfoxide (DMSO) treatment killed approximately 65% (% viability: 36.89 ± 3.13) of cells (fig. S1, A and B).
Fig. 1. LNP characterization and in vitro mRNA transfection.
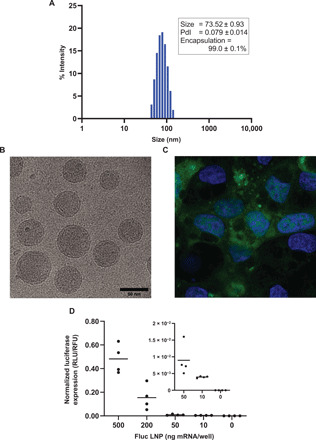
(A) Dynamic light scattering analysis of LNP size distribution and mRNA encapsulation of the LNP encapsulating αmutENaC mRNA (n = 3). (B) Cryo-EM imaging of the LNPs encapsulating FLuc mRNA. (C) Imaging expression of EGFP transfection in CFBE41o- cells using LNPs encapsulating EGFP mRNA. (D) Measuring luminescence (FLuc)–based expression of firefly luciferase in CFBE41o- cells, with LNPs encapsulating FLuc mRNA (n = 4). Inset: Rescaled y axis for visualization of luminescence with 50, 10, and 0 ng per well transfection of FLuc mRNA. RLU, relative luminescence units; RFU, relative fluorescent units.
αmutENaC mRNA transfection reduces amiloride-sensitive current in CFBE41o- cell
The α ENaC subunit is cleaved at two sites by furin, a protease that resides in the trans-Golgi network, to release an imbedded inhibitory peptide that leads to activation of ENaC (18). To generate a channel-inactivating αmutENaC mutant, we engineered an mRNA containing two sets of modifications. First, the conserved arginines at the furin cleavage sites (R175, 177, 178 and R204, 202, 201) in the extracellular finger domain of human ENaC α subunit were mutated to alanines (Fig. 2A; also see fig. S2, A and B) (19). These mutations prevent furin from excising an 8-mer imbedded inhibitory peptide in the α subunit that is critical for ENaC activation (19). The resultant channel will have a low open probability. Second, a single Ser→Ala substitution mutation was made on the conserved serine residue at the degenerin site in the second transmembrane domain of α ENaC subunit (Fig. 2A). Serine residues are prone to undergo enzymatic and nonenzymatic posttranslational modifications such as acetylation, methylation, N-linked or O-linked glycosylation, pyrophosphorylation, and carboxymethylation that entail addition of a bulky residue on the amino acid side chain (20, 21). Previous studies have shown that modification or substitution of the conserved serine with a bulky amino acid residue at the degenerin site leads to a significant increase in channel open probability even after furin modification (22). To prevent such bulky posttranslational modifications of the serine that might compromise the desired effects of the engineered mutations in the extracellular domain, we conservatively substituted serine with alanine that is resistant to modifications (23). Together, the mutant α subunit carrying these two sets of mutations will drive assembled ENaC to an inactivated state.
Fig. 2. αmutENaC transfection reduces amiloride-sensitive ENaC current in airway cell.
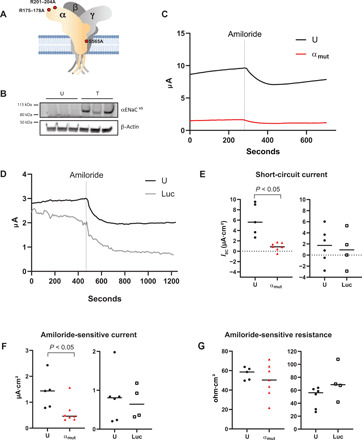
(A) α ENaC subunit mRNA was mutated (red dots) at the furin cleavage sites (R175, 177, 178 and R204, 202, 201) and the degenerin site (S565) to alanine. Trimeric ENaC channel is shown. The α subunit is colored yellow; the β and γ subunits are in gray. Each subunit is shown to have cytosolic, transmembrane, and extracellular domains. (B) Western blot of CFBE41o- cell extracts 72 hours after transfection (T), with LNPs encapsulating 750 ng of C-terminal V5 epitope–tagged αmutENaCV5 mRNA (n = 3). Untransfected (U) cells were used as controls (n = 3). The blots were probed using anti-V5 antibody. β-Actin was used as a gel loading control. Polarized CFBE41o- cells were transfected with lipid nanoparticles containing (C) αmutENaC (U, n = 5; αmut, n = 7) [untransfected (U), black line; αmut transfected (αmut), red line] or (D) luciferase [luciferase-transfected (Luc), gray line] (U, n = 6; Luc, n = 4) mRNAs, and short-circuit currents (ISC) were recorded after 72 hours. Statistical analyses were performed using experimental values of (E) short circuit currents (ISC) before amiloride treatment, (F) amiloride-sensitive ISC, and (G) amiloride-sensitive resistance. P values are indicated in the panels. U, untransfected (solid black circle); αmut, αmutENaC-transfected (red triangle); and Luc, luciferase-transfected (open square).
Expression of αmutENaC protein was confirmed 72 hours after transfection of C-terminal V5 epitope–tagged αmutENaCV5 mRNA by LNP in CFBE41o- cell lysates by immunoblotting (Fig. 2B). Polarized CFBE41o- cells grown on polycarbonate supports were transfected with αmutENaC mRNA–LNP, while the luciferase-transfected or untransfected cells served as controls. Short-circuit currents (ISC) of αmutENaC-transfected cells were recorded 72 hours after transfection before and after amiloride, a specific ENaC inhibitor, was added to the apical side and compared with untransfected controls (Fig. 2, C and E). In short-circuit current experiments, we noted that the overall current is significantly lower (P < 0.05) in αmutENaC-transfected (0.9 ± 0.78 μA) cells compared to untreated cells (6.09 ± 3.1 μA) (Fig. 2, C and E) at rest, whereas transfection of luciferase mRNA LNP did not alter ISC compared to the control group at rest (Fig. 2, D and E). Addition of amiloride led to rapid depolarization of ISC in the controls (Fig. 2, C and D). Amiloride-sensitive current likely reflects ENaC activity and is measured by calculating the difference between ISC pre- and post-amiloride treatment (24). Amiloride responses of luciferase-transfected controls were similar to untreated controls (Fig. 2F). However, our data show that αmutENaC transfection (0.61 ± 0.44 μA) led to a significant decrease (P < 0.05) in the amiloride-sensitive current compared to controls (1.39 ± 0.66 μA), suggesting an inhibition of ENaC activity (Fig. 2F). The cell viability upon transfection with the mutant ENaC mRNA was not compromised (fig. S1, A and B).
In parallel to ISC measures, we also recorded transepithelial resistance (TER) of the monolayers during short-circuit current experiments in αmutENaC mRNA–transfected and control groups before and after addition of amiloride (table S1). As expected, amiloride treatment increased resistance across the inserts in all groups due to a decrease in electrogenic ENaC activity, thus ensuring that the epithelial tight junction integrity was preserved after treatment with either αmutENaC or the luciferase mRNA-LNP (Fig. 2G). αmutENaC transfection did not alter TER in polarized CFBE41o- cells (fig. S3), suggesting that one or more of the apical, basolateral, or paracellular resistances that contribute to the TER may be altered to keep the overall TER unchanged. Last, change in amiloride-sensitive resistance, measured as the difference in resistances before and after addition of amiloride, was similar in αmutENaC-transfected and untransfected controls. Because other ionic pathways such as that mediated by CFTR also regulate airway epithelial resistance (25), our data may indicate alteration in such pathways that additionally influence TER in response to acute and chronic ENaC inhibition in αmutENaC-transfected cells.
αmutENaC transfection increases ASL height in CFBE41o- cells
The ASL lines the mucosal surface of airway cells facilitating mucociliary clearance when at the normal ciliary height. Mucus moves above the watery periciliary liquid fraction of the ASL because of ciliary movement (26). In CF, dehydration of the periciliary liquid reduces ASL height, impairing mucociliary clearance (27). We used dextran beads conjugated with rhodamine (red) to label the ASL and calcein (green), a membrane-permeable live cell marker to demarcate the cell (Fig. 3A). Using confocal microscopy, representative three-dimensional (3D)–rendered images from the ASL to the basolateral surface of the cells were generated (Fig. 3A). The vertical height of labeled dextran from the cellular surface was considered as measure of ASL height. Confocal z-section images on live cells were obtained from six relatively fixed points on Transwell inserts. For measuring ASL height, average vertical height of rhodamine-dextran was computed from three randomly selected points in a single image representing one of the six fixed points. This measurement was repeated on images of all six points on the insert for each experimental group for obtaining average ASL height on each membrane. We hypothesized that reduction of ENaC activity will decrease mucosal Na+ and water hyperabsorption and result in an increase in the ASL height. CFBE-WT cells that stably overexpress wild-type human CFTR (WT) were used as a positive control. Expectedly, the ASL height of CFBE-WT cells (39.05 ± 1.40 μm) was greater (P < 0.01) than that of the untransfected CFBE41o- cells (24.51 ± 2.83 μm) (Fig. 3B). αmutENaC mRNA–LNP transfection in CFBE41o- cells led to a significant increase (P < 0.001) in ASL height (35.16 ± 4.06 μm) compared to untransfected controls (23.03 ± 2.73 μm) (Fig. 3C), suggesting that LNP transfection of αmutENaC led to increased ASL height in CF patient–derived cell lines.
Fig. 3. αmutENaC transfection increases ASL height in airway cells.

ASL height was measured in polarized CFBE41o- or WT CFTR stably overexpressing CFBE-WT cells. (A) Representative 3D-rendered confocal images of polarized CFBE-WT (WT, top), untransfected (U, middle), and αmutENaC-transfected CFBE41o- cells (αmut, bottom). Cells were labeled with calcein (green), and the ASL was labeled with cell-impermeant rhodamine-dextran (red). ASL height of (height of rhodamine-dextran) untransfected CFBE41o- (U) cells was compared with that of (B) CFBE-WT (U_WT, n = 3) and (C) αmutENaC-transfected CFBE41o- cells (αmut, n = 6). Colored shapes indicate average ASL height of each of the six points on an insert: Statistical analyses were performed between mean ASL height of untransfected [U_Mean, solid black circles (B) or solid black diamonds (C)] and CFBE-WT (U_WT_Mean, solid gray circles) or αmutENaC-transfected CFBE41o- cells (U_αmut_Mean, solid gray diamonds). The P values are indicated.
αmutENaC transfection reduced amiloride-sensitive NPD in CFTRKO mice
Having observed αmutENaC-mediated inhibition of endogenous ENaC activity in vitro, we proceeded to assess these effects in vivo using mouse models. We used intranasal instillation to transfect murine nasal epithelium with a luciferase reporter gene and imaged luciferase activity. Five hours after administration of LNP-delivered luciferase mRNA, a strong bioluminescence was observed in the nasal cavity indicating effective gene delivery in the nasal epithelium (Fig. 4A). A previous study from our group has shown that Luc mRNA transfection does not alter electrical properties of nasal epithelia in mouse (28).
Fig. 4. αmutENaC transfection reduced amiloride-sensitive NPD in CFTRKO mice.
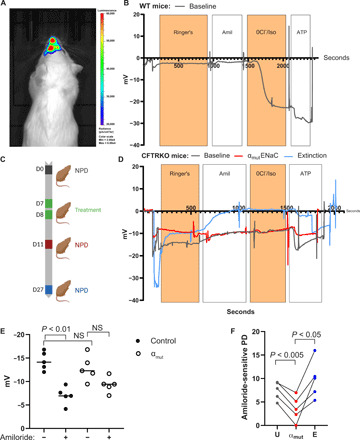
(A) In vivo luciferase expression was detected in a mouse treated with LNP encapsulating FLuc mRNA by intranasal instillation. Photo credit: Jeonghwan Kim, Oregon State University. (B) Representative NPD trace of WT mouse. (C) Timeline of LNP dosing and NPD recordings on CFTRKO mice. Color of each NPD recording time marker corresponds to the color of the NPD traces in the next panel. (D) Representative NPD traces of untransfected (gray line) and 72-hour αmutENaC-transfected (red line) and extinction of dosed LNP (blue line) on CFTRKO mice. (E) NPD values of untransfected (solid circles) and 72-hour αmutENaC-transfected (open circles) before and after amiloride treatment were graphically plotted. NS, not significant. (F) Amiloride-sensitive NPD traces of untransfected (gray circles) and 72-hour αmutENaC-transfected (red circles) and extinction recordings (blue circles) were measured and tracked in the same animal. The P values are indicated in the panels.
In vivo efficacy of αmutENaC mRNA was evaluated by measurement of NPD. NPD measurement is a well-established procedure performed clinically on patients for a definitive diagnosis of CF, based on net transepithelial ion transport activities of CFTR and ENaC (29). Nasal respiratory epithelium can be used as a substitute for measurements of electrophysiological activities of the lower airways, because they share common bioelectric properties (29).
NPD measurements were performed on WT control mice (Fig. 4B) as a baseline, by sequential perfusion with the following buffers (table S2): (i) Ringer’s: modified Ringer’s buffer to measure the net potential difference arising from the baseline activity of all channels in the nasal airway epithelium; (ii) Amil: modified Ringer’s buffer containing amiloride, which targets ENaC activity; (iii) 0Cl−/Iso: modified zero [Cl−] Ringer’s buffer containing isoproterenol, which raises adenosine 3′,5′-monophosphate (cAMP) in the cells, leading to CFTR-mediated Cl− secretion along with amiloride for continuing ENaC inhibition; (iv) ATP (adenosine 5′-triphosphate): modified zero [Cl−] Ringer’s buffer containing ATP for stimulating Cl− efflux by activation of calcium-activated chloride channels and isoproterenol and amiloride (28). We found that the baseline potential difference in WT mice was −1.58 ± 1.23 mV, n = 4 (table S3). Expectedly, WT mice showed a typically small amiloride response (amiloride-sensitive NPD: 1.40 ± 0.58 mV, n = 4) (table S3), whereas the magnitude of CFTR-mediated isoproterenol response was 22.11 ± 4.95 mV (n = 4) (table S3), suggesting a subdued ENaC and normal CFTR activity that is expected in WT mice (30). Additional calcium-activated chloride channel–mediated hyperpolarization response induced by ATP served as an internal control, indicating both the intactness of the nasal epithelium and correct positioning of the nasal bridge (Fig. 4B).
Next, CFTRKO mice were used to perform electrophysiological studies for measuring the effect of αmutENaC mRNA–LNP transfection in the nasal respiratory epithelia. Baseline NPD recording was performed in CFTRKO mice on day 0, before any treatment, following the same in vivo perfusion protocol used for WT mice (Fig. 4C). After a rest period, the same animals were transfected with αmutENaC mRNA–LNP in the nasal epithelia on days 7 and 8; thus, each animal was its own control. NPD recording assessing the effect of LNP transfection was performed after 72 hours on day 11. After 2 weeks of rest, NPD recording was repeated on the same animal to confirm extinction of the effect of LNP transfection on day 27 (Fig. 4C). NPD recording before transfection shows a hyperpolarized baseline (−14.41 ± 1.97 mV) followed by a characteristically large depolarization after perfusion with amiloride-containing Ringer’s buffer (amiloride-sensitive NPD: 7.42 ± 1.92 mV) (Fig. 4, D and E) (31). As expected, the CFTRKO mouse exhibited no isoproterenol response. Large hyperpolarizing response was observed after ATP treatment, indicating intact epithelia in these animal models (Fig. 4D, gray line). After gene transfection of αmutENaC mRNA, we found that the amiloride-mediated depolarization (amiloride-sensitive NPD: 3.57 ± 1.92 mV) was blunted (P < 0.005) in animals (Fig. 4, D and E). Amiloride-mediated depolarization regained perhaps due to clearance of the exogenous mutant mRNA after 2 weeks (Fig. 4D, blue line). The overall potential difference before amiloride treatment in the transfected (−12.86 ± 2.74 mV) and untransfected (−14.41 ± 1.97 mV) groups remained similar (Fig. 4E). Addition of amiloride in the untransfected group led to a significant decrease (P < 0.01) in overall potential difference (−Amil versus +Amil: −14.41 ± 1.97 mV versus −6.99 ± 1.88 mV), whereas in the αmutENaC-transfected animals, the amiloride response did not change significantly (−Amil versus +Amil: −12.86 ± 2.74 mV versus −9.29 ± 1.66 mV) (Fig. 4E). The amiloride-sensitive potential difference was greater in untransfected animals compared to animals treated with mutant mRNA, signifying that ENaC activity was inhibited (Fig. 4F). The third NPD recording performed after 2 weeks showed a restoration of the amiloride-sensitive potential difference to the levels of untransfected animals, suggesting that the mutant protein was cleared (Fig. 4F).
DISCUSSION
In this study, we have shown that engineered ENaC α mRNA mutants decreased amiloride-sensitive currents as a measure of ENaC activity. Unopposed or excessive ENaC channel activity is a long-standing proposed mechanism in CF pathophysiology (Fig. 5, A and B) (32). ENaC mutations in the α and β ENaC subunits that result in increased channel activity cause a CF-like phenotype (33, 34). Lung-specific β ENaC subunit–overexpressing mice develop thick inspissated mucus and lung disease similar to CF (35). In contrast, patients with an ENaC-inactivating mutation showed increased ASL height and greater mucociliary clearance (36). Heightened amiloride responses in CF patients and patient-derived cell lines suggest enhanced ENaC activity (1, 37) such that the channel function is either increased or unopposed. The exact mechanism of ENaC regulatory activity in the context of a functional decrease in CFTR activity is unclear. However, it is conceivable that in CF, a decrease in either the expression or functional CFTR activity alters inhibitory influences on ENaC. ENaC is activated by specific proteases (38) and conversely inhibited by extracellular Cl− (39). In CF, mucosal [Cl−] is decreased (37). Moreover, contraction of ASL volume leads to increased concentration of extracellular proteases that cleave ENaC and result in its activation (38). Previous studies have shown that inhibiting ENaC-activating proteases results in an increase in ASL height in cultured cells (38). Thus, ENaC-mediated sodium absorption and attendant mucus dehydration is an independent therapeutic target in the treatment of CF.
Fig. 5. Reduction of ENaC activity by αmutENaC in CF epithelium.
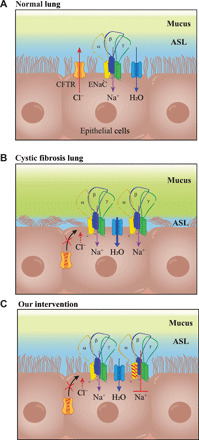
(A) Normal lung: Channels and transporters involved in maintaining ASL liquid height. (B) CF lung: Defect in CFTR protein leads to excessive sodium absorption by ENaC, resulting in excessive transcellular water absorption by aquaporin channels, leading to dehydration of ASL, mucus buildup, and obstruction of ciliary movement. (C) Our intervention: Reduction of ENaC activity by mutant α ENaC subunit restores ASL height in CF epithelia.
Early approaches that used inhaled ENaC-specific inhibitors such as benzamil and amiloride were ineffective because of rapid clearance from the lung (10). More complete ENaC inhibition by candidate compounds—GS-9411, P-552-02, and compound A—that were purported to have better pharmacokinetics and potency exhibited detrimental side effects (40, 41). Chronic and complete inhibition of ENaC and ENaC-dependent renal K+ excretion by these compounds resulted in hyperkalemia (40, 41). Thus, to reduce but not completely attenuate ENaC activity, several groups have attempted targeted gene knockdown of the ENaC subunits (10, 42). Prevailing antisense oligonucleotides and RNAi-driven, knockdown-based approach reduced ENaC activity both in vitro and in vivo by predominantly silencing of the a ENaC subunit. Both approaches led to an increase in ASL height in cultured cells (42). Although currently a promising inhalable siRNA formulation known as ARO-ENaC is being tested in a phase 1/2 clinical trial (43), past attempts have not succeeded in yielding an RNAi-based therapeutic for CF (10). Moreover, these approaches are inherently prone to off-target silencing of unintended genes (44). ENaC is part of the larger degenerin family of proteins that include acid-sensing ion channels (45). These channels share large homologies with ENaC and play an important role in lung physiology (46) and can be knocked down (7). However, mRNAs can be engineered to incorporate chemical modifications and regulatory elements to minimize immunogenicity and off-target protein expression, respectively (47). Last, indirect anti-ENaC approaches such as inhibition of SPLUNC1 facilitated ENaC degradation, which led to decreased channel activity (48). Although effective in vitro, clinical trials of SPLUNC1 inhibitors were not successful, leaving ENaC inhibition an unmet target in CF therapeutics.
In this study, we used mRNA as an ion channel inhibitor (Fig. 5C). Our mRNA therapeutics have the advantage of offering versatility in regulating and fine-tuning both αβγ and δβγ channel activities. The δβγ channels have been shown to be expressed widely in human airway epithelial cells (49). These channels have significantly higher open probability and sodium conductance compared to the typical αβγ channels (50). We proposed that our engineered αmutENaC might compete with both endogenous α and δ subunits to form inactivated αmutβγ channels to effectively reduce overall ENaC activity. Future studies using exogenously transfected WT and mutant ENaC subunits that carry different epitope tags will be used to conclusively show mutant αmutENaC integration into WT αβγ and δβγ ENaC channels expressed at the surface of cultured cells. Appropriately designed mutant subunits of ENaC will also be able to regulate the degree of ENaC inhibition. For example, mutating the furin cleavage site of the γ ENaC subunit has been shown to have a lesser inhibitory impact on channel activity when compared to mutations in furin cleavage sites in the α ENaC subunit (18). Other mutations such as those in the cysteine-rich domains or the thumb domain of β ENaC subunit reduce channel cell surface expression and channel half-life (51). Such mutations can be taken advantage of for targeting different aspects of channel biogenesis and function, thereby finely regulating ENaC activity by modifying channel expression, gating, or response of the channel to modulators such as proteases (7, 52, 53). Furthermore, because mutant α ENaC subunit alone cannot form functional channels, transfection of αmutENaC mRNA will likely not alter ion transport profile in cells that do not express endogenous ENaC. Thus, even without using targeted delivery mechanisms, our strategy will effectively alter ion transport specifically in ENaC-expressing cells and thus minimize off-target effect on ion transport profiles in nontarget cells (54).
On the basis of the previously discussed studies, we engineered α ENaC mRNA that lacks furin sites in the extracellular domain, while the conserved serine residue in the degenerin site was mutated to alanine (55). The furin site mutations inhibit ENaC activity, while the precautionary mutation at degenerin site helps maintain ENaC in an inhibited state. Our mutant αENaC mRNA used in this study was effective in reducing amiloride-sensitive current (by 85.22%) in CFBE41o- (untreated 6.09 ± 3.1 versus αmutENaC-transfected 0.9 ± 0.78 cells; Fig. 2, B and D), which is in comparable level to that achieved by RNAi-based methods (10, 42).
Improvement in ASL height is a precursor to improved mucociliary clearance in the airways. ASL height is maintained by the coordinated action of CFTR, ENaC, and aquaporins in the airway cells (56, 57). Transcellular water transport is driven largely by the movement of Na+ and Cl− ions across the epithelia (58). We tested whether αmutENaC-mediated suppression results in increasing the height of ASL. In our study, the immortalized cells had been grown to confluence before treatment that resembles the state of the airway in vivo. Our data show that ENaC inhibition led to an increase in ASL height in CFBE41o- cells by 34%. This is similar to a recent in vitro ASL change using siRNA ENaC targeting (42). In that study, Tagalakis et al. demonstrated that the change in ASL was enough to increase ciliary beat frequency. Our future studies will focus on determining whether delivery of CFTR or αmutENaC mRNA alone or in combination can increase ciliary beat frequency in polarized cultured primary CF patient–derived lung or nasal epithelial cells.
We have previously shown that LNP-delivered CFTR mRNA restores Cl− secretion in vitro and in vivo (28). As suggested by previous reports, we observed that amiloride-sensitive potential difference is elevated in CFTRKO mice, suggesting an increase in ENaC activity (30, 35). In our experiments, intranasal delivery of αmutENaC mRNA–LNP over two consecutive days resulted in a decrease in amiloride response, which is regained after 2 weeks in CFTRKO mice. A key challenge to be addressed in future studies will be to overcome the characteristic thick mucus barrier of CF airways for efficient delivery of mRNA via LNPs for which we will use a β-ENaC–overexpressing mouse model that replicates inspissated mucus phenotype observed in CF patients (35). In this model, LNPs can be delivered by aerosol and deposition to the lung studied. Last, mucus barriers will be applied ex vivo to epithelial monolayers to demonstrate LNP penetration. Nanoparticles coated with non–mucus-adhesive polymers such as PEG have been shown to readily penetrate CF sputum. Thus, sputum-penetrating drug- and gene-carrier nanoparticles can be developed for CF. We have not examined whether ENaC inhibition by αmutENaC transfection alters chloride conductance in cells. However, previous studies have shown that correction of CFTR chloride efflux by gene therapy does not significantly alter dysregulated sodium absorption in human and murine studies (4, 28, 59). Amiloride-sensitive NPD did not alter significantly after CFTR transfection in two studies (28, 59). Therefore, complementary treatment with αmutENaC and CFTR mRNA could lead to optimized ion transport and ASL height regulation. Lipid nanoparticles can be used to deliver these mRNA in combination (60). Similarly, ENaC inhibition in combination with partial correction of chloride transport achieved by the current triple combination CFTR modulator therapy may prove to be beneficial for nonresponders as well as patients who respond modestly to the drug regimen (3, 5). Through these studies, we have shown that engineered mRNA can be used to inactivate the ENaC channel selectively in vitro and in vivo. Our results showcase the promise of mRNA therapeutics as a potential universal treatment of CF regardless of underlying CFTR mutation.
MATERIALS AND METHODS
Materials
Engineered mRNAs were produced at TriLink BioTechnologies Inc. (San Diego, CA). 1,2-Distearoyl-sn-glycero-3-phosphocholine (DSPC) was obtained from Avanti Polar Lipids Inc. (Alabaster, AL). Cholesterol (catalog number C8667) was purchased from Sigma-Aldrich Corp. (St. Louis, MO), DMG-PEG2K was obtained from NOF America, and DLin-MC3-DMA was custom-synthesized by Biofine Inc. (Vancouver, BC, Canada).
LNP formulation and characterization
Our LNPs are composed of DLin-MC3-DMA, cholesterol, DSPC, and DMG-PEG2K along with the engineered mRNA and are assembled using microfluidic mixing as described previously (28). Briefly, all lipids were dissolved in pure ethanol at 50:38.5:10:1.5 molar ratios, and mRNA was diluted in 50 mM citrate buffer solution. The amount of ENaC mRNA ranged from 150 to 200 μg per batch. The N/P ratio between mRNA and DLin-MC3-DMA was 5:30 in all LNPs. Lipid solution and mRNA solution were mixed by microfluidics using NanoAssemblr Benchtop (Precision NanoSystems Inc., Vancouver, BC, Canada) at a 1:3 ratio, followed by dialysis against sterile phosphate-buffered saline (PBS) overnight using Slide-A-Lyzer dialysis cassettes (Thermo Fisher Scientific, Waltham, MA) with 10,000-Da molecular weight cutoff (MWCO). The resulting LNP solutions were concentrated using Amicon Ultra centrifugal filter units with 10,000-Da MWCO (Millipore). Hydrodynamic size and polydispersity index of LNPs were characterized by dynamic light scattering using Zetasizer Nano ZSP (Malvern Instruments, Malvern, Worcestershire, UK). mRNA encapsulation was assayed using a Quant-iT RiboGreen RNA Assay kit (Thermo Fisher Scientific, Waltham, MA) and a multimode microplate reader (Tecan Trading AG, Männedorf, Switzerland).
Cell culture
CFBE41o- and hCFTR-expressing CFBE-WT cell cultures, gifts from J. P. Clancy and D. Gruenert, were maintained in vented T75 tissue culture flasks (Corning Inc., Corning, NY) using minimum essential medium culture medium (Corning Inc., Corning, NY), supplemented with 10% fetal bovine serum and penicillin/streptomycin/glutamine (Corning Inc., Corning, NY) in humidified 5% CO2 incubator. For electrophysiological studies, 200,000 cells were seeded onto collagen I–coated 12-mm Snapwells/Transwells (Corning Inc., Corning, NY). Cultures were maintained with medium change every other day. Transepithelial electrical resistance (TEER) measurements were performed with Millicell ERS-2 (EMD Millipore, Burlington, MA) 30 min after medium change for checking tight junction formation. Cells were transfected when TEER values stabilized to >200 ohm∙cm2 (ERS-2) after 5 to 6 days of polarization (TEER of empty inserts approximately 80 to 85 ohm∙cm2) (61).
In vitro transfection assay
For luciferase transfection assay, CFBE41o- parental cells were seeded on a black 96-well plate at 4 × 103 cells per well and allowed to attach overnight. Cells were transfected with the LNP encapsulating firefly luciferase mRNA in various doses and incubated for 24 hours. Cell viability and luciferase expression were assayed using the ONE-Glo + Tox Luciferase Reporter and Cell Viability Assay Kit (Promega Corp., Madison, WI).
For cell viability assay, CFBE41o- parental cells were seeded and cultured in a 24-well plate. When confluent, the cells were treated with the LNP encapsulating ENaC mutant mRNA in various doses or 10% DMSO and incubated for 72 hours. Cell viability after the ENaC LNP treatment was assayed using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega Corp., Madison, WI).
Immunoblotting
CFBE41o- cells were solubilized using a NE-PER kit (Thermo Fisher Scientific) following the manufacturer’s protocol. Reduced and denatured cell extracts were electrophoresed in 4 to 12% Bolt Bis-Tris Plus (Thermo Fisher Scientific) gels in MES (Thermo Fisher Scientific) running buffer followed by dry transfer to polyvinylidene difluoride membrane. The membranes were probed with anti-V5 [Rabbit (Rb), 1:1000] (Cell Signaling Technology) and β-actin (Thermo Fisher Scientific) [Mouse (Ms), 1:10,000] primary antibodies.
Cryo–electron microscopy
Cryo–electron microscopy (Cryo-EM) acquisition was performed at 300 kV using Titan Krios with a Falcon III camera and Direct Electron Detector (DED). The sample (3 μl) was dispensed on a plasma-cleaned grid (Quantifoil R 1.2/1.3, 300 mesh, Ted Pella, Redding, CA) in the Vitrobot chamber at ~95% relative humidity and allowed to rest for 5 s. Then, the grid was blotted for 3 s with filter paper and plunged into liquid ethane cooled by liquid nitrogen. The frozen grids were then checked for visible defects and assembled into cassettes. The collected images were then processed and analyzed using ImageJ.
Fluorescent microscopy
For viable cell imaging, CFBE41o- parental cells were seeded and cultured in a 24-well plate. When confluent, the cells were treated with the αmutENaC-LNP mRNA in various doses or 10% DMSO and incubated for 72 hours. Live or dead cells were stained using the LIVE/DEAD Cell Imaging Kit (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol, and the center areas of each well were automatically scanned using an EVOS FL Auto fluorescent microscope (Invitrogen, Carlsbad, CA).
Animals
All animal studies were conducted at the Oregon Health & Science University and approved by the Institutional Animal Care and Use Committee (IP00001707). Female BALB/c mice were sedated using isoflurane, and the LNP encapsulating FLuc mRNA was pipetted onto a single nostril with spontaneous inhalation (0.25 mg/kg). After 5 hours, 200 μl of d-luciferin substrate (150 mg/kg; Thermo Fisher Scientific, Waltham, MA) was intraperitoneally injected to the mice 10 min before bioluminescent imaging. Image acquisition and analysis were performed using IVIS Lumina XRMS and the manufacturer’s software (PerkinElmer, MA). Cftr−/−tm1Unc Tg(FABPCFTR)1 Jaw/J double-transgenic CFTRKO mice were obtained from The Jackson Laboratory (JAX 002364) and maintained as described previously (62). For LNP administration, mice were anesthetized with an intraperitoneal injection of a mixture of ketamine and xylazine (100 μg/10 μg per kilogram body weight). LNPs were administered on two consecutive days to a single nostril (2 μl per application, 10 applications over 20 min, 0.5 mg/kg per day) (28).
Short-circuit current recording
Confluent CFBE41o- monolayers were transfected with 750 ng of αmutENaC or luciferase mRNA LNP on day 6 (TEER values stabilized to >200 ohm∙cm2, ERS-2, seeding day considered as day 0). ISC recording was performed on untransfected and transfected cells 72 hours after transfection. Snapwell inserts were mounted in EasyMount Ussing Chamber (P2302, Physiological Instruments, San Diego, CA) equipped with a heat block for temperature control. The apical and basolateral hemichambers contained 4 ml of Krebs buffer solution (135 mM NaCl, 1.6 mM MgCl2.6H2O, 2.4 mM K2HPO4, 0.4 mM KH2PO4, and 2.25 mM CaCl2.2H2O). The chamber temperature was maintained at 37°C. The hemichambers were continuously bubbled with air, which maintained the pH at 7.4. The apical and basolateral hemichambers were connected to Ag/AgCl electrodes via 3 M KCl agar bridges for voltage sensing and current passage. A VCC MC6 (multichannel voltage/current clamp) (Physiological Instruments, San Diego, CA) was used to generate current to maintain voltage at 0 mV. Data generated were recorded by Acquire and Analyze 2.3 software (Physiological Instruments, San Diego, CA). After an equilibration period to achieve a stable ISC (∼10 to 15 min), the amiloride-sensitive component of the ISC was then determined by adding 100 μM amiloride to the apical hemichamber.
Confocal microscopy and measurement of ASL height
For EGFP mRNA transfection, CFBE41o- parental cells were seeded on the collagen-coated three-well chambered slide (iBidi USA Inc., Fitchburg, WI). When cells became a complete monolayer, LNP encapsulating EGFP mRNA was added to the cells at a dose of 500 ng of mRNA per chamber. After incubating the specimen for 24 hours, cells were fixed in 4% paraformaldehyde, and nuclei were stained with Hoechst 33342 (Thermo Fisher Scientific, Waltham, MA). Fixed specimens were mounted with ProLong Diamond Antifade Mountant (Thermo Fisher Scientific, Waltham, MA) and imaged under an LSM 880 confocal microscope (Carl Zeiss AG).
CFBE41o- and WT-CFBE41o- cells were polarized on (TEER >200 ohm∙cm2 ERS-2), Transwell (Corning) inserts for 6 days. The CFBE41o- cells were transfected with 750 ng of αmutENaC-LNP on day 6 polarization or were left untreated as controls. ASL depth (i.e., depth of both the mucus and periciliary liquid layers) was measured 72 hours after transfection (day 9 of polarization) using a confocal microscope. Untransfected WT-CFBE41o- (400,000) were seeded on collagen-coated Transwell and also imaged at day 9 of polarization. The ASL in each Transwell was washed with PBS and then labeled with 50 μl of PBS containing rhodamine B isothiocyanate-dextran (2 mg/ml) (10 kDa; Sigma-Aldrich, Dorset, UK) by apical application 1 hour before the experiment. The cells were stained using 5 μM calcein AM (Thermo Fisher Scientific, Northumberland, UK) dissolved in culture medium for at least 60 min and introduced to the basolateral compartment of the insert. Perfluorocarbon (200 μl, FC-770; Sigma-Aldrich) was added to the apical compartment of the insert to prevent ASL evaporation. Fluorescent images of the epithelial layer and ASL height were obtained using an LSM 880 confocal microscope (LBRB 478)–Zeiss Laser-Scanning Confocal Fast Airy (Water Objective C-Apochromat 40×/1.2 W Corr M27, Coverglass Thickness = 0.14 to 0.19 mm). Samples were imaged in a #1.5 glass-bottom dish (FD35, Fluorodish, World Precision Instruments, Hitchin, UK). Z-stack acquisition used the optimal z-step as calculated in the Zen software. For each Transwell, six predetermined points were chosen such that these points are way from the meniscus and the center of the insert. Each point was XZ-scanned. XZ height of three randomly selected points in a field was measured. The average height of each image field for all six points of XZ scans for one insert was pooled for analysis.
Measurement of NPD
Experimental setup
NPD was measured using a modification of the previously described methods (28). Briefly, CFTR null or WT mice were anesthetized with an intraperitoneal injection of a ketamine and xylazine mixture (100 μg/10 μg per gram body weight). Anesthetized animals were intubated orally beginning with direct visualization of the vocal folds using an otoscope with a 2-mm speculum (model no. 20200; Welch Allyn, Skaneateles Falls, NY). A flexible guide wire was advanced through the vocal folds (catalog number RA-04020, Arrow International Inc., Reading, PA), and a 20-gauge intravenous catheter was passed over the wire (BD Medical, Franklin, NJ). Mice were placed head down on a 15° incline. Body temperature was monitored rectally (TH-5; Physitemp, Clifton, NJ) and maintained with a heating pad and heat lamp as needed. A high-impedance voltmeter (World Precision Instruments, Sarasota, FL) was connected by silver-chloride pellet electrodes to an exploring nasal bridge and a reference subcutaneous bridge. The voltmeter offset was adjusted to zero. The nasal bridge, a single polyethylene tube (PE10, 0.28-mm inner diameter; Clay-Adams, BD, Sparks, MD), was pulled to approximately one-half its original diameter and cut at an acute angle to maximize surface area. The resulting orifice was ∼0.5 mm in diameter. The tubing was marked at 3 and 5 mm from the tip. To ensure placement of the tubing in the nasal respiratory epithelia, the tubing was inserted into the naris to 3 mm and, after steady state, was advanced to the point of maximum voltage but never beyond 5 mm. The subcutaneous bridge was a 25-gauge butterfly-type (Abbott, Chicago, IL) needle containing Ringer’s solution inserted subcutaneously in the right abdominal wall. Each solution was warmed to 37°C and perfused to the naris at 7.5 nl/min using a perfusion pump.
Experimental protocol
Buffers were perfused sequentially through the nasal cavity for measuring baseline potential difference (PD), amiloride-sensitive PD, isoproterenol-sensitive PD, and ATP-sensitive PD. The buffer compositions are listed in table S2.
Experimental timeline
Sequential PDs were measured in untransfected animals by placing the nasal bridge in one nostril for establishing baseline NPD profile for an individual mouse. The animals were allowed to rest for 10 to 14 days, following which two consecutive days of nasal instillation of mRNA-LNP (2 μl per application, 10 applications over 20 min, 0.5 mg/kg per day) was performed. Care was taken to instill mRNA-LNP on the nostril that has not been used for the placement of the nasal bridge for baseline recording, on both days. NPD on the transfected nostril was performed 72 hours after the second day of nasal instillation, followed by a 2-week recovery period. The NPD was then performed on the same nostril to confirm the extinction of the effect of dosed mRNA and return of the NPD profile to baseline values.
Statistics
All experiments were performed at least in triplicate, and significance was determined by Student’s t test using GraphPad Prism for all analyses.
Supplementary Material
Acknowledgment
We thank T. Haley and A. Paolo for technical support. We would also like to thank the OHSU confocal imaging facility. EM was performed at the Multiscale Microscopy Core (MMC) with technical support from the Oregon Health & Science University (OHSU). Funding: This project was supported through funding from the National Heart Lung and Blood Institute (1R01HL146736-01 to G.S.) and the Cystic Fibrosis Foundation (SAHAY18G0 to G.S. and SAHAY19XX0 to G.S.). Author contributions: G.S. directed the project. A.M., G.S., and K.D.M. conceived and designed studies. J.K. formulated and characterized LNPs with mRNA. A.M. and K.D.M. performed Ussing chamber and NPD studies. A.M. did cell culturing and performed TEER and Western blot studies with M.I.H.’s assistance. A.M. performed confocal microscopy studies. Y.E. performed EM studies. All authors contributed in data analysis arising from experiments that they were involved in. A.M. and G.S. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/47/eabc5911/DC1
REFERENCES AND NOTES
- 1.Jiang C., Finkbeiner W. E., Widdicombe J. H., McCray P. B. Jr., Miller S. S., Altered fluid transport across airway epithelium in cystic fibrosis. Science 262, 424–427 (1993). [DOI] [PubMed] [Google Scholar]
- 2.Rowntree R. K., Harris A., The phenotypic consequences of CFTR mutations. Ann. Hum. Genet. 67 ( Pt 5), 471–485 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Heijerman H. G. M., McKone E. F., Downey D. G., Van Braeckel E., Rowe S. M., Tullis E., Mall M. A., Welter J. J., Ramsey B. W., McKee C. M., Marigowda G., Moskowitz S. M., Waltz D., Sosnay P. R., Simard C., Ahluwalia N., Xuan F., Zhang Y., Taylor-Cousar J. L., McCoy K. S., McCoy K., Donaldson S., Walker S., Chmiel J., Rubenstein R., Froh D. K., Neuringer I., Jain M., Moffett K., Taylor-Cousar J. L., Barnett B., Mueller G., Flume P., Livingston F., Mehdi N., Teneback C., Welter J., Jain R., Kissner D., Patel K., Calimano F. J., Johannes J., Daines C., Keens T., Scher H., Chittivelu S., Reddivalam S., Klingsberg R. C., Johnson L. G., Verhulst S., Macedo P., Downey D., Connett G., Nash E., Withers N., Lee T., Bakker M., Heijerman H., Vermeulen F., Van Braeckel E., Knoop C., De Wachter E., van der Meer R., Merkus P., Majoor C., Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 394, 1940–1948 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alton E. W. F. W., Boyd A. C., Porteous D. J., Davies G., Davies J. C., Griesenbach U., Higgins T. E., Gill D. R., Hyde S. C., Innes J. A.; UK Cystic Fibrosis Gene Therapy Consortium , A phase I/IIa safety and efficacy study of nebulized liposome-mediated gene therapy for cystic fibrosis supports a multidose trial. Am. J. Respir. Crit. Care Med. 192, 1389–1392 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies J. C., Moskowitz S. M., Brown C., Horsley A., Mall M. A., McKone E. F., Plant B. J., Prais D., Ramsey B. W., Taylor-Cousar J. L., Tullis E., Uluer A., McKee C. M., Robertson S., Shilling R. A., Simard C., Van Goor F., Waltz D., Xuan F., Young T., Rowe S. M.; VX16-659-101 Study Group , VX-659–tezacaftor–ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N. Engl. J. Med. 379, 1599–1611 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanukoglu I., Hanukoglu A., Epithelial sodium channel (ENaC) family: Phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene 579, 95–132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kashlan O. B., Kleyman T. R., ENaC structure and function in the wake of a resolved structure of a family member. Am. J. Physiol. Renal Physiol. 301, F684–F696 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stutts M. J., Canessa C. M., Olsen J. C., Hamrick M., Cohn J. A., Rossier B. C., Boucher R. C., CFTR as a cAMP-dependent regulator of sodium channels. Science 269, 847–850 (1995). [DOI] [PubMed] [Google Scholar]
- 9.Ho L. P., Samways J. M., Porteous D. J., Dorin J. R., Carothers A., Greening A. P., Innes J. A., Correlation between nasal potential difference measurements, genotype and clinical condition in patients with cystic fibrosis. Eur. Respir. J. 10, 2018–2022 (1997). [DOI] [PubMed] [Google Scholar]
- 10.Shei R.-J., Peabody J. E., Kaza N., Rowe S. M., The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis. Curr. Opin. Pharmacol. 43, 152–165 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsui H., Grubb B. R., Tarran R., Randell S. H., Gatzy J. T., Davis C. W., Boucher R. C., Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 95, 1005–1015 (1998). [DOI] [PubMed] [Google Scholar]
- 12.Matsui H., Randell S. H., Peretti S. W., Davis C. W., Boucher R. C., Coordinated clearance of periciliary liquid and mucus from airway surfaces. J. Clin. Invest. 102, 1125–1131 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canessa C. M., Schild L., Buell G., Thorens B., Gautschi I., Horisberger J.-D., Rossier B. C., Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367, 463–467 (1994). [DOI] [PubMed] [Google Scholar]
- 14.Waldmann R., Champigny G., Bassilana F., Voilley N., Lazdunski M., Molecular cloning and functional expression of a novel amiloride-sensitive Na+ channel. J. Biol. Chem. 270, 27411–27414 (1995). [DOI] [PubMed] [Google Scholar]
- 15.Haerteis S., Krueger B., Korbmacher C., Rauh R., The δ-subunit of the epithelial sodium channel (ENaC) enhances channel activity and alters proteolytic ENaC activation. J. Biol. Chem. 284, 29024–29040 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu M., Echeverri F., Kalabat D., Laita B., Dahan D. S., Smith R. D., Xu H., Staszewski L., Yamamoto J., Ling J., Hwang N., Kimmich R., Li P., Patron E., Keung W., Patron A., Moyer B. D., Small molecule activator of the human epithelial sodium channel. J. Biol. Chem. 283, 11981–11994 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Akinc A., Maier M. A., Manoharan M., Fitzgerald K., Jayaraman M., Barros S., Ansell S., Du X., Hope M. J., Madden T. D., Mui B. L., Semple S. C., Tam Y. K., Ciufolini M., Witzigmann D., Kulkarni J. A., van der Meel R., Cullis P. R., The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 14, 1084–1087 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Carattino M. D., Hughey R. P., Kleyman T. R., Proteolytic processing of the epithelial sodium channel γ subunit has a dominant role in channel activation. J. Biol. Chem. 283, 25290–25295 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheng S., Carattino M. D., Bruns J. B., Hughey R. P., Kleyman T. R., Furin cleavage activates the epithelial Na+ channel by relieving Na+ self-inhibition. Am. J. Physiol. Renal Physiol. 290, F1488–F1496 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Walsh C. T., Garneau-Tsodikova S., Gatto G. J. Jr., Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. Int. Ed. Engl. 44, 7342–7372 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Mukherjee S., Hao Y.-H., Orth K., A newly discovered post-translational modification—The acetylation of serine and threonine residues. Trends Biochem. Sci. 32, 210–216 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Carattino M. D., Sheng S., Bruns J. B., Pilewski J. M., Hughey R. P., Kleyman T. R., The epithelial Na+ channel is inhibited by a peptide derived from proteolytic processing of its α subunit. J. Biol. Chem. 281, 18901–18907 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Sheng S., Li J., McNulty K. A., Kieber-Emmons T., Kleyman T. R., Epithelial sodium channel pore region: Structure and role in gating. J. Biol. Chem. 276, 1326–1334 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Kieber-Emmons T., Lin C., Foster M. H., Kleyman T. R., Antiidiotypic antibody recognizes an amiloride binding domain within the alpha subunit of the epithelial Na+ channel. J. Biol. Chem. 274, 9648–9655 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Mall M., Bleich M., Greger R., Schreiber R., Kunzelmann K., The amiloride-inhibitable Na+ conductance is reduced by the cystic fibrosis transmembrane conductance regulator in normal but not in cystic fibrosis airways. J. Clin. Invest. 102, 15–21 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verkman A. S., Song Y., Thiagarajah J. R., Role of airway surface liquid and submucosal glands in cystic fibrosis lung disease. Am. J. Physiol. Cell Physiol. 284, C2–C15 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Knowles M. R., Boucher R. C., Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Invest. 109, 571–577 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson E., MacDonald K. D., Slaughter K., McKinney M., Patel S., Sun C., Sahay G., Lipid nanoparticle-delivered chemically modified mrna restores chloride secretion in cystic fibrosis. Mol. Ther. 26, 2034–2046 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rowe S. M., Clancy J. P., Wilschanski M., Nasal potential difference measurements to assess CFTR ion channel activity. Methods Mol. Biol. 741, 69–86 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grubb B. R., Vick R. N., Boucher R. C., Hyperabsorption of Na+ and raised Ca(2+)-mediated Cl- secretion in nasal epithelia of CF mice. Am. J. Physiol. 266 ( 5 Pt 1), C1478–C1483 (1994). [DOI] [PubMed] [Google Scholar]
- 31.Knowles M., Gatzy J., Boucher R., Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N. Engl. J. Med. 305, 1489–1495 (1981). [DOI] [PubMed] [Google Scholar]
- 32.Greger R., Mall M., Bleich M., Ecke D., Warth R., Riedemann N., Kunzelmann K., Regulation of epithelial ion channels by the cystic fibrosis transmembrane conductance regulator. J. Mol. Med. 74, 527–534 (1996). [DOI] [PubMed] [Google Scholar]
- 33.Rauh R., Diakov A., Tzschoppe A., Korbmacher J., Azad A. K., Cuppens H., Cassiman J.-J., Dötsch J., Sticht H., Korbmacher C., A mutation of the epithelial sodium channel associated with atypical cystic fibrosis increases channel open probability and reduces Na+ self inhibition. J. Physiol. 588 ( Pt 8), 1211–1225 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rauh R., Soell D., Haerteis S., Diakov A., Nesterov V., Krueger B., Sticht H., Korbmacher C., A mutation in the β-subunit of ENaC identified in a patient with cystic fibrosis-like symptoms has a gain-of-function effect. Am. J. Physiol. Lung Cell. Mol. Physiol. 304, L43–L55 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Mall M., Grubb B. R., Harkema J. R., O’Neal W. K., Boucher R. C., Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat. Med. 10, 487–493 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Kerem E., Bistritzer T., Hanukoglu A., Hofmann T., Zhou Z., Bennett W., MacLaughlin E., Barker P., Nash M., Quittell L., Boucher R., Homolya V., Keenan B., Knowles M. R., Pulmonary epithelial sodium-channel dysfunction and excess airway liquid in pseudohypoaldosteronism. N. Engl. J. Med. 341, 156–162 (1999). [DOI] [PubMed] [Google Scholar]
- 37.Boucher R. C., Cotton C. U., Gatzy J. T., Knowles M. R., Yankaskas J. R., Evidence for reduced Cl- and increased Na+ permeability in cystic fibrosis human primary cell cultures. J. Physiol. 405, 77–103 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Myerburg M. M., Butterworth M. B., McKenna E. E., Peters K. W., Frizzell R. A., Kleyman T. R., Pilewski J. M., Airway surface liquid volume regulates ENaC by altering the serine protease-protease inhibitor balance: A mechanism for sodium hyperabsorption in cystic fibrosis. J. Biol. Chem. 281, 27942–27949 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Collier D. M., Snyder P. M., Extracellular chloride regulates the epithelial sodium channel. J. Biol. Chem. 284, 29320–29325 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirsh A. J., Zhang J., Zamurs A., Fleegle J., Thelin W. R., Caldwell R. A., Sabater J. R., Abraham W. M., Donowitz M., Cha B., Johnson K. B., St. George J. A., Johnson M. R., Boucher R. C., Pharmacological properties of N-(3,5-diamino-6-chloropyrazine-2-carbonyl)-N′-4-[4-(2,3-dihydroxypropoxy)phenyl] butyl-guanidine methanesulfonate (552-02), a novel epithelial sodium channel blocker with potential clinical efficacy for cystic fibrosis lung disease. J. Pharmacol. Exp. Ther. 325, 77–88 (2008). [DOI] [PubMed] [Google Scholar]
- 41.O’Riordan T. G., Donn K. H., Hodsman P., Ansede J. H., Newcomb T., Lewis S. A., Flitter W. D., White V. S., Johnson M. R., Montgomery A. B., Warnock D. G., Boucher R. C., Acute hyperkalemia associated with inhalation of a potent ENaC antagonist: Phase 1 trial of GS-9411. J. Aerosol Med. Pulm. Drug Deliv. 27, 200–208 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Tagalakis A. D., Munye M. M., Ivanova R., Chen H., Smith C. M., Aldossary A. M., Rosa L. Z., Moulding D., Barnes J. L., Kafetzis K. N., Jones S. A., Baines D. L., Moss G. W. J., O’Callaghan C., McAnulty R. J., Hart S. L., Effective silencing of ENaC by siRNA delivered with epithelial-targeted nanocomplexes in human cystic fibrosis cells and in mouse lung. Thorax 73, 847–856 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Study of ARO-ENaC in Healthy Volunteers and in Patients With Cystic Fibrosis, Arrowhead Pharmaceuticals, ClinicalTrials.gov Identifier: NCT04375514 (2020), https://clinicaltrials.gov/ct2/show/NCT04375514.
- 44.Jackson A. L., Burchard J., Schelter J., Chau B. N., Cleary M., Lim L., Linsley P. S., Widespread siRNA "off-target" transcript silencing mediated by seed region sequence complementarity. RNA 12, 1179–1187 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.García-Añoveros J., Samad T. A., Žuvela-Jelaska L., Woolf C. J., Corey D. P., Transport and localization of the DEG/ENaC ion channel BNaC1α to peripheral mechanosensory terminals of dorsal root ganglia neurons. J. Neurosci. 21, 2678–2686 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trac P. T., Thai T. L., Linck V., Zou L., Greenlee M., Yue Q., Al-Khalili O., Alli A. A., Eaton A. F., Eaton D. C., Alveolar nonselective channels are ASIC1a/α-ENaC channels and contribute to AFC. Am. J. Physiol. Lung Cell. Mol. Physiol. 312, L797–L811 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaczmarek J. C., Kowalski P. S., Anderson D. G., Advances in the delivery of RNA therapeutics: From concept to clinical reality. Genome Med. 9, 60 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Couroux P., Farias P., Rizvi L., Griffin K., Hudson C., Crowder T., Tarran R., Tullis E., First clinical trials of novel ENaC targeting therapy, SPX-101, in healthy volunteers and adults with cystic fibrosis. Pulm. Pharmacol. Ther. 58, 101819 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Zhao R.-Z., Nie H.-G., Su X.-F., Han D.-Y., Lee A., Huang Y., Chang Y., Matalon S., Ji H.-L., Characterization of a novel splice variant of δ ENaC subunit in human lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 302, L1262–L1272 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rauh R., Hoerner C., Korbmacher C., δβγ-ENaC is inhibited by CFTR but stimulated by cAMP in Xenopus laevis oocytes. Am. J. Physiol. Lung Cell. Mol. Physiol. 312, L277–L287 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Sheng S., Chen J., Mukherjee A., Yates M. E., Buck T. M., Brodsky J. L., Tolino M. A., Hughey R. P., Kleyman T. R., Thumb domains of the three epithelial Na+ channel subunits have distinct functions. J. Biol. Chem. 293, 17582–17592 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mukherjee A., Mueller G. M., Kinlough C. L., Sheng N., Wang Z., Mustafa S. A., Kashlan O. B., Kleyman T. R., Hughey R. P., Cysteine palmitoylation of the γ subunit has a dominant role in modulating activity of the epithelial sodium channel. J. Biol. Chem. 289, 14351–14359 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mukherjee A., Wang Z., Kinlough C. L., Poland P. A., Marciszyn A. L., Montalbetti N., Carattino M. D., Butterworth M. B., Kleyman T. R., Hughey R. P., Specific palmitoyltransferases associate with and activate the epithelial sodium channel. J. Biol. Chem. 292, 4152–4163 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Y., Jiang B.-J., Zhao R.-Z., Ji H.-L., Epithelial sodium channels in pulmonary epithelial progenitor and stem cells. Int. J. Biol. Sci. 12, 1150–1154 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carattino M. D., Passero C. J., Steren C. A., Maarouf A. B., Pilewski J. M., Myerburg M. M., Hughey R. P., Kleyman T. R., Defining an inhibitory domain in the α-subunit of the epithelial sodium channel. Am. J. Physiol. Renal Physiol. 294, F47–F52 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Verkman A. S., Matthay M. A., Song Y., Aquaporin water channels and lung physiology. Am. J. Physiol. Lung Cell. Mol. Physiol. 278, L867–L879 (2000). [DOI] [PubMed] [Google Scholar]
- 57.Gianotti A., Melani R., Caci E., Sondo E., Ravazzolo R., Galietta L. J. V., Zegarra-Moran O., Epithelial sodium channel silencing as a strategy to correct the airway surface fluid deficit in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 49, 445–452 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Kis A., Krick S., Baumlin N., Salathe M., Airway hydration, apical K+ Secretion, and the large-conductance, Ca2+-activated and voltage-dependent potassium (BK) Channel. Ann. Am. Thorac. Soc. 13 ( Suppl 2), S163–S168 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Caplen N. J., Alton E. W. F. W., Mddleton P. G., Dorin J. R., Stevenson B. J., Gao X., Durham S. R., Jeffery P. K., Hodson M. E., Coutelle C., Huang L., Porteous D. J., Williamson R., Geddes D. M., Liposome-mediated CFTR gene transfer to the nasal epithelium of patients with cystic fibrosis. Nat. Med. 1, 39–46 (1995). [DOI] [PubMed] [Google Scholar]
- 60.Barba A. A., Bochicchio S., Dalmoro A., Lamberti G., Lipid delivery systems for nucleic-acid-based-drugs: From production to clinical applications. Pharmaceutics 11, 360 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Srinivasan B., Kolli A. R., Esch M. B., Abaci H. E., Shuler M. L., Hickman J. J., TEER measurement techniques for in vitro barrier model systems. J. Lab. Autom. 20, 107–126 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou L., Dey C. R., Wert S. E., DuVall M. D., Frizzell R. A., Whitsett J. A., Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science 266, 1705–1708 (1994). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/47/eabc5911/DC1