

Supplemental Digital Content is available in the text.
Keywords: acidosis, brain ischemia, mice, neurons, proton
Background and Purpose:
Brain acidosis is prevalent in stroke and other neurological diseases. Acidosis can have paradoxical injurious and protective effects. The purpose of this study is to determine whether a proton receptor exists in neurons to counteract acidosis-induced injury.
Methods:
We analyzed the expression of proton-sensitive GPCRs (G protein-coupled receptors) in the brain, examined acidosis-induced signaling in vitro, and studied neuronal injury using in vitro and in vivo mouse models.
Results:
GPR68, a proton-sensitive GPCR, was present in both mouse and human brain, and elicited neuroprotection in acidotic and ischemic conditions. GPR68 exhibited wide expression in brain neurons and mediated acidosis-induced PKC (protein kinase C) activation. PKC inhibition exacerbated pH 6-induced neuronal injury in a GPR68-dependent manner. Consistent with its neuroprotective function, GPR68 overexpression alleviated middle cerebral artery occlusion–induced brain injury.
Conclusions:
These data expand our knowledge on neuronal acid signaling to include a neuroprotective metabotropic dimension and offer GPR68 as a novel therapeutic target to alleviate neuronal injuries in ischemia and multiple other neurological diseases.
Parenchymal acidification of the brain occurs in multiple neurological diseases, including ischemic stroke.1 The magnitude of such acidosis ranges from a few tenths of pH units to below pH 6.0 (eg, in severe ischemia) and can last from seconds to hours.2 In previous studies, the pH range of 7.1 to 6.6 (mild acidosis) can lead to protection against ischemic injury while a reduction down to 6.0 or lower (severe acidosis) leads to neuronal injury.1,3–7 These results underline the importance and complexity of acid signaling in the brain.
Protons can directly gate acid-sensing ion channels (ASICs) and proton-activated chloride channel (PAC); both contribute to acidosis-mediated neuronal injury in ischemia and other diseases.5,7–11 These studies greatly advanced our understanding of how acidosis leads to neuronal injury. However, 2 fundamental biological questions remain unanswered about neuronal proton signaling. First, both ASICs and PAC mediate an injurious effect. It remains unknown whether there exists a proton receptor that mediates the protective branch as described in the pH paradox.12 Second, previous studies mainly focused on ion channels. It remains unclear whether a general acid-sensing metabotropic receptor exists in brain neurons. Answering these questions will provide novel insights into our understanding of proton signaling in health and disease.
Four proton-sensitive GPCRs (G protein coupled receptors): GPR4, GPR65, GPR68 (also termed OGR1 which stands for Ovarian cancer GPCR 1), and GPR132, exhibit acid-dependent activation within the pH range (7.4–6) normally observed in physiological and disease conditions.13 Although previous studies reported that some of these GPCRs are present in specific types of neurons,14–16 it is unclear whether metabotropic acid signaling is a general mechanism in brain neurons. Nor is it clear whether such signaling counterbalances acidosis-induced neuronal injury. Here, we started with a miniscale screening against the 4 known proton-sensitive GPCRs. Our result indicates that deleting GPR68 exacerbated pH 6-induced neuronal injury. Next, we determined the expression of GPR68 in the brain, investigated its role in acid-mediated signaling, and examined how GPR68 deletion or overexpression alters ischemia outcome.
Methods
Data Availability
All data supporting the findings of this study are available within the paper and in the Data Supplement. Additional inquires can be directed to the corresponding author.
Human Cortical Tissue
Human cortical tissue was obtained with consent. Obtaining and using of human tissue were approved by the IRB and IBC committees at the University of South Alabama (see the Data Supplement).
Mice
All procedures were approved by the University of South Alabama Animal Care and Use Committee (see the Data Supplement).
Reagents and Methods
Table I in the Data Supplement lists the antibodies and primers used. To determine gene expression, brain RNA was isolated and analyzed with standard reverse-transcription-polymerase chain reaction (RT-PCR). Immunostaining of a transgenic (Gpr68-GFP [green fluorescent protein]) mouse was performed to localize GPR68 expression in the brain. Flow cytometry analysis and sorting was used to isolated neurons from neonatal brain. Organotypic brain slices were used to study neuronal injury in vitro. Acid-induced signaling was examined by immunostaining and Western blot. Transient middle cerebral artery occlusion (MCAO) (tMCAO) in mice was performed as described earlier.5,17 Stereotaxic injection was used to deliver bicarbonate or overexpressing GPR68 in brain. See Methods in the Data Supplement for details.
Statistical Analysis
Statistics was performed in GraphPad Prism and Microsoft Excel. For 2 groups, we used 2-tailed Student t test or Mann-Whitney U test. For phospho-PKC substrates (pPKCSS) time course, we used Wilcoxon signed-rank test. For multiple comparisons, we used ANOVA followed by Tukey HSD post hoc correction. For histogram comparison, we used the Kolmogorov-Smirnov test. Data were reported as mean±SD. Differences were considered significant if P<0.05.
Results
GPR68 Mediates a Protective Effect Against Acidosis-Induced Neuronal Injury
To determine the expression of proton-sensitive GPCRs in mouse and human brains, we isolated total RNA from mouse brain and acutely resected human cortical tissue and performed RT-PCR analysis. At 35 cycles, we detected the expression of GPR4, -65, and -68 (Figure 1A and 1B). GPR68 expression was evident at 30 cycles (Figure I in the Data Supplement). Consistent with previous reports,18,19 we did not detect GPR132 expression in either mouse or human brain tissues. For this reason, we focused on GPR4, -65, and -68 in our functional screening, using an in vitro slice injury model. We cultured organotypic cortical slices from wild-type (WT) and these 3 GPCR knockout mice, treated the slices with pH 6 for 2 hours, and analyzed neuronal injury 24 hour later with propidium iodide staining. We used pH 6 here because this is a well-established condition to study acidosis-induced neuronal injury.5,6,10,20 In WT cortical slices, pH 6 treatment increased propidium iodide staining, indicating an increase in neuronal injury (Figure 1C). Compared with the WT, deleting GPR4 or GPR65 had no effect while deleting GPR68 significantly increased pH 6-induced neuronal injury. This result suggests that GPR68 mediates a protective function in acidotic condition.
Figure 1.
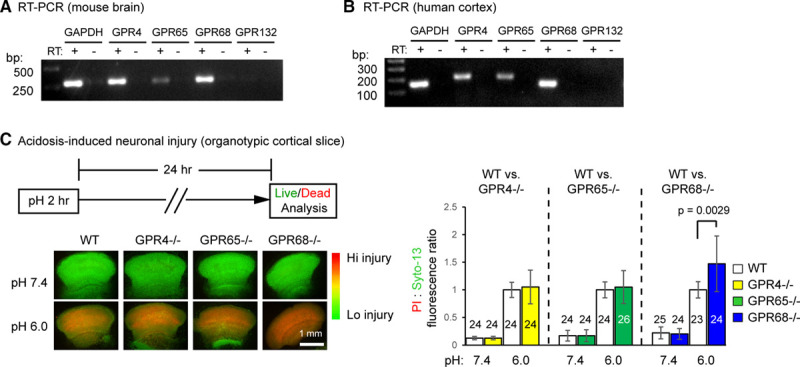
Functional screening identifies GPR68 as a regulator of pH 6-induced neuronal injury. A and B, Reverse transcription (RT)-polymerase chain reaction (PCR) result on the expression of proton-sensitive GPCRs (G protein-coupled receptor) in mouse brain (A) and human cortical tissue (B). −RT has no reverse transcriptase added into RT. Images shown were from products of 35 cycles of PCR. C, Representative fluorescence images and quantification showing pH 6-induced neuronal injury in organotypic cortical slices from wild-type (WT) and corresponding GPCR knockouts. Organotypic cortical slices were treated with pH medium buffered at pH 7.4 or 6.0 for 2 h and stained for propidium iodide (PI, red) and Syto-13 (green) 24 h later. Relative fluorescence intensity of PI and Syto-13 was quantified. Increased red/green ratio indicates increased neuronal injury. Dashed lines on the bar graph indicate that the WT controls for the 3 sets were different. For all experiments, the knockouts were compared with the WT that was cultured and treated in parallel. To better compare different experiments, injury (PI:Syto-13 ratio) in pH 6 of WT in a given experiment was normalized to 1. N on the bars indicate total number of slices quantified from 6 different experiments.
GPR68 Exhibits Ubiquitous Pattern of Expression in Brain Neurons
To understand how GPR68 exerts its protective effect, we first asked where is GPR68 expressed in the brain. RT-PCR analysis detected GPR68 expression across all brain regions examined (Figure 2A). Next, we examined GPR68 expression at the cellular level. The GPR68 antibodies which we tested were unspecific in detecting endogenous GPR68 (with negative controls using the GPR68−/− brain, not shown). Therefore, we studied a reporter mouse line, a transgenic Gpr68-GFP mouse.21 This mouse carries a transgene of a bacterial artificial chromosome, which contains the Gpr68 gene locus but with the eGFP coding region inserted after the first methionine of GPR68 (Figure 2B; diagram). Hence, the expression of GFP reflects endogenous GPR68 expression. We prepared coronal sections of brains isolated from Gpr68-GFP and WT mice and performed immunostaining using anti-GFP antibodies. Gpr68-GFP brain exhibited widespread GFP immunofluorescence, which was absent in WT brain (Figure 2B). In cortex, superficial and deeper layers exhibited higher GFP immunofluorescence. At higher magnification, the majority of cortical neurons and essentially all striatal neurons exhibited positive GFP immunofluorescence (Figure 2C). As an alternative approach to determine whether neurons preferentially express GPR68, we dissociated neocortex and striatum from WT mice, labeled neurons with an APC-conjugated anti-Thy1 antibody, sorted APC (Thy1)-positive cells with flow cytometry analysis and sorting, and performed RT-PCR analysis. Compared with unsorted cells (total), Thy1+ cells exhibited higher GPR68 and diminished GPR4 expression (Figure 2D).
Figure 2.
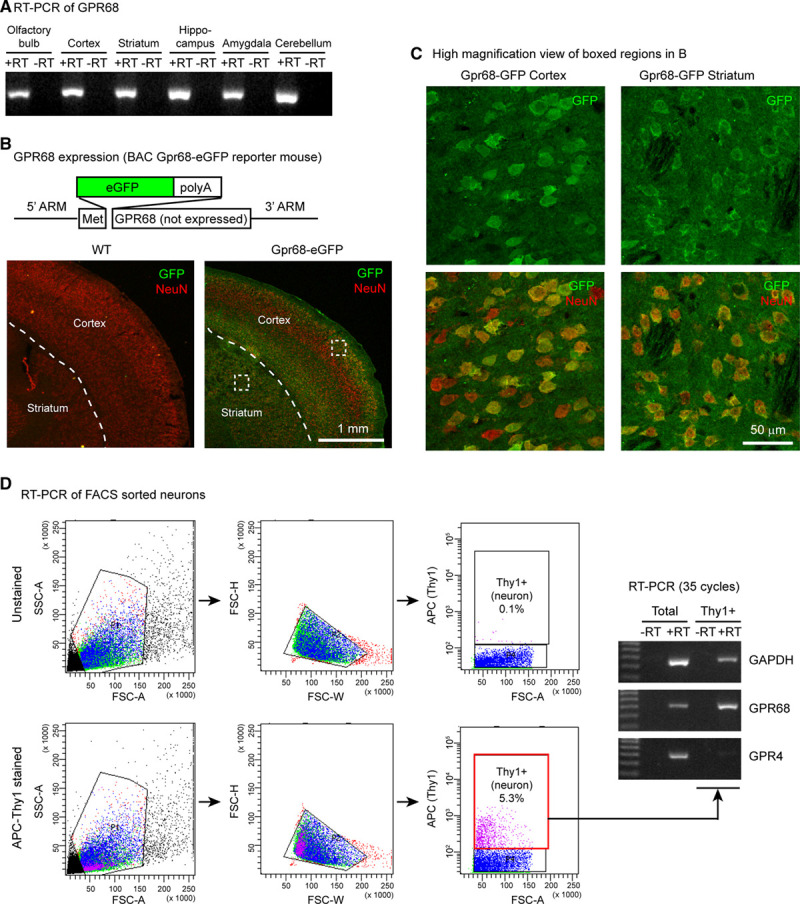
GPR68 expression in brain neurons. A, Reverse-transcription-polymerase chain reaction (RT-PCR) result on GPR68 expression in different regions of mouse brain. −RT has no reverse transcriptase added into RT. B, GFP expression in the Gpr68-eGFP reporter mouse line. Diagram illustrates the organization of the BAC transgene, in which eGFP was inserted after the first methione of GPR68. The GPR68 gene in the BAC chromosome was not expressed because of the addition of poly-A after eGFP coding. To visualize GFP signal, we performed immunofluorescence staining with an anti-GFP antibody, together with a NeuN antibody to identify neurons. Dashed lines outline the boundary between cortex and striatum. C, High magnification view of the boxed cortical and striatal regions in Gpr68-GFP image in (B). D, GPR68 expression in FACS-sorted neurons. Neurons were labeled with APC-conjugated Thy1 antibody. Left panels illustrate the gates and cell distribution of unstained (top) and antibody-stained samples. Gel images show RT-PCR result of total (unsorted) and Thy1+ (neuron) population. APC indicates allophycocyanin; BAC, bacterial artificial chromosome; eGFP, enhanced GFP; FACS, fluorescence-activated cell sorting; FSC, forward scatter; GFP, green fluorescent protein; and SSC, side scatter.
GPR68 Mediates Acidosis-Induced PKC Activation
GPR68 primarily couples to Gq, which increases inositol triphosphate and diacylglycerol,22 resulting in PKC (protein kinase C) activation. Therefore, to determine whether GPR68 mediates acid-induced signaling in brain slices, we probed for PKC-dependent activities using an antibody recognizing pPKCSS. First, we performed immunostaining in organotypic brain slices. Acidic pH (pH 6.5, 15-minute treatment) increased pPKCSS staining in WT cortical slices (Figure 3A). Essentially, all pPKCSS-positive cells were NeuN positive. Go6983, a selective inhibitor of several members of the conventional and novel PKCs: α, β, γ, δ, and ξ,23 largely abolished the response. In GPR68−/− slices, acidosis had diminished effect on pPKCSS staining.
Figure 3.
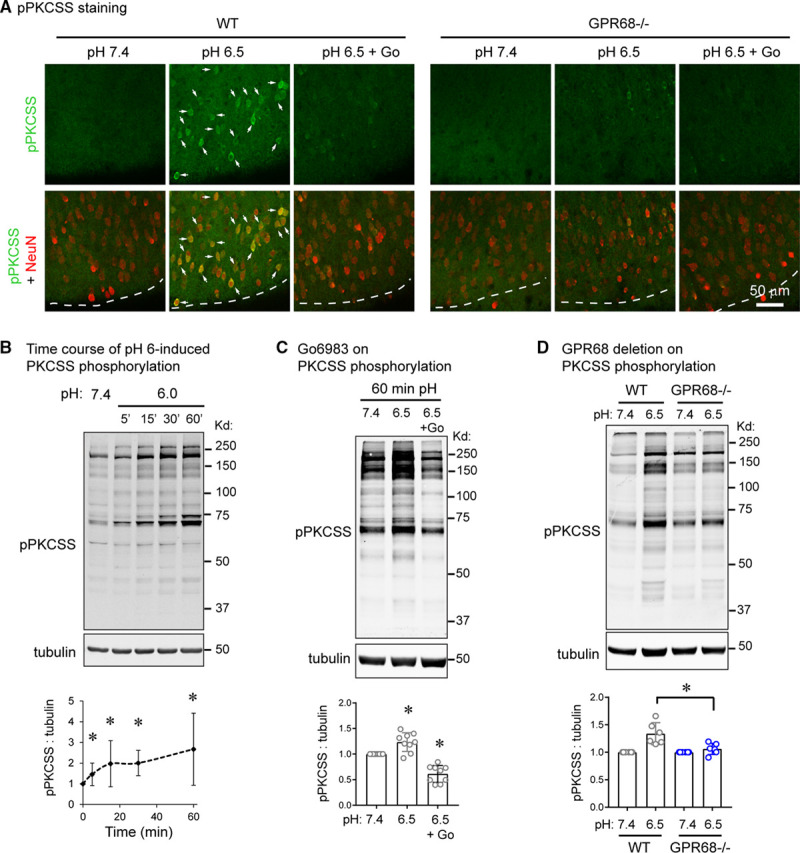
GPR68 mediates acidosis-induced signaling in slice neurons. A, Immunofluorescence of acidosis-induced phospho-PKC substrate (pPKCSS) increase in cortical slices. Wild-type (WT) and GPR68−/− slices were treated with pH 7.4, 6.5, or 6.5+Go6983 for 15 min and immunostained with a pPKCSS antibody together with a NeuN antibody. Arrows indicated neurons, which exhibited an increase in pPKCSS signal in WT slices. Dashed lines indicate the outside (superficial) boundary of the cortex. B, Representative Western blot and quantification of acidosis-induced PKC (protein kinase C) activation. Organotypic brain slices were treated with pH media as indicated, lysed, and analyzed by Western blot. To probe for PKC activation, a pPKCSS antibody was used. C, Representative Western blot and quantification showing the effect of Go6983 on acid-induced phosphorylation of PKC substrate. D, GPR68 deletion on acid-induced PKCSS phosphorylation. WT and GPR68−/− cortical slices were treated with pH media as indicated. Blots and images were representative from 4 to 7 different experiments.
To gain more quantitative measurement of acidosis-induced signaling, we performed Western blot analysis. We treated organotypic cortical or hippocampal slices with acidic medium and blotted for pPKCSS. Acidosis increased pPKCSS signals in a time-dependent manner (Figure 3B). Go6983 abolished acidosis-induced increase of pPKCSS signal (Figure 3C). Besides activated PKC, acidosis also induced CaMKII phosphorylation on Thr 286 but had little effect on phosphorylation of Akt, Erk, or JNK (Figure II in the Data Supplement). Deleting GPR68 attenuated acidosis-induced phosphorylation of PKCSS (Figure 3D) but had no significant effect on acidosis-induced CaMKII phosphorylation (Figure IIA in the Data Supplement). To determine whether PKC activity mediates the prosurvival effect of GPR68, we inhibited PKC with Go6983 and analyzed acidosis-induced injury. Go6983 worsened pH 6-induced injury in WT but not GPR68−/− slices (Figure 4A).
Figure 4.
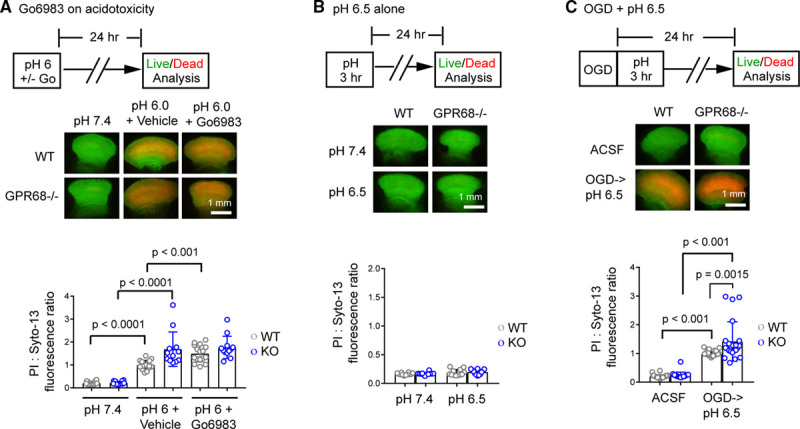
PKC (protein kinase C) inhibition or GPR68 deletion on neuronal injury in vitro. A, Organotypic cortical slices were treated with pH 7.4, 6.0+vehicle, or 6.0+5 µmol/L Go6983 as indicated for 120 min. Neuronal injury was determined by propidium iodide (PI) stain as described in Methods. B, pH 6.5 alone did not induce neuronal injury in organotypic cortical slices. C, Neuronal injury following oxygen-glucose deprivation (OGD) and pH 6.5 postincubation in vitro. Cortical slices were subjected for 30 min OGD, changed to medium buffered at pH 6.5 for 3 h, and examined for neuronal injury 24 h after OGD. Each circle indicates one slice from a total of 4 (A), 3 (B), or 7 (C) different experiments. ACSF indicates artificial cerebrospinal fluid; and WT, wild-type.
GPR68 Deletion Worsens Ischemic Neuronal Injury
Next, we investigated ischemia-induced neuronal injury. We first examined the effect of oxygen-glucose deprivation, an in vitro ischemia-reperfusion model. In mice, 60-minute tMCAO reduces brain pH to ≈6.5 for hours.24 Therefore, to better mimic in vivo ischemia paradigm, we added 3-hour pH 6.5 treatment after oxygen-glucose deprivation (Figure 4B diagram) and analyzed neuronal injury 24 hour after oxygen-glucose deprivation. As a control, we treated the slices with pH 6.5 alone for 3 hours, which did not induce neuronal injury (Figure 4B). This lack of injurious effect by pH 6.5 is consistent with previous studies.25 Following oxygen-glucose deprivation-pH 6.5 incubation, GPR68−/− slices exhibited larger injury than WT slices (Figure 4C).
To determine ischemia outcome in vivo, we performed 45-minute tMCAO, sectioned the brain 24 hours later, and stained with vital dye: 2,3,5-triphenyltetrazolium chloride.17 Infarct volume was 24.4±7.1% in WT mice. In GPR68−/− mice, infarct volume was increased significantly (P=0.0020, Mann-Whitney U test) to 38.3±11.1% (Figure 5A). Since brain infarct may continue to develop during the first 48 to 72 hours after reperfusion, we further analyzed the outcome on the third day. In this experiment, we also performed behavioral assessment. In the absence of tMCAO, the 2 genotypes did not differ in baseline locomotor activities or the corner test (Figure III in the Data Supplement). Following tMCAO, GPR68−/− mice exhibited a trend (P=0.0861, Mann-Whitney U test) of higher total travel distance in home cage monitoring (Figure 5B), which fits with previously described poststroke hyperactivity.26 Next, we quantified the number of left versus right rotations. To factor in the variation in distance traveled, we calculated a rotation index, which is defined as the number of rotations per 10 m distance traveled. This normalized index allows us to better compare animals with different activity levels. As expected for post-tMCAO animals, both WT and GPR68−/− animals exhibited more left (ipsilateral) than right rotations (Figure 5B, middle, upper bar graph, P from paired 2-tailed t test). However, the knockout mice showed a larger imbalance. When we compared the net rotation between the 2 genotypes, GPR68−/− exhibited significantly more left rotations (P=0.0287, Mann-Whitney U test) than the WT animals (Figure 5B, middle, lower bar graph). Next, we performed the corner test.27 Both genotypes exhibited a similar preferential turning to the ipsilateral (surgery) side (Figure 5B, right). Thus, both rotation analysis and the corner test confirmed that tMCAO resulted in sensorimotor deficit on the lesion/surgery side in both genotypes. However, the differences in rotation in home cage monitoring indicate that GPR68 deletion led to stronger left/right imbalance in locomotor function. Last, we performed 2,3,5-triphenyltetrazolium chloride staining at 72-hour after tMCAO. The percentage of infarct in WT was 31.71±14.3% (Figure 5C). Deletion of GPR68 increased the infarct percentage to 41.69±12.6% (P=0.0392, Mann-Whiney U test).
Figure 5.
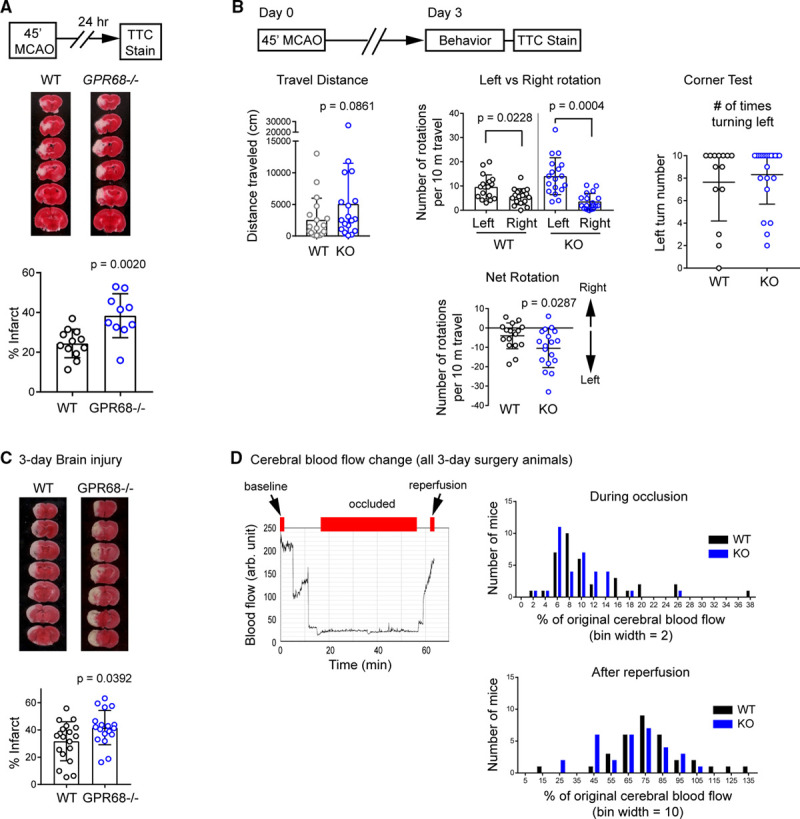
GPR68 deletion worsens ischemia outcome in vivo. A, Brain injury after middle cerebral artery occlusion (MCAO). Diagram on top right illustrates the MCAO procedure. Bottom panels show representative images and quantification of brain infarct in wild-type (WT) and GPR68−/− (KO) at 24 h after MCAO. WT and KO animals were subjected to 45 min MCAO. Brain injury was analyzed by TTC staining. B, Behavioral outcome at 3 d after MCAO. Diagram illustrates the experimental outline. On the third day after MCAO, locomotion of the mouse in a home cage was recorded for 90 min with an infrared-based SmartCage, followed by the corner test. The animals were euthanized at 72 h after MCAO, and brain injury was analyzed by TTC staining. Left panel shows the quantification of travel distance during the 60 (from 31 to 90 min) min recording. Middle shows rotation (entire 90 min) per 10 m traveled. For net rotation plot, positive and negative values indicate right and left rotations, respectively. See Methods in the Data Supplement for details. Right shows the average number of left turns (in 10 trials) in the corner test. C, Representative TTC stain and quantification of infarct percentage in WT and KO animals. D, Cerebral blood flow during occlusion and after reperfusion. Graph on the left shows a typical blood flow trace from the MCA territory, monitored with a laser doppler flowmetry. Red bars on top illustrate the window used to calculate the starting (0–1 min, set as 100%), occluded (sixth min—the end of occlusion), and reperfusion (last 1 min) of the blood flow. Histograms on the right show the distribution of blood flow changes for all animals where a successful occlusion was achieved (ie, suture reached the MCA position), which include both the animals met and those which did not meet the inclusion criteria for subsequent injury analysis (see Materials and Methods). Histograms were not different when compared for mean (Mann-Whitney U test) or frequency distribution (Kolmogorov-Smirnov test). P were from 2-tailed t test or Mann-Whitney U test as described in text. MCA indicates middle cerebral artery; and TTC, 2,3,5-triphenyltetrazolium chloride.
In this experiment, we noticed that brain infarct exhibited a large variation. We speculated that this may reflect the fact that some animals started to recover at 72 hours. However, this wide range of injury raised a question of whether GPR68 deletion alters collateral blood flow. To answer this question, we analyzed cerebral blood flow (CBF) changes during occlusion and after reperfusion. For this analysis, we included all the animals which underwent a successful surgery, which also include mice which did not meet the inclusion criteria for 2,3,5-triphenyltetrazolium chloride study and mice which met the inclusion criteria but died before the 72-hour time point. Figure 5D shows the histogram of the CBF during occlusion and after reperfusion. For either the occluded or reperfused CBF, there were no significant differences in either the mean (Mann-Whitney U test) or the frequency distribution (Kolmogorov-Smirnov test) between the occluded or reperfused CBF between the 2 genotypes.
Attenuating Acidosis or GPR68 Overexpression Alleviates Ischemia-Induced Brain Injury
Together with the literature, our data suggest the following model: a mild acidosis is sufficient to activate GPR68 while severe acidosis further recruits the injurious ASICs and PAC (Figure 6A). To assess this model, we first examined whether intracerebroventricular bicarbonate injection, which is a previously established protocol to attenuate brain acidosis,24 leads to protection. In this and the next experiment, as we were assessing protection, we increased MCAO duration to 60 minutes, which would increase the initial injury and thus facilitate the detection of a protective effect. Mice receiving saline had an average infarct of 32.6±11.2% (Figure 6B). Similar to the previous report,24 bicarbonate (2 mg/kg) reduced brain infarct to 18.7±10.2% (P=0.0289, Mann-Whitney U test). Next, we assessed the effect of GPR68 overexpression. We performed stereotaxic injection into mouse brain with adenoassociated virus (AAV)2/1 which expresses either eGFP (control) or GPR68 (Figure 6C, diagram). To ensure that we can target a large enough MCA territory, we performed injection at 2 depth and used a higher titer of AAV particles. Figure 6C, left illustrates the injection sites. At second week after injection, GFP expression was apparent in a large area in the MCA territory (Figure 6C, middle). To verify that the AAV-GPR68 virus correctly expresses GPR68, we infected organotypic slices and blotted the lysates using a custom-made GPR68 antibody, which detected overexpressed GPR68 (Figure 6C, right, top blot). Further, immunofluorescence showed that, at 3 to 4 weeks following AAV infection in vivo, the majority of GFP-positive cells were neurons (Figure 6C, right, images). To determine ischemia outcome, we performed MCAO at 3 to 5 weeks following AAV injection and analyzed brain infarction 24 hour later. Mice receiving AAV-GPR68 exhibited an average infarct of 21.97±12.4%, significantly (P=0.0022, Mann-Whitney U test) smaller than those receiving AAV-GFP (37.2±6.8%; Figure 6D).
Figure 6.
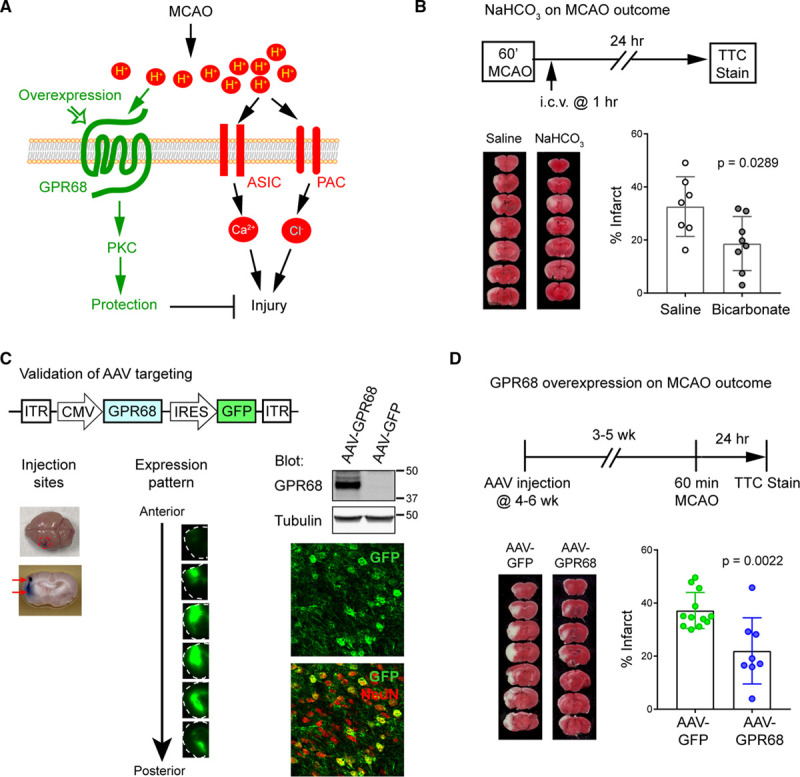
Bicarbonate or GPR68 overexpression offers protection in vivo. A, A model illustrating the protective and injurious pathways of acidosis on ischemic injury. B, Effect of bicarbonate (2 mg/kg) on ischemic injury. i.c.v. injection was performed at 1 h after transient middle cerebral artery occlusion (MCAO) (tMCAO). C, Adenoassociated virus (AAV)-mediated targeting of brain neurons. Top diagram illustrates the design of the AAV-GPR68. Left panel shows a brain injected with trypan blue; arrows indicate positions for the 2-site injection. Middle panel shows a typical set of GFP images at second week after AAV-CMV-eGFP injection. To help visualization, boundaries of the slices are traced with dashed lines. Right: Blots shown were from AAV-infected organotypic slices, blotted with a custom made GPR68 antibody (upper blot) and tubulin (lower blot). Confocal images show immunofluorescence of cryosections prepared from an AAV-GPR68 infected mouse brain. The majority of GFP-positive cells were positive for NeuN. D, GPR68 overexpression on MCAO-induced injury. Top diagram illustrates the experimental design: at 3 to 5 wks after AAV injection, 60 min MCAO was performed followed by TTC staining 24 h later. Lower panel shows representative images for TTC staining and quantification of % infarct. P were from Mann-Whitney U test. ASIC indicates acid-sensing ion channel; CMV, cytomegalovirus promoter; eGFP, enhanced GFP; GFP, green fluorescent protein; i.c.v., intracerebroventricular; IRES, internal ribosome entry site; ITR, inverted terminal repeat; PAC, proton-activated chloride channel; PKC, protein kinase C; and TTC, 2,3,5-triphenyltetrazolium chloride.
Discussion
Up until now, the literature on neuronal proton signaling focuses mostly on ion channels, including ASICs and PAC. Our data introduce a new metabotropic dimension, mediated by GPR68, in neuronal proton signaling. Both ASICs and PAC contribute to acidosis- and ischemia-induced neuronal injury.5–7,9,10,17,24 In contrast, GPR68 mediates a novel protective pathway in neurons. This finding provides new insights into our inquiry of acidosis in ischemic brain injuries. The expression of GPR68 in human cortical tissue further suggests that the similar protective mechanism may apply to human brain.
In previous studies, pH reduction to the range of 7.1 to 6.6 results in protection, further reduction down to 6.2 does not have major injurious effect by itself, and a severe acidosis to pH 6.0 or below is clearly deleterious.3–5 This pH paradox12 suggests that while acidosis is a well-established perpetrator of neuronal injury, a mild acidosis, probably in the range around 6.8, can have a protective effect. GPR68 starts to activate at pH 7.4, reaches maximal activation at ≈6.8 to 6.5, and does not exhibit rapid desensitization.22 In contrast, ASICs and PAC start to open at pH 7.2 to 7 with a pH50 of ≈6.4 (ASICs) and 5.0 (PAC).9,28,29 These data together suggest that the magnitude of acidosis determines the balance between the protective GPR68 and the injurious ASICs and PAC (see Figure 6A). One caution is that acidosis is not the only contributor to neuronal injury in vivo. The exact effect of a specific pH will depend on additional factors. For example, while pH ≈6.5 itself is not injurious, pairing it with ischemic insult can exacerbates the injury. In one study, 60-minute MCAO reduced brain pH to ≈6.5 at 4 hours; and 2.5 mg/kg bicarbonate injection raised pH to ≈7.2.24 Although we did not measure brain pH here, our result is consistent with the previous finding on bicarbonate-induced protection. Given that acidosis is a prominent feature accompanying multiple diseases, targeting GPR68 has its advantage to achieve the specificity (re: location and timing) needed for therapeutic interventions.
Previous studies have examined the expression of proton-sensitive GPCRs in the brain. GPR4 is predominantly present in cerebrovessels and in neurons within the retrotrapezoid nucleus and dorsal raphe nucleus; GPR65 exhibits restricted expression in microglia within circumventricular organs; and GPR132 was undetectable at the messenger level.14,16,18,19,30 Our RT-PCR results are in good agreement with these reports. Nevertheless, we cannot rule out a low level of GPR132 or GPR65 expression in brain neurons. About GPR68, one study shows that GPR68 is present in cerebellar granule cells and contributes to calcium signaling.15 Here, we showed that GPR68 is present throughout the brain. Further, results from 3 different experiments—immunostaining of Gpr68-GFP mouse, RT-PCR of sorted neurons, and pPKCSS immunostaining in slice neurons—all support a primary neuronal role of GPR68 in the brain. Together, these data suggest that GPR68 serves as a ubiquitously expressed proton-sensitive receptor in brain neurons. Together with previous studies on proton-sensitive ion channels, our finding suggests a more dynamic picture of neuronal proton signaling through the activation of cationic ASICs, anionic PAC, and metabotropic GPR68.
Although our result here indicates that neurons are the main cell type which expresses GPR68, immune cells and endothelial cells have been reported to express GPR68 as well.31–33 It is likely that GPR68, depending on its expression, will have differential vascular impacts in different systems. For example, GPR68 is present in high percentage in third-order mesenteric vessels but sparsely in cerebral arterial endothelial cells.31 Similarly, we did not observe apparent GFP expressing cerebral vessels in the Gpr68-GFP mouse brain (not shown). These data suggest that GPR68 may not have as big an impact on cerebral vessel as compared with mesenteric vessels.31 Our CBF result supports this speculation. Nevertheless, systematic analysis of PcomA plasticity in the GPR68−/− warrants a future study.
Our results on Go6983 indicated that GPR68-dependent protection requires, at least in part, PKC activities. It will be important to determine the specific subtype of PKC which GPR68 activates, and its downstream targets. Poststroke functional outcome is another important question. Our 3-day behavioral assessment, although remains at the acute phase, showed that GPR68−/− animals exhibited larger left:right imbalance and a trend of hyperactivity, which previously correlates with stroke-induced neurodegeneration.26,34,35 These data together suggest a larger functional deficit in GPR68−/− animals. It will be important to determine whether GPR68 deletion worsens or whether enhancing GPR68 expression or activity improves long-term functional outcome after stroke.
While the functional study here focuses on ischemic injury, pH reduction also occurs in physiological conditions. Previous studies have shown that protons regulate spine remodeling and plasticity.36–38 The high pH sensitivity makes GPR68 one excellent mediator of neuronal proton signaling. It will be interesting to examine whether GPR68 contributes to synaptic function and/or learning. Given the prevalence of pH reduction in physiology and disease, results obtained will generate essential information to better interpret how protons regulate normal brain function and pathological processes.
Acknowledgments
We thank Nan Jiang, Lan Jing, Junjun Wu (all from University of South Alabama), Tao Yang (Morehouse School of Medicine), and University South Alabama Flow Cytometry Core for technical assistance. Dr Wang performed reverse-transcription-polymerase chain reaction (RT-PCR) and in vivo experiments. G. Zhou performed immunostaining of GPR68-GFP brain, slice survival, and phosphorylation studies. Dr He performed middle cerebral artery occlusion-24 hour outcome and pilot phosphorylation analysis. Yuanyuan Xu performed RT-PCR and pilot slice survival experiments. Dr Rusyniak provided patient cortical sample. Yan Xu provided the GPR68−/− mice. Drs Ji, Simon, and Xiong provided important discussion and/or technical advice on ischemia models. Dr Zha designed the study, performed pilot experiments, and wrote the manuscript. All authors reviewed the manuscript.
Sources of Funding
The study was supported by National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NINDS) grants R01NS104349 (Dr Xiong), R01NS102495, and R21NS093522 and an intramural grant no. 1341 from University of South Alabama College of Medicine (Dr Zha). The Nikon A1 microscope was funded by an NIH/ARRA equipment grant no. S10RR027535.
Disclosures
None.
Supplemental Materials
Expanded Materials and Methods
Figures I–III
References 39–44
Supplementary Material
{kind=link}
Nonstandard Abbreviations and Acronyms
- AAV
- adenoassociated virus
- ASIC
- acid-sensing ion channel
- BAC
- bacterial artificial chromosome
- CBF
- cerebral blood flow
- GPCR
- G protein-coupled receptor
- MCAO
- middle cerebral artery occlusion
- PAC
- proton-activated chloride channel
- PKC
- protein kinase C
- pPKCSS
- phospho-PKC substrate
- RT-PCR
- reverse-transcription-polymerase chain reaction
- tMCAO
- transient MCAO
- WT
- wild-type
Dr Wang and G. Zhou contributed equally.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/STROKEAHA.120.031479.
For Sources of Funding and Disclosures, see page 3699.
Contributor Information
Tao Wang, Email: taowang@southalabama.edu.
Guokun Zhou, Email: gkzhou@shu.edu.cn.
Mindi He, Email: hemindi@tmmu.edu.cn.
Yuanyuan Xu, Email: xyuan_1216@163.com.
W.G. Rusyniak, Email: wgrusyniak@health.southalabama.edu.
Yan Xu, Email: xu2@iupui.edu.
Yonghua Ji, Email: yhji@staff.shu.edu.cn.
Roger P. Simon, Email: rsimon@msm.edu.
Zhi-Gang Xiong, Email: zxiong@msm.edu.
References
- 1.Siesjö BK, Katsura K, Mellergård P, Ekholm A, Lundgren J, Smith ML. Acidosis-related brain damage. Prog Brain Res. 1993;96:23–48. [PubMed] [Google Scholar]
- 2.Huang Y, Jiang N, Li J, Ji YH, Xiong ZG, Zha XM. Two aspects of ASIC function: synaptic plasticity and neuronal injury. Neuropharmacology. 2015;94:42–48. doi: 10.1016/j.neuropharm.2014.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tombaugh GC, Sapolsky RM. Mild acidosis protects hippocampal neurons from injury induced by oxygen and glucose deprivation. Brain Res. 1990;506:343–345. doi: 10.1016/0006-8993(90)91277-n [DOI] [PubMed] [Google Scholar]
- 4.Simon RP, Niro M, Gwinn R. Brain acidosis induced by hypercarbic ventilation attenuates focal ischemic injury. J Pharmacol Exp Ther. 1993;267:1428–1431. [PubMed] [Google Scholar]
- 5.Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–698. doi: 10.1016/j.cell.2004.08.026 [DOI] [PubMed] [Google Scholar]
- 6.Sherwood TW, Askwith CC. Dynorphin opioid peptides enhance acid-sensing ion channel 1a activity and acidosis-induced neuronal death. J Neurosci. 2009;29:14371–14380. doi: 10.1523/JNEUROSCI.2186-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friese MA, Craner MJ, Etzensperger R, Vergo S, Wemmie JA, Welsh MJ, Vincent A, Fugger L. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat Med. 2007;13:1483–1489. doi: 10.1038/nm1668 [DOI] [PubMed] [Google Scholar]
- 8.Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature. 1997;386:173–177. doi: 10.1038/386173a0 [DOI] [PubMed] [Google Scholar]
- 9.Yang J, Chen J, Del Carmen Vitery M, Osei-Owusu J, Chu J, Yu H, Sun S, Qiu Z. PAC, an evolutionarily conserved membrane protein, is a proton-activated chloride channel. Science. 2019;364:395–399. doi: 10.1126/science.aav9739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yermolaieva O, Leonard AS, Schnizler MK, Abboud FM, Welsh MJ. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci U S A. 2004;101:6752–6757. doi: 10.1073/pnas.0308636100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin T, Lindley TE, Albert GW, Ahmed R, Schmeiser PB, Grady MS, Howard MA, Welsh MJ. Loss of acid sensing ion channel-1a and bicarbonate administration attenuate the severity of traumatic brain injury. PLoS One. 2013;8:e72379 doi: 10.1371/journal.pone.0072379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LaManna JC. Hypoxia/ischemia and the pH paradox. Adv Exp Med Biol. 1996;388:283–292. doi: 10.1007/978-1-4613-0333-6_36 [DOI] [PubMed] [Google Scholar]
- 13.Okajima F. Regulation of inflammation by extracellular acidification and proton-sensing GPCRs. Cell Signal. 2013;25:2263–2271. doi: 10.1016/j.cellsig.2013.07.022 [DOI] [PubMed] [Google Scholar]
- 14.Kumar NN, Velic A, Soliz J, Shi Y, Li K, Wang S, Weaver JL, Sen J, Abbott SB, Lazarenko RM, et al. PHYSIOLOGY. Regulation of breathing by CO2 requires the proton-activated receptor GPR4 in retrotrapezoid nucleus neurons. Science. 2015;348:1255–1260. doi: 10.1126/science.aaa0922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei WC, Jacobs B, Becker EB, Glitsch MD. Reciprocal regulation of two G protein-coupled receptors sensing extracellular concentrations of Ca2+ and H. Proc Natl Acad Sci U S A. 2015;112:10738–10743. doi: 10.1073/pnas.1506085112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hosford PS, Mosienko V, Kishi K, Jurisic G, Seuwen K, Kinzel B, Ludwig MG, Wells JA, Christie IN, Koolen L, et al. CNS distribution, signalling properties and central effects of G-protein coupled receptor 4. Neuropharmacology. 2018;138:381–392. doi: 10.1016/j.neuropharm.2018.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang N, Wu J, Leng T, Yang T, Zhou Y, Jiang Q, Wang B, Hu Y, Ji YH, Simon RP, et al. Region specific contribution of ASIC2 to acidosis-and ischemia-induced neuronal injury. J Cereb Blood Flow Metab. 2017;37:528–540. doi: 10.1177/0271678X16630558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weng Z, Fluckiger AC, Nisitani S, Wahl MI, Le LQ, Hunter CA, Fernal AA, Le Beau MM, Witte ON. A DNA damage and stress inducible G protein-coupled receptor blocks cells in G2/M. Proc Natl Acad Sci U S A. 1998;95:12334–12339. doi: 10.1073/pnas.95.21.12334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zohn IE, Klinger M, Karp X, Kirk H, Symons M, Chrzanowska-Wodnicka M, Der CJ, Kay RJ. G2A is an oncogenic G protein-coupled receptor. Oncogene. 2000;19:3866–3877. doi: 10.1038/sj.onc.1203731 [DOI] [PubMed] [Google Scholar]
- 20.Wang YZ, Wang JJ, Huang Y, Liu F, Zeng WZ, Li Y, Xiong ZG, Zhu MX, Xu TL. Tissue acidosis induces neuronal necroptosis via asic1a channel independent of its ionic conduction. Elife. 2015;4:e05682 doi: 10.7554/eLife.05682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033 [DOI] [PubMed] [Google Scholar]
- 22.Ludwig MG, Vanek M, Guerini D, Gasser JA, Jones CE, Junker U, Hofstetter H, Wolf RM, Seuwen K. Proton-sensing G-protein-coupled receptors. Nature. 2003;425:93–98. doi: 10.1038/nature01905 [DOI] [PubMed] [Google Scholar]
- 23.Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marmé D, Schächtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- 24.Pignataro G, Simon RP, Xiong ZG. Prolonged activation of ASIC1a and the time window for neuroprotection in cerebral ischaemia. Brain. 2007;130:151–158. doi: 10.1093/brain/awl325 [DOI] [PubMed] [Google Scholar]
- 25.Kaku DA, Giffard RG, Choi DW. Neuroprotective effects of glutamate antagonists and extracellular acidity. Science. 1993;260:1516–1518. doi: 10.1126/science.8389056 [DOI] [PubMed] [Google Scholar]
- 26.Balkaya M, Kröber JM, Rex A, Endres M. Assessing post-stroke behavior in mouse models of focal ischemia. J Cereb Blood Flow Metab. 2013;33:330–338. doi: 10.1038/jcbfm.2012.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Schallert T, Zhang ZG, Jiang Q, Arniego P, Li Q, Lu M, Chopp M. A test for detecting long-term sensorimotor dysfunction in the mouse after focal cerebral ischemia. J Neurosci Methods. 2002;117:207–214. doi: 10.1016/s0165-0270(02)00114-0 [DOI] [PubMed] [Google Scholar]
- 28.Zha XM. Acid-sensing ion channels: trafficking and synaptic function. Mol Brain. 2013;6:1 doi: 10.1186/1756-6606-6-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gründer S, Pusch M. Biophysical properties of acid-sensing ion channels (ASICs). Neuropharmacology. 2015;94:9–18. doi: 10.1016/j.neuropharm.2014.12.016 [DOI] [PubMed] [Google Scholar]
- 30.Vollmer LL, Ghosal S, McGuire JL, Ahlbrand RL, Li KY, Santin JM, Ratliff-Rang CA, Patrone LG, Rush J, Lewkowich IP, et al. Microglial acid sensing regulates carbon dioxide-evoked fear. Biol Psychiatry. 2016;80:541–551. doi: 10.1016/j.biopsych.2016.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu J, Mathur J, Vessières E, Hammack S, Nonomura K, Favre J, Grimaud L, Petrus M, Francisco A, Li J, et al. GPR68 Senses flow and is essential for vascular physiology. Cell. 2018;173:762–775.e16. doi: 10.1016/j.cell.2018.03.076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D’Souza CA, Zhao FL, Li X, Xu Y, Dunn SE, Zhang L. OGR1/GPR68 Modulates the severity of experimental autoimmune encephalomyelitis and regulates nitric oxide production by macrophages. PLoS One. 2016;11:e0148439 doi: 10.1371/journal.pone.0148439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H, Wang D, Singh LS, Berk M, Tan H, Zhao Z, Steinmetz R, Kirmani K, Wei G, Xu Y. Abnormalities in osteoclastogenesis and decreased tumorigenesis in mice deficient for ovarian cancer G protein-coupled receptor 1. PLoS One. 2009;4:e5705 doi: 10.1371/journal.pone.0005705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kilic E, Kilic U, Bacigaluppi M, Guo Z, Abdallah NB, Wolfer DP, Reiter RJ, Hermann DM, Bassetti CL. Delayed melatonin administration promotes neuronal survival, neurogenesis and motor recovery, and attenuates hyperactivity and anxiety after mild focal cerebral ischemia in mice. J Pineal Res. 2008;45:142–148. doi: 10.1111/j.1600-079X.2008.00568.x [DOI] [PubMed] [Google Scholar]
- 35.Winter B, Juckel G, Viktorov I, Katchanov J, Gietz A, Sohr R, Balkaya M, Hörtnagl H, Endres M. Anxious and hyperactive phenotype following brief ischemic episodes in mice. Biol Psychiatry. 2005;57:1166–1175. doi: 10.1016/j.biopsych.2005.02.010 [DOI] [PubMed] [Google Scholar]
- 36.Zha XM, Wemmie JA, Green SH, Welsh MJ. Acid-sensing ion channel 1a is a postsynaptic proton receptor that affects the density of dendritic spines. Proc Natl Acad Sci U S A. 2006;103:16556–16561. doi: 10.1073/pnas.0608018103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du J, Reznikov LR, Price MP, Zha XM, Lu Y, Moninger TO, Wemmie JA, Welsh MJ. Protons are a neurotransmitter that regulates synaptic plasticity in the lateral amygdala. Proc Natl Acad Sci U S A. 2014;111:8961–8966. doi: 10.1073/pnas.1407018111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kreple CJ, Lu Y, Taugher RJ, Schwager-Gutman AL, Du J, Stump M, Wang Y, Ghobbeh A, Fan R, Cosme CV, et al. Acid-sensing ion channels contribute to synaptic transmission and inhibit cocaine-evoked plasticity. Nat Neurosci. 2014;17:1083–1091. doi: 10.1038/nn.3750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang LV, Radu CG, Roy M, Lee S, McLaughlin J, Teitell MA, Iruela-Arispe ML, Witte ON. Vascular abnormalities in mice deficient for the G protein-coupled receptor GPR4 that functions as a pH sensor. Mol Cell Biol. 2007;27:1334–1347. doi: 10.1128/MCB.01909-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radu CG, Nijagal A, McLaughlin J, Wang L, Witte ON. Differential proton sensitivity of related G protein-coupled receptors T cell death-associated gene 8 and G2A expressed in immune cells. Proc Natl Acad Sci U S A. 2005;102:1632–1637. doi: 10.1073/pnas.0409415102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kahle MP, Bix GJ. Successfully climbing the “STAIRs”: surmounting failed translation of experimental ischemic stroke treatments. Stroke Res Treat. 2012;2012:374098 doi: 10.1155/2012/374098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stroke Therapy Academic Industry R. Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke. 1999;30:2752–2758. doi: 10.1161/01.str.30.12.2752 [DOI] [PubMed] [Google Scholar]
- 43.Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47 [DOI] [PubMed] [Google Scholar]
- 44.Xu Y, Jiang YQ, Li C, He M, Rusyniak WG, Annamdevula N, Ochoa J, Leavesley SJ, Xu J, Rich TC, et al. Human ASIC1a mediates stronger acid-induced responses as compared with mouse ASIC1a. FASEB J. 2018;32:3832–3843. doi: 10.1096/fj.201701367R [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the paper and in the Data Supplement. Additional inquires can be directed to the corresponding author.