

Mycobacterial F1Fo-ATP synthases (α3:β3:γ:δ:ε:a:b:b′:c9) are incapable of ATP-driven proton translocation due to their latent ATPase activity. This prevents wasting of ATP and altering of the proton motive force, whose dissipation is lethal to mycobacteria. We demonstrate that the mycobacterial C-terminal extension of nucleotide-binding subunit α contributes mainly to the suppression of ATPase activity in the recombinant mycobacterial F1-ATPase.
KEYWORDS: Mycobacterium, tuberculosis, F-ATP synthase, subunit α, ATP hydrolysis, bioenergetics
ABSTRACT
Mycobacterial F1Fo-ATP synthases (α3:β3:γ:δ:ε:a:b:b′:c9) are incapable of ATP-driven proton translocation due to their latent ATPase activity. This prevents wasting of ATP and altering of the proton motive force, whose dissipation is lethal to mycobacteria. We demonstrate that the mycobacterial C-terminal extension of nucleotide-binding subunit α contributes mainly to the suppression of ATPase activity in the recombinant mycobacterial F1-ATPase. Using C-terminal deletion mutants, the regions responsible for the enzyme’s latency were mapped, providing a new compound epitope.
INTRODUCTION
F1Fo-ATP synthase is essential in Mycobacterium tuberculosis isolates for the formation of ATP (1, 2) and has become a drug target to fight tuberculosis (3–7). The F1 domain contains subunits α3:β3:γ:ε, the proton-translocating Fo domain (a:c9), and subunits b:b′:δ holding both domains together (8–10). Rotation of γ:ε connects H+ conduction and ATP formation within subunits α3:β3. Interestingly, mycobacterial F1Fo-ATP synthase does not perform ATP hydrolysis-driven proton translocation because of latent ATPase activity (11, 12). Different structural features in the mycobacterial nucleotide-binding subunit α (13, 14) and γ:ε have been proposed to be linked to suppress ATPase activity (11, 12, 15, 16). These features include the extended 3.5-kDa C terminus of subunit α (αCTD) (14) (Fig. 1), the extra 14-amino-acid γ-loop (12), and the C terminus of subunit ε (15, 16). Understanding these mycobacterial entities resulted in discovery of the mycobacterial F1Fo-ATP synthase inhibitors GaMF1 (17), epigallocatechin gallate (2) and EpNMF1 (16).
FIG 1.

Amino acid sequence alignment of subunit α of different mycobacterial organisms in comparison with Homo sapiens, Escherichia coli, and G. stearothermophilus. The sequence alignment of subunit α of the following organisms: H. sapiens (UniProt ID P25705-2), E. coli (UniProt ID P0ABB0), G. stearothermophilus (UniProt ID P42005), M. tuberculosis (UniProt ID P9WPU7), M. smegmatis (UniProt ID A0R202), and Mycobacterium bovis (UniProt ID A1KI96) were obtained from the UniProt database (30) and imported into Jalview (31). Alignment of the sequences was performed using ClustalWS (32). Thereafter, the calculation of the percentage of identity was performed and presented in darker to lighter shades of blue, representing the most homologous to the least homologous. As highlighted in red, the C-terminal extension was observed specifically in mycobacteria and not in other species. As previously studied, the α-helix is present from V525 to V538 (according to M. tuberculosis amino acid numbering). For reference, the α-helix present in the C terminus is presented by a green cylinder, and the region showing no secondary structure is denoted by a single black line.
The mycobacterial αCTD was unresolved in the crystallographic structure (PDB ID 6FOC) (18). Residues 514 to 549 and 540 to 549 of the M. tuberculosis αCTD were predicted to form a random coil, whereby residues 526 to 539 were determined to form an α-helix (13). The chromosomal deletion mutation of the αCTD mutant Δα(514–548) stimulated ATP hydrolysis of inverted membrane vesicles (IMVs) (13), whereas fusing the M. tuberculosis αCTD at the C terminus of subunit α of the Geobacillus stearothermophilus (formerly Bacillus PS3) F1-ATPase decreased ATPase activity of the hybrid enzyme (13). These data suggest that the mycobacterial αCTD may play a role in latency.
The 8-fold decrease of recombinant Mycobacterium smegmatis F1-ATPase compared with its ε-free form α3:β3:γ demonstrated an inhibitory effect of subunit ε in ATP hydrolysis (16). However, the α3:β3:γ ATPase activity is still significantly lower than the nonlatent α3:β3:γ complex, e.g., of G. stearothermophilus [4.9 ± 0.04 μmol min−1 (mg of protein)−1] (13), highlighting a significant contribution of another mycobacterial F1-ATPase element to latency. To fully comprehend the mechanical system of latency and the role of the αCTD and its three regions, a systematic assessment using recombinant M. smegmatis F1-ATPase mutants at the αCTD was performed.
First, the αCTD-deleted M. smegmatis F1-ATPase mutant, MsF1-αΔ514-549βγε, was engineered using the recently generated template of the atp genes AGDC, encoding subunits α:β:γ:ε within the pYUB1049 vector (16, 18) and the primers listed in Table S1 in the supplemental material. The linearized pYUB1049 vector was amplified (19), and the two DNA fragments were incorporated as previously published (16). To ease purification, a His6 tag was added to the N terminus of the β subunit (18). Protein purification was performed as mentioned previously published (16), with an MsF1-αΔ514-549βγε in proper stoichiometry and an αΔ514-549 band running faster than its wild-type (WT) counterpart, revealing the successful deletion (Fig. 2A and B). Subsequently, continuous ATP hydrolysis assay was performed according to previously published methods (16, 20, 21). ATPase activity of 0.05 ± 0.001 μmol min−1 (mg of protein)−1 was calculated for WT MsF1-ATPase (Fig. 2C, Table 1) and 3.31 ± 0.2 μmol min−1 (mg of protein)−1 for mutant MsF1-αΔ514-549βγε, reflecting a >60-fold increase in ATP hydrolysis. The 60-fold increase compared to the 1.7-fold increase of the chromosomal deletion Δα(514–548) mutant in IMVs (13) underlines the need for a defined enzyme to be presented, since endogenous MsF1Fo ATP synthase and ATP-driven translocators within IMVs effect accurate measures. Interestingly, the ATP hydrolysis rate of MsF1-αΔ514-549βγε was significantly higher than that of the ε-free complex [0.63 ± 0.003 μmol min−1 (mg of protein)−1] (16) but comparable to that of the nonlatent G. stearothermophilus F1-ATPase [4.9 ± 0.04 μmol min−1 (mg of protein)−1] (13). The results suggest that subunit α is a major contributing factor in latent ATP hydrolysis of mycobacterial F1-ATPase.
FIG 2.

Characterization of the recombinant MsF1-αΔCTD mutants. (A) Fractions from ion exchange were pooled and subjected to size-exclusion chromatography. The recombinant proteins showed consistency in elution at ∼11.6 ml, and their integrity and constituents were confirmed on a 12% SDS-PAGE gel (inset). The subunits are labeled, where α* refers to subunit α and its mutants β, γ, and ε, which correspond to ∼60, 54 , 35, and 10 kDa, respectively. The corresponding proteins are as labeled: lane 1, MsF1-ATPase; lane 2, MsF1-αΔ514-549βγε; lane 3, MsF1-αΔ523-549βγε; and lane 4, MsF1-αΔ538-549βγε. The purification protocol and 12% SDS-PAGE gel were replicated at least three times, and results represented in the elution diagram and gel remained consistent. (B) Densitometric analysis of the γ to ε ratio of MsF1-αΔ514-549βγε revealed a 1:0.3 ratio, identical to that of the WT enzyme (16) and demonstrating the correct stoichiometric subunit ratio. (C) Recombinant mutants were tested for their ATP hydrolysis rate. The decrease in NADH absorption at 340 nm is plotted against the progressing time. MsF1-αΔ514-549βγε showed a significant increase in ATP hydrolysis (red triangle). On the other hand, MsF1-αΔ523-549βγε (purple diamond) and MsF1-αΔ538-549βγε (green asterisk) showed lesser ATP hydrolysis than MsF1-αΔ514-549βγε. To calculate the specific activity, the initial rate was used (solid lines), and their calculated specific activities and standard error of regression slope (Sb1) were 3.31 ± 0.18, 1.54 ± 0.03, and 1.33 ± 0.01 μmol min−1 (mg of protein)−1.
TABLE 1.
Summary of specific enzyme activities of the MsF1-ATPase and its mutants
MsF1-αΔ523-549βγε and MsF1-αΔ538-549βγε were designed to identify whether the random coil regions 514 to 522 and 538 to 549 (18) or the α-helix region 523 to 537 (13) are/is critical for latent ATP hydrolysis (Table S1). Protein purification and ATP hydrolysis assay were performed as described previously. MsF1-αΔ523-549βγε and MsF1-αΔ538-549βγε (Fig. 2A) showed a similar ∼30-fold increase to that of the WT enzyme (Fig. 2C, Table 1). Compared to the ∼60-fold ATP hydrolysis increase of MsF1-αΔ514-549βγε, the 30-fold increase in ATP hydrolysis of MsF1-αΔ538-549βγε suggests that the αCTD residues 538 to 549 and the 514-to-522 region contribute to the suppression of ATPase activity. In contrast, the comparable enzymatic increase in MsF1-αΔ523-549βγε reflects that the 523-to-537 region has no major impact on latency. The two-step increase (30- to 60-fold) of MsF1-αΔ538-549βγε and MsF1-αΔ514-549βγε suggests that both regions may interact with two different mechanistic epitopes of the enzyme. The F1-ATPase structure of the pathogen Trypanosoma brucei (22), also consisting of an extended subunit of the α C terminus, might illustrate these aspects. As shown in Fig. 3, residues 536 to 539 of this extension form one α-helical turn, followed by a random region (540 to 544) and an α-helix (546 to 558) that come within 7.1 Å of the ADP within the nucleotide binding site. Either a small conformational change may bring R558 in close proximity to ADP or one of the remaining C-terminal residues not resolved in the structure may interact with the nucleotide, thereby stabilizing the ADP-inhibiting state (23). In analogy, we propose that the very C-terminal residues 538 to 549 of mycobacterial subunit α, whose deletion led to a 30-fold ATPase activity increase, come close to the ADP and trap the nucleotide.
FIG 3.
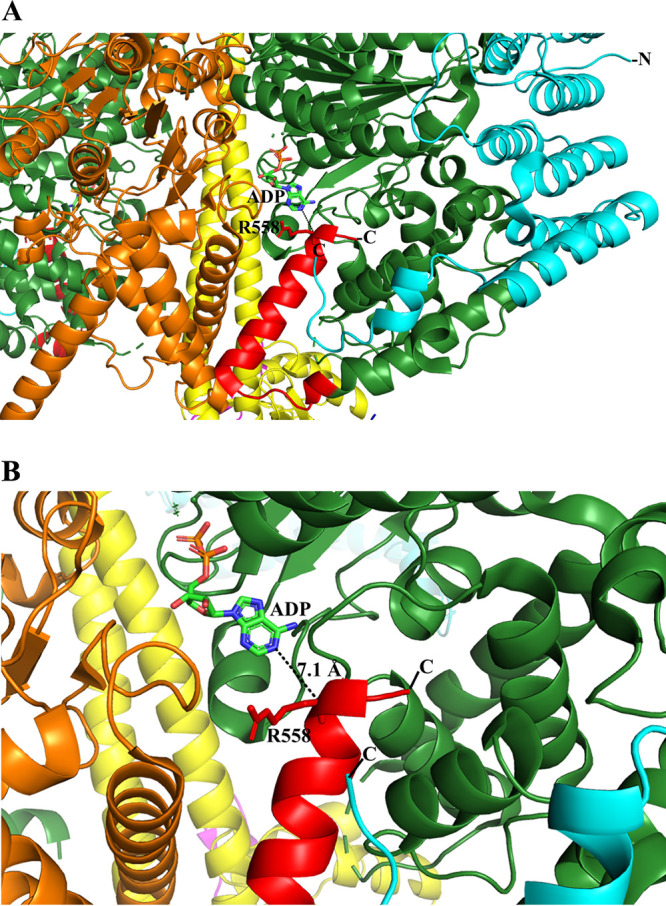
A proposed mechanism of ATP hydrolysis inhibition. (A) Part of the T. brucei F1-ATPase crystal structure (PDB ID 6F5D ) (22) and a further zoom to highlight the proximity of its extended subunit α C terminus and ADP. The T. brucei C-terminal residues 536 to 539 (red) form an α-helical turn, followed by a random region (540 to 544) and an α-helix (546 to 558) that come close to the ADP. A conformational alteration could bring R558 closer to ADP to generate a hydrogen bond with ADP, or one of the C-terminal residues, not resolved in the structure, could come in proximity to the nucleotide. We predict that the C-terminal residues 538 to 549 of mycobacterial subunit α may come in close proximity to the ADP to stabilize the inhibited state. Subunits α, β, and γ and the T. brucei-specific p18 are shown in green, orange, yellow, and cyan, respectively. The figure was generated via PyMOL (33).
Concerning the second epitope interaction leading to the final 60-fold ATPase activity increase, residue 522 of the mycobacterial stretch 514 to 522 was described to come in proximity with polar residues of γ of the hybrid αchi3:β3:γ complex, thereby decreasing the angular velocity of the power stroke after ATP binding (13).
In conclusion, during evolution, F-ATP synthases have evolved various mechanisms regulating ATP hydrolysis inhibition, including additional features, such as the inhibitory protein (24), subunit ζ (25), the extended C terminus of subunit ε (26–28), or the species-specific extra loop in γ (12, 29). Recent studies proposed cumulative effects of mycobacterial subunits α, γ, and ε to be responsible for suppressed ATP hydrolysis (12, 13, 15). Using defined enzyme complexes, the data presented demonstrate that the mycobacterial αCTD of subunit α is the major regulator of latent ATP hydrolysis activity, preventing wastage of ATP. Together with the inhibitory mechanisms proposed, the data may contribute to the design of molecules disrupting the interactions of subunit α’s unique C terminus to activate ATPase hydrolysis.
Supplementary Material
ACKNOWLEDGMENTS
This work and the research scholarship of C.F.W. were supported by the National Research Foundation (NRF) Singapore, NRF Competitive Research Program (CRP) (grant NRF–CRP18–2017–01).
We declare that we have no conflicts of interest.
G.G. conceptualized and supervised the study, in addition to acquiring funding. C.-F.W. performed the investigation. G.G. and C.F.W. wrote, reviewed, and edited the manuscript.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Cook GM, Greening C, Hards K, Berney M. 2014. Energetics of pathogenic bacteria and opportunities for drug development, p 1–62. Poole RK. (ed), Advances in microbial physiology, 1st ed, vol 65 Academic Press, San Diego, CA. [DOI] [PubMed] [Google Scholar]
- 2.Saw WG, Wu ML, Ragunathan P, Biuković G, Lau AM, Shin J, Harikishore A, Cheung CY, Hards K, Sarathy JP, Bates RW, Cook GM, Dick T, Grüber G. 2019. Disrupting coupling within mycobacterial F-ATP synthases subunit ε causes dysregulated energy production and cell wall biosynthesis. Sci Rep 9:16759. doi: 10.1038/s41598-019-53107-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andries K, Verhasselt P, Guillemont J, Göhlmann HWH, Neefs J-M, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 4.Sutherland HS, Tong AST, Choi PJ, Conole D, Blaser A, Franzblau SG, Cooper CB, Upton AM, Lotlikar MU, Denny WA, Palmer BD. 2018. Structure-activity relationships for analogs of the tuberculosis drug bedaquiline with the naphthalene unit replaced by bicyclic heterocycles. Bioorg Med Chem 26:1797–1809. doi: 10.1016/j.bmc.2018.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blaser A, Sutherland HS, Tong AST, Choi PJ, Conole D, Franzblau SG, Cooper CB, Upton AM, Lotlikar M, Denny WA, Palmer BD. 2019. Structure-activity relationships for unit C pyridyl analogues of the tuberculosis drug bedaquiline. Bioorg Med Chem 27:1283–1291. doi: 10.1016/j.bmc.2019.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tantry SJ, Markad SD, Shinde V, Bhat J, Balakrishnan G, Gupta AK, Ambady A, Raichurkar A, Kedari C, Sharma S, Mudugal NV, Narayan A, Naveen Kumar CN, Nanduri R, Bharath S, Reddy J, Panduga V, Prabhakar KR, Kandaswamy K, Saralaya R, Kaur P, Dinesh N, Guptha S, Rich K, Murray D, Plant H, Preston M, Ashton H, Plant D, Walsh J, Alcock P, Naylor K, Collier M, Whiteaker J, McLaughlin RE, Mallya M, Panda M, Rudrapatna S, Ramachandran V, Shandil R, Sambandamurthy VK, Mdluli K, Cooper CB, Rubin H, Yano T, Iyer P, Narayanan S, Kavanagh S, Mukherjee K, Balasubramanian V, Hosagrahara VP, Solapure S, Ravishankar S, Hameed PS. 2017. Discovery of imidazo[1,2-a]pyridine ethers and squaramides as selective and potent inhibitors of mycobacterial adenosine triphosphate (ATP) synthesis. J Med Chem 60:1379–1399. doi: 10.1021/acs.jmedchem.6b01358. [DOI] [PubMed] [Google Scholar]
- 7.Kumar S, Mehra R, Sharma S, Bokolia NP, Raina D, Nargotra A, Singh PP, Khan IA. 2018. Screening of antitubercular compound library identifies novel ATP synthase inhibitors of Mycobacterium tuberculosis. Tuberculosis (Edinb) 108:56–63. doi: 10.1016/j.tube.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 8.Lu P, Lill H, Bald D. 2014. ATP synthase in mycobacteria: special features and implications for a function as drug target. Biochim Biophys Acta 1837:1208–1218. doi: 10.1016/j.bbabio.2014.01.022. [DOI] [PubMed] [Google Scholar]
- 9.Preiss L, Langer JD, Yildiz O, Eckhardt Strelau L, Guillemont JE, Koul A, Meier T. 2015. Structure of the mycobacterial ATP synthase Fo rotor ring in complex with the anti-TB drug bedaquiline. Sci Adv 1:e1500106. doi: 10.1126/sciadv.1500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamariah N, Ragunathan P, Shin J, Saw WG, Wong CF, Dick T, Grüber G. 2020. Unique structural and mechanistic properties of mycobacterial F-ATP synthases: implications for drug design. Prog Biophys Mol Biol 152:64–73. doi: 10.1016/j.pbiomolbio.2019.11.006. [DOI] [PubMed] [Google Scholar]
- 11.Haagsma AC, Driessen NN, Hahn MM, Lill H, Bald D. 2010. ATP synthase in slow- and fast-growing mycobacteria is active in ATP synthesis and blocked in ATP hydrolysis direction. FEMS Microbiol Lett 313:68–74. doi: 10.1111/j.1574-6968.2010.02123.x. [DOI] [PubMed] [Google Scholar]
- 12.Hotra A, Suter M, Biuković G, Ragunathan P, Kundu S, Dick T, Grüber G. 2016. Deletion of a unique loop in the mycobacterial F-ATP synthase γ subunit sheds light on its inhibitory role in ATP hydrolysis-driven H+ pumping. FEBS J 283:1947–1961. doi: 10.1111/febs.13715. [DOI] [PubMed] [Google Scholar]
- 13.Ragunathan P, Sielaff H, Sundararaman L, Biuković G, Manimekalai MSS, Singh D, Kundu S, Wohland T, Frasch W, Dick T, Grüber G. 2017. The uniqueness of subunit α of mycobacterial F-ATP synthases: an evolutionary variant for niche adaptation. J Biol Chem 292:11262–11279. doi: 10.1074/jbc.M117.784959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bogdanović N, Sundararaman L, Kamariah N, Tyagi A, Bhushan S, Ragunathan P, Shin J, Dick T, Grüber G. 2018. Structure and function of Mycobacterium-specific components of F-ATP synthase subunits alpha and epsilon. J Struct Biol 204:420–434. doi: 10.1016/j.jsb.2018.10.006. [DOI] [PubMed] [Google Scholar]
- 15.Joon S, Ragunathan P, Sundararaman L, Nartey W, Kundu S, Manimekalai MSS, Bogdanović N, Dick T, Grüber G. 2018. The NMR solution structure of Mycobacterium tuberculosis F-ATP synthase subunit epsilon provides new insight into energy coupling inside the rotary engine. FEBS J 285:1111–1128. doi: 10.1111/febs.14392. [DOI] [PubMed] [Google Scholar]
- 16.Wong CF, Lau AM, Harikishore A, Saw WG, Shin J, Ragunathan P, Bhushan S, Ngan SC, Sze SK, Bates RW, Dick T, Grüber G. 2020. A systematic assessment of mycobacterial F1-ATPase subunit epsilon's role in latent ATPase hydrolysis. FEBS J doi: 10.1111/febs.15440. [DOI] [PubMed] [Google Scholar]
- 17.Hotra A, Ragunathan P, Ng PS, Seankongsuk P, Harikishore A, Sarathy JP, Saw WG, Lakshmanan U, Sae-Lao P, Kalia NP, Shin J, Kalyanasundaram R, Anbarasu S, Parthasarathy K, Pradeep CN, Makhija H, Dröge P, Poulsen A, Tan JHL, Pethe K, Dick T, Bates RW, Grüber G. 2020. Discovery of a novel mycobacterial F-ATP synthase inhibitor and its potency in combination with diarylquinolines. Angew Chem Int Ed Engl 59:13295–13304. doi: 10.1002/anie.202002546. [DOI] [PubMed] [Google Scholar]
- 18.Zhang AT, Montgomery MG, Leslie AGW, Cook GM, Walker JE. 2019. The structure of the catalytic domain of the ATP synthase from Mycobacterium smegmatis is a target for developing antitubercular drugs. Proc Natl Acad Sci U S A 116:4206–4211. doi: 10.1073/pnas.1817615116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bashiri G, Rehan AM, Greenwood DR, Dickson JM, Baker EN. 2010. Metabolic engineering of cofactor F420 production in Mycobacterium smegmatis. PLoS One 5:e15803. doi: 10.1371/journal.pone.0015803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lötscher HR, deJong C, Capaldi RA. 1984. Interconversion of high and low adenosinetriphosphatase activity forms of Escherichia coli F1 by the detergent lauryldimethylamine oxide. Biochemistry 23:4140–4143. doi: 10.1021/bi00313a020. [DOI] [PubMed] [Google Scholar]
- 21.Ho J, Sielaff H, Nadeem A, Svanborg C, Grüber G. 2015. The molecular motor F-ATP synthase is targeted by the tumoricidal protein HAMLET. J Mol Biol 427:1866–1874. doi: 10.1016/j.jmb.2015.01.024. [DOI] [PubMed] [Google Scholar]
- 22.Montgomery MG, Gahura O, Leslie AGW, Zíková A, Walker JE. 2018. ATP synthase from Trypanosoma brucei has an elaborated canonical F1-domain and conventional catalytic sites. Proc Natl Acad Sci U S A 115:2102–2107. doi: 10.1073/pnas.1720940115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirono-Hara Y, Noji H, Nishiura M, Muneyuki E, Hara KY, Yasuda R, Kinosita K, Yoshida M. 2001. Pause and rotation of F1-ATPase during catalysis. Proc Natl Acad Sci U S A 98:13649–13654. doi: 10.1073/pnas.241365698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gledhill JR, Montgomery MG, Leslie AGW, Walker JE. 2007. How the regulatory protein, IF1, inhibits F1-ATPase from bovine mitochondria. Proc Natl Acad Sci U S A 104:15671–15676. doi: 10.1073/pnas.0707326104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.García-Trejo JJ, Zarco-Zavala M, Mendoza-Hoffmann F, Hernández-Luna E, Ortega R, Mendoza-Hernández G. 2016. The inhibitory mechanism of the ζ subunit of the F1FO-ATPase nanomotor of Paracoccus denitrificans and related α-proteobacteria. J Biol Chem 291:538–546. doi: 10.1074/jbc.M115.688143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cingolani G, Duncan TM. 2011. Structure of the ATP synthase catalytic complex (F(1)) from Escherichia coli in an autoinhibited conformation. Nat Struct Mol Biol 18:701–707. doi: 10.1038/nsmb.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shirakihara Y, Shiratori A, Tanikawa H, Nakasako M, Yoshida M, Suzuki T. 2015. Structure of a thermophilic F1-ATPase inhibited by an ε-subunit: deeper insight into the ε-inhibition mechanism. FEBS J 282:2895–2913. doi: 10.1111/febs.13329. [DOI] [PubMed] [Google Scholar]
- 28.Sobti M, Smits C, Wong AS, Ishmukhametov R, Stock D, Sandin S, Stewart AG. 2016. Cryo-EM structures of the autoinhibited E. coli ATP synthase in three rotational states. Elife 5:e21598. doi: 10.7554/eLife.21598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hahn A, Vonck J, Mills DJ, Meier T, Kuhlbrandt W. 2018. Structure, mechanism, and regulation of the chloroplast ATP synthase. Science 360:eaat4318. doi: 10.1126/science.aat4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.The UniProt Consortium. 2012. Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res 40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. 2009. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schrödinger Inc. 2017. The PyMOL molecular graphics system, version 2.0. https://www.schrodinger.com/pymol/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.