

Abstract
Understanding the mechanism of small molecules is a critical challenge in chemical biology and drug discovery. Medicinal chemistry is essential for elucidating drug mechanism, enabling variation of small molecule structure to gain structure–activity relationships (SARs). However, the development of complementary approaches that systematically vary target protein structure could provide equally informative SARs for investigating drug mechanism and protein function. Here we explore the ability of CRISPR–Cas9 mutagenesis to profile the interactions between lysine-specific histone demethylase 1 (LSD1) and chemical inhibitors in the context of acute myeloid leukemia (AML). Through this approach, termed CRISPR-suppressor scanning, we elucidate drug mechanism of action by showing that LSD1 enzyme activity is not required for AML survival and that LSD1 inhibitors instead function by disrupting interactions between LSD1 and the transcription factor GFI1B on chromatin. Our studies clarify how LSD1 inhibitors mechanistically operate in AML and demonstrate how CRISPR-suppressor scanning can uncover novel aspects of target biology.
Genome-editing technologies provide new capabilities to systematically alter protein sequence in situ by directly manipulating the endogenous coding sequence (CDS). One such technology, clustered regularly interspaced short palindromic repeats (CRISPR) scanning, is a novel method, which has been applied to identify functionally important protein domains1,2. In this approach, the programmable nuclease, CRISPR-associated protein 9 (Cas9) and single-guide RNAs (sgRNAs) spanning the CDS of the protein of interest are used to generate cells containing heterogeneous insertion or deletion (indel) mutations formed on DNA repair. If the protein target is essential for cell growth, cells harboring frameshift indels will drop out from the population due to knockout of the essential target protein. By contrast, cells harboring in-frame indels can often form nearly full-length protein with alterations at the site targeted by a particular sgRNA. In-frame variants in a functional region of an essential protein frequently result in loss-of-function mutations that impair cell fitness. However, in-frame variants might also result in gain-of-function mutations that confer a fitness advantage in the context of a selection pressure, such as a small-molecule drug3–5. Whether these in-frame drug-suppressor mutations can be employed to reveal precise structural information on the binding site of a small molecule, effectively discriminate between structurally related inhibitors, and clarify the mechanism of action of a molecule remains to be fully explored.
To examine these possibilities, we explored the ability of CRISPR scanning to interrogate structure–activity relationships (SARs) between lysine-specific histone demethylase 1 (LSD1) and its small-molecule inhibitors in the context of acute myeloid leukemia (AML). First identified through its association in histone deacetylase-containing corepressor complexes6–8, LSD1 is a flavin adenine dinucleotide (FAD)-dependent histone lysine demethylase, which demethylates monomethyl and dimethyl histone H3 lysine 4 (H3K4)9,10. Several small-molecule LSD1 inhibitors are now in clinical development for the treatment of various cancers, including AML and small-cell lung cancer11–20. The development of LSD1 inhibitors was motivated by the premise that the demethylase activity of LSD1 is necessary for AML proliferation10. However, a recent report suggests that the demethylase activity of LSD1 is not essential in this context15. Instead, LSD1 inhibitors may disrupt the interaction of LSD1 with GFI1 and/or GFI1B, two highly related transcription factors13–15,19,20. GFI1/GFI1B bind directly to the LSD1 catalytic site via their N-terminal SNAG domains to recruit LSD1 to their cognate genome-binding sites15,21–23. Consequently, the relative contributions of the enzymatic and nonenzymatic functions of LSD1 are convoluted, and the precise mechanistic role of LSD1 in AML remains ambiguous.
By combining CRISPR scanning with small-molecule-inhibitor treatments, an approach we refer to as CRISPR-suppressor scanning, we systematically identify mutations in LSD1 that confer resistance to LSD1 inhibitors in AML. By comparing the mutations differentially enriched by structurally related as well as mechanistically distinct compounds, we highlight the ability of this approach to interrogate SAR and essential features of the small-molecule binding site. Many of the drug-resistance mutations identified reside in the LSD1 catalytic site and inactivate enzymatic function, indicating that LSD1 demethylase activity is not required for AML survival. By rescuing growth using a drug-resistant GFI1B allele that only binds to LSD1 in the presence of the drug, we show that drug-mediated disruption of a LSD1–GFI1B complex is sufficient to block AML proliferation. Moreover, by comparing the effects of LSD1 inhibitors on wild-type versus mutant AML cells containing enzymatically inactive LSD1, we distinguish the relative roles of LSD1 enzymatic versus nonenzymatic function in controlling gene expression. We demonstrate that drug-induced disruption of the LSD1–GFI1B complex on chromatin may result in the activation of GFI1B-occupied enhancers by potentiating PU.1 activity. Our studies showcase the ability of CRISPR mutagenesis to both investigate small-molecule SAR and provide critical information on the molecular mechanisms of target biology.
Results
CRISPR-suppressor scanning identifies functional regions of LSD1.
To identify regions within LSD1 that modulate sensitivity to LSD1 inhibitors in AML, we conducted Streptococcus pyogenes Cas9 (SpCas9) CRISPR scanning of KDM1A, which encodes LSD1, using a pool of all possible 360 NGG PAM-restricted sgRNAs that tile the LSD1 CDS (Fig. 1a) and 81 control sgRNAs5 (Supplementary Dataset 1). The pool of sgRNAs was transduced into SET-2 and MV4;11, two AML cell lines highly sensitive to LSD1 knockout and to treatment with LSD1 inhibitors, including GSK-LSD1 (1)—a derivative of tranylcypromine (TCP) that forms a covalent bond with the FAD cofactor of LSD1 through a suicide-inhibition mechanism17,24 (Supplementary Fig. 1a). After transduction, the cells were split and then treated with vehicle (DMSO) or GSK-LSD1 (100 nM). Genomic DNA was isolated from the surviving cell populations at multiple time points and sequenced to deconvolute sgRNA identities enriched under each condition. Given the essential nature of LSD1 in these cell lines, most sgRNAs targeting LSD1 were depleted under both conditions as compared with the functionally neutral genome-targeting control sgRNAs (Fig. 1b,c, Supplementary Fig. 1b–e and Supplementary Datasets 2 and 3). However, several sgRNAs were enriched after prolonged GSK-LSD1-treatment, leading to diverging sgRNA enrichment profiles between vehicle and GSK-LSD1 conditions over time (Fig. 1b–d, Supplementary Fig. 1b–e and Supplementary Datasets 2 and 3). These observations are consistent with the emergence and expansion of drug-resistant populations.
Fig. 1 |. CRISPR-suppressor scanning identifies regions of LSD1 that mediate its function and susceptibility to pharmacological inhibitors.
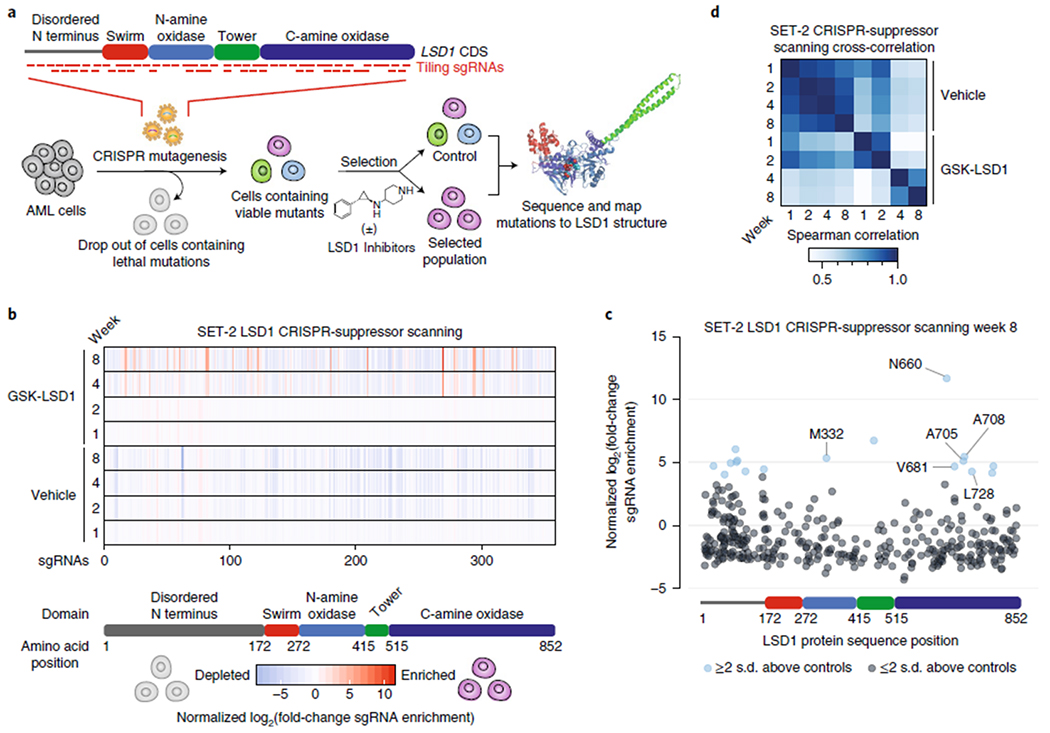
a, Schematic of CRISPR-suppressor scanning workflow to profile SARs of LSD1 small-molecule inhibitors. b, Heat maps depicting log2(fold-change sgRNA enrichment) in SET-2 at the indicated time points and conditions versus week 0 normalized against functionally neutral genome-targeting control sgRNAs. The sgRNAs are arrayed on the x axis by the LSD1 CDS. Color represents mean values across three replicate transductions. c, Scatter plot showing log2(fold-change sgRNA enrichment) in SET-2 under GSK-LSD1 treatment at week 8 versus week 0 normalized against functionally neutral genome-targeting control sgRNAs. The sgRNAs are arrayed on the x axis by the LSD1 CDS. Data represent mean values across three replicate transductions. d, Heat map showing cross-correlation of overall sgRNA enrichment at the specified time points during the CRISPR-suppressor scanning in SET-2. Data represent mean values across three replicate transductions.
Depleted and enriched sgRNAs were asymmetrically distributed across the different protein domains of the LSD1 CDS (Fig. 1b,c). Consequently, it was not immediately apparent whether these sgRNAs clustered in particular regions of LSD1 by only considering the linear CDS1. To identify potential regions of LSD1 critical for AML fitness, we considered whether sgRNAs, enriched either through negative or positive selection, may be spatially clustered in three-dimensional space. To assess this phenomenon, we adapted the clustering of mutations in protein structures algorithm to analyze sgRNA enrichment25. First, each sgRNA was assigned to the closest amino acid residue overlapping the predicted Cas9 cleavage site. Then, all pairwise combinations of sgRNAs were scored by (1) their combined enrichment and (2) the Euclidean distance between their assigned amino acid residues based on reported LSD1 structural data21 (Methods and Fig. 2a). To identify hotspots, the proximity-weighted enrichment score (PWES) matrix of all pairwise sgRNA interactions was grouped by hierarchical clustering, leading to the identification of 12 clusters of proximal sgRNAs that define distinct regions of LSD1 characterized by similar enrichment profiles (Fig. 2b).
Fig. 2 |. Spatial clustering of CRISPR-suppressor scanning data reveals potential functional hotspots of LSD1 that mediate drug action.
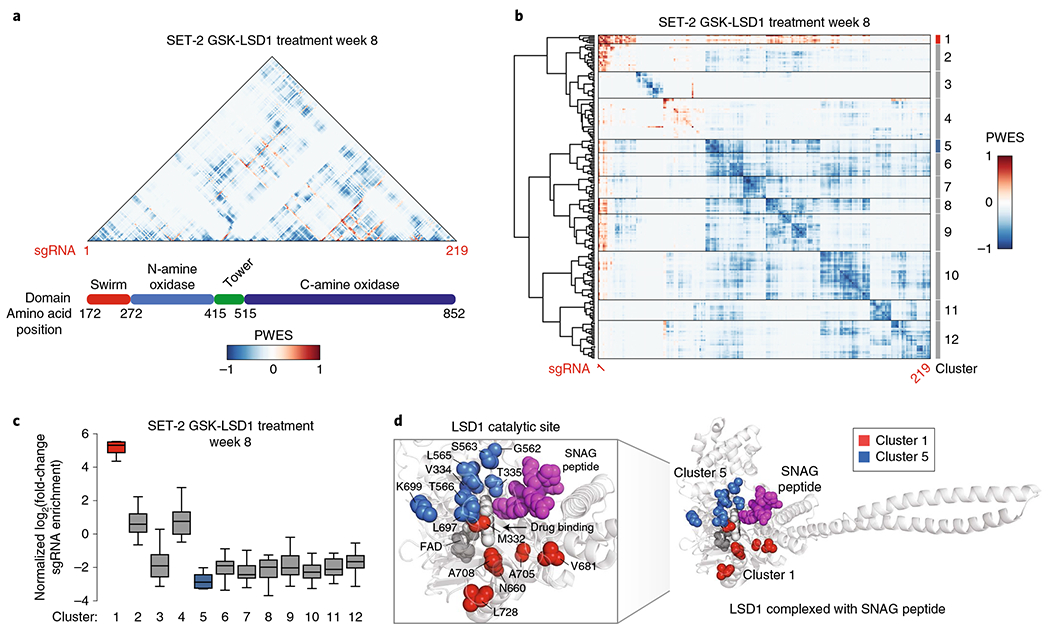
a,b, Heat maps depicting the PWES between sgRNAs ordered by the CDS (a) or by hierarchical clustering (b). sgRNAs targeting the disordered LSD1 N terminus were omitted in this analysis because this domain was truncated in structural studies. c, Box plot showing the enrichment of sgRNA groups as defined by hierarchical clustering in b for SET-2 under GSK-LSD1 treatment at week 8. The y axis represents mean log2(fold-change sgRNA enrichment) at week 8 versus week 0 across three replicate transductions. In the plot, bars represent the median, the box represents the interquartile range (IQR) and the whiskers represent 1.5× IQR. d, Structural view of LSD1 complexed with a SNAG peptide (magenta) showing the location of clusters 1 (red) and 5 (blue) defined in b (Protein Data Bank (PDB): 2Y48).
Consistent with our previous observations, most clusters identified corresponded to sgRNAs depleted during the selection (Fig. 2b,c and Supplementary Fig. 2a–c). By contrast, cluster 1 stood out as the most highly enriched cluster in the presence of GSK-LSD1 (Fig. 2c). When mapped to the LSD1 structure, this enriched cluster was located within the catalytic site (Fig. 2d), which is also the binding site of GSK-LSD1. To test whether clustering of enriched sgRNAs is statistically significant, a summed PWES was calculated for sgRNAs in a particular group, and this value was compared with the simulated distribution of summed PWES determined by randomizing the positions of these sgRNAs throughout the protein (Methods and Supplementary Fig. 2d). Comparing the summed PWES for sgRNAs in cluster 1 to the simulated distribution indicated that the spatial clustering of these sgRNAs was significant (P=2 × 10−5, Supplementary Fig. 2d). Notably, the targeted amino acids comprising cluster 1 (M332, N660, V681, A705, A708, L728) are close to the FAD cofactor, which is bound by GSK-LSD1, but more distal to the substrate-binding site21 (Fig. 2d, SNAG peptide). This suggests that drug-resistance mutations are selected such that they potentially alter drug binding while permitting essential binding interactions of LSD1 to its substrate(s), which may be highly important for AML proliferation1. In agreement with this notion, we observed that the most highly depleted cluster, cluster 5, comprises sgRNAs targeting residues involved in substrate binding21,26 (Fig. 2c,d). Similarly to cluster 1, the spatial clustering of sgRNAs for cluster 5 was found to be statistically significant (P < 10−5, Supplementary Fig. 2d). Altogether, our analysis demonstrates that CRISPR-suppressor scanning data can be used to identify spatially clustered hotspots in protein targets that may modulate inhibitor mechanism of action and protein function.
CRISPR-suppressor scanning reveals SARs.
We next considered whether CRISPR scanning could discriminate among different classes of LSD1 inhibitors as well as structurally related analogs by investigating how variations of the small-molecule inhibitor might change the overall sgRNA enrichment profile and resulting LSD1 mutation signature. We began by synthesizing five structural analogs of GSK-LSD1 (AW1 (2), AW2 (3), AW3 (4), AW4 (5) and AW5 (6)), each possessing a different substituent on the phenyl group of GSK-LSD1 (Fig. 3a). We measured the in vitro and cellular activities of these GSK-LSD1 analogs along with two previously described reversible LSD1 inhibitors, GSK690 (7) and NV93 (8)27,28 (Fig. 3a and Supplementary Fig. 3a–c). We subsequently subjected the CRISPR-suppressor scanning transduced pool of SET-2 cells to these compounds dosed at 500 nM. We sequenced genomic DNA to determine sgRNA enrichment under each drug treatment. Focusing the analysis on positively enriched sgRNAs, we normalized the enrichment scores of the inhibitor-treated samples to those of the vehicle-treated samples (Fig. 3b and Supplementary Dataset 4).
Fig. 3 |. CRISPR-suppressor scanning enables profiling of LSD1 SARs.
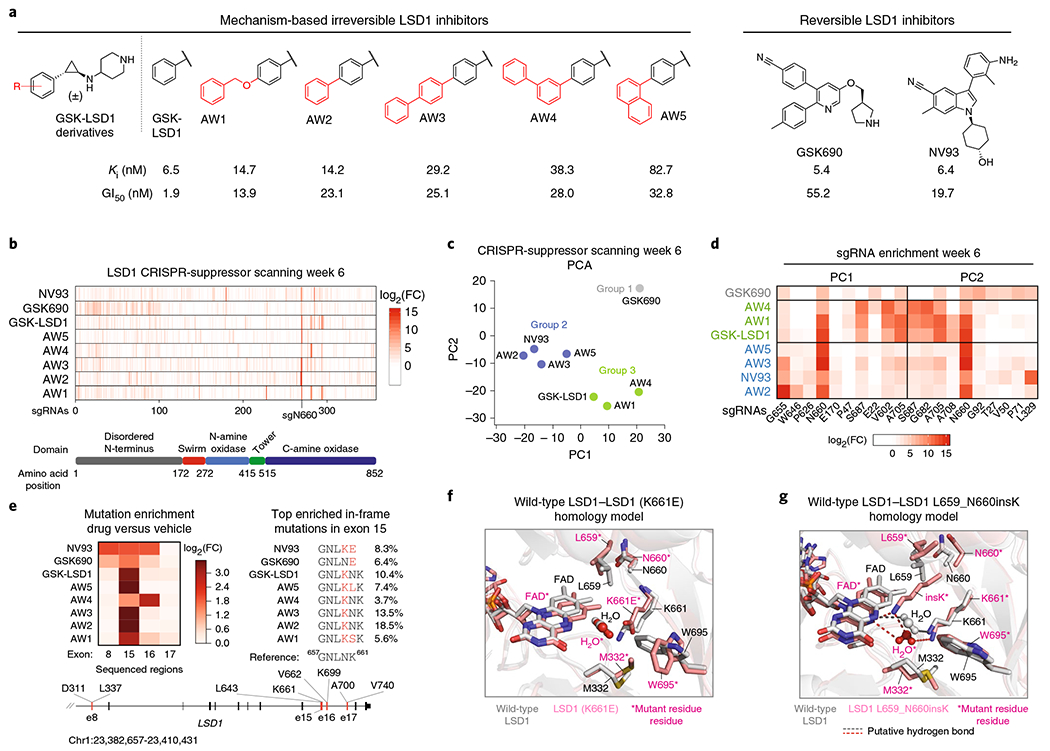
a, Structures of LSD1 inhibitors used in CRISPR-suppressor scanning. For each compound, Ki and GI50 values were measured across three replicates, where Ki is the equilibrium dissociation constant and GI50 is the drug concentration that results in half-maximal inhibition of cell proliferation. See Supplementary Fig. 3a–c for further details. b, Heat maps showing sgRNA enrichment measured in SET-2 at week 6. The sgRNAs are arrayed on the x axis by the LSD1 CDS. Color represents mean log2(fold-change sgRNA enrichment normalized to vehicle treatment) across three replicate transductions. FC = fold-change. c, Scatter plot showing projection of compound sgRNA enrichment profiles at week 6 onto principal component space. Groups of compounds were identified by k-means clustering (k = 3) and are marked. Data represent mean values across three replicate transductions. d, Heat map showing sgRNA enrichment measured in SET-2 at week 6 for the top five sgRNAs with largest principal component coefficient loadings and bottom five sgRNAs with smallest principal component coefficient loadings. The sgRNAs are ordered on the x axis by principal component coefficient, increasing from left to right, and are labeled according to the residues they target in the LSD1 CDS. Color represents mean log2(fold-change) of sgRNA enrichment normalized to vehicle treatment across three replicate transductions. e, Left, heat map showing mutation enrichment in sequenced exons across different drug treatment conditions. Mutated reads are defined as any read containing a modification in a coding region. Color represents log2(fold-change mutated reads) of drug-treatment relative to vehicle treatment at week 6. DNA from three replicate transductions were pooled at equal concentrations before sequencing to obtain an average log2(fold-change) of mutated reads. Right, schematic shows the genotypes of the most abundant mutation in exon 15 for each treatment condition at week 6. Percentages indicate the allele frequency as determined through sequencing. Bottom, schematic depicts the LSD1 gene locus and the regions sequenced. f, Homology model of LSD1 (K661E) overlaid with wild-type LSD1 is shown. W695, involved in a hydrophobic interaction with GSK690, is rotated out of plane in the model of the mutant structure (PDB: 2HKO). g, Homology model of LSD1 L659_N660insK overlaid with wild-type LSD1 is shown. The N5 atom of FAD and K661 are involved in a water-mediated hydrogen-bonding network (PDB: 2HKO).
To investigate whether sgRNAs were differentially enriched depending on the inhibitor treatment, we performed principal component analysis (PCA) on the various drug treatments. Clustering the transformed data suggests that sgRNA enrichment broadly classifies the compounds into three groups (Fig. 3c, group 1: GSK690; group 2: AW2, AW3, AW5, NV93; group 3: GSK-LSD1, AW1, AW4). This compound classification was mostly maintained when considering each treatment replicate separately, suggesting that the groupings are not driven by technical noise between replicates (Supplementary Fig. 3d). GSK690, which led to the weakest enrichment of active site sgRNAs and growth inhibition, was distinctly separated from the other compounds by PCA (Fig. 3c,d). GSK-LSD1 analogs in group 2 were distinguished by strong enrichment of sgRNAs targeting the residues N660 (sgN660) and G655 (sgG655) (Fig. 3d); NV93 was also marked by enrichment of sgL329 (Fig. 3d). Notably, these sgRNAs target residues directly adjacent to the FAD cofactor (Supplementary Fig. 3e). While compounds in group 3 were also characterized by strong enrichment of sgN660 (Supplementary Fig. 3e), sgRNAs targeting peripheral residues within the C-terminal amine oxidase domain were also enriched (Supplementary Fig. 3e, sgA705, sgA708, sg682, sgS687). These results suggest that CRISPR-suppressor scanning can discriminate between structurally similar and diverse inhibitors.
We next considered whether the underlying mutations, resulting from selection by each inhibitor treatment, could afford structure–function information on the LSD1 pocket (Fig. 1a). To investigate this possibility, we sequenced four exons of LSD1 encompassing the most highly enriched sgRNAs, including many identified through PCA (Fig. 3e)29. The majority of mutations were observed in exon 15 surrounding the most highly enriched sgRNA, sgN660 (Fig. 3e). Examination of the mutations in exon 15 revealed that GSK690 and NV93 selected LSD1 mutants containing K661E point substitutions. Modeling the K661E mutation onto a LSD1 structure suggests that the mutation rotates W695 and removes a hydrogen bond between K661 and the drug’s nitrile group (Fig. 3f and Supplementary Fig. 3f), perturbing interactions known to be essential for GSK690 and probably, NV93 binding30. By contrast, treatment with GSK-LSD1 analogs led to similar mutations that insert a K residue after LSD1 L659 (Fig. 3e). Modeling the L659_N660insK mutation onto the LSD1 structure suggests that the inserted K projects into the binding pocket (Fig. 3g and Supplementary Fig. 3g,h), potentially altering a hydrogen bond network involving K661, a water molecule and the FAD cofactor, which is necessary for enzyme activity31.
Aside from mutations in exon 15, different drugs selected heterogeneous mutations in exon 8 and exon 16 to varying extents (Fig. 3e and Supplementary Fig. 3i–l). Notably, deletion mutations in exon 16 were selectively enriched by AW4 (Supplementary Fig. 3i,j), causing the mutation profile of AW4 to be substantially different from those of other GSK-LSD1 analogs (including its isomer, AW3) (Fig. 3e). Altogether, these data support the notion that CRISPR-suppressor scanning can effectively discriminate between mechanistically distinct inhibitors and even more closely related chemical analogs, at the level of both sgRNA enrichment and protein-coding mutations.
Drug-resistant LSD1 mutants are enzymatically inactive.
Many of the mutations identified by CRISPR-suppressor scanning are predicted to potentially disrupt LSD1 enzyme activity, prompting us to investigate whether LSD1 enzyme activity is necessary for AML proliferation. To characterize these in-frame LSD1 mutations and the subsequent resistance mechanism(s) in an isolated setting, the top-enriched sgRNAs, sgN660 and sgG655, were transduced individually into AML cells, which were then treated with vehicle or GSK-LSD1. In the presence of GSK-LSD1, AML cells transduced with the sgRNAs (GFP+ cells) gained a competitive growth advantage versus non-transduced cells (GFP− cells), consistent with the emergence of drug resistance observed in the pooled screens (Fig. 4a and Supplementary Fig. 4a).
Fig. 4 |. Identification of enzyme-inactivated LSD1 alleles that maintain AML proliferation and GFI1B binding in the presence of GSK-LSD1.
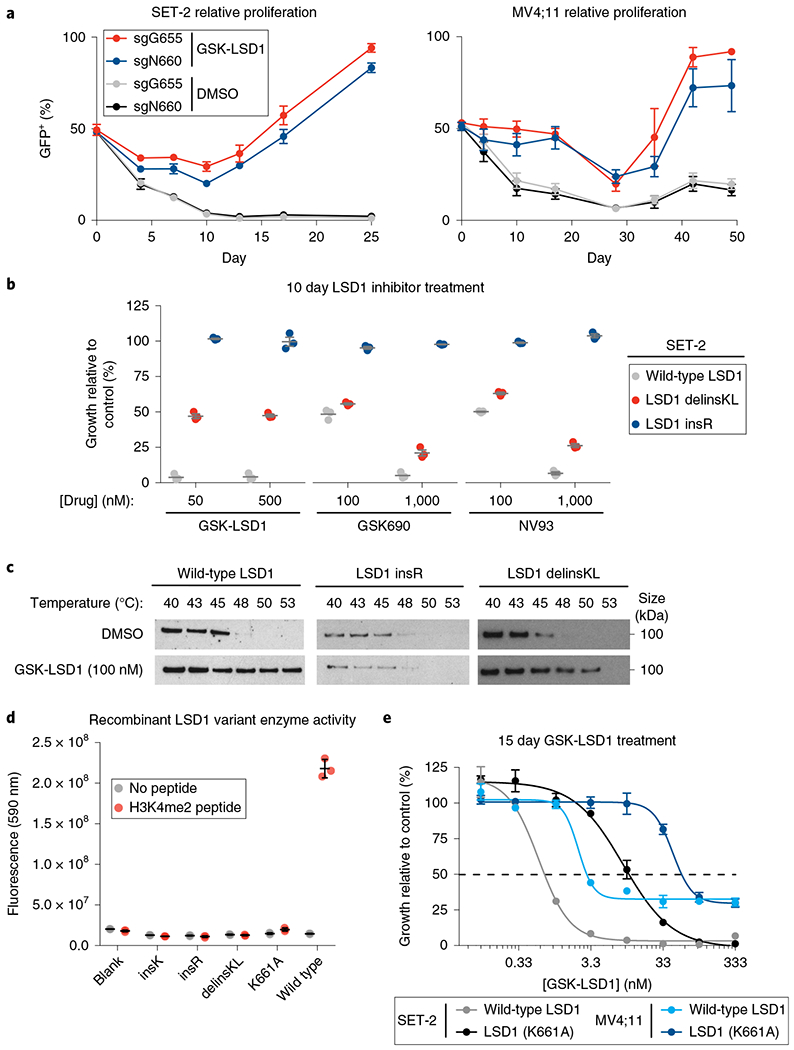
a, Line graph showing the fraction of GFP+ cells (y axis) over a time course following lentiviral transduction in SET-2 and MV4;11 cells. Under GSK-LSD1 treatment, mutagenesis by sgN660 leads to enrichment of GFP+ cells. Data represent mean±s.d. across three technical replicates. One of two independent replicates is shown. b, Scatter plots for relative cell growth (y axis) of SET-2 cells treated with GSK-LSD1, GSK690 and NV93 are shown. Lines represent mean±s.e.m. across three replicates. One of two independent replicates is shown. c, Immunoblots showing levels of LSD1 protein after heat treatment of SET-2 cells at the different temperatures indicated during the CETSA protocol. One of two independent replicates is shown. See Supplementary Fig. 7b for uncropped blot. d, Scatter plot showing enzyme activities (y axis) of mutant LSD1 variants identified (x axis) on a H3K4me2 peptide substrate. Gray lines represent mean±s.d. across three replicates. One of two independent replicates is shown. e, Dose-response curves for GSK-LSD1 tested in SET-2 and MV4;11 cells overexpressing either wild-type LSD1 or LSD1(K661A) are shown. Data represent mean±s.e.m. across three technical replicates. One of two independent replicates is shown.
We next expanded and genotyped clonal drug-resistant SET-2 cell lines. The mutations identified were largely similar to those obtained in CRISPR-suppressor scanning, including LSD1 N660_delinsKL and LSD1 L659_N660insR, abbreviated delinsKL and insR, respectively (Supplementary Fig. 4b–d). In-frame mutations were often paired with frameshift mutations in the drug-resistant clones at equal frequencies29 (Supplementary Fig. 4c), suggesting that the clones express a single in-frame LSD1 variant. Notably, whereas SET-2 LSD1 insR displayed complete resistance to all LSD1 inhibitors tested at the doses employed, LSD1 delinsKL only displayed resistance to GSK-LSD1 (Fig. 4b). Using cellular thermal shift assays (CETSA), we found that treatment with GSK-LSD1 led to LSD1 stabilization in wild-type SET-2 but not SET-2 LSD1 insR, suggesting that GSK-LSD1 binding to the insR LSD1 mutant is altered (Fig. 4c). By contrast, CETSA indicated that there is partial stabilization of LSD1 delinsKL in the presence of GSK-LSD1 relative to the vehicle control, suggesting that GSK-LSD1 may still bind this specific mutant at some level (Fig. 4c). This result is consistent with the partial drug-resistance phenotype of SET-2 LSD1 delinsKL (Fig. 4b). These observations support the premise that CRISPR-suppressor scanning can generate protein variants that can resolve mechanistically different classes of inhibitors.
Owing to the mechanism-based binding of GSK-LSD1 to the LSD1 FAD cofactor, we considered whether the LSD1 mutations may potentially alter GSK-LSD1 binding by compromising enzyme activity. This notion is supported by the frequent occurrence of mutations identified by CRISPR-suppressor scanning that potentially perturb the FAD cofactor-binding pocket, which is necessary for LSD1 activity31. Indeed, testing recombinant LSD1 mutant proteins indicated that these mutations lead to undetectable demethylation activity against an H3K4me2 peptide (Fig. 4d and Supplementary Fig. 4e–g). Additionally, we profiled these LSD1 variants using ThermoFAD, an approach that measures the shift in fluorescence of FAD as it is released from proteins upon heating32. These experiments revealed that LSD1 insR and LSD1 delinsKL release FAD at lower temperatures compared with wild-type LSD1 (Supplementary Fig. 4h), supporting the notion that these mutations potentially alter the binding of the FAD cofactor and consequently enzymatic activity. Furthermore, overexpression of enzyme-inactive LSD1 (K661A), but not wild-type LSD1, confers resistance to GSK-LSD1 (Fig. 4e)15. Altogether, these observations suggest that perturbing interactions with the FAD cofactor, even at the expense of destroying enzyme activity, is a viable strategy to promote drug resistance and that the enzyme activity of LSD1 is not required for AML proliferation.
The LSD1-GFI1B interaction is sufficient for AML growth.
The CRISPR-suppressor scanning data and the identification of drug-resistant, enzyme-inactive mutants support the notion that the anti-proliferative effects of LSD1 inhibitors in AML are not due to the inhibition of enzyme activity but instead are due to the perturbation of another function of the catalytic site. In particular, the CRISPR-suppressor scanning data suggest that protein binding interactions are critical for LSD1 function in AML (Fig. 2c,d and Supplementary Fig. 2a–c). In agreement with this hypothesis, recent studies have demonstrated that LSD1 inhibitors disrupt the interaction of LSD1 with GFI1/GFI1B, which is involved in AML proliferation13–15,19,20. If GFI1/GFI1B are the relevant protein partners, CRISPR-suppressor scanning should select mutants that maintain binding with GFI1/GFI1B in the presence of GSK-LSD1. In agreement, co-immunoprecipitation (Co-IP) experiments for GFI1B and LSD1 demonstrated that while the wild-type LSD1–GFI1B complex is disrupted by GSK-LSD1, the GFI1B complex with LSD1 delinsKL and LSD1 insR mutants were unperturbed by drug treatment (Fig. 5a). The drug-induced disruption of the LSD1–GFI1B interaction for wild-type LSD1 but not for the drug-resistant LSD1 mutants implicates this interaction in AML survival.
Fig. 5 |. An orthogonal drug-complementary GFI1B allele establishes sufficiency of the LSD1-GFI1B interaction for AML survival.
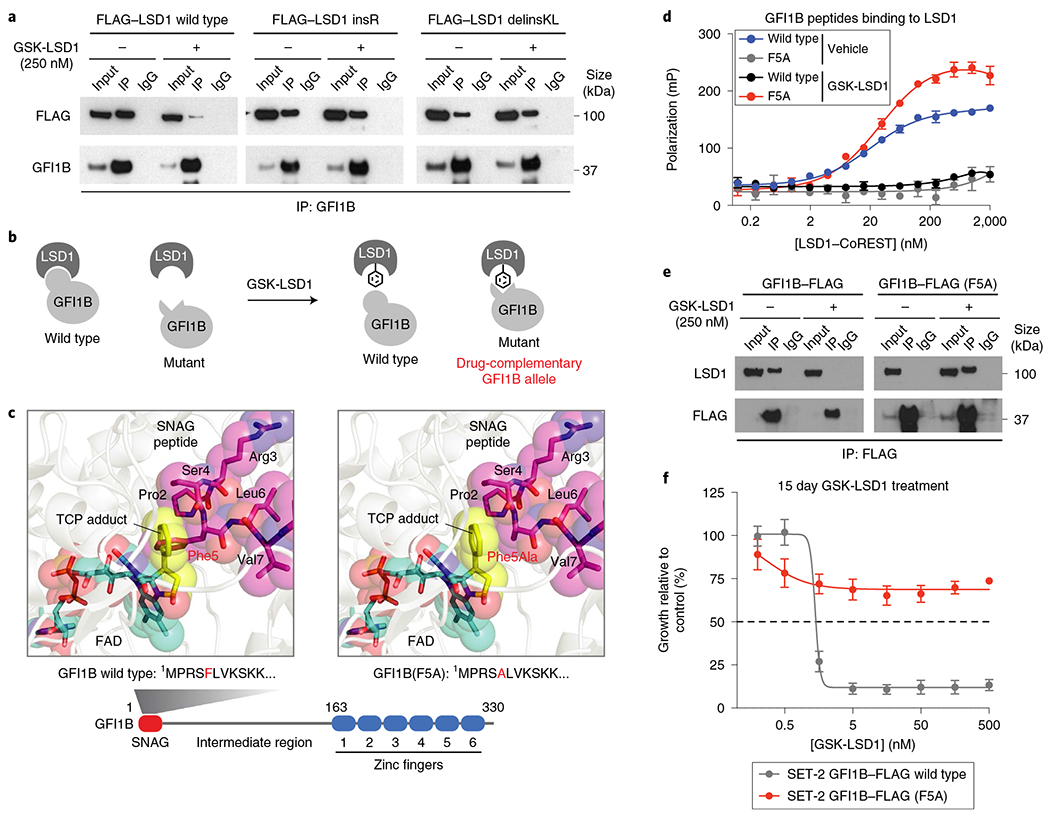
a, Co-IP of wild-type FLAG-LSD1 and FLAG-LSD1 mutant variants with GFI1B was performed after vehicle or GSK-LSD1 treatment (250 nM, 48 h) in transiently transfected HEK 293T cells. Co-IP was performed using an anti-GFI1B antibody. LSD1 was detected using an anti-FLAG antibody. One of two independent replicates is shown. See Supplementary Fig. 7c for uncropped blot. b, Illustration depicting the concept of an orthogonal drug-complementary GFI1B allele that can only bind LSD1 in the presence of GSK-LSD1. c, Structural views showing the LSD1 catalytic site labeled by TCP and bound to either a wild-type SNAG peptide (left) or a modeled SNAG (F5A) peptide (right). Schematic of the protein domains of GFI1B highlighting the SNAG domain sequence (bottom). d, Binding curves showing fluorescence polarization (y axis) for increasing concentrations of LSD1-CoREST complex (x axis) in the presence of either wild-type GFI1B 2-10 peptide or GFI1B (F5A) 2-10 peptide (100nM) and/or GSK-LSD1 (50 μM). Data represent mean ± s.e.m. across three technical replicates. One of two independent replicates is shown. e, Co-IP immunoblots showing levels of wild-type GFI1B-FLAG, GFI1B-FLAG (F5A) and LSD1 after anti-FLAG IP in SET-2 overexpressing wild-type GFI1B-FLAG or GFI1B-FLAG (F5A) after vehicle or GSK-LSD1 treatment (250 nM, 48 h). One of two independent replicates is shown. See Supplementary Fig. 7d for uncropped blot. f, Dose-response curves for SET-2 cells expressing wild-type GFI1B-FLAG or GFI1B-FLAG (F5A) treated with GSK-LSD1 are shown. Expression of GFI1B-FLAG (F5A) compared with wild-type GFI1B-FLAG significantly rescues growth of SET-2 cells. Data represent mean ± s.e.m. across three replicates. One of two independent replicates is shown.
To demonstrate that the specific LSD1–GFI1B interaction is necessary for AML proliferation, we considered whether an orthogonal GFI1B mutant allele could be identified that only binds to LSD1 covalently modified by GSK-LSD1 but not the unmodified enzyme (Fig. 5b). On the basis of protein modeling, we hypothesized that the GFI1B (F5A) mutant could selectively bind to LSD1 covalently modified by GSK-LSD1, as the mutation might restore a hydrophobic interaction between the two proteins21,22,33 (Fig. 5c and Supplementary Fig. 5a). We confirmed that a wild-type GFI1B peptide only binds purified LSD1, complexed with its binding partner CoREsT, in the absence of GSK-LSD133 (Fig. 5d). Conversely, a odified GFI1B (F5A) peptide only binds LSD1–CoREST in the presence of GSK-LSD1 (Fig. 5d). Consistent with this in vitro data, Co-IP experiments demonstrated that overexpressed wild-type GFI1B–FLAG only interacts with LSD1 in the absence of GSK-LSDl while overexpressed GFI1B–FLAG (F5A) only interacts with LSD1 in the presence of GSK-LSD1 (Fig. 5e). These data demonstrate that GFI1B (F5A) is an orthogonal drug-complementary allele34.
Critically, overexpression of GFI1B–FLAG (F5A) but not wild-type GFI1B–FLAG rescued the growth of AML cells in the presence of GSK-LSD1 (Fig. 5f and Supplementary Fig. 5b). By contrast, treatment with the bulkier AW2 analog abrogated the protective effect of GFI1B–FLAG (F5A) on growth (Supplementary Fig. 5c). Overexpression of GFI1–FLAG (F5A) was also sufficient to rescue growth (Supplementary Fig. 5d), indicating that GFI1 and GFI1B may be redundant for AML proliferation. Altogether, our data provide compelling evidence that the LSD1–GFI1B interaction, complexed in its native context, is necessary for the growth of AML cells.
LSD1–GFI1B suppresses enhancer activation.
The LSD1–GFI1B interaction may serve to recruit LSD1 and corepressor complexes to GFI1B-target enhancers, thus suppressing their activity15,23,35. By disrupting LSD1 recruitment, GSK-LSD1 may induce the activation of GFI1B-target genes and promote differentiation13,15. However, these changes in gene expression could be induced by the disruption of a LSD1–GFI1B complex, the inhibition of enzyme activity, or both. Given the importance of the LSD1–GFI1B complex in AML survival irrespective of LSD1 enzymatic activity, we sought to employ the drug-resistant LSD1 mutant cell lines to deconvolute the relative contributions of enzyme inhibition versus complex disruption on influencing gene expression upon LSD1 inhibitor treatment.
We first performed RNA sequencing (RNA-seq) on wild-type SET-2 and enzyme-inactive SET-2 LSD1 insR treated with vehicle or GSK-LSD1 (250 nM, 48 h), revealing 1,290 differentially expressed genes across all conditions (Fig. 6a and Supplementary Fig. 6a). Treatment of wild-type SET-2 with GSK-LSD1 induced substantial changes in gene expression: 244 genes were upregulated while 119 genes were downregulated (|log2(fold-change)| > 2, P<0.01). By contrast, using the same statistical thresholds, no genes were found to be differentially expressed on treatment of SET-2 LSD1 insR with GSK-LSD1, consistent with its fully drug-resistant phenotype (Fig. 6a). Genes differentially expressed in wild-type SET-2 upon treatment with GSK-LSD1 were enriched in gene signatures associated with myeloid cells and erythroid cells by gene set enrichment analysis13,36 (Supplementary Fig. 6b), in agreement with past studies suggesting that LSD1 inhibition induces AML transdifferentiation13. Overall, these data are consistent with our previous observations that enzyme-inactive LSD1 insR suppresses the effects of GSK-LSD1 and indicate that most changes in gene expression, including those in genes involved in AML differentiation, are caused by on-target inhibition of LSD1.
Fig. 6 |. Drug-resistant AML cells maintain LSD1-GFI1B binding on chromatin and fail to activate GFI1B-bound enhancers in the presence of GSK-LSD1.
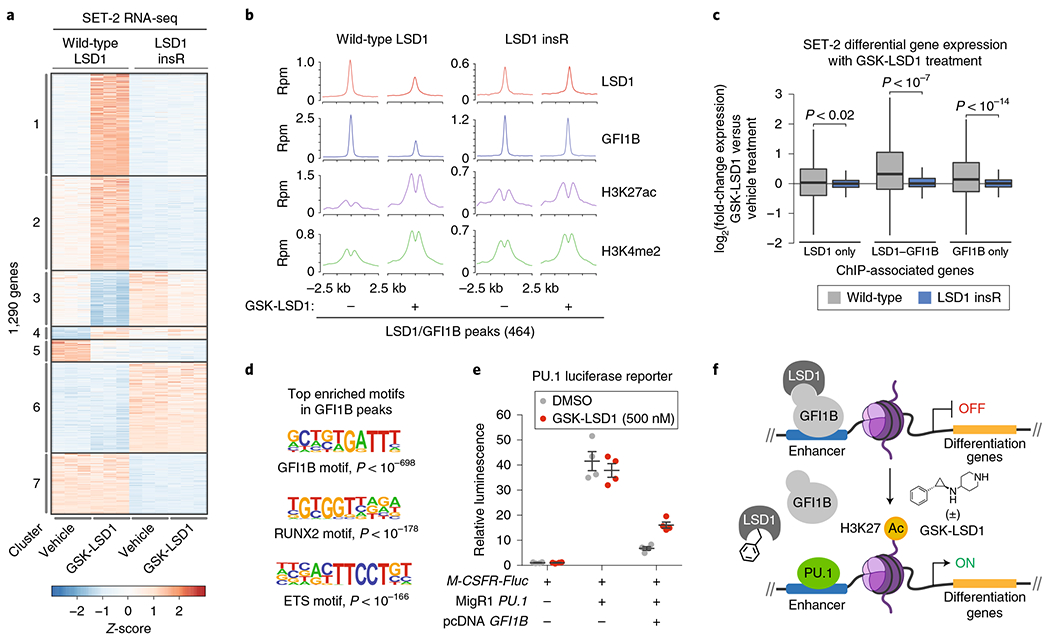
a, Heat map showing expression profiles of the 1,290 most variably expressed genes (|log2(fold-change)|> 2, Padj<0.01) across wild-type SET-2 and SET-2 LSD1 insR after treatment with vehicle or GSK-LSD1 (250 nM, 48 h) for three replicates per condition, each shown independently. The adjusted P value for testing significance of differential expression was calculated using the Benjamini–Hochberg correction. Genes exhibiting correlated patterns of expression were grouped by k-means clustering (k = 7). Color represents Z-scores of gene expression across rows. b, ChIP-seq composite plots showing average signal (y axis, reads per million (r.p.m.)) for LSD1, GFI1B, H3K27ac and H3K4me2 in wild-type SET-2 and SET-2 LSD1 insR cells treated with vehicle or GSK-LSD1 (250 nM, 48 h) centered around LSD1-GFI1B co-occupied peaks identified in wild-type SET-2. The x axis shows flanking regions of ±2.5 kb around the peak center. Experiment performed once. c, Box plot showing differential gene expression of ChIP-seq-associated gene subsets upon GSK-LSD1 treatment (250 nM, 48 h) in wild-type SET-2 or SET-2 LSD1 insR cells. In the plot, bars represent the median, the box represents the IQR and the whiskers represent 1.5× IQR. P values were calculated using the Wilcoxon test (two-sided). d, Consensus GFI1B, RUNX2 and ETS motif logos detected in GFI1B ChIP-seq peaks in wild-type SET-2 cells. The corresponding P values for enrichment are calculated as previously described38. e, Scatter plots showing relative firefly luciferase (Fluc) expression in the absence or presence of a PU.1 reporter plasmid (M-CSFR-Fluc), PU.1 expression plasmid (MigR1 PU.1), GFI1B expression plasmid (pcDNA GFI1B), or GSK-LSD1 (500 nM, 48 h). Gray lines represent mean±s.e.m. across four replicates. One of two independent replicates is shown. f, Schematic of a proposed model, whereby GSK-LSD1 induces displacement of a LSD1-GFI1B complex from enhancers allows PU.1-mediated activation of genes involved in AML differentiation.
We next considered whether we could deconvolute the relative roles of the enzymatic and nonenzymatic functions of LSD1 in mediating the response of cells to GSK-LSD1. To investigate this phenomenon, we identified seven gene sets that exhibit correlated changes in expression using k-means clustering (Fig. 6a). Genes in clusters 1–3 are differentially expressed when wild-type SET-2 are treated with GSK-LSD1, but otherwise display comparable expression levels across the other samples. Since LSD1 insR is enzyme-inactive yet does not phenocopy the effects of GSK-LSD1 for clusters 1–3, these genes are likely differentially expressed in wild-type SET-2 due to perturbing a nonenzymatic function of LSD1. By contrast, genes in clusters 4 and 5 are differentially expressed specifically due to loss of LSD1 enzymatic activity because enzyme-inactive LSD1 insR phenocopies the effects of drug treatment on wild-type SET-2 gene expression. Finally, genes in clusters 6 and 7 are differentially expressed between the cell lines independent of inhibitor treatment. Notably, there are many more genes in clusters 1–3 than in clusters 4 and 5, suggesting that inhibition of the nonenzymatic functions of LSD1 are driving the majority of changes in gene expression observed upon acute drug treatment. Altogether, by comparing the responses of wild-type SET-2 and SET-2 LSD1 insR to GSK-LSD1, we distinguish between genes that change expression owing to the inhibition of LSD1 enzymatic versus nonenzymatic functions.
The observation that SET-2 LSD1 insR suppresses drug-mediated gene expression changes prompted us to investigate whether retention of GFI1B DNA binding underlies these effects. Consequently, we mapped GFI1B, LSD1, H3K4me2 and H3K27ac in wild-type SET-2 and SET-2 LSD1 insR by chromatin immunoprecipitation followed by sequencing (ChIP-seq) after vehicle or GSK-LSD1 treatment. To restrict our analysis to drug-induced effects in the context of wild-type AML cells, we focused on ChIP-seq peaks called in wild-type SET-2 cells. In wild-type SET-2 cells, we identified 3,391 GFHB and 3,324 LSD1 high-confidence binding sites, which significantly overlap (424 LSD1-GFI1B co-occupied sites, hypergeometric P < 10−905). In agreement with a recent report, GSK-LSD1 treatment led to reduced LSD1 and GFI1B binding compared with vehicle treatment15 (Fig. 6b and Supplementary Fig. 6c,d). While the reduction of LSD1 binding was mostly confined to LSD1 binding sites co-occupied by GFI1B, reduction of GFI1B binding occurred more indiscriminately.
To investigate how drug-induced disruption of GFI1B binding may impact gene expression, we considered the expression of the closest gene to each LSD1–GFI1B ChIP–seq binding site. The median expression of these genes in wild-type SET-2 cells significantly increased upon treatment with GSK-LSD1 (Fig. 6c) and was accompanied by increases in levels of H3K27ac and H3K4me2, histone modifications associated with active cis-regulatory regions, at LSD1–GFI1B peaks in wild-type SET-2 (Fig. 6b and Supplementary Fig. 6d). Notably, the drug-induced increase in H3K27ac, but not H3K4me2, extended beyond GFI1B-bound sites, suggesting a broader role of LSD1 inhibition in enhancer activation in SET-2 cells (Supplementary Fig. 6e,f). By contrast, increased expression of LSD1–GFI1B-associated genes and accompanying changes in chromatin state were suppressed in drug-resistant SET-2 LSD1 insR upon GSK-LSD1 treatment (Fig. 6c and Supplementary Fig. 6c,d). The necessity of the LSD1–GFI1B interaction may stem from its ability to recruit histone deacetylase complexes to silence LSD1–GFI1B target genes15,37. In support, the LSD1 drug-resistance mutations confer no protective advantage against vorinostat, a histone deacetylase inhibitor (Supplementary Fig. 6g). Altogether, our observations are consistent with the notion that GSK-LSD1 leads to the dissociation of LSD1–GFI1B complexes from chromatin, inducing subsequent enhancer activation and upregulation of many LSD1–GFI1B-associated genes.
To gain an understanding of what factors may drive enhancer activation after GFI1B eviction, we interrogated the DNA sequences underlying GFI1B peaks for over-represented motifs. Aside from GFI1B-binding motifs, this analysis identified binding motifs for RUNX and ETS factors as highly enriched (Fig. 6d)38, nominating them as candidate activators. In particular, PU.1 is an ETS factor that is antagonistic to GFI1/GFI1B, and previous work has demonstrated that LSD1 inhibitors can activate PU.1 target genes11,13,15. To establish a possible connection between PU.1, GFI1B and LSD1 inhibitors, we employed a PU.1 luciferase reporter driven by the M-CSFR promoter39. Overexpression of wild-type GFI1B diminished PU.1 activity in the reporter, consistent with earlier studies showing that GFI1 can antagonize PU.1 transactivation39 (Fig. 6e). Significantly, GSK-LSD1 treatment partially suppressed the inhibitory effects of GFI1B on PU.1 transactivation, demonstrating a role of LSD1 in this mechanism. Altogether, our studies support a model where a chromatin-associated LSD1–GFI1B complex, independent of LSD1 enzyme activity, represses acetylation and activation of enhancers by potentially suppressing PU.1 activity (Fig. 6f).
Discussion
The identification of cancer mutations that alter the function of chromatin-modifying enzymes has brought increasing attention to them as drug targets40–43. Although the notion that inhibitors targeting chromatin modifiers exert their anti-proliferative activity by correcting aberrant chromatin states is often invoked, whether changes in chromatin are directly responsible for their efficacy is often unclear. Motivated by these questions, we have employed CRISPR-suppressor scanning to dissect the mechanism of action of chemical inhibitors directly in cells. Evaluating a small panel of LSD1 inhibitors, our studies show that CRISPR-suppressor scanning can discriminate between structurally similar and distinct molecules at both the level of sgRNA enrichment and coding mutations. These data can be integrated with structural information and computational methods developed here to provide structure-function relationships that may reveal critical regions of proteins, modes of drug binding, as well as hotspots essential for drug potency. Although exact molecular interactions cannot be described using this approach, we expect that further computational modeling in addition to advancements that increase the resolution of CRISPR mutagenesis may improve its ability to resolve these interactions. Moreover, the mutations identified through this approach may predict how easily cells develop resistance to small molecules, which could have significant clinical implications. In particular, in the context of LSD1, our data suggest that resistance mutations may be easier to achieve since they are not constrained to the selection pressure of maintaining enzymatic function.
Using CRISPR-suppressor scanning, we independently uncover and corroborate the unexpected mode by which LSD1 inhibitors block AML cell growth, in which inhibition of demethylase activity is not directly responsible for blocking proliferation15. Instead, CRISPR mutagenesis identified drug-resistant LSD1 mutants that are enzyme-inactive but bind to GFI1B in the presence of GSK-LSD1. To show that the LSD1–GFI1B complex is sufficient for AML proliferation, we identified a compensatory GFI1B mutant allele that structurally complements LSD1 specifically modified by GSK-LSD1. The bump-hole rescue, afforded by this orthogonal drug-complementary GFI1B allele, establishes the essential nature of the LSD1–GFI1B interaction in its native context. This orthogonal drug-complementary allele could prove critical for interrogating this important protein–protein interaction with precise chemical control.
Leveraging drug-resistant mutants, we show that LSD1 inhibitors operate by disrupting a complex of LSD1 with GFI1B on chromatin, inducing the activation of GFI1B-target genes in a demethylase independent fashion. This activation may in part be mediated by PU.1. Although PU.1 had previously been implicated in the anti-proliferative mechanism of LSD1 inhibitors11,13,15, its direct connection to GFI1/GFI1B has not been established in this context. We show that LSD1 inhibitors suppress the ability of GFI1B to repress PU.1 transactivation, establishing a possible direct link between GFI1B and PU.1 in AML39. More broadly, our results suggest that inhibition of LSD1’s scaffolding as opposed to its enzymatic activity is the major driver underlying drug-induced alterations in gene expression. Instead, LSD1 inhibitors potentially offset an opposing relationship between the two transcription factors, GFI1B and PU.111,39, which may reflect aspects of their antagonism during normal hematopoiesis35. These findings highlight the nuanced mechanisms by which chromatin modifiers regulate gene expression outside of their archetypal functions.
In summary, we demonstrate the versatility of CRISPR-suppressor scanning to interrogate target protein–small molecule interactions through deciphering the mechanism of action of LSD1 inhibitors. Drug-resistance alleles are often generated through rational design with the explicit goal of validating a small-molecule target44,45. By systematically identifying drug-resistance alleles that span a target, many of which occur through unexpected mutations, CRISPR-suppressor scanning can be used as a hypothesis generator to discover new aspects of target biology and drug mechanism of action. More broadly, our work raises important questions regarding the precise roles of the enzymatic versus scaffolding activities of chromatin regulators in the diverse functions these modifiers play in gene regulation.
Methods
Cell culture and lentivirus production.
MV4;11, CMK-86 and K562 were obtained from ATCC; SET-2 was a gift from M.D. Shair (Harvard University); HEK 293T was a gift from B.E. Bernstein (Massachusetts General Hospital). All cell lines were authenticated by Short Tandem Repeat profiling (Genetica) and routinely tested for mycoplasma (Sigma-Aldrich). All media were supplemented with 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (Life Technologies) and FBS (Peak Serum). All cell lines were cultured in a humidified 5% CO2 incubator at 37 °C. MV4;11, CMK-86 and K562 were cultured in RPMI-1640 (Life Technologies), 10% FBS. SET-2 were cultured in RPMI-1640, 20% FBS. HEK 293T cells were cultured in DMEM (Life Technologies), 10% FBS. For lentivirus production, plasmids were co-transfected with GAG/POL and VSVG plasmids into HEK 293T using FuGENE HD (Promega). Media was exchanged after 8 h and the viral supernatant was collected 72 h after transfection and filtered (0.45 μm). Cell lines were transduced by spinfection at 1,800g for 2 h at 37 °C with 8 μg ml−1 polybrene (Santa Cruz Biotechnology). After 48 h post-transduction, puromycin (Thermo Fisher Scientific) selection was carried out for 5 d at 1.5 μg ml−1 for SET-2 and 1 μg ml−1 for MV4;11.
Chemical reagents.
Compounds were stored at −20 °C in 100% DMSO. The vehicle condition represents 0.1% DMSO treatment. GSK-LSD1 and GSK690 was used as purchased from Sigma-Aldrich (≥98% purity by HPLC) and Aobious (98% purity by HPLC), respectively. AW1–5 were synthesized as described in the Supplementary Notes 1 and 2, and NV93 was synthesized according to a previously described protocol28.
Cell growth assays.
Cell lines were plated in triplicate with drug or vehicle treatments (2,500 cells per well for MV4;11 and 5,000 cells per well for all other cell lines). An equal volume of cells from each well was split and re-plated with fresh media containing vehicle or inhibitor at day 5 and day 10. Cell viability was monitored at day 10 and 15 by measuring end point luminescence using CellTiter-Glo (Promega) on a SpectraMax i3x platereader. Dose–response curves were determined through interpolation using GraphPad Prism v.7 nonlinear regression fit ([inhibitor] versus normalized response–variable slope). All growth assays were performed at least twice.
sgRNA pooled cloning and CRISPR-suppressor scanning experiments.
The LSD1 tiling library included every sgRNA with an NGG PAM and a cleavage site within the LSD1 coding sequence (NP_055828.2). These sgRNA sequences are listed in Supplementary Dataset 1. Oligos containing the sgRNA sequences were cloned into pLentiCRISPR.v2 in a pooled fashion as previously described5. pLentiCRISPR.v2 was a gift from F. Zhang (Addgene plasmid number 52961, Broad Institute). Lentiviral particles carrying the resultant LSD1 tiling library were generated as described above and titered according to published procedure46. Cells (1 × 107) were transduced such that the multiplicity of infection < 0.3 and subsequently selected with puromycin. After puromycin selection, cells were split into pools and treated with the drug(s) or vehicle. Genomic DNA was isolated at specified time points using the QIAamp DNA Mini Kit (Qiagen). All CRISPR-suppressor scanning experiments were performed with three separate transductions. To measure the composition of the population, sgRNA sequences for all replicates were amplified with PCR primers (Supplementary Dataset 5) and sequenced as previously described5.
Generation of clonal drug-resistant LSD1 mutant cell lines.
sgRNA sequences targeting LSD1, sgG655 (GGGATTTGGCAACCTTAACA) and sgN660 (TAGGGCAAGCTACCTTGTTA), were cloned into pLentiCRISPR-eGFP.v1. SET-2 and MV4;11 cells were transduced with the resultant plasmids as indicated above and subsequently treated with 100 nM GSK-LSD1 for 50 d to enrich for drug-resistant mutant cells. Surviving cells were FACS-sorted as single cells and then expanded. pLentiCRISPR-eGFP.v1 was a gift from B. Bornhauser (Addgene plasmid number 75159, University Children’s Hospital Zurich).
Genotype determination.
Genomic DNA was isolated as mentioned above. For library preparation, genomic PCR primers (Supplementary Dataset 5) with Illumina adapter sequences were used to amplify specified regions of LSD1 as previously described47. Samples were sequenced on a MiSeq genome analyzer (Illumina). The sequencing reads were analyzed using CRISPResso (v.1.0.13)29.
Mammalian overexpression constructs.
The LSD1 and LSD1 (K661A) open reading frames (ORFs) were subcloned from pCMV LSD1 and pCMV LSD1 (K661A), which were gifts from R. Shiekhattar (University of Miami Miller School of Medicine). Site-directed mutagenesis was performed using Q5 Site-Directed Mutagenesis Kit (New England Biolabs) to generate LSD1 N660delinsKL and LSD1 L659_N660insR. All modified LSD1 ORFS were subcloned into pSMALP and pcDNA.3 with an N-terminal FLAG tag using Gibson cloning (New England Biolabs). GFI1 and GFI1B were subcloned from pENTR GFI1 and pENTR GFI1B, respectively, which were gifts from H. Zoghbi (Addgene plasmid numbers 16168 and 16169, respectively; Baylor College of Medicine), and subcloned into pcDNA.3 or pSMALP with a C-terminal FLAG tag using Gibson cloning. Site-directed mutagenesis was performed using Gibson cloning to generate GFI1 (F5A) and GFI1B (F5A). The Renilla luciferase ORF was subcloned into pcDNA.3 using Gibson cloning. pSMALP was generated from pSMAL through introduction of a puromycin cassette into pSMAL (a gift from J. E. Dick, University of Toronto).
Immunoblotting.
Cells were lysed on ice using RIPA buffer (Boston BioProducts) supplemented with fresh HALT Protease Inhibitor (Thermo Fisher Scientific) and the lysates were cleared. The protein concentration of the lysate was determined using the BCA Protein Assay Kit (Thermo Fisher Scientific). Immunoblotting was performed according to standard procedures. The primary antibodies used are as follows: LSD1 (Bethyl Laboratories A300-215A, LOT number 2); GFI1B (B-7) X (Santa Cruz Biotechnology sc-28356X, LOT number D1615); monoclonal anti-FLAG M2 (Sigma-Aldrich F1804, LOT number SLBW3851); GAPDH (Santa Cruz Biotechnology sc-477724, LOT number B0210); and GFI1B (Santa Cruz Biotechnology sc-28356X, LOT number D1615).
Co-immunoprecipitation.
SET-2 cells (2 × 107) expressing wild-type GFI1B–FLAG or GFI1B-FLAG (F5A) were washed twice with cold PBS and flash frozen. Cells were thawed, lysed on ice in N450 buffer (50 mM Tris-HCl pH 7.5, 450 mM NaCl, 1 mM MgCl2, 1% NP-40 alternative, 5% glycerol) supplemented with 1:10,000 benzonase (Sigma-Aldrich), and the lysates were cleared. The protein concentration was quantified as above and diluted to 1 mg ml−1 in lysis buffer. Supernatants were immunoprecipitated overnight at 4 °C with 2.5 μg anti-FLAG M2 antibody and 40 μl Protein G Dynabeads (Thermo Fisher Scientific). Beads were washed twice with lysis buffer, eluted in SDS-PAGE loading buffer, and carried forward to immunoblotting as described above. For Co-IPs performed in HEK 293T cells, cells were plated at 70–80% confluency and treated with 250 nM GSK-LSD1 or vehicle. 24 h after plating, cells were co-transfected with 1 μg pcDNA.3 FLAG–LSD1 plasmid (wild-type, L659_N660insR, or N660delinsKL) and 2 μg pcDNA.3 wild-type GFI1B–FLAG using FuGENE HD. 48 h post-transfection, cells were washed with PBS then lysed with N450 buffer. Co-IP was performed using 4 μg GFI1B antibody (Santa Cruz Biotechnology sc-28356X).
Cellular thermal shift assay.
CETSA was performed on the basis of a modified protocol48. Cells (2 × 106) were treated with vehicle or 100 nM GSK-LSD1 for 1 h at 37 °C, then washed with PBS and resuspended in 500 μl PBS supplemented with Roche cOmplete Protease Inhibitor Cocktail Tablets, EDTA-free. Aliquots of 25 μl from each condition were distributed into PCR strip tubes and heated at 40, 43, 45, 47, 50, or 53 °C for 3 min before being cooled to room temperature for another 3 min. The samples were lysed by three freeze-thaw cycles. The lysate was clarified by centrifugation at 17,000g for 20 min at 4 °C. The samples were analyzed by immunoblotting as described above.
Luciferase reporter assay.
According to a modified procedure, HEK 293T cells were plated in quadruplicate at a density of 1,000–1,500 cells per well and treated with vehicle or 500 nM GSK-LSD139. After 24 h, the cells were transfected with 200 ng pcDNA.3 GFI1B–FLAG using FuGENE HD. 24 h post-transfection, 15 ng pcDNA.3 Renilla luciferase, 50 ng M-CSFR firefly luciferase and 100 ng MigR1 PU.1 were co-transfected. 48 h after the second transfection, the Dual-Luciferase Reporter Assay System (Promega) was used to measure end point luminescence (SpectraMax i3x). M-CSFR firefly luciferase and MigR1 PU.1 plasmids were gifts from M. Simon (University of Pennsylvania School of Medicine) and R. Dahl (Indiana University School of Medicine).
Protein purification.
For bacterial constructs, the LSD1 (Δ1–150) gene was codon optimized and synthesized as two fragments from Integrated DNA Technologies and Quintara Biosciences. The fragments were cloned into pET28b containing an N-terminal His6-tag using Gibson cloning. Mutations were introduced with Q5 Site-Directed Mutagenesis Kit. The constructs were expressed in NiCo21(DE3) competent Escherichia coli (New England Biolabs) using a previously described protocol49. Protein fractions with >95% purity as assessed by SDS-PAGE were pooled and stored at −80 °C in 25 mM sodium phosphate pH 7.4, 150 mM NaCl, 0.3 mM TCEP and 5% glycerol (storage buffer). CoREST(305–482) was codon optimized for bacterial expression and assembled into pET28b containing an N-terminal GST-tag followed by a TEV cleavage site from a synthesized fragment purchased from GeneWiz. Recombinant CoREST expression and purification were carried out according to a modified literature procedure50. Recombinant protein was purified by GST affinity chromatography using a linear gradient of 0–50 mM reduced glutathione in lysis buffer and the buffer was exchanged to TEV protease cleavage buffer (50 mM Tris-HCl pH 8.0, 75 mM NaCl, 1 mM DTT and 0.5 mM EDTA). The GST-tag was removed by incubation with TEV protease overnight at 4 °C. The cleaved protein was purified using a GSTrap column followed by Superdex 200 10/300 GL column (GE Healthcare) in storage buffer. Purified CoREST was incubated with LSD1 in a 2:1 molar ratio for 2 h and gel-filtered on a Superdex 200 10/300 GL column equilibrated in storage buffer. The purity of the complex was verified by SDS-PAGE and fractions with 90–95% purity were pooled and stored at −80 °C. Fast protein liquid chromatography traces for proteins purified by size-exclusion chromatography are included in Supplementary Fig. 8.
LSD1 enzyme assays.
LSD1 enzymatic activity assays were performed in triplicate using Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (Invitrogen) with recombinant LSD1 and a synthetic peptide corresponding to the first 21 amino acids of H3K4me2 (Anaspec). LSD1 (500 ng per well) was incubated with 20 μM peptide at room temperature for 30 min. The end point fluorescence was measured on a microplate reader (excitation: 530 nm; emission: 590 nm) after 60 min following the addition of the Amplex Red/HRP mixture. Inhibition assays were performed as described above. In brief, LSD1 (75 ng per well) and inhibitors at the appropriate concentration were incubated at room temperature for 10 min in reaction buffer with 0.01% BRIJ35 (Thermo Fisher Scientific) before the addition of peptide. Ki values were determined in GraphPad Prism v.7 by nonlinear regression analysis (one site-fit Ki) of the concentration/inhibition data.
ThermoFAD assay.
ThermoFAD was performed in quintets as previously described32. Thermal curves were obtained using a quantitative PCR with reverse transcription detection system (BioRad C1000 Touch Thermal Cycler) with a temperature gradient from 20 to 95 °C. The melting temperature was determined by fitting the thermal curves to a Boltzmann sigmoid and calculating the inflection point (GraphPad Prism v.7).
UV-vis spectroscopy.
UV-vis spectra were collected every 20 nm from 300 nm to 600 nm on a microplate reader (SpectraMax i3x) at a protein concentration of 10 μM.
Circular dichroism.
CD spectra were acquired using a quartz cuvette with a 1 mm path length on a Jasco J-815 CD spectrometer collecting five scans (1 s averaging time) for each spectrum. Mean residue ellipticity (degrees cm2 dmol−1) values were calculated using the following equation, where θ is ellipticity (mdeg), l is path length (cm), C is peptide concentration (M), Nis number of residues.
Fluorescence polarization assay.
Binding assays were performed in two independent experiments with three technical replicates. The change in fluorescence polarization of fluorescently labeled GFI1B and GFI1B (F5A) peptides on binding to LSD1–CoREST was monitored using a previously described protocol37. LSD1–CoREST (2 μM) was incubated with the labeled peptides (2 nM) for 1 h on ice. After incubation, the samples were prepared by a two-fold serial dilution in the assay buffer (15 mM KH2PO4, pH 7.2, 5% glycerol and 1 mg ml−1 BSA) containing 2 nM labeled peptide.
For assays in the presence of 10 μM GSK-LSD1, the protein complex was incubated with the inhibitor for 1 h on ice and then with the labeled peptides for 1 h. Fluorescence polarization was measured using a microplate reader in 384-well black microplates at 25 °C. The G-factor on the microplate reader was adjusted to 35 mP for the reference well containing labeled peptide. The binding curves were fit by nonlinear regression analysis in GraphPad Prism v.7 as described previously37.
Structural modeling analyses.
MODELLER was used to generate homology models of the in-frame mutants of LSD1 by using PDB structure 2HKO as a template. One thousand models were generated per structure. The best model for each variant, as judged by DOPE score, was refined using the Relax application in Rosetta 2018 (release 7111c54)51. The scoring function was used with harmonic constraints on the backbone atom positions. These restraints were ramped down towards the end of the optimization using the -relax:constrain_relax_to_start_coords option.
To assess the water-mediated hydrogen bonding network between FAD and K661, semi-empirical models were built of the active site for selected structures. Complete residues within 7 Å of atom C4X in the FAD ligand or 4 Å of the water oxygen were exported to Gaussian 16 with all connections capped by hydrogens. Each structure was optimized with the PDDG semi-empirical quantum mechanical method with the protein residues fixed while the FAD and water ligands remained unconstrained52.
SNAG peptide mutations were generated in Chimera suite and modeled in Maestro Schrödinger using Protein Preparation Wizard (OPLS3 force field).
ChIP–seq.
ChIP was performed as previously described in singlicate on SET-2 cells (wild type and LSD1 L659_N660insR) treated with GSK-LSD1 (250 nM) or vehicle for 48 h53. The following antibodies were used: LSD1 (Bethyl Laboratories A300–215A, LOT number 2); GFI1B (Abcam ab26132, LOT number GR318193–8); H3K4me2 (Active Motif 39141, LOT number 01008001); H3K27ac (Active Motif 39133, LOT number 31814008). Libraries were sequenced on the NextSeq 500 sequencer (Illumina) for 75 bases in paired-end mode.
RNA-seq library preparation.
In triplicate separate cell cultures, total RNA from SET-2 cells (wild type and LSD1 L659_N660insR) treated with GSK-LSD1 (250 nM) or vehicle for 48 h was isolated using the RNeasy Plus Mini Kit (Qiagen). Library preparation was performed using the QuantSeq 3′ mRNA-Seq Library Prep Kit FWD (Lucigen) according to the manufacturers protocol. The samples were sequenced on the NextSeq 500 (Illumina) for 42 bases in single-end mode.
Analysis of sgRNA enrichment and clustering method.
All data processing was performed in Python v.3.5.5 (www.python.org) and R (version ≥ 3.4.2) (www.Rproject.org). sgRNA enrichment was calculated as previously described5. In brief, reads uniquely assigned to each sgRNA sequence were read-count normalized, log2 + 1 transformed, averaged across three replicates, and were then normalized by subtracting the average of the log2-transformed counts across three replicates of that sgRNA at week 0. Finally, for each condition the mean of the transformed counts for all functionally neutral genome-targeting control sgRNAs (averaged across replicates and normalized as described above) was subtracted from each the enrichment value of each sgRNA for that specific condition. This value is referred to as the normalized log2(fold-change sgRNA enrichment), or log2(enrichment). Libraries that did not receive adequate sequencing depth (> 1,000× coverage) and sgRNAs with zero counts at week 0 were excluded from the analysis.
Pairwise distances between LSD1 amino acid residues were obtained by determining the Euclidean distance between the centroids of each residue using PyMOL (v.1.8.4.2) (PDB: 2Y48). To calculate the PWES, sgRNAs were first assigned to the amino acid closest to the predicted Cas9 cleavage site. The PWES score was then determined as follows:
An analogous approach was employed in the clustering of mutations in protein structures algorithm except that mutation frequencies instead of sgRNA normalized log2(enrichment) were used25. To group the sgRNAs according to their pairwise PWES, hierarchical clustering was employed with the Ward clustering method and Euclidean distance as a metric. Clusters were called using a cophenetic distance cutoff.
To test the significance of the clustering of enriched sgRNAs in three-dimensional space, the absolute values of all possible pairwise combinations of the PWES were summed for the sgRNAs in each specified category. An empirical P value was calculated by scrambling the positions of the sgRNAs, while keeping the enrichment scores constant, and recalculating the summed PWES for each of 100,000 random permutations.
Data processing for CRISPR-suppressor scanning analysis.
Data processing was performed using the same languages as in the CRISPR-suppressor scanning experiments specified above. sgRNA counts were normalized with the following modification: the sgRNA read counts were normalized relative to the corresponding count values of the vehicle treatment at the time point of interest. For the principal component analysis, the top five sgRNAs that contributed both positively and negatively to each component were selected based on their coefficient loadings. The sequencing reads from genotyping exons 8, 15, 16 and 17 for the compound CRISPR-suppressor scanning experiment were analyzed using CRISPResso (v.1.0.13)29.
RNA-seq data processing and differential gene expression analysis.
RNA-seq data was processed according to the QuantSeq 3′ mRNA-Seq Library Prep recommended analysis pipeline through alignment to the Ensembl transcriptome (GRCh37.75). DESeq2 with R (v.3.5.1) was employed for the differential expression analysis54. Hierarchical clustering was performed on the regularized log2-transformed counts using Euclidean distance as a metric and the complete clustering method. To identify variable genes across cell types and conditions, all six possible pairwise comparisons of conditions were performed. All genes with both an adjusted P < 0.01, calculated using the Benjamini-Hochberg correction, and |log2(fold-change)| > 2 in at least one of the comparisons was considered to be differentially expressed. The regularized log2-transformed counts for the variable genes were grouped by k-means clustering (k = 7) and the Z-scores for the variable genes, ordered according to their cluster identity, are depicted. A table of significantly differentially expressed genes is provided in Supplementary Dataset 6. Gene set enrichment analysis (v.3.0)13 was carried out on log2 + 1 transformed count values and the P values for gene set enrichment analysis were calculated as previously described13. Gene signatures used for gene set enrichment analysis are listed in Supplementary Dataset 7.
ChIP-seq data analysis.
ChIP-seq data was analyzed as previously described53. Peaks were called with HOMER (v.4.10) using matched inputs with the following parameters: H3K4me2, -histone -tagThreshold 30; H3K27ac, -histone -tagThreshold 50; LSD1 -style factor -tagThreshold 10 -P 0.000001 -L 5 -F 10; GFI1B -style factor -tagThreshold 10 -P 0.000001. ChIP-seq peaks were annotated to the closest transcription start site using HOMER (v.4.10). Transcription factor motif enrichment analysis was performed using HOMER (v.4.10) centered on previously called GFI1B peaks (parameters: -size 200 -mask). HOMER (v.4.10) was used to identify overlapping and unique peaks for LSD1 and GFI1B. ChIP-seq profile plots were generated using ngs.plot55.
Statistical methods.
Statistical parameters including the exact value and definition of n, the definition of center, dispersion, precision measures (mean ± s.d. or s.e.m.) and statistical significance are reported in figures and figure legends.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
We thank members of the Liau lab, B. E. Bernstein, M. D. Shair and J. Kim for helpful discussions. We thank Q. Yao for assistance on computational analysis. We thank C. Lee for assistance with figures. We thank S. Miller, K. Zhao, R. Boursiquot, H. Rees and C. Daly for their assistance in high-throughput DNA sequencing. We thank J. Nelson and Z. Niziolek for their assistance with FACS sorting. We thank R. Dahl and M. Simon for providing the M-CSFR-Fluc reporter and MigR1 PU.1 expression plasmid. A.P.S. was supported by the Herchel Smith Graduate Fellowship Program. A.M.F. was supported by award number T32GM007753 from the National Institute of General Medical Sciences. This research was supported by startup funds from Harvard University.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of data availability and associated accession codes are available at https://doi.org/10.1038/s41589-019-0263-0.
Supplementary information is available for this paper at https://doi.org/10.1038/s41589-019-0263-0.
Competing interests
The authors declare no competing interests.
Data availability
ChIP-seq and RNA-seq data have been deposited to NCBI GEO (GSE121426). Transformed CRISPR-suppresor scanning reads (log2 + 1) used for Figs. 1–3 and Supplementary Figs. 1–3 are supplied in Supplementary Datasets 2–4.
Code availability
Code employed in Fig. 2 and Supplementary Fig. 2 is available upon reasonable request.
References
- 1.Shi J et al. Discovery of cancer drug targets by CRISPR–Cas9 screening of protein domains. Nat. Biotechnol 33, 661–667 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shen C et al. NSD3-Short is an adaptor protein that couples BRD4 to the CHD8 chromatin remodeler. Mol. Cell 60, 847–859 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neggers JE et al. Target identification of small molecules using large-scale CRISPR–Cas mutagenesis scanning of essential genes. Nat. Commun 9, 502–515 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ipsaro JJ et al. Rapid generation of drug-resistance alleles at endogenous loci using CRISPR–Cas9 indel mutagenesis. PLoS One 12, e0172177 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donovan KF et al. Creation of novel protein variants with CRISPR/Cas9-mediated mutagenesis: turning a screening by-product into a discovery tool. PLoS One 12, e0170445 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hakimi M-A et al. A core–BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc. Natl Acad. Sci. USA 99, 7420–7425 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi Y et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Tong JK, Hassig CA, Schnitzler GR, Kingston RE & Schreiber SL Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature 395, 917–921 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Forneris F, Battaglioli E, Mattevi A & Binda C New roles of flavoproteins in molecular cell biology: histone demethylase LSD1 and chromatin. FEBS J. 276, 4304–4312 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Maiques-Diaz A & Somervaille TC LSD1: biologic roles and therapeutic targeting. Epigenomics 8, 1103–1116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cusan M et al. LSD1 inhibition exerts its antileukemic effect by recommissioning PU.1- and C/EBPα-dependent enhancers in AML. Blood 131, 1730–1742 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris WilliamJ. et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 21, 473–487 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Ishikawa Y et al. A novel LSD1 inhibitor T-3775440 disrupts GFI1B-containing complex leading to transdifferentiation and impaired growth of AML cells. Mol. Cancer Ther 16, 273–284 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Maes T et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell 33, 495–511.e412 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Maiques-Diaz A et al. Enhancer activation by pharmacologic displacement of LSD1 from GFI1 induces differentiation in acute myeloid leukemia. Cell Rep. 22, 3641–3659 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGrath JP et al. Pharmacological inhibition of the histone lysine demethylase KDM1A suppresses the growth of multiple acute myeloid leukemia subtypes. Cancer Res. 76, 1975–1988 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Mohammad HP et al. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell 28, 57–69 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Schenk T et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med 18, 605–611 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takagi S et al. LSD1 inhibitor T-3775440 inhibits SCLC cell proliferation by disrupting LSD1 interactions with SNAG domain proteins INSM1 and GFI1B. Cancer Res. 77, 4652–4662 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto R et al. Selective dissociation between LSD1 and GFI1B by a LSD1 inhibitor NCD38 induces the activation of ERG super-enhancer in erythroleukemia cells. Oncotarget 9, 21007–21021 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baron R, Binda C, Tortorici M, McCammon JA & Mattevi A Molecular mimicry and ligand recognition in binding and catalysis by the histone demethylase LSD1-CoREST complex. Structure 19, 212–220 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin Y et al. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 29, 1803–1816 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saleque S, Kim J, Rooke HM & Orkin SH Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b Is mediated by the cofactors CoREST and LSD1. Mol. Cell 27, 562–572 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Lee MG, Wynder C, Schmidt DM, McCafferty DG & Shiekhattar R Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem. Biol 13, 563–567 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Kamburov A et al. Comprehensive assessment of cancer missense mutation clustering in protein structures. Proc. Natl Acad. Sci. USA 112, E5486–E5495 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang M et al. Structural basis of histone demethylation by LSD1 revealed by suicide inactivation. Nature Struct. Mol. Biol 14, 535–539 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Dhanak D Drugging the cancer epigenome. In Proc. 104th Annual Meeting of the American Association for Cancer Research, AACR, Washington DC 6–10 (2013). [Google Scholar]
- 28.Du-Cuny L, Xiao Q, Xun G, Zheng Q & He F Cyano-substituted indole compounds and uses thereof as lsd1 inhibitors. Patent WO/2017/149463 (2017).
- 29.Pinello L et al. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat.Biotechnol 34, 695–697 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niwa H, Sato S, Hashimoto T, Matsuno K & Umehara T Crystal structure of LSD1 in complex with 4-[5-(piperidin-4-ylmethoxy)-2-(p-tolyl) pyridin-3-yl]benzonitrile. Molecules 23, E1538 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karasulu B, Patil M & Thiel W Amine oxidation mediated by lysine-specific demethylase 1: quantum mechanics/molecular mechanics insights into mechanism and role of lysine 661. J. Am. Chem. Soc 135, 13400–13413 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Forneris F, Orru R, Bonivento D, Chiarelli LR & Mattevi A ThermoFAD, a Thermofluor®-adapted flavin ad hoc detection system for protein folding and ligand binding. FEBS J. 276, 2833–2840 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Tortorici M et al. Protein recognition by short peptide reversible inhibitors of the chromatin-modifying LSD1/CoREST lysine demethylase. ACS Chem. Biol 8, 1677–1682 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Clackson T et al. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc. Natl Acad. Sci. USA 95, 10437–10442 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moroy T, Vassen L, Wilkes B & Khandanpour C From cytopenia to leukemia: the role of Gfi1 and Gfi1b in blood formation. Blood 126, 2561–2569 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pilotto S et al. Interplay among nucleosomal DNA, histone tails, and corepressor CoREST underlies LSD1-mediated H3 demethylation. Proc. Natl Acad. Sci. USA 112, 2752–2757 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dahl R, Iyer SR, Owens KS, Cuylear DD & Simon MC The transcriptional repressor GFI-1 antagonizes PU.1 activity through protein–protein interaction. J. Biol. Chem 282, 6473–6483 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cai SF, Chen CW & Armstrong SA Drugging chromatin in cancer: recent advances and novel approaches. Mol. Cell 60, 561–570 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dawson MA The cancer epigenome: concepts, challenges, and therapeutic opportunities. Science 355, 1147–1152 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Dawson MA & Kouzarides T Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Helin K & Dhanak D Chromatin proteins and modifications as drug targets. Nature 502, 480–488 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Kasap C, Elemento O & Kapoor TM DrugTargetSeqR: a genomics- and CRISPR–Cas9-based method to analyze drug targets. Nat. Chem. Biol 10, 626–628 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schenone M, Dancik V, Wagner BK & Clemons PA Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol 9, 232–240 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Joung J et al. Genome-scale CRISPR–Cas9 knockout and transcriptional activation screening. Nat. Protoc 12, 828–863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pattanayak V et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol 31, 839–843 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schulz-Fincke J et al. Structure-activity studies on N-substituted tranylcypromine derivatives lead to selective inhibitors of lysine specific demethylase 1 (LSD1) and potent inducers of leukemic cell differentiation. Eur. J. Med. Chem 144, 52–67 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Burg JM, Gonzalez JJ, Maksimchuk KR & McCafferty DG Lysine-specific demethylase 1A (KDM1A/LSD1): product recognition and kinetic analysis of full-length histones. Biochemistry 55, 1652–1662 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Forneris F, Binda C, Adamo A, Battaglioli E & Mattevi A Structural basis of LSD1–CoREST selectivity in histone H3 recognition. J. Biol. Chem 282, 20070–20074 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Conway P, Tyka MD, DiMaio F, Konerding DE & Baker D Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci. 23, 47–55 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dral PO, Wu X, Sporkel L, Koslowski A & Thiel W Semiempirical quantum-chemical orthogonalization-corrected methods: benchmarks for ground-state properties. J. Chem. Theory Comput 12, 1097–1120 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liau BB et al. Adaptive chromatin remodeling drives glioblastoma stem cell plasticity and drug tolerance. Cell Stem Cell 20, 233–246 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shen L, Shao N, Liu X & Nestler E ngs.plot: quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15, 284–298 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.