

A novel system is developed for high-throughput multiplexed biomarker detection in single extracellular vesicles.
Abstract
MicroRNAs (miRNAs), mRNA, and proteins in/on extracellular vesicles (EVs) represent potential cancer biomarkers. Concurrent detection of multiple biomarkers at a single-EV level would greatly improve prognosis and/or diagnosis and understanding of EV phenotypes, biogenesis, and functions. Here, we introduced a High-throughput Nano-bio Chip Integrated System for Liquid Biopsy (HNCIB) system for simultaneous detection of proteins and mRNA/miRNA in a single EV. Validated through systematic control experiments, HNCIB showed high reliability, sensitivity, and specificity. In a panel of 34 patients with lung adenocarcinoma (LUAD) and 35 healthy donors, HNCIB detected an up-regulated expression of programmed death-ligand 1 mRNA and protein and miR-21 in EVs derived from patients with LUAD compared to those from healthy donors. HNCIB has low sample requirement (~90 μl), fast assay time (~6 hours), and high throughput (up to 384 samples per assay) and would have great potential in the study of EVs and their clinical applications.
INTRODUCTION
Extracellular vesicles (EVs) are generic particles typically <1000 nm in size and naturally released from the cell that are delimited by a lipid bilayer. EVs include endosomal-derived exosomes (30 to 150 nm) and plasma membrane–derived microvesicles (1). EVs contain proteins and nucleic acids including double-stranded DNA, mRNAs, and noncoding RNAs such as microRNAs (miRNAs) and long noncoding RNAs. EVs represent their originating cells and are the carriers for cellular communication (2). Emerging evidence has shown that EVs are highly associated with the initiation, development, and metastasis of tumors, making EVs potential cancer biomarkers.
Lung cancer is one of the most important causes of cancer-related death, with lung adenocarcinoma (LUAD) being the most common subtype (3). The 5-year survival rate of patients with LUAD is low because of late diagnosis; thus, it is critical to identify and discover novel diagnostic biomarkers for early lung cancer detection (3). As a liquid biopsy, EVs have multiple advantages, for example, abundance in biofluids and protecting the proteins and nucleic acids in EVs from degradation by their lipid bilayer membrane (2). However, it is still challenging to quantitate the proteins and nucleic acids contained in EVs due to their small size (ranging from 50 to 1000 nm) (4). Flow cytometry (FCM), Western blotting, and enzyme-linked immunosorbent assay are the standard methods for measuring the proteins, whereas reverse transcription quantitative polymerase chain reaction (RT-qPCR) for measuring the mRNA of EVs. However, these traditional techniques require a large amount of sample (normally >500 μl) to isolate enough EVs for analysis. In addition, most of these techniques are “bulk assays” and the obtained results are the average of the overall molecular contents from all EVs. They are incapable of providing information on the molecular content of individual EVs and heterogeneity within the EV population. Furthermore, the sensitivity of the methods is low and could not detect the expression of protein or mRNA at a single-EV level (5), which is critical to identify the contents in EV subtypes. Several recent studies showed that it was possible to detect miRNA (6) or multiplexed proteins at a single-EV level (7). Using a microarray-based chip, digital counting of individual exosomes was reported using a novel single-particle interferometric reflectance imaging sensor (8). Nevertheless, concurrent analyses of nucleic acids and proteins have not been reported.
Immunotherapy is a very promising therapy for lung cancer; however, cancer cells can express membrane proteins such as programmed death-ligand 1 (PD-L1) to escape the immune attack by inactivating immune cells such as CD8+ T cells (9), which could be reversed by anti–PD-L1 antibodies that inhibit PD-L1 on cancer cells (10). However, recent studies demonstrated that exosomal PD-L1 released by cancer cells could promote tumor progression by remotely blocking PD-1 on the CD8+ T cells (11, 12). Thus, it is critical to monitor the expression levels of exosomal PD-L1 during treatment with immunotherapy.
In this study, we report on simultaneous detection of proteins and mRNA/miRNA at a single-vesicle level using a newly developed High-throughput Nano-bio Chip Integrated System for Liquid Biopsy (HNCIB) system. The system uses a high-throughput nano-biochip for high-efficiency, targeted EV capture and total internal reflective fluorescence microscopy (TIRFM) for rapid and high-resolution detection. A deep learning algorithm was developed to automate the analysis to acquire semiquantitative to quantitative information on the distribution of mRNA/miRNA and membrane proteins, as well as the colocation of multiple proteins and their ratios. The technology allows for rapid single-EV analysis (total assay time of ~6 hours) and requires very small sample quantity (~90 μl plasma). To our knowledge, this is the first technology that enables simultaneous detection and analysis of multiple types of biomolecules (e.g., nuclei acids and proteins) from both the surface and lumen of the EVs as biomarkers. Using lung cancer as a case study, we demonstrated the capability of the HNCIB system for the simultaneous detection of PD-L1 protein and mRNA/miRNA expression of EVs derived from patients with LUAD and healthy donors. The HNCIB system could be used to reliably evaluate the miRNA, mRNA, and protein expression in EVs from clinical samples.
In principle, the technology has the potential to become a novel platform for multiplexed, simultaneous detection of disease-specific EV surface proteins and internal molecular cargo. This would allow for greater detection accuracy and provide more comprehensive information on EVs phenotypes and disease state and further improve the sensitivity, specificity, and accuracy for disease prognosis, diagnosis, and monitoring of patient treatment progress.
RESULTS
In previous studies, we have demonstrated the feasibility of measuring miRNA or mRNA in EVs (13, 14). In this study, we developed an HNCIB system and show that the system is capable of highly efficient capture of EVs and simultaneous detection of proteins and RNA in a single EV. The design of our system is illustrated in Fig. 1. Using the HNCIB system, we first captured the nano-biochip EVs and detected RNA with molecular beacons (MBs), and antibodies were then applied to visualize the protein (Fig. 1A). High-throughput high-sensitivity detection and analysis can be realized, with a total assay time of around 6 hours. Four biomarkers can be analyzed concurrently per sample (~90 μl), and up to 384 samples can be analyzed in one assay run.
Fig. 1. Design of HNCIB system.
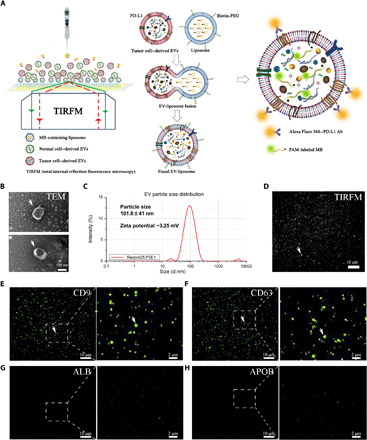
(A) Illustration of the HNCIB system for simultaneous detection of PD-L1 membrane protein and mRNA in a single EV. Photo credit: Ming Wang, Hangzhou Dixiang Co. Ltd., Hangzhou, China. (B) Electron micrograph showing the EVs isolated from human plasma. (C) Particle size and zeta potential measurement of the EVs isolated from human plasma by nanoparticle tracking analysis technique. d.nm, diameter (nm). (D) Bright-field TIRFM images showing the EVs isolated from human plasma. (E) EV marker CD9 and CD63 (F) membrane protein measured by the HNCIB system. (G) Non-EV markers ALB and APOB (H) protein measured by the HNCIB system.
Proof-of-principle validation of single-EV analysis by HNCIB system
We first demonstrated the effectiveness of the system for EV capture. EVs were isolated from human plasma, and their morphology was characterized using a transmission electron microscope (TEM). The EM images demonstrated that the EVs from human plasma had a typical round shape with a diameter of roughly 100 nm (Fig. 1B), which is consistent with previous studies (1). Furthermore, particle size analysis by dynamic light scattering (DLS) showed that the sizes of the EVs from human plasma were distributed around 101.8 ± 41.0 nm, and zeta potential measurements revealed that EVs were negatively charged, with a potential value of ~3.25 mV (Fig. 1C). Consistent with the TEM observation, these results further confirmed that the EVs isolated using the current isolation protocol are dominantly exosomes. The isolated EVs were analyzed using the HNCIB system. A representative bright-field TIRFM image (Fig. 1D) showed individual EVs (bright spots in the image) with a high number density. This indicates that the HNCIB system has a high efficiency to capture the EVs. Using our HNCIB system, we were also able to detect CD9 and CD63, two well-known EV markers, on EVs isolated from human plasma by staining EVs with CD9 and CD63 antibodies, respectively (Fig. 1, E and F). Each single green fluorescent spot represents EVs containing CD9 membrane protein (Fig. 1E) or EVs containing CD63 membrane protein (Fig. 1F). On the other hand, very low fluorescence signals were detected from albumin (ALB) or apolipoprotein B (APOB), two non-EV markers, on EVs isolated from human plasma by staining EVs with ALB or APOB antibodies, respectively (Fig. 1, G and H). Together, these results confirmed the successful and highly efficient capture of EVs by the HBNIC system and the capability of high-resolution imaging at a single-EV level.
Validation of specificity, sensitivity, and detection of protein colocation in EVs by HNCIB system
To verify that our HNCIB system could reliably and unequivocally detect specific protein expression on EVs, several control experiments, both positive and negative, were conducted. We first evaluated the protein expression of EV marker CD63 on EVs isolated from human plasma. There were no detectable fluorescence signals in phosphate-buffered saline (PBS) only (no EVs and no antibodies) or EV only (no antibodies) samples, thus ruling out the possible autofluorescence from background or from EVs, respectively (Fig. 2A).
Fig. 2. Verification of PD-L1 protein expression on EVs.
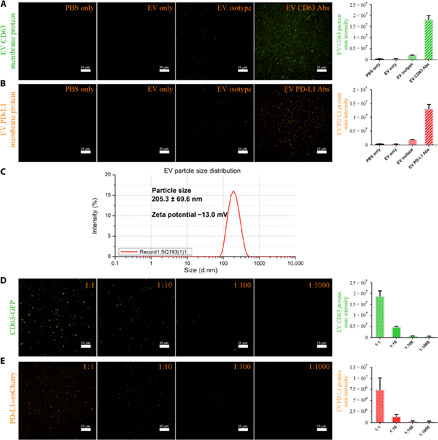
(A) Fluorescence signals of PBS only (no EVs and no antibodies), EV only (EVs without staining with any antibodies), EV isotype (EVs stained with isotype control antibody), and EV CD63 antibodies (Abs) (EVs stained with CD63 antibody). (B) Fluorescence signals of PBS only, EV only, EV isotype, and EV PD-L1 Abs (EVs stained with PD-L1 antibody). (C) Particle size and zeta potential measurement of the EVs isolated from A549 cells overexpressed with CD63-GFP by nanoparticle tracking analysis technique. d.nm, diameter (nm). (D) EVs isolated from human A549 cells overexpressed with CD63-GFP or PD-L1–mCherry (E) and diluted to different concentrations. 1:1, initial concentration; 1:10, 10-fold dilution; 1:100, 100-fold dilution; 1:1000, 1000-fold dilution.
To evaluate the specificity of the HNCIB system, we then applied an isotype control, a nonspecific immunoglobulin G, and CD63 antibodies to the EVs and found that very weak fluorescence signals on EVs stained with the isotype control antibody were observed, whereas EVs stained with CD63 antibody showed very high and substantially stronger fluorescence signals (nine times that of the EV isotype; Fig. 2A), suggesting that the HNCIB system could specifically detect the CD63 membrane protein on EVs. Similar to CD63 staining, for PD-L1 detection, there were no detectable fluorescence signals in PBS-only or EV-only samples, relatively low signals on EVs stained with isotype control antibody, whereas high fluorescence signals from EVs stained with PD-L1 antibody were observed (seven times that of the EV isotype; Fig. 2B). For the two non-EV markers ALB and APOB detection, there were no detectable fluorescence signals in PBS-only or EV-only samples and relatively low signals on EVs stained with ALB or APOB antibody, whereas high fluorescence signals from EVs stained with CD9 or CD63 antibody were observed (at least 10 times that of the EV stained with ALB or APOB; fig. S1, A and B, and table S1).
We next evaluated the sensitivity of the HNCIB system by measuring the signals from CD63-GFP (green fluorescent protein)–expressed EVs with a series of dilution. The EVs (from the supernatant of A549 cells that were overexpressed with CD63-GFP) had a particle size of around 205.3 ± 69.6 nm and a zeta potential of ~13.0 mV (Fig. 2C). We found that the fluorescent intensity was proportional to concentration of CD63-GFP–expressed EVs when diluted 10, 100, or 1000 times (Fig. 2D). Signals can be detected even at a 1:1000 dilution ratio, demonstrating that the HNCIB system is highly sensitive and is capable of detecting the changes of CD63-GFP–expressed EVs. In a similar trend, the fluorescent intensity was also proportional to the EV concentration in PD-L1–mCherry–expressed EVs (Fig. 2E), further confirming the system’s capabilities.
Next, to further confirm that PD-L1 is expressed on CD63-positive EVs, we performed CD63 and PD-L1 double immunofluorescence staining on A549 cells and found that CD63 and PD-L1 were both distributed as single spots in cytoplasm [fig. S2, A (confocal image) and B (TIRFM image)]. In some instances, CD63 and PD-L1 colocated at the same locations, as illustrated by white cycles in the merged images (the rightmost image in fig. S2B). To ascertain whether CD63 and PD-L1 coexist on single EVs and whether the HNCIB system is capable of concurrent detection of multiple proteins in a single EV, we then stained EVs with both CD63 and PD-L1 antibodies and analyzed them by HNCIB (EVs were from both cell supernatant and human plasma). In both cases, colocation of CD63 and PD-L1 on the same EV particle was observed in a portion of the EV population, visualized as the spots of yellow color (blend of orange and green) and orange color (blend of red and green), respectively, in the merged images (Fig. 3, A and B). We further quantitatively analyzed the CD63 and PD-L1–colocated EVs. The fluorescence images for both proteins were first converted to gray ones separately, after which background noise reduction algorithm (15) was applied. Thereafter, the Sobel edge detection operator (also called the Sobel-Feldman operator or Sobel filter) (16, 17), which has been widely used in image processing and computer vision such as edge detection of medical images (18), volume detection (19), and image mining (20), was implemented in the EV edge detection, by which edges were determined at those points where the gradient of the image is maximum. This results in well-resolved EV particles with clear boundaries. After the determination of the EV areas, the fluorescence intensity in and out of the areas can be calculated, and the overlapped areas can also be determined on the basis of the comparison of the two images. The overlapped areas of the two images (one for CD63 and the other for PD-L1) were determined to be the EVs for which both proteins were colocated. The processed binary images revealing the colocation of both proteins are shown in Fig. 3 (A and B) (rightmost images). By applying the algorithm to the CD63-only image and the colocated image, the fraction of the EV subpopulation that expresses both proteins in the entire EV population can be calculated. Figure 3C shows the results.
Fig. 3. Double immunofluorescence staining of CD63 and PD-L1 membrane protein.
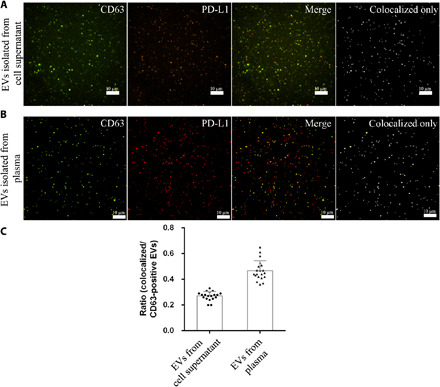
(A) EVs isolated from 1000 μl of cell supernatant of human A549 cells overexpressed with CD63-GFP and PD-L1–mCherry. The dots in the colocalized only image represent EVs expressing both CD63 and PD-L1 protein. (B) EVs isolated from 90 μl of human plasma are stained with CD63-AF488 and PD-L1–AF647 antibodies. The dots in the colocalized only image represent EVs expressing both CD63 and PD-L1 protein. (C) Ratio of CD63- and PD-L1–positive EVs to CD63-positive EVs isolated from human A549 cells overexpressed with CD63-GFP and PD-L1–mCherry or isolated from plasma of 20 patients with LUAD.
Simultaneous detection of PD-L1 mRNA and protein expressed on EVs in vitro
To validate that our technique could detect the changes of PD-L1 mRNA and protein expressed on EVs in vitro, we then overexpressed PD-L1 in A549 cells and measured the expression of PD-L1 mRNA and protein for EVs isolated from PD-L1–overexpressed or vector-overexpressed A549 cells. RT-qPCR and Western blotting analysis confirmed that the mRNA and protein expression levels of PD-L1–A549 cells were substantially higher than those of vector-A549 cells (Fig. 4, A and B). HNCIB system was then used to analyze the PD-L1 mRNA in EVs and protein on EVs, and Fig. 4 (C and D) shows the results. EVs from PD-L1–A549 cells showed a significantly increased expression of PD-L1 mRNA and protein in comparison with vector-A549 cells. This agrees well with the RT-qPCR and Western blotting analysis and confirms the HBNIC system’s capability for simultaneous detection of lumen RNA and surface membrane proteins.
Fig. 4. Measurement of EV mRNA and protein expression in vitro.
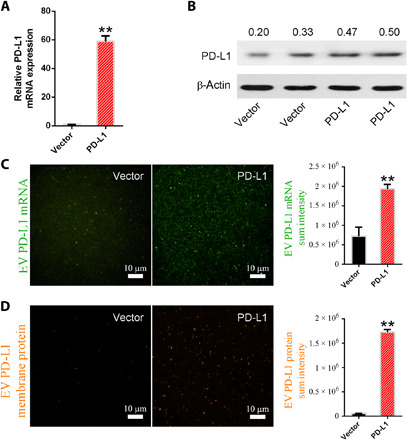
(A) RT-qPCR and (B) Western blotting analysis of PD-L1 expression of vector and PD-L1–overexpressed A549 cells. (C) HNCIB analysis of PD-L1 mRNA and protein expression of EVs isolated from supernatant of A549 cells overexpressing vector and PD-L1 (D). **P < 0.01 versus vector control.
Clinical application
Last, we demonstrated the HNCIB system’s detection ability of EVs in plasma from patients with lung cancer. A panel of 34 patients with LUAD and 35 healthy donors was recruited for the study. The demographic characteristic of patients with LUAD and healthy donors are demonstrated in table S2, and there is no significant difference in age, gender, and smoking status between the two groups.
Previous studies have demonstrated that exosomal miR-21 was a miRNA biomarker widely reported for many types of cancers including lung cancer (21); we therefore first used it as the benchmark to verify the detection of EV cargo contents and their changes using the HNCIB system. We found that, consistent with previous studies (21), the expression levels of EV miR-21 derived from patients with LUAD (9.93 × 106 ± 4.87 × 106) were significantly higher than those from healthy donors (6.83 × 106 ± 1.92 × 106) (Fig. 5, A and B). It should be noted that the result represents the true difference between the groups and is not the result of the lower EV abundance in the healthy groups than the LUAD groups. First, HNCIB used a nano-biochip to capture the EVs through electrostatic interaction to realize high-efficiency capture. The EV density of the chip surface is dependent on the density of the EV-targeting groups on the nano-biochip surface, which are similar across all experiments because of the use of the same nano-biochip. It is not influenced by the disparity in the amount of EV in the samples. As a result, EVs with similar density were immobilized on the surface of the nano-biochip despite the difference of abundance in the sample. It should also be noted that high efficiency of EV capture, as realized in the HNCIB, is critically important for capturing EVs from healthy donors who have low EV abundance. The nano-biochip in HNCIB can achieve an average particle density of ~106/mm2, an order of magnitude higher than that achieved by other technologies. Such high-density capture ensures the similar surface density of EVs in both groups. Second, the measurements were performed using TIRFM, which collects signals only from a sample in the near vicinity of the nano-biochip surface (~100 nm), one of the main reasons that TIRFM can achieve a high signal-to-background ratio. As a result, the measured signals were from the surface-immobilized EVs, which had similar density in both healthy and LUAD groups.
Fig. 5. Measurement of miRNA, mRNA, and protein expression of EVs isolated from human plasma.
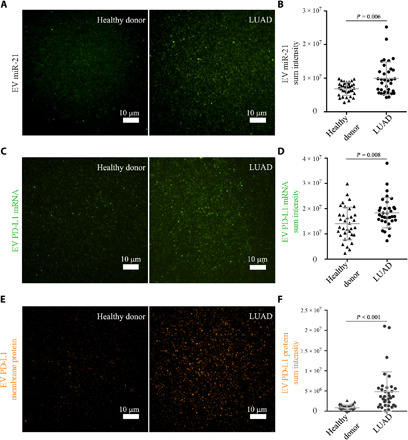
Samples were from 35 healthy donors and 34 patients with LUAD. (A and B) Representative images and statistical analysis of EV miR-21. (C and D) Representative images and statistical analysis of EV PD-L1 mRNA. (E and F) Representative images and statistical analysis of EV PD-L1 membrane protein. Note that in (F), P = 1 × 10−6; it is shown as P < 0.001 because only three decimal digits were shown.
Last, we demonstrate the ability of the HNCIB system to detect the changes of both PD-L1 mRNA and PD-L1 protein of EVs in plasma from patients with lung cancer. We found that the PD-L1 mRNA expression levels of EVs derived from patients with LUAD (1.84 × 107 ± 5.95 × 106) were significantly higher than those from healthy donors (1.41 × 107 ± 6.46 × 106) (Fig. 5, C and D). In a similar trend, we found that the PD-L1 protein expression levels of EVs derived from patients with LUAD (4.81 × 106 ± 4.93 × 106) were significantly higher than those from healthy donors (0.84 × 106 ± 0.66 × 106) (Fig. 5, E and F).
It is noteworthy that although statistically significant differences exist between the healthy donor group and LUAD patient group for all three biomarkers, the PD-L1 protein had a more appreciable separation between the two groups. Overlap between the groups was observed for the PD-L1 mRNA and was more pronounced for miR-21. It is plausible that the specificity of different biomarkers varies for the same diseases, and single biomarker may not always be sufficient for proper diagnosis/prognosis. Simultaneous detection of multiple biomarkers, enabled by HNCIB, thus would greatly facilitate improving diagnosis/prognosis accuracy.
Despite being bulk measurements, FCM and RT-qPCR remain standard methods to measure the overall protein and mRNA expression on EVs (22, 23). We thus applied FCM and RT-qPCR to determine the overall EV PD-L1 protein and mRNA expression and compared the results with those from single-EV measurement by HNCIB. We first used CD63 exosome capture beads to isolated EVs in plasma from healthy donors and patients with LUAD and then stained EVs with CD63-AF488 and PD-L1–AF647 fluorescent antibodies and analyzed the signals using two-color FCM. We found that there was no CD63 and PD-L1 protein expression in the control group (PBS only, without EV), ruling out the false-positive fluorescence signals from the background (Fig. 6A). The CD63 protein expression on EVs in plasma from healthy donors was lower compared to that in plasma from patients with LUAD (Fig. 6B). In a similar trend, the PD-L1 protein expression on EVs in plasma from healthy donors (2.41 ± 1.74%) was significantly lower compared to that in plasma from patients with LUAD (26.95 ± 20.04%) (Fig. 6, A, C, and D). In addition, RT-qPCR analysis showed that the PD-L1 mRNA expression on EVs in plasma from healthy donors (0.86 ± 0.49) was significantly lower compared to that in plasma from patients with LUAD (2.49 ± 1.92) (Fig. 6E). The PD-L1 protein and mRNA results, obtained separately by bulk assays FCM and RT-qPCR, agree very well with those obtained simultaneously from single-EV measurements by the HNCIB system (Fig. 5, C to F).
Fig. 6. Simultaneous detection of EV CD63 and PD-L1 membrane protein by FCM and detection of EV PD-L1 mRNA by RT-qPCR.
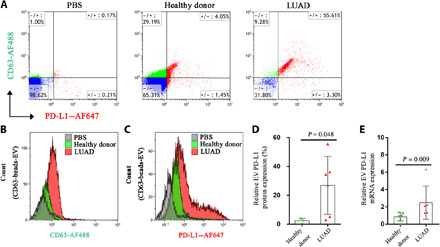
(A) Double staining with CD63-AF488 and PD-L1–AF647 fluorescent antibodies of EVs in plasma from healthy donors and patients with LUAD. (B) EVs in plasma samples were captured with CD63 exosome capture beads and incubated with CD63-AF488 antibody, and FCM was used to detect the expression of EV membrane protein CD63. (C) EVs in plasma samples were captured with CD63 exosome capture beads and incubated with PD-L1–AF647 antibody, and FCM was used to detect EV membrane protein PD-L1 expression. (D) Statistic analysis of the differential membrane protein expression of PD-L1+ EVs in plasma from three healthy donors and six patients with LUAD. (E) Statistic analysis of the differential mRNA expression of PD-L1 in EVs from plasma of six healthy donors and six patients with LUAD.
DISCUSSION
Liquid biopsy monitors tumor development through noninvasive sampling, making it much more attractive than traditional tissue biopsy, which requires patients to undergo surgery or bronchial biopsy to obtain tissue. Abundant in cellular components of the cells of origin, e.g., nucleic acids (mRNAs and miRNAs) and proteins, EVs are excellent sources of biomarkers for diseases. They emerge as one of the most promising liquid biopsies, and a number of studies have demonstrated that EVs could reflect tumor initiation and development.
Because of their small size (<1000 nm) and complicated genesis, it is still very challenging to extract sufficient clinical information from EVs using current methods. Traditional methods for protein and mRNA measurement such as Western blotting and RT-qPCR suffer from the requirement of a large amount of samples and the accurate reflection of information from all the EVs examined. Thus, there is still a lack of technique to reliably measure the expression of a specific protein and nucleic acid at a single-EV level.
In this study, we introduced an integrated HNCIB system and demonstrated their capability for single-EV capture and visualization and simultaneous detection of both surface and cargo components within the EVs. Currently, there is no established standard for single-EV measurement, and such unique capability would be of great value to the general field of EV study. The system uses a high-throughput nano-biochip for EV capture, TIRFM for fast and high-resolution detection, and deep learning algorithm for automated data processing and improved accuracy of the results. We first demonstrated the system’s capability for individual EV detection, which was verified by comparing the characteristics of the captured EVs with those from standard methods such as TEM, DLS, and zeta potential measurement. The high-resolution detection was facilitated by the excellent signal-to-noise ratio that the system achieved. For example, the signal-to-noise ratio can be as high as ~60 when using either PBS or EV as negative controls (table S1). We then evaluated the specificity of the HNCIB system by comparing the fluorescent signals from EVs stained with CD63 (or PD-L1) antibodies with those from isotype control antibodies. The ratio, which can be considered a quantitative index for specificity, was as high as 9 (table S1), indicating excellent specificity. Next, we evaluated the sensitivity of the HNCIB system by using EVs from a series of solution of increasing dilution from supernatants of CD63-GFP– or PD-L1–mCherry–overexpressed A549 cells. We found that the fluorescent intensity was proportional to concentration of CD63-GFP– or PD-L1–mCherry–expressed EVs, and the system was capable of single-EV detection at very low concentration. We next demonstrated the system’s utility in concurrent detection of both CD63 and PD-L1 proteins and their colocation on single EVs and the quantification of the subpopulation with such colocation characteristic. Next, we demonstrated the HNCIB’s utility in detection of both cargo and surface constituents in/on single EVs simultaneously, by successfully detecting both PD-L1 mRNA and protein in/on EVs derived from PD-L1–overexpressed A549 cells and by using EVs from vector-overexpressed A549 cells as control.
To evaluate its clinical applicability and effectiveness, we next applied the HNCIB to analyze PD-L1 mRNA and protein in/on single EVs isolated from clinical samples. Increase in both PD-L1 mRNA and protein was observed in patients with LUAD in comparison to those from healthy donors. The results from the HNCIB system were in excellent agreement with those from FCM and RT-qPCR, the current standard methods for measuring the protein and mRNA expression on EVs (22, 23), respectively. It is also worthy to note that while FCM and RT-qPCR each require at least 200 μl of human plasma sample to separately measure protein or mRNA expression, HNCIB analysis only needs ~90 μl of human plasma samples to complete the measurements of both proteins and mRNA.
EVs are present in all types of body fluids and contain both membrane and luminal constituents that represent the cells that they originate from, including proteins (membrane, cytosolic, nuclear, extracellular matrix, etc.), metabolites, and nucleic acids (DNA, mRNA, and noncoding RNA species such as miRNAs). EVs may come from different sources and are inherently heterogeneous in size, content, and functions. Current standard methods and most EV liquid biopsy development, to date, are bulk assays, and the results are characteristics of the ensemble of the heterogeneous EV populations. Analysis at a single-EV level would provide much wealthier information such as potential biomarker expression heterogeneity and EV subpopulation. It is particularly powerful to help identify disease-derived EVs in which the biomarkers may be up-/down-regulated from healthy cell–derived EVs in which the biomarkers are also present at normal levels. This would be of tremendous value for the early detection of diseases.
EV analysis as liquid biopsy can be substantially more informative and valuable by simultaneous detection of multiple EV constituents as biomarkers, e.g., mRNAs, miRNAs, proteins, and other metabolites. These constituents may reflect distinct and different aspects of disease-generating/relating EVs and thus would lead to greatly improved sensitivity, specificity, and accuracy for EV-based diagnosis, particularly at the early state of diseases when the lower quantity of the biomarkers may prevent correct diagnosis by a single biomarker. Multicomponent analyses at a single-EV level would also facilitate the detection of specific EV subpopulations and phenotypes and understanding of their biogenesis and functions in both healthy and disease cells and tissues.
To date, there are only limited studies on single-EV analysis. Daaboul and colleagues (8) reported the detection and counting of individual EVs based on interferometric imaging of EVs immune-captured on a substrate. Multiplexed protein profiling and phenotyping were reported using difference in the expression of surface protein markers CD9, CD63, and CD81. Detection of the EV cargo contents was not reported. He et al. (6) reported the detection of a single cargo constituent (miR-21). Profiling of multiple proteins in a single EV was reported recently using repeated antibody staining–imaging–washing cycles in a microfluidic device (7).
Compared to these studies, the HNCIB technology for single-EV analysis presented in this study allows for simultaneous analysis of multiple cargo and surface biomarkers, e.g., proteins, RNAs, and their combination, thus greatly improving the usefulness of single-EV analysis. The technology also can achieve excellent signal-to-noise ratio, high sensitivity, and specificity (table S1) and is capable of EV subpopulation identification by protein colocation. The highly resolved images also would allow for accurate counting of EVs. Results consistent with conventional, standard analysis can be achieved much more rapidly using a substantially less amount of clinical sample (~90 μl). High-throughput analyses can be achieved, and the technology is particularly amenable for clinical adoption because the chip can be made to the same multiwell plate form commonly used in standard bioassays.
HNCIB realize these technology advantages through effective EV capture mechanism, high-resolution visualization technique, and deep learning algorithm–based data analysis. The chip surface is covalently functionalized with a densely packed molecular brush layer. The molecular brush enables subsequent immobilization of lipoplex nanoparticles in high density. The covalently linked high-density surface layers also serve as a cushion to avoid disruption and/or disintegration of the nanoparticles when attached to a hard, solid substrate (24) and to improve the particle integrity. Via electrostatic interaction, the negatively charged EV particles are captured by the positively charged lipoplex nanoparticles in a more selective and target manner. Together, these enable the high-efficiency capture and enrichment of the EV nanoparticles. As an example, in Fig. 1F, the average particle density is ~1.6 × 106/mm2 by image analysis, about an order of magnitude higher than that captured by simple coating of antibodies targeting the tetraspanin proteins such as CD9, CD63, and CD81, the prevalent method for immunocapture of EVs (8).
For single-EV analysis, while surface proteins can be detected directly by immunoblotting, detection of EV RNAs and other cargos require access to the EV lumen while preserving its overall structure integrity. Previous studies used lysis to achieve accessibility (6, 7), and the probes were delivered from solution. Such an approach may pose several challenges. As a lysing agent was used to partially disrupt/dissolve the EV membrane to generate pores, the possibility of removing potential surface biomarkers of interest cannot be always avoided. In addition, the process is highly sensitive to the type of EV particles being lysed, the type and concentration of the lysing agent, and the lysing conditions used (time, temperature, type of buffer used, etc.) (25). The pore generation thus is inherently prone to large variation. The probe was delivered in solution by diffusion through the lysed nanosized pores. The diffusion-controlled process may be time consuming for large-sized probes such as antibodies. The appropriate balance between acceptable diffusion time and surface membrane integrity may be difficult to maintain, and consistency across all experimental runs may be a concern. During solution delivery, the probes were also akin to be adsorbed onto the EV surface by a nonspecific interaction. This would lead to a large variation in the probe delivered between experiments and reduced repeatability, blurred EV boundaries, and reduced image quality. Leakage of cargo content through the lysed pores may also be a possibility, particularly in sequential staining-washing-imaging-washing cycles for multiplex profiling. Such leakage not only leads to reduced signals but also contributes to increased background and decreased signal-to-noise ratio. Although these challenges may be overcome by elaborate experiment techniques (6, 7), a more robust approach to deliver probe molecules to the interior of the EVs for cargo content detection is highly desired for improved repeatability and ease of operation.
HNCIB technology achieves access to the lumen by fusion of the lipid bilayer membranes (26) of the lipoplex and EV particles. This leads to the formation of single fused particles, accompanied by combination and redistribution of their respective membrane constituents and mixing of the cargo content (27, 28). This enables the detection of the cargo content, e.g., EV RNA by MBs and, in principle, also proteins by antibodies. The fused particle retains the surface biomarkers of interest from the EV for subsequent detection by immunoblotting. Such an approach avoids the use of lysis and potential damage to the membrane structures, as well as the potential leakage of cargo content during probe delivery. As the probes are compartmentalized in the lipoplex nanoparticles and delivered by fusion, nonspecific adsorption is anticipated to be minimal and reduced interference from the sample environment is expected. These would result in improved image quality and result accuracy.
HNCIB analysis also makes effective use of TIRFM (29, 30) to achieve single-EV imaging. TIRFM uses the evanescent electromagnetic field induced on the sample side when the incident beam goes through total internal reflection at the glass-sample interface. This takes place when the incident angle exceeds a critical angle, θc = sin−1(n1/n2), where n1 and n2 are the refractive indices of the sample and the glass coverslip, respectively; n1 = 1.333 and n2 = 1.523. The depth of the evanescent field is calculated by , where λ0 is the wavelength of the incident light and θ is the incident angle. In the current study, the incident angle was 65°, and resulting evanescent field depth (for all incident lights used) is ~100 nm, same order of the size of a single EV. This results in low background and enhanced signal-to-noise ratio and substantially improves image quality because only the molecules within the evanescent field were excited to emit fluorescent signals.
The HNCIB system also includes an efficient algorithm for automated processing and analysis of a vast number of images. Using the experimentally acquired images as the training set, we developed, trained, and improved a deep learning–based image screening module to achieve intelligent screening of the acquired fluorescence images. Additional modules were developed for effective differentiation of EV particle fluorescence spots from those of contaminant origin and/or noise. These facilitate the improved signal quality. It is understood that the ideal approach to improve the signal-to-noise ratio is through the optimization of assays or platform, as such improvement is achieved on a biological level. Several improvements were conducted in the study to improve the HNCIB signal-to-noise ratio on the biological level, e.g., optimization of the nano-biochip’s EV capture capability, improving washing efficiency to remove impurity, enhancing the specificity of the MB, and fine tuning of the working condition of the fluorescent antibody.
Meanwhile, image analysis is also a critical part of any assay involving images with convoluted features. HNCIB applied a deep learning algorithm that we developed to achieve more truthful analysis and better signal-to-noise ratio than existing methods reported in the literature. Such algorithm transfers the data/image analyses from a rules-based algorithm to a training system, resulting in greatly more objective decision-making. The methodology provides a framework for designing and implementing deep neural networks with algorithms and pretrained models. This resulted in vastly improved signal quality comparing to existing methods that only use single-threshold background removal. Convolutional neural networks (ConvNets, CNNs) were used to perform classification on image. CNNs take advantage of local spatial coherence in the images, which allow them to have higher efficiency. This process, taking the form of convolutions, makes them especially well suited to analyze relevant information at a low computational cost. The algorithms can realize automatic and high-throughput image screening, optimal thresholding and fluorescent object-background separation for substantially improved signal-to-noise ratio, and quantitation of the detected target markers. These advantages are particularly important in improving diagnostic accuracy for clinical adoption. It should also be noted that the effectiveness of the deep learning algorithm affects the size of the training set. Because of resource limitation, current image category contains only 1 million images (1000 samples with 1000 images for each sample). In the future, more images will be included in the training set to achieve more comprehensive validation and further improve the fidelity of analysis.
Despite the great progress in EV-based liquid biopsy, many issues are still yet to be resolved before miRNAs, mRNA, and proteins in/on EVs become a reliable cancer biomarker. For example, what is the difference between EVs derived from healthy donors and from patients with lung cancer? Are there any changes at different tumor stages? Are there any specific markers for EVs originated from normal lung tissue or lung cancer tissue? Methodologies and techniques that enable the characterization of EVs at a single-EV level would greatly facilitate answering these questions. In this regard, the advantages and unique capabilities of the HNCIB may facilitate the advancement of EV liquid biopsy to address these issues. For example, immunohistochemical detection of PD-L1 expression in tumor tissue samples is currently the standard diagnostic test for immunotherapy for advanced cancer including non–small cell lung cancer (NSCLC). However, PD-L1 expression in cancer cells are dynamic, and it is impossible to obtain lung tissue to examine the PD-L1 expression multiple times (31); thus, it is very challenging to monitor the change in PD-L1 expression during the period of immunotherapy treatment. Therefore, liquid biopsy could provide an alternative to the PD-L1 immunohistochemistry. Furthermore, exosomal PD-L1 released by cancer cells remotely blocks PD-1 on the CD8+ T cells to help cancer cells escape immune attack (11, 12, 32). Thus, it is fundamental to monitor the expression levels of exosomal PD-L1 during the immunotherapy. Our technique could detect the changes of PD-L1 mRNA and protein in EVs from healthy donors and patients with LUAD. Although we used the PD-L1 mRNA and protein in EVs from lung cancer as a case study, by applying different antibodies or mRNA/miRNA probes, we could measure any miRNA, mRNA, and protein of EVs from patients with various cancer types.
Additional vital information may be gained from HNCIB analysis that is currently not available from other techniques. In this study, we quantitated the fraction of CD63+ and PD-L1+ EV subpopulation in the entire EV population from both cell supernatant and from human plasma and found that about 27% EVs were both CD63+ and PD-L1+ in the EV population from cell supernatant and about 47% from human plasma (Fig. 3), suggesting that the HNCIB system is powerful in detecting the difference in the fraction of CD63+ and PD-L1+ EV between the two groups. Such capability may have broad implications. For example, clinically, it is still very difficult to determine which subpopulation of patients with NSCLC could benefit from immunotherapy treatments. Although the KEYNOTE-024 study showed that patients with NSCLC with PD-L1 expression of more than 50% of tumor cells in the lung tissue could respond to immune checkpoint inhibitors (33), a subpopulation of patients with NSCLC with PD-L1 negative could be also beneficial from the treatment, suggesting that other biomarkers are needed to predict the efficacy of immunotherapy treatments (31). The HNCIB system not only could monitor the EV PD-L1 during immunotherapy treatment but also provides the opportunity to identify the subpopulation of patients who could benefit from PD-L1 antibodies on the basis of the fraction of EV PD-L1 and/or other EV biomarkers including miRNA, mRNA, and proteins.
In conclusion, we report a newly developed HNCIB system that is capable of capture and simultaneous detection of EV proteins and mRNA from a single EV. The system showed excellent signal-to-noise ratio, sensitivity, and specificity in detecting both surface and cargo biomarkers of EVs. The system is capable of high-throughput analysis, and the accuracy was improved by implementing machine learning in image processing. The HNCIB system contributes to the advancement of the field of liquid biopsy and has potential clinical applications to enhance cancer care. Although only demonstrated in the clinical use for patients with LUAD, it is anticipated that the system can be applied across the spectrum of cancer care, including early cancer detection, delivering precision therapy, evaluating response to treatment, and early detection of disease recurrence.
MATERIALS AND METHODS
Study populations
A total of 35 healthy donors and 34 patients with LUAD were enrolled in the study at the Department of Pulmonary Medicine, Zhongshan Hospital, Fudan University. Patients with LUAD were only chosen because PD-L1 was the targeted detection marker for the study, and previous studies (34, 35) showed that PD-L1 expression does not correlate with outcomes in patients with lung squamous cell carcinoma but does correlate with outcomes in patients with LUAD; we then randomly selected patients with LUAD. The inclusion criteria for this study included the following: (i) aged above 18 years; (ii) male or female; (iii) male smoker (≥400 cigarettes/year), female smoker, or nonsmoker; and (iv) healthy donor with matched age, gender, and smoking history, or patients without cancer other than LUAD confirmed by pathology or histology. The exclusion criteria included the following: (i) the absence of histopathological diagnosis and (ii) patients with lung squamous cell carcinoma. All study participants provided informed consent, and the study protocol was approved by the institutional ethics committee (approval numbers B2016-154R and B2012-069). Plasma samples were collected in accordance with the relevant guidelines.
Sample collection
A venous blood sample (4000 μl) was collected from patients with LUAD and healthy donors using an EDTA-coated plastic whole-blood tube (BD Biosciences, Franklin Lakes, NJ, USA). The sample was centrifuged at room temperature at 820g for 10 min to separate the plasma, and then the plasma was centrifuged at 4°C at 16,000g for 10 min. The plasma was aliquoted and frozen at 4°C for 2 hours and −20°C for 30 min and stored at −80°C for later use. The demographic characteristics of patients with LUAD and healthy donors are listed in table S2.
Isolation and accumulation of EVs from plasma of human blood
The human plasma was first thawed and vortexed. A volume of 90 μl of sample was thawed, and the vortexed supernatant was placed in a new 1.5-ml Eppendorf (EP) tube and centrifuged at room temperature at 2000g for 10 min to remove residual cells and cell fragments. The supernatant was transferred into a new EP tube and centrifuged at 4°C at 10,000g for 20 min to remove large vesicles. The supernatant was transferred into a new EP tube, and 30 μl of 1× PBS was added and vortexed. Then, 3 μl of proteinase K (1245680100; Sigma-Aldrich, St. Louis., MO, USA) was added and vortexed again. The sample was then incubated at 37°C for 10 min. Following incubation, 18 μl of Exosome Precipitation Agent (from plasma; 4484450; Thermo Fisher Scientific, Waltham, MA, USA) was added and vortexed. The sample was then incubated at 4°C for 30 min. The incubated sample was filtered with a 0.8-μm filter into a new EP tube and centrifuged at 4°C at 10,000g for 5 min. The supernatant was carefully removed and discarded. PBS (1×) was then added to resuspend the EV precipitates for later use.
Cell culture
Human LUAD cell line A549 was purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). A549 was cultured as a monolayer at 37°C in a humidified atmosphere of 5% CO2 and in RPMI 1640 medium (Thermo Fisher Scientific) supplemented with 10% heat-inactivated fetal bovine serum and penicillin/streptomycin (100 U/ml).
Isolation and accumulation of EV from supernatant from cell culture media
The supernatant was first thawed and vortexed. Such supernatant (1000 μl) was placed in a new 2.0-ml EP tube and centrifuged at room temperature at 2000g for 30 min to remove residual cells and cell fragments. The supernatant was transferred into a new EP tube, and 500 μl of 1× PBS was added and vortexed. Then, 50 μl of proteinase K (1245680100, Sigma-Aldrich) was added and vortexed again. The sample was then incubated at 37°C for 20 min. Following incubation, 300 μl of Exosome Precipitation Agent (from cell culture media; 4478359, Thermo Fisher Scientific) was added and vortexed. The sample was then incubated at 4°C overnight. The incubated sample was then filtered with a 0.8-μm filter into a new EP tube and centrifuged at 4°C at 10,000g for 1 hour. The supernatant was carefully removed and discarded. PBS (1×) was then added to resuspend the EV precipitates for later use.
Transmission electron microscope
EM was performed to characterize the EVs. The EV pellet was first fixed with 4% paraformaldehyde and then incubated with 1% glutaraldehyde. After 5 min, the uranyl acetate staining was performed, and the sample was imaged with a TEM (Hitachi H7500 TEM, Japan).
EV particle size and zeta potential measurement
Measurement of the EV particle size and zeta potential was conducted on a Malvern Zetasizer Nano ZS90 (Malvern Instruments, Worcestershire, UK). The isolated EV was resuspended in 1× PBS for the measurement.
HNCIB system
Figure 1A illustrates the overall HNCIB system principles. The HNCIB system consists of the following subsystems: (i) a scalable and high-throughput nano-biochip with immobilized lipoplex nanoparticles for EV capture and enrichment, (ii) an imaging system using TIRFM to realize high-resolution imaging on a single EV with multiple analytes, and (iii) a deep learning algorithm for rapid and automated image analysis at a single-EV level. Details of the system were discussed in detail elsewhere (15, 36, 37). The basic principles of the HNCIB system is briefly described below. Additional details can be found in the Supplementary Materials.
Nano-biochip for EV capture
HNCIB captures and detects the target biomolecules, e.g., RNAs and membrane proteins using a nano-biochip (36), which was previously used for detection of mRNA/miRNA (14). The glass-based nano-biochip was fabricated using a cleanroom-free process (36, 37) and can be prepared from a glass coverslip and multiwell plates (e.g., 6, 24, 96, or 384) commonly used in cell culture and bioassays. A series of agents were linked onto the glass substrate. Subsequently, cationic lipoplex nanoparticles, which were used for EV capture, were immobilized onto the substrate.
RNA and protein detection
RNAs were detected using MBs. The MBs typically have a hairpin-like structure consisting of a stem and loop region. A fluorescent dye and quencher are located at either end of the stem (38). In the absence of the target nucleotide sequence, such structure is thermodynamically stable and there is no fluorescent signal as the dye and quencher are in close proximity. When targets are present with matching sequence with of the MBs, hybridization takes place, leading to the separation of the fluorescent dye and quencher and the generation of fluorescent signals (38). In this study, two MBs were used for the detection of PD-L1 mRNA transcript and the miRNA miR-21 with the following sequences: 5′-6FAM-CGTTCAGCAAATGCCAGTAGGTCACATTTGCTGA-BHQ1-3′ (for PD-L1 mRNA) and 5′-6FAM-TCAACATCAGTCTGATAAGCTATTATCAGACTGA-BHQ1-3′ (for miR-21 miRNA) (Sangon Biotech, Shanghai, China). Both MBs were labeled with FAM as the fluorophore and BHQ as the quencher. The MBs were encapsulated in the cationic lipoplex nanoparticles that were tethered on the biochip, as described in the above section. The detection assay started with the addition of suspension of isolated EVs in PBS to the chip followed by incubation. During incubation, the negatively charged EVs were attracted via electrostatic force and fused with the cationic nanoparticles, allowing for the mixing of the cargo content of both particles and the hybridization of MB with the targeted RNAs. The EV surface membrane proteins were detected using immunostaining (see the “Staining of EV-positive and EV-negative markers and EV-associated protein” section).
TIRFM for single-EV detection
Biomarker detection and imaging were conducted via “through-the-objective” type of TIRFM using a Nikon Eclipse Ti-E inverted microscope system equipped with four laser beams for simultaneous multichannel image acquisitions. Images were acquired using Nikon NIS-Elements AR software.
Image processing and data analysis
A custom-developed, big-data–based image analysis algorithm was applied for imaging processing and data analysis (15). The deep learning algorithm enabled rapid and automatic image screening and selection of valid images, background reduction, and calculation of fluorescent intensity from valid images (15). The selection of valid images is based on a deep learning model. A CNN, trained using a minimum of 1000 categorized images in a 512 × 512 size operated as a filter to classify the input images into two categories: normal and abnormal. Images with unexpected size or intensity of fluorescence were categorized as abnormal and rejected and were analyzed further. Background noise reduction was realized using the steepest descent method and applied to normal images. The value of each processed image was determined according to the fluorescence intensity of the image after the threshold was determined by the background noise reduction algorithm.
Immunofluorescence staining for the membrane protein of cells
To stain CD63 or PD-L1 membrane protein, A549 cells were fixed with 4% paraformaldehyde and stained with CD63-AF488 (NBP2-42225AF488; Novus Biologicals, Littleton, CO, USA) or PD-L1–AF647 (ab209960; Abcam, Cambridge, MA, USA) fluorescent antibodies, respectively (table S3). Before imaging, 4,6-diamidino-2-phenylindole was applied to visualize cell nuclei. After staining, the samples were imaged with a total internal reflective fluorescence microscope (Nikon, Japan) and a confocal microscope (Nikon, Japan).
Staining of EV-positive and EV-negative markers and EV-associated protein
The following procedure was used to stain the CD9, CD63, and PD-L1 membrane protein and ALB and APOB protein in EVs. EVs were captured and immobilized onto the nano-biochip, as discussed earlier. The membrane proteins may be detected independently or together with RNA detection. In the latter case, RNA detection was carried out first. Samples were then washed thoroughly, after which CD9-AF488 (NB500-327AF488, Novus Biologicals), CD63-AF488 (NBP2-42225AF488, Novus Biologicals), or isotype control (IC002G; R&D Systems, Minneapolis, MN, USA), PD-L1–AF647 (ab209960, Abcam), primary PD-L1 (NBP1-76769, Novus Biologicals), or isotype control (NBP2-24891, Novus Biologicals), and secondary Alexa Fluor 568 (A-11011, Thermo Fisher Scientific), ALB-AF488 (IC1455G, Novus Biologicals), and APOB-FITC (ab27637, Abcam) antibodies were applied followed by incubation. Incubation took place at 37°C for both primary and secondary antibodies, and the incubation time was 60 min for the former and 30 min for the latter.
FCM detection of CD63 and PD-L1 on EVs
CD63 exosome capture beads (ab239686, Abcam) were used to isolate EVs according to the manufacturer’s instruction. Briefly, 200 μl of plasma from healthy donors or patients with LUAD was centrifuged at 500g at room temperature for 10 min to remove residual cells. The supernatant was transferred to a new EP tube and centrifuged at 2000g at room temperature for 10 min to remove cell fragments. The supernatant was then transferred into a new EP tube and centrifuged at room temperature at 14,000g for 30 min to remove large vesicles. Last, the supernatant was transferred into a new EP tube and diluted with same volume of 1× PBS. Then, a volume of 200 μl of diluted plasma was aspirated into a new EP tube, and 100 μl of CD63 exosome capture beads was added and vortexed again. The sample was incubated overnight at room temperature in the dark. Following incubation, CD63-AF488 (NBP2-42225AF488, Novus Biologicals) and PD-L1–AF647 (ab209960, Abcam) fluorescent antibodies were added to the mixture at a volume ratio of 1:100 and incubated at 4°C for 2 hours in the dark. Stained EVs were sorted with a Navios cytometer (Beckman Coulter, CA, USA).
Lentivirus construction and infection
Lentiviral vector overexpressing human CD63 (CD63-GFP) (GenBank, NM_001267698.1), overexpressing human PD-L1 (GenBank, NM_014143.3), or empty vector was purchased from GeneCopoeia (Guangzhou, China). Lentiviral vector overexpressing human PD-L1–mCherry (GenBank, NM_014143.3) or empty vector was purchased from OBiO Technology (Shanghai, China). Lentiviral particles were produced in human embryonic kidney–293T cells (ATCC). A549 cells grown to 50 to 70% confluence in six-well plates were then infected at multiplicity of infection with lentiviral particles and suspended in polybrene (4 μg/ml).
Reverse transcription quantitative polymerase chain reaction
For A549 cells, total RNA was extracted using TRIzol (Thermo Fisher Scientific) and reverse-transcribed into complementary DNA (cDNA) using a reverse transcriptase (Toyobo, Osaka, Japan). Subsequently, 20 ng of cDNA was used as a qPCR template to quantify PD-L1 with 5′-ACGCCCCATACAACAAAATC-3′ and 5′-GGTCACTGCTTGTCCAGATG-3′ and β-actin with 5′-CTGGCACCCAGCACAATG-3′ and 5′-CCGATCCACACGGAGTA CTTG-3′. Targets were amplified over 40 cycles at 95°C for 15 s, 60°C for 15 s, and 72°C for 45 s. Gene expression was normalized to β-actin according to the cycle threshold (2−ΔΔCT) method.
For EVs, total RNA was extracted using TRIzol and reverse-transcribed into the cDNA using a cDNA synthesis kit (K1622, Thermo Fisher Scientific). After the cDNA product was diluted five times, 2.5 ml of cDNA was prepared for each preamplification reaction using the KAPA Hyper Prep Kit (KAPA Biosystems, Shanghai, China). The preamplification conditions were as follows: 95°C for 3 min; 20 cycles of 95°C for 15 s, 60°C for 15 s, 72°C for 15 s, and, last, 72°C for 1 min. After diluted 10 times, the preamplification product were amplified over 40 cycles at 95°C for 2 min, 95°C for 5 s, and 60°C for 10 s. Gene expression was normalized to U6 with 5′-CTCGCTTCGGCAGCACA-3′ and 5′-AACGCTTCACGAATTTGCGT-3′ according to the cycle threshold (2−ΔΔCT) method.
Western blotting
Total protein was extracted in radioimmunoprecipitation buffer (Beyotime Institute of Biotechnology, Shanghai, China), separated on polyacrylamide gels, transferred to polyvinylidene difluoride, and probed overnight at 4°C with antibodies against PD-L1 (Novus Biologicals) and actin (Cell Signaling Technology, Danvers, MA, USA). Membranes were then labeled at room temperature for 1 hour with goat anti-rabbit or anti-mouse immunoglobulin G conjugated to horseradish peroxidase, visualized by enhanced chemiluminescence (Pierce, Rockford, IL, USA), and analyzed in Quantity One version 4.6 (Bio-Rad, Hercules, CA, USA).
Statistical analysis
Data are analyzed using GraphPad Prism 6 and reported as means ± SD of at least three independent experiments with three replicates. The normality test was performed for each group of the data. Differences between two groups were evaluated by Mann-Whitney test. Differences were considered statistically significant at P < 0.05.
Supplementary Material
Acknowledgment
Funding: This work was supported by the National Science and Technology Major Project “Key New Drug Creation and Manufacturing Program” (no. 2018ZX09201002-006), National Natural Science Foundation of China (nos. 81770039, 81570028, 81490533, and 81400018), National Key R&D Plan (2016YFC1304104), Zhongshan Hospital Clinical Research Foundation (nos. 2016ZSLC05 and 2019ZSGG15), Shanghai Municipal Key Clinical Specialty (shslczdzk02201), and Shanghai Top-Priority Clinical Key Disciplines Construction Project (2017ZZ02013). Author contributions: C.B. and H.Z. had full access to all the data in the study and take responsibility for the integrity of the data and accuracy of the data analysis. Study concept and design: H.Z., J.Z., Z.W., and C.B. Clinical sample collection: J.Z., J.H., D.Y., and M.D. Experiments and data collection: J.Z., Z.W., W.W., C.X., M.W., X.C., Q.W., J.L., W.P., and Y.W. Data interpretation and statistical analysis: J.Z., Z.W., and H.Z. Drafting of the manuscript: J.Z. and Z.W. Study supervision: Y.S., H.Z., and C.B. Competing interests: Z.W., H.Z., W.W., C.X., and M.W. are employees of Hangzhou Dixiang Co. Ltd. The remaining authors declared that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/47/eabc1204/DC1
REFERENCES AND NOTES
- 1.Théry C., Witwer K. W., Aikawa E., Alcaraz M. J., Anderson J. D., Andriantsitohaina R., Antoniou A., Arab T., Archer F., Atkin-Smith G. K., Ayre D. C., Bach J.-M., Bachurski D., Baharvand H., Balaj L., Baldacchino S., Bauer N. N., Baxter A. A., Bebawy M., Beckham C., Bedina Zavec A., Benmoussa A., Berardi A. C., Bergese P., Bielska E., Blenkiron C., Bobis-Wozowicz S., Boilard E., Boireau W., Bongiovanni A., Borràs F. E., Bosch S., Boulanger C. M., Breakefield X., Breglio A. M., Brennan M. Á., Brigstock D. R., Brisson A., Broekman M. L., Bromberg J. F., Bryl-Górecka P., Buch S., Buck A. H., Burger D., Busatto S., Buschmann D., Bussolati B., Buzás E. I., Byrd J. B., Camussi G., Carter D. R., Caruso S., Chamley L. W., Chang Y.-T., Chen C., Chen S., Cheng L., Chin A. R., Clayton A., Clerici S. P., Cocks A., Cocucci E., Coffey R. J., Cordeiro-da-Silva A., Couch Y., Coumans F. A., Coyle B., Crescitelli R., Criado M. F., D’Souza-Schorey C., Das S., Datta Chaudhuri A., de Candia P., De Santana E. F. Junior, De Wever O., Del Portillo H. A., Demaret T., Deville S., Devitt A., Dhondt B., Di Vizio D., Dieterich L. C., Dolo V., Dominguez Rubio A. P., Dominici M., Dourado M. R., Driedonks T. A., Duarte F. V., Duncan H. M., Eichenberger R. M., Ekström K., El Andaloussi S., Elie-Caille C., Erdbrügger U., Falcón-Pérez J. M., Fatima F., Fish J. E., Flores-Bellver M., Försönits A., Frelet-Barrand A., Fricke F., Fuhrmann G., Gabrielsson S., Gámez-Valero A., Gardiner C., Gärtner K., Gaudin R., Gho Y. S., Giebel B., Gilbert C., Gimona M., Giusti I., Goberdhan D. C., Görgens A., Gorski S. M., Greening D. W., Gross J. C., Gualerzi A., Gupta G. N., Gustafson D., Handberg A., Haraszti R. A., Harrison P., Hegyesi H., Hendrix A., Hill A. F., Hochberg F. H., Hoffmann K. F., Holder B., Holthofer H., Hosseinkhani B., Hu G., Huang Y., Huber V., Hunt S., Ibrahim A. G.-E., Ikezu T., Inal J. M., Isin M., Ivanova A., Jackson H. K., Jacobsen S., Jay S. M., Jayachandran M., Jenster G., Jiang L., Johnson S. M., Jones J. C., Jong A., Jovanovic-Talisman T., Jung S., Kalluri R., Kano S.-I., Kaur S., Kawamura Y., Keller E. T., Khamari D., Khomyakova E., Khvorova A., Kierulf P., Kim K. P., Kislinger T., Klingeborn M., Klinke D. J. II, Kornek M., Kosanović M. M., Kovács Á. F., Krämer-Albers E.-M., Krasemann S., Krause M., Kurochkin I. V., Kusuma G. D., Kuypers S., Laitinen S., Langevin S. M., Languino L. R., Lannigan J., Lässer C., Laurent L. C., Lavieu G., Lázaro-Ibáñez E., Le Lay S., Lee M.-S., Lee Y. X. F., Lemos D. S., Lenassi M., Leszczynska A., Li I. T., Liao K., Libregts S. F., Ligeti E., Lim R., Lim S. K., Linē A., Linnemannstöns K., Llorente A., Lombard C. A., Lorenowicz M. J., Lörincz Á. M., Lötvall J., Lovett J., Lowry M. C., Loyer X., Lu Q., Lukomska B., Lunavat T. R., Maas S. L., Malhi H., Marcilla A., Mariani J., Mariscal J., Martens-Uzunova E. S., Martin-Jaular L., Martinez M. C., Martins V. R., Mathieu M., Mathivanan S., Maugeri M., McGinnis L. K., McVey M. J., Meckes D. G. Jr., Meehan K. L., Mertens I., Minciacchi V. R., Möller A., Møller Jørgensen M., Morales-Kastresana A., Morhayim J., Mullier F., Muraca M., Musante L., Mussack V., Muth D. C., Myburgh K. H., Najrana T., Nawaz M., Nazarenko I., Nejsum P., Neri C., Neri T., Nieuwland R., Nimrichter L., Nolan J. P., Nolte-’t Hoen E. N., Noren Hooten N., O’Driscoll L., O’Grady T., O’Loghlen A., Ochiya T., Olivier M., Ortiz A., Ortiz L. A., Osteikoetxea X., Østergaard O., Ostrowski M., Park J., Pegtel D. M., Peinado H., Perut F., Pfaffl M. W., Phinney D. G., Pieters B. C. H., Pink R. C., Pisetsky D. S., Pogge von Strandmann E., Polakovicova I., Poon I. K. H., Powell B. H., Prada I., Pulliam L., Quesenberry P., Radeghieri A., Raffai R. L., Raimondo S., Rak J., Ramirez M. I., Raposo G., Rayyan M. S., Regev-Rudzki N., Ricklefs F. L., Robbins P. D., Roberts D. D., Rodrigues S. C., Rohde E., Rome S., Rouschop K. M. A., Rughetti A., Russell A. E., Saá P., Sahoo S., Salas-Huenuleo E., Sánchez C., Saugstad J. A., Saul M. J., Schiffelers R. M., Schneider R., Schøyen T. H., Scott A., Shahaj E., Sharma S., Shatnyeva O., Shekari F., Shelke G. V., Shetty A. K., Shiba K., Siljander P. R. M., Silva A. M., Skowronek A., Snyder O. L. II, Soares R. P., Sódar B. W., Soekmadji C., Sotillo J., Stahl P. D., Stoorvogel W., Stott S. L., Strasser E. F., Swift S., Tahara H., Tewari M., Timms K., Tiwari S., Tixeira R., Tkach M., Toh W. S., Tomasini R., Torrecilhas A. C., Tosar J. P., Toxavidis V., Urbanelli L., Vader P., van Balkom B. W., van der Grein S. G., Van Deun J., van Herwijnen M. J., van Keuren-Jensen K., van Niel G., van Royen M. E., van Wijnen A. J., Vasconcelos M. H., Vechetti I. J. Jr., Veit T. D., Vella L. J., Velot É., Verweij F. J., Vestad B., Viñas J. L., Visnovitz T., Vukman K. V., Wahlgren J., Watson D. C., Wauben M. H., Weaver A., Webber J. P., Weber V., Wehman A. M., Weiss D. J., Welsh J. A., Wendt S., Wheelock A. M., Wiener Z., Witte L., Wolfram J., Xagorari A., Xander P., Xu J., Yan X., Yáñez-Mó M., Yin H., Yuana Y., Zappulli V., Zarubova J., Žėkas V., Zhang J.-Y., Zhao Z., Zheng L., Zheutlin A. R., Zickler A. M., Zimmermann P., Zivkovic A. M., Zocco D., Zuba-Surma E. K., Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 7, 1535750 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Niel G., D’Angelo G., Raposo G., Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 19, 213–228 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Siegel R. L., Miller K. D., Jemal A., Cancer statistics, 2019. CA Cancer J. Clin. 69, 7–34 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Jeppesen D. K., Fenix A. M., Franklin J. L., Higginbotham J. N., Zhang Q., Zimmerman L. J., Liebler D. C., Ping J., Liu Q., Evans R., Fissell W. H., Patton J. G., Rome L. H., Burnette D. T., Coffey R. J., Reassessment of exosome composition. Cell 177, 428–445.e18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Im H., Lee K., Weissleder R., Lee H., Castro C. M., Novel nanosensing technologies for exosome detection and profiling. Lab Chip 17, 2892–2898 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He D., Wang H., Ho S.-L., Chan H.-N., Hai L., He X., Wang K., Li H.-W., Total internal reflection-based single-vesicle in situ quantitative and stoichiometric analysis of tumor-derived exosomal microRNAs for diagnosis and treatment monitoring. Theranostics 9, 4494–4507 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee K., Fraser K., Ghaddar B., Yang K., Kim E., Balaj L., Chiocca E. A., Breakefield X. O., Lee H., Weissleder R., Multiplexed profiling of single extracellular vesicles. ACS Nano 12, 494–503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daaboul G. G., Gagni P., Benussi L., Bettotti P., Ciani M., Cretich M., Freedman D. S., Ghidoni R., Ozkumur A. Y., Piotto C., Prosperi D., Santini B., Ünlü M. S., Chiari M., Digital detection of exosomes by interferometric imaging. Sci. Rep. 6, 37246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen D. S., Irving B. A., Hodi F. S., Molecular pathways: Next-generation immunotherapy—Inhibiting programmed death-ligand 1 and programmed death-1. Clin. Cancer Res. 18, 6580–6587 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Brahmer J. R., Drake C. G., Wollner I., Powderly J. D., Picus J., Sharfman W. H., Stankevich E., Pons A., Salay T. M., McMiller T. L., Gilson M. M., Wang C., Selby M., Taube J. M., Anders R., Chen L., Korman A. J., Pardoll D. M., Lowy I., Topalian S. L., Phase I study of single-agent anti–programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 28, 3167–3175 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen G., Huang A. C., Zhang W., Zhang G., Wu M., Xu W., Yu Z., Yang J., Wang B., Sun H., Xia H., Man Q., Zhong W., Antelo L. F., Wu B., Xiong X., Liu X., Guan L., Li T., Liu S., Yang R., Lu Y., Dong L., McGettigan S., Somasundaram R., Radhakrishnan R., Mills G., Lu Y., Kim J., Chen Y. H., Dong H., Zhao Y., Karakousis G. C., Mitchell T. C., Schuchter L. M., Herlyn M., Wherry E. J., Xu X., Guo W., Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 560, 382–386 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poggio M., Hu T., Pai C.-C., Chu B., Belair C. D., Chang A., Montabana E., Lang U. E., Fu Q., Fong L., Blelloch R., Suppression of exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell 177, 414–427.e13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou J., Qu G., Zhang G., Wu Z., Liu J., Yang D., Li J., Chang M., Zeng H., Hu J., Fang T., Song Y., Bai C., Glycerol kinase 5 confers gefitinib resistance through SREBP1/SCD1 signaling pathway. J. Exp. Clin. Cancer Res. 38, 96 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou J., Kwak K. J., Wu Z., Yang D., Li J., Chang M., Song Y., Zeng H., Lee L. J., Hu J., Bai C., PLAUR confers resistance to gefitinib through EGFR/P-AKT/Survivin signaling pathway. Cell. Physiol. Biochem. 47, 1909–1924 (2018). [DOI] [PubMed] [Google Scholar]
- 15.H. D. C. Ltd, P. National Intellectual Property Administration (Hangzhou Dixion Co. Ltd, 2019).
- 16.I. Sobel, in Machine Vision for Three-Dimensional Scences, H. Freeman, Ed. (Academic Press, 1990), pp. 376–379.
- 17.Gupta S., Mazumdar S. G., Sobel edge dection algorithm. Int. J. Comp. Sci. Manage. Res. 2, 1578–1583 (2013). [Google Scholar]
- 18.Y.-q. Zhao, W.-h. Gui, Z.-c. Chen, J.-t. Tang, L.-y. Li, 2005 IEEE Engineering in Medicine and Biology 27th Annual Conference (IEEE, 2005), pp. 6492–6495. [Google Scholar]
- 19.N. Kazakova, M. Margala, N. G. Durdle, 2004 IEEE International Symposium on Circuits and Systems (IEEE Cat. No.04CH37512) (IEEE, 2004), vol. 2, pp. II-913.
- 20.Vijayarani S., Vinupriya M., Performance analysis of canny and sobel edge detection algorithms in image mining. Int. J. Innov. Res. Comp. Commun. Eng. 1, 1760–1767 (2013). [Google Scholar]
- 21.Shen J., Todd N. W., Zhang H., Yu L., Lingxiao X., Mei Y., Guarnera M., Liao J., Chou A., Lu C. L., Jiang Z., Fang H., Katz R. L., Jiang F., Plasma microRNAs as potential biomarkers for non-small-cell lung cancer. Lab. Invest. 91, 579–587 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lacroix R., Robert S., Poncelet P., Kasthuri R. S., Key N. S., Dignat-George F.; ISTH SSC Workshop , Standardization of platelet-derived microparticle enumeration by flow cytometry with calibrated beads: Results of the International Society on Thrombosis and Haemostasis SSC Collaborative workshop. J. Thromb. Haemost. 8, 2571–2574 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Zeringer E., Li M., Barta T., Schageman J., Pedersen K. W., Neurauter A., Magdaleno S., Setterquist R., Vlassov A. V., Methods for the extraction and RNA profiling of exosomes. World J. Methodol. 3, 11–18 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.P. Vermette, Methods in Enzymology (Academic Press, 2009), vol. 465, pp. 43–73. [DOI] [PubMed]
- 25.Osteikoetxea X., Sódar B., Németh A., Szabó-Taylor K., Pálóczi K., Vukman K. V., Tamási V., Balogh A., Kittel Á., Pállinger É., Buzás E. I., Differential detergent sensitivity of extracellular vesicle subpopulations. Org. Biomol. Chem. 13, 9775–9782 (2015). [DOI] [PubMed] [Google Scholar]
- 26.White J., Membrane fusion. Science 258, 917–924 (1992). [DOI] [PubMed] [Google Scholar]
- 27.Diao J., Su Z., Ishitsuka Y., Lu B., Lee K. S., Lai Y., Shin Y.-K., Ha T., A single-vesicle content mixing assay for SNARE-mediated membrane fusion. Nat. Commun. 1, 54 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diao J., Ishitsuka Y., Lee H., Joo C., Su Z., Syed S., Shin Y.-K., Yoon T.-Y., Ha T., A single vesicle-vesicle fusion assay for in vitro studies of SNAREs and accessory proteins. Nat. Protoc. 7, 921–934 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Axelrod D., Total internal reflection fluorescence microscopy in cell biology. Traffic 2, 764–774 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Sako Y., Minoghchi S., Yanagida T., Single-molecule imaging of EGFR signalling on the surface of living cells. Nat. Cell Biol. 2, 168–172 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Meyers D. E., Bryan P. M., Banerji S., Morris D. G., Targeting the PD-1/PD-L1 axis for the treatment of non-small-cell lung cancer. Curr. Oncol. 25, e324–e334 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Y., Li C.-W., Chan L.-C., Wei Y., Hsu J.-M., Xia W., Cha J.-H., Hou J., Hsu J. L., Sun L., Hung M.-C., Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 28, 862–864 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reck M., Rodríguez-Abreu D., Robinson A. G., Hui R., Csőszi T., Fülöp A., Gottfried M., Peled N., Tafreshi A., Cuffe S., O’Brien M., Rao S., Hotta K., Leiby M. A., Lubiniecki G. M., Shentu Y., Rangwala R., Brahmer J. R.; KEYNOTE-024 Investigators , Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N. Engl. J. Med. 375, 1823–1833 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Brahmer J., Reckamp K. L., Baas P., Crinò L., Eberhardt W. E. E., Poddubskaya E., Antonia S., Pluzanski A., Vokes E. E., Holgado E., Waterhouse D., Ready N., Gainor J., Arén Frontera O., Havel L., Steins M., Garassino M. C., Aerts J. G., Domine M., Paz-Ares L., Reck M., Baudelet C., Harbison C. T., Lestini B., Spigel D. R., Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 373, 123–135 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borghaei H., Paz-Ares L., Horn L., Spigel D. R., Steins M., Ready N. E., Chow L. Q., Vokes E. E., Felip E., Holgado E., Barlesi F., Kohlhäufl M., Arrieta O., Burgio M. A., Fayette J., Lena H., Poddubskaya E., Gerber D. E., Gettinger S. N., Rudin C. M., Rizvi N., Crinò L., Blumenschein G. R. Jr., Antonia S. J., Dorange C., Harbison C. T., Graf Finckenstein F., Brahmer J. R., Nivolumab versus Docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 373, 1627–1639 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.H. D. C. Ltd, P. National Intellectual Property Administration (Hangzhou Dixion Co. Ltd, 2018), chap. PCT/CN2019/089478.
- 37.H. D. C. Ltd, P. National Intellectual Property Administration (Hangzhou Dixion Co. Ltd, 2018).
- 38.Tan W., Wang K., Drake T. J., Molecular beacons. Curr. Opin. Chem. Biol. 8, 547–553 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/47/eabc1204/DC1