

Abstract

Herein we present the catalytic activation of N2O at a BiI⇄BiIII redox platform. The activation of such a kinetically inert molecule was achieved by the use of bismuthinidene catalysts, aided by HBpin as reducing agent. The protocol features remarkably mild conditions (25 °C, 1 bar N2O), together with high turnover numbers (TON, up to 6700) and turnover frequencies (TOF). Analysis of the elementary steps enabled structural characterization of catalytically relevant intermediates after O-insertion, namely a rare arylbismuth oxo dimer and a unique monomeric arylbismuth hydroxide. This protocol represents a distinctive example of a main-group redox cycling for the catalytic activation of N2O.
Nitrous oxide (N2O) is known to be a potent greenhouse gas, with much greater warming potential than CO2.1 The concentration of this gaseous molecule in the atmosphere has significantly increased in the modern era as a result of human activities, thus generating an environmental threat.1b In this sense, the development of catalytic strategies for the decomposition of N2O has recently drawn much attention.2 From the chemical standpoint, N2O is a thermodynamically powerful O atom transfer reagent, and indeed, Nature has evolved a powerful enzymatic pathway for converting N2O into N2 in microbial denitrification processes.3 Yet, N2O is kinetically inert, which poses challenges for its catalytic activation with artificial systems.2c In contrast to heterogeneous catalysts,2a,2b,2d a handful of examples are known to be capable of activating N2O based on homogeneous catalysts (Figure 1A),2e mainly consisting of transition metals (Ru,4 Rh,5 Co,6 and others7).
Figure 1.

(A) Homogeneous N2O activation at centers of elements: red (stoichiometric), blue (catalytic). (B) Catalytic N2O deoxygenation at a Bi(I)⇄Bi(III) redox platform.
With the aim of emulating transition-metal-like reactivity, a growing number of organo-main-group compounds have been shown to activate small molecules.8 Among them, many low-valent p-block compounds circumvented the kinetic barrier for N2O activation and formed structurally unique O-containing compounds.9 Additionally, frustrated Lewis pairs (FLPs)10 and N-heterocyclic carbenes (NHCs)11 have also been shown to capture N2O and eventually cleave the N–O bond. Recently, degradation of N2O with disilanes initiated by a fluoride anion has also been achieved,12 representing alternative pathways based on main-group systems. However, despite these precedents, catalytic redox processes for the activation of N2O by a main group compound still remain elusive.
Based on earlier precedents, activation of N2O could be facilitated by low-valent main group species, through the formation of an M–O bond with release of N2.9 If catalytic turnover is to be achieved, such M–O species should be reduced satisfactorily back to the starting low-valent main group catalyst. Although this process is well-established for high-valent oxo compounds,13 access to low-valent counterparts is nontrivial. Nevertheless, bismuth presents itself as an interesting candidate to address such a challenging idea, due to its demonstrated ability to access various oxidation states.14 One-electron BiII⇄BiIII has been described by Coles15 and recently by Lichtenberg,16 whereas, two-electron Bi redox catalysis (BiI⇄BiIII and BiIII⇄BiV) has recently been reported by us.17−19 We have previously demonstrated that the N,C,N-chelated bismuthinidene complexes, originally reported by Dostál,20 provide a privileged platform for BiI⇄BiIII redox cycling. Herein, we demonstrate that N,C,N-chelated bismuthinidenes are able to catalyze N2O deoxygenation in the presence of pinacolborane (HBpin) (Figure 1B). The catalytic system features the activation of N2O at remarkably mild conditions (25 °C, 1 bar) with high TON (up to 6700) and TOF. Ligand design and structural analysis on the bismuthinidene catalysts enabled the full characterization of key BiIII–oxo intermediates by NMR, X-ray, and HRMS.
To interrogate the reactivity of bismuthinidenes with N2O, we initially subjected complex 1 to a N2O atmosphere (1 bar) in THF-d8 at −78 °C (Scheme 1). The green solution slowly turned pale yellow with concomitant evolution of gas. Analysis of the head space by GC-TCD identified the formation of N2 during the reaction.211H NMR analysis at −40 °C revealed complete consumption of 1 after 45 min and the formation of a major species containing intact N,C,N-ligand scaffold and a C–Bi bond.21 ESI-HRMS analysis of this mixture clearly suggested the formation of dimeric arylbismuth oxides [(ArBiO)2+H+, calcd 937.33012, found 937.33070]. Such species was found to be dynamic in solution and thermally unstable, preventing its characterization by crystallographic techniques. The observed behavior for this species is consistent with other related Ar–BiIII oxo or sulfido dimers.22−24
Scheme 1. Oxidation of Bismuthinidene 1 with N2O.

In order to shed light on the possible structure of this species, the tBu on imines was replaced with m-terphenyl (m-Tp, 4 and 5, Figure 2A), which has been previously utilized to stabilize reactive organobismuth compounds such as Bi–H,25 Bi=Bi,26 and Bi–OH.27 Due to the sensitivity of aryl-(ket)imines to metal hydrides,28 we developed a facile and scalable procedure to obtain 4 and 5: reduction of the parent arylbismuth dichlorides 2 and 3 with Cp2Co afforded 4 and 5 after simple filtration as dark purple and red-purple solids respectively in very high yields (Figure 2A).21 X-ray crystallography revealed that, in spite of the steric bulkiness of the m-Tp groups, the bond lengths and angles resemble those reported for 1 and related ketimine-N,C,N-complexes of bismuth (Figure 2B).20
Figure 2.

(A) Preparation of bismuthinidenes 4 and 5. (B) ORTEP drawing of 4 and 5, with ellipsoids drawn at the 50% probability level. H atoms of 4 and 5 as well as distortions of 4 are omitted for clarity. Ar = m-Tp. Selected bond lengths (Å): for 4, Bi1–C1 2.1487(19), Bi1–N1 2.4601(15), Bi1–N2 2.5066(15), N1–C7 1.301(3), N2–C8 1.300(2); for 5, Bi1–C1 2.1503(18), Bi1–N1 2.4621(15), Bi1–N2 2.4552(16), N1–C7 1.301(2), N2–C9 1.305(2).
When 4 was exposed to a N2O atmosphere at room temperature, the color slowly changed from dark purple to pale yellow and evolution of N2 was observed (Figure 3A). 1H NMR analysis at −50 °C confirmed the full consumption of 4 after 40 min and indicated the formation of a single species with an asymmetric N,C,N-pincer backbone. Crystals suitable for X-ray crystallography were obtained by slow diffusion of n-pentane into a concentrated toluene solution of the reaction mixture at −78 °C. The crystal structure unequivocally determined the presence of the dimeric mono-organobismuth oxide 6, which features two μ-oxo bridge moieties (Figure 3B).
Figure 3.

(A) Oxidation of bismuthinidine 4 with N2O. (B) ORTEP drawing of 6, with ellipsoids drawn at the 50% probability level. H atoms, the toluene molecules and the enantiomer of 6 in the unit cell are omitted for clarity. Selected bond lengths (Å) and angles (deg): Bi1–C1 2.300(5), Bi1–O1 2.103(3), Bi1–O2 2.120(3), Bi1–N1 2.672(4), O1–Bi1–O2 79.70(13). (C) Dynamic imine coordination.21
Examples of mono-organobismuth(III) oxides are rare.22,23,29 Due to the high polarity of the Bi–O bond and the large difference in orbital size between Bi and O, these oxides readily undergo dimerization or polymerization.30 To the best of our knowledge, only two crystal structures of dimeric mono-organobismuth oxides have been reported: a syn-23 and an anti-isomer.22,31 Here, 6 represents an anti-isomer with a slightly asymmetric Bi2O2 core (Figure 3B). As a result of the weak coordination of N1 to Bi1 [Bi1–N1, 2.672(4) Å], the Bi1–O2 distance [2.120(3) Å] is marginally longer than Bi1–O1 [2.103(3) Å]. Interestingly, one m-Tp group in each half of the complex points away from the central Bi. Although H27 could not be refined unambiguously, the short C27–O2 distance (3.104 Å) strongly indicated a hydrogen bonding between H27 (and its symmetric H) and O2. 1H NMR at −50 °C reveals dramatically different chemical shifts for both imines (8.11 and 10.04 ppm), thus endorsing the hydrogen-bonding proposed. Yet, the dynamic imine coordination was indicated by the exchange peaks in ROESY-NMR and convergence of these imine peaks at higher temperatures as shown in VT-NMR data (Figure 3C). On the other hand, DOSY-NMR experiments suggested that the Bi2O2 ring of 6 was preserved in solution and no dissociation occurred.21 The structure of 6 suggests that similar species are formed when 1 is oxidized with N2O (Scheme 1).
At this point, we speculated that the dimeric nature of 6 could be the result of a rapid dimerization of a monomeric terminal Ar–Bi=O compound. Based on previous examples,32 we speculated that replacement of the imines with ketimines would favor the isolation of a monomeric species via tautomerization processes. To entertain this hypothesis, 5 was subjected to N2O in THF-d8 (Figure 4A). Similar to 6, 1H NMR of the resulting orange-red solution indicated the formation of one single species. X-ray crystallography unequivocally determined that 7 was a monomeric organobismuth hydroxide (Figure 4B). The high quality of the crystals allowed the unambiguous assignment of the positions of the H1, H8a, H8b, and H10 (3 H). One of the Me groups in the ketimines converted into a CH2, resulting in a reduction of the C–C length in C7–C8 [1.3552(16) Å], consistent with a double bond.33 The longer C7–N1 and Bi1–N2 distances [1.3832(15) and 2.6117(9) Å] compared to C9–N2 and Bi1–N1 [1.2848(14) and 2.2319(9) Å] also manifest the presence of an amido bond in one of the arms of the pincer.34 Interestingly, the OH points to a phenyl group of a m-Tp with a short H–phenyl centroid distance of 2.622 Å, indicative of a weak OH···π interaction.27 Due to the high tendency to form oxides or clusters through dehydration, reports on well-defined organobismuth hydroxides are limited;24a,27,35 yet, 7 is noticeably stable.
Figure 4.

(A) Oxidation of bismuthinidine 5 with N2O; (B) ORTEP drawing of 7, with ellipsoids drawn at the 50% probability level. H atoms except H1, H8s, and H10s and the enantiomer of 7 in the unit cell are omitted for clarity. Selected bond lengths (Å) and angles (deg): Bi1–C1 2.1869(11), Bi1–O1 2.0984(10), Bi1–N1 2.2319(9), Bi1–N2 2.6117(9), N1–C7 1.3832(15), N2–C9 1.2848(14), C7–C8 1.3552(16), C9–C10 1.4969(16), C1–Bi1–O1 94.68(4).
The monomeric compound 7 represents a tautomeric form of a monomeric Ar–Bi=O, an elusive species which has yet to be reported. In the same way, 6 can be conceived as the result of a fast dimerization process of two molecules of monomeric Ar–Bi=O. The formation of hydroxide 7 highlights the high basicity of the O atom in Ar–Bi=O, which could be better described as a polarized Bi=O bond: Ar–Bi+–O–. Therefore, it is reasonable to assume that both 6 and 7 are fingerprints for the transient generation of such elusive species (int-I, both resonance structures depicted; Scheme 2), which rapidly dimerizes or tautomerizes to the more stable compounds 6 and 7.
Scheme 2. Postulated Intermediates during Oxidation of Bi(I) with N2O.

Having identified the intermediacy of Bi–O bonds after N2O activation, we explored the reduction of 6 and 7 to Bi(I) to sustain a putative catalytic cycle. Among other uses,36 HBpin has been utilized as a deoxygenation agent for amine and phosphine oxides37 as well as for the catalytic reduction of CO2.38 Inspired by this reactivity, we treated 6 with 2.0 equiv of HBpin, which resulted in immediate formation of a dark purple solution (Scheme 3). Bismuthinidene 4 formed in 79% yield judging by 1H NMR. Similarly, the reduction of 7 gave 78% of 5. Meanwhile, ca. 1 equiv of HBpin was converted to a mixture of HO–Bpin (8) and (pinB)2O (9).39
Scheme 3. Reduction of 6 and 7 with HBpin.

Reaction conditions: 6 (17.8 μmol) or 7 (35.7 μmol), HBpin (71.4 μmol), mesitylene (35.7 μmol, 1.0 equiv) in 1.25 mL of THF-d8 at 25 °C.
At this point, we decided to merge this reactivity to unfold a catalytic system for the activation of N2O with Bi(I) compounds. Blank experiments demonstrated that no reaction occurs in the absence of Bi(I) (Table 1, entry 1). Catalytic N2O deoxygenation with HBpin proceeded smoothly at room temperature in the presence of 1 mol % of 4 or 5, with the TON reaching 54 and 89, respectively (entries 2 and 3). The higher efficiency of 5 over 4 could be ascribed to the higher stability of oxobismuth species 7 compared to 6. To our delight, when 1 was revisited as catalyst, a dramatic rate enhancement was observed; the reaction was complete in 3 min with vigorous release of N2 gas (entry 4). The high reactivity of 1 permitted lowering the catalyst loading to 0.01 mol % (entries 4 to 7). Unprecedentedly, the TON reached to 6700 at 0.01 mol % catalyst loading (entry 7). In addition, the TOF was estimated to be 52 min–1 at 0.1 mol % catalyst loading (entry 5).21 Since dimerization and tautomerization have already been shown to proceed really fast, we believe that species similar to 6 and 7 could probably be involved in the catalytic cycle. The mild conditions and high catalytic efficiency contrast with the elevated temperatures, high catalyst loadings, and/or prolonged reaction times usually required for transition metals.
Table 1. Bi(I)-Catalyzed N2O Deoxygenation with HBpin.

Based on HBpin.
Calculated by 1H NMR using mesitylene as internal standard.
Determined by disappearance of the characteristic color of Bi(I).
In conclusion, this work demonstrates the capacity of bismuthinidenes to catalytically activate N2O in a Bi(I)⇄Bi(III) redox platform. The synthesis of sterically congested bismuthinidenes using m-Tp substituents on the imines permitted isolation and characterization of catalytically relevant species such as bismuth oxide dimer 6 and bismuth hydroxide 7. Bis-imine and bis-ketimine N,C,N-chelated bismuthinidenes provide the first main-group redox platform for catalytic N2O decomposition. The ambient conditions and the very high catalytic efficiency make this system akin to transition-metal counterparts, unveiling an alternative opportunity for catalytic N2O transformations.
Acknowledgments
Financial support for this work was provided by Max-Planck-Gesellschaft, Max-Planck-Institut für Kohlenforschung and Fonds der Chemischen Industrie (FCI-VCI). This project has received funding from European Union’s Horizon 2020 research and innovation programme under Agreement No. 850496 (ERC Starting Grant, J.C.). We thank Prof. Dr. A. Fürstner for insightful discussions and generous support. We thank the MS, GC, and X-ray departments of Max-Planck-Institut für Kohlenforschung for analytic support. We thank Dr. R. Goddard for X-ray crystallographic analysis. Y.P. thanks CSC for a PhD scholarship.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c10092.
Experimental procedures and analytical data (1H, 13C, and 11B NMR, HRMS) for new compounds (PDF)
The authors declare no competing financial interest.
Notes
Crystallographic data for compounds 2–7 can be obtained free of charge from www.ccdc.cam.ac.uk under reference numbers 2031447, 2031446, 2031445, 2031448, 2031449, and 2031450, respectively.
Supplementary Material
References
- a Prather M. J. Time Scales in Atmospheric Chemistry: Coupled Perturbations to N2O, NOy and O3. Science 1998, 279, 1339–1341. 10.1126/science.279.5355.1339. [DOI] [PubMed] [Google Scholar]; b Hansen J.; Sato M. Greenhouse Gas Growth Rates. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 16109–16114. 10.1073/pnas.0406982101. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ravishankara A. R.; Daniel J. S.; Portmann R. W. Nitrous Oxide (N2O): The Dominant Ozone-Depleting Substance Emitted in the 21st Century. Science 2009, 326, 123–125. 10.1126/science.1176985. [DOI] [PubMed] [Google Scholar]; d Dameris M. Depletion of the Ozone Layer in the 21st Century. Angew. Chem., Int. Ed. 2010, 49, 489–491. 10.1002/anie.200906334. [DOI] [PubMed] [Google Scholar]
- For examples of stoichiometric coordination, activation and functionalization of N2O based on homogeneous and heterogeneous transition metal systems, see:; a Leont’ev A. V.; Fomicheva O. A.; Proskurnina M. V.; Zefirov N. S. Modern Chemistry of Nitrous Oxide. Russ. Chem. Rev. 2001, 70, 91–104. 10.1070/RC2001v070n02ABEH000631. [DOI] [Google Scholar]; b Parmon V. N.; Panov G. I.; Uriarte A.; Noskov A. S. Nitrous Oxide in Oxidation Chemistry and Catalysis: Application and Production. Catal. Today 2005, 100, 115–131. 10.1016/j.cattod.2004.12.012. [DOI] [Google Scholar]; c Tolman W. B. Binding and Activation of N2O at Transition-Metal Centers: Recent Mechanistic Insights. Angew. Chem., Int. Ed. 2010, 49, 1018–1024. 10.1002/anie.200905364. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Konsolakis M. Recent Advances on Nitrous Oxide (N2O) Decomposition over Non-Noble-Metal Oxide Catalysts: Catalytic Performance, Mechanistic Considerations, and Surface Chemistry Aspects. ACS Catal. 2015, 5, 6397–6421. 10.1021/acscatal.5b01605. [DOI] [Google Scholar]; e Severin K. Synthetic Chemistry with Nitrous Oxide. Chem. Soc. Rev. 2015, 44, 6375–6386. 10.1039/C5CS00339C. [DOI] [PubMed] [Google Scholar]; and references therein.
- Lehnert N.; Dong H. T.; Harland J. B.; Hunt A. P.; White C. J. Reversing Nitrogen Fixation. Nat. Rev. Chem. 2018, 2, 278–289. 10.1038/s41570-018-0041-7. [DOI] [Google Scholar]
- a Yamada T.; Hashimoto K.; Kitaichi Y.; Suzuki K.; Ikeno T. Nitrous Oxide Oxidation of Olefins Catalyzed by Ruthenium Porphyrin Complexes. Chem. Lett. 2001, 30, 268–269. 10.1246/cl.2001.268. [DOI] [Google Scholar]; b Zeng R.; Feller M.; Ben-David Y.; Milstein D. Hydrogenation and Hydrosilylation of Nitrous Oxide Homogeneously Catalyzed by a Metal Complex. J. Am. Chem. Soc. 2017, 139, 5720–5723. 10.1021/jacs.7b02124. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zeng R.; Feller M.; Diskin-Posner Y.; Shimon L. J. W.; Ben-David Y.; Milstein D. CO Oxidation by N2O Homogeneously Catalyzed by Ruthenium Hydride Pincer Complexes Indicating a New Mechanism. J. Am. Chem. Soc. 2018, 140, 7061–7064. 10.1021/jacs.8b03927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianetti T. L.; Annen S. P.; Santiso-Quinones G.; Reiher M.; Driess M.; Grützmacher H. Nitrous Oxide as a Hydrogen Acceptor for the Dehydrogenative Coupling of Alcohols. Angew. Chem., Int. Ed. 2016, 55, 1854–1858. 10.1002/anie.201509288. [DOI] [PubMed] [Google Scholar]
- a Yamamoto A.; Kitazume S.; Pu L. S.; Ikeda S. Synthesis and Properties of Hydridodinitrogentris(triphenylphosphine)cobalt(I) and the Related Phosphine-cobalt Complexes. J. Am. Chem. Soc. 1971, 93, 371–380. 10.1021/ja00731a012. [DOI] [Google Scholar]; b Gianetti T. L.; Rodríguez-Lugo R. E.; Harmer J. R.; Trincado M.; Vogt M.; Santiso-Quinones G.; Grützmacher H. Zero-Valent Amino-Olefin Cobalt Complexes as Catalysts for Oxygen Atom Transfer Reactions from Nitrous Oxide. Angew. Chem., Int. Ed. 2016, 55, 15323–15328. 10.1002/anie.201609173. [DOI] [PubMed] [Google Scholar]; c Corona T.; Company A. Nitrous Oxide Activation by a Cobalt(II) Complex for Aldehyde Oxidation under Mild Conditions. Dalton Trans. 2016, 45, 14530–14533. 10.1039/C6DT01704E. [DOI] [PubMed] [Google Scholar]
- a Yamada T.; Suzuki K.; Hashimoto K.; Ikeno T. N2O Oxidation of Phosphines Catalyzed by Low-Valent Nickel Complexes. Chem. Lett. 1999, 28, 1043–1044. 10.1246/cl.1999.1043. [DOI] [Google Scholar]; b Yonke B. L.; Reeds J. P.; Zavalij P. Y.; Sita L. R. Catalytic Degenerate and Nondegenerate Oxygen Atom Transfers Employing N2O and CO2 and a MII/MIV Cycle Mediated by Group 6 MIV Terminal Oxo Complexes. Angew. Chem., Int. Ed. 2011, 50, 12342–12346. 10.1002/anie.201106074. [DOI] [PubMed] [Google Scholar]; c Kiefer G.; Jeanbourquin L.; Severin K. Oxidative Coupling Reactions of Grignard Reagents with Nitrous Oxide. Angew. Chem., Int. Ed. 2013, 52, 6302–6305. 10.1002/anie.201302471. [DOI] [PubMed] [Google Scholar]; d Saito S.; Ohtake H.; Umezawa N.; Kobayashi Y.; Kato N.; Hirobe M.; Higuchi T. Nitrous Oxide Reduction-coupled Alkene-alkene Coupling Catalysed by Metalloporphyrins. Chem. Commun. 2013, 49, 8979–8981. 10.1039/c3cc43912g. [DOI] [PubMed] [Google Scholar]
- a Power P. P. Main-group Elements as Transition Metals. Nature 2010, 463, 171–177. 10.1038/nature08634. [DOI] [PubMed] [Google Scholar]; b Martin D.; Soleilhavoup M.; Bertrand G. Stable Singlet Carbenes as Mimics for Transition Metal Centers. Chem. Sci. 2011, 2, 389–399. 10.1039/C0SC00388C. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Chu T.; Nikonov G. I. Oxidative Addition and Reductive Elimination at Main-Group Element Centers. Chem. Rev. 2018, 118, 3608–3680. 10.1021/acs.chemrev.7b00572. [DOI] [PubMed] [Google Scholar]; d Weetman C.; Inoue S. The Road Travelled: After Main-Group Elements as Transition Metals. ChemCatChem 2018, 10, 4213–4228. 10.1002/cctc.201800963. [DOI] [Google Scholar]; e Melen R. L. Frontiers in Molecular p-block Chemistry: From Structure to Reactivity. Science 2019, 363, 479–484. 10.1126/science.aau5105. [DOI] [PubMed] [Google Scholar]
- a Xiong Y.; Yao S.; Driess M. Chemical Tricks To Stabilize Silanones and Their Heavier Homologues with E = O Bonds (E = Si-Pb): From Elusive Species to Isolable Building Blocks. Angew. Chem., Int. Ed. 2013, 52, 4302–4311. 10.1002/anie.201209766. [DOI] [PubMed] [Google Scholar]; b Zhong M.; Sinhababu S.; Roesky H. W. The Unique β-Diketiminate Ligand in Aluminum(I) and Gallium(I) Chemistry. Dalton Trans. 2020, 49, 1351–1364. 10.1039/C9DT04763H. [DOI] [PubMed] [Google Scholar]; c Hicks J.; Vasko P.; Goicoechea J. M.; Aldridge S.. The Aluminyl Anion: A New Generation of Aluminium Nucleophile. Angew. Chem., Int. Ed. 2020, 10.1002/anie.202007530. [DOI] [PubMed] [Google Scholar]; d Loh Y. K.; Aldridge S.. Acid-Base Free Main Group Carbonyl Analogues. Angew. Chem., Int. Ed. 2020, 10.1002/anie.202008174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Otten E.; Neu R. C.; Stephan D. W. Complexation of Nitrous Oxide by Frustrated Lewis Pairs. J. Am. Chem. Soc. 2009, 131, 9918–9919. 10.1021/ja904377v. [DOI] [PubMed] [Google Scholar]; b Neu R. C.; Otten E.; Stephan D. W. Bridging Binding Modes of Phosphine-Stabilized Nitrous Oxide to Zn(C6F5)2. Angew. Chem., Int. Ed. 2009, 48, 9709–9712. 10.1002/anie.200905650. [DOI] [PubMed] [Google Scholar]; c Kelly M. J.; Gilbert J.; Tirfoin R.; Aldridge S. Frustrated Lewis Pairs as Molecular Receptors: Colorimetric and Electrochemical Detection of Nitrous Oxide. Angew. Chem., Int. Ed. 2013, 52, 14094–14097. 10.1002/anie.201308475. [DOI] [PubMed] [Google Scholar]; d Ménard G.; Hatnean J. A.; Cowley H. J.; Lough A. J.; Rawson J. M.; Stephan D. W. C-H Bond Activation by Radical Ion Pairs Derived from R3P/Al(C6F5)3 Frustrated Lewis Pairs and N2O. J. Am. Chem. Soc. 2013, 135, 6446–6449. 10.1021/ja402964h. [DOI] [PubMed] [Google Scholar]; e Mo Z.; Kolychev E. L.; Rit A.; Campos J.; Niu H.; Aldridge S. Facile Reversibility by Design: Tuning Small Molecule Capture and Activation by Single Component Frustrated Lewis Pairs. J. Am. Chem. Soc. 2015, 137, 12227–12230. 10.1021/jacs.5b08614. [DOI] [PubMed] [Google Scholar]
- a Tskhovrebov A. G.; Solari E.; Wodrich M. D.; Scopelliti R.; Severin K. Covalent Capture of Nitrous Oxide by N-Heterocyclic Carbenes. Angew. Chem., Int. Ed. 2012, 51, 232–234. 10.1002/anie.201106589. [DOI] [PubMed] [Google Scholar]; b Tskhovrebov A. G.; Solari E.; Wodrich M. D.; Scopelliti R.; Severin K. Sequential N-O and N-N Bond Cleavage of N-Heterocyclic Carbene-Activated Nitrous Oxide with a Vanadium Complex. J. Am. Chem. Soc. 2012, 134, 1471–1473. 10.1021/ja210976a. [DOI] [PubMed] [Google Scholar]; c Tskhovrebov A. G.; Vuichoud B.; Solari E.; Scopelliti R.; Severin K. Adducts of Nitrous Oxide and N-Heterocyclic Carbenes: Syntheses, Structures, and Reactivity. J. Am. Chem. Soc. 2013, 135, 9486–9492. 10.1021/ja4030287. [DOI] [PubMed] [Google Scholar]; d Tskhovrebov A. G.; Naested L. C. E.; Solari E.; Scopelliti R.; Severin K. Synthesis of Azoimidazolium Dyes with Nitrous Oxide. Angew. Chem., Int. Ed. 2015, 54, 1289–1292. 10.1002/anie.201410067. [DOI] [PubMed] [Google Scholar]
- Anthore-Dalion L.; Nicolas E.; Cantat T. Catalytic Metal-free Deoxygenation of Nitrous Oxide with Disilanes. ACS Catal. 2019, 9, 11563–11567. 10.1021/acscatal.9b04434. [DOI] [Google Scholar]
- a O’Brien C. J.; Tellez J. L.; Nixon Z. S.; Kang L. J.; Carter A. L.; Kunkel S. R.; Przeworski K. C.; Chass G. A. Recycling the Waste: The Development of a Catalytic Wittig Reaction. Angew. Chem., Int. Ed. 2009, 48, 6836–6839. 10.1002/anie.200902525. [DOI] [PubMed] [Google Scholar]; b van Kalkeren H. A.; Leenders S. H. A. M.; Hommersom C. R. A.; Rutjes F. P. J. T.; van Delft F. L. In Situ Phosphine Oxide Reduction: A Catalytic Appel Reaction. Chem. - Eur. J. 2011, 17, 11290–11295. 10.1002/chem.201101563. [DOI] [PubMed] [Google Scholar]; c Buonomo J. A.; Aldrich C. C. Mitsunobu Reactions Catalytic in Phosphine and a Fully Catalytic System. Angew. Chem., Int. Ed. 2015, 54, 13041–13044. 10.1002/anie.201506263. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zhao W.; Yan P. K.; Radosevich A. T. A Phosphetane Catalyzes Deoxygenative Condensation of α-Keto Esters and Carboxylic Acids via PIII/PV-O Redox Cycling. J. Am. Chem. Soc. 2015, 137, 616–619. 10.1021/ja511889y. [DOI] [PubMed] [Google Scholar]; e Nykaza T. V.; Harrison T. S.; Ghosh A.; Putnik R. A.; Radosevich A. T. A Biphilic Phosphetane Catalyzes N-N Bond-Forming Cadogan Heterocyclization via PIII/PV-O Redox Cycling. J. Am. Chem. Soc. 2017, 139, 6839–6842. 10.1021/jacs.7b03260. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Nykaza T. V.; Cooper J. C.; Li G.; Mahieu N.; Ramirez A.; Luzung M. R.; Radosevich A. T. Intermolecular Reductive C-N Cross Coupling of Nitroarenes and Boronic Acids by PIII/PV-O Catalysis. J. Am. Chem. Soc. 2018, 140, 15200–15205. 10.1021/jacs.8b10769. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Nykaza T. V.; Ramirez A.; Harrison T. S.; Luzung M. R.; Radosevich A. T. Biphilic Organophosphorus-Catalyzed Intramolecular Csp2-H Amination: Evidence for a Nitrenoid in Catalytic Cadogan Cyclizations. J. Am. Chem. Soc. 2018, 140, 3103–3113. 10.1021/jacs.7b13803. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Ghosh A.; Lecomte M.; Kim-Lee S.-H.; Radosevich A. T. Organophosphorus-Catalyzed Deoxygenation of Sulfonyl Chlorides: Electrophilic (Fluoroalkyl)sulfenylation by PIII/PV=O Redox Cycling. Angew. Chem., Int. Ed. 2019, 58, 2864–2869. 10.1002/anie.201813919. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Longwitz L.; Werner T. Reduction of Activated Alkenes by PIII/PV Redox Cycling Catalysis. Angew. Chem., Int. Ed. 2020, 59, 2760–2763. 10.1002/anie.201912991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ruffell K.; Ball L. T. Organobismuth Redox Manifolds: Versatile Tools for Synthesis. Trends in Chemistry 2020, 2, 867–869. 10.1016/j.trechm.2020.07.008. [DOI] [Google Scholar]; b Kundu S. Pincer Type Ligand Assisted Catalysis and Small Molecules Activation by non-VSEPR Main-group Compounds. Chem. - Asian J. 2020, 15, 3209–3224. 10.1002/asia.202000800. [DOI] [PubMed] [Google Scholar]
- Schwamm R. J.; Lein M.; Coles M. P.; Fitchett C. M. Catalytic Oxidative Coupling Promoted by Bismuth TEMPOxide Complexes. Chem. Commun. 2018, 54, 916–919. 10.1039/C7CC08402A. [DOI] [PubMed] [Google Scholar]
- Ramler J.; Krummenacher I.; Lichtenberg C.. Well-defined, Molecular Bismuth Compounds: Catalysts in Photochemically-induced Radical Dehydrocoupling Reactions. Chem. Eur. J. 2020, 10.1002/chem.202002219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Planas O.; Cornella J. Bi(I)-Catalyzed Transfer-Hydrogenation with Ammonia-Borane. J. Am. Chem. Soc. 2019, 141, 4235–4240. 10.1021/jacs.9b00594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Planas O.; Wang F.; Leutzsch M.; Cornella J. Fluorination of Arylboronic Esters Enabled by Bismuth Redox Catalysis. Science 2020, 367, 313–317. 10.1126/science.aaz2258. [DOI] [PubMed] [Google Scholar]; b Planas O.; Peciukenas V.; Cornella J. Bismuth-Catalyzed Oxidative Coupling of Arylboronic Acids with Triflate and Nonaflate Salts. J. Am. Chem. Soc. 2020, 142, 11382–11387. 10.1021/jacs.0c05343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For an electrochemical H2 generation catalyzed by Dostal’s bismuthinidene, see:; Xiao W.-C.; Tao Y.-W.; Luo G.-G. Hydrogen Formation Using a Synthetic Heavier Main-group Bismuth-based Electrocatalyst. Int. J. Hydrogen Energy 2020, 45, 8177–8185. 10.1016/j.ijhydene.2020.01.152. [DOI] [Google Scholar]
- a Šimon P.; de Proft F.; Jambor R.; Růžička A.; Dostál L. Monomeric Organoantimony(I) and Organobismuth(I) Compounds Stabilized by an NCN Chelating Ligand: Syntheses and Structures. Angew. Chem., Int. Ed. 2010, 49, 5468–5471. 10.1002/anie.201002209. [DOI] [PubMed] [Google Scholar]; b Vránová I.; Alonso M.; Lo R.; Sedlák R.; Jambor R.; Růžička A.; Proft F. D.; Hobza P.; Dostál L. From Dibismuthenes to Three- and Two-Coordinated Bismuthinidenes by Fine Ligand Tuning: Evidence for Aromatic BiC3N Rings through a Combined Experimental and Theoretical Study. Chem. - Eur. J. 2015, 21, 16917–16928. 10.1002/chem.201502724. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for details.
- a Tokitoh N.; Arai Y.; Okazaki R.; Nagase S. Synthesis and Characterization of a Stable Dibismuthene: Evidence for a Bi-Bi Double Bond. Science 1997, 277, 78–80. 10.1126/science.277.5322.78. [DOI] [Google Scholar]; b Sasamori T.; Arai Y.; Takeda N.; Okazaki R.; Furukawa Y.; Kimura M.; Nagase S.; Tokitoh N. Syntheses, Structures and Properties of Kinetically Stabilized Distibenes and Dibismuthenes, Novel Doubly Bonded Systems between Heavier Group 15 Elements. Bull. Chem. Soc. Jpn. 2002, 75, 661–675. 10.1246/bcsj.75.661. [DOI] [Google Scholar]
- Strîmb G.; Pöllnitz A.; Raţ C. I.; Silvestru C. A General Route to Monoorganopnicogen(III) (M = Sb, Bi) Compounds with a Pincer (N,C,N) Group and Oxo Ligands. Dalton Trans. 2015, 44, 9927–9942. 10.1039/C5DT00603A. [DOI] [PubMed] [Google Scholar]
- a Breunig H. J.; Königsmann L.; Lork E.; Nema M.; Philipp N.; Silvestru C.; Soran A.; Varga R. A.; Wagner R. Hypervalent Organobismuth(III) Carbonate, Chalcogenides and Halides with the Pendant Arm Ligands 2-(Me2NCH2)C6H4 and 2,6-(Me2NCH2)2C6H3. Dalton Trans. 2008, 1831–1842. 10.1039/b717127g. [DOI] [PubMed] [Google Scholar]; b Chovancová M.; Jambor R.; Růžička A.; Jirásko R.; Císařová I.; Dostál L. Synthesis, Structure, and Reactivity of Intramolecularly Coordinated Organoantimony and Organobismuth Sulfides. Organometallics 2009, 28, 1934–1941. 10.1021/om801194h. [DOI] [Google Scholar]
- Hardman N. J.; Twamley B.; Power P. P. (2,6-Mes2H3C6)2BiH, a Stable, Molecular Hydride of a Main Group Element of the Sixth Period, and Its Conversion to the Dibismuthene (2,6-Mes2H3C6)BiBi(2,6-Mes2C6H3). Angew. Chem., Int. Ed. 2000, 39, 2771–2773. . [DOI] [PubMed] [Google Scholar]
- Twamley B.; Sofield C. D.; Olmstead M. M.; Power P. P. Homologous Series of Heavier Element Dipnictenes 2,6-Ar2H3C6E = EC6H3-2,6-Ar2 (E = P, As, Sb, Bi; Ar = Mes = C6H2-2,4,6-Me3; or Trip = C6H2-2,4,6-iPr3) Stabilized by m-Terphenyl Ligands. J. Am. Chem. Soc. 1999, 121, 3357–3367. 10.1021/ja983999n. [DOI] [Google Scholar]
- Breunig H. J.; Haddad N.; Lork E.; Mehring M.; Mügge C.; Nolde C.; Raţ C. I.; Schürmann M. Novel Sterically Congested Monoorganobismuth(III) Compounds: Synthesis, Structure, and Bismuth-Arene π Interaction in ArBiXY (X, Y = Br, I, OH, 2,6-Mes2-4-t-Bu-C6H2PHO2). Organometallics 2009, 28, 1202–1211. 10.1021/om800934c. [DOI] [Google Scholar]
- Vránová I.; Alonso M.; Jambor R.; Růžička A.; Turek J.; Dostál L. Different Products of the Reduction of (N),C,N-Chelated Antimony(III) Compounds: Competitive Formation of Monomeric Stibinidenes versus 1H-2,1-Benzazastiboles. Chem. - Eur. J. 2017, 23, 2340–2349. 10.1002/chem.201604142. [DOI] [PubMed] [Google Scholar]
- Organobismuth(III) Compounds. In Organobismuth Chemistry, 1st ed.; Suzuki H., Matano Y., Eds.; Elsevier Science: Amsterdam, 2001; pp 21–245. [Google Scholar]
- a Matano Y.; Nomura H. Dimeric Triarylbismuthane Oxide: A Novel Efficient Oxidant for the Conversion of Alcohols to Carbonyl Compounds. J. Am. Chem. Soc. 2001, 123, 6443–6444. 10.1021/ja010584k. [DOI] [PubMed] [Google Scholar]; b Matano Y.; Nomura H.; Hisanaga T.; Nakano H.; Shiro M.; Imahori H. Diverse Structures and Remarkable Oxidizing Ability of Triarylbismuthane Oxides. Comparative Study on the Structure and Reactivity of a Series of Triarylpnictogen Oxides. Organometallics 2004, 23, 5471–5480. 10.1021/om0494115. [DOI] [Google Scholar]
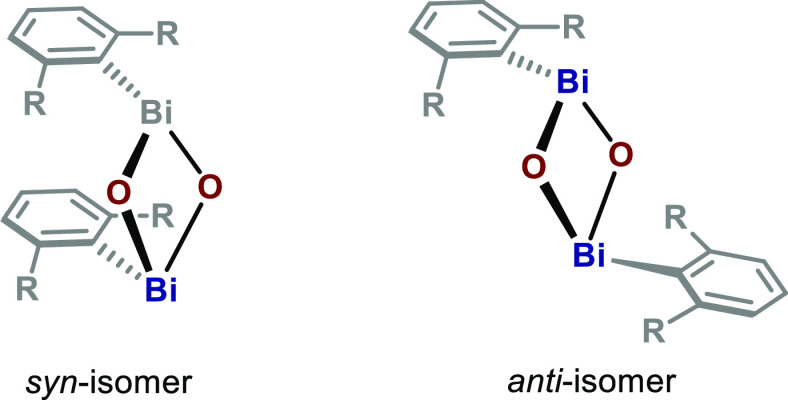
- The denomination of syn- and anti- for explaining the orientation of arylbismuth
oxo dimers is adopted from a previous report (ref (23)) on these types of structures.
A schematic explanation of this nomenclature is shown below.
- Wang Y.; Hu H.; Zhang J.; Cui C. Comparison of Anionic and Lewis Acid Stabilized N-Heterocyclic Oxoboranes: Their Facile Synthesis from a Borinic Acid. Angew. Chem., Int. Ed. 2011, 50, 2816–2819. 10.1002/anie.201007417. [DOI] [PubMed] [Google Scholar]
- Smith M. B.; March J.. March’s Advanced Organic Chemistry, 6th ed.; Wiley: Hoboken, NJ, 2007. [Google Scholar]
- a Pineda L. W.; Jancik V.; Nembenna S.; Roesky H. W. Synthetic and Structural Studies of Lead and Bismuth Organohalides Bearing a β-Diketiminato Ligand. Z. Anorg. Allg. Chem. 2007, 633, 2205–2209. 10.1002/zaac.200600325. [DOI] [Google Scholar]; b Knispel C.; Limberg C. C-H Bond Activation in a Molybdenumoxo-Bismuth Compound. Organometallics 2011, 30, 3701–3703. 10.1021/om2004223. [DOI] [Google Scholar]; c Vránová I.; Jambor R.; Růžička A.; Hoffmann A.; Herres-Pawlis S.; Dostál L. Antimony(III) and Bismuth(III) Amides Containing Pendant N-Donor Groups-a Combined Experimental and Theoretical Study. Dalton Trans. 2015, 44, 395–400. 10.1039/C4DT02692F. [DOI] [PubMed] [Google Scholar]; d Bresien J.; Hinz A.; Schulz A.; Villinger A. Trapping of Transient, Heavy Pnictogen-centred Biradicals. Dalton Trans. 2018, 47, 4433–4436. 10.1039/C8DT00487K. [DOI] [PubMed] [Google Scholar]; e Hanft A.; Lichtenberg C. Aminotroponiminates: Ligand-centred, Reversible Redox Events under Oxidative Conditions in Sodium and Bismuth Complexes. Dalton Trans. 2018, 47, 10578–10589. 10.1039/C8DT01019F. [DOI] [PubMed] [Google Scholar]; f Turner Z. R. Bismuth Pyridine Dipyrrolide Complexes: a Transient Bi(II) Species Which Ring Opens Cyclic Ethers. Inorg. Chem. 2019, 58, 14212–14227. 10.1021/acs.inorgchem.9b02314. [DOI] [PubMed] [Google Scholar]; g Kindervater M. B.; Hynes T.; Marczenko K. M.; Chitnis S. S.. Squeezing Bi: PNP and P2N3 Pincer Complexes of Bismuth. Dalton Trans. 2020, 10.1039/d0dt01413c. [DOI] [PubMed] [Google Scholar]
- a Battaglia L. P.; Bonamartini Corrradi A.; Pelizzi C.; Pelosi G.; Tarasconi P. Chemical and Structural Investigations on Bismuth Complexes of 2,6-Di-acetylpyridine Bis(2-thenoylhydrazone) and 2,6-Diacetylpyridine Bis(thiosemicarbazone). J. Chem. Soc., Dalton Trans. 1990, 3857–3860. 10.1039/dt9900003857. [DOI] [Google Scholar]; b Yin S. F.; Maruyama J.; Yamashita T.; Shimada S. Efficient Fixation of Carbon Dioxide by Hypervalent Organobismuth Oxide, Hydroxide, and Alkoxide. Angew. Chem., Int. Ed. 2008, 47, 6590–6593. 10.1002/anie.200802277. [DOI] [PubMed] [Google Scholar]; c Fridrichová A.; Svoboda T.; Jambor R.; Padělková Z.; Růžička A.; Erben M.; Jirásko R.; Dostál L. Synthesis and Structural Study on Organoantimony(III) and Organobismuth(III) Hydroxides Containing an NCN Pincer Type Ligand. Organometallics 2009, 28, 5522–5528. 10.1021/om900607n. [DOI] [Google Scholar]; d Roggan S.; Limberg C.; Ziemer B.; Siemons M.; Simon U. Reactivity and Properties of [-O-BiIII...O = Mo-]n Chains. Inorg. Chem. 2006, 45, 9020–9031. 10.1021/ic061198c. [DOI] [PubMed] [Google Scholar]
- Ramachandran P. V.; Chandra J. S.; Ros A.; Fernández R.; Lassaletta J. M.; Aggarwal V. K.; Blair D. J.; Myers E. L.. Pinacolborane. In Encyclopedia of Reagents for Organic Synthesis; Wiley: 2017. [Google Scholar]
- a Köster R.; Morita Y. Oxidation of Organoboranes with Amine Oxides. Angew. Chem., Int. Ed. Engl. 1966, 5, 580–580. 10.1002/anie.196605801. [DOI] [Google Scholar]; b Hawkeswood S.; Stephan D. W. Syntheses and Reactions of the Bis-boryloxide O(Bpin)2 (pin = O2C2Me4). Dalton Trans. 2005, 2182–2187. 10.1039/b504246a. [DOI] [PubMed] [Google Scholar]
- a Bontemps S.; Vendier L.; Sabo-Etienne S. Borane-Mediated Carbon Dioxide Reduction at Ruthenium: Formation of C1 and C2 Compounds. Angew. Chem., Int. Ed. 2012, 51, 1671–1674. 10.1002/anie.201107352. [DOI] [PubMed] [Google Scholar]; b Bontemps S.; Sabo-Etienne S. Trapping Formaldehyde in the Homogeneous Catalytic Reduction of Carbon Dioxide. Angew. Chem., Int. Ed. 2013, 52, 10253–10255. 10.1002/anie.201304025. [DOI] [PubMed] [Google Scholar]; c Bontemps S.; Vendier L.; Sabo-Etienne S. Ruthenium-Catalyzed Reduction of Carbon Dioxide to Formaldehyde. J. Am. Chem. Soc. 2014, 136, 4419–4425. 10.1021/ja500708w. [DOI] [PubMed] [Google Scholar]; d Bagherzadeh S.; Mankad N. P. Catalyst Control of Selectivity in CO2 Reduction Using a Tunable Heterobimetallic Effect. J. Am. Chem. Soc. 2015, 137, 10898–10901. 10.1021/jacs.5b05692. [DOI] [PubMed] [Google Scholar]
- HO-Bpin (8) reacted further with HBpin to yield (pinB)2O (9). See ref (38d) for an example of this reactivity.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.