

Summary
Alzheimer's disease (AD) is a progressive neurodegenerative disorder of the brain and the most common form of dementia among the elderly. The single-cell RNA-sequencing (scRNA-Seq) and single-nucleus RNA-sequencing (snRNA-Seq) techniques are extremely useful for dissecting the function/dysfunction of highly heterogeneous cells in the brain at the single-cell level, and the corresponding data analyses can significantly improve our understanding of why particular cells are vulnerable in AD. We developed an integrated database named scREAD (single-cell RNA-Seq database for Alzheimer's disease), which is as far as we know the first database dedicated to the management of all the existing scRNA-Seq and snRNA-Seq data sets from the human postmortem brain tissue with AD and mouse models with AD pathology. scREAD provides comprehensive analysis results for 73 data sets from 10 brain regions, including control atlas construction, cell-type prediction, identification of differentially expressed genes, and identification of cell-type-specific regulons.
Subject Areas: Neuroscience, Bioinformatics, Biological Database
Graphical Abstract
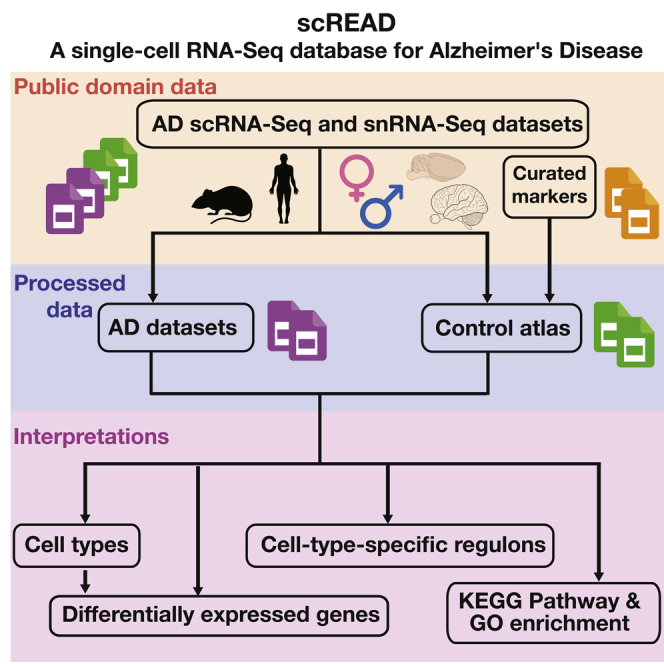
Highlights
-
•
First-of-its-kind database dedicated to Alzheimer's disease sc/snRNA-Seq data sets
-
•
Control atlas and disease data sets construction for major cell types in the brain
-
•
User-friendly web server to provide comprehensive analysis interpretations
Neuroscience; Bioinformatics; Biological Database
Introduction
Alzheimer's disease (AD) is the most common cause of dementia. Currently, there are an estimated 5.8 million Americans aged 65 yeas or older suffering from AD (Claxton et al., 2015). AD is a slowly progressive brain disease that only after years of brain changes do individuals experience noticeable symptoms, such as difficulty in remembering recent conversations, names or events, and language problems (Shinagawa, 2016). Symptoms occur because neurons in parts of the brain involved in thinking, learning, and memory have been damaged or destroyed, probably by the accumulation of amyloid beta (Aβ) protein and tau protein aggregates and the neuroinflammation (Dolgin, 2018; Mucke, 2009). Unfortunately, there is no effective therapeutics that can cure or alter the disease process (Gao et al., 2016). Furthermore, molecular mechanisms underlying AD, especially the cellular vulnerability, are poorly understood.
Single-cell RNA sequencing (scRNA-Seq) examines the dynamic transcriptomic profile of individual cells with next-generation sequencing technologies and hence provides a higher resolution of cellular differences and a better understanding of the function of an individual cell in the context of its microenvironment (Seweryn et al., 2020; Wang et al., 2020; Wu et al., 2014). For frozen brain samples, using the single-nucleus RNA sequencing (snRNA-Seq) is also an important strategy. It addresses these samples that cannot be readily dissociated into a single-cell suspension and minimizes the alteration of gene expression caused by the procedure of dissociation. Previous studies have demonstrated that AD pathology differs in age, gender, brain regions, and cell types (Ewers et al., 2011; Mucke, 2009; Sala Frigerio et al., 2019). In order to study the cellular heterogeneity of the brain and reveal the complex cellular changes in the AD brain by profiling tens of thousands of individual cells, scRNA-Seq provides an alternative method (Mathys et al., 2017). The scRNA-Seq can reveal complex and rare cell populations, uncover regulatory relationships between genes, and track the trajectories of distinct cell lineages in development (Grubman et al., 2019; Qi et al., 2020). The single-cell view of AD pathology paints a unique cellular-level view of transcriptional alterations associated with AD pathology and significantly improves our understanding of the pathogenesis of AD (Del-Aguila et al., 2019; Mathys et al., 2019).
Here, we developed a database called scREAD (single-cell RNA-Seq database for Alzheimer's disease), which provides comprehensive analysis results of all the existing scRNA-Seq and snRNA-Seq data sets collected from Gene Expression Omnibus (GEO) (Barrett et al., 2013) and Synapse databases. The scREAD has several key features, namely, (i) it is the first-of-its-kind database with a collection of all the 17 existing human and mouse AD scRNA-Seq and snRNA-Seq data sets from the public domain (Table S1); (ii) it re-defines the 17 data sets into 73 data sets, each of which corresponds to a specific species (human or mouse), gender (male or female), brain region (entorhinal cortex, prefrontal cortex, superior frontal gyrus, cortex, cerebellum, subventricular zone, superior parietal lobe, or hippocampus) (Table S2), disease or control, and age stage (7 months, 15 months, or 20 months for mice and 50–100+ years old for human) (Table S3); (iii) it provides comprehensive analysis results for each of the 73 data sets, including but not limited to the construction of control atlas, cell clustering, prediction of cell types, identification of differentially expressed genes (DEGs), and identification of cell-type-specific regulons (CTSRs) in support of the in-depth analysis of heterogeneous regulatory mechanisms; (iv) all these analysis results are visualized through a one-stop and user-friendly interface to free AD biologists from programming burdens (Data S1); (v) the backend workflow enabling all the above computational analyses is freely accessible as stand-alone one-line-command scripts in R (Data S2).
Results
Overview Functionalities of scREAD Database
There are four major functionalities in scREAD: (i) construction of control atlas for different human and mouse brain regions based on the 23 control data sets; (ii) identification of human and mouse disease cell types by projecting the AD data sets onto the control atlases; (iii) identification of DEGs for each cell type among different conditions and functional enrichment analysis of DEGs; and (iv) identification of CTSRs for each cell type among different conditions. These four functions and the schematic workflow of scREAD are shown in Figure 1.
Figure 1.

The Workflow of scREAD
Construction of Control Atlas for Different Human and Mouse Brain Regions
We constructed 23 human and mouse control cell atlases based on 17 scRNA-Seq and snRNA-Seq data sets, which cover 10 brain regions, two genders, and different mouse and human ages, totally 713,640 cells (Figure 2A and Transparent Methods). These 17 data sets were redefined into 73 data sets according to species, gender, brain region, disease or control, and age (Figure 2B). The number of cells and the statistical distribution of these 73 data sets are shown in Figures S1 and S2. Not all the data sets in scREAD are available to download for users; data sets from the GEO database are available to download, but data sets from Synapse are not available to download.
Figure 2.

General Information about scREAD Data sets
(A) General statistical distribution of all the 73 data sets. The pie charts represent four factors of distribution: species, control/disease condition, brain region, and gender from the left side to the right side, respectively. Each pie chart represents one factor, and each color in each pie chart represents one element, and the number represents the number of data sets for each element under each factor for 73 data sets.
(B) General information table on the homepage, which includes nine factors (species, gender, condition, region, Braak stage, age, mice model, GEO/synapse ID, and #cells).
Cell types of these 23 control atlases were assigned using Seurat (Stuart et al., 2019) and Semi-supervised Category Identification and Assignment (SCINA) (Zhang et al., 2019), and the known cell-type marker genes used in this process were collected from literature and PanglaoDB (Franzen et al., 2019) (Table S4 and Transparent Methods). scREAD contains eight major cell types of the human and mouse brain, i.e. astrocytes, endothelial cells, excitatory neurons, inhibitory neurons, microglia, oligodendrocytes, oligodendrocyte precursor cells, and pericytes. These 23 control atlases were then visualized using Uniform Manifold Approximation and Projection (UMAP) (Becht et al., 2018) and can be downloaded on the “Browse control atlas” page.
Identification of Human and Mouse Disease Cell Types Based on the Control Atlas
Not all cells collected from AD patient samples are malignant, and there are heterogeneous cells within individual patients, i.e., normal control cells are included. In Granja et al.’s research (Granja et al., 2019), they defined these control cells as control-like cells. Here, we applied this concept to AD data sets in our scREAD. These control cells maintain distinct regulatory mechanisms and gene expression patterns compared to AD cells, and they will disturb the accurate identification of AD cell types. Thus, we removed these control cells from AD data sets to identify real AD-associated cells. Using the human and mouse control atlas, we sought to project AD-associated cells onto the control atlas at single-cell resolution to identify human and mouse disease cell types (Transparent Methods). The general information is located at the top of the result page for retrieving an overview of data set description, source, and other data sets from the same experiment in scREAD (Figure 3A). The cell types and subclusters can be visualized interactively on the UMAP plot (Figure 3B) and can be exported in Portable Network Graphics (PNG) format by clicking on the “save” button at the right corner. The Adjusted Rand Index or silhouette score is also listed next to the UMAP plots for evaluating the clustering performance (see Transparent Methods) (Lovmar et al., 2005; Steinley et al., 2016). For each gene, the gene expression values are visualized interactively overlaid onto the same UMAP coordinates. For example, MBP is the marker gene of oligodendrocyte cell type, and it has higher expression in oligodendrocytes than in other cell types as expected (Figure 3C).
Figure 3.

Overall Information of an AD Disease Data set (AD00103) and UMAP Plot of the Cell Types and Expression Distribution of Gene MBP in This Data set
(A) Overall information on an AD disease data set (AD00103). It includes the information of species, brain region, condition, gender, age, number of control-like and AD-associated cells, data set source, and data sets from the same experiment.
(B) UMAP plot colored by cell type on this AD disease data set. We identified seven cell types, i.e. astrocytes, endothelial cells, excitatory neurons, inhibitory neurons, microglia, oligodendrocytes, and oligodendrocyte precursor cells.
(C) UMAP plot of expression distribution of oligodendrocyte marker gene MBP in the same data set. The darker the color is in this UMAP, the higher the expression value of the gene. Adj. p-value: wilcoxon rank sum test, Bonferroni corrected.
Transcriptional alterations seemed to stem from changes in cell state, with certain cell-type subpopulations more readily captured in AD pathology. To dissect disease-associated cellular subpopulations and cell-type heterogeneity, subclusters were identified for each cell type. For each cell type, we have carried out the subcluster finding analysis for investigating subcluster-specific changes and functional diversity occurring in AD. For different brain regions, enforcing an annotation to the closest cell type is likely to result in misannotation of such regions, but we are aware that subtypes could finely resolve and characterize this problem. Therefore, the subcluster function of our scREAD would provide a more comprehensive cell-type annotation considering cross-region heterogeneity. Due to no standard or consistent annotations available, we have not annotated those subclusters.
Differential Gene Expression and Functional Enrichment Analysis
Differential gene expression analyses include cell-type-specific genes, subcluster-specific genes, and cell-type-pairwise DEGs within one data set or between data sets based on diverse conditions (Table S5) (Monier et al., 2019). All the conditions are in the same species under the same gender, brain region, and age. The DEGs are presented based on the selections of the comparison group and the cell type of interest, and users can drag or type on the panel to apply different parameter cutoffs. The log2 fold-change can be adjusted ranging from 0 to 5, and the adjusted p value range can be adjusted from 10−6 to 1. All DEGs are scaled by cell types or conditions and are presented in the tables, allowing users to explore the differential expression of interesting genes across different conditions (Figure 4A).
Figure 4.

The DEGs and Functional Enrichment Analysis of Selected DEGs on an AD Disease Data set (AD00103)
(A) The DEG panel from the astrocytes cell type using the default parameters (left) and the DEG result table (right). Adjusted p-value: wilcoxon rank sum test, Bonferroni corrected.
(B) The functional enrichment analysis of DEGs. For each functional enrichment analysis, the top five most functional enrichment analysis results are shown. Adjusted p-value: hypergeometric test, Benjamini-Hochberg corrected.
Functional enrichment analysis is a computational method for inferring knowledge about an input gene set by comparing it to annotated gene sets representing prior biological knowledge. scREAD provides enrichment analysis of the DEGs against Kyoto Encyclopedia of Genes and Genomes pathways and Gene Ontology databases (Figure 4B) (Gene Ontology, 2015; Kanehisa and Goto, 2000). The enrichment analysis is performed and displayed in real time from the DEG list based on the input of the current DEG cutoffs. All of the DEG tables and functional enrichment tables are available to be downloaded by users.
Identification of CTSRs
CTSRs are defined as a group of genes, which receive similar regulatory signals in a specific cell type, hence tending to have similar expression patterns and share conserved motifs in this cell type (Wan et al., 2019; Yang et al., 2019). A successful elucidation of CTSRs will substantially improve the identification of transcriptionally co-regulated gene modules, realistically allowing reliable prediction of global transcription regulation networks encoded in a specific cell type (Ma et al., 2020b; Xie et al., 2020).
scREAD provides both the CTSR result table and the visualization detail information of each CTSR across each cell type for each data set (Figure 5). Taking an AD disease data set (AD00103) as an example, scREAD shows all the identified CTSRs in the table based on the index of cell types and allows users to download this table (Figure 5A). We also display an interactive visualization of all the CTSRs below the result table (Figure 5B). For CT3-R1 (the first regulon in cell type 3), this regulon includes 64 genes co-regulated by the same transcription factor (TF), MXI1. CT3-R1 is marked as a CTSR based on a significant regulon specificity score (RSS) of 0.77. Of all the 64 genes, 25 are differentially expressed in CT3 (marked with stars), according to the differential expression analysis using Seurat. Details of each gene and motif can be found by clicking on the gene name and TF logo, respectively. More detailed motif finding results including positions, sequences, and position weight matrix information can be found by clicking the “Open”. For each gene in this regulon, the UMAP plot of its expression value across all cell types can be achieved by clicking the “Display” button.
Figure 5.

The CTSR Result Table and Details of the CT3-R1 on an AD Disease Data set (AD00103)
(A) The result table of CTSRs on AD00103.
(B) The details of the top one CTSR of cell type three. (1) A regulon is named as CTn-Rm with n representing the index of cell type and m represents the regulon rank. (2) Asterisks indicate marker genes, that is, the differential expressed gene, identified in each cluster using Seurat. (3) Gene symbols and links to the GeneCards (Human) or the Mouse Genome Informatics (MGI) website. (4) Corresponding gene Ensembl ID columns link to the website. (5) Gene expression UMAP and comparison to the cell types. (6) The corresponding TF with a corresponding link to the HOCOMOCO database. (7) Detailed motif finding results from including positions, sequences, position weight matrix, etc. (8) Motif details linking to the TOMTOM database. Regulon p-value: wilcoxon rank sum test, Bonferroni corrected.
Discussion
In this paper, we described the first release of scREAD, which is as far as we know the first database that collects all existing human and mouse scRNA-Seq and snRNA-Seq data sets with AD pathology and provides a one-stop interactive visualization of the control atlas and analysis results based on these data sets. These data sets have been published and freely accessible in the public domain as of September 22nd, 2020. With the development and application of scRNA-Seq technology, scREAD will continue to be enriched and expanded to be a big database such as SC2disease (Zhao et al., 2020). Furthermore, scREAD allows users to submit a new data set through the submit page to reproduce all the analysis results showcased in scREAD in support of their AD research. We will ask for users' permission if we want to store the data uploaded by users into our database. We believe that our database will benefit the AD researchers particularly through studying the data and corresponding analysis results in scREAD.
scREAD provides comprehensive analysis results for those 73 scRNA-Seq data sets collected so far, including the construction of control cell atlas, cell clustering and subclustering, prediction of cell types, identification of DEGs, and identification of CTSRs. Based on the constructed control cell atlas, we can identify those AD-associated cells at specific brain regions and disease stages. Further analysis of the function/dysfunction of highly heterogeneous cells in the brain at the single-cell level via cell clustering and subclustering, as well as DEG and functional enrichment analysis, can help us understand subcluster-specific changes in the transcriptomic profile and functional diversity occurring in AD. The identification of CTSRs will substantially improve the reliable prediction of global transcription regulation networks encoded in a specific cell type. Thus, scREAD will greatly help the AD community by supporting the in-depth analysis of heterogeneous regulatory mechanisms in AD and identifying the potential therapeutic targets for the prevention and/or treatment of AD.
Limitations of the Study
Currently, scREAD only contains the scRNA-Seq and snRNA-Seq data sets as of September 22nd, 2020, and have not included other omics and spatial transcriptomics data. In the future, we will collect more AD scRNA-Seq and snRNA-Seq data from more brain regions and build up healthy atlas in diverse brain regions of human, mouse and extend to other species. Meanwhile, we will collect AD single-cell omics data, such as scATAC-seq and proteomics data, and achieve more comprehensive analysis results based on single-cell multiple omics data (Li et al., 2020; Ma et al., 2020a). In addition, spatial transcriptomics and in situ sequencing have been recently used in studying AD (Chen et al., 2020). The transcriptome-scale spatial gene expression data sets can further provide insights into answering the regional and cellular vulnerability in AD. Thus, we will specifically add the spatial transcriptomics and in situ sequencing data sets from human AD and AD-like animals to the current scREAD to enable more functional interpretation. Currently, scREAD only contains nine cell types across 10 human and mouse brain regions; we will provide a more comprehensive cell-type annotation considering more brain regions. In this study, we have only removed the control-like cells in the AD data sets. However, the individuals in the control group may be patients with potential AD, and thus, control samples might also have AD-like cells. Therefore, we will use the same strategy on the control data sets to construct control atlases in the future.
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Qin Ma (qin.ma@osumc.edu) or Hongjun Fu (hongjun.fu@osumc.edu).
Materials Availability
This study did not generate new unique data.
Data and Code Availability
All data sets used in this work are available from publicly available sources as cited in the manuscript. scREAD is a one-stop and user-friendly interface and freely available at https://bmbls.bmi.osumc.edu/scread/. The backend workflow can be downloaded from https://github.com/OSU-BMBL/scread/tree/master/script to enable more discovery-driven analyses.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by the National Institutes of Health [R01 GM131399-01, Q.M.; K01 AG056673, H.F.], the Department of Defense [W81XWH1910309, H.F.], the Alzheimer's Association [AARF-17-505009, H.F.], and the Neuroscience Research Institute Pilot Award and the Chronic Brain Injury Pilot Award from The Ohio State University [H.F.]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Science Foundation and the National Institutes of Health.
Author Contributions
Q.M. and H.F. designed the manuscript contents and experiments. J.J. contributed to data analysis and the initial draft. C.W. developed and implemented the database and the API. Q.R. tested the database and curated part of the data. All authors revised the final manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: November 20, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101769.
Contributor Information
Hongjun Fu, Email: hongjun.fu@osumc.edu.
Qin Ma, Email: qin.ma@osumc.edu.
Supplemental Information
References
- Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E., McInnes L., Healy J., Dutertre C.A., Kwok I.W.H., Ng L.G., Ginhoux F., Newell E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018;37:38–44. doi: 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- Chen W.T., Lu A., Craessaerts K., Pavie B., Sala Frigerio C., Corthout N., Qian X., Lalakova J., Kuhnemund M., Voytyuk I. Spatial transcriptomics and in situ sequencing to study Alzheimer's disease. Cell. 2020;182:976–991. doi: 10.1016/j.cell.2020.06.038. [DOI] [PubMed] [Google Scholar]
- Claxton A., Baker L.D., Hanson A., Trittschuh E.H., Cholerton B., Morgan A., Callaghan M., Arbuckle M., Behl C., Craft S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer's disease dementia. J. Alzheimer's Dis. 2015;44:897–906. doi: 10.3233/JAD-141791. [DOI] [PubMed] [Google Scholar]
- Del-Aguila J.L., Li Z., Dube U., Mihindukulasuriya K.A., Budde J.P., Fernandez M.V., Ibanez L., Bradley J., Wang F., Bergmann K. A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimer's Res. Ther. 2019;11:71. doi: 10.1186/s13195-019-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolgin E. Alzheimer's disease is getting easier to spot. Nature. 2018;559:S10–S12. doi: 10.1038/d41586-018-05721-w. [DOI] [PubMed] [Google Scholar]
- Ewers M., Sperling R.A., Klunk W.E., Weiner M.W., Hampel H. Neuroimaging markers for the prediction and early diagnosis of Alzheimer's disease dementia. Trends Neurosciences. 2011;34:430–442. doi: 10.1016/j.tins.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzen O., Gan L.M., Bjorkegren J.L.M. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database. 2019;2019:baz046. doi: 10.1093/database/baz046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L.B., Yu X.F., Chen Q., Zhou D. Alzheimer's Disease therapeutics: current and future therapies. Minerva Med. 2016;107:108–113. [PubMed] [Google Scholar]
- Gene Ontology C. Gene Ontology consortium: going forward. Nucleic Acids Res. 2015;43:D1049–D1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granja J.M., Klemm S., McGinnis L.M., Kathiria A.S., Mezger A., Corces M.R., Parks B., Gars E., Liedtke M., Zheng G.X.Y. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat. Biotechnol. 2019;37:1458–1465. doi: 10.1038/s41587-019-0332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubman A., Chew G., Ouyang J.F., Sun G., Choo X.Y., McLean C., Simmons R.K., Buckberry S., Vargas-Landin D.B., Poppe D. A single-cell atlas of entorhinal cortex from individuals with Alzheimer's disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019;22:2087–2097. doi: 10.1038/s41593-019-0539-4. [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Ma A., Mathe E.A., Li L., Liu B., Ma Q. Elucidation of biological networks across complex diseases using single-cell omics. Trends Genetics. 2020 doi: 10.1016/j.tig.2020.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovmar L., Ahlford A., Jonsson M., Syvanen A.C. Silhouette scores for assessment of SNP genotype clusters. BMC Genomics. 2005;6:35. doi: 10.1186/1471-2164-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A., McDermaid A., Xu J., Chang Y., Ma Q. Integrative methods and practical challenges for single-cell multi-omics. Trends Biotechnol. 2020;38:1007–1022. doi: 10.1016/j.tibtech.2020.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A., Wang C., Chang Y., Brennan F.H., McDermaid A., Liu B., Zhang C., Popovich P.G., Ma Q. IRIS3: integrated cell-type-specific regulon inference server from single-cell RNA-Seq. Nucleic Acids Res. 2020;48:W275–W286. doi: 10.1093/nar/gkaa394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H., Adaikkan C., Gao F., Young J.Z., Manet E., Hemberg M., De Jager P.L., Ransohoff R.M., Regev A., Tsai L.H. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. 2017;21:366–380. doi: 10.1016/j.celrep.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H., Davila-Velderrain J., Peng Z., Gao F., Mohammadi S., Young J.Z., Menon M., He L., Abdurrob F., Jiang X. Single-cell transcriptomic analysis of Alzheimer's disease. Nature. 2019;570:332–337. doi: 10.1038/s41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monier B., McDermaid A., Wang C., Zhao J., Miller A., Fennell A., Ma Q. IRIS-EDA: an integrated RNA-Seq interpretation system for gene expression data analysis. PLoS Comput. Biol. 2019;15:e1006792. doi: 10.1371/journal.pcbi.1006792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L. Neuroscience: Alzheimer's disease. Nature. 2009;461:895–897. doi: 10.1038/461895a. [DOI] [PubMed] [Google Scholar]
- Qi R., Ma A., Ma Q., Zou Q. Clustering and classification methods for single-cell RNA-sequencing data. Brief. Bioinform. 2020;21:1196–1208. doi: 10.1093/bib/bbz062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala Frigerio C., Wolfs L., Fattorelli N., Thrupp N., Voytyuk I., Schmidt I., Mancuso R., Chen W.T., Woodbury M.E., Srivastava G. The major risk factors for Alzheimer's disease: age, sex, and genes modulate the microglia response to abeta Plaques. Cell Rep. 2019;27:1293–1306.e6. doi: 10.1016/j.celrep.2019.03.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seweryn M.T., Pietrzak M., Ma Q. Application of information theoretical approaches to assess diversity and similarity in single-cell transcriptomics. Comput. Struct. Biotechnol. J. 2020;18:1830–1837. doi: 10.1016/j.csbj.2020.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinagawa S. [Language symptoms of Alzheimer's disease] Brain and nerve = Shinkei kenkyu no shinpo. 2016;68:551–557. doi: 10.11477/mf.1416200437. [DOI] [PubMed] [Google Scholar]
- Steinley D., Brusco M.J., Hubert L. The variance of the adjusted Rand index. Psychol. Methods. 2016;21:261–272. doi: 10.1037/met0000049. [DOI] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902 e1821. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan C., Chang W., Zhang Y., Shah F., Lu X., Zang Y., Zhang A., Cao S., Fishel M.L., Ma Q. LTMG: a novel statistical modeling of transcriptional expression states in single-cell RNA-Seq data. Nucleic Acids Res. 2019;47:e111. doi: 10.1093/nar/gkz655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Ma A., Chang Y., Gong J., Jiang Y., Fu H., Wang C., Qi R., Ma Q., Xu D. scGNN: a novel graph neural network framework for single-cell RNA-Seq analyses. bioRxiv. 2020 doi: 10.1038/s41467-021-22197-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A.R., Neff N.F., Kalisky T., Dalerba P., Treutlein B., Rothenberg M.E., Mburu F.M., Mantalas G.L., Sim S., Clarke M.F. Quantitative assessment of single-cell RNA-sequencing methods. Nat. Methods. 2014;11:41–46. doi: 10.1038/nmeth.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J., Ma A., Zhang Y., Liu B., Cao S., Wang C., Xu J., Zhang C., Ma Q. QUBIC2: a novel and robust biclustering algorithm for analyses and interpretation of large-scale RNA-Seq data. Bioinformatics. 2020;36:1143–1149. doi: 10.1093/bioinformatics/btz692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Ma A., Hoppe A.D., Wang C., Li Y., Zhang C., Wang Y., Liu B., Ma Q. Prediction of regulatory motifs from human Chip-sequencing data using a deep learning framework. Nucleic Acids Res. 2019;47:7809–7824. doi: 10.1093/nar/gkz672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Luo D., Zhong X., Choi J.H., Ma Y., Wang S., Mahrt E., Guo W., Stawiski E.W., Modrusan Z. SCINA: a semi-supervised subtyping algorithm of single cells and bulk samples. Genes. 2019;10:531. doi: 10.3390/genes10070531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T., Lyu S., Lu G., Juan L., Zeng X., Wei Z., Hao J., Peng J. SC2disease: a manually curated database of single-cell transcriptome for human diseases. Nucleic Acids Res. 2020:gkaa838. doi: 10.1093/nar/gkaa838. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data sets used in this work are available from publicly available sources as cited in the manuscript. scREAD is a one-stop and user-friendly interface and freely available at https://bmbls.bmi.osumc.edu/scread/. The backend workflow can be downloaded from https://github.com/OSU-BMBL/scread/tree/master/script to enable more discovery-driven analyses.