

Abstract
Telomere maintenance via telomerase reactivation is a nearly universal hallmark of cancer cells which enables replicative immortality. In contrast, telomerase activity is silenced in most adult somatic cells. Thus, telomerase represents an attractive target for highly selective cancer therapeutics. However, development of telomerase inhibitors has been challenging and thus far there are no clinically approved strategies exploiting this cancer target. The discovery of prevalent mutations in the TERT promoter region in many cancers and recent advances in telomerase biology has led to a renewed interest in targeting this enzyme. Here we discuss recent efforts targeting telomerase, including immunotherapies and direct telomerase inhibitors, as well as emerging approaches such as targeting TERT gene expression driven by TERT promoter mutations. We also address some of the challenges to telomerase-directed therapies including potential therapeutic resistance and considerations for future therapeutic applications and translation into the clinical setting. Although much work remains to be done, effective strategies targeting telomerase will have a transformative impact for cancer therapy and the prospect of clinically effective drugs is boosted by recent advances in structural models of human telomerase.
Telomerase has been considered an attractive target for cancer therapy since the discovery over 20 years ago that reactivation of this enzyme in cancer cells mediates immortalization via telomere extension [1]. Telomerase represents a highly specific target for transformed cells, as its reverse transcriptase activity is silenced in most normal adult somatic cells, except in some stem-like cells and T-cells which transiently activate telomerase during proliferation [2]. Furthermore, upregulation of telomerase is a nearly universal feature across diverse cancer types, suggesting that strategies targeting telomerase could have broad therapeutic applicability. Additionally, whereas oncogenic signalling pathways typically exhibit substantial redundancy, facilitating therapeutic resistance, thus far only a single alternative pathway for telomere maintenance has been identified. Tumour cells are therefore expected to possess a limited capacity for resistance to telomerase therapies. Accordingly, significant effort has been directed towards developing drugs that target telomerase for cancer therapy. Herein we discuss the status of telomerase as a cancer target, focusing on recent advances, challenges to translate promising preclinical results, and opportunities for future directions.
Telomerase and telomere maintenance
Vertebrate telomeres consist of an array of TTAGGG nucleotide repeats at the chromosome termini, which are bound by a six-member protein complex known as shelterin. These structures preserve genomic integrity, protecting chromosomes from unchecked degradation and preventing aberrant activation of a DNA damage response (DDR) that could lead to inappropriate processing of telomeres as sites for double-strand break repair [3]. Telomeres terminate with a 50–200 nucleotide single-stranded 3’ overhang that can invade preceding telomeric dsDNA to form a stable telomere loop (T-loop) structure with shelterin [4]. Each cell division results in the loss of 50–100 bp from telomeres due to the inability of DNA polymerases to replicate the end of the lagging strand, oxidative damage, and exonuclease resection [5] [6].
Telomere shortening can be counteracted by the telomerase ribonucleoprotein complex, which extends the 3’ overhang via telomerase reverse transcriptase (TERT) catalytic activity [7]. TERT uses an RNA template (TERC) to synthesize single-stranded TTAGGG repeats. TERT and TERC are sufficient to reconstitute telomerase activity in vitro, although additional factors such as H/ACA RNPs and TCAB1 regulate assembly and localization of the human telomerase holoenzyme in vivo (reviewed in [8]). TERT expression is silenced during development, unlike TERC and other telomerase components which are constitutively expressed. Consequently, TERT levels typically act as the limiting factor for telomerase activity in somatic human cells, although TERC can be limiting in some cancers and stem cells [9] [10] [11]. TERC levels have been found to be upregulated in certain cancer types, such as carcinomas of the cervix, ovary, head and neck, and lung, thereby providing a potential anti-tumour target [10] [11].
Telomerase and telomere dysfunction in cancer
Silencing of TERT expression results in gradual telomere shortening with each cell division. Eventually, critical telomere attrition elicits a DDR that mediates cell cycle arrest leading to replicative senescence or apoptosis via the p53 or Rb tumour suppressor pathways [12]. Thus, telomere attrition acts as a barrier to replicative immortality. Neoplastic alterations can permit replication beyond this checkpoint. However, continued telomere erosion eventually elicits telomere crisis, a process characterized by telomere dysfunction driving extensive genomic instability and cell death. Rare viable clones may escape from crisis via reactivation of telomere maintenance mechanisms [13].
The vast majority of cancers overcome replicative senescence by upregulating TERT expression and hence telomerase activity; telomerase activity has been reported in ~90% of cancers [1]. A recent pan-cancer genomics study detected TERT expression in ~75% of tumour samples [14], with 31% of TERT-expressing samples harbouring point mutations in the TERT promoter and 53% exhibiting TERT promoter methylation. However, this may not fully reflect the prevalence of telomerase reactivation in cancer, as minimal TERT expression is sufficient to maintain telomeres [15].
Aberrant expression of TERT in approximately 15–25% of tumours [14] [16] is driven by mutually exclusive mutations in the TERT promoter (−57 A>C; −124 C>T; −138/−139 CC>TT; −146 C>T) that generate de novo binding sites for ETS family transcription factors, such as GABP [17] [18]. TERT promoter mutations (TPMs) are predominantly heterozygous and lead to the allele-specific re-expression of TERT from the mutant promoter via recruitment of GABP, promoting an epigenetic shift from a repressed to active chromatin conformation [19]. Notably, TPMs constitute the most common non-coding driver mutations in cancer [20]. For example, ~85% of cutaneous melanomas harbour TPMs [21]. Importantly, TPMs are associated with elevated TERT expression and worse overall survival in many cancers including glioblastoma [22], cutaneous melanoma [23] and meningioma [24]. Furthermore, tumours harbouring TPMs may have a higher risk of recurrence [25]. Cancers with high TPM incidence generally have low rates of self-renewal and/or are associated with exposure to specific mutagenic factors such as ultraviolet radiation [26].
Other mechanisms that promote TERT expression in cancer include TERT gene amplification, chromosomal rearrangement, and promoter hypermethylation [14] [27]. Additionally, telomere shortening has been implicated in re-expression of TERT in cancer via an epigenetic looping mechanism whereby the 5p sub-telomeric region forms a chromatin loop with the TERT locus that represses telomerase transcription when telomeres are long, but is disengaged upon telomere shortening [28].
Telomere length in cancer
Telomere length in tumours is generally shorter than that of matched normal tissue [14]. This is likely to be a consequence of tumour cells having undergone more cell divisions than non-malignant cells. Although prospective studies have found little or no association between telomere length and cancer risk [29], Mendelian randomization analyses, which are less susceptible to confounding effects and reverse causation, indicate that germline genetic variants associated with long telomeres increase cancer risk for most cancer types [30] [31]. This may be due to longer telomeres permitting more cell divisions before the onset of replicative senescence and thereby increasing the likelihood of non-malignant cells acquiring oncogenic lesions [32].
Telomere length can impact the efficacy of telomerase-directed therapy. Time taken for telomerase inhibitors to exert anticancer effects is expected to depend on initial telomere length since the length of the shortest telomere in a cell dictates the onset of telomere dysfunction [33]. Consequently, determining the distribution of telomere lengths rather than mean telomere length is advisable when monitoring response to telomerase therapies [34]. A phase II trial of the telomerase inhibitor imetelstat identified a trend towards increased survival in patients with short telomeres, indicating that telomerase inhibition could be most beneficial in this subpopulation [35]. Cancer cells typically have shorter telomeres than normal somatic cells and, conversely, telomere length in most telomerase-expressing stem cells is longer than in the corresponding differentiated somatic cells [36]. This differential telomere length may provide a suitable therapeutic window for telomerase inhibitors to selectively kill cancer cells without eliciting excessive toxicity due to effects on telomerase-expressing stem cells. Nonetheless, the predictive value of telomere length to cancer risk and response to therapy requires further evaluation.
Non-canonical functions of telomerase
Pro-survival roles for telomerase have been proposed beyond its canonical role in telomere maintenance. These non-canonical functions include modulation of chromatin state, DNA damage responses, oxidative stress protection, and proliferative gene activity [37] [38] [39]. For example, telomerase may promote MYC-driven tumourigenesis independently of its reverse transcriptase activity. Homozygous deletion of TERT, but not TERC, delays MYC-induced lymphogenesis in mice [39]. Short-term depletion of endogenous TERT mediates telomere dysfunction independently of telomere shortening by destabilizing a telomere protective complex containing the nuclease Snm1B/Apollo and TRF2 [40]. Although still debatable, these non-canonical functions may explain the rapid cell death and apparent reverse transcriptase activity-independent effects observed upon telomerase inhibition in some models [41]. Nonetheless, these findings are provocative and suggest the value of developing novel strategies inhibiting extra-telomeric functions.
Telomerase as a cancer target: challenges and opportunities
Although telomerase possesses many desirable properties as a cancer target, development of successful clinical therapies has been hampered by significant challenges including limitations of preclinical models, lack of high resolution structure of human telomerase, and adaptive drug resistance, as summarized below.
Therapies based on inhibiting telomerase reverse transcriptase activity require an extended period of treatment before anti-cancer effects are exerted due to their reliance on the gradual attrition of telomeres with each cell division. This may make them unsuitable for use as first-line therapy and increase the potential for evolution and outgrowth of resistant clones. Furthermore, side effects of telomerase-directed therapies could potentially arise due to expression of telomerase in stem and precursor cells such as hematopoietic lineages. Additionally, telomerase inhibitor efficacy is likely to be constrained by the ability of low levels of telomerase activity to maintain short telomeres and sustain tumour cell proliferation, indicating a need for highly potent inhibitors. Nevertheless, telomerase is an attractive cancer target in terms of its near universality, high specificity to cancer cells and ability to confer replicative immortality.
Preclinical modelling of telomerase-directed therapies
Preclinical studies often rely upon the availability of mouse models that closely recapitulate human pathology. However, mice have several shortcomings as models of human telomere biology. Established inbred mouse strains have far longer telomeres (5–10 times) than humans or more recently derived strains and do not appear to rely upon telomere erosion-induced replicative senescence as a protective mechanism against cancer [42]. Unlike humans, mouse telomerase is widely expressed in adult tissues [43]. Thus, telomerase activation does not present a comparable barrier to replicative immortality in mice as it does in humans. This likely contributes to the greater susceptibility of mouse cells to transformation on a per-cell basis [44]. Defects in telomerase function or telomere maintenance elicit distinct phenotypes in mice vs. humans. For instance, mice tolerate complete loss of telomere extension due to TERC knockout for several generations before telomeres become dysfunctional and mice exhibit haematopoietic deficiencies [45] [46], whereas heterozygous telomerase point mutations are sufficient to predispose to regenerative diseases in humans due to haploinsufficiency [47].
The functional and phenotypic differences between mouse and human telomere biology hinder preclinical evaluation of telomerase-directed therapies. Mouse models are unlikely to closely recapitulate side effects of telomerase inhibition on telomerase-expressing cells considering their tolerance for telomerase ablation. Furthermore, testing therapies based on telomere attrition in mice bearing fast-growing tumours can be problematic, as animals may need to be euthanized before the anti-tumour effects of telomerase inhibition become evident. Hence, better models to bolster the predictive value of preclinical telomerase therapy studies are sorely needed. Early passage patient-derived xenografts, which closely match the genetic complexity of human cancers, may prove such models.
Structural models of telomerase
Until recently, the structure and composition of the human telomerase holoenzyme were poorly characterized, hampering drug design and mechanistic analysis. This is partly a consequence of the low cellular abundance of telomerase hindering purification and crystallization of active telomerase. Initial low-resolution (30 Å) negative-stain electron microscopy reconstruction of human telomerase revealed a bilobal structure interpreted as a TERT dimer [48]. Our understanding of human telomerase architecture has been guided by high-resolution structures from the flour beetle Tribolium castaneum (2.7 Å) [49] and the protozoan Tetrahymena thermophila (4.8 Å) [50], which revealed that the TERT domains form a ring structure. Although not yet at atomic resolution, a recent cryo-electron microscopy study indicates that substrate-bound human telomerase forms a monomeric structure (7–8 Å) [51] consisting of two lobes linked by an extended hTR RNA scaffold; a catalytic lobe contains TERT and its associated hTR motifs, while an H/ACA ribonucleoprotein (RNP) lobe harbours two sets of heterotetrameric H/ACA proteins bound to RNA hairpins plus a single copy of the nuclear trafficking regulator, TCAB1 [52]. Ultimately, these improvements to structural resolution should facilitate the design of more effective small molecule inhibitors targeting human telomerase.
Activation of adaptive mechanisms of telomere maintenance
Telomeres are maintained by telomerase-independent homologous recombination mechanisms known as alternative lengthening of telomeres (ALT) in ~10% of cancers that lack telomerase expression [53] [54]. ALT is uncommon in epithelial malignancies, but highly prevalent in cancers of a mesenchymal origin such as certain sarcomas [55]. ALT-positive cells are characterized by long, heterogeneous telomere length, extrachromosomal telomeric DNA, telomeric-sister chromatid exchange (T-SCE) and PML nuclear bodies containing telomeric DNA (ALT-associated PML bodies; APBs). Although ALT mechanisms are not well-defined, cancers with high ALT prevalence frequently harbour loss-of-function mutations in the ATRX/DAXX chromatin remodelling complex, which may promote telomeric recombination by impairing resolution of sister telomere cohesion [56]. ATRX suppresses ALT, however, additional genetic alterations are required to activate ALT [57]. In p53/pRb-deficient glioma models, oncogenic IDH1 point mutation cooperates with ATRX loss to promote ALT [58]. Likewise, depletion of ASF1a and ASF1b histone chaperones is sufficient to trigger ALT in cell lines with long telomeres that may be predisposed to ALT such as HeLa LT, highlighting the key role of chromatin disruption to ALT activation [59].
The existence of telomerase-independent mechanisms of telomere maintenance raises the question of whether telomerase inhibition could be circumvented by switching telomere maintenance to ALT pathways. This would be a concern if ALT activity pre-exists in clonal populations prior to treatment. Studies of the prevalence of ALT in non-neoplastic tissues or following telomerase ablation in model systems indicate the rarity of this phenomenon in humans. ALT activity was not detected in benign neoplasms or normal human tissue samples monitored for APBs [53]. Although the emergence of ALT as a resistance mechanism to genetic ablation of telomerase activity was identified in rare (~2.5×10−7) clones following TERC knockout from telomerase-positive human cells [60]. In mice, ALT switching has been identified in a conditional TERT-driven ATM-null model of T-cell lymphoma [61]. However, an ATM-null model may be predisposed to ALT considering ATM has dual roles at telomeres; involved in signalling telomere dysfunction and regulating telomere elongation [62]. Moreover, susceptibility to ALT switching may be distinct between mice and humans, considering intrinsic differences in telomere biology, tumour suppression and immortalization rates.
Anticancer strategies targeting telomerase
Approaches to targeting telomerase range from immunotherapies that recognize TERT tumour-associated antigens, to small molecule inhibitors or oligonucleotides that directly bind telomerase and suppress telomere extension, to indirect methods of disrupting telomerase regulation or function such as G-quadruplex stabilization, targeting TERT gene expression or inducing telomere dysfunction through the incorporation of nucleoside analogues into newly extended telomeres (Figure 1).
Figure 1 –
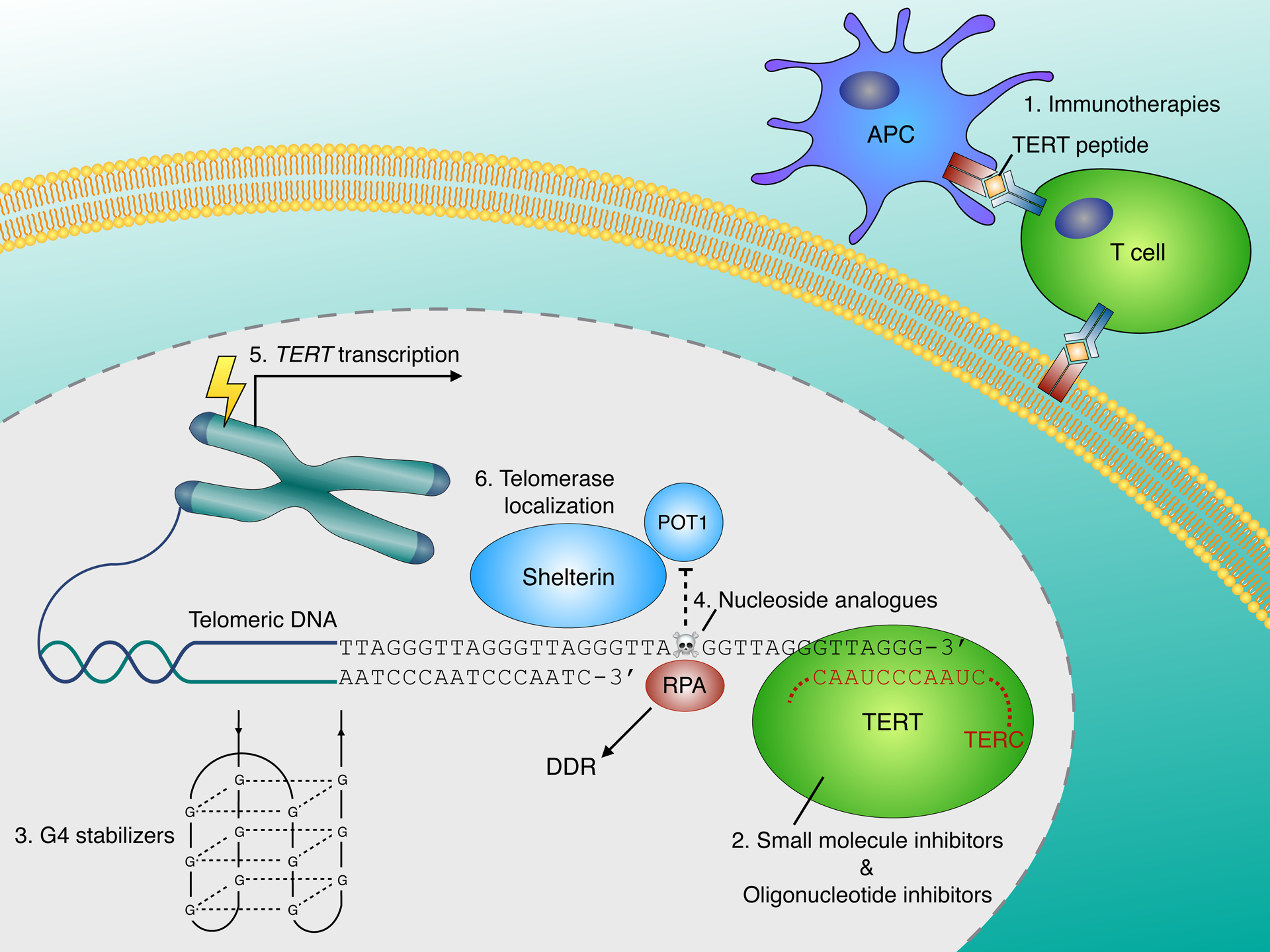
Therapeutic strategies for targeting telomerase
Approaches to targeting telomerase include: 1. Immunotherapies - Peptide or DNA vaccines supply immunogenic TERT epitopes that stimulate immune responses against telomerase-expressing cancer cells. Adoptive cell transfer therapies entail the infusion of telomerase-specific cytotoxic T-cells. 2. Direct telomerase inhibitors – small molecules can bind to TERT and inhibit its catalytic activity resulting in gradual telomere attrition. Alternatively, oligonucleotides complementary to the TERC template region can act as competitive telomerase inhibitors. 3. G-quadruplex (G4) stabilizers disrupt telomerase function by blocking the resolution of telomeric G-quadruplex DNA. 4. Incorporation of nucleoside analogues into newly synthesized telomeres impairs POT1 binding, causing telomere dysfunction that elicits a DNA damage response and cell death. 5. Targeting TERT gene expression - TERT promoter mutations (TPMs) generate novel binding sites for ETS transcription factors that reactivate TERT expression in cancer (see Figure 2). Targeting regulation of the mutant TERT promoter represents an emerging approach. 6. Disrupting telomerase localization – interference with telomerase recruitment mediated by TCAB1 and shelterin subunits elicits telomere dysfunction.
Immunotherapies
The development of immunotherapies targeting telomerase was prompted by its identification as a widely expressed tumour-associated antigen [63]. Endogenous TERT peptides produced by cancer cells can be recognized by major histocompatibility complex (MHC) class I or II molecules and trigger adaptive immune responses. Telomerase-directed immunotherapies include vaccines, adoptive cell transfer and arguably oncolytic virotherapy.
Numerous TERT peptide vaccines have progressed to early stage clinical trials, typically eliciting few adverse events despite concerns over potential autoimmune responses against haematopoietic cells that express telomerase during clonal expansion. Therapeutic TERT-based vaccines are capable of mediating specific T-cell responses in a high proportion of cancer patients. For example, the TERT peptide vaccine UV1 elicited an immune response in 86% of patients with metastatic hormone-naive prostate cancer enrolled in a phase I/IIa trial [64]. However, immune responses to TERT vaccines have proved insufficient to control disease progression. Four vaccines have progressed to phase II trials, of which one TERT vaccine (GV1001) has advanced to phase III (see [65] for a summary of TERT immunotherapy trials). The phase III trial of GV1001 in patients with advanced pancreatic cancer failed to demonstrate any survival advantage over chemotherapy [66]. In contrast to vaccines, immune checkpoint inhibitors unleash potent anti-tumour immune responses in a subset of patients, dramatically improving patient outcomes in many cancers [67]. Thus, TERT vaccines have been evaluated in combination with immune checkpoint blockade in preclinical studies. A synthetic TERT DNA vaccine synergized with anti-CTLA-4 therapy to suppress tumour growth and prolong survival in a mouse model weakly responsive to single immune checkpoint inhibitors, providing a strong rationale to support further development of immunotherapies combining TERT vaccines with immune checkpoint inhibitors [68].
While TERT vaccination has hitherto achieved limited anti-tumour efficacy, clinical trials have not selected for patients or cancer types most likely to respond. Responses to TERT immunotherapy may be boosted by enrolling patients with TERT promoter mutations and/or high TERT expression, as high TERT expression may enhance TERT antigen presentation. Vaccine efficacy may be limited by immune-tolerance processes selecting against T-cells expressing T-cell receptors with high avidity for wildtype TERT antigens. In light of this, adoptive cell transfer (ACT) has been evaluated in preclinical studies. High avidity telomerase-specific cytotoxic T-cells impaired tumour growth and enhanced survival in mouse cancer models, but also caused transient B-cell depletion due to autoimmunity [69]. T-cells transduced with a high avidity T-cell receptor for human TERT suppressed acute myeloid leukaemia or acute lymphoblastic leukaemia progression following ACT in humanized mouse models [70].
The elevated telomerase expression characteristic of cancer has been exploited by oncolytic virotherapies that target telomerase-expressing cells. Telomelysin is an oncolytic adenovirus designed to selectively replicate in cancer cells via E1 gene expression under the control of the hTERT promoter. This strategy enriched, but did not confine, viral replication to cancer cells versus untransformed cells in vitro [71]. Telomelysin induced cell death in cancer cells, suppressed xenograft growth, and sensitized non-immunogenic gastrointestinal tumours to anti-PD1 immunotherapy in preclinical mouse models [72]. Intratumoural injection of telomelysin was well tolerated in a phase I trial, however there was limited evidence for an anti-tumour response [73]. Questions remain over the selectivity of an oncovirus regulated by the wildtype hTERT promoter considering that TERT reactivation in cancer cells is commonly mediated by alterations to the TERT locus rather than upstream factors.
Direct inhibitors
Oligonucleotide inhibitors
Despite the extensive history of telomerase as a cancer target, only a single direct telomerase inhibitor, imetelstat, has progressed to clinical trials. Imetelstat is a lipidated 13-mer thiophosphoramidate oligonucleotide complementary to the TERC template region, which competitively inhibits telomerase activity, suppressing cancer cell viability in vitro and tumour growth in mouse xenograft models [74]. Imetelstat promotes gradual telomere attrition resulting in activation of a DNA damage response and cell death following a prolonged lag period. Clinical trials on patients with solid tumours uncovered dose-limiting toxicity due to haematological side effects including thrombocytopenia and neutropenia that can necessitate treatment lapse [75]. Moreover, no improvement in progression-free survival (PFS) or overall survival (OS) was evident in a phase II trial of imetelstat on patients with advanced non-small cell lung cancer, although a trend towards improved survival was observed in patients with the shortest telomeres [35]. Based on these results, imetelstat is being repurposed for myeloproliferative disorders. Imetelstat has elicited robust response rates in phase II trials involving patients with myelofibrosis or essential thrombocytopenia [76] [77]. However, responses neither correlated with baseline telomere length nor was telomere shortening observed in responders, raising concerns that the mechanism of action of imetelstat in responders may be due to sequence-independent side effects of phosphoramidates on immunostimulation and not due to telomerase inhibition [78].
Small molecule inhibitors
While oligonucleotide and immunotherapeutic approaches to targeting telomerase have progressed furthest in clinical development, small molecule inhibitors such as BIBR1532 have generated promising preclinical results. BIBR1532 is a non-competitive small molecule inhibitor of telomerase that mediates progressive telomere shortening in cancer cells and replicative senescence following extended treatment [79]. Structural analysis using Tribolium castaneum TERT has revealed that BIBR1532 impairs telomerase assembly by binding to a conserved hydrophobic pocket (FVYL motif) of TERT and disrupting interactions with the activation domain of TERC (CR4/5) [80]. Although BIBR1532 has poor pharmacokinetic properties that restrict its clinical applicability, resolution of the BIBR1532 binding site should assist design of more potent and bioavailable telomerase inhibitors. It is important to keep in mind that the efficacy of inhibitors of telomerase catalytic activity may be limited by the prolonged treatment period required before anti-tumour effects caused by critical telomere attrition are exerted. However, high doses of BIBR1532 administered to leukaemia cells rapidly elicit cytotoxicity independent of telomere shortening, which has been attributed to acute induction of telomere dysfunction involving a p53-mediated DDR [41]. This highlights the potential value of developing strategies that acutely trigger anticancer responses such as telomere ‘uncapping’.
Lastly, natural compounds have been reported to act as telomerase inhibitors through diverse poorly-defined mechanisms. These compounds are invariably pan-assay interference compounds (PAINS) with promiscuous activity across unrelated bioassays and are not considered optimizable clinical prospects. For example, epigallocatechin (EGCG) and its derivative MST-312 have been declared telomerase inhibitors despite EGCG being a notorious pan-assay interference compound. EGCG contains a catechol motif responsible for PAINS behaviour due to redox activity, metal chelation and non-specific membrane perturbation [81].
Indirect inhibitors
G-quadruplex stabilizers
G-quadruplex secondary structures can form in guanine-rich DNA or RNA sequences including telomeres [82]. These structures contain stacks of guanine tetrads formed by Hoogsteen hydrogen bonding between four guanine bases in a square planar arrangement. Telomeric G-quadruplexes are resolved by DNA helicases prior to telomere extension [83]. Consequently, small molecules that stabilize G-quadruplexes can disrupt telomere extension by telomerase, triggering a DNA damage response and cell death [84] [85]. G-quadruplex stabilizers have elicited anticancer effects in preclinical studies, but have undergone limited clinical development. For example, the G-quadruplex stabilizer telomestatin suppresses telomerase activity and tumour growth in leukaemia xenograft models [84]. However, affinity for non-telomeric G-quadruplexes may lead to unacceptable toxicity. Although G-quadruplex motifs are most over-represented at telomeres, computational analysis predicts potential G-quadruplex formation at over 300,000 sites in the human genome [86]. Thus, it is essential to establish which conformations of G-quadruplex stably form at telomeres in vivo in order to identify more telomere-specific G-quadruplex ligands [87]. It remains to be seen whether the vast number of G-quadruplexes in the genome and structural similarity between G-quadruplexes at telomeres and non-telomeric sites will ultimately preclude sufficient selectivity of G-quadruplex ligands for telomeres.
Nucleoside analogues
Inhibitors of telomerase catalytic activity rely upon gradual telomere attrition with successive rounds of DNA replication, until critical telomere erosion triggers a DNA damage response mediated by ATM and ATR, replicative senescence and cell death. Prolonged telomerase inhibition can cause haematological toxicities requiring treatment lapse, hence undermining treatment efficacy by permitting telomere length recovery. Furthermore, adaptations that overcome progressive telomere shortening, such as alternative lengthening of telomeres, may be selected for during prolonged telomerase inhibition. Accordingly, alternative strategies have been developed that co-opt telomerase activity to acutely target cancer cells. Nucleoside analogues such as 6-thio-2’-deoxyguanosine (6-thio-dG) or 5-fluoro-2’-deoxyuridine (5-FdU) triphosphate rapidly induce telomere dysfunction and cell death in telomerase-expressing cells following their incorporation into newly synthesized telomeres [88] [89]. These nucleoside analogues act as telomerase-dependent ‘uncapping agents’ that impede the binding of the shelterin complex to telomeric DNA and activate a DDR.
Telomere uncapping agents have minimal effects on non-transformed and telomerase-negative cells, however they have proved effective at mediating telomerase-expressing cancer cell cytotoxicity and impairing tumour growth in mouse xenografts. For example, 6-thio-dG induces shrinkage of non-small cell lung cancer xenografts resistant to EGFR inhibitors or chemotherapy [90] and impairs growth of therapy-resistant medulloblastoma [91] and melanoma xenografts [92]. In contrast, therapeutically relevant doses of 6-thio-dG do not elicit significant toxicity in non-tumour-bearing mice besides minor neutropenia [88]. Notably, telomere uncapping agents acutely induce cell death in telomerase-expressing tumour cells independently of initial telomere length. Consequently, efficacy of this method is not restricted by telomere length heterogeneity and it is possible that toxicities associated with prolonged telomerase inhibition may be avoided. Telomere uncapping agents may be effective across a range of cancers including melanoma [92]. However, treatment with 6-thio-dG can trigger adaptive responses to oxidative stress that counteract cell death in melanoma models [93]. In this scenario, the anti-melanoma effects of 6-thio-dG are potentiated by combination with the mitochondrial Hsp90 inhibitor Gamitrinib, which blunts the SOD2-mediated antioxidant response. Evidence for the anticancer efficacy and limited toxicity of 6-thio-dG in vivo supports further development of this strategy, although the mechanistic basis of telomere uncapping by nucleoside analogues requires further investigation.
Targeting TERT gene expression
TERT expression is regulated by an atypical GC-rich promoter that harbours multiple binding sites for SP1 and c-Myc transcription factors, but lacks TATA and CAAT boxes (Figure 2). Repressive chromatin remodelling and epigenetic modifications silence TERT expression in non-transformed cells [94] [95]. In contrast, the vast majority of cancers acquire replicative immortality through telomerase re-expression. Approximately 15–25% of cancers appear to reactivate telomerase via mutations in the TERT promoter that generate de novo binding motifs for ETS transcription factors. Genome editing reveals that TERT promoter mutations are sufficient to prevent TERT silencing and maintain telomere length upon differentiation of human embryonic stem cells without concomitant oncogenic mutations [96]. However, it has not been demonstrated that acquisition of TPMs is sufficient to activate telomerase in the context of a differentiated cell and reactivation likely has other requirements such as concomitant mutations in the MAPK pathway [97]. TPMs emerge early in tumour evolution [98] [99]; for example, in melanoma, TPMs arise at the transition from a benign naevus with activated MAPK signalling to malignant melanoma [100]. If TPMs are necessary to sustain replicative immortality, then targeting the regulators that bind to the de novo ETS binding sites may represent an effective therapeutic strategy.
Figure 2 –
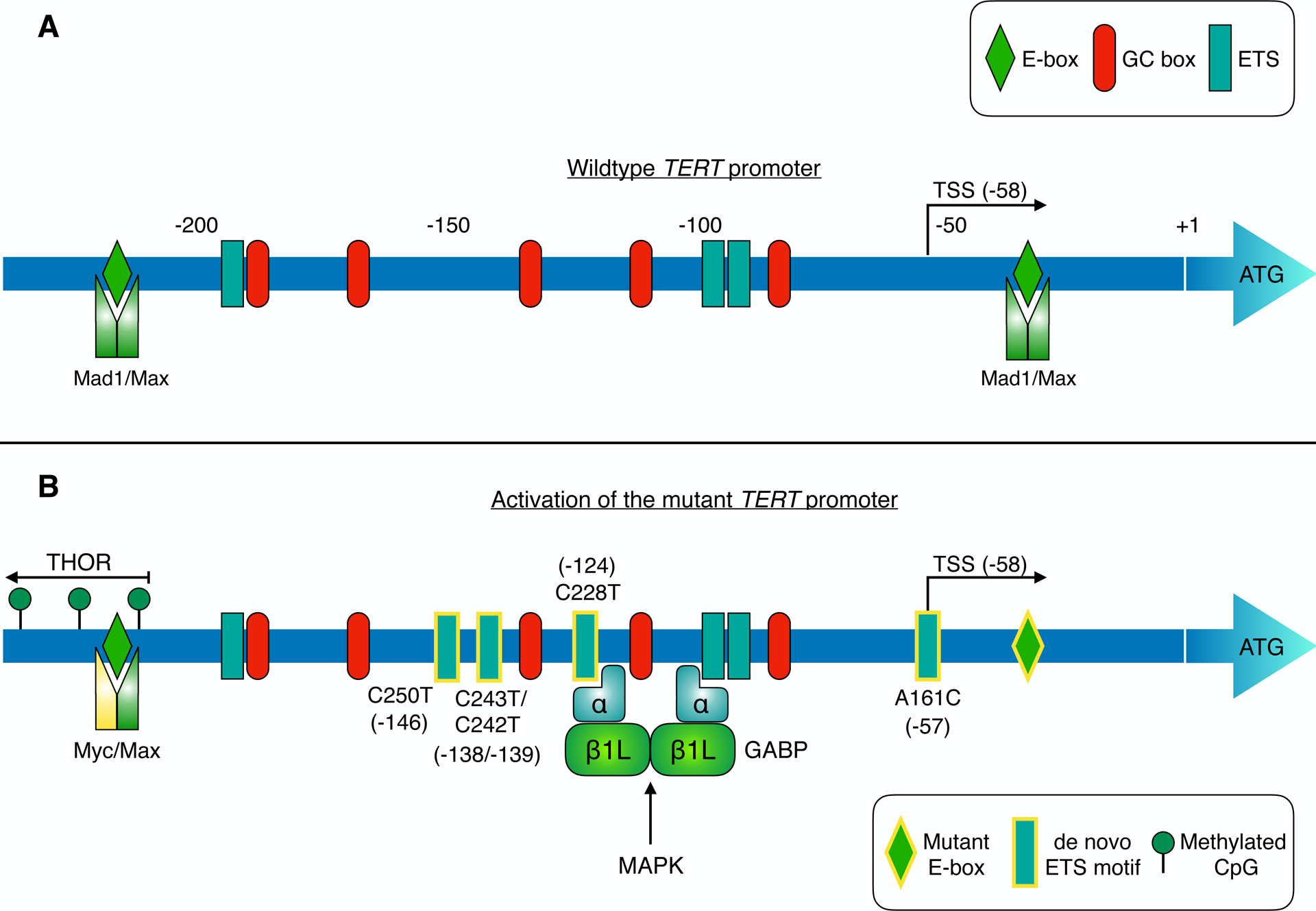
TERT promoter regulation: a new therapeutic avenue in cancer?
The TERT core promoter region harbours multiple binding sites for SP1, Myc-Max-Mad1 and ETS family transcription factors (corresponding to GC boxes, E-boxes and ETS motifs respectively; panel A). TERT transcription is suppressed in telomerase-negative cells by different mechanisms including repressive chromatin modifications and binding of Mad1/Max to E-boxes in the core promoter [95] [120]. Telomerase expression is reactivated in cancers (panel B) via diverse mechanisms, such as TERT promoter mutation, hypermethylation, and TERT gene amplification [14] [27]. Mutually exclusive point mutations in the TERT promoter generate de novo binding sites (depicted with yellow-bordered rectangles) for ETS transcription factors in ~15–25% of tumours. These de novo ETS motifs can be bound by distinct ETS family members in different tumour types, thereby enhancing TERT transcription (see Table 1). The activity of these TFs can be regulated by oncogenic pathways such as MAPK. For instance, in glioblastoma, heterotetrameric GABP is recruited to de novo ETS sites in the mutant promoter, enhancing TERT expression [18]. GABP activation of the mutant TERT promoter is responsive to MAPK stimulation [121]. Similarly, ETS1 activation of TERT is suppressed by MEK inhibition in BRAF-mutant melanoma models [102]. Methylation of CpG sites upstream of the core promoter upregulates TERT expression in cancer. This TERT hypermethylated oncological region (THOR) spans from approximately −217 to −649, relative to the ATG start codon [27]. Point mutation of a Myc-Max-Mad1 binding site (yellow-bordered diamond) in clear cell renal cell carcinoma may impair repression of TERT transcription by Mad1 [122].
Reversion of TERT promoter mutations in cancer cell lines to wildtype via CRISPR genome editing depletes active chromatin marks and suppresses, but does not completely eliminate, TERT expression and telomerase activity [101]. Thus, it would be important to determine the efficacy of targeting regulators of TPMs, which could circumvent the side effects of telomerase inhibition on telomerase-expressing stem cells harbouring wildtype TERT promoters. The dependence of TERT expression on individual ETS family transcription factors following the acquisition of TPMs appears to differ between cancer types (Table 1) [102] [18] [103]. ETS1 is implicated in reactivating TERT expression at TPMs in melanoma [102], but may act as an indirect regulator of the mutant TERT promoter in glioblastoma [18]. In contrast, the ETS transcription factor GABP has been shown to directly activate the mutant TERT promoter in glioblastoma cell lines via a GABPα2β2 tetramer containing the dispensable GABPβ1L isoform [104]. Functionally, GABPβ1L depletion promotes telomere shortening and induces cell death in glioblastoma cells and xenografts harbouring TPMs but not in those with wildtype TERT promoters. Loss of viability was rescued by TERT overexpression indicating that cell death was dependent on loss of TERT expression (and likely telomerase activity). Genetic ablation of GABPβ1L incompletely suppresses TERT expression, indicating that although the GABPβ1L tetramer is the primary trans factor responsible for regulating TERT expression at TPMs, it may not be the sole player regulating TPM-dependent TERT expression. Residual TERT expression may be mediated by other ETS family members or GABP isoforms. This raises the possibility that a drug capable of targeting GABPβ1L would leave residual TERT expression sufficient for maintaining the shortest telomeres, which would require elimination with a secondary drug. Nevertheless, GABPβ1L seems a viable, albeit challenging target for reversing TPM-driven cellular immortality. GABP functions as an obligate multimer of DNA-binding GABPα and transactivating GABPβ subunits, either as a heterodimer composed of one GABPα and one GABPβ1S subunit or as a heterotetramer of two GABPα and two GABPβ1L/β2 subunits. Notably, whereas GABPα is essential for mouse development, GABPβ1L is not [105] [106]. Hence, to develop a well-tolerated inhibitor it will be important to isolate the essential heterodimeric functions of GABP from the dispensable GABPβ1L heterotetrameric functions, potentially by targeting the GABPα2β1L2 tetramerization interface.
Table 1 -.
Regulation of the mutant TERT promoter by ETS transcription factors
| Trans-acting factor | Cell type | Reference | Notes |
|---|---|---|---|
| GABP | Glioblastoma | [18] | GABPA knockdown suppressed TERT expression/activity. GABPA binding to mutant TERT promoter also identified in melanoma, hepatocellular carcinoma and neuroblastoma cell lines. |
| Glioblastoma | [104] | Disruption of tetramer-forming GABPB1L isoform depleted TERT expression, leading to telomere shortening and loss of replicative immortality. | |
| Melanoma | [123] | GABP binding to mutant TERT promoter excluded ELF1, promoting TERT expression. | |
| Melanoma & glioblastoma | [101] | CRISPR reversion of TPMs to wildtype suppressed GABPA binding and TERT expression in isogenic cell lines. | |
| Thyroid cancer & melanoma | [121] | BRAFmut. MAPK activation of FOS enhanced GABPB expression, GABP recruitment to mutant TERT promoter, and TERT expression. | |
| Thyroid cancer | [124] | GABPA knockdown suppressed TERT in TERT promoter mutant and wildtype cells. | |
| ETS1 | Melanoma | [102] | BRAFmut. MEK inhibition suppressed phospho-ETS1 (Thr38) and TERT expression/activity. |
| Glioma | [125] | BRAFmut. BRAF inhibition attenuated TERT expression/activity. | |
| Glioblastoma | [103] | ETS1/2, in cooperation with non-canonical NF-κB signalling, enhanced TERT expression/activity selectively in cells with C250T, but not C228T TERT promoter mutations. | |
| ETV5 | Thyroid cancer | [126] | ETV5 transactivated TERT in thyroid cancer cells lacking GABP activity. |
| Thyroid cancer | [127] | MAPK pathway inhibition suppressed TERT in TPM cells and the expression and binding of ETV1, 4 & 5 to the mutant TERT promoter. |
Point mutations in the TERT promoter generate novel ETS motifs in ~15–25% of tumours. Binding and transactivation at these sites by ETS family transcription factors displays a distinct pattern between tumour types.
Inhibition of oncogenic signalling pathways that impinge on TERT transcription may concomitantly suppress both TERT expression and telomerase activity. MEK or BRAF inhibitors decrease TERT expression and telomerase activity in BRAF-mutant melanoma cells harbouring TERT promoter mutations [107]. Similarly, NRAS knockdown diminishes TERT expression and activity in NRAS-mutant melanoma cells harbouring TPMs and, to a lesser extent, in those without a TPM [93]. The core TERT promoter contains GC-boxes that are bound by SP1; thus, regulation of TERT transcription by SP1 may be targetable via MAPK pathway inhibition. MAPK inhibition is proposed to suppress recruitment of activated ERK to the TERT promoter where ERK phosphorylation of SP1 facilitates dissociation of the histone deacetylase 1 repressor complex, although modulation of SP1 levels at the TERT promoter was not evident in this system [107]. Taken together, these findings suggest that FDA-approved MAPK pathway inhibitors could be leveraged to downregulate telomerase activity in combination with other methods. Likewise, non-canonical NF-κB signalling has been implicated in driving TERT re-expression specifically from the C250T TERT promoter mutant, which generates a p52 half-site binding motif. Non-canonical NF-κB stimuli are reported to mediate p52 recruitment to the C250T TPM, enhancing TERT transcription in cooperation with ETS transcription factors [103]. This suggests that inhibiting non-canonical NF-κB signalling could selectively target cancer cells harbouring this TPM, although further investigation of TERT regulation by non-canonical NF-κB signalling is required.
TERT pre-mRNA can be spliced into multiple isoforms, only one of which encodes catalytically active telomerase. The splicing factors required for production of full-length TERT mRNA have been identified from an RNAi screen [108]. Notably, knockdown of the lead candidate, NOVA1, impaired full-length telomerase production and telomerase activity, and suppressed cancer cell growth in vitro and in xenografts. Similarly, knockdown of the NOVA1-dependent splicing factor PTBP1 promoted telomere shortening [109]. Considering that splicing factors are likely to have pleiotropic effects, it is important to clarify whether the effects of NOVA1/PTBP1 depletion on cancer cell growth are primarily due to reduced telomerase activity. For instance, rescue experiments with ectopic TERT would help to establish whether the splicing machinery required for active telomerase production is a viable anticancer target.
Regulation of telomerase localization and catalysis
The telomerase holoenzyme complex incorporates an RNA scaffold protein, TCAB1, which controls nuclear trafficking of telomerase and stimulates telomerase catalysis by regulating conformation of the TERC CR4/5 RNA domain [110]. Loss of TCAB1 disrupts telomerase localization and impairs telomere extension [111]. TCAB1 depletion suppresses growth of xenografted tumours, indicating that TCAB1 may be a potential anticancer target [112].
The shelterin complex consists of six subunits (TRF1, TRF2, POT1, RAP1, TIN2 and TPP1) that regulate telomerase activity and prevent chromosome ends from erroneously engaging a DDR by sequestering the 3’ telomeric overhang and compacting chromatin [113]. Genetic ablation of shelterin complex components elicits telomere dysfunction via telomere uncapping. For instance, systemic depletion of the negative regulator of telomere extension TRF1 in a p53-null KrasG12V lung adenocarcinoma mouse model induced telomere dysfunction and impaired tumour development, without overtly compromising viability of control mice [114]. Phosphorylation of TRF1 by BRAF and ERK2 has recently been shown to regulate TRF1 telomere localization [115]. Accordingly, inhibition of MEK/ERK mimics TRF1 depletion and induces telomeric DNA damage, indicating that inhibition of the MAPK pathway could be used to enhance telomere uncapping strategies. Additionally, phosphorylation of TRF1 by ATM promotes the dissociation of TRF1 from telomeres and impairs its ability to inhibit telomere extension [116] [62]. The shelterin component TPP1 mediates telomerase recruitment to telomeres and stimulates telomerase repeat addition processivity. These activities depend on interactions between the TEL patch of TPP1 and telomerase. Mutation of key TEL patch residues disrupts telomerase recruitment and inhibits telomere length maintenance [117]. Thus, this interface could serve as a therapeutic target. Accordingly, concomitant targeting of TPP1 and telomerase has been explored. Inhibition of telomerase activity by BIBR1532 synergistically induces cell death and telomere shortening in combination with TEL patch mutation [118]. Although targeting shelterin components elicits telomere dysfunction, questions remain as to how specific cytotoxic effects are likely to be for telomerase-expressing cancer cells.
Recent research has provided proof of concept that synthetic RNA-binding pentatricopeptide repeat protein (PPR) mimics of POT1 can bind to telomeric single-stranded DNA (ssDNA) and inhibit telomerase [119]. The shelterin subunit POT1 binds telomeric ssDNA, thereby antagonizing RPA accumulation and activation of the ATR DDR checkpoint. Native PPRs consist of arrays of ~35 amino acid repeats that recognize specific RNA repeat sequences. PPRs engineered to mimic POT1 recognition of telomeric ssDNA block primer extension by immunopurified telomerase. Although this approach is far from clinical translation, it is interesting to note that PPRs were able to bind ssDNA targets independently of G-quadruplex forming potential.
Conclusions and future directions
Tumours rely on the reactivation of telomerase to maintain telomeres and enable replicative immortality. In the absence of telomere maintenance, continued telomere attrition triggers replicative senescence, acting as a barrier to the indefinite expansion of neoplastic cells. The identification of this crucial dependence supports the therapeutic value of targeting telomerase and has driven the development of strategies targeting telomerase-expressing cells for cancer therapy.
Although vaccines (e.g. GV1001) and oligonucleotide inhibitors (e.g. imetelstat) of telomerase have advanced to early stage clinical trials [66] [35], neither approach has yet demonstrated clinical efficacy despite their well-founded therapeutic rationale, raising questions over their failure to translate. Indeed, the primary mechanistic basis of imetelstat is still not fully resolved. This is an important consideration as it would provide further insight into the clinical relevance of direct telomerase inhibitors. While imetelstat promotes telomere shortening in cell culture and xenograft studies, leading to replicative senescence, a correlation between baseline telomere length or telomere shortening and patient benefit has not been established in clinical trials for solid tumours. Imetelstat and most telomerase-directed approaches rely upon cumulative telomere shortening before anticancer effects are exerted. However, minimal residual telomerase activity can extend and protect the shortest telomeres, sustaining tumour cell proliferation. This implies that highly potent telomerase inhibitors are required to fully deplete telomerase activity and maintain selective pressure on cancer cells. Consequently, the current generation of telomerase inhibitors may be insufficiently potent to control disease progression in humans. However, recent advances in structural models of human telomerase should facilitate the rational design of more effective telomerase inhibitors that may prove clinically effective [51].
Ultimately, strategies relying on telomere attrition may be more effective as maintenance therapy to control cancer recurrence than as frontline therapy because the lag period may permit disease progression before critical telomere erosion. To maximize therapeutic benefit, telomerase inhibitors could be introduced as adjuvants following initial debulking surgery on solid tumours. Furthermore, inhibitors of telomere maintenance should be preferentially directed towards tumours with the shortest telomeres that are likely to respond more rapidly. In contrast, approaches based on telomere uncapping, such as nucleoside analogues that acutely induce telomere dysfunction, can rapidly trigger cancer cell death [90], warranting further investigation of tolerability and their potential clinical translatability. Although TERT vaccines have elicited high immunological response rates, the effects have proved insufficient to control cancer progression. In this context, it will be important to evaluate potential synergy between TERT vaccines and immune checkpoint inhibitors which can markedly extend survival in responsive patients.
The discovery that telomerase is frequently reactivated in cancer by non-coding mutations in the TERT promoter has sparked renewed interest in therapeutic means to target telomerase [17]. Emerging efforts are attempting to specifically target cancer cells harbouring these TERT promoter mutations; for example, by suppressing GABPβ1L-driven transcription at these de novo ETS binding sites [104]. A key theoretical advantage of TPM-based approaches is that they should discriminate between normal and transformed telomerase-expressing cells. However, the incidence and nature of TPMs differs vastly between cancer types. The underlying reasons for this remain a significant question in the field, as understanding the pattern of occurrence may reveal important regulatory dynamics of TERT in cancer cells with TPMs versus those without these mutations.
Overall, and despite significant challenges, telomerase remains an attractive target for cancer therapy. For therapies to achieve clinical efficacy, studies should focus on developing improved inhibitors in tandem with higher resolution structural models of human telomerase. Additionally, it is critical to investigate approaches that acutely induce telomere dysfunction and determine potential synergy between TERT vaccines and immune checkpoint blockade. Finally, considering the heterogeneity of tumours and ability of most cancer cells to rapidly adapt to pharmacological challenges, successful strategies targeting telomerase will likely need to be combined with either targeted therapies or immunotherapies to achieve optimal anti-tumour effects.
Acknowledgments
Work in our laboratory is supported by NIH grants R01CA215733, R01CA226888, P01CA114046, P50CA174523, P30CA010815, the Department of Defense Melanoma Research Program (W81XWH-20-1-0356), the PA Department of Health and Martha W. Rogers Trust.
Footnotes
Ethics declarations
The authors declare that they have no conflict of interest.
References
- 1.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–5. [DOI] [PubMed] [Google Scholar]
- 2.Morrison SJ, Prowse KR, Ho P, Weissman IL. Telomerase activity in hematopoietic cells is associated with self-renewal potential. Immunity. 1996;5:207–16. [DOI] [PubMed] [Google Scholar]
- 3.de Lange T Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. Cold Spring Harbor Lab; 2005;19:2100–10. [DOI] [PubMed] [Google Scholar]
- 4.Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, et al. Mammalian telomeres end in a large duplex loop. Cell. 1999;97:503–14. [DOI] [PubMed] [Google Scholar]
- 5.Levy MZ, Allsopp RC, Futcher AB, Greider CW, Harley CB. Telomere end-replication problem and cell aging. J. Mol. Biol 1992;225:951–60. [DOI] [PubMed] [Google Scholar]
- 6.Hackett JA, Greider CW. End resection initiates genomic instability in the absence of telomerase. Mol. Cell. Biol American Society for Microbiology Journals; 2003;23:8450–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–13. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt JC, Cech TR. Human telomerase: biogenesis, trafficking, recruitment, and activation. Genes Dev. Cold Spring Harbor Lab; 2015;29:1095–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. American Association for the Advancement of Science; 1998;279:349–52. [DOI] [PubMed] [Google Scholar]
- 10.Baena-Del Valle JA, Zheng Q, Esopi DM, Rubenstein M, Hubbard GK, Moncaliano MC, et al. MYC drives overexpression of telomerase RNA (hTR/TERC) in prostate cancer. Journal of Pathology. John Wiley & Sons, Ltd; 2018;244:11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao Y, Bryan TM, Reddel RR. Increased copy number of the TERT and TERC telomerase subunit genes in cancer cells. Cancer Sci John Wiley & Sons, Ltd; 2008;99:1092–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobs JJL, de Lange T. Significant role for p16INK4a in p53-independent telomere-directed senescence. Curr. Biol 2004;14:2302–8. [DOI] [PubMed] [Google Scholar]
- 13.Chiba K, Lorbeer FK, Shain AH, McSwiggen DT, Schruf E, Oh A, et al. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science. American Association for the Advancement of Science; 2017;357:1416–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barthel FP, Wei W, Tang M, Martinez-Ledesma E, Hu X, Amin SB, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet Nature Publishing Group; 2017;49:349–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ouellette MM, Liao M, Herbert BS, Johnson M, Holt SE, Liss HS, et al. Subsenescent telomere lengths in fibroblasts immortalized by limiting amounts of telomerase. J. Biol. Chem 2000;275:10072–6. [DOI] [PubMed] [Google Scholar]
- 16.Vinagre J, Almeida A, Pópulo H, Batista R, Lyra J, Pinto V, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun. Nature Publishing Group; 2013;4:959–6. [DOI] [PubMed] [Google Scholar]
- 17.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. American Association for the Advancement of Science; 2013;339:957–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bell RJA, Rube HT, Kreig A, Mancini A, Fouse SD, Nagarajan RP, et al. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science. American Association for the Advancement of Science; 2015;348:1036–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stern JL, Theodorescu D, Vogelstein B, Papadopoulos N, Cech TR. Mutation of the TERT promoter, switch to active chromatin, and monoallelic TERT expression in multiple cancers. Genes Dev. Cold Spring Harbor Lab; 2015;29:2219–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weinhold N, Jacobsen A, Schultz N, Sander C, Lee W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet Nature Publishing Group; 2014;46:1160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, et al. Whole-genome landscapes of major melanoma subtypes. Nature. 2017;545:175–80. [DOI] [PubMed] [Google Scholar]
- 22.Labussière M, Boisselier B, Mokhtari K, Di Stefano A-L, Rahimian A, Rossetto M, et al. Combined analysis of TERT, EGFR, and IDH status defines distinct prognostic glioblastoma classes. Neurology. Wolters Kluwer Health, Inc on behalf of the American Academy of Neurology; 2014;83:1200–6. [DOI] [PubMed] [Google Scholar]
- 23.Griewank KG, Murali R, Puig-Butille JA, Schilling B, Livingstone E, Potrony M, et al. TERT promoter mutation status as an independent prognostic factor in cutaneous melanoma. J. Natl. Cancer Inst 2014;106:949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spiegl-Kreinecker S, Lötsch D, Neumayer K, Kastler L, Gojo J, Pirker C, et al. TERT promoter mutations are associated with poor prognosis and cell immortalization in meningioma. Neuro-oncology. 2018;20:1584–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sahm F, Schrimpf D, Olar A, Koelsche C, Reuss D, Bissel J, et al. TERT Promoter Mutations and Risk of Recurrence in Meningioma. J. Natl Cancer Inst. 2016;108:djv377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. U.S.A National Academy of Sciences; 2013;110:6021–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee DD, Leão R, Komosa M, Gallo M, Zhang CH, Lipman T, et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J. Clin. Invest American Society for Clinical Investigation; 2019;129:223–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim W, Ludlow AT, Min J, Robin JD, Stadler G, Mender I, et al. Regulation of the Human Telomerase Gene TERT by Telomere Position Effect-Over Long Distances (TPE-OLD): Implications for Aging and Cancer Durocher D, editor. PLoS Biol. Public Library of Science; 2016;14:e2000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu X, Han W, Xue W, Zou Y, Xie C, Du J, et al. The association between telomere length and cancer risk in population studies. Sci Rep. ; 2016;6:518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Telomeres Mendelian Randomization Collaboration, Haycock PC, Burgess S, Nounu A, Zheng J, Okoli GN, et al. Association Between Telomere Length and Risk of Cancer and Non-Neoplastic Diseases: A Mendelian Randomization Study. JAMA Oncol. American Medical Association; 2017;3:636–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang C, Doherty JA, Burgess S, Hung RJ, Lindström S, Kraft P, et al. Genetic determinants of telomere length and risk of common cancers: a Mendelian randomization study. Hum. Mol. Genet 2015;24:5356–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aviv A, Anderson JJ, Shay JW. Mutations, Cancer and the Telomere Length Paradox. Trends Cancer. 2017;3:253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hemann MT, Strong MA, Hao LY, Greider CW. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell. 2001;107:67–77. [DOI] [PubMed] [Google Scholar]
- 34.Lai T-P, Zhang N, Noh J, Mender I, Tedone E, Huang E, et al. A method for measuring the distribution of the shortest telomeres in cells and tissues. Nat Commun. Nature Publishing Group; 2017;8:1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiappori AA, Kolevska T, Spigel DR, Hager S, Rarick M, Gadgeel S, et al. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Ann. Oncol 2015;26:354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Notaro R, Cimmino A, Tabarini D, Rotoli B, Luzzatto L. In vivo telomere dynamics of human hematopoietic stem cells. Proceedings of the National Academy of Sciences. 1997;94:13782–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masutomi K, Possemato R, Wong JMY, Currier JL, Tothova Z, Manola JB, et al. The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proceedings of the National Academy of Sciences. National Academy of Sciences; 2005;102:8222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nitta E, Yamashita M, Hosokawa K, Xian M, Takubo K, Arai F, et al. Telomerase reverse transcriptase protects ATM-deficient hematopoietic stem cells from ROS-induced apoptosis through a telomere-independent mechanism. Blood. American Society of Hematology; 2011;117:4169–80. [DOI] [PubMed] [Google Scholar]
- 39.Koh CM, Khattar E, Leow SC, Liu CY, Muller J, Ang WX, et al. Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J. Clin. Invest 2015;125:2109–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perera ON, Sobinoff AP, Teber ET, Harman A, Maritz MF, Yang SF, et al. Telomerase promotes formation of a telomere protective complex in cancer cells. Sci Adv American Association for the Advancement of Science; 2019;5:eaav4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.El-Daly H, Kull M, Zimmermann S, Pantic M, Waller CF, Martens UM. Selective cytotoxicity and telomere damage in leukemia cells using the telomerase inhibitor BIBR1532. Blood. American Society of Hematology; 2005;105:1742–9. [DOI] [PubMed] [Google Scholar]
- 42.Hemann MT, Greider CW. Wild-derived inbred mouse strains have short telomeres. Nucleic Acids Res. 2000;28:4474–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greenberg RA, Allsopp RC, Chin L, Morin GB, DePinho RA. Expression of mouse telomerase reverse transcriptase during development, differentiation and proliferation. Oncogene. Nature Publishing Group; 1998;16:1723–30. [DOI] [PubMed] [Google Scholar]
- 44.Epidemiology Peto R., multistage models, and short-term mutagenicity tests. Int J Epidemiol. 2016;45:621–37. [DOI] [PubMed] [Google Scholar]
- 45.Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, et al. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91:25–34. [DOI] [PubMed] [Google Scholar]
- 46.Raval A, Behbehani GK, Nguyen LXT, Thomas D, Kusler B, Garbuzov A, et al. Reversibility of Defective Hematopoiesis Caused by Telomere Shortening in Telomerase Knockout Mice. Saretzki G, editor. PLoS ONE Public Library of Science; 2015;10:e0131722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N. Engl. J. Med 2005;352:1413–24. [DOI] [PubMed] [Google Scholar]
- 48.Sauerwald A, Sandin S, Cristofari G, Scheres SHW, Lingner J, Rhodes D. Structure of active dimeric human telomerase. Nat. Struct. Mol. Biol Nature Publishing Group; 2013;20:454–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gillis AJ, Schuller AP, Skordalakes E. Structure of the Tribolium castaneum telomerase catalytic subunit TERT. Nature. Nature Publishing Group; 2008;455:633–7. [DOI] [PubMed] [Google Scholar]
- 50.Jiang J, Wang Y, Sušac L, Chan H, Basu R, Zhou ZH, et al. Structure of Telomerase with Telomeric DNA. Cell. 2018;173:1179–1190.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nguyen THD, Tam J, Wu RA, Greber BJ, Toso D, Nogales E, et al. Cryo-EM structure of substrate-bound human telomerase holoenzyme. Nature. Nature Publishing Group; 2018;557:190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Venteicher AS, Abreu EB, Meng Z, McCann KE, Terns RM, Veenstra TD, et al. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science. American Association for the Advancement of Science; 2009;323:644–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heaphy CM, Subhawong AP, Hong S-M, Goggins MG, Montgomery EA, Gabrielson E, et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol 2011;179:1608–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dilley RL, Greenberg RA. ALTernative Telomere Maintenance and Cancer. Trends Cancer. 2015;1:145–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Henson JD, Hannay JA, McCarthy SW, Royds JA, Yeager TR, Robinson RA, et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin. Cancer Res 2005;11:217–25. [PubMed] [Google Scholar]
- 56.Ramamoorthy M, Smith S. Loss of ATRX Suppresses Resolution of Telomere Cohesion to Control Recombination in ALT Cancer Cells. Cancer Cell. 2015;28:357–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Napier CE, Huschtscha LI, Harvey A, Bower K, Noble JR, Hendrickson EA, et al. ATRX represses alternative lengthening of telomeres. Oncotarget. Impact Journals; 2015;6:16543–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mukherjee J, Johannessen T-C, Ohba S, Chow TT, Jones L, Pandita A, et al. Mutant IDH1 Cooperates with ATRX Loss to Drive the Alternative Lengthening of Telomere Phenotype in Glioma. Cancer Res. American Association for Cancer Research; 2018;78:2966–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Sullivan RJ, Arnoult N, Lackner DH, Oganesian L, Haggblom C, Corpet A, et al. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol Nature Publishing Group; 2014;21:167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Min J, Wright WE, Shay JW. Alternative lengthening of telomeres can be maintained by preferential elongation of lagging strands. Nucleic Acids Res. 2017;45:2615–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu J, Hwang SS, Liesa M, Gan B, Sahin E, Jaskelioff M, et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell. 2012;148:651–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tong AS, Stern JL, Sfeir A, Kartawinata M, de Lange T, Zhu X-D, et al. ATM and ATR Signaling Regulate the Recruitment of Human Telomerase to Telomeres. Cell Rep. 2015;13:1633–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vonderheide RH. Telomerase as a universal tumor-associated antigen for cancer immunotherapy. Oncogene. Nature Publishing Group; 2002;21:674–9. [DOI] [PubMed] [Google Scholar]
- 64.Lilleby W, Gaudernack G, Brunsvig PF, Vlatkovic L, Schulz M, Mills K, et al. Phase I/IIa clinical trial of a novel hTERT peptide vaccine in men with metastatic hormone-naive prostate cancer. Cancer Immunol. Immunother Springer Berlin Heidelberg; 2017;66:891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zanetti M A second chance for telomerase reverse transcriptase in anticancer immunotherapy. Nat Rev Clin Oncol. Nature Publishing Group; 2017;14:115–28. [DOI] [PubMed] [Google Scholar]
- 66.Middleton G, Silcocks P, Cox T, Valle J, Wadsley J, Propper D, et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): an open-label, randomised, phase 3 trial. Lancet Oncol. 2014;15:829–40. [DOI] [PubMed] [Google Scholar]
- 67.Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front Oncol. Frontiers; 2018;8:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Duperret EK, Wise MC, Trautz A, Villarreal DO, Ferraro B, Walters J, et al. Synergy of Immune Checkpoint Blockade with a Novel Synthetic Consensus DNA Vaccine Targeting TERT. Mol. Ther 2018;26:435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ugel S, Scarselli E, Iezzi M, Mennuni C, Pannellini T, Calvaruso F, et al. Autoimmune B-cell lymphopenia after successful adoptive therapy with telomerase-specific T lymphocytes. Blood. American Society of Hematology; 2010;115:1374–84. [DOI] [PubMed] [Google Scholar]
- 70.Sandri S, Bobisse S, Moxley K, Lamolinara A, De Sanctis F, Boschi F, et al. Feasibility of Telomerase-Specific Adoptive T-cell Therapy for B-cell Chronic Lymphocytic Leukemia and Solid Malignancies. Cancer Res American Association for Cancer Research; 2016;76:2540–51. [DOI] [PubMed] [Google Scholar]
- 71.Kawashima T, Kagawa S, Kobayashi N, Shirakiya Y, Umeoka T, Teraishi F, et al. Telomerase-specific replication-selective virotherapy for human cancer. Clin. Cancer Res 2004;10:285–92. [DOI] [PubMed] [Google Scholar]
- 72.Kanaya N, Kuroda S, Morihiro T, Kakiuchi Y, Kubota T, Kakiuchi S, et al. Abstract 2744: Telomelysin-induced immunogenic cell death synergizes with anti-PD-1 antibody in non-immunogenic gastrointestinal tumors. Cancer Res. 2018;78:2744–4. [Google Scholar]
- 73.Nemunaitis J, Tong AW, Nemunaitis M, Senzer N, Phadke AP, Bedell C, et al. A phase I study of telomerase-specific replication competent oncolytic adenovirus (telomelysin) for various solid tumors. Mol. Ther 2010;18:429–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Asai A, Oshima Y, Yamamoto Y, Uochi T-A, Kusaka H, Akinaga S, et al. A novel telomerase template antagonist (GRN163) as a potential anticancer agent. Cancer Res. 2003;63:3931–9. [PubMed] [Google Scholar]
- 75.Thompson PA, Drissi R, Muscal JA, Panditharatna E, Fouladi M, Ingle AM, et al. A phase I trial of imetelstat in children with refractory or recurrent solid tumors: a Children’s Oncology Group Phase I Consortium Study (ADVL1112). Clin. Cancer Res American Association for Cancer Research; 2013;19:6578–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tefferi A, Lasho TL, Begna KH, Patnaik MM, Zblewski DL, Finke CM, et al. A Pilot Study of the Telomerase Inhibitor Imetelstat for Myelofibrosis. N. Engl. J. Med 2015;373:908–19. [DOI] [PubMed] [Google Scholar]
- 77.Baerlocher GM, Oppliger Leibundgut E, Ottmann OG, Spitzer G, Odenike O, McDevitt MA, et al. Telomerase Inhibitor Imetelstat in Patients with Essential Thrombocythemia. N. Engl. J. Med 2015;373:920–8. [DOI] [PubMed] [Google Scholar]
- 78.Armanios M, Greider CW. Treating Myeloproliferation--On Target or Off? N. Engl. J. Med Massachusetts Medical Society; 2015;373:965–6. [DOI] [PubMed] [Google Scholar]
- 79.Damm K, Hemmann U, Garin-Chesa P, Hauel N, Kauffmann I, Priepke H, et al. A highly selective telomerase inhibitor limiting human cancer cell proliferation. EMBO J. EMBO Press; 2001;20:6958–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bryan C, Rice C, Hoffman H, Harkisheimer M, Sweeney M, Skordalakes E. Structural Basis of Telomerase Inhibition by the Highly Specific BIBR1532. Structure. 2015;23:1934–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baell JB. Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). Journal of Natural Products. American Chemical Society and American Society of Pharmacognosy; 2016;79:616–28. [DOI] [PubMed] [Google Scholar]
- 82.Biffi G, Tannahill D, McCafferty J, Balasubramanian S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat Chem. Nature Publishing Group; 2013;5:182–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Drosopoulos WC, Kosiyatrakul ST, Schildkraut CL. BLM helicase facilitates telomere replication during leading strand synthesis of telomeres. J. Cell Biol Rockefeller University Press; 2015;210:191–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tauchi T, Shin-ya K, Sashida G, Sumi M, Okabe S, Ohyashiki JH, et al. Telomerase inhibition with a novel G-quadruplex-interactive agent, telomestatin: in vitro and in vivo studies in acute leukemia. Oncogene. Nature Publishing Group; 2006;25:5719–25. [DOI] [PubMed] [Google Scholar]
- 85.Liu W, Zhong Y-F, Liu L-Y, Shen C-T, Zeng W, Wang F, et al. Solution structures of multiple G-quadruplex complexes induced by a platinum(II)-based tripod reveal dynamic binding. Nat Commun Nature Publishing Group; 2018;9:3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huppert JL, Balasubramanian S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005;33:2908–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moye AL, Porter KC, Cohen SB, Phan T, Zyner KG, Sasaki N, et al. Telomeric G-quadruplexes are a substrate and site of localization for human telomerase. Nat Commun. Nature Publishing Group; 2015;6:7643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mender I, Gryaznov S, Dikmen ZG, Wright WE, Shay JW. Induction of telomere dysfunction mediated by the telomerase substrate precursor 6-thio-2’-deoxyguanosine. Cancer Discov. American Association for Cancer Research; 2015;5:82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zeng X, Hernandez-Sanchez W, Xu M, Whited TL, Baus D, Zhang J, et al. Administration of a Nucleoside Analog Promotes Cancer Cell Death in a Telomerase-Dependent Manner. Cell Rep. 2018;23:3031–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mender I, LaRanger R, Luitel K, Peyton M, Girard L, Lai T-P, et al. Telomerase-Mediated Strategy for Overcoming Non-Small Cell Lung Cancer Targeted Therapy and Chemotherapy Resistance. Neoplasia. 2018;20:826–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sengupta S, Sobo M, Lee K, Senthil Kumar S, White AR, Mender I, et al. Induced Telomere Damage to Treat Telomerase Expressing Therapy-Resistant Pediatric Brain Tumors. Mol. Cancer Ther American Association for Cancer Research; 2018;17:1504–14. [DOI] [PubMed] [Google Scholar]
- 92.Zhang G, Wu LW, Mender I, Barzily-Rokni M, Hammond MR, Ope O, et al. Induction of Telomere Dysfunction Prolongs Disease Control of Therapy-Resistant Melanoma. Clin. Cancer Res American Association for Cancer Research; 2018;24:4771–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Reyes-Uribe P, Adrianzen-Ruesta MP, Deng Z, Echevarria-Vargas I, Mender I, Saheb S, et al. Exploiting TERT dependency as a therapeutic strategy for NRAS-mutant melanoma. Oncogene. Nature Publishing Group; 2018;37:1–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Atkinson SP, Hoare SF, Glasspool RM, Keith WN. Lack of telomerase gene expression in alternative lengthening of telomere cells is associated with chromatin remodeling of the hTR and hTERT gene promoters. Cancer Res. American Association for Cancer Research; 2005;65:7585–90. [DOI] [PubMed] [Google Scholar]
- 95.Cong YS, Bacchetti S. Histone deacetylation is involved in the transcriptional repression of hTERT in normal human cells. J. Biol. Chem American Society for Biochemistry and Molecular Biology; 2000;275:35665–8. [DOI] [PubMed] [Google Scholar]
- 96.Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM, Hockemeyer D. Cancer-associated TERT promoter mutations abrogate telomerase silencing. Elife. eLife Sciences Publications Limited; 2015;4:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shain AH, Joseph NM, Yu R, Benhamida J, Liu S, Prow T, et al. Genomic and Transcriptomic Analysis Reveals Incremental Disruption of Key Signaling Pathways during Melanoma Evolution. Cancer Cell. 2018;34:45–55.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac-Sage P, Laurent C, et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun. Nature Publishing Group; 2013;4:2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kinde I, Munari E, Faraj SF, Hruban RH, Schoenberg M, Bivalacqua T, et al. TERT promoter mutations occur early in urothelial neoplasia and are biomarkers of early disease and disease recurrence in urine. Cancer Res. 2013;73:7162–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med 2015;373:1926–36. [DOI] [PubMed] [Google Scholar]
- 101.Akıncılar SC, Khattar E, Boon PLS, Unal B, Fullwood MJ, Tergaonkar V. Long-Range Chromatin Interactions Drive Mutant TERT Promoter Activation. Cancer Discov. American Association for Cancer Research; 2016;6:1276–91. [DOI] [PubMed] [Google Scholar]
- 102.Vallarelli AF, Rachakonda PS, André J, Heidenreich B, Riffaud L, Bensussan A, et al. TERT promoter mutations in melanoma render TERT expression dependent on MAPK pathway activation. Oncotarget. Impact Journals; 2016;7:53127–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li Y, Zhou Q-L, Sun W, Chandrasekharan P, Cheng HS, Ying Z, et al. Non-canonical NF-κB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat. Cell Biol Nature Publishing Group; 2015;17:1327–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mancini A, Xavier-Magalhães A, Woods WS, Nguyen K-T, Amen AM, Hayes JL, et al. Disruption of the β1L Isoform of GABP Reverses Glioblastoma Replicative Immortality in a TERT Promoter Mutation-Dependent Manner. Cancer Cell. 2018;34:513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ristevski S, O’Leary DA, Thornell AP, Owen MJ, Kola I, Hertzog PJ. The ETS transcription factor GABPalpha is essential for early embryogenesis. Mol. Cell. Biol American Society for Microbiology Journals; 2004;24:5844–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xue H-H, Jing X, Bollenbacher-Reilley J, Zhao D-M, Haring JS, Yang B, et al. Targeting the GA binding protein beta1L isoform does not perturb lymphocyte development and function. Mol. Cell. Biol American Society for Microbiology Journals; 2008;28:4300–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li Y, Cheng HS, Chng WJ, Tergaonkar V. Activation of mutant TERT promoter by RAS-ERK signaling is a key step in malignant progression of BRAF-mutant human melanomas. Proc. Natl. Acad. Sci. U.S.A National Academy of Sciences; 2016;113:14402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ludlow AT, Wong MS, Robin JD, Batten K, Yuan L, Lai T-P, et al. NOVA1 regulates hTERT splicing and cell growth in non-small cell lung cancer. Nat Commun. Nature Publishing Group; 2018;9:3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sayed ME, Yuan L, Robin JD, Tedone E, Batten K, Dahlson N, et al. NOVA1 directs PTBP1 to hTERT pre-mRNA and promotes telomerase activity in cancer cells. Oncogene. Nature Publishing Group; 2019;38:2937–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen L, Roake CM, Freund A, Batista PJ, Tian S, Yin YA, et al. An Activity Switch in Human Telomerase Based on RNA Conformation and Shaped by TCAB1. Cell. 2018;174:218–230.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stern JL, Zyner KG, Pickett HA, Cohen SB, Bryan TM. Telomerase recruitment requires both TCAB1 and Cajal bodies independently. Mol. Cell. Biol American Society for Microbiology Journals; 2012;32:2384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sun C-K, Luo X-B, Gou Y-P, Hu L, Wang K, Li C, et al. TCAB1: a potential target for diagnosis and therapy of head and neck carcinomas. Mol. Cancer. BioMed Central; 2014;13:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bandaria JN, Qin P, Berk V, Chu S, Yildiz A. Shelterin Protects Chromosome Ends by Compacting Telomeric Chromatin. Cell. 2016;164:735–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.García-Beccaria M, Martínez P, Méndez-Pertuz M, Martínez S, Blanco-Aparicio C, Cañamero M, et al. Therapeutic inhibition of TRF1 impairs the growth of p53-deficient K-RasG12V-induced lung cancer by induction of telomeric DNA damage. EMBO Mol Med. EMBO Press; 2015;7:930–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bejarano L, Bosso G, Louzame J, Serrano R, Gómez-Casero E, Martínez-Torrecuadrada J, et al. Multiple cancer pathways regulate telomere protection. EMBO Mol Med. John Wiley & Sons, Ltd; 2019;:e10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.McKerlie M, Lin S, Zhu X-D. ATM regulates proteasome-dependent subnuclear localization of TRF1, which is important for telomere maintenance. Nucleic Acids Res. 2012;40:3975–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nandakumar J, Bell CF, Weidenfeld I, Zaug AJ, Leinwand LA, Cech TR. The TEL patch of telomere protein TPP1 mediates telomerase recruitment and processivity. Nature. Nature Publishing Group; 2012;492:285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nakashima M, Nandakumar J, Sullivan KD, Espinosa JM, Cech TR. Inhibition of telomerase recruitment and cancer cell death. J. Biol. Chem American Society for Biochemistry and Molecular Biology; 2013;288:33171–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Spåhr H, Chia T, Lingford JP, Siira SJ, Cohen SB, Filipovska A, et al. Modular ssDNA binding and inhibition of telomerase activity by designer PPR proteins. Nat Commun. Nature Publishing Group; 2018;9:2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Oh S, Song YH, Yim J, Kim TK. Identification of Mad as a repressor of the human telomerase (hTERT) gene. Oncogene. Nature Publishing Group; 2000;19:1485–90. [DOI] [PubMed] [Google Scholar]
- 121.Liu R, Zhang T, Zhu G, Xing M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat Commun. Nature Publishing Group; 2018;9:579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mitchell TJ, Turajlic S, Rowan A, Nicol D, Farmery JHR, O’Brien T, et al. Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell. 2018;173:611–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Makowski MM, Willems E, Fang J, Choi J, Zhang T, Jansen PWTC, et al. An interaction proteomics survey of transcription factor binding at recurrent TERT promoter mutations. Imhof A, Rappsilber J, editors. Proteomics. John Wiley & Sons, Ltd; 2016;16:417–26. [DOI] [PubMed] [Google Scholar]
- 124.Yuan X, Mu N, Wang N, Strååt K, Sofiadis A, Guo Y, et al. GABPA inhibits invasion/metastasis in papillary thyroid carcinoma by regulating DICER1 expression. Oncogene. 4 ed Nature Publishing Group; 2019;38:965–79. [DOI] [PubMed] [Google Scholar]
- 125.Gabler L, Lötsch D, Kirchhofer D, van Schoonhoven S, Schmidt HM, Mayr L, et al. TERT expression is susceptible to BRAF and ETS-factor inhibition in BRAFV600E/TERT promoter double-mutated glioma. Acta Neuropathol Commun. BioMed Central; 2019;7:128–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bullock M, Lim G, Zhu Y, Åberg H, Kurdyukov S, Clifton-Bligh R. ETS Factor ETV5 Activates the Mutant Telomerase Reverse Transcriptase Promoter in Thyroid Cancer Thyroid. Mary Ann Liebert, Inc, publishers 140 Huguenot Street, 3rd Floor New Rochelle, NY 10801; USA; 2019;29:1623–33. [DOI] [PubMed] [Google Scholar]
- 127.Song YS, Yoo S-K, Kim HH, Jung G, Oh A-R, Cha J-Y, et al. Interaction of BRAF-induced ETS factors with mutant TERT promoter in papillary thyroid cancer. Endocr. Relat. Cancer Bioscientifica Ltd; 2019;26:629–41. [DOI] [PubMed] [Google Scholar]