

Abstract

In this article, we investigated the I2-promoted cyclic dialkyl ether formation from 6-membered oxanickelacycles originally reported by Hillhouse. A detailed mechanistic investigation based on spectroscopic and crystallographic analysis revealed that a putative reductive elimination to forge C(sp3)–OC(sp3) using I2 might not be operative. We isolated a paramagnetic bimetallic NiIII intermediate featuring a unique Ni2(OR)2 (OR = alkoxide) diamond-like core complemented by a μ-iodo bridge between the two Ni centers, which remains stable at low temperatures, thus permitting its characterization by NMR, EPR, X-ray, and HRMS. At higher temperatures (>−10 °C), such bimetallic intermediate thermally decomposes to afford large amounts of elimination products together with iodoalkanols. Observation of the latter suggests that a C(sp3)–I bond reductive elimination occurs preferentially to any other challenging C–O bond reductive elimination. Formation of cyclized THF rings is then believed to occur through cyclization of an alcohol/alkoxide to the recently forged C(sp3)–I bond. The results of this article indicate that the use of F+ oxidants permits the challenging C(sp3)–OC(sp3) bond formation at a high-valent nickel center to proceed in good yields while minimizing deleterious elimination reactions. Preliminary investigations suggest the involvement of a high-valent bimetallic NiIII intermediate which rapidly extrudes the C–O bond product at remarkably low temperatures. The new set of conditions permitted the elusive synthesis of diethyl ether through reductive elimination, a remarkable feature currently beyond the scope of Ni.
Introduction
Dialkyl ethers constitute one of the most valuable functional groups, and their synthesis represents one of the oldest strategies to build chemical complexity. As a result, formation of C–O bonds through the union of two organic fragments has prevailed one of the most powerful technologies, finding application across the chemical sciences: from covalent linkages and solid supports to crucial motifs in biologically active compounds.1 From a synthetic point of view, formation of the C–O bond has largely relied on the venerable Williamson ether synthesis,2 which involves the union of an alcohol and an alkyl halide through a SN2 reaction in the presence of a strong base (Scheme 1A). The high practicality and scalability of this transformation has placed it as a cornerstone reaction in both academic and industrial settings.3 Yet, the nucleophilic mechanism of the reaction is in turn its Achilles heel: the reaction efficiency is largely affected by the competitive alkoxide- or base-promoted E1 and E2 processes when secondary and tertiary alkyl halides are utilized. To circumvent these limitations, organic chemists have devoted their efforts in developing many strategies to produce highly coveted ethers—electrochemistry,4 organocatalysis,5 Lewis acid/base catalysis,6 among others. Nevertheless, one of the most promising alternatives in the literature to forge C–O bonds relies on the mediation of transition metals. Exploiting their redox properties, transition metal catalysis has been demonstrated to be one of the pillars in the construction of these linkages.7 For example, Chan-Lam or Ullmann couplings have experienced great success and found broad application for the synthesis of a myriad of highly relevant ethers.8 Despite the advantages associated with these methods, they have been largely dominated by linkages such as C(sp2)–O–C(sp2) and C(sp2)–O–C(sp3). In addition, Pd catalysis has also experienced tremendous development in this front, providing catalytic methods for C(sp2)–O bond formation.9−11 Yet, Pd complexes that permit construction of dialkylethers (C(sp3)–O–C(sp3)) through reductive elimination still present severe challenges.12 A remarkable example of the formation of cyclic ethers is the Co-catalyzed radical cyclization of alkenols from Mukaiyama,13 which has found ample success in various synthetic endeavors.13c
Scheme 1. (A) Williamson Ether Synthesis (Advantages and Pitfalls); (B) Existing Methods for C(sp2)–O Bond Formation Using Ni Catalysts; (C) C(sp3)–O–C(sp3) Bond Formation from High-Valent Ni.

Other first-row transition metals (Cu, Fe, Ni)14,15 have also been demonstrated to excel as catalysts in various C–O coupling strategies.16 In particular, Ni-catalyzed transformations have gained tremendous momentum for their enormous capabilities in forging C–heteroatom bonds.17 A seminal work by Hartwig described the possibility to forge C(sp2)–O–C(sp3) bonds using Ni(COD)2 and dppf as the optimal catalytic system.18 Encompassing Hartwig’s precedent, Stradiotto described a general C(sp2)–O bond formation from L2NiII complexes capitalizing on a newly designed set of phosphine-based ligands (L = CyPAd-DalPhos)19 (Scheme 1B). More recently, methods that replace the phosphine by simple diamine ligands have been reported, which rely on access to high-valent20 or high-energy21 Ni complexes through light irradiation, which rapidly forge the C(sp2)–O bond. Mechanistic investigations on these latter approaches revealed that C(sp2)–O bond formation can proceed either via a NiIII intermediate or via an excited NiII complex after energy transfer (Scheme 1B).22 In 2020, Ackermann and co-workers developed a nickel-catalyzed electrochemical C(sp2)–H alkoxylation, which proceeds through a NiIII intermediate.23 Recently, Nocera reported a NiI-catalyzed etherification protocol that mimics the reactivity of photoredox-catalyzed couplings without the use of a light source or photocatalyst.24 Similarly to the metallaphotoredox protocols, it is believed that the high oxidation state of the NiIII intermediate provides the necessary driving force to undergo C(sp2)–O bond linkage upon reductive elimination. Whereas realization of C(sp2)–O–C(sp3) through reductive elimination at a Ni center is well precedented and studied,25 a fundamental mechanistic understanding of the analogous process to forge dialkyl ethers through C(sp3)–OC(sp3) reductive elimination still remains elusive with virtually no systematic studies on their feasibility (Scheme 1C).26
In the 1990s, Hillhouse provided one of the first examples of C(sp3)–O–C(sp3) bond formation from well-defined oxanickelacycles (1a–e) bearing bipyridine ligands and using stoichiometric I2 as oxidant (Scheme 2A).27 In these seminal reports, involvement of high-valent nickel species such as NiIII–I or NiIV–I complexes was suggested; yet, minimal evidence was provided, and such intermediates remained purely speculative. Interestingly, when I2 was replaced by other oxidants such as O2 or ferrocenium (Fc+) the C(sp3)–OC(sp3) bond formation was not observed.27d In addition, higher yields were obtained for those complexes where β-hydride elimination is hampered by the limited conformations of the oxanickelacycles (Scheme 2A). As a result, formation of the C(sp3)–OC(sp3) bond was limited to cyclic products, as exemplified by the incapacity of 1f to deliver the corresponding acyclic dialkyl ether.27b On the basis of these early results, Love and co-workers capitalized on the I2-promoted C–O bond formation and applied it to the oxanickelacyclobutane 1g bearing a 1,2-bis(di-tert-butylphosphino)ethane (dtbpe) as the ligand (Scheme 2B).28 The authors observed rapid and clean formation of the corresponding epoxide in good yield along with almost quantitative formation of the corresponding (dtbpe)NiI2 (3). In this case, deleterious β-hydride elimination is not operative due to the presence of a ketone. To the best of our knowledge, these reports represent solitary examples present in the literature regarding formation of dialkyl ethers from well-defined organometallic species. Despite the powerful reactivity observed, no evidence of the intermediates involved has been reported. Yet, fundamental understanding of the key parameters that govern this particular transformation would provide tremendous insights for the design of future catalytic C(sp3)–O–C(sp3) ether syntheses. Herein, we report a comprehensive mechanistic study on the transformation originally described by Hillhouse: characterization of the reaction intermediates revealed formation of a robust and paramagnetic NiIII dimer, which thermally decomposes to afford primarily elimination products. Additional mechanistic data suggests that direct C(sp3)–OC(sp3) reductive elimination from such NiIII intermediate to forge simple THF rings is highly unlikely. On the contrary, experimental evidence supports an alternative mechanism based on a preferential C(sp3)–I reductive elimination followed by an intramolecular SN2 reaction. Yet, all of these drawbacks were overridden by the replacement of I2 by fluorine-containing oxidants which prevent not only competitive C–X reductive elimination but also deleterious elimination side reactions. In this manner, high yields of the cyclized tetrahydrofurans were obtained.
Scheme 2. I2-Promoted C(sp3)–O–C(sp3) Bond Formation: (A) Hillhouse’s Seminal Work with Bipyridine Oxanickelacycles; (B) Love’s Example Using Strained Oxanickelacycle with Bidentate Phosphine.

Results and Discussion
Initially, we considered that oxanickelacyles 1a and 1b provided an excellent platform to investigate an oxidative C(sp3)–OC(sp3) bond formation (Scheme 3). Both complexes represent challenging substrates to undergo intramolecular C–O bond formation (Scheme 2A) due to the dynamic behavior of the alkoxide anion as a result of the fluxionality of the alkyl backbone. Subsequently, diverse conformations of the CH and CH2 groups can be adopted which could lead to unproductive β-hydride elimination or simple E2. With these potential drawbacks in mind, we set out to synthesize nickelacyclopentanes 4a and 4b as described in the literature:27,29 the corresponding 1,4-alkyldibromides (1.0 equiv) were reacted with an excess of Ni(COD)2 (2.0 equiv) and bipyridine (bipy, 4.0 equiv) at −78 °C in THF. After warming up to 25 °C, the mixture is filtered and the dark-green complexes 4a and 4b are obtained in 69% and 50% yield, respectively (step 1, Scheme 3). Subsequently, 4a and 4b were subjected to O-atom insertion using N2O following Hillhouse’s procedure.27 After exposing 4a and 4b to N2O atmosphere (1 atm) in THF, oxanickelacyclohexane complexes 1a and 1b were obtained as deep purple solids in 53% and 75% yield, respectively (step 2, Scheme 3). It is important to mention that to access high-purity 1a and 1b, further filtration is required through Avicel30 in order to remove some colloidal nickel particles.31 Complexes 1a and 1b exhibit remarkable stability in the solid state and can be stored in the freezer of the glovebox. Yet, solutions of 1a and 1b slowly degrade, probably through decoordination of the alkoxide ligand, which complicates purification. Indeed, purities that ranged from 85% to 93% could be achieved for 1a and 1b after a series of crystallizations and filtrations. Such NiII complexes are square planar and easily characterized by NMR in the diamagnetic region.31 Whereas 1a offers inherent symmetry, 1b is not symmetric and different products can arise in the O-atom insertion step (step 2, Scheme 3). Compound 1b has both a 1° and 2° carbons, and the selectivity for the O insertion was found to be 85:15, favoring isomer 1b over 1b-isomer.32 However, considering that the reductive elimination of both isomers would lead to the same THF product 2b, no further separation was attempted. For clarity, only structure 1b will be used in the following schemes.
Scheme 3. Synthetic Route to Oxanickelacyclohexanes31.

This yield comprises a mixture of 85:15 of 1b and 1b-isomer.
With these complexes in hand, cyclic voltammetry studies were performed in order to gain insight into their redox properties. As shown in Figure 1, the cyclic voltammogram of complex 1b in CD3CN revealed two oxidative waves at −0.70 and +0.31 V against Fc/Fc+. Interestingly, the first oxidative wave is not reversible, whereas the second wave is quasi-reversible. On the basis of other similar CVs for well-defined cyclic (bipy)NiII(alkyl)(aryl) and (terpy)NiII(C4F8) complexes,33 we tentatively assigned the redox potentials to the corresponding single-electron oxidation NiII/NiIII and NiIII/NiIV couples, respectively. It is worth pointing out that the low values for both processes manifest the facility of 1b to access high-valent Ni intermediates.34
Figure 1.

Cyclic voltammogram of 1b (1.0 mM) in CD3CN, recorded versus Ag/AgNO3 electrode, using n-Bu4NPF6 (0.2 M) as electrolyte, under argon, with a scan rate of 100 mV·s–1. Potentials are then converted to the Fc/Fc+ couple.31
On the basis of the oxidation potential obtained for 1b (E1/2(NiII/NiIII) = −0.70 V, E1/2(NiIII/NiIV) = +0.31 V vs Fc/Fc+ in CD3CN), it is reasonable to propose that oxidation of this complex to the corresponding NiIII should be feasible using Fc+ or O2.27d However, Hillhouse already noticed that Fc+ and O2 did not lead to any C–O bond formation in good yield. Indeed, when 1b was oxidized with FcBF4, mainly the elimination product was obtained (6b) with only trace amounts of C(sp3)–OC(sp3) bond formation (Scheme 4). Other single-electron oxidants were tested, such as photocatalyst 4CzIPN (2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile (5),35 which would also lead to cationic NiIII intermediates. Yet, no desired THF ring 2b could be detected in the crude mixture. These results suggest that a C(sp3)–OC(sp3) reductive elimination from cationic NiIII intermediates is unlikely (vide infra).
Scheme 4. Attempts to C–O Bond Formation after One-Electron Oxidation of 1b To Access High-Valent NiIII.

To gain further insight into the C–O bond-forming event, the reaction reported by Hillhouse and co-workers was repeated: complex 1b (1.0 equiv) was reacted with I2 (1.1 equiv) in CD2Cl2 at 25 °C.36 To our surprise, upon addition of I2 to a solution of 1b, the color of the reaction mixture quickly changed from deep purple to orange. After 20 min of stirring at 25 °C, brown solids precipitated from the solution. At this point, the solids were filtered off and the filtrate was analyzed by 1H NMR and GC-MS. Surprisingly, the desired cyclic ether 2b was not observed (Scheme 5A). On the other hand, significant amounts of pent-4-en-2-ol (6b, 45%) were obtained, presumably through side-elimination reactions. More importantly, peaks relative to 5-iodopentan-2-ol (7b, 10%) were detected and successfully assigned.31 The presence of 7b was further confirmed by GC-MS. After solubilizing the solids, analysis by 1H NMR revealed a set of broad paramagnetic peaks, suggesting the presence of ligated bipy–Ni species. HRMS analysis of the solid indicated that a possible structure could be (bipy)NiI2,28 although dimeric compounds could not be ruled out.37 Slow crystallization in CD2Cl2 at −20 °C led to crystals suitable for X-ray analysis, which unequivocally confirmed formation of a polymeric (bipy)NiI2 (8) consisting of octahedral Ni complexes linked together by μ-iodo bridges (Scheme 5B).
Scheme 5. (A) Hillhouse I2-Promoted C(sp3)–O–C(sp3) Bond Formation Reaction (Analysis of the Fate of the Organic and Inorganic Compounds); (B) X-ray Structure of Complex 8;31 (C) NiIII Bimetallic Intermediate 9a–b; (D) 1H NMR of Paramagnetic Complex 9a(31).
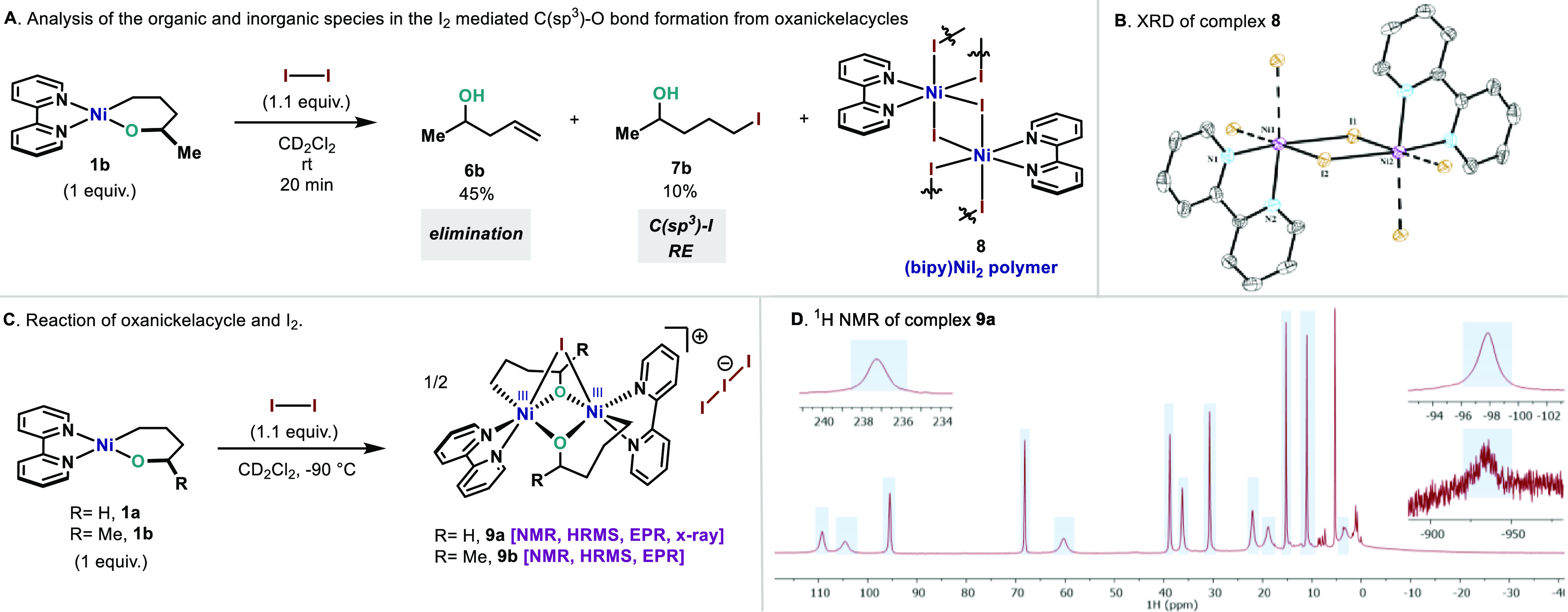
Disordered iodine atoms are omitted for clarity. Selected distances (Angstroms): Ni1–I1 = N2–I2 = 2.78; Ni1–I2 = Ni2–I1 = 2.83; Ni1–N1 = Ni1–N2 = 2.07. Selected angles (degrees): I1–Ni–I2 = 89.2; Ni1–I1–Ni2 = 94.2.
Insets correspond to peaks at 237, −98, and −930 ppm.
Considering the redox potential of iodine (E1/2(I2/I2–•) = +0.21 V vs Fc+/Fc),38 it is reasonable to assume formation of a high-valent nickel NiIII–I complex upon mixing 1a and 1b with I2. However, similarly to the case of Fc+, formation of NiIV is highly unlikely. To investigate whether NiIII–I intermediates are formed in the reaction system, we decided to monitor the reaction by 1H NMR at low temperature. Upon adding a cold solution of I2 (1.1 equiv) to a solution of 1a in a J-Young tube at −90 °C, a rapid color change was observed from deep purple to deep orange (Scheme 5C). Complete conversion of 1a to a new set of well-defined signals was observed. The 1H NMR spectrum at −90 °C exhibits peaks ranging from −930 to +237 ppm, pointing to a paramagnetic complex (Scheme 5D).31 Upon storing a concentrated solution at −35 °C, good-quality crystals formed. X-ray analysis unambiguously determined that the intermediate consisted of a symmetric cationic NiIII bimetallic complex (9a). This complex represents a unique and unprecedented structure of a dinuclear NiIII with several structural and electronic interesting features (Figure 2). First, 9a contains two NiIII atoms in an octahedral arrangement with a large Ni–Ni distance of 2.84 Å, thus suggesting no metal–metal interaction.39 In addition, 9a features a rather unique μ-iodo bridge unifying both Ni atoms with a highly strained Ni1–I1–Ni2 angle of 58.07°. As a result, the I is bridging both NiIII centers through an elongated Ni–I bond (Ni1–I1 = Ni2–I1, 2.92 Å). It is important to mention that NiIII dimers with one bridging halogen atom have been recently proposed as intermediates in C(sp2)–X (X = Br, Cl, I) bond formation.39a,40 However, the most striking feature of 9a is the diamond-like core formed by the Ni atoms and the alkoxide ligands. Two μ2-bridging alkoxide anions join the two Ni centers in a highly symmetric environment (Ni1–O1 = Ni1–O2 = Ni2–O1 = Ni2–O2, 2.00 Å) with angles of Ni1–O1–Ni2 = 92.72° and O1–Ni1–O2 = 79.57°. The Ni–O distances for 9a are in the range of other bis(μ2-oxide)-bridging NiIII complexes known in the literature.41 The complex is complemented by the bipyridine ligands with similar Ni–N distances for both N (Ni1–N1, 1.99 Å; Ni1–N2, 2.05 Å). Finally, the remaining position of the octahedron is occupied by the alkyl residue with Ni–C(sp3) distances resembling those reported for other NiIII–C(sp3) bonds (Ni1–C1, 2.013 Å).42 A parallel behavior was observed when complex 1b was reacted with I2. However, attempts to obtain crystals of the NiIII intermediate were unsuccessful. Bimetallic complexes 9a and 9b were further characterized by HRMS both in ESI+ and in ESI– modes.31 Finally, due to the paramagnetic nature of complexes 9a and 9b, further structural and electronic characterization was attempted by electron paramagnetic resonance (EPR).
Figure 2.
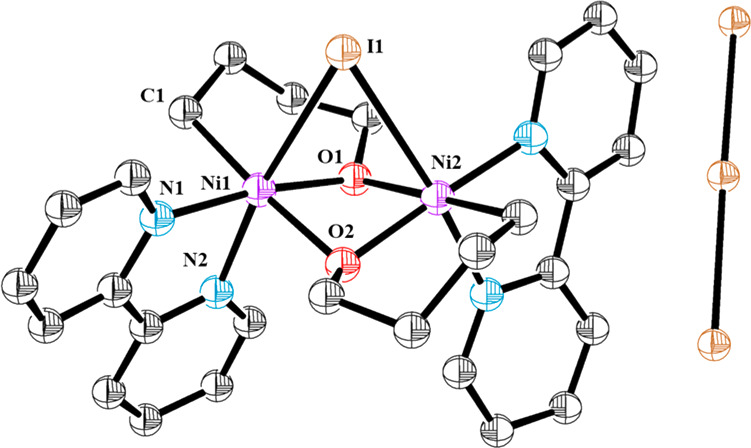
X-ray structure of compound 9a.31 Hydrogen atoms and disordered iodide atoms in I3– counterion are omitted for clarity. Selected distances (Angstroms): Ni1–Ni2 = 2.84; Ni1–I1 = Ni2–I1 = 2.92; Ni1–O1 = Ni1–O2 = Ni2–O1 = Ni2–O2 = 2.00; Ni1–N1 = 1.99; Ni1–N2 = 2.05; Ni1–C1 = 2.013. Selected angles (degrees): Ni1–I1–Ni2 = 58.07; Ni1–O1–Ni2 = 92.72; O1–Ni1–O2 = 79.57.
The EPR spectra of complexes 9a and 9b were recorded at the X-band, 30 K, and are depicted in Figure 3. As expected, the EPR spectra of 9a and 9b are very similar but show subtle differences in line splitting and intensity ratio between the different spectral features. The multiple line splitting suggests an electron–electron spin–spin interaction between the two NiIII centers. This means that the dimer structure found in the solid state (Figure 2) is retained in solution for both complexes. Making use of the symmetry properties of the dimer complex,31 we were able to simulate the EPR spectra as shown in Figure 3. The g-matrix principal values obtained for 9a (2.081, 2.155, 2.279) and 9b (2.084, 2.144, 2.287) are similar to what has been observed for a similar N,N-ligand-coordinated NiIII monomer complex (2.03, 2.14, 2.20).40 The magnetic interaction between the two NiIII centers is dominated by the dipolar contribution found to be (0.9, 1.1, −2)*550 MHz for 9a and (0.9, 1.1, −2)*517 MHz for 9b. The J coupling between the two NiIII centers is very small (50 MHz), and its effect on the EPR is only visible as a small splitting at the center of the spectrum. As confirmed by DFT analysis,31 the two NiIII centers effectively behave as isolated S = 1/2 systems. This is in agreement with NMR analysis, estimating the magnetic susceptibility of the dimer complex using the Evans method to be S = 1/2 for each NiIII center.31
Figure 3.
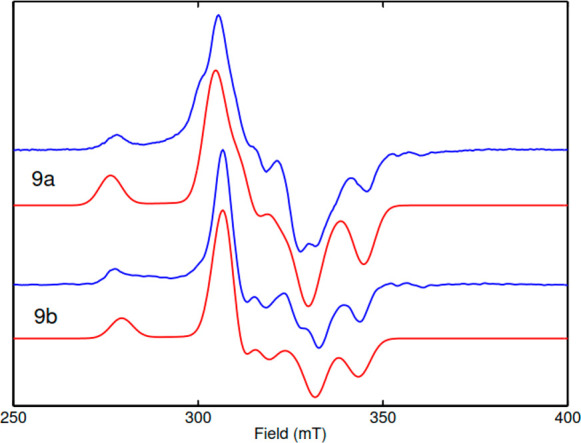
X-band EPR (9.623 GHz) of complexes 9a and 9b recorded at 30 K (blue traces).31 Experimental parameters: 1 mW, 100 kHz, 7.5 G field modulation. Red traces represent the Easyspin43 (esfit) simulation with the following parameters: g(9a) = (2.081, 2.155, 2.279); g(9b) = (2.084, 2.144, 2.287). Dipolar interaction D(9b) = 517 MHz; D(9a) = 550 MHz. J coupling < 50 MHz.
Having identified and characterized 9, we set out to explore its reactivity. Upon slowly warming solutions of 9a and 9b in CD2Cl2 from −90 to 25 °C, several interesting observations were made. First, the chemical shifts of complexes 9a and 9b are highly dependent on the temperature, which further confirms the paramagnetic nature of 9.31 Moreover, 9a and 9b have a remarkable stability across a wide range of temperatures, from −90 to −10 °C. Beyond −10 °C, rapid evolution of 9a and 9b to terminal alkenes 6a and 6b and iodoalcohols 7a and 7b is observed. While traces of THF could be detected in the case of 9a, no detectable amount of the C(sp3)–OC(sp3) bond formation product 2b was observed for 9b.44 This last observation indicates that the C(sp3)–I reductive elimination is kinetically more favorable to any other C–O bond-forming event at Ni. Whereas such C(sp3)–I bond formation proceeds via a reductive elimination from NiIII–I or direct attack of the I counterion to the Ni–C(sp3) bond in a SN2 fashion is currently unknown.42,45 However, a similar system was recently reported by Diao, suggesting that C(sp3)–I bond formation could proceed through monomeric square pyramid NiIII complexes.40 Hence, it is plausible to think that after dissociation of 9a and 9b, a similar process could be operative.
The absence of 2b upon warming 9b to 25 °C together with rapid consumption of oxanickelacycle 1b at −90 °C toward preferential formation of 6b and 7b led us to consider that the C(sp3)–OC(sp3) bond formation pathway accounting for the ca. 10–14% yield of 2b may arise from a slow intramolecular cyclization of the iodoalkoxide/iodoalcohol in a SN2 fashion.46 Indeed, after 48 h of reaction time in the NMR tube without stirring, the iodoalcohol formed initially slowly evolves to form 2b (Scheme 6).31 It is well established that 5-iodoalkoxides can undergo intramolecular 5-exo-tet cyclization to afford cyclic ethers.47 This experimental evidence supports the lack of reactivity when attempting formation of open-chain ethers such as Et2O (2f) due to the much slower rates for intermolecular SN2 reactions.48
Scheme 6. Thermal Decomposition of Complex 9b and Hypothetical SN2 Reaction from 7b.

With these results in hand, we addressed such a defying and elusive reductive elimination. It was clear that other oxidants that enable access to high-valent Ni species should be scrutinized.49 When I2 was replaced by Umemoto’s reagent (S-(trifluoromethyl)dibenzothiophenium triflate, TDTT, 10a),33a a low yield of 2b was observed (10%). A reduced amount of side product 6b was obtained when CD3CN was used instead. During monitoring studies at variable temperatures, HCF3 (boiling point = −82.1 °C) was detected. Formation of fluoroform suggests the involvement of CF3–Ni–H intermediates and points to alkenol 6b being formed through β-hydride elimination pathways. In addition to alkenol 6b and HCF3, other byproducts containing C(sp3)–CF3 were also identified by 19F NMR, which was consistent with formation of high-valent Ni intermediates.50 Despite the low yields, to the best of our knowledge, this challenging C(sp3)–CF3 bond formation is unprecedented at a Ni center.51 At this point, it was quite evident that competitive C(sp3)–X reductive eliminations (X = I, CF3) should be suppressed if the challenging C(sp3)–O–C(sp3) is to be achieved. Hence, we speculated that the presence of F ligands in the coordination sphere of a high-valent Ni intermediate would dramatically reduce the observed side reactions due to the high kinetic barrier to forge C(sp3)–F bonds.52 When 1b was mixed with 1.05 equiv of XeF2 in CD2Cl2 or CD3CN, immediate reaction took place and the desired ether 2b was observed as the major product in 60% or 47% yield, respectively.50a,52f,53 Interestingly, formation of 6b remained minor in CD2Cl2 and could be largely suppressed in CD3CN (8%). It is important to mention that products derived from a putative C(sp3)–F reductive elimination were only observed in trace amounts.31 In this line, when XeF2 was replaced by SelectFluor (10c) in CD3CN a similar outcome was obtained with a 45% yield of 2b along with a minimal amount of 6b (10%). We then investigated several commercially available substituted 1-fluoro-pyridinium salts (10d–f) as they have increased solubility in CH3CN.54 Using 10d, 45% 2b and <5% 6b were obtained. Gratifyingly, when using NFTPB (10e, N-fluoro-2,4,6-trimethylpyridinium tetrafluoroborate) and NFTPT (10f, N-fluoro-2,4,6-trimethylpyridinium triflate) an increase in the yield of 2b was observed (61% and 63%, respectively). Notice that when using BF4 as counterion, formation of 6b could be minimized to a residual 8%. As expected, 2a was also obtained in a satisfactory 56% and 63% yield when using 10e and 10f, respectively (Scheme 7B).
Scheme 7. (A) Screening of Oxidants for the Oxidatively Induced C(sp3)–OC(sp3) Bond Formation; (B) Application to the Synthesis of THF.

Reaction conditions: oxanickelacycle 1b (1 equiv), oxidant 10 (1.05 equiv) in CD3CN or CD2Cl2 at 25 °C.
Having identified a set of conditions that enable formation of cyclic C(sp3)–O–C(sp3) bonds at high-valent Ni centers, we speculated that a similar pathway should be valid for formation of acyclic ethers. In order to study this possibility, an acyclic precursor was synthesized through a two-step sequence (Scheme 8A). Following the protocol from Ikeda,55 we initially synthesized (bipy)NiEt2 (11) by reacting bipy, Ni(acac)2, and (EtO)AlEt2 in Et2O for 50 h at 25 °C. After isolation via filtration (87%), the deep green solid 11 is subjected to N2O atmosphere at 25 °C, allowing smooth O insertion into the Ni–C(sp3) bond,27a,27b leading to (bipy)Ni(Et)(OEt) (1f) in 63% yield. At this point, 1f was subjected to our optimized oxidation conditions using 10f in CD3CN at 25 °C (Scheme 8B). Gratifyingly, rapid formation of Et2O (2f) was observed in 20% yield at 25 °C. A similar result was obtained using 10e. Formation of 2f from 1f represents a unique example of C(sp3)–OC(sp3) bond formation and contrasts with the results obtained from Hillhouse, where C(sp3)–OC(sp3) bond formation from acyclic NiII complexes could not be achieved (see Scheme 2A). This unprecedented result for unbiased, acyclic substrates combined with the use of simple pyridinium salt 10e and 10f as a mild oxidant provides an interesting proof-of-concept for the development of new strategies based on Ni and may open the door to new avenues for catalytic dialkyl ether syntheses in the future.
Scheme 8. (A) Synthesis of (bipy)NiEt211 and (bipy)Ni(Et)(OEt) 1f; (B) Oxidatively Induced Synthesis of Diethyl Ether from Nickel Complex 1f.

Reaction conditions: (step 1) Ni(acac)2 (1 equiv), bipy (1 equiv) Et2AlOEt (3 equiv) in Et2O, from −50 to 25 °C, 50 h, 11 87% isolated yield; (step 2) 11 (1 equiv), N2O (1 atm) in THF at 25 °C, 1f 63% isolated yield.
Reaction conditions: oxanickelacycle 1f (1 equiv), 10e and 10f (1.05 equiv) in CD3CN at 25 °C, 1 min. Yields determined by 1H NMR using mesitylene as internal standard.
Successful formation of C(sp3)–O–C(sp3) bonds using N-fluoropyridinium reagents posed the question of what is the exact nature of the high-valent Ni species involved in the process. On the basis of the CV results for 2b (Figure 1) together with the oxidation results using Fc+, O2, and 4CzIPN photocatalyst (Scheme 4),27 it is evident that access to NiIII species is facile;49 yet, cationic NiIII are not capable of C–O bond formation due to fast elimination side reactions. It is important to point out that owing to the extremely fast reaction rates for C–O bond formation, mechanistic investigations on this particular system pose an experimental challenge. Indeed, 1a, 1b, 10e, and 10f react at −90 °C in <1 min, and no Ni intermediates could be detected spectroscopically (1H and 19F NMR). Attempts to stabilize high-valent Ni species using tripodal ligands (tris(pyrazolyl)borate56 or tris(pyridyl)methane)33a,49 resulted in degradation or failed to incorporate O to the NiII–C(sp3) bond. To our delight however, a signal could be detected by EPR from reaction of 1b with 10f. Such species were very short lived, but a sufficient amount could be trapped after rapid (1 s) mixing at −95 °C (melting point of PhMe) and subsequent freezing in liquid N2 (Scheme 9A). The EPR spectrum showed multiple splitting features consistent with a NiIII dimer species (int-I, Scheme 9B). The width of the spectrum, however, is reduced with respect to that of 9a and 9b, suggesting a reduction in the dipolar interaction between the two NiIII centers. Indeed, spectral fits resulted in a smaller value (299 vs 517 and 550 MHz), whereas the g-tensor only differed slightly.31 It is important to point out that although the fitting parameters for int-I are probably not unique due to the few spectral features and large number of free parameters, the current fits would be consistent with a NiIII dimer with symmetry properties resembling 9a.57 Additional information about the putative intermediates was provided by mass spectrometry analysis. When a low-temperature mixture of 1b and 10f was analyzed by mass spectrometry, a m/z corresponding to int-I could be detected. In addition, m/z consistent with structures such as int-II (or 1b) and int-IV were also identified.58 Although the nature of the exact species prior to reductive elimination still remains elusive, several possibilities are envisaged. On one hand, dissociation of int-I would afford int-II and int-III (path a).59 An alternative pathway would involve a disproportionation of int-I into starting oxanickelacycle 1b and int-IV (path c).60 Reductive elimination could then occur from either int-III or int-IV. The high degree of elimination obtained in Scheme 4 when using Fc+ or 4CzIPN would argue against path a as a major contributor. Moreover, another possible pathway could involve direct reductive elimination from the dinuclear complex int-I (path b), which has been postulated for certain C–heteroatom and C–C bond-forming events.40,61 In an attempt to discern between mechanistic pathways, we carried out the oxidation of 1b with only 0.5 equiv of 10f. In this case, one-half of the yield of 2b observed in Scheme 7A was obtained (35–40%) without trace amounts of 6b.31 The absence of elimination byproducts suggests that cationic NiIII species might not be present and adds additional evidence about path a not being operative. However, the variety of pathways by which the C–O bond could be formed manifests the need for further mechanistic investigations to fully elucidate the nature of the intermediates involved.
Scheme 9. (A) EPR of int-I at 20 K; (B) Mass Spectrometry Results and Postulated Mechanistic Pathways.

Experimental conditions: Power = 2.0 mW, modulation (100 kHz) amplitude 7.5 G. Dotted red trace represents the Easyspin43 (esfit) simulation with parameters g = (2.103, 2.200, 2.227). Dipolar interaction D(int-I) = 299 MHz. J coupling = 100 MHz.
Conclusions
In conclusion, we studied the seminal I2-promoted C–O bond formation reported by Hillhouse toward formation of cyclic ethers (2a and 2b). A detailed mechanistic investigation revealed formation of 9a and 9b, an unprecedented NiIII bimetallic structure as a cationic intermediate, which was fully characterized by NMR, EPR, HRMS, and X-ray in the case of 9a. These paramagnetic complexes feature a unique Ni2(OR)2 (OR = alkoxide) diamond-like core complemented by a μ-iodo bridge between the two Ni centers. The anionic counterion of the complexes consists of the linear I3–, which remains in the outer sphere of the robust bimetallic cation. Thermal decomposition of 9 beyond −10 °C leads primarily to elimination products (6). In addition, substantial amounts of iodoalkanols (7) were detected through preferential C(sp3)–I reductive elimination. The low yields obtained for 2a and 2b are postulated to arise from an intramolecular SN2 reaction from 7 over long periods of time. This manifests that the original mechanistic picture for direct C(sp3)–OC(sp3) bond formation through reductive elimination is extremely challenging for simple THF rings. Cyclic voltammetry studies as well as a survey of oxidants identified the use of fluoropyridinium reagents 10e and 10f as excellent candidates to afford good yields of 2 while minimizing formation of elimination byproducts (6). In addition, this new set of conditions was successfully applied in the elusive synthesis of acyclic diethyl ether (2f) from a well-defined NiII complex. Preliminary mechanistic studies revealed that upon oxidation of 1 with 10, a highly reactive intermediate could be detected in solution by EPR and HRMS, which is consistent with a NiIII dimeric structure (int-I). Efforts to fully characterize the high-valent species involved after oxidation and prior to reductive elimination are currently under investigation in our laboratory. We believe the findings reported here open the door to new avenues for Ni catalysis and could aid practitioners in the field to unravel novel metal-catalyzed ether synthesis.
Acknowledgments
Financial support for this work was provided by the Max-Planck-Gesellschaft, Max-Planck-Institut für Kohlenforschung, and Fonds der Chemischen Industrie (VCI-FCI). F.L.V. thanks the Swiss National Science Foundation for an Early Mobility Postdoctoral Fellowship (Grant no. 184406). Sigrid Lutz is acknowledged for help in the synthesis of (bipy)Ni(Et)2. We thank M. Kochius for technical support with low-temperature NMR measurements. The analytical departments (X-ray crystallography, NMR spectroscopy, and Mass spectrometry) at the MPI für Kohlenforschung are gratefully acknowledged for support in characterization of the compounds. We thank Prof. Dr. T. Ritter for providing access to the photoredox equipment in his laboratory. E.J.R. thanks Frank Reikowski and Dr. Leonid Rapatskiy for technical assistance with the EPR measurements. We thank Petra Hoefer and Dr. Christophe Werlé from the Max-Planck-Institut für Chemische Energiekonversion (MPI-CEC) for assistance in the cyclic voltammetry studies. We are thankful to Prof. Dr. A. Fürstner for insightful discussions and generous support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c07381.
Experimental procedures and analytical data (1H, 19F, and 13C NMR, EPR, X-ray, HRMS, CV) (PDF)
CIF file for compound 8, also available free of charge from the www.ccdc.cam.ac.uk under CCDC number 2014828 (CIF)
CIF file for compound 9a, also available free of charge from the www.ccdc.cam.ac.uk under CCDC number 2014830 (CIF)
CIF file for compound S1, also available free of charge from the www.ccdc.cam.ac.uk under CCDC number 2014829 (CIF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Roughley S. D.; Jordan A. M. The medicinal chemist’s toolbox: an analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 2011, 54, 3451. 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]; b Brown A.; Brown L.; Brown T. B.; Calabrese A.; Ellis D.; Puhalo N.; Smith C. R.; Wallace O.; Watson L. Triazole oxytocin antagonists: identification of aryl ether replacements for a biaryl substituent. Bioorg. Med. Chem. Lett. 2008, 18, 5242. 10.1016/j.bmcl.2008.08.066. [DOI] [PubMed] [Google Scholar]; c Backes B. J.; Ellman J. A. Solid support linker strategies. Curr. Opin. Chem. Biol. 1997, 1, 86. 10.1016/S1367-5931(97)80113-5. [DOI] [PubMed] [Google Scholar]; d Ganesan A.Wang resin. Encyclopedia of reagents for organic synthesis; Wiley: Hoboken, NJ, 2003. [Google Scholar]; e Harris J. M.Poly(ethylene glycol) Chemistry: Biotechnical and Biomedical Applications; Plenum Press: New York, 1992. [Google Scholar]
- Williamson A. W. J. Chem. Soc. 1852, 4, 229. 10.1039/QJ8520400229. [DOI] [Google Scholar]; Alexander William Williamson (1824–1904) discovered this reaction in 1850 at University College, London
- For selected reviews on Williamson ether synthesis, see:; a Li J. J.Williamson ether synthesis. Name Reactions; Springer:Cham, 2014. [Google Scholar]; b Wang Z.Williamson Ether Synthesis. Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons: Hoboken, NJ, 2010; pp 3026–3030. [Google Scholar]
- Xiang J.; Shang M.; Kawamata Y.; Lundberg H.; Reisberg S.; Chen M.; Mykhailiuk P.; Beutner G.; Collins M.; Davies A.; Del Bel M.; Gallego G.; Spangler J.; Starr J. T.; Yang S.; Blackmond D.; Baran P. S. Hindered Dialkyl Ether Synthesis via Electrogenerated Carbocations. Nature 2019, 573, 398. 10.1038/s41586-019-1539-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibutani S.; Kodo T.; Takeda M.; Nagao K.; Tokunaga N.; Sasaki Y.; Ohmiya H. Organophotoredox-Catalyzed Decarboxylative C(sp3)–O Bond Formation. J. Am. Chem. Soc. 2020, 142, 1211. 10.1021/jacs.9b12335. [DOI] [PubMed] [Google Scholar]
- a Ghosh A. K.; Tomaine A. J.; Cantwell K. E. Lewis Acid Mediated Cyclizations: Diastereoselective Synthesis of Six- to Eight-Membered Substituted Cyclic Ethers. Synthesis 2017, 49, 4229. 10.1055/s-0036-1589054. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Trost B. M.; Mata G. Enantioselective Palladium-Catalyzed [3 + 2] Cycloaddition of Trimethylenemethane and Fluorinated Ketones. Angew. Chem., Int. Ed. 2018, 57, 12333. 10.1002/anie.201807308. [DOI] [PubMed] [Google Scholar]; c De Nanteuil F.; Serrano E.; Perrotta D.; Waser J. Dynamic Kinetic Asymmetric [3 + 2] Annulation Reactions of Aminocyclopropanes. J. Am. Chem. Soc. 2014, 136, 6239. 10.1021/ja5024578. [DOI] [PubMed] [Google Scholar]; d Parsons A. T. M.; Johnson J. S. Catalytic Enantioselective Synthesis of Tetrahydrofurans: A Dynamic Kinetic Asymmetric [3 + 2] Cycloaddition of Racemic Cyclopropanes and Aldehydes. J. Am. Chem. Soc. 2009, 131, 3122. 10.1021/ja809873u. [DOI] [PubMed] [Google Scholar]; e Tsuji N.; Kennemur J. L.; Buyck T.; Lee S.; Prevost S.; Kaib P. S. J.; Bykov D.; Fares C.; List B. Activation of olefins via asymmetric Brønsted acid catalysis. Science 2018, 359, 1501. 10.1126/science.aaq0445. [DOI] [PubMed] [Google Scholar]; f Luo C.; Bandar J. S. Superbase-Catalyzed anti-Markovnikov Alcohol Addition Reactions. J. Am. Chem. Soc. 2018, 140, 3547. 10.1021/jacs.8b00766. [DOI] [PubMed] [Google Scholar]; g Shigeno M.; Hayashi K.; Nozawa-Kumada K.; Kondo Y. Phosphazene Base tBu-P4 Catalyzed Methoxy–Alkoxy Exchange Reaction on (Hetero)Arenes to Aryl Alkenes. Chem. - Eur. J. 2019, 25, 6077. 10.1002/chem.201900498. [DOI] [PubMed] [Google Scholar]
- a Vedernikov A. N.C–O Reductive Elimination from High Valent Pt and Pd Centers. In C-X Bond Formation. Topics in Organometallic Chemistry; Vigalok A., Ed.; Springer, Berlin, Heidelberg, 2010; Vol. 31. [Google Scholar]; b Stambuli J. P.New Trends in Cross-Coupling: Theory and Application. In Transition Metal-Catalyzed Formation of C–O and C–S Bonds; Colacot T. J., Eds.; Royal Society of Chemistry: Cambridge, UK, 2014; Vol. 254. [Google Scholar]
- a Munir I.; Zahoor A. F.; Rasool N.; Naqvi S. A. R.; Zia K. M.; Ahmad R. Synthetic applications and methodology development of Chan–Lam coupling: a review. Mol. Diversity 2019, 23, 215. 10.1007/s11030-018-9870-z. [DOI] [PubMed] [Google Scholar]; b Qiao J. X.; Lam P. Y. S. In Boronic Acids:Preparation and Applications in Organic Synthesis, Medicine and Materials; Hall D. G., Ed.; Wiley-VCH: Weinheim, 2011; pp 315–361. [Google Scholar]; c Sambiagio C.; Marsden S. P.; Blacker A. J.; McGowan P. C. Copper catalysed Ullmann type chemistry: from mechanistic aspects to modern development. Chem. Soc. Rev. 2014, 43, 3525. 10.1039/C3CS60289C. [DOI] [PubMed] [Google Scholar]; d Monnier F.; Taillefer M. Catalytic C–C, C–N, and C–O Ullmann-Type Coupling Reactions. Angew. Chem., Int. Ed. 2009, 48, 6954. 10.1002/anie.200804497. [DOI] [PubMed] [Google Scholar]
- For seminal Pd-catalyzed C(sp2)–O bond formation, see:; a Mann G.; Hartwig J. F. Palladium Alkoxides: Potential Intermediacy in Catalytic Amination, Reductive Elimination of Ethers, and Catalytic Etheration. Comments on Alcohol Elimination from Ir(III). J. Am. Chem. Soc. 1996, 118, 13109. 10.1021/ja963273w. [DOI] [Google Scholar]; b Palucki M.; Wolfe J. P.; Buchwald S. L. Palladium-Catalyzed Intermolecular Carbon-Oxygen Bond Formation: A New Synthesis of Aryl Ethers. J. Am. Chem. Soc. 1997, 119, 3395. 10.1021/ja9640152. [DOI] [Google Scholar]; c Torraca K. E.; Huang X.; Parrish C. A.; Buchwald S. L. An Efficient Intermolecular Palladium-Catalyzed Synthesis of Aryl Ethers. J. Am. Chem. Soc. 2001, 123, 10770. 10.1021/ja016863p. [DOI] [PubMed] [Google Scholar]
- For recent works on Pd-catalyzed C(sp2)–O bond formation, see:; a Mikus M. S.; Sanchez C.; Fridrich C.; Larrow J. F. Palladium Catalyzed C–O Coupling of Amino Alcohols for the Synthesis of Aryl Ethers. Adv. Synth. Catal. 2020, 362, 430. 10.1002/adsc.201901302. [DOI] [Google Scholar]; b Zhang H.; Ruiz-Castillo P.; Schuppe A. W.; Buchwald S. L. Improved Process for the Palladium-Catalyzed C–O Cross-Coupling of Secondary Alcohols. Org. Lett. 2020, 22, 5369. 10.1021/acs.orglett.0c01668. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhang H.; Ruiz-Castillo P.; Buchwald S. L. Palladium-Catalyzed C–O Cross-Coupling of Primary Alcohols. Org. Lett. 2018, 20, 1580. 10.1021/acs.orglett.8b00325. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Gowrisankar S.; Sergeev A. G.; Anbarasan P.; Spannenberg A.; Neumann H.; Beller M. A General and Efficient Catalyst for Palladium-Catalyzed C–O Coupling Reactions of Aryl Halides with Primary Alcohols. J. Am. Chem. Soc. 2010, 132, 11592. 10.1021/ja103248d. [DOI] [PubMed] [Google Scholar]; e Maligres P. E.; Li J.; Krska S. W.; Schreier J. D.; Raheem I. T. C–O Cross-Coupling of Activated Aryl and Heteroaryl Halides with Aliphatic Alcohols. Angew. Chem., Int. Ed. 2012, 51, 9071. 10.1002/anie.201203460. [DOI] [PubMed] [Google Scholar]; f Sawatzky R. S.; Hargreaves B. K. V.; Stradiotto M. A Comparative Ancillary Ligand Survey in Palladium-Catalyzed C–O Cross-Coupling of Primary and Secondary Aliphatic Alcohols. Eur. J. Org. Chem. 2016, 2016, 2444. 10.1002/ejoc.201600198. [DOI] [Google Scholar]
- a Canty A. J.; Jin H.; Skelton B. W.; White A. H. Oxidation of Complexes by (O2CPh)2 and (ER)2 (E = S, Se), Including Structures of Pd(CH2CH2CH2CH2)(SePh)2(bpy) (bpy = 2,2′-Bipyridine) and MMe2(SePh)2(L2) (M = Pd, Pt; L2 = bpy, 1,10-Phenanthroline) and C–O and C–E Bond Formation at Palladium(IV). Inorg. Chem. 1998, 37, 3975. 10.1021/ic9715005. [DOI] [PubMed] [Google Scholar]; b Dick A. R.; Hull K. L.; Sanford M. S. A Highly Selective Catalytic Method for the Oxidative Functionalization of C–H Bonds. J. Am. Chem. Soc. 2004, 126, 2300. 10.1021/ja031543m. [DOI] [PubMed] [Google Scholar]; c Desai L. V.; Hull K. L.; Sanford M. S. Palladium-Catalyzed Oxygenation of Unactivated sp3 C–H Bonds. J. Am. Chem. Soc. 2004, 126, 9542. 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]; d Giri R.; Liang J.; Lei J.-G.; Li J.-J.; Wang D.-H.; Chen X.; Naggar I. C.; Guo C.; Foxman B. M.; Yu J.-Q. Pd-Catalyzed Stereoselective Oxidation of Methyl Groups by Inexpensive Oxidants under Mild Conditions: A Dual Role for Carboxylic Anhydrides in Catalytic C–H Bond Oxidation. Angew. Chem., Int. Ed. 2005, 44, 7420. 10.1002/anie.200502767. [DOI] [PubMed] [Google Scholar]; e Canty A. J.; Ariafard A.; Camasso N. M.; Higgs A. T.; Yates B. F.; Sanford M. S. Computational study of C(sp3)–O bond formation at a PdIV centre. Dalton Trans. 2017, 46, 3742. 10.1039/C7DT00096K. [DOI] [PubMed] [Google Scholar]; f Zhuang Z.; Yu J.-Q. Lactonization as a General Route to β-C(sp3)–H Functionalization. Nature 2020, 577, 656. 10.1038/s41586-019-1859-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Zhuang Z.; Herron A. N.; Fan Z.; Yu J.-Q. Ligand-Enabled mono-Selective β-C(sp3)–H Acyloxylation of Free Carboxylic Acids Using a Practical Oxidant. J. Am. Chem. Soc. 2020, 142, 6769. 10.1021/jacs.0c01214. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Whitehurst W. G.; Gaunt M. J. Synthesis and Reactivity of Stable Alkyl-Pd(IV) Complexes Relevant to Monodentate N-Directed C(sp3)–H Functionalization Processes. J. Am. Chem. Soc. 2020, 142, 14169. 10.1021/jacs.0c04732. [DOI] [PubMed] [Google Scholar]
- Recent examples of Pd-catalyzed alkoxylations suggested the intermediacy of PdIV complexes prior to the reductive elimination, see:; a Zhang S.-Y.; He G.; Zhao Y.; Wright K.; Nack W. A.; Chen G. Efficient Alkyl Ether Synthesis via Palladium-Catalyzed, Picolinamide Directed Alkoxylation of Unactivated C(sp3)–H and C(sp2)–H Bonds at Remote Positions. J. Am. Chem. Soc. 2012, 134, 7313. 10.1021/ja3023972. [DOI] [PubMed] [Google Scholar]; b Chen Y.-Q.; Wu Y.; Wang Z.; Qiao J. X.; Yu J.-Q. Transient Directing Group Enabled Pd-catalyzed gamma-C(sp3)–H Oxygenation of Alkyl Amines. ACS Catal. 2020, 10, 5657. 10.1021/acscatal.0c01310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Inoki S.; Mukaiyama T. A Convenient Method for the Stereoselective Preparation of trans-2-Hydroxymethyltetrahydrofurans by the Oxidative Cyclization of 5-Hydroxy-1-alkenes with Molecular Oxygen Catalyzed by Cobalt(II) Complex. Chem. Lett. 1990, 19, 67. 10.1246/cl.1990.67. [DOI] [Google Scholar]; b Morra B.; Morra N. A.; MacDonald D. G.; Pagenkopf B. L. Gram-Scale Synthesis of the Co(nmp)2 Catalyst to Prepare trans-2,5-Disubstituted Tetrahydrofurans by the Aerobic Oxidative Cyclization of Pent-4-en-1-ols. Synthesis 2020, 52, 847. 10.1055/s-0039-1690730. [DOI] [Google Scholar]; c Heinrich M.; Murphy J. J.; Ilg M. K.; Letort A.; Flasz J. T.; Philipps P.; Fürstner A. Chagosensine: A Riddle Wrapped in a Mystery Inside an Enigma. J. Am. Chem. Soc. 2020, 142, 6409. 10.1021/jacs.0c01700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent work on Cu catalyzed C(sp3)–O bond formation, see:; a Hu H.; Chen S.-J.; Mandal M.; Pratik S. M.; Buss J. A.; Krska S. W.; Cramer C. J.; Stahl S. S. Copper-Catalysed Benzylic C–H Coupling with Alcohols via Radical Relay Enabled by Redox Buffering. Nature Catal. 2020, 3, 358. 10.1038/s41929-020-0425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cheng Y. F.; Liu J.-R.; Gu Q.-S.; Yu S.-L.; Wang J.; Li Z.-L.; Bian J.-Q.; Wen W.-T.; Wang X.-J.; Hong X.; Liu X.-Y. Catalytic Enantioselective Desymmetrizing Functionalization of Alkyl Radicals via Cu(I)/CPA Cooperative Catalysis. Nat. Catal 2020, 3, 401. 10.1038/s41929-020-0439-8. [DOI] [Google Scholar]; c Mao R.; Balon J.; Hu X. Decarboxylative C(sp3)–O cross-coupling. Angew. Chem., Int. Ed. 2018, 57, 13624. 10.1002/anie.201808024. [DOI] [PubMed] [Google Scholar]; d Chen Z.; Jiang Y.; Zhang L.; Guo Y.; Ma D. Oxalic Diamides and tert-Butoxide: Two Types of Ligands Enabling Practical Access to Alkyl Aryl Ethers via Cu-Catalyzed Coupling Reaction. J. Am. Chem. Soc. 2019, 141, 3541. 10.1021/jacs.8b12142. [DOI] [PubMed] [Google Scholar]; For recent work on Cu-catalyzed C(sp2)–O bond formation, see:; e Sang R.; Korkis S. E.; Su W.; Ye F.; Engl P. S.; Berger F.; Ritter T. Site-Selective C–H Oxygenation via Aryl Sulfonium Salts. Angew. Chem., Int. Ed. 2019, 58, 16161. 10.1002/anie.201908718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For previous work on Fe catalyzed C–O bond formation, see:; a Xia N.; Taillefer M. Copper- or Iron-Catalyzed Arylation of Phenols from respectively Aryl Chlorides and Aryl Iodides. Chem. - Eur. J. 2008, 14, 6037. 10.1002/chem.200800436. [DOI] [PubMed] [Google Scholar]; b Bistri O.; Correa A.; Bolm C. Iron-Catalyzed C–O Cross-Couplings of Phenols with Aryl Iodides. Angew. Chem., Int. Ed. 2008, 47, 586. 10.1002/anie.200704018. [DOI] [PubMed] [Google Scholar]
- Beletskaya I. P.; Cheprakov A. V. Copper in Cross-Coupling Reactions: The Post-Ullmann Chemistry. Coord. Chem. Rev. 2004, 248, 2337. 10.1016/j.ccr.2004.09.014. [DOI] [Google Scholar]
- For recent reviews on Ni catalysis, see:; a Tasker S. Z.; Standley E. A.; Jamison T. F. Recent Advances in Homogeneous Nickel Catalysis. Nature 2014, 509, 299. 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Standley E. A.; Tasker S. Z.; Jensen K. L.; Jamison T. F. Nickel Catalysis: Synergy between Method Development and Total Synthesis. Acc. Chem. Res. 2015, 48, 1503. 10.1021/acs.accounts.5b00064. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Diccianni J. B.; Diao T. Mechanisms of Nickel-Catalyzed Cross-Coupling Reactions. Trends in Chemistry 2019, 1, 830. 10.1016/j.trechm.2019.08.004. [DOI] [Google Scholar]; d Cavedon C.; Seeberger P. H.; Pieber B. Photochemical Strategies for Carbon–Heteroatom Bond Formation. Eur. J. Org. Chem. 2020, 2020, 1379. 10.1002/ejoc.201901173. [DOI] [Google Scholar]; e Lavoie C. M.; Stradiotto M. Bisphosphines: A Prominent Ancillary Ligand Class for Application in Nickel-catalyzed C–N Cross-coupling. ACS Catal. 2018, 8, 7228. 10.1021/acscatal.8b01879. [DOI] [Google Scholar]
- a Mann G.; Hartwig J. F. Nickel- vs Palladium-Catalyzed Synthesis of Protected Phenols from Aryl Halides. J. Org. Chem. 1997, 62, 5413. 10.1021/jo970453v. [DOI] [Google Scholar]; For an example using vinyl iodides and Ni(COD)2 without ligand, see:; b Han S.-J.; Doi R.; Stoltz B. M. Nickel-Catalyzed Intramolecular C–O Bond Formation: Synthesis of Cyclic Enol Ethers. Angew. Chem., Int. Ed. 2016, 55, 7437. 10.1002/anie.201601991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacQueen P. M.; Tassone J. P.; Diaz C.; Stradiotto M. Exploiting Ancillary Ligation To Enable Nickel-Catalyzed C–O Cross Couplings of Aryl Electrophiles with Aliphatic Alcohols. J. Am. Chem. Soc. 2018, 140, 5023. 10.1021/jacs.8b01800. [DOI] [PubMed] [Google Scholar]
- a Terrett J. A.; Cuthbertson J. D.; Shurtleff V. W.; MacMillan D. W. C. Switching on Elusive Organometallic Mechanisms with Photoredox Catalysis. Nature 2015, 524, 330. 10.1038/nature14875. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cavedon C.; Madani A.; Seeberger P. H.; Pieber B. Semiheterogeneous Dual Nickel/Photocatalytic (Thio)etherification Using Carbon Nitrides. Org. Lett. 2019, 21, 5331. 10.1021/acs.orglett.9b01957. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yang L.; Lu H.-H.; Lai C.-H.; Li G.; Zhang W.; Cao R.; Liu F.; Wang C.; Xiao J.; Xue D. Light-Promoted Nickel Catalysis: Etherification of Aryl Electrophiles with Alcohols Catalyzed by a NiII-Aryl Complex. Angew. Chem., Int. Ed. 2020, 59, 12714. 10.1002/anie.202003359. [DOI] [PubMed] [Google Scholar]
- Welin E. R.; Le C.; Arias-Rotondo D. M.; McCusker J. K.; MacMillan D. W. C. Photosensitized, Energy Transfer-Mediated Organometallic Catalysis Through Electronically Excited Nickel(II). Science 2017, 355, 380. 10.1126/science.aal2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhu B.; Yan L.-K.; Geng Y.; Ren H.; Guan W.; Su Z.-M. IrIII/NiII-Metallaphotoredox Catalysis: the Oxidation State Modulation Mechanism Versus the Radical Mechanism. Chem. Commun. 2018, 54, 5968. 10.1039/C8CC03550D. [DOI] [PubMed] [Google Scholar]; b Tian L.; Till N. A.; Kudisch B.; MacMillan D. W. C.; Scholes G. D. Transient Absorption Spectroscopy Offers Mechanistic Insights for an Iridium/Nickel-Catalyzed C–O Coupling. J. Am. Chem. Soc. 2020, 142, 4555. 10.1021/jacs.9b12835. [DOI] [PubMed] [Google Scholar]; c Ma P.; Wang S.; Chen H. Reactivity of Transition-Metal Complexes in Excited States: C–O Bond Coupling Reductive Elimination of a Ni(II) Complex Is Elicited by the Metal-to-Ligand Charge Transfer State. ACS Catal. 2020, 10, 1. 10.1021/acscatal.9b03827. [DOI] [Google Scholar]
- Zhang S.-K.; Struwe J.; Hu L.; Ackermann L. Nickela-electrocatalyzed C–H Alkoxylation with Secondary Alcohols: Oxidation-Induced Reductive Elimination at Nickel(III). Angew. Chem., Int. Ed. 2020, 59, 3178. 10.1002/anie.201913930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R.; Qin Y.; Nocera D. G. General Paradigm in Photoredox Ni-Catalyzed Cross-Coupling Allows for Light-Free Access to Reactivity. Angew. Chem., Int. Ed. 2020, 59, 9527. 10.1002/anie.201916398. [DOI] [PubMed] [Google Scholar]
- For an example from well-defined NiIII complex, see:; Zhou W.; Schultz J. W.; Rath N. P.; Mirica L. M. Aromatic Methoxylation and Hydroxylation by Organometallic High-Valent Nickel Complexes. J. Am. Chem. Soc. 2015, 137, 7604. 10.1021/jacs.5b04082. [DOI] [PubMed] [Google Scholar]
- Smith S. M.; Planas O.; Gómez L.; Rath N. P.; Ribas X.; Mirica L. M. Aerobic C–C and C–O Bond Formation Reactions Mediated by High-Valent Nickel Species. Chem. Sci. 2019, 10, 10366. 10.1039/C9SC03758F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Matsunaga P. T.; Hillhouse G. L.; Rheingold A. L. Oxygen-Atom Transfer from Nitrous Oxide to a Nickel Metallacycle. Synthesis, Structure, and Reactions of [cyclic] (2,2′-bipyridine)Ni(OCH2CH2CH2CH2). J. Am. Chem. Soc. 1993, 115, 2075. 10.1021/ja00058a085. [DOI] [Google Scholar]; b Matsunaga P. T.; Mavropoulos J. C.; Hillhouse G. L. Oxygen-Atom Transfer from Nitrous oxide (N=N=O) to Nickel Alkyls. Syntheses and Reactions of Nickel(II) Alkoxides. Polyhedron 1995, 14, 175. 10.1016/0277-5387(94)00330-H. [DOI] [Google Scholar]; c Koo K.; Hillhouse G. L.; Rheingold A. L. Oxygen-Atom Transfer from Nitrous Oxide to an Organonickel(II) Phosphine Complex. Syntheses and Reactions of New Nickel(II) Aryloxides and the Crystal Structure of (Me2PCH2CH2PMe2)Ni(0-o-C6H4CMe2CH2). Organometallics 1995, 14, 456. 10.1021/om00001a062. [DOI] [Google Scholar]; d Koo K.; Hillhouse G. L. Formation of a Substituted Tetrahydrofuran by Formal [2 + 2 + 1] Coupling of an Oxygen Atom with Two Olefins at a Nickel Center. Organometallics 1998, 17, 2924. 10.1021/om980182u. [DOI] [Google Scholar]
- Desnoyer A. N.; Bowes E. G.; Patrick B. O.; Love J. A. Synthesis of 2-Nickela(II)oxetanes from Nickel(0) and Epoxides: Structure, Reactivity, and a New Mechanism of Formation. J. Am. Chem. Soc. 2015, 137, 12748. 10.1021/jacs.5b06735. [DOI] [PubMed] [Google Scholar]
- a Binger P.; Doyle M. J.; Krüger C.; Tsay Y.-H. Z. Metallacycloalkane, III Darstellung und Charakterisierung von a.a′-Dipyridyl-Nickelacyclopentan /Preparation and Characterisation of a,a′-Bipyridyl-nickelacyclopentane. Z. Naturforsch., B: J. Chem. Sci. 1979, 34B, 1289. 10.1515/znb-1979-0926. [DOI] [Google Scholar]; b Cámpora J. Nickel–Carbon σ-Bonded Complexes. Comprehensive Organometallic Chemistry III 2007, 8, 27–132. 10.1016/B0-08-045047-4/00100-X. [DOI] [Google Scholar]; c Echavarren A. M.; Castaño A. M. Oxa-and Azametallacycles of Nickel: Fundamental Aspects and Synthetic Applications. Adv. Met.-Org. Chem. 1998, 6, 1–47. 10.1016/S1045-0688(98)80003-8. [DOI] [Google Scholar]
- Avicel is a powder composed by small particles of cellulose. It is efficient for the removal of Ni black nanoparticles through filtration.
- See Supporting Information for more details.
- Selectivity issues in the O-insertion step were not considered in the early work from Hillhouse, and preference for the secondary carbon was always obtained. See ref (27b).
- a Camasso N. M.; Sanford M. S. Design, Synthesis, and Carbon-Heteroatom Coupling Reactions of Organometallic Nickel(IV) Complexes. Science 2015, 347, 1218. 10.1126/science.aaa4526. [DOI] [PubMed] [Google Scholar]; b Yu S.; Dudkina Y.; Wang H.; Kholin K. V.; Kadirov M. K.; Budnikova Y. H.; Vicic D. A. Accessing Perfluoroalkyl Nickel(II), (III), and (IV) Complexes Bearing a Readily attached [C4F8] ligand. Dalton Trans. 2015, 44, 19443. 10.1039/C5DT01771H. [DOI] [PubMed] [Google Scholar]
- Moreover, two reductive waves were also observed at −2.1 and −2.71 V vs Fc/Fc+, corresponding to NiII/NiI and NiI/Ni0 couples, respectively. See SI for details.
- a Luo J.; Zhang J. Donor–Acceptor Fluorophores for Visible-Light-Promoted Organic Synthesis: Photoredox/Ni Dual Catalytic C(sp3)–C(sp2) Cross-Coupling. ACS Catal. 2016, 6, 873. 10.1021/acscatal.5b02204. [DOI] [Google Scholar]; b Le Vaillant F.; Garreau M.; Nicolai S.; Gryn’ova G.; Corminboeuf C.; Waser J. Fine-Tuned Organic Photoredox Catalysts for Fragmentation-Alkynylation Cascades of Cyclic Oxime Ethers. Chem. Sci. 2018, 9, 5883. 10.1039/C8SC01818A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The reaction was also tested in the original solvent used by Hillhouse (benzene-d6) with similar results as for CD2Cl2. In order to be able to follow the reaction at low temperature, CD2Cl2 was preferred due to its low melting point and single peak in NMR
- Kalvet I.; Guo Q.; Tizzard G. J.; Schoenebeck F. When Weaker Can Be Tougher: The Role of Oxidation State (I) in P- vs N-Ligand-Derived Ni-Catalyzed Trifluoromethylthiolation of Aryl Halides. ACS Catal. 2017, 7, 2126. 10.1021/acscatal.6b03344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanbury D. M.; Sykes A. G.. Advances in Inorganic Chemistry; Academic Press: New York, 1989; Vol. 33; p 69. [Google Scholar]
- a Diccianni J. B.; Hu C.; Diao T. Binuclear, High-Valent Nickel Complexes: Ni–Ni Bonds in Aryl–Halogen Bond Formation. Angew. Chem., Int. Ed. 2017, 56, 3635. 10.1002/anie.201611572. [DOI] [PubMed] [Google Scholar]; b Dürr A. B.; Fisher H. C.; Kalvet I.; Truong K.-N.; Schoenebeck F. Divergent Reactivity of a Dinuclear (NHC) Nickel(I) Catalyst versus Nickel(0) Enables Chemoselective Trifluoromethylselenolation. Angew. Chem., Int. Ed. 2017, 56, 13431. 10.1002/anie.201706423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.; Diccianni J. B.; Katigbak J.; Hu C.; Zhang Y.; Diao T. Bimetallic C–C Bond Forming Reductive Elimination from Nickel. J. Am. Chem. Soc. 2016, 138, 4779. 10.1021/jacs.6b00016. [DOI] [PubMed] [Google Scholar]
- a Shiren K.; Ogo S.; Fujinami S.; Hayashi H.; Suzuki M.; Uehara A.; Watanabe Y.; Moro-oka Y. Synthesis, Structures, and Properties of Bis(μ-oxo)nickel(III) and Bis(μ-superoxo)nickel(II) Complexes: An Unusual Conversion of a NiIII2(μ-O)2 Core into a NiII2(μ-OO)2 Core by H2O2 and Oxygenation of Ligand. J. Am. Chem. Soc. 2000, 122, 254. 10.1021/ja990311d. [DOI] [Google Scholar]; b Morimoto Y.; Takagi Y.; Saito T.; Ohta T.; Ogura T.; Tohnai N.; Nakano M.; Itoh S. A Bis(μ-oxido)dinickel(III) Complex with a Triplet Ground State. Angew. Chem., Int. Ed. 2018, 57, 7640. 10.1002/anie.201802779. [DOI] [Google Scholar]; c Padamati S. K.; Angelone D.; Draksharapu A.; Primi G.; Martin D. J.; Tromp M.; Swart M.; Browne W. R. Transient Formation and Reactivity of a High-Valent Nickel(IV) Oxido Complex. J. Am. Chem. Soc. 2017, 139, 8718. 10.1021/jacs.7b04158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Roy P.; Bour J. R.; Kampf J. W.; Sanford M. S. Catalytically Relevant Intermediates in the Ni-Catalyzed C(sp2)–H and C(sp3)–H Functionalization of Aminoquinoline Substrates. J. Am. Chem. Soc. 2019, 141, 17382. 10.1021/jacs.9b09109. [DOI] [PubMed] [Google Scholar]; b Smith A. K. In Comprehensive Organometallic Chemistry II; Pudephatt R. J., Ed.; Elsevier, 1995; Vol. 9, Chapter 2. [Google Scholar]
- Stoll S.; Schweiger A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42. 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Notice that compounds 2, 6, and 7 are volatile, and therefore, calculation of the yield was challenging after filtration of the paramagnetic solids. See ref (31).
- a Sladek M. I.; Braun T.; Neumann B.; Stammler H.-G. 3-Fluoropyridyl Nickel Complexes as Useful Tools for the Selective Synthesis of new 2,4,5,6-Tetrafluoropyridines: a Route Complementing the Established Methods to Access Fluorinated Pyridines. New J. Chem. 2003, 27, 313. 10.1039/b207943g. [DOI] [Google Scholar]; b Bennett M. A.; Glewis M.; Hockless D. C. R.; Wenger E. Successive Insertion of Tetrafluoroethylene and CO and of Tetrafluoroethylene and Acetylenes into Aryne–Nickel(0) bonds. J. Chem. Soc., Dalton Trans. 1997, 3105. 10.1039/a702375h. [DOI] [Google Scholar]; c Coronas J. M.; Muller G.; Rocamora M. Decomposition of [NiRR′L2] Complexes Induced by Bromine or Anodic Oxidation. J. Organomet. Chem. 1986, 301, 227. 10.1016/0022-328X(86)80013-4. [DOI] [Google Scholar]
- Aihara Y.; Chatani N. Nickel-Catalyzed Reaction of C–H Bonds in Amides with I2: ortho-Iodination via the Cleavage of C(sp2)–H Bonds and Oxidative Cyclization to β-Lactams via the Cleavage of C(sp3)–H Bonds. ACS Catal. 2016, 6, 4323. 10.1021/acscatal.6b00964. [DOI] [Google Scholar]
- a Timmons C.; Chen D.; Cannon J. F.; Headley A. D.; Li G. New Asymmetric Halo Aldol Reaction Provides a Novel Approach to Biologically Important Chiral Cyclothers and Cycloamines. Org. Lett. 2004, 6, 2075. 10.1021/ol049255y. [DOI] [PubMed] [Google Scholar]; For several examples of applications of such cyclization in total syntheses, see:; b Murata Y.; Kamino T.; Aoki T.; Hosokawa S.; Kobayashi S. Highly Efficient Total Synthesis of (+)-Citreoviral. Angew. Chem., Int. Ed. 2004, 43, 3175. 10.1002/anie.200454212. [DOI] [PubMed] [Google Scholar]; c Snyder S. A.; Brucks A. P.; Treitler D. S.; Moga I. Concise Synthetic Approaches for the Laurencia Family: Formal Total Syntheses of (±)-Laurefucin and (±)-E- and (±)-Z-Pinnatifidenyne. J. Am. Chem. Soc. 2012, 134, 17714. 10.1021/ja3076988. [DOI] [PubMed] [Google Scholar]; d Yakura T.; Sato S.; Yoshimoto Y. Enantioselective Synthesis of Pachastrissamine (Jaspin B) Using Dirhodium(II)-Catalyzed C–H Amination and Asymmetric Dihydroxylation as Key Steps. Chem. Pharm. Bull. 2007, 55, 1284. 10.1248/cpb.55.1284. [DOI] [PubMed] [Google Scholar]
- a Brown R. F.; Van Gulick N. M. The Geminal Alkyl Effect on the Rates of Ring Closure of Bromobutylamines. J. Org. Chem. 1956, 21, 1046. 10.1021/jo01115a616. [DOI] [Google Scholar]; b Anslyn E. V.; Dougherty D. A.. Modern Physical Organic Chemistry; University Science: Sausalito, CA, 2006. [Google Scholar]
- a Nebra N. High-Valent NiIII and NiIV Species Relevant to C–C and C–Heteroatom Cross-Coupling Reactions: State of the Art. Molecules 2020, 25, 1141. 10.3390/molecules25051141. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mirica L. M.; Smith S. M.; Griego L.. Organometallic Chemistry of High-Valent Ni(III) and Ni(IV) Complexes. In Nickel Catalysis in Organic Synthesis: Methods and Reactions; Ogoshi S., Ed.; Wiley-VCH Verlag GmbH & Co., 2019; pp 223–248. [Google Scholar]
- a D’Accriscio F.; Borja P.; Saffon-Merceron N.; Fustier-Boutignon M.; Mézailles M.; Nebra N. C–H Bond Trifluoromethylation of Arenes Enabled by a Robust, High-Valent Nickel(IV) Complex. Angew. Chem., Int. Ed. 2017, 56, 12898. 10.1002/anie.201706237. [DOI] [PubMed] [Google Scholar]; b Meucci E. A.; Nguyen S. N.; Camasso N. M.; Chong E.; Ariafard A.; Canty A.; Sanford M. S. Nickel(IV)-Catalyzed C–H Trifluoromethylation of (Hetero)arenes. J. Am. Chem. Soc. 2019, 141, 12872. 10.1021/jacs.9b06383. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bour J. R.; Camasso N. M.; Meucci E. A.; Kampf J. W.; Canty A. J.; Sanford M. S. Carbon–Carbon Bond-Forming Reductive Elimination from Isolated Nickel(III) Complexes. J. Am. Chem. Soc. 2016, 138, 16105. 10.1021/jacs.6b10350. [DOI] [PubMed] [Google Scholar]
- For a rare example of C(sp3)–CF3 bond formation mediated by high-valent copper complexes, see:; Liu S.; Liu H.; Liu S.; Lu Z.; Lu C.; Leng X.; Lan Y.; Shen Q. C(sp3)–CF3 Reductive Elimination from a Five-Coordinate Neutral Copper(III) Complex. J. Am. Chem. Soc. 2020, 142, 9785. 10.1021/jacs.0c03304. [DOI] [PubMed] [Google Scholar]
- a Furuya T.; Kamlet A. S.; Ritter T. Catalysis for Fluorination and Trifluoromethylation. Nature 2011, 473, 470. 10.1038/nature10108. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lin X.; Weng Z. Transition metal complex assisted Csp3–F bond formation. Dalton Trans. 2015, 44, 2021. 10.1039/C4DT03410D. [DOI] [PubMed] [Google Scholar]; c Chen Y.-Q.; Singh S.; Wu Y.; Wang Z.; Hao W.; Verma P.; Qiao J. X.; Sunoj R. B.; Yu J.-Q. Pd-Catalyzed γ-C(sp3)–H Fluorination of Free Amines. J. Am. Chem. Soc. 2020, 142, 9966. 10.1021/jacs.9b13537. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Racowski J. M.; Gary J. B.; Sanford M. S. Carbon(sp3)–Fluorine Bond-Forming Reductive Elimination from Palladium(IV) Complexes. Angew. Chem., Int. Ed. 2012, 51, 3414. 10.1002/anie.201107816. [DOI] [PubMed] [Google Scholar]; e Hull K. L.; Anani W. Q.; Sanford M. S. Palladium-Catalyzed Fluorination of Carbon-Hydrogen Bonds. J. Am. Chem. Soc. 2006, 128, 7134. 10.1021/ja061943k. [DOI] [PubMed] [Google Scholar]; f Mankad N. P.; Toste F. D. C(sp3)–F Reductive Elimination from Alkyl Gold(III) Fluoride Complexes. Chem. Sci. 2012, 3, 72. 10.1039/C1SC00515D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tramšek M.; Žemva B. Synthesis, Properties and Chemistry of Xenon(II) Fluoride. Acta Chim. Slov. 2006, 53, 105. [Google Scholar]; b Planas O.; Wang F.; Leutzsch M.; Cornella J. Fluorination of Arylboronic Esters Enabled by Bismuth Redox Catalysis. Science 2020, 367, 313. 10.1126/science.aaz2258. [DOI] [PubMed] [Google Scholar]; c Ball N. D.; Sanford M. S. Synthesis and Reactivity of a Mono-σ-Aryl Palladium (IV) Fluoride Complex. J. Am. Chem. Soc. 2009, 131, 3796. 10.1021/ja8054595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Rozatian N.; Ashworth I. W.; Sandford G.; Hodgson D. R. W. Chem. Sci. 2018, 9, 8692. 10.1039/C8SC03596B. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lal G. S.; Pez G. P.; Syvret R. G. Electrophilic Fluorinating Agents. Chem. Rev. 1996, 96, 1737. 10.1021/cr941145p. [DOI] [PubMed] [Google Scholar]
- Saito T.; Uchida Y.; Misono A.; Yamamoto A.; Morifuji K.; Ikeda S. Diethyldipyridylnickel. Preparation, Characterization, and Reactions. J. Am. Chem. Soc. 1966, 88, 5198. 10.1021/ja00974a030. [DOI] [Google Scholar]
- a Bour J. R.; Camasso N. M.; Sanford M. S. Oxidation of Ni(II) to Ni(IV) with Aryl Electrophiles Enables Ni-Mediated Aryl–CF3 Coupling. J. Am. Chem. Soc. 2015, 137, 8034. 10.1021/jacs.5b04892. [DOI] [PubMed] [Google Scholar]; b Meucci E. A.; Ariafard A.; Canty A. J.; Kampf J. W.; Sanford M. S. Aryl–Fluoride Bond-Forming Reductive Elimination from Nickel(IV) Centers. J. Am. Chem. Soc. 2019, 141, 13261. 10.1021/jacs.9b06896. [DOI] [PubMed] [Google Scholar]; c Bour J. R.; Ferguson D. M.; McClain E. J.; Kampf J. W.; Sanford M. S. Connecting Organometallic Ni(III) and Ni(IV): Reactions of Carbon-Centered Radicals with High-Valent Organonickel Complexes. J. Am. Chem. Soc. 2019, 141, 8914. 10.1021/jacs.9b02411. [DOI] [PubMed] [Google Scholar]; d Meucci E. A.; Camasso N. M.; Sanford M. S. An Organometalllic NiIV Complex That Participates in Competing Transmetalation and C(sp2)–O Bond-Forming Reductive Elimination Reactions. Organometallics 2017, 36, 247. 10.1021/acs.organomet.6b00810. [DOI] [Google Scholar]; e See also refs (33a) , (49), (50b), and (50c).
- It is important to note that fitting of multifrequency EPR experiments on the same compound should certainly enhance the reliability of the obtained parameters. Such multifrequency experiments could probably provide insights into the expected and currently not observed HFI coupling for Ni–F and its magnitude.
- Detection of m/z for int-II and int-IV from the reaction mixture can also be attributed to fragmentation of a putative dimer during analysis. In addition, a m/z corresponding to dimeric NiIII–NiIII corresponding to the defluorinated int-I could also be observed. See Supporting Information for details.
- For NiIII-F complexes, see:; a Mondal P.; Lovisari M.; Twamley B.; McDonald A. R. Fast Hydrocarbon Oxidation by a High-Valent Nickel–Fluoride Complex. Angew. Chem., Int. Ed. 2020, 59, 13044. 10.1002/anie.202004639. [DOI] [PubMed] [Google Scholar]; b Lee H.; Börgel J.; Ritter T. Carbon–Fluorine Reductive Elimination from Nickel(III) Complexes. Angew. Chem., Int. Ed. 2017, 56, 6966. 10.1002/anie.201701552. [DOI] [PubMed] [Google Scholar]; c Zhou W.; Zheng S.; Schultz J. W.; Rath N. P.; Mirica L. M. Aromatic Cyanoalkylation through Double C–H Activation Mediated by NiIII. J. Am. Chem. Soc. 2016, 138, 5777. 10.1021/jacs.6b02405. [DOI] [PubMed] [Google Scholar]; d See also ref (50a).
- For NiIV–F complexes, see:; a Roberts C. C.; Chong E.; Kampf J. W.; Canty A. J.; Ariafard A.; Sanford M. S. Nickel(II/IV) Manifold Enables Room-Temperature C(sp3)–H Functionalization. J. Am. Chem. Soc. 2019, 141, 19513. 10.1021/jacs.9b11999. [DOI] [PubMed] [Google Scholar]; b Chong E.; Kampf J. W.; Ariafard A.; Canty A. J.; Sanford M. S. Oxidatively Induced C–H Activation at High Valent Nickel. J. Am. Chem. Soc. 2017, 139, 6058. 10.1021/jacs.7b02387. [DOI] [PubMed] [Google Scholar]; c Kosobokov M. D.; Sandleben A.; Vogt N.; Klein A.; Vicic D. A. Nitrogen–Nitrogen Bond Formation via a Substrate-Bound Anion at a Mononuclear Nickel Platform. Organometallics 2018, 37, 521. 10.1021/acs.organomet.7b00887. [DOI] [Google Scholar]; d See also refs (50a) and (56b).
- a Wolf W. J.; Winston M. S.; Toste F. D. Exceptionally Fast Carbon–Carbon Bond Reductive Elimination from Gold(III). Nat. Chem. 2014, 6, 159. 10.1038/nchem.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Powers D. C.; Benitez D.; Tkatchouk E.; Goddard W. A. III; Ritter T. Bimetallic Reductive Elimination from Dinuclear Pd(III) Complexes. J. Am. Chem. Soc. 2010, 132, 14092. 10.1021/ja1036644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.