

SUMMARY
The RNA isoform repertoire is regulated by splicing-factor (SF) expression, and alterations in SF-levels are associated with disease. SFs contain ultraconserved poison exon (PE) sequences, which exhibit greater identity across species than nearby coding exons, yet their physiological role and molecular regulation is incompletely understood. We show that PEs in SR proteins, a family of 14 essential SFs, are differentially spliced during iPSC differentiation and in tumors vs. normal tissues. We uncover an extensive cross-regulatory network of SR proteins controlling their expression via alternative splicing coupled to nonsense-mediated decay. We define sequences that regulate PE inclusion and protein expression of the oncogenic SF TRA2β using an RNA-targeting CRISPR screen. We demonstrate location-dependency of RS domain activity on the regulation of the TRA2β-PE using CRISPR-artificial SFs. Finally, we develop splice-switching antisense oligonucleotides to reverse the increased skipping of TRA2β-PE detected in breast tumors, altering breast cancer cell viability, proliferation, and migration.
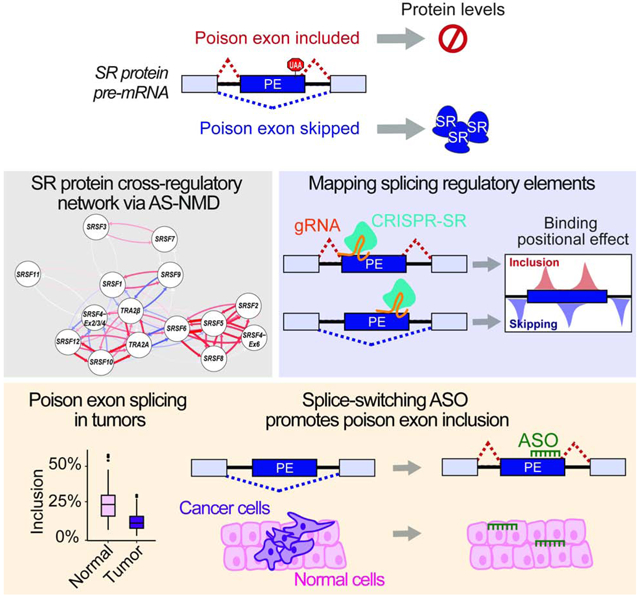
Graphical Abstract
eTOC Blurb Molecular Cell
Leclair et al. demonstrate that the expression of 14 SR proteins is coordinated through splicing of their poison-exons during cell differentiation and tumorigenesis. Using TRA2β poison exon as an example, they identify splicing regulatory sequences and design splice-switching antisense oligonucleotides that decrease TRA2β protein expression and exhibit anti-cancer effects.
INTRODUCTION
Alternative RNA splicing (AS) is a key step in gene expression regulation and a source of proteomic and functional diversity (Manning and Cooper, 2017). Regulation of AS is essential for normal development and defects in AS are implicated in diseases, including cancer (Scotti and Swanson, 2016). AS is regulated by the core splicing machinery along with regulatory splicing factors (SFs) that bind specific RNA sequences and act in a dose-dependent manner (Long and Caceres, 2009). SFs are frequently altered in human diseases, leading to downstream changes in RNA isoforms (Dvinge et al., 2016; Urbanski et al., 2018).
Serine-arginine rich (SR) proteins are a family of 14 essential SFs (SRSF1 to 12, and SR-like members TRA2α and TRA2β) that evolved from a common ancestor and contain at least one RNA recognition motif (RRM), responsible for binding specific pre-mRNA sequences, and at least one arginine-serine rich (RS) domain that coordinates protein interactions (Long and Caceres, 2009). SR proteins are involved in both constitutive and alternative splicing, promoting either exon skipping or inclusion (Bradley et al., 2015; Pandit et al., 2013; Park et al., 2019). In addition, several SR proteins have splicing-independent roles, including mRNA export, mRNA decay, or translation regulation (Huang et al., 2003; Maslon et al., 2014; Michlewski et al., 2008; Muller-McNicoll et al., 2016; Sanford et al., 2004; Sato et al., 2008; Swanson et al., 2010; Zhang and Krainer, 2004). Therefore, changes in SR protein levels can affect a wide network of downstream targets and AS events.
SR proteins are critical for normal development, and defects in SR proteins have causatively been implicated in several pathologies, including cardiac and liver dysfunction, brain abnormalities, diabetes, lupus, and cancer (Dichmann et al., 2015; Ding et al., 2004; Grellscheid et al., 2011; Kaminska et al., 2016; Kumar et al., 2019; Lu et al., 2014; Neugebauer et al., 2000; Ortiz-Sanchez et al., 2019; Ratnadiwakara et al., 2018; Roberts et al., 2014; Sen et al., 2013; Storbeck et al., 2014; Urbanski et al., 2018; Xu et al., 2005). Solid tumors often exhibit altered levels of SR proteins which subsequently promote formation of RNA isoforms involved in tumor biology (Urbanski et al., 2018). Few studies have investigated the molecular mechanisms driving alterations in SR protein levels, which in tumors frequently occurs without genomic copy number changes (Best et al., 2013; Karni et al., 2007; Park et al., 2019; Sebestyen et al., 2016), arguing for misregulation at the post-transcriptional level. Transcriptional activation of SFs, for example by the Myc oncogene, occurs in a subset of tumors (Anczukow et al., 2012; Caggiano et al., 2019; Das et al., 2012; David et al., 2010; Koh et al., 2015; Park et al., 2019). Therefore, investigation into the fundamental mechanisms that regulate the AS machinery is imperative.
Post-transcriptionally, SR proteins are auto-regulated by AS coupled to nonsense-mediated decay (AS-NMD). SR protein genes comprise ultraconserved regions (UCR) containing noncoding exons, called “poison exons” (PE), or 3’UTR poison sequences, that when included or spliced, respectively, introduce a premature termination codon (PTC) and target the mRNA for degradation (Lareau et al., 2007; Ni et al., 2007). For example, binding of the TRA2β protein to the TRA2β-PE enhances its inclusion (Stoilov et al., 2004). Similar auto-regulatory patterns that link PE splicing to changes in protein levels have been experimentally demonstrated for only five other SR proteins (Jumaa and Nielsen, 1997; Konigs et al., 2020; Sun et al., 2010; Sureau et al., 2001; Yang et al., 2018). A few examples of cross-regulation have been uncovered whereby an SR protein influences the inclusion of PEs in other SFs, including for TRA2β and its paralog TRA2α (Best et al., 2014), and for SRSF3 on SRSF5 or SRSF7 (Anko et al., 2012; Yang et al., 2018). However, the extent or sequence determinants of cross-regulation within the SR protein family is unknown. Thus, we sought to better understand how all 14 SR proteins interact through AS-NMD, providing insight into the mechanisms that maintain SR protein homeostasis during development and how these become dysregulated in disease.
RESULTS
SR protein PEs are differentially spliced during cell differentiation and tumorigenesis
SR proteins coordinate AS events in the development of several tissue types and their expression in stem cells helps maintain pluripotency (Ye and Blelloch, 2014). We examined the role of PEs in regulating SR protein levels through AS-NMD during the differentiation of induced human pluripotent stem cells (iPSC) into distinct cell types. We profiled SR protein RNA isoforms using published RNA-sequencing (RNA-seq) data from iPSCs differentiated into neurons, pancreatic β-cells, or lung alveolar type II epithelial cells (Burke et al., 2020; Jacob et al., 2017; Xie et al., 2013). Additionally, we performed RNA-seq on iPSCs differentiated into intermediate mesoderm progenitors or cardiomyocytes. Each member of the SR protein family contains at least one highly conserved sequence, hereafter called SR-PEs, located in the coding sequence or 3’UTR, that can potentially elicit AS-NMD (Figure 1A, Table S1A). Inclusion of each SR-PE during iPSC differentiation was quantified as ‘percent spliced-in’ (PSI) using rMATS (Shen et al., 2014) (Figure 1B-F). Low levels of SRSF8, SRSF10 and SRSF12 transcripts prevented us from accurately quantifying their PE inclusion. SRSF5-PE and SRSF1-3UTR are included in multiple isoforms, making it difficult to accurately reconstruct these transcripts from short reads. In all models, most PEs exhibit low inclusion in iPSCs and are more included during differentiation (Figure 1B-F, S1A,B). For example, SRSF3-PE inclusion increases in human cardiomyocytes (Figure 1C,D), a pattern that follows the decrease in SRSF3 coding mRNA in embryonic mouse heart development (Ortiz-Sanchez et al., 2019). TRA2β, critical for brain development (Grellscheid et al., 2011; Roberts et al., 2014; Storbeck et al., 2014), exhibits increased PE inclusion in mature neurons (Figure 1E,F). Finally, AS patterns of SRSF3-PE and TRA2β-PE impact SRSF3 and TRA2β protein levels during cardiac cell differentiation (Figure S1C,D), suggesting that PEs may play an important role in the regulation of SF-levels during cell differentiation.
Figure 1. SR-PEs are differentially spliced during cell differentiation and tumorigenesis.

(A) Schematic of SR protein genes with coding exons, non-coding regions, and internal cassette PEs or 3’UTR poison sequences (not at scale). (B) SR-PE inclusion (mean PSI of RNA-seq replicates) in human iPSCs differentiated to neurons, pancreatic β-cells, type II alveolar lung epithelial cells, or cardiomyocytes. (C,D) SRSF3-PE inclusion in iPSCs differentiated into mesoderm or cardiomyocytes shown as RNA-seq read coverage and evolutionary conservation across 100 vertebrates (C), and plotted as PSI (D) (n=4, median±interquartile range; *P<0.05, ns- not significant). (E,F) TRA2β-PE inclusion in iPSCs differentiated into neurons shown as RNA-seq read coverage and evolutionary conservation (E), and plotted as PSI (F) (n>4, median±interquartile range; **P<0.01, ****P<0.0001). (G) SR-PE inclusion in tumors vs. matched adjacent normal tissues in 13 TCGA tumor types (n≥30; P<0.05, ns- not significant). (H) SR-PE inclusion in TCGA breast tumors vs. matched adjacent normal tissues (n=213; *P<0.05, **P<0.01, ***P<0.001, ****p<0.0001, ns- not significant). (I-J) Inclusion of SRSF3-PE (I) and TRA2β-PE (J) in TCGA tumors and matched adjacent normal tissues (*P<0.05, **P<0.01, ***P<0.001, ****p<0.0001, ns- not significant). See also Table S1 and Figure S1.
Reasoning that a decrease in SR-PE inclusion would increase SR protein levels as detected in tumors (Urbanski et al., 2018), we examined SR-PE AS patterns in human tumors and adjacent normal tissues from The Cancer Genome Atlas (TCGA). To minimize inter-patient variation, we selected 13 tumor types for which ≥15 tumors and patient-matched normal tissue were available (Table S1B-D). Most SR-PEs are differentially spliced in at least one tumor type vs. normal tissue (Figure 1G-J and S1E). In breast tumors, less inclusion of all SR-PEs (Figure 1H) would lead to increased levels of known breast cancer oncogenes SRSF4, SRSF6 or TRA2β (Park et al., 2019). Conversely, SR-PEs are more included in kidney clear cell carcinoma (Figure 1G), which exhibits low SR protein levels (Piekielko-Witkowska et al., 2010).
Unlike solid tumors, hematological cancers harbor recurrent mutations in specific SFs, including SRSF2 in myelodysplastic syndrome (MDS) (Liang et al., 2018; Yoshida et al., 2011). To define the impact of SF mutations on SR-PE splicing, we analyzed RNA-seq data from CD34+ cells of 82 MDS patients with or without mutations in SF3B1, SRSF2, or U2AF1, and 8 healthy controls (Figure S1F) (Pellagatti et al., 2018). Overall, most SR-PEs are included at similar levels in SF-mutant vs. wild-type MDS samples. The exceptions are: i) SRSF2-3UTR and SRSF7-PE which are more skipped in SF3B1Mut; ii) SRSF11-PE1 more included in SRSF2Mut; iii) and TRA2α-PE more included in U2AF1Mut.
Overall, SR-PE splicing is disrupted in tumors vs. normal tissues, and distinct PEs are included or skipped across tumor types. A better understanding of how PEs are regulated is needed for the development of therapeutic strategies targeting SFs.
SR proteins bind PEs in other members of the SR protein family and influence their splicing
We investigated to what extent and how SR proteins use PE or 3’UTR sequences to cross-regulate levels of other SR proteins. To determine how changes in the levels of a given SR protein impact AS of other PEs, we analyzed AS events in ENCODE RNA-seq data from HepG2 and K562 cell lines with knockdown (KD) of ~230 RNA binding proteins (RBPs) (Van Nostrand et al., 2020), including 7 SR proteins (Figure 2A and S2A,B). First, inclusion of each SR-PE increases following KD of UPF1, UPF2, or XRN2, three components of the NMD pathway (Figure S2C), consistent with reports that inclusion of these PEs elicits NMD (Lareau et al., 2007; Ni et al., 2007). Second, our analysis detects the autoregulation of SRSF3 and SRSF7 in HepG2 or K562 cells (Figure 2A), as in other cell types (Jumaa and Nielsen, 1997; Pervouchine et al., 2019). Importantly, KD of individual SR proteins alters AS of not just their own PEs but also those of other SR proteins (Figure 2A). For example, SRSF1 is a positive regulator of SRSF2-, SRSF3-, SRSF6-, SRSF7-, and TRA2β-PE inclusion, and a negative regulator of SRSF4- and SRSF11-PE inclusion. The strongest changes in PE inclusion are detected for SRSF3-KD which increases TRA2β-PE inclusion by 38%. Additionally, SRSF6-PE inclusion is positively regulated by SRSF4 and SRSF5 in HepG2 or K562, SR proteins closely related to SRSF6 (Lareau and Brenner, 2015).
Figure 2. Splicing of SR-PEs is cross-regulated in a coordinated fashion.

(A) SR-PE inclusion in HepG2 or K562 cells with SR protein KD, normalized as ΔPSI relative to KD control (n=2; P<0.05). (B) Schematic of the splicing reporter minigene for cassette PEs and resulting processed isoforms. (C-F) PE splicing and protein levels from SRSF3-PE (C), TRA2β-PE (D), SRSF7-PE (E), or TRA2α-PE (F) minigenes co-transfected with HA-SR-CDS in HEK293 cells. PE inclusion is measured by RT-PCR with minigene-specific primers that amplify included and skipped isoforms, normalized as ΔPSI relative to empty vector control (CTL) (n=3, mean±SD; *P<0.05, **P<0.01). Protein level is quantified by western blot using a minigene-specific Myc-tag antibody and tubulin loading control, normalized as Log2 fold change (FC) to empty vector (n=3, mean±SD; *P<0.05, **P<0.01). (G) Heatmap representation of PE inclusion and protein levels for 15 SR-PE minigenes co-transfected with 14 HA-SR-CDS quantified as in (C-F) (n=3, mean±SD). (H-I) Clustering of SR proteins based on coding sequence similarity (H) or PE inclusion (I). See also Figures S2-S5 and Tables S2-3.
To define whether changes in AS are due to direct or indirect effects of SR protein binding to the pre-mRNA, we examined SR-PE sequences for evidence of SR protein binding. First, we compiled RNA binding motifs previously identified in vitro and in vivo (Table S2A) and mapped them to exonic and intronic sequences. Second, to complement this analysis which does not evaluate binding site occupation, we examined available crosslinking and immunoprecipitation (CLIP-seq) datasets for experimental evidence of SF binding. PE sequences and surrounding introns contain both binding motifs and CLIP binding peaks for multiple SR proteins (Figure S3A-C). For example, TRA2β-PE flanking introns contain SRSF3 binding motifs, supporting the increased TRA2β-PE inclusion upon SRSF3-KD.
Finally, AS of SR-PEs can be regulated by non-SR RBPs, including heterogeneous nuclear ribonucleoproteins (HNRNPs) (Figure S2D-G and Table S2), suggesting a large cross-regulatory network that controls SF-levels. Binding motifs for several other RBPs, including HNRNPC, MBLN1, PTBP1, or TIA, are found in intronic sequences surrounding SR-PEs (Table S2). Twenty other RBPs can both impact SR-PE inclusion and directly bind SR-PE sequences as evidenced by CLIP-binding peaks in either HepG2 or K562 (Figure S2E-G).
In sum, SR proteins directly interact with PEs of other SR proteins, suggesting widespread cross-regulation of each other’s expression through AS-NMD. However, PE-containing isoforms are degraded and difficult to quantify accurately without NMD inhibitors (Lareau et al., 2007). Thus, the ENCODE data analysis likely underestimates changes in PE splicing. Additionally, the incomplete ENCODE binding and SR protein KD data limits the ability to comprehensively characterize how the SR family interacts as a broad network, prompting us to design targeted experiments to characterize SR-PE cross-regulation.
SR-PEs form a cross-regulatory network that coordinates the levels of related family members
To experimentally define a network of SR protein cross-regulation, we constructed PE splicing reporter minigenes, that contain: i) the PE along with upstream and downstream intron and exon sequences; ii) a Myc protein tag; and iii) a DsRed protein for detection in live cells. All minigenes create truncated proteins that do not feedback on their own transcripts, thus improving sensitivity of detecting PE splicing changes without NMD inhibition. Additionally, the Myc-tag enables quantification of SR proteins for which reliable antibodies are not available. For SR proteins containing a cassette PE, exon skipping leads to a transcript that encodes a Myc-SR-dsRed fusion protein, while inclusion introduces a PTC leading to transcript degradation and no protein expression (Figure 2B). For SR proteins that undergo splicing of their 3’UTRs, the minigene contains the Myc-tag followed by a dsRed sequence and the 3’UTR of the SR protein transcript (Figure S4A). Splicing of the 3’UTR sequence creates a splice junction downstream of the canonical stop codon that leads to transcript degradation, while an unspliced UTR sequence encodes a stable transcript that produces Myc-dsRed protein. We did not detect any smaller Myc-tag fusion proteins corresponding to translation of the PTC-containing transcript (Figure S4B).
To validate that the minigenes recapitulate endogenous AS profiles and determine how SR proteins influence AS of their own PEs, we co-transfected PE minigenes with plasmids containing hemagglutinin (HA)-tagged coding sequences (CDS) of their respective SR protein (HA-SR-CDS) or an empty vector. All HA-SR-CDS proteins were primarily localized in the nucleus as previously described (Caceres et al., 1997), consistent with their role in AS (Figure S4C). The minigenes recapitulate established examples of SR protein autoregulation at the AS level for SRSF1, SRSF2, SRSF3, SRSF5, SRSF7, or TRA2β (Jumaa and Nielsen, 1997; Pervouchine et al., 2019; Sun et al., 2010; Sureau et al., 2001; Yang et al., 2018), and demonstrate an effect at the protein level (Figure S4D,E). We also uncovered autoregulatory events in each of the remaining 7 SR proteins (Figure S4D,E), except SRSF11, thus demonstrating this regulation is conserved across the whole family.
To determine how SR proteins cross-regulate AS of PEs in other SR proteins, we co-transfected each of the 15 reporter minigenes along with each of the 14 HA-SR-CDS and measured changes in PE splicing and protein levels (Figures 2C-G and S5). We uncover a well-orchestrated network of SR protein cross-regulation. For example, SRSF3-PE inclusion is positively regulated by SRSF3 and SRSF8, whereas all other SR proteins promote SRSF3-PE skipping and increase protein levels (Figure 2C). We also validate know positive regulators of TRA2β-PE inclusion TRA2α and TRA2β (Best et al., 2014). Interestingly, almost all other SR proteins cause TRA2β-PE skipping and increase protein levels by >2-fold (Figure 2D). Notably for all minigenes, small changes in PSI (∣ΔPSI∣ <10%) lead to >2-fold changes in protein levels (Figure 2C-G). For all SR-PEs, except SRSF4 and SRSF11 which contain multiple PEs, PE splicing is significantly negatively correlated with protein levels (Figure S5B and Table S3). Hierarchical clustering based on ΔPSI reveals that SR protein cross-regulation follows coding sequence evolutionary similarity (Figure 2H,I). Closely related SR proteins (e.g., SRSF4, SRSF5, SRSF6) induce PE inclusion in each other, decreasing protein levels; while those that are less related (e.g., SRSF1, SRSF3, and TRA2β) promote PE skipping, increasing protein levels. SR proteins could not be clustered efficiently by their motifs (Figure S5C), likely because their binding motifs are similar and short (Dominguez et al., 2018).
SR proteins compete or cooperate to regulate AS of TRA2β-PE in a dose-dependent manner
We next sought to uncover how SR proteins interact together to alter AS of a target PE, focusing on TRA2β, an oncogene overexpressed in multiple cancer types (Best et al., 2013; Ji et al., 2014; Park et al., 2019). The TRA2β-PE minigene was co-transfected into HeLa cells, in which the baseline inclusion of TRA2β-PE is 50%, along with various amounts and combinations of HA-SR-CDS. First, TRA2β-PE is cross-regulated in a dose-dependent manner by all other SR proteins at both the AS and protein levels (Figures 3A,B and S5D). Increased levels of a negative regulator of TRA2β-PE inclusion (e.g., SRSF1, SRSF3, SRSF4) leads to a dose-dependent protein increase, while increased levels of positive PE regulator (e.g., TRA2α, TRA2β) leads to a dose-dependent protein decrease. Next, we co-transfected two HA-SR-CDS together with the TRA2β-PE minigene, focusing on the cooperation between TRA2β-PE negative or positive regulators identified above; we selected three negative regulators (SRSF1, SRSF3, SRSF4) and one positive regulator (TRA2β) from separate SR protein subfamilies that when overexpressed led to significant changes in PSI. The effects of TRA2β, which induces inclusion of its PE, can be outcompeted by co-transfecting an SR protein that promotes PE skipping, an effect common to all tested negative regulators of TRA2β-PE inclusion (e.g., SRSF1, SRSF3, or SRSF4) (Figure 3C-E). Additionally, co-expression at similar levels of two negative regulators of TRA2β-PE inclusion, SRSF1 and SRSF3, increases protein levels by log2 5-fold, which is more than either of them alone, suggesting a synergistic effect (Figure 3F). In sum, SR protein cross-regulation is titratable and dependent on the level of individual SR proteins and their coordinated ability to regulate PE splicing.
Figure 3. SR proteins compete or cooperate in the cross-regulation of TRA2β-PE in a dose-dependent manner.

(A) PE splicing and protein levels from the TRA2β-PE minigene in HeLa cells co-transfected with increasing concentration of HA-SR-CDS. TRA2β-PE inclusion is measured by RT-PCR with minigene-specific primers that amplify included and skipped isoforms, normalized to empty vector. Protein level is quantified by western blotting using minigene-specific Myc-tag antibody and tubulin loading control, normalized to empty vector. HA-SR-CDS proteins are detected using HA-tag antibody and GAPDH loading control. (B) Heatmap representation of PE splicing and protein levels from the TRA2β-PE minigene co-transfected with increasing concentration of 14 HA-SR-CDS in HeLa cells quantified as in (A). (C-F) PE splicing and protein levels from the TRA2β-PE minigene co-transfected with varying concentration of indicated pairs of HA-SR-CDS in HeLa cells, quantified as in (A). See also Figure S5D.
UCRs contain sequences required to respond to distinct SR proteins
To further characterize the elements that control TRA2β-PE splicing, we generated mutant minigenes in which either intronic or exonic sequences were swapped between SRSF3 and TRA2β. We focused on these two SR proteins as they: i) are more evolutionary distant within the SR protein family; ii) have opposing baseline inclusion of their PEs; iii) regulate each other’s PEs in opposite ways; and iv) their PEs are both regulated by SRSF1 (Figure 2-3). We swapped either i) the PE and ~100bp of intronic 5’ and 3’ UCR; ii) the PE alone; iii) ~100bp of intronic 5’ and 3’ UCR together; or iv) ~100bp of intronic 5’ or 3’ UCR alone (Figure 4A).
Figure 4. Exonic and intronic regions of SR proteins carry PE splicing regulatory sequences.

(A) Schematic of TRA2β-PE and SRSF3-PE regions swapped in mutant minigenes, and location of UCRs. (B) Domain structure of wild-type and mutant domain swapped SRSF3 and TRA2β proteins. (C) Exonic or intronic regions of the TRA2β-PE minigene (purple) are replaced by regions from the SRSF3-PE minigene (green), along with indicated 5’ or 3’ splice site sequences (SS) (top panel). Wild-type and mutant TRA2β-PE minigenes are co-transfected in HeLa cells with wild-type or mutant HA-SR-CDS shown in B (middle panel). TRA2β-PE inclusion is quantified as PSI by RT-PCR with minigene-specific primers (bottom panel). Native SSs are maintained in intron swaps (Lane 19-36). (D) AS analysis of mutant SRSF3-PE minigenes as in (C). (E) PE splicing and protein expression from the SRSF3-PE minigene in HeLa cells co-transfected with wild-type or mutant HA-SR-CDS. PE inclusion is measured by RT-PCR with minigene specific primers, normalized to empty vector control. Protein expression (Log2FC) is quantified by western blotting using a minigene specific Myc-tag antibody and tubulin loading control, normalized to empty vector control. (F) Same as in (E) with the TRA2β-PE minigene. See also Figure S4F-H.
We compared the baseline of PE inclusion from each minigene and its response to AS regulators. First, each PE maintains its native regulation even when surrounded by heterologous intronic sequences. All tested HA-SR-CDS that promote PE inclusion or skipping in the wild type minigene also promote inclusion or skipping in the minigene with swapped introns, indicating that exonic regions are sufficient for this regulation. Specifically, SRSF1- or SRSF3-CDS leads to TRA2β-PE skipping, while TRA2β-CDS leads to inclusion (Figure 4C, first 4 lanes of each gel). SRSF3-PE also maintains its regulation in that SRSF1- or TRA2β-CDS lead to SRSF3-PE skipping, while SRSF3-CDS leads to inclusion (Figure 4D, first 4 lanes of each gel).
Interestingly, intronic sequences impact the baseline inclusion of either TRA2β-PE or SRSF3-PE. Specifically, adding SRSF3 intronic sequences to the TRA2β minigene decreases TRA2β-PE baseline inclusion (Figure 4C, lanes 1 vs. 19, 25 or 31), while surrounding SRSF3-PE with TRA2β intronic sequences increases SRSF3-PE baseline inclusion (Figure 4D, lanes 1 vs. 19, 25 or 31). Furthermore, changing only one of the two intronic regions is sufficient to guide this effect (Figure 4C,D, lanes 1-6 vs. 25-30 or 31-36), with a more pronounced effect obtained by swapping only the 5’ intronic region.
To further interrogate the role of SR protein domains, we constructed HA-SR-CDS mutants in which the RRM domains are swapped (Figure 4B), or the RRM or RS domains are deleted (Figure S4F). All mutant SR proteins are mostly localized to the nucleus (Figure S4G). Swapping the RRM between SRSF3 and TRA2β is sufficient to swap the PE regulation. Specifically, SRSF3 with the TRA2β-RRM regulates the TRA2β-PE and SRSF3-PE to the same extent as wild-type TRA2β; similarly TRA2β with the SRSF3-RRM now behaves like wild-type SRSF3 (Figure 4C,D, last 2 lanes of each gel). This suggests that AS changes are due to direct binding specificity and locations guided by these RRMs. Finally, SRSF3 AS activity is dependent on the presence of both an RRM and RS domain, as deleting either domain abrogates its activity on both PEs wild-type minigenes (Figure 4E,F). Interestingly, TRA2β deletion mutants still maintain AS activity on TRA2β-PE, with mutants lacking the RRM exhibiting the most dampened effect (Figure 4F). Deleting either of the two TRA2β RS domains alone had no effect on TRA2β-PE splicing and suggests a redundant function for these two RS domains (Figure 4F).
In sum, UCRs contain sequences important for PE regulation, with exonic sequences conferring directionality to effects on SR proteins via the location-dependent RNA-recognition of RRMs, and intronic sequences fine tuning regulatory effects.
Exonic and intronic elements differentially affect TRA2β-PE splicing
To map the sequences that influence TRA2β-PE splicing, we created mutant minigenes Δ1 to Δ30 by deleting ~25bp regions in the exon or introns, covering the UCR, and excluding 10bp around the splice sites (Figure 5A). Mutant minigenes were transfected into HeLa cells and their impact on AS and protein levels was measured by RT-PCR and western blotting. Validating our approach, deleting two sites known to be important for TRA2β autoregulation (Δ11, Δ18-19) (Stoilov et al., 2004) decreases TRA2β-PE inclusion and increases protein levels. We identify additional sequences that when deleted cause exon skipping and increase protein levels, specifically a region in the upstream intron (Δ9-10) that partially overlaps a predicted branch point (Desmet et al., 2009), and two regions in the downstream intron (Δ21-22 and Δ26-27). Furthermore, several deletions promote TRA2β-PE inclusion, including three regions in the PE (Δ12, Δ15-16 and Δ20) and one in the downstream intron (Δ23). Interestingly, deletions with the strongest effect are positioned in the UCR (Δ8–23), suggesting a functional selection that may drive the conservation of sequences involved in AS regulation.
Figure 5. TRA2β-PE inclusion is regulated by discrete exonic and intronic UCR sequences.

(A) Locations of deletions Δ1 to Δ30 (grey boxes) and UCR in the TRA2β-PE minigene. Wild-type or mutant TRA2β-PE minigenes are transfected into HeLa cells and TRA2β-PE inclusion is quantified by RT-PCR with minigene-specific primers and normalized as ΔPSI to wild-type minigene (n=3, mean±SD; *P<0.05, **P<0.01). Protein level is quantified by western blotting using a minigene-specific Myc-tag antibody and tubulin loading control, normalized as Log2FC to wild-type minigene (n=3, mean±SD; *P<0.05, **P<0.01). (B) Positions of gRNAs tiled across TRA2β-PE and upstream or downstream introns. HEK293T cells are transfected with the wild-type TRA2β-PE minigene along with dCasRx and gRNA. TRA2β-PE inclusion is assessed by RT-PCR with minigene specific primers and normalized to non-targeting control (CTL) gRNA. (C) Locations of the deletions from (A) and gRNAs binding positions from (B) along with their effects on TRA2β-PE inclusion, and RBP motifs positions in two regions of interest. See also Figures S2F-G and S6A-C.
To further characterize regions required for TRA2β-PE splicing, we used an RNA-targeting catalytically inactive RfxCas13d (dCasRx) (Konermann et al., 2018) to sterically block specific SF binding sites while keeping the native sequence intact. We designed a series of forty 22nt-long dCasRx guide RNAs (gRNAs) that target sequences in TRA2β-PE and 100nt of surrounding introns and tiled with an 11nt overlap (Figure 5B). Most of the exon-targeting gRNAs promoted dCasRx-mediated PE inclusion compared to non-targeting control gRNA, suggesting these exonic sequences are bound by SFs that prevent exon inclusion. Additionally, three gRNAs induced exon skipping, including i1-7 that targets the branch point near the 5’ splice site, and e2-3 that overlaps a known TRA2β-PE autoregulatory site (Stoilov et al., 2004).
Overall, several genomic regions exhibit similar effects on TRA2β-PE splicing when deleted or when targeted with dCasRx (Figure 5C). First, two adjacent deletions, Δ11 and Δ12, that have opposing effects on TRA2β-PE inclusion, are mirrored by adjacent gRNAs, e2-3 and e2-4, that target similar sequences. These two deletion mutants, Δ11 and Δ12, differentially disrupt local RNA secondary structures when removed, but do not have a large impact on the overall secondary structure of TRA2β-PE and surrounding introns (Figure S6A-C). Therefore, in addition to their effect on predicted RBP motifs and binding sites, these differences in local secondary structure could affect the accessibility and binding of an RBP (Dominguez et al., 2018; Taliaferro et al., 2016). Second, Δ15-16 and overlapping e2-11 through −15 all promote exon inclusion. These regions contain binding motifs and peaks for additional RBPs (Figure S2F,G) that may be involved in regulation of TRA2β-PE and should be investigated in the future.
SR proteins binding location differentially influences TRA2β-PE splicing
Given the redundant function of RS domains on AS regulation (Graveley and Maniatis, 1998; Shin et al., 2005) and our own experiments demonstrating that swapping of RRMs between SRSF3 and TRA2β mirror the effects of the wild-type protein, we hypothesized that their opposing regulation of TRA2β-PE splicing is dependent on binding location rather than differences in RS domain activity. We developed CRISPR Artificial Splicing Factors (CASFx) (Du et al., 2020) for each SR protein (CASFx-SR) by fusing the RS domain, which confers splicing activity, to a dCasRx protein that replaces the RRM and carries the RNA binding activity (Figure 6A,B). All CASFx-SR proteins are nuclear localized with an SV40 nuclear localization signal (Figure S6D,G).
Figure 6. CASFx-SRs reveal location preferences for RS domain activity.

(A,B) Domain structure of CASFx-SR compared to SR protein (A), and gRNA-guided AS modulation principle (B). (C) TRA2β-PE minigene along with CASFx-SRs and TRA2β targeting gRNAs are co-transfected in HEK293T cells. Heatmap summary of TRA2β-PE inclusion measured by RT-PCR with minigene specific primers and normalized to non-targeting CTL gRNA. (D) PE inclusion in TRA2β endogenous transcript in HEK293T cells transfected with CASFx-SRs and gRNAs measured by RT-PCR using gene-specific primers that amplify included and skipped isoforms (n=3, mean±SD; *P<0.05, **P<0.01). (E) A multi-gRNA CRISPR array targets both TRA2β-PE and SRSF3-PE. (F) PE inclusion in TRA2β and SRSF3 endogenous transcripts in HEK293T cells transfected with CASFx-SRSF3 and gRNAs individually targeting TRA2β-PE, SRSF3-PE, a multi-gRNA array targeting both, or CTL gRNA. TRA2β-PE and SRSF3-PE PSI is quantified by RT-PCR using gene specific primers that amplify included and skipped isoforms (n=3, mean±SD; *P<0.05, **P<0.01). See also Figure S6D-M.
To define their positional effects, CASFx-SR are targeted to specific locations along the TRA2β UCR (Figure 6B) using the forty 22nt gRNAs from the dCasRx experiment. We first focus on positional effects of CASFx-SRSF1, -SRSF3, -SRSF4, and -TRA2β as these SFs show strong effects on TRA2β-PE AS in our previous experiments. CASFx-SR and gRNA-encoding plasmids are transfected along with the TRA2β-PE minigene in HEK293T cells and inclusion of TRA2β-PE is measured by RT-PCR (Figure 6C and S6F). First, the RS domain from CASFx-SR exhibits AS activity that is not solely due to dCasRx blocking a SF binding site, as shown by opposite effects of CASFx-SR vs. dCasRx with the same gRNA. For example, e2-3 induces exon skipping with dCasRx (Figure 5B), while inducing inclusion with CASFx-SRSF3 (Figure 6C). Second, we compare TRA2β-PE inclusion for each of the 40 gRNAs with either CASFx-SRSF1, -SRSF3, -SRSF4, or -TRA2β, and identify a location preference for some RS domains. For example, CASFx-SRSF4 has its strongest effect when localized with e2-7 or e2-11, whereas CASFx-SRSF1 and CASFx-TRA2β induce their strongest inclusion with e2-13. CASFx-TRA2β has opposing effects when positioned only 11nt apart with e2-12 and e2-13 (Figure 6C). Furthermore, CASFx-TRA2β mostly induces exon inclusion when targeted to the exon center but promotes skipping when targeted to either of the surrounding introns. Finally, we compare the AS efficiency of 9 other CASFx-SR with e2-12 and e2-13 (Figure S6G). Interestingly, apart from CASFx-SRSF11, all SR domains promote exon inclusion (ΔPSI>5) with e2-13 while only some can promote inclusion with e2-12. Together these results suggest both redundancy and functional independence of SR protein RS domains.
We then use CASFx-SR to target AS of the endogenous TRA2β transcript. We examined endogenous TRA2β-PE splicing in HEK293T cells co-transfected with CASFx-SRSF1, -SRSF3, -SRSF4 or TRA2β along with selected gRNAs that promote exon inclusion in the TRA2β-PE minigene for each respective CASFx-SR. Targeting CASFx-SRSF1, -SRSF3 or - TRA2β to the center of the TRA2β-PE with e2-13 has the strongest effect on exon inclusion (Figure 6D), similar to the patterns detected in the minigene. We demonstrate that this method can manipulate AS of two additional SP-PEs by designing gRNAs targeting SRSF3-PE and SRSF4-PE2. Both CASFx-SRSF1 and -SRSF3 promote SRSF3-PE inclusion with e4-2 and e4-3 which are designed to target predicted SRSF3 and TRA2β motifs in contrast to dCasRx which only promoted inclusion with e4-3 and to a lesser extent (Figure S6J,L). Additionally, we identified gRNAs that promote either skipping or inclusion of endogenous SRSF4-PE2 with either CASFx-SRSF1 and -SRSF3 (Figure S6K,M), although their effect is weaker than for SRSF3-PE and more similar to that of dCasRx alone.
Lastly, to target multiple endogenous PEs simultaneously, we created a Cas13/CRISPR multi-guide array (mgRNA) consisting of one gRNA targeting TRA2β-PE (e2-13) and one targeting SRSF3-PE (e4-3) (Figure 6E). CASFx-SRSF3 targeted with the multi-guide array increases both TRA2β-PE and SRSF3-PE inclusion simultaneously (Figure 6F), to similar levels as either of the gRNAs alone, demonstrating the utility of this system to target multiple AS events. Interestingly, the SRSF3 targeting e4-3 gRNA alone only increases SRSF3-PE inclusion, whereas the TRA2β targeting e2-13 gRNA alone increases inclusion of both TRA2β-PE and SRSF3-PE. Thus, altering TRA2β-PE inclusion could indirectly affect SRSF3-PE, supporting the PE-mediated cross-regulatory feedback between SRSF3 and TRA2β described above.
ASO-mediated TRA2β-PE inclusion increases cell death and decreases proliferation in breast cancer models
Given the decreased inclusion of TRA2β-PE in multiple tumor types including breast, we reasoned that forcing its inclusion could decrease TRA2β protein levels in cancer cells. We tested this approach using splice-switching antisense oligonucleotides (ASOs), a class of FDA-approved RNA-based therapeutics that bind to reverse complementary regions on target pre-mRNA and alter AS by blocking regulatory sequences (Figure 7A) (Lieberman, 2018).
Figure 7. ASO-mediated TRA2β-PE inclusion alters breast cancer cell viability, proliferation, and migration.

(A) Principle of ASOs blocking intronic silencer sequences (ISS) to promote TRA2β-PE inclusion. (B) TRA2β-PE splicing in MDA-MB231 and SUM159 cells transfected with TRA2β-targeting (1570) or non-targeting control (CTL) 2’MOE ASOs. TRA2β-PE inclusion and protein levels are measured by RT-PCR and western blot (n=3, mean±SD; *P<0.05, **P<0.01). (C) Cell proliferation in MDA-MB231 and SUM159 cells transfected with 2’MOE ASO-1570 vs. −CTL quantified as percent 5-ethynyl-2´-deoxyuridine (EdU)+ to total Hoechst+ cells (n=3, mean±SD; *P<0.05, **P<0.01). Representative stains are shown. (D) Cell death in MDA-MB231 and SUM159 cells transfected with 2’MOE ASO-1570 vs. −CTL quantified as percent AnnexinV+ to total Hoechst+ cells normalized to ASO-CTL (n=3, mean±SD; *P<0.05, **P<0.01). Representative stains are shown. (E) AS of known TRA2β targets measured by RT-PCR in MDA-MB231 and SUM159 cells transfected with 2’MOE ASO-1570 vs. −CTL (n=3, mean±SD; *P<0.05, **P<0.01). (F,G) Same as in (B,D) for MCF-10A and AR7 human mammary epithelial cells (HMEC) non-transformed mammary epithelial cells. See also Figure S7.
We combined our prior results identifying regulatory regions for TRA2β-PE splicing with a computational method that incorporates RNA secondary structure and SF binding motifs (Tabaglio et al., 2018) to design six ASOs targeting the TRA2β UCR (Figure S7A). We examined endogenous TRA2β-PE splicing and TRA2β protein levels in MDA-MB231 or HS578T breast cancer cell lines transfected with TRA2β-targeting or non-targeting control 2’-O-methyl phosphorothioated (2’OMe) ASOs. Two ASOs decrease TRA2β-PE inclusion and increase protein levels (ASO-1565 and −1567), and two increase exon inclusion and decrease protein levels (ASO-1566 and −1570) (Figure S7B,C).
ASO-1570, which has the strongest effect on TRA2β-PE inclusion (Figure S7B,C), targets a downstream intronic region containing predicted splice silencer motifs and hnRNPA1 binding sites which overlap a region deleted in the minigene Δ23 that enhanced TRA2β-PE inclusion (Figure S7A), suggesting these sites are functionally important for regulating TRA2β-PE inclusion. Overexpression of hnRNPA1-CDS along with the TRA2β-PE minigene decreases PE inclusion and increases TRA2β protein levels (Figure S7D). Finally, co-transfection of an increasing amount of hnRNPA1-CDS along with ASO-1570 can off-compete the ASO splice-switching effects and increase TRA2β protein levels (Figure S7E), suggesting that ASO-1570 functions, at least in part, by blocking hnRNPA1 intronic silencer motifs.
We further assay how ASO-1570-mediated inclusion of TRA2β-PE affects cancer-relevant phenotypes in vitro using 2’-O-methoxyethyl phosphorothioated (2’MOE) chemistry, which enhances ASO stability while decreasing off-target effects and toxicity observed with 2’OMe chemistry (Scharner et al., 2020). The 2’MOE ASO-1570 increases TRA2β-PE inclusion and decreases TRA2β protein levels by ~30-50% when transfected into MDA-MB231, SUM159, or HS578T cells (Figure 7B and S7F). Breast cancer cells treated with 2’MOE ASO-1570 exhibit decreased cell proliferation (Figure 7C) and increased cell death (Figure 7D) compared to ASO-CTL. The strongest effects are detected in MDA-MB231 cells which also exhibit the largest splicing switch and TRA2β protein KD. ASO-1570-treated HS578T cells display increased cell death, but no changes in cell proliferation (Figure S7G,H), possibly because of their slower growth rate. We then assayed the effect of 2’MOE ASO-1570 on SUM159 and HS578T cell migration in wound healing assays, excluding MDA-MB231 in which cell death would be a confounding factor. ASO-1570 decreases wound closure in both SUM159 and HS578T cells compared to ASO-CTL, which likely reflects a reduction in the migratory and proliferative potential of these cancer cells (Figure S7I). Interestingly, even a small change in TRA2β-PE inclusion ΔPSI<10%) in SUM159 or HS578T cells results in ~30% decrease of TRA2β protein levels and impacts both cell death and migration (Figure 7D and S7H,I).
Next, we investigate whether 2’MOE ASO-1570 exhibits on-target effects by examining the AS of TRA2β target transcripts. We quantified exon inclusion in a set of six known TRA2β splicing targets, previously identified in MDA-MB231 and SUM159 cells with short-hairpin RNA (shRNA)-mediated TRA2β protein KD (Park et al., 2019). MDA-MB231 cells treated with 2’MOE ASO-1570 exhibit increased exon inclusion in GOLGA5 and PFKM, and increased exon skipping in AZIN1, IFI44, KIF23, and TRA2α-PE compared to ASO-CTL (Figure 7E), validating 6 out of 6 TRA2β-regulated splicing events in this cell line. Similarly, we validate 5 out of 6 of these events in SUM159 and HS578T (Figure 7E and S7J). Neither TRA2β-targeting shRNA or ASO-1570 alter PFKM splicing in these cells (Park et al., 2019).
Finally, we determine the effect of ASO-1570 on two non-transformed mammary epithelial cell lines. Similar to its effects in breast cancer cells, 2’MOE ASO-1570 increases TRA2β-PE inclusion and decreases TRA2β protein levels in both MCF-10A and AR7-HMEC cells (Figure 7F). However, 2’MOE ASO-1570-treated non-transformed cells do not exhibit increased cell death (Figure 7G) or decreased cell migration (Figure S7K). We then co-cultured GFP-tagged MDA-MB231 cells together with MCF-10A cells and simultaneously transfected them with ASO-1570 or −CTL. The proportion of GFP-tagged MDA-MB231 to total cells is measured by fluorescent imaging at different timepoints after transfection. Starting at day 5, the percent of MDA-MB231 cells decreases in the ASO-1570 vs. −CTL treated populations (Figure S7L), confirming that breast cancer cells are more sensitive to changes in TRA2β levels compared to non-transformed mammary epithelial cells.
Altogether, these data suggest that targeting PE splicing events in oncogenic SFs could represent a viable strategy to decrease SF levels and limit the survival of SF-dependent cancer cells while leaving a therapeutic window limiting effects on normal cells.
DISCUSSION
We experimentally validate an extensive cross-regulatory network, which through AS-NMD of PEs, controls expression of all 14 SR protein family members. Our data directly link changes in SR-PE inclusion with changes in SR protein levels and reveal that small changes in splicing can lead to >2-fold changes in protein. The degree of cross-regulation between any two SR proteins correlates with their sequence similarity, suggesting that PEs evolved to coordinately control the levels of closely related SR proteins, possibly to appropriately dose spliced isoforms regulated by each family member. While we focus on SR proteins, our analysis reveals that SR-PEs are also bound and regulated by other RBPs, including members of the HNRNP family, QKI, and RBFOX2, a SF that has been shown to regulate AS of ~44 RBPs (Jangi et al., 2014). We demonstrate that HNRNPA1 can regulate AS of TRA2β-PE, highlighting that the regulation of SR and HNRNP protein families is likely to be coordinated. While our study represents the largest experimentally validated RBP cross-regulatory network to date, further studies are needed to explore the cross-regulation across the >300 RBPs implicated in AS.
PEs regulate SF levels across multiple cell differentiation models, suggesting a post-transcriptional regulatory program that decreases SR protein levels as the tissue differentiates. Our findings also implicate PE splicing in controlling changes in SR protein levels previously described during heart or brain development (Grellscheid et al., 2011; Ortiz-Sanchez et al., 2019; Roberts et al., 2014; Storbeck et al., 2014). While we are unable to detect SRSF9-PE splicing in iPSC, the low levels of PE-containing transcript limits our ability to detect differences in its inclusion and mask its role in cell differentiation. Additionally, SRSF9-PE could play a role in differentiation in other cell types besides the ones examined, as changes in SRSF9 protein influence skeletal muscle differentiation (Bjorkman et al., 2019). Further elucidating the function of PEs in coordinating RBP levels during development is needed to advance our understanding of developmental disorders associated with RBP alterations.
Our findings suggest differences in baseline PE inclusion between cell types, and in how PEs respond to SR protein levels. PE inclusion varies between iPSC differentiation models; for example, SRSF11-PE2 is included in all analyzed differentiated cell types except in neuronal cells. Similarly, HepG2 and K562 cells display differences in baseline PE inclusion, and differences in their AS patterns in cells with SF-KD. Additionally, while SRSF10 induces TRA2β-PE skipping in HEK293 cells, it promotes inclusion in HeLa cells. These differences are likely due to variation in RBP levels between cell types. While the majority of RBPs are ubiquitously expressed in normal tissues (Gerstberger et al., 2014), their levels and activity vary between cell types, states and in response to external stimuli (Preussner et al., 2017). In cancer cells, many RBPs are differentially expressed (Galante et al., 2009; Kechavarzi and Janga, 2014) and could, in addition to SR proteins, impact PE splicing. While this cross-regulatory network likely expands beyond the SR protein family and represents a general mechanism that fine-tunes RBP levels, its universality should be further investigated experimentally across diverse cell types.
PE sequences correspond to UCRs, exhibiting greater sequence identity across species than nearby coding exons, and are hypothesized to contain critical binding regions for regulatory RBPs (Lareau et al., 2007; Ni et al., 2007). We demonstrate that UCRs of either SRSF3 or TRA2β contain sequences required to respond to their SR protein regulators. Furthermore, cross-regulation of SRSF3-PE and TRA2β-PE by SRSF3 and TRA2β proteins is likely mediated by direct interaction with the pre-mRNA, as exchanging the RRMs of these two SR proteins switches their regulation of each other’s PEs. Interestingly, the TRA2β-RRM only mutant localized both to the nucleus and peri-nuclear cytoplasmic inclusions. Specific RBPs play a role in stress granule formation (Markmiller et al., 2018) or trafficking of NMD transcripts to the endoplasmic reticulum (Longman et al., 2020), yet whether TRA2β plays a similar role or functions in the cytoplasm remains unknown. Additionally, our study demonstrates that TRA2β influences AS of its own PE both with and without direct binding from its RRM domain, which may provide additional regulatory activity in addition to binding specificity.
We identify sequences in the TRA2β-PE and flanking introns that regulate exon inclusion by combining deletion mutagenesis and an RNA-targeting dCasRx gRNA screen that does not alter the sequence of the transcript. Remarkably, these two approaches identify overlapping regulatory sequences and reveal that sequences with the strongest AS effect are located in the UCR, suggesting a regulatory role for their conservation. We cannot, however, discount the role of secondary structure in our analysis as our TRA2β splicing minigene contains truncated introns. Further analysis of these sequences in the endogenous context is warranted to identify regulatory intronic sequences located more than 250bp from the PE.
We demonstrate that RNA-targeting CRISPR screens are useful to explore the relationship between binding location of SR proteins and their AS activity. The CASFx-SR system reveals that the RS domain binding location influences its AS activity, as previously described for a few SR proteins (Caceres et al., 1997; Chandler et al., 1997; Graveley et al., 1998; van Der Houven Van Oordt et al., 2000). Our comparative analysis of four SR proteins reveals that not all SR domains are equally active at the same position. A CASFx-SR protein can alter exon AS when tethered to a specific location while other CASFx-SRs exhibit no effect or sometimes opposite effects with the same gRNA. Finally, CASFx can alter AS of endogenous transcripts and provides a powerful tool to interrogate AS regulatory elements in their native context.
Human cancers that express high SR protein levels are often associated with poor prognosis (Park et al., 2019). Our analysis reveals that SR-PEs are differentially spliced in tumor samples compared to adjacent normal biopsies, suggesting AS changes could underlie the overexpression or downregulation of SR proteins in cancer. For example, TRA2β is an oncogene in breast, lung, and prostate cancers (Urbanski et al., 2018), and its PE is skipped in these tumor types. Other tumor types exhibit increased inclusion of SR-PEs, potentially suggesting tissue-specific differences in AS-NMD regulation of SR protein levels. We focused on SR proteins, but AS of PEs in other SFs, such as SMNDC1 (Thomas et al., 2020), is likely a common mechanism for post-transcriptional alterations in RBPs in tumors. Tumors exhibit differences in NMD efficiency, likely due to alterations in core NMD factors (Popp and Maquat, 2018). Our analysis therefore would overestimate differences in exon inclusion when NMD efficiency is high, and underestimate exon inclusion when it NMD efficiency is low. Additionally, mRNAs containing PTCs have recently been shown to trigger genomic compensation responses in related genes (El-Brolosy et al., 2019; Ma et al., 2019). Further work is required to fully elucidate how this complex interplay between NMD and genetic compensation influences SR protein levels in cancer.
A recent study demonstrates the tumor-suppressive potential of PEs through removal of genomic sequences in oncogenic SF genes (Thomas et al., 2020). Our study extends these findings by targeting the TRA2β-PE directly in the pre-mRNA to demonstrate its role as a tumor suppressor, overcoming potential off-target effects of CRISPR editing on a genomic locus, including changes in chromatin, genome architecture or long-range interactions. Our use of ASOs unequivocally ascribes this cancer cell growth phenotype to PE splicing, and suggests a therapeutic window for ASO-mediated cell death in breast cancer but not in non-transformed mammary cells. PEs are promising targets for future therapies targeting SF alterations in disease, and this strategy can be applied to PEs in other genes, including other RBPs (Inoue et al., 2019; Guo et al., 2018; Parra et al., 2020; Sun et al., 2019). A better understanding of how SF-levels are regulated during development and diseases is needed for the success of any future therapies targeting the splicing machinery to treat complex genetic diseases or cancer.
LIMITATIONS
We cannot exclude that some of the AS effects observed when expressing SR-CDS are due, at least in part, to indirect changes in endogenous levels of other RBPs. While we observe some differences between cell lines, we also find strong interactions that persist across models and highlight the conservation of SR-PE cross-regulation. Across HepG2, HEK293, and K562 cells, SRSF6-PE inclusion is regulated by SRSF4 and SRSF5; and SRSF4 promotes TRA2α-PE inclusion. SRSF2-3UTR is regulated by SRSF3, SRSF4, SRSF7, and TRA2α in the same way in at least two tested cell lines.
While our study focuses on the role of SR proteins in AS of their PEs, we do not rule out their role in other RNA processing steps that may influence their cross-regulation such as mRNA export, stability, and translation (Caceres et al., 1998; Huang and Steitz, 2001; Maslon et al., 2014; Muller-McNicoll et al., 2016; Sanford et al., 2004; Zhang and Krainer, 2004). Indeed, protein levels from our minigene reporter did not always strongly correlate with the magnitude of AS changes. For example, SRSF1 or SRSF3 increase TRA2β protein to similar levels, although SRSF3 mediates this effect by promoting a stronger increase in exon skipping compared to SRSF1. This could be due to the role of SRSF1 in stabilizing its target transcripts and promoting their translation (Maslon et al., 2014; Sanford et al., 2004).
STAR+METHODS
Resource availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Olga Anczukow (olga.anczukow@jax.org).
Materials Availability
Plasmid and cell lines generated are available from the Lead Contact without restrictions with reasonable compensation by requestor for its processing and shipping.
Data and Code Availability
The RNA-seq data from iPSC to cardiomyocyte differentiation generated during this study is available from GEO (GSE158578).
The original/source data for Figures 2-7 and Figures S1-7 in the paper is available from Mendeley https://data.mendeley.com/datasets/smf5hf7t68/1.
Experimental model and subject details
Human cell lines
HEK293, HEK293T, and HeLa cell lines were obtained from Adrian Krainer (CSHL) maintained in DMEM (Gibco) supplemented with 10% FBS, 1% penicillin streptomycin (Sigma). MDA-MB231 shRNA-TRA2β (Park et al., 2019), SUM159 (a gift from Steve Ethier, MUSC), and HS578T (a gift from Adrian Krainer, CSHL) cells were maintained in DMEM (Gibco) supplemented with 20% FBS, 1% penicillin streptomycin (Sigma). MCF-10A (ATCC) were maintained in DMEM/F12 supplemented with 5% horse serum (Gibco), 100 ug/mL epidermal growth factor, 1 mg/mL hydrocortisone, 1mg/mL cholera toxin, 10mg/mL insulin, and 1% penicillin streptomycin (Sigma). HMEC AR-7 (a gift from Shailja Pathania, UMB) were grown in mammary epithelial cell basal medium (PromoCell) supplemented with 0.4% bovine pituitary extract, 10ng/mL epidermal growth factor, 5ug/mL insulin, 0.5ug/mL hydrocortisone All cell lines were grown at 37°C under a humidified atmosphere with 5% CO2. Cells were routinely tested negative for mycoplasma using the MycoAlert™ Mycoplasma Detection Kit (Lonza), and cell aliquots from early passages were used. All cell lines used here where established from female subjects.
iPSC culture
The human iPSC PGP1 cell line obtained from Coriell Institute (GM23338) was maintained on Matrigel (Corning)-coated tissue culture plates in mTeSR1 media (Stemcell) as described (Cohn et al., 2019; Hinson et al., 2017; Hinson et al., 2015). Media was replenished daily, and cells were passaged at a 1:6 ratio using Accutase (BD) and 10 μM ROCK inhibitor Y-27632 (Tocris) once they reached 80-90% confluency.
Method details
Plasmids
SR-CDS plasmids (pCI-neo-HA-SR-CDS):
The coding sequence (CDS) of each SR or SR-like human protein along with an sequence encoding the HA-tag was cloned into a pCI-neo mammalian expression plasmid (Hua et al., 2007) using sequence and ligation independent cloning (SLIC) with primers listed in Table S4A. Empty vector control contains the HA-tag but no CDS sequence. SRSF1-, SRSF2-, SRSF3-, SRSF4-, SRSF5-, SRSF6-, SRSF7-, SRSF9-, SRSF10-, SRSF12-, TRA2α-, TRA2β-, or hnRNPA1-CDS were amplified from MCF-10A cDNA; SRSF8- and SRSF11-CDS were amplified from commercially available open reading frame clones (GenScript; NM_032102.3 and NM_004768.4 respectively).
SR-CDS domain mutant plasmids (pCI-neo-HA-SRmutant-CDS):
SRSF3 and TRA2β protein domain deletion mutants and domain swaps were constructed with SLIC and primers listed in Table S4A using wild-type pCI-neo-HA-SRSF3-CDS and pCI-neo-HA-SRSF3-CDS plasmids. Amino acid position for the SRSF3 and TRA2β mutants are indicated in Figure S4F; domain sequences and locations are from Uniprot.
SR-PE minigenes (pCDNA5/FRT-Myc-SR-PE-DsRed):
SR protein splicing reporter minigenes were constructed with SLIC using a modified pcDNA5/FRT vector in which a Myc-tag and the dsRed coding sequence were introduced in the cloning site between nucleotide position 923 (immediately following the KpnI site) using primers listed in Table S4A.
For SR-PEs splicing reporters the genomic sequence containing the PE along with the flanking introns and exons and the second downstream exon and intron was amplified from MCF-10A genomic DNA using the primers listed in Table S4A, and introduced between the Myc-tag and dsRed sequences. For SRSF4 which contains four PEs, two minigenes were created to model the two splicing events detected for the endogenous gene(Lareau et al., 2007): i) inclusion of exons 2, 3 and 4 together (SRSF4-PE1) which create a stop codon in exon 4; or ii) inclusion of exon 6 (SRSF4-PE2). All minigenes contained full length genomic DNA except for TRA2α-PE, TRA2β-PE, SRSF4-PE2 and SRSF12-PE, in which, due to large intron sizes, intronic sequences were truncated to 250bp of the 5’ and 3’ sequences to improve cloning and downstream transfection efficiency. Specifically, TRA2β intron 1 and 2; TRA2α intron 1 and 2; TRA2α intron 4 and 5; SRSF4 intron 1, 2 and 4; and SRSF12 intron 1, were truncated. The SRSF4-PE2, SRSF6-PE, and SRSF12-PE minigenes were synthesized as geneblocks (Genscript) and subcloned into the pcDNA5/FRT vector by restriction enzyme digestion and ligation; SRSF6-PE utilized NheI and BamHI sites, SRSF4-PE2 utilized NheI and NotI sites, and SRSF12-PE utilized HindII and NotI sites.
For SR protein 3’UTR splicing reporter minigenes, the 3’UTR sequence was amplified from MCF-10A genomic DNA using the primers listed in Table S4A and introduced directly 3’ of the dsRed sequence. For SRSF1 and SRSF8, the UTR sequence was truncated to 2046bp and 826bp, respectively to increase cloning and transfection efficiency.
All PCR products with unexpected sizes were sanger sequenced to verify the product, and only validated PCR bands that contained the PE or NMD inducing frameshift (SRSF9 and SRSF11) were used in the quantification of PSI.
TRA2β-PE deletion minigenes (pCDNA5/FRT-Myc-TRA2β-PEdeletions-DsRed):
we used PCR-based mutagenesis to construct 30 minigenes containing 20-25bp deletions, covering the full exonic sequence as well as 250bp of each flanking intron, but excluding 10bp near the splice sites to avoid impairing splicing by disrupting the splice sites. Primers (Table S4B) were design using the Agilent web-software for Quick-Change mutagenesis. Each deletion mutagenesis PCR reaction used 20 ng of the wild-type pCDNA5/FRT-Myc-TRA2β-PE-DsRed minigene, 0.8μM of each primer and 25μl of 2X Phusion Flash High-Fidelity PCR Master Mix (Thermo Fisher F548L), following manufacturer instructions. Samples were amplified for 25 cycles, and incubated at 37C overnight with 2 μl of DpnI (200 U/ul, NEB R0176).
Mutant TRA2β-PE and SRSF3-PE minigenes:
Exonic and intronic sequences were swapped between pCDNA5/FRT-Myc-TRA2β-PE-DsRed and pCDNA5/FRT-Myc-SRSF3-PE-DsRed minigenes using SLIC with primers listed in Table S4A. For the intron swaps, we swapped i) intron 1 sequences located −113bp to −10bp relative to 5’SS, excluding 10bp closest to 5’SS; and/or ii) intron 2 sequences located +10bp to +106bp relative to the 3’SS, excluding 10nt closest to the 3’SS (Figure 4A). The swapped sequences of SRSF3 are 100% conserved between human and mouse genome(Lareau et al., 2007), while TRA2β sequences in intron 1 and 2 are 90% and 100% conserved respectively. Total intron length in mutant and wild-type minigenes was kept constant.
CASFx plasmids (pMAX-CASFx-SR):
CRISPR Artificial Splicing Factors SR proteins were constructed by subcloning RS domains from the pCI-neo-HA-SR protein plasmids in place of RBFOX1 domains in the pMAX-RBFOX1N-dCasRx-C plasmid (Du et al., 2020), using SLIC or restriction enzyme digestion and ligation with primers listed in Table S4C. The dCasRx domain and RS domain are separate by a glycine-rich hinge region.
CasRx-gRNA plasmids (pCR8-gRNA):
Spacer sequences were ordered as forward and reverse DNA oligos (IDT), containing a 5’AAAC or 5’AAAA overhang sequence respectively (Table S4D). 20pmol of forward and reverse oligos were annealed in 1x annealing buffer (10mM TRIS pH8.0, 50nM NaCl, 1mM EDTA). Annealed oligos were ligated into BbsI digested pCR8-Cas13d-DR-ccdB directly 3’ of the Cas13d Direct Repeat (DR).
All vectors and inserts were verified and authenticated by Sanger sequencing (Eton Bioscience).
Transfections
Co-transfection of splicing reporter minigenes and HA-SR-CDS plasmids:
24h prior to transfections HeLa or HEK293 cells were seeded in a 12 well plate at 200,000 or 400,000 cells per well, respectively. 250ng of SR splicing reporter minigene (pCDNA5/FRT-Myc-SR-PE-DsRed) and 500ng of SR coding (pCI-neo-HA-SR-CDS) or control (pCI-neo-HA) plasmids were transfected using lipofectamine 3000 (Invitrogen) as per manufacturer’s protocol. Cells were collected 48h after transfection by lifting with 2mM EDTA in PBS and analyzed by RT-PCR and western blotting. For titration experiments, the total concentration of pCI-neo plasmids was kept constant by supplementing up to 1000ng of DNA with pCI-neo-HA control empty vector.
Co-transfections of splicing reporter minigenes and CASFx plasmids:
48h prior to transfection HEK293T cells were seeded in a 12 well plate at 100,000 cells per well. 25ng of SR splicing reporter minigene (pCDNA5/FRT-Myc-SR-PE-DsRed), 300ng of Cas effector plasmid (pMAX-CASFx-SR), and 300ng of gRNA plasmid (pCR8-gRNA) were transfected using lipofectamine 3000 (Invitrogen) as per manufacturer’s protocol. Cells were collected 72h after transfection by lifting with 2mM EDTA in PBS and analyzed by RT-PCR.
CASFx transfections to alter endogenous PE splicing:
24h prior to transfection HEK293T cells were seeded into a 12 well plate at 400,000 cells per well. 1000ng of Cas effector plasmid (pMAX-CASFx-SR) and 1000ng of gRNA plasmid (pCR8-gRNA) were transfected using lipofectamine 3000 (Invitrogen) as per manufacturer’s protocol. Cells were collected 48h after transfection by lifting with 2mM EDTA in PBS and analyzed by RT-PCR and western blotting.
ASO transfections:
24h prior to transfection, MDA-MB231, SUM159 or HS578T cells were seeded into a 12 well plate at 250,000 cells per well. Cells were transfected using lipofectamine 3000 with i) 50nM of 2’-O-methly-phosphorothioated TRA2β-targeting (TechNOA tNOA-2OM-hTRA2B-01 #1565, hTRA2B-01 #1566, hTRA2B-01 #1567, hTRA2B-02 #1568, hTRA2B-02 #1569, hTRA2B-02 #1570) or non-targeting control (TechNOA tNOA-2OM-NC) ASOs ; or ii) 50nM of 2’-O-methoxyethyl-phosphorothioated TRA2β-targeting ASO-1570 (IDT) Table S4F) or non-targeting control ASO (Sahashi et al., 2012). All ASOs were purified using RNAse free high performance liquid chromatography. Cells were collected 48h after transfection by lifting with 2mM EDTA in PBS and analyzed by RT-PCR and western blot.
RT-PCR analysis
Cells were harvested as described above and RNA was extracted using an RNAeasy kit (Qiagen) including DNAse I treatment. 500-1000ng of RNA was reverse transcribed using Superscript III reverse transcriptase (Invitrogen). Semi-quantitative PCR was used to amplify 20-200ng cDNA with Phusion hot start II DNA polymerase (Thermo Fisher) and primers listed in Table S4E,G. PCR products were separated in 1% or 2% agarose gel stained with SYBR Safe (Invitrogen) and imaged using ChemiDoc MP Imaging System (Bio-rad). PCR bands were quantified using ImageLab 6.0 software (Bio-rad) and the percent spliced in (PSI) ratio of each exon-containing transcript was calculated as the exon-included isoform band intensity divided by the intensity of included and skipped isoform bands. ΔPSI is calculated as PSIcase - PSIcontrol (HA-empty vector for minigene experiments, non-targeting gRNA for CASFx experiments, non-targeting ASO-CTL for ASO experiments).
Western blot analysis
Cells were harvested as described above and lysed in Laemmli buffer (50mM Tris-HCl pH 6.2, 5% (v/v) β-mercaptoethanol, 10% (v/v) glycerol, 3% (w/v) SDS). Cell lysate was ran on 8-16% gradient gels (Biorad), transferred onto 0.2 um nitrocellulose membranes (Millipore) and blocked in 5% (w/v) milk in Tween 20-TBST (50mM Tris pH 7.5, 150mM NaCl, 0.05% (v/v) Tween 20). Blots were incubated with anti-Myc Tag (Cell Signaling #2276), anti-HA Tag (Cell Signaling #3724), anti-β Tubulin (GenScript #A01203-40), anti-TRA2β (Abcam #ab31353), anti-GAPDH (GenScript #A01622). Anti-mouse IR-Dye 680 or IR-Dye 800 conjugated anti-mouse or rabbit IgG secondary antibodies (LICOR) were used for infrared detection with a ChemiDoc MP Imaging System (Bio-rad). Protein expression was quantified using ImageLab 6.0 software (Biorad) and normalized first to a lane loading control and then expressed as fold change (FC) or Log2FC to experimental controls.
Immunofluorescence
48 hours after transfection with 500ng of pCI-neo-HA-SR-CDS or pMAX-CASFx-SR, HeLa cells were fixed using 5% formalin in PBS, washed with IF buffer (7.6g/L NaCl, 1.896g/L Na2HPO4, 0.414g/L NaH2PO4, 0.5g/L NaN3, 1g/L BSA, 0.2% Triton X-100, 0.05% Tween-20, pH 7.4), permeabilized with 0.5% TritonX-100, and blocked with 10% goat serum (Sigma). HA primary antibody (Cell signaling #3724) was used at 1:500 dilution and Alexa-fluor-568 conjugated secondary antibody (Invitrogen) was used at 1:500 dilution. Imaging was performed on Ti Eclipse Widefield Nikon fluorescent microscope.
Cell death assays
48h after 2’MOE ASO transfections, MDA-MB231, SUM159, and HS578T cells were incubated with AnnexinV-Alexa647 (Invitrogen; 1:100) and Hoechst (Life Technologies; 5ng/mL) in 1x AnnexinV binding buffer (Invitrogen; 10mM HEPES, 140mM NaCl, 2.5mM CaCl2, pH 7.4) for 15 minutes at 37°C. Pre-warmed media was added to wells and plates were imaged with using a Phenix high content confocal imaging system (Perkin Elmer). Four fields of view per replicate were captured using a 10x objective. AnnexinV+ and total Hoescht+ cells were counted using the Columbus analysis software (Perkin Elmer). Data is represented as fold change in AnnexinV+/Total cells between targeting and control ASO treated samples.
Cell proliferation assays
24h prior to transfection with 2’MOE ASOs, MDA-MB231, SUM159 or HS578T cells were seeded into a 48 well plate at 50,000 cells per well. 48h after 2’MOE ASO transfections, MDA-MB231, SUM159, or HS578T cells were incubated with 10μM EdU for 24h. EdU labelling was detected using the Click-iT cell proliferation kit (Thermo Fisher C10340). Briefly, cells were fixed in 5% formalin then permeabilized using 0.5% tritonX-100. EdU was detected with an alexa-647 azide, and total cell DNA was stained with Hoescht 33342 5μg/mL). Plates were imaged with using a Phenix high content confocal imaging system (Perkin Elmer). 25 fields of view per replicate were captured using a 10x objective. EdU+ and total Hoescht+ cells were counted using the Columbus analysis software (Perkin Elmer). Data is represented as percentage of EdU+ cells for both ASO-1570 and the control ASO.
Wound Closure Assays
24h prior to transfection with 2’MOE ASOs, SUM159 and HS578T cells were seeded into a 48 well plate at 50,000 cells per well. 48h after transfection the cell monolayer was scratched with a P200 tip, then washed with PBS and refreshed with culture media. Plates were imaged with brightfield using a Phenix high content confocal imaging system and a 10x objective (Perkin Elmer) at 0h, 12h, 24h, and 48h after the initial scratch. To obtain a representative image of the scratch, four fields of view were stitched together using a 10% overlap on the Phenix Harmony system and exported for analysis in ImageJ (https://imagej.nih.gov/ij/). Wound closure was measured using the MRI wound healing tool plugin for ImageJ (https://github.com/MontpellierRessourcesImagerie/imagej_macros_and_scripts/wiki/Wound-Healing-Tool). All measurements were normalized to the initial scratch area (0h measurement) for each replicate to generate a percent wound closure.
Co-culture assay
24h prior to transfection with 2’MOE ASOs, MCF-10A and EGFP expressing MDA-MB231 cells were seeded together into a 48 well plate at 30,000 and 20,000 cells, respectively, in MCF-10A growth media. Prior to imaging, cells were stained with Hoechst (Life Technologies; 5ng/mL). Plates were imaged at 0h (day of transfection), 2d, 3d, 5d, 10d, and 15d after transfection using a Phenix high content confocal imaging system and a 10x objective (Perkin Elmer). Cell segmentation and counting on the EGFP channel was done using the Phenix Harmony system and used to quantify the number of MDA-MB231 cells in the culture. Total cell populations per well were quantified by counting Hoechst+ nuclei using the Phenix Harmony system. Proportion of MDA-MB231 cells to total cells in each well was quantified by dividing the number of EGFP+cells by total Hoechst+ cells.
iPSC cardiomyocyte differentiation
Human iPSC PGP1 cells were differentiated into cardiomyocytes through modulation of Wnt/β-catenin signaling as previously described (Cohn et al., 2019; Hinson et al., 2017; Hinson et al., 2015; Lian et al., 2012). Briefly, on day 0 of differentiation 12 μM CHIR99021 (Tocris) in RPMI 1640 (Gibco) supplemented with B27 minus insulin (Gibco) and GlutaMAX (Gibco) were added for 24 hours. On day 3, cells were treated with 5 μM IWP-4 (Tocris) for 48 hours. On Day 9, cells were maintained in RPMI with B27 supplement (Gibco). On Day 13, cardiomyocytes were enriched by metabolic selection by previously described methods (Tohyama et al., 2013). Following selection, cardiomyocytes were re-plated onto fibronectin-coated tissue culture plates (Gibco). Cardiomyocytes were replenished with RPMI-B27 every other day.
RNA-seq from iPSC to cardiomyocyte differentiation model
Total RNA was isolated from iPSCs at day-2 (mesoderm) or days 25-30 (cardiomyocytes) of differentiation using Trizol (ThermoFisher). For each sample, RNA from biological replicates (n=4) was collected and libraries were generated using the TruSeq Stranded mRNA library preparation kit (Illumina). RNA sequencing libraries were sequenced on an Illumina NextSeq 500/550 v2.5 platform (Illumina) to generate single-end reads.
RBP protein motif analysis
SR protein RNA binding motifs compiled from literature (Table S2A) (Ajiro et al., 2016; Anczukow et al., 2015; Anko et al., 2012; Cartegni et al., 2003; Clery et al., 2011; Dominguez et al., 2018; Jensen et al., 2014; Kim et al., 2015; Paradis et al., 2007; Ray et al., 2013; Samatanga et al., 2017) and from the RBPmap default list (Paz et al., 2014) were mapped to the PE sequences and surrounding 250nt of intron sequences using the RBPmap webtool (http://rbpmap.technion.ac.il/) with medium stringency and conservation filter cutoffs. The resulting motifs were visualized in the UCSC genome browser.
RNA binding motifs for other 84 RBPs (Table S2B) in the SR-PEs and surrounding intron sequences, were mapped with medium stringency and conservation filter cutoffs, and their motif density is calculated as number of motifs per 100bp.
SR protein CLIP analysis
ENCODE HepG2 eCLIP (Van Nostrand et al., 2020Van Nostrand, 2016 #397) (ENCSR989VIY, ENCSR513NDD, ENCSR773KRC, ENCSR314UMJ) and published HeLa (Krchnakova et al., 2019) (GSE113813) or MDA-MB231 (Best et al., 2014) (GSE59335) iCLIP normalized datasets were visualized as unique reads compared to control in the UCSC genome browser. ENCODE eCLIP data for HepG2 and K562 used in Figures S2F and S2G was visualized on the UCSC browser as peaks for each RBP indicated.
Differential splicing analysis ENCODE RBP KD
RNA-seq data for 237 RBPs KD using either RNAi or CRISPR in both HepG2 and K562 cells (Sundararaman et al., 2016) was downloaded from ENCODE (www.encode.org) and processed using rMATS (v4.0.1) (Phillips et al., 2020). For each alternative splicing event, the inclusion level is calculated as PSI for RBP-KD and control samples and averaged across the two replicates for each condition. We then limited these results to highly-confident ΔPSI values using a cutoff of P<0.05 for rMATS reproducibility p-value (likelihood ratio) when comparing RBP-KD to control.
Differential splicing analysis iPSC models
Raw fastq files for published RNA-seq datasets of iPSC differentiation to neurons (Burke et al., 2020) (PRJNA596331), pancreatic β-cells (Xie et al., 2013) (GSE9664), and type II alveolar epithelial cells (Jacob et al., 2017) (PRJEB1195) were downloaded from NCBI and analyzed along with in-house RNA-seq from iPSC to cardiomyocyte differentiation. Paired or single-end reads were filtered for quality and cropped with Trimmomatic (v 0.3.9) (Bolger et al., 2014) to the same length within each dataset. Reads were mapped to the human reference genome using STAR in 2-pass mode (v 2.7.3a) (Dobin et al., 2013) and the Gencode GRCh38 v.32 reference transcript annotation. We utilized an in-house pipeline that implemented rMATS (v 4.0.2) (Phillips et al., 2020) to detect splicing events using both splice junction read counts and exon body counts and calculated a percent spliced in (PSI) score for each splicing event.
Differential splicing analysis TCGA tumor and matched normal tissue samples
Tumor and corresponding normal tissue samples were downloaded as bam files from the NCI Genomic Data Commons and processed on the used Google Cloud Platform. Sample IDs are listed in Table S1B. PSI values for each splicing event were quantified using the rMATS turbo v 4.0.2 (Phillips et al., 2020) docker (https://github.com/nuno-agostinho/rmats4/blob/master/Dockerfile) with GENCODE v32 reference transcriptome. Each sample was run against itself, and PSI values from each sample were then extracted from the unfiltered rMATS output for further analysis in R. Patient matched ΔPSI values (Figure 1G) were calculated as PSITumor – PSINormal by matching TCGA IDs.
Differential splicing analysis MDS patient samples
RNA-seq of CD34+ cells from MDS patients (Pellagatti et al., 2018) (GSE114922) was downloaded from NCBI as Raw fastq files as 100bp paired-end reads and were filtered for quality by Trimmomatic (v 0.3.9) (Bolger et al., 2014). Sample IDs are listed in Table S1E. Reads were mapped to the human reference genome using STAR in 2-pass mode (v 2.7.3a) (Dobin et al., 2013) and the Gencode GRCh38 v.32 reference transcript annotation. We utilized an in-house pipeline that implemented rMATS (v 4.1.0) (Phillips et al., 2020) to detect splicing events using both splice junction read counts and exon body counts and calculated a percent spliced in (PSI) score for each splicing event. PSI values were extracted from the unfiltered rMATS output for plotting and analysis in R.
Clustering of SR proteins
SR protein coding sequences and SR protein binding motifs were clustered using the Clustal Omega web server (https://www.ebi.ac.uk/Tools/msa/clustalo/) and dendrograms were drawn using the ggTree wrapper for ggplot2 in R. Clustering of SR proteins based on PE inclusion is quantified as the Pearson correlation of ΔPSI values using the pheatmap package for R.
RNA secondary structure analysis
Predicted minimum free energy structures for wild-type, Δ11 and Δ12 mutant sequences were generated using RNAfold web server (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) (Lorenz et al., 2011). Input sequences contained the TRA2β-PE and 250bp of flanking intron sequences.
Graphs and figures
Plots were generated using Microsoft Excel and R. Figures were generated using Adobe CC 2019 Illustrator and Photoshop software in compliance with the Nature Publishing Group policy concerning image integrity.
Quantification and statistical analysis
For iPSC and TCGA analysis, the data is presented as median±interquartile range as indicated, and quantified using a Wilcoxon rank test. For RT-PCR, western blot, immunofluorescence quantifications, and wound healing the data is presented as the mean±standard deviation and significant differences to a control are assessed using an unpaired two-tailed Student t-test. For RNA-seq analysis, significance is assessed with an unpaired Wilcoxon rank sum test using the ggpubr add on to ggplot2 in R.
Additional resources
RNA-seq data from iPSC to cardiomyocyte differentiation has been deposited to GEO (GSE158578). Published RNA-seq datasets of iPSC differentiation are available on GEO under accession numbers: PRJNA596331 (neurons, Burke et al., 2020), GSE9664 (pancreatic β-cells, Xie et al., 2013), and PRJEB1195 (type II alveolar epithelial cells, Jacob et al., 2017).
RNA-sequencing data from TCGA breast tumors (Cancer Genome Atlas, 2012) is available via ISB-CGC cloud. Sample IDs are listed in Table S1B. RNA-seq data from MDS patients (Pellagatti et al., 2018) is available on GEO (GSE114922). Sample IDs are listed in Table S1E.
ENCODE RNA-seq and eCLIP-seq datasets are available from https://www.encodeproject.org/. iCLIP-seq for TRA2β (Best et al., 2014) (GSE59335), as well as SRSF2, SRSF5, and SRSF6 (Krchnakova et al., 2019) (GSE113813) were previously described and are available on GEO.
Supplementary Material
Table S1. Computational analysis of differential splicing in human tumor samples (Related to Figure 1 and S1)
Table S2. Analysis of RBP binding to SR protein poison exon sequences (Related to Figures 2 and S2-3)
Table S3. Correlation of SR protein minigene splicing and protein expression (Related to Figure 2 and S5)
Table S4. Sequence based reagents used in this study (Related to STAR Methods)
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-HA rabbit | Cell Signaling | #3724 |
| Anti-MYC-Tag mouse | Cell Signaling | #2276 |
| Anti-TRA2β rabbit | Abcam | #AB31353 |
| Anti-SRSF3 rabbit | MBLI | #RN080PW |
| Anti-β Tubulin III rabbit | Genscript | #A01203-40 |
| anti-GAPDH mouse | Genscript | #A01622 |
| Alexa Fluor 568 anti-mouse | Invitrogen | #A-11031 |
| Alexa Fluor 488 anti-rabbit | Invitrogen | #A-11034 |
| IRDye 800CW Goat anti-Rabbit IgG (H + L) | Licor | #926-32211 |
| IRDye 680 Goat anti-Mouse IgG (H + L) | Licor | #926-3220 |
| AnnexinV-Alexa 647 | Invitrogen | #A23204 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Hoechst 33342, Trihydrochloride, Trihydrate | Life Technologies | #H3570 |
| Goat Serum | Sigma | #G6767 |
| Matrigel | Corning | #354277 |
| Accutase | BD Biosciences | #561527 |
| ROCK inhibitor Y-27632 | Tocris | #1254 |
| CHIR99021 | Tocris | #4423 |
| B27 | Gibco | #A1895601 |
| GlutaMAX | Gibco | #35050061 |
| IWP-4 | Tocris | #5214 |
| Critical Commercial Assays | ||
| RNAeasy kit | Qiagen | #74106 |
| Lipofectamine 3000 | Invitrogen | #L3000075 |
| Phusion flash high fidelity master mix | Thermo Fisher | #F548L |
| Phusion hot start II DNA polymerase | Thermo Fisher | #F549L |
| Click-iT EdU Alexa 647 Imaging Kit | Thermo Fisher | #C10340 |
| Deposited Data | ||
| RNA-seq iPS to cardiomyocytes | This paper | GSE158578 |
| Experimental Models: Cell Lines | ||
| HEK293 | Krainer Lab (CSHL) | N/A |
| HEK293T | Krainer Lab (CSHL) | N/A |
| HeLa | Krainer Lab (CSHL) | N/A |
| MDA-MB231 TRMPV-neo shTRA2β | Park, et al. 2019 | N/A |
| SUM159 | Ethier Lab (MUSC) | N/A |
| HS578T | Krainer Lab (CSHL) | N/A |
| PGP1 iPSC | Coriell Institute | GM23338 |
| MCF-10A | ATCC | |
| AR7-HMEC | Pathania Lab (UMB) | N/A |
| Experimental Models: Organisms/Strains | ||
| None | ||
| Recombinant DNA | ||
| pCI-neo | Krainer Lab (CSHL) | (Hua et al., 2007) |
| pCI-neo-HA-SRSF1 | This paper | N/A |
| pCI-neo-HA-SRSF2 | This paper | N/A |
| pCI-neo-HA-SRSF3 | This paper | N/A |
| pCI-neo-HA-SRSF4 | This paper | N/A |
| pCI-neo-HA-SRSF5 | This paper | N/A |
| pCI-neo-HA-SRSF6 | This paper | N/A |
| pCI-neo-HA-SRSF7 | This paper | N/A |
| pCI-neo-HA-SRSF8 | This paper | N/A |
| pCI-neo-HA-SRSF9 | This paper | N/A |
| pCI-neo-HA-SRSF10 | This paper | N/A |
| pCI-neo-HA-SRSF11 | This paper | N/A |
| pCI-neo-HA-SRSF12 | This paper | N/A |
| pCI-neo-HA-TRA2A | This paper | N/A |
| pCI-neo-HA-TRA2B | This paper | N/A |
| pCI-neo-HA-SRSF3ΔRRM | This paper | N/A |
| pCI-neo-HA-SRSF3ΔRS | This paper | N/A |
| pCI-neo-HA-TRA2BΔRRM | This paper | N/A |
| pCI-neo-HA-TRA2BΔRS1 | This paper | N/A |
| pCI-neo-HA-TRA2BΔRS2 | This paper | N/A |
| pCI-neo-HA-TRA2B-RRM | This paper | N/A |
| pCI-neo-HA-TRA2B-RS1 | This paper | N/A |
| pCI-neo-HA-TRA2B-RS2 | This paper | N/A |
| pCI-neo-HA-TRA2B+SRSF3-RRM | This paper | N/A |
| pCI-neo-HA-SRSF3+TRA2B-RRm | This paper | N/A |
| pCI-neo-HA-hnRNPA1 | This paper | N/A |
| pcDNA5/FRT | Krainer Lab (CSHL) | Wong et al. 2018 |
| pcDNA5/FRT-MYC-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-SRSF1-PE-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-SRSF2-PE-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-SRSF3-PE-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-SRSF4-PE-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-SRSF5-PE-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-SRSF6-PE-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-SRSF7-PE-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-SRSF8-PE-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-SRSF9-PE-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-SRSF10-PE-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-SRSF11-PE-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-SRSF12-PE-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-TRA2A-PE-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-TRA2B-PE-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-SRSF3+TRA2B-PE+i-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-SRSF3+TRA2B-PE - DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-SRSF3+TRA2B-i-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-SRSF3+TRA2B-5’i-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-SRSF3+TRA2B-3’i-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-TRA2B+SRSF3-PE+i-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-TRA2B+SRSF3-PE-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-TRA2B+SRSF3-i-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-TRA2B+SRSF3-5’i-DsRed | This paper | N/A |
| pcDNA5/FRT-MYC-TRA2B+SRSF3-3’i-DsRed | This paper | N/A |
| pMAX-dCasRx | Addgene | 18634 |
| pMAX-RBFOX1N-dCasRx-C | Addgene | 118635 |
| pMAX-dCasRx-SRSF1-RS-C | This paper | N/A |
| pMAX-dCasRx-SRSF2-RS-C | This paper | N/A |
| pMAX-dCasRx-SRSF3-RS-C | This paper | N/A |
| pMAX-dCasRx-SRSF4-RS-C | This paper | N/A |
| pMAX-dCasRx-SRSF5-RS-C | This paper | N/A |
| pMAX-dCasRx-SRSF6-RS-C | This paper | N/A |
| pMAX-dCasRx-SRSF7-RS-C | This paper | N/A |
| pMAX-dCasRx-SRSF8-RS-C | This paper | N/A |
| pMAX-dCasRx-SRSF9-RS-C | This paper | N/A |
| pMAX-dCasRx-SRSF10-RS-C | This paper | N/A |
| pMAX-dCasRx-SRSF11-RS-C | This paper | N/A |
| pMAX-dCasRx-SRSF12-RS-C | This paper | N/A |
| pMAX-N-TRA2β-RS1-dCasRx-TRA2β-RS2-C | This paper | N/A |
| pCR8-Cas13d-DR-ccdB | Cheng Lab (JAX) | N/A |
| pCR8-Cas13d-DR-gRNA-CTL | Addgene | 118645 |
| pCR8-Cas13d-DR-gRNA-TRA2β | This paper | N/A |
| Sequence-Based Reagents | ||
| SLIC cloning primer sequences | IDT | Sequences Table S4A-C |
| gRNA sequences | IDT | Sequences Table S4D |
| RT-PCR primer sequences | IDT | Sequences Table S4E,G |
| 2OMe Antisense oligonucleotides | TechNOA | tNOA-2OM-hTRA2B-01 #1565 |
| 2OMe Antisense oligonucleotides | TechNOA | tNOA-2OM-hTRA2B-01 #1566 |
| 2OMe Antisense oligonucleotides | TechNOA | tNOA-2OM-hTRA2B-01 #1567 |
| 2OMe Antisense oligonucleotides | TechNOA | tNOA-2OM-hTRA2B-02 #1568 |
| 2OMe Antisense oligonucleotides | TechNOA | tNOA-2OM-hTRA2B-02 #1569 |
| 2OMe Antisense oligonucleotides | TechNOA | tNOA-2OM-hTRA2B-02 #1570 |
| 2MOE Antisense oligonucleotide 1570 | IDT | Sequences Table S4F |
| ORF NM_032102.3 | GenScript – SRSF8 | #OHu09269 |
| ORF NM_004768.4 | GenScript – SRSF11 | #OHu04591 |
| Software and Algorithms | ||
| ISB-CGC | (Reynolds et al., 2017) | https://isb-cgc.appspot.com/ |
| dockerhub | N/A | https://hub.docker.com/ |
| STAR v.2.7.3a | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| Stringtie | ||
| rMATS v.4.0.2 | (Shen et al., 2014) | http://rnaseq-mats.sourceforge.net/ |
| Nikon imaging software | Nikon | N/A |
| GraphPad Prism | GraphPad | N/A |
| ImageJ Digital Image Processing Software | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| MRI Wound Healing Plugin for ImageJ | https://github.com/MontpellierRessourcesImagerie/imagej_macros_and_scripts/wiki/Wound-Healing-Tool | |
| Harmony Image Analysis Software | Perkin Elmer | N/A |
| Photoshop and Illustrator CC2019 | Adobe | N/A |
Highlights.
SR proteins levels are coordinated through splicing of their poison exons
SR protein poison exon splicing is altered during cell differentiation and tumors
ASOs promoting TRA2β poison exon inclusion have anti-cancer effects in vitro
ACKNOWLEDGMENTS
We thank T. Helenius and G. Carmichael for comments on the manuscript, E. Guccione for discussion and help with 2’OMe ASO work, D. Wee, R. Lee and Tom. Tabaglio from TechNOA for assistance with 2’OMe ASO design, the Ethier lab for the gift of SUM159PT, the Krainer lab for HeLa, HEK293 and HS578T, the Pathania lab for AR7-HMEC. We acknowledge assistance from Microscopy, Single Cell Biology Laboratory and Genome Technologies Sequencing Services at The Jackson Laboratory (JAX). This work was supported by NIH grants R00CA178206 and R01GM138541 to OA, R35GM118140 to BRG, R01HG009900 to AC, R01HL142787 and U01EB028898 to JTH, and NCI P30CA034196 to JAX. We acknowledge the use of data generated by TCGA, managed by NCI and NHGRI, downloaded using ISB Cancer Genome Cloud and processed using Google Cloud Platform Research Credit award. The content is solely the responsibility of the authors and does not necessarily represent NIH official views.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
A patent application concerning this work is in preparation.
REFERENCES
- Ajiro M, Tang S, Doorbar J, and Zheng ZM (2016). Serine/Arginine-Rich Splicing Factor 3 and Heterogeneous Nuclear Ribonucleoprotein A1 Regulate Alternative RNA Splicing and Gene Expression of Human Papillomavirus 18 through Two Functionally Distinguishable cis Elements. J Virol 90, 9138–9152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anczukow O, Akerman M, Clery A, Wu J, Shen C, Shirole NH, Raimer A, Sun S, Jensen MA, Hua Y, et al. (2015). SRSF1-Regulated Alternative Splicing in Breast Cancer. Mol Cell 60, 105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anczukow O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, Muthuswamy SK, and Krainer AR (2012). The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol 19, 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anko ML, Muller-McNicoll M, Brandl H, Curk T, Gorup C, Henry I, Ule J, and Neugebauer KM (2012). The RNA-binding landscapes of two SR proteins reveal unique functions and binding to diverse RNA classes. Genome Biol 13, R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best A, Dagliesh C, Ehrmann I, Kheirollahi-Kouhestani M, Tyson-Capper A, and Elliott DJ (2013). Expression of Tra2 beta in Cancer Cells as a Potential Contributory Factor to Neoplasia and Metastasis. Int J Cell Biol 2013, 843781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best A, James K, Dalgliesh C, Hong E, Kheirolahi-Kouhestani M, Curk T, Xu Y, Danilenko M, Hussain R, Keavney B, et al. (2014). Human Tra2 proteins jointly control a CHEK1 splicing switch among alternative and constitutive target exons. Nat Commun 5, 4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkman KK, Buvoli M, Pugach EK, Polmear MM, and Leinwand LA (2019). miR-1/206 downregulates splicing factor Srsf9 to promote C2C12 differentiation. Skelet Muscle 9, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley T, Cook ME, and Blanchette M (2015). SR proteins control a complex network of RNA-processing events. RNA 21, 75–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke EE, Chenoweth JG, Shin JH, Collado-Torres L, Kim SK, Micali N, Wang Y, Colantuoni C, Straub RE, Hoeppner DJ, et al. (2020). Dissecting transcriptomic signatures of neuronal differentiation and maturation using iPSCs. Nat Commun 11, 462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres JF, Misteli T, Screaton GR, Spector DL, and Krainer AR (1997). Role of the modular domains of SR proteins in subnuclear localization and alternative splicing specificity. J Cell Biol 138, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres JF, Screaton GR, and Krainer AR (1998). A specific subset of SR proteins shuttles continuously between the nucleus and the cytoplasm. Genes Dev 12, 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caggiano C, Pieraccioli M, Panzeri V, Sette C, and Bielli P (2019). c-MYC empowers transcription and productive splicing of the oncogenic splicing factor Sam68 in cancer. Nucleic Acids Res 47, 6160–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas, N. (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Wang J, Zhu Z, Zhang MQ, and Krainer AR (2003). ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res 31, 3568–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler SD, Mayeda A, Yeakley JM, Krainer AR, and Fu XD (1997). RNA splicing specificity determined by the coordinated action of RNA recognition motifs in SR proteins. Proc Natl Acad Sci U S A 94, 3596–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clery A, Jayne S, Benderska N, Dominguez C, Stamm S, and Allain FH (2011). Molecular basis of purine-rich RNA recognition by the human SR-like protein Tra2-beta1. Nat Struct Mol Biol 18, 443–450. [DOI] [PubMed] [Google Scholar]
- Cohn R, Thakar K, Lowe A, Ladha FA, Pettinato AM, Romano R, Meredith E, Chen YS, Atamanuk K, Huey BD, et al. (2019). A Contraction Stress Model of Hypertrophic Cardiomyopathy due to Sarcomere Mutations. Stem Cell Reports 12, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Anczukow O, Akerman M, and Krainer AR (2012). Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep 1, 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, Chen M, Assanah M, Canoll P, and Manley JL (2010). HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463, 364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, and Beroud C (2009). Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 37, e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichmann DS, Walentek P, and Harland RM (2015). The alternative splicing regulator Tra2b is required for somitogenesis and regulates splicing of an inhibitory Wnt11b isoform. Cell Rep 10, 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding JH, Xu X, Yang D, Chu PH, Dalton ND, Ye Z, Yeakley JM, Cheng H, Xiao RP, Ross J, et al. (2004). Dilated cardiomyopathy caused by tissue-specific ablation of SC35 in the heart. EMBO J 23, 885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M , and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez D, Freese P, Alexis MS, Su A, Hochman M, Palden T, Bazile C, Lambert NJ, Van Nostrand EL, Pratt GA, et al. (2018). Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol Cell 70, 854–867 e859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M, Jillette N, Zhu JJ, Li S, and Cheng AW (2020). CRISPR artificial splicing factors. Nat Commun 11, 2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, Kim E, Abdel-Wahab O, and Bradley RK (2016). RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer 16, 413–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Brolosy MA, Kontarakis Z, Rossi A, Kuenne C, Gunther S, Fukuda N, Kikhi K, Boezio GLM, Takacs CM, Lai SL, et al. (2019). Genetic compensation triggered by mutant mRNA degradation. Nature 568, 193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galante PA, Sandhu D, de Sousa Abreu R, Gradassi M, Slager N, Vogel C, de Souza SJ, and Penalva LO (2009). A comprehensive in silico expression analysis of RNA binding proteins in normal and tumor tissue: Identification of potential players in tumor formation. RNA Biol 6, 426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstberger S, Hafner M, and Tuschl T (2014). A census of human RNA-binding proteins. Nat Rev Genet 15, 829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, Hertel KJ, and Maniatis T (1998). A systematic analysis of the factors that determine the strength of pre-mRNA splicing enhancers. EMBO J 17, 6747–6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, and Maniatis T (1998). Arginine/serine-rich domains of SR proteins can function as activators of pre-mRNA splicing. Mol Cell 1, 765–771. [DOI] [PubMed] [Google Scholar]
- Grellscheid S, Dalgliesh C, Storbeck M, Best A, Liu Y, Jakubik M, Mende Y, Ehrmann I, Curk T, Rossbach K, et al. (2011). Identification of evolutionarily conserved exons as regulated targets for the splicing activator tra2beta in development. PLoS Genet 7, e1002390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Che X, Wang X, and Jia R (2018). Inhibition of the expression of oncogene SRSF3 by blocking an exonic splicing suppressor with antisense oligonucleotides. Royal Society of Chemistry 8, 7159–7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinson JT, Chopra A, Lowe A, Sheng CC, Gupta RM, Kuppusamy R, O'Sullivan J, Rowe G, Wakimoto H, Gorham J, et al. (2017). Integrative Analysis of PRKAG2 Cardiomyopathy iPS and Microtissue Models Identifies AMPK as a Regulator of Metabolism, Survival, and Fibrosis. Cell Rep 19, 2410. [DOI] [PubMed] [Google Scholar]
- Hinson JT, Chopra A, Nafissi N, Polacheck WJ, Benson CC, Swist S, Gorham J, Yang L, Schafer S, Sheng CC, et al. (2015). HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 349, 982–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Vickers TA, Baker BF, Bennett CF, and Krainer AR (2007). Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol 5, e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Gattoni R, Stevenin J, and Steitz JA (2003). SR splicing factors serve as adapter proteins for TAP-dependent mRNA export. Mol Cell 11, 837–843. [DOI] [PubMed] [Google Scholar]
- Huang Y, and Steitz JA (2001). Splicing factors SRp20 and 9G8 promote the nucleocytoplasmic export of mRNA. Mol Cell 7, 899–905. [DOI] [PubMed] [Google Scholar]
- Inoue D, Chew GL, Liu B, Michel BC, Pangallo J, D'Avino AR, Hitchman T, North K, Lee SC, Bitner L, et al. (2019). Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature 574, 432–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob A, Morley M, Hawkins F, McCauley KB, Jean JC, Heins H, Na CL, Weaver TE, Vedaie M, Hurley K, et al. (2017). Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell 21, 472–488 e410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jangi M, Boutz PL, Paul P, and Sharp PA (2014). Rbfox2 controls autoregulation in RNA-binding protein networks. Genes Dev 28, 637–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MA, Wilkinson JE, and Krainer AR (2014). Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat Struct Mol Biol 21, 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji L, Ni T, Shen Y, Xue Q, Liu Y, Chen B, Cui X, Lv L, Yu X, Cui Y, et al. (2014). Transformer 2beta (Tra2beta/SFRS10) positively regulates the progression of NSCLC via promoting cell proliferation. J Mol Histol 45, 573–582. [DOI] [PubMed] [Google Scholar]
- Jumaa H, and Nielsen PJ (1997). The splicing factor SRp20 modifies splicing of its own mRNA and ASF/SF2 antagonizes this regulation. EMBO J 16, 5077–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminska D, Kakela P, Nikkola E, Venesmaa S, Ilves I, Herzig KH, Kolehmainen M, Karhunen L, Kuusisto J, Gylling H, et al. (2016). Regulation of alternative splicing in human obesity loci. Obesity (Silver Spring) 24, 2033–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, and Krainer AR (2007). The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol 14, 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kechavarzi B, and Janga SC (2014). Dissecting the expression landscape of RNA-binding proteins in human cancers. Genome Biol 15, R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. (2015). SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 27, 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CM, Bezzi M, Low DH, Ang WX, Teo SX, Gay FP, Al-Haddawi M, Tan SY, Osato M, Sabo A, et al. (2015). MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 523, 96–100. [DOI] [PubMed] [Google Scholar]
- Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, and Hsu PD (2018). Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 173, 665–676 e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konigs V, de Oliveira Freitas Machado C, Arnold B, Blumel N, Solovyeva A, Lobbert S, Schafranek M, Ruiz De Los Mozos I, Wittig I, McNicoll F, et al. (2020). SRSF7 maintains its homeostasis through the expression of Split-ORFs and nuclear body assembly. Nat Struct Mol Biol 27, 260–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krchnakova Z, Thakur PK, Krausova M, Bieberstein N, Haberman N, Muller-McNicoll M, and Stanek D (2019). Splicing of long non-coding RNAs primarily depends on polypyrimidine tract and 5' splice-site sequences due to weak interactions with SR proteins. Nucleic Acids Res 47, 911–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D, Das M, Sauceda C, Ellies LG, Kuo K, Parwal P, Kaur M, Jih L, Bandyopadhyay GK, Burton D, et al. (2019). Degradation of splicing factor SRSF3 contributes to progressive liver disease. J Clin Invest 130, 4477–4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau LF, and Brenner SE (2015). Regulation of splicing factors by alternative splicing and NMD is conserved between kingdoms yet evolutionarily flexible. Mol Biol Evol 32, 1072–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau LF, Inada M, Green RE, Wengrod JC, and Brenner SE (2007). Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 446, 926–929. [DOI] [PubMed] [Google Scholar]
- Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, Raval KK, Zhang J, Kamp TJ, and Palecek SP (2012). Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A 109, E1848–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Tebaldi T, Rejeski K, Joshi P, Stefani G, Taylor A, Song Y, Vasic R, Maziarz J, Balasubramanian K, et al. (2018). SRSF2 mutations drive oncogenesis by activating a global program of aberrant alternative splicing in hematopoietic cells. Leukemia 32, 2659–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman J (2018). Tapping the RNA world for therapeutics. Nat Struct Mol Biol 25, 357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JC, and Caceres JF (2009). The SR protein family of splicing factors: master regulators of gene expression. Biochem J 417, 15–27. [DOI] [PubMed] [Google Scholar]
- Longman D, Jackson-Jones KA, Maslon MM, Murphy LC, Young RS, Stoddart JJ, Hug N, Taylor MS, Papadopoulos DK, and Caceres JF (2020). Identification of a localized nonsense-mediated decay pathway at the endoplasmic reticulum. Genes Dev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, and Hofacker IL (2011). ViennaRNA Package 2.0. Algorithms Mol Biol 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Loh YH, Li H, Cesana M, Ficarro SB, Parikh JR, Salomonis N, Toh CX, Andreadis ST, Luckey CJ, et al. (2014). Alternative splicing of MBD2 supports self-renewal in human pluripotent stem cells. Cell Stem Cell 15, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Zhu P, Shi H, Guo L, Zhang Q, Chen Y, Chen S, Zhang Z, Peng J, and Chen J (2019). PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature 568, 259–263. [DOI] [PubMed] [Google Scholar]
- Manning KS, and Cooper TA (2017). The roles of RNA processing in translating genotype to phenotype. Nat Rev Mol Cell Biol 18, 102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo EC, Krach F, Yang D, Sen A, et al. (2018). Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 172, 590–604 e513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslon MM, Heras SR, Bellora N, Eyras E, and Caceres JF (2014). The translational landscape of the splicing factor SRSF1 and its role in mitosis. Elife, e02028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michlewski G, Sanford JR, and Caceres JF (2008). The splicing factor SF2/ASF regulates translation initiation by enhancing phosphorylation of 4E-BP1. Mol Cell 30, 179–189. [DOI] [PubMed] [Google Scholar]
- Muller-McNicoll M, Botti V, de Jesus Domingues AM, Brandl H, Schwich OD, Steiner MC, Curk T, Poser I, Zarnack K, and Neugebauer KM (2016). SR proteins are NXF1 adaptors that link alternative RNA processing to mRNA export. Genes Dev 30, 553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathaniel Jillette AC (2018). CRISPR Artificial Splicing Factors. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer KM, Merrill JT, Wener MH, Lahita RG, and Roth MB (2000). SR proteins are autoantigens in patients with systemic lupus erythematosus. Importance of phosphoepitopes. Arthritis Rheum 43, 1768–1778. [DOI] [PubMed] [Google Scholar]
- Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O'Brien G, Shiue L, Clark TA, Blume JE, and Ares M Jr. (2007). Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev 21, 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Sanchez P, Villalba-Orero M, Lopez-Olaneta MM, Larrasa-Alonso J, Sanchez-Cabo F, Marti-Gomez C, Camafeita E, Gomez-Salinero JM, Ramos-Hernandez L, Nielsen PJ, et al. (2019). Loss of SRSF3 in Cardiomyocytes Leads to Decapping of Contraction-Related mRNAs and Severe Systolic Dysfunction. Circ Res 125, 170–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit S, Zhou Y, Shiue L, Coutinho-Mansfield G, Li H, Qiu J, Huang J, Yeo GW, Ares M Jr., and Fu XD (2013). Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol Cell 50, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis C, Cloutier P, Shkreta L, Toutant J, Klarskov K, and Chabot B (2007). hnRNP I/PTB can antagonize the splicing repressor activity of SRp30c. RNA 13, 1287–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Brugiolo M, Akerman M, Das S, Urbanski L, Geier A, Kesarwani AK, Fan M, Leclair N, Lin KT, et al. (2019). Differential Functions of Splicing Factors in Mammary Transformation and Breast Cancer Metastasis. Cell Rep 29, 2672–2688 e2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra MK, Zhang W, Vu J, DeWitt MA, and Conboy JG (2020). Antisense targeting of decoy exons can reduce intron retention and increase protein expression in human erythroblasts. RNA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz I, Kosti I, Ares M Jr., Cline M, and Mandel-Gutfreund Y (2014). RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res 42, W361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellagatti A, Armstrong RN, Steeples V, Sharma E, Repapi E, Singh S, Sanchi A, Radujkovic A, Horn P, Dolatshad H, et al. (2018). Impact of spliceosome mutations on RNA splicing in myelodysplasia: dysregulated genes/pathways and clinical associations. Blood 132, 1225–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervouchine D, Popov Y, Berry A, Borsari B, Frankish A, and Guigo R (2019). Integrative transcriptomic analysis suggests new autoregulatory splicing events coupled with nonsense-mediated mRNA decay. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JW, Pan Y, Tsai BL, Xie Z, Demirdjian L, Xiao W, Yang HT, Zhang Y, Lin CH, Cheng D, et al. (2020). Pathway-guided analysis identifies Myc-dependent alternative pre-mRNA splicing in aggressive prostate cancers. Proc Natl Acad Sci U S A 117, 5269–5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piekielko-Witkowska A, Wiszomirska H, Wojcicka A, Poplawski P, Boguslawska J, Tanski Z, and Nauman A (2010). Disturbed expression of splicing factors in renal cancer affects alternative splicing of apoptosis regulators, oncogenes, and tumor suppressors. PLoS One 5, e13690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp MW, and Maquat LE (2018). Nonsense-mediated mRNA Decay and Cancer. Curr Opin Genet Dev 48, 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preussner M, Goldammer G, Neumann A, Haltenhof T, Rautenstrauch P, Muller-McNicoll M, and Heyd F (2017). Body Temperature Cycles Control Rhythmic Alternative Splicing in Mammals. Mol Cell 67, 433–446 e434. [DOI] [PubMed] [Google Scholar]
- Ratnadiwakara M, Archer SK, Dent CI, Ruiz De Los Mozos I, Beilharz TH, Knaupp AS, Nefzger CM, Polo JM, and Anko ML (2018). SRSF3 promotes pluripotency through Nanog mRNA export and coordination of the pluripotency gene expression program. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, et al. (2013). A compendium of RNA-binding motifs for decoding gene regulation. Nature 499, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds SM, Miller M, Lee P, Leinonen K, Paquette SM, Rodebaugh Z, Hahn A, Gibbs DL, Slagel J, Longabaugh WJ, et al. (2017). The ISB Cancer Genomics Cloud: A Flexible Cloud-Based Platform for Cancer Genomics Research. Cancer Res 77, e7–e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JM, Ennajdaoui H, Edmondson C, Wirth B, Sanford JR, and Chen B (2014). Splicing factor TRA2B is required for neural progenitor survival. J Comp Neurol 522, 372–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahashi K, Hua Y, Ling KK, Hung G, Rigo F, Horev G, Katsuno M, Sobue G, Ko CP, Bennett CF, et al. (2012). TSUNAMI: an antisense method to phenocopy splicing-associated diseases in animals. Genes Dev 26, 1874–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samatanga B, Clery A, Barraud P, Allain FH, and Jelesarov I (2017). Comparative analyses of the thermodynamic RNA binding signatures of different types of RNA recognition motifs. Nucleic Acids Res 45, 6037–6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford JR, Gray NK, Beckmann K, and Caceres JF (2004). A novel role for shuttling SR proteins in mRNA translation. Genes Dev 18, 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Hosoda N, and Maquat LE (2008). Efficiency of the pioneer round of translation affects the cellular site of nonsense-mediated mRNA decay. Mol Cell 29, 255–262. [DOI] [PubMed] [Google Scholar]
- Scharner J, Ma WK, Zhang Q, Lin KT, Rigo F, Bennett CF, and Krainer AR (2020). Hybridization-mediated off-target effects of splice-switching antisense oligonucleotides. Nucleic Acids Res 48, 802–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotti MM, and Swanson MS (2016). RNA mis-splicing in disease. Nat Rev Genet 17, 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebestyen E, Singh B, Minana B, Pages A, Mateo F, Pujana MA, Valcarcel J, and Eyras E (2016). Large-scale analysis of genome and transcriptome alterations in multiple tumors unveils novel cancer-relevant splicing networks. Genome Res 26, 732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, Jumaa H, and Webster NJ (2013). Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat Commun 4, 1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A 111, E5593–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, Kleiman FE, and Manley JL (2005). Multiple properties of the splicing repressor SRp38 distinguish it from typical SR proteins. Mol Cell Biol 25, 8334–8343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoilov P, Daoud R, Nayler O, and Stamm S (2004). Human tra2-beta1 autoregulates its protein concentration by influencing alternative splicing of its pre-mRNA. Hum Mol Genet 13, 509–524. [DOI] [PubMed] [Google Scholar]
- Storbeck M, Hupperich K, Gaspar JA, Meganathan K, Martinez Carrera L, Wirth R, Sachinidis A, and Wirth B (2014). Neuronal-specific deficiency of the splicing factor Tra2b causes apoptosis in neurogenic areas of the developing mouse brain. PLoS One 9, e89020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Zhang Z, Sinha R, Karni R, and Krainer AR (2010). SF2/ASF autoregulation involves multiple layers of post-transcriptional and translational control. Nat Struct Mol Biol 17, 306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Yan L, Guo J, Shao J, and Jia R (2019). Downregulation of SRSF3 by antisense oligonucleotides sensitizes oral squamous cell carcinoma and breast cancer cells to paclitaxel treatment. Cancer Chemother Pharmacol 84, 1133–1143. [DOI] [PubMed] [Google Scholar]
- Sundararaman B, Zhan L, Blue SM, Stanton R, Elkins K, Olson S, Wei X, Van Nostrand EL, Pratt GA, Huelga SC, et al. (2016). Resources for the Comprehensive Discovery of Functional RNA Elements. Mol Cell 61, 903–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureau A, Gattoni R, Dooghe Y, Stevenin J, and Soret J (2001). SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EmBO J 20, 1785–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson CM, Sherer NM, and Malim MH (2010). SRp40 and SRp55 promote the translation of unspliced human immunodeficiency virus type 1 RNA. J Virol 84, 6748–6759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabaglio T, Low DH, Teo WKL, Goy PA, Cywoniuk P, Wollmann H, Ho J, Tan D, Aw J, Pavesi A, et al. (2018). MBNL1 alternative splicing isoforms play opposing roles in cancer. Life Sci Alliance 1, e201800157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliaferro JM, Lambert NJ, Sudmant PH, Dominguez D, Merkin JJ, Alexis MS, Bazile C, and Burge CB (2016). RNA Sequence Context Effects Measured In Vitro Predict In Vivo Protein Binding and Regulation. Mol Cell 64, 294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Polaski JT, Feng Q, De Neef EJ, Hoppe ER, McSharry MV, Pangallo J, Gabel AM, Belleville AE, Watson J, et al. (2020). RNA isoform screens uncover the essentiality and tumor-suppressor activity of ultraconserved poison exons. Nat Genet 52, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, Hashimoto H, Suzuki T, Yamashita H, Satoh Y, et al. (2013). Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 12, 127–137. [DOI] [PubMed] [Google Scholar]
- Urbanski LM, Leclair N, and Anczukow O (2018). Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip Rev RNA 9, e1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Der Houven Van Oordt W, Newton K, Screaton GR, and Caceres JF (2000). Role of SR protein modular domains in alternative splicing specificity in vivo. Nucleic Acids Res 28, 4822–4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Pratt GA, Yee BA, Wheeler EC, Blue SM, Mueller J, Park SS, Garcia KE, Gelboin-Burkhart C, Nguyen TB, et al. (2020). Principles of RNA processing from analysis of enhanced CLIP maps for 150 RNA binding proteins. Genome Biol 21, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MS, Kinney JB, and Krainer AR (2018). Quantitative Activity Profile and Context Dependence of All Human 5' Splice Sites. Mol Cell 71, 1012–1026 e1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie R, Everett LJ, Lim HW, Patel NA, Schug J, Kroon E, Kelly OG, Wang A, D'Amour KA, Robins AJ, et al. (2013). Dynamic chromatin remodeling mediated by polycomb proteins orchestrates pancreatic differentiation of human embryonic stem cells. Cell Stem Cell 12, 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Yang D, Ding JH, Wang W, Chu PH, Dalton ND, Wang HY, Bermingham JR Jr., Ye Z, Liu F, et al. (2005). ASF/SF2-regulated CaMKIIdelta alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell 120, 59–72. [DOI] [PubMed] [Google Scholar]
- Yang S, Jia R, and Bian Z (2018). SRSF5 functions as a novel oncogenic splicing factor and is upregulated by oncogene SRSF3 in oral squamous cell carcinoma. Biochim Biophys Acta Mol Cell Res 1865, 1161–1172. [DOI] [PubMed] [Google Scholar]
- Ye J, and Blelloch R (2014). Regulation of pluripotency by RNA binding proteins. Cell Stem Cell 15, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. (2011). Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69. [DOI] [PubMed] [Google Scholar]
- Zhang Z, and Krainer AR (2004). Involvement of SR proteins in mRNA surveillance. Mol Cell 16, 597–607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Computational analysis of differential splicing in human tumor samples (Related to Figure 1 and S1)
Table S2. Analysis of RBP binding to SR protein poison exon sequences (Related to Figures 2 and S2-3)
Table S3. Correlation of SR protein minigene splicing and protein expression (Related to Figure 2 and S5)
Table S4. Sequence based reagents used in this study (Related to STAR Methods)
Data Availability Statement
The RNA-seq data from iPSC to cardiomyocyte differentiation generated during this study is available from GEO (GSE158578).
The original/source data for Figures 2-7 and Figures S1-7 in the paper is available from Mendeley https://data.mendeley.com/datasets/smf5hf7t68/1.