

Abstract
Esophageal squamous cell carcinoma accounts for a large proportion of cancer-associated mortalities in both men and women. Melittin is the major active component of bee venom, which has been reported to possess anti-inflammatory, antibacterial and anti-cancer properties. The aim of the present study was to construct a tumor targeted recombinant plasmid [pc-telomerase reverse transcriptase (TERT)-melittin] containing a human TERT promoter followed by a melittin coding sequence and to explore the effects of this plasmid in esophageal cell carcinoma and investigate preliminarily the underlying mechanisms of this effect. TE1 cells were transfected with pcTERT-melittin and the resulting apoptosis was subsequently examined. The viability of TE1 cells transfected with pcTERT-melittin was measured using a Cell Counting Kit-8 assay, which indicated inhibited proliferation. The disruption of mitochondrial membranes and the concomitant production of reactive oxygen species demonstrated an inducible apoptotic effect of melittin in TE1 cells. Apoptotic cells were also counted using an Annexin V-FITC and PI double-staining assay. The upregulation of cleaved caspase-9, cleaved caspase-3, Bax and poly(ADP-ribose) polymerase 1 in pcTERT-melittin transfected TE1 cells, suggested that pcTERT-melittin-induced apoptosis was associated with the mitochondrial pathway. TE1 cells were also arrested in the G0/G1 phase when transfected with pcTERT-melittin, followed by the decline of CDK4, CDK6 and cyclin D1 expression levels. As cell invasion and metastasis are common in patients with esophageal cancer, a cell migration assay was conducted and it was found that pcTERT-melittin transfection reduced the migratory and invasive abilities of TE1 cells. The findings of the present study demonstrated that pcTERT-melittin may induce apoptosis of esophageal carcinoma cells and inhibit tumor metastasis.
Keywords: melittin, telomerase reverse transcriptase promoter, esophageal cancer, gene therapy
Introduction
Esophageal squamous cell carcinoma (ESCC) is a common form of aggressive malignancy (1), with a poor prognosis and high mortality rate, 455,800 new esophageal cancer cases and 400,200 deaths occurred in 2012 worldwide and most of the histological subtype were ESCC (2). Predisposing factors for ESCC include smoking and alcohol (3). The development and progression of ESCC is very complex, with multiple genetic mutations and disorders involved, such as mutations in TP53, CDKN2A or a loss of RB gene (4,5). Currently, surgery, radiotherapy and chemotherapy are commonly used singly or in combination to treat ESCC. However, these methods may also damage healthy cells and have limited efficacy in cancer infiltration and metastasis (6,7). Therefore, novel and curative agents for ESCC are urgently required. Gene therapy, which delivers therapeutic genes into cells to alter the gene expression of a patient, may be a promising strategy for cancer treatment (8). Numerous cancer gene therapy approaches have reported significant progress, including gene immunization, suppressor genes or gene replacements, gene directed enzyme-prodrug or suicide gene therapies and oncolytic virus therapies (9–11).
In its mature form, melittin is a 26 amino acid peptide with strong hemolytic and antimicrobial activity, and is the principal active component of bee venom (12). Melittin is in routine use as a non-steroidal anti-inflammatory reagent for the relief of pain and the treatment of chronic inflammatory diseases (13,14). Moreover, melittin has demonstrated the ability to inhibit cancer cell proliferation and induce apoptosis (15,16). Compared with other pro-apoptotic proteins such as p53, Bak and Bax, melittin is not selective, damaging both healthy and cancer cells. To overcome the non-specific cytotoxicity of melittin, targeted expression is a necessary prerequisite for successful cancer gene therapy (17).
Human telomerase reverse transcriptase (hTERT) is the catalytic subunit of telomerase and is the rate-limiting step in the activation of telomerase (18). Furthermore, it has been reported that hTERT expression is common to most cancer cells (18).
The present study constructed a recombinant plasmid (pcTERT-melittin) containing the hTERT promoter followed by melittin, and investigated the ability of this pcTERT-melittin construct to act as a therapeutic agent able to inhibit proliferation and induce apoptosis in TE1 cells.
Materials and methods
Materials
The human esophageal carcinoma TE1 and normal esophageal epithelial cells Het-1a cell lines were obtained from the American Type Culture Collection. DMEM and FBS were obtained from Hyclone (Cytiva). BEGM BulletKit containing basal medium (BEBM) plus additives (growth factors, hEGF, VEGF, hFGF, IGF-1; FBS, hydrocortisone, ascorbic acid, heparin and transferrin) (BEGM SingleQuots) was obtained Lonza Group, Ltd. Opti-MEM reduced serum medium, pcDNA3.1+ plasmid, TRIzol RNA Isolation Reagents and Lipofectamine® 2000 transfection reagent were purchased from Invitrogen (Thermo Fisher Scientific, Inc.). Takara Ex Taq DNA Polymerase was from Takara Biotechnology. The Cell Counting Kit (CCK)-8 (cat. no. C0037), reactive oxygen species (ROS) assay kit (cat. no. S0033S), mitochondrial membrane potential (∆Ψm) assay kit with JC-1 (cat. no. C2006), caspase-3 activity assay kit (cat. no. C1115) and caspase-9 activity assay kit (cat. no. C1158) were purchased from Beyotime Institute of Biotechnology. Annexin V-FITC staining kit and complete protease inhibitor cocktail tablets were purchased from Roche Diagnostics.
Antibodies against tubulin (1:1,000; cat. no. sc-80005) and horseradish peroxidase-conjugated secondary antibodies to rabbit, mouse and goat primary antibodies were purchased from Santa Cruz Biotechnology, Inc. Antibodies against poly(ADP-ribose) polymerase 1 (PARP) (1:1,000; cat. no. 9542), cleaved caspase-9 (1:1,000; cat. no. 20750), caspase-3 (1:1,000; cat. no. 9662), cleaved caspase-3 (1:1,000; cat. no. 9661) and Bax (1:100; cat. no. 2772) were purchased from Cell Signaling Technology, Inc. Antibodies against CDK4 (1:1,000; cat. no. A0366), CDK6 (1:10,00l; cat. no. A1545), cyclin D1 (1:1,000; cat. no. A19038) and p53 (1:1,000; cat. no. A11232) were purchased from Abclonal Biotech Co., Ltd.
EasySee® western blotting kit was purchased from Beijing Transgen Biotech Co., Ltd. Transwell chambers were purchased from Corning, Inc. A Primescript™ Reverse Transcription reagent kit was purchased from Takara Biotechnology Co., Ltd. SYBR® premix ex Taq™ II, ROX plus reagent kit was purchased from Roche Diagnostics. All other chemicals were analytical reagent grade.
Construction of pcTERT-melittin vector
A recombinant plasmid encoding melittin was constructed using a pcDNA3.1+ plasmid containing the hTERT promoter. The enhancer sequence of pcDNA3.1+ was first amplified using PCR. The pcDNA3.1 plasmid was used as template. The thermocycling conditions were as follows: 30 cycles at 95°C for 10 sec, 56°C for 30 sec and 72°C for 1 min. Forward Primer: 5′-CTGACGCGTGGAGTTCCGCGTTAC−3′; Reverse Primer: 5′-CGCGCTAGCCAAAACAAACTCCCATTG−3′)with the MluI and NheI restriction sites engineered at the ends of primer, then four different hTERT promoter DNAs, each with NheI and HindIII restriction sties, were amplified from human genomic DNA. The melittin coding gene was synthesized by Sangon Biotech Co., Ltd. The enhancer, hTERT promoter and melittin gene sequence (ATGGGCATCGGCGCCATCCTGAAGGTGCTGAGCACCGGCCTGCCCGCCCTGATCAGCTGGATCAAGAGAAAGAGACAGGAGTAA) were inserted into the pcDNA3.1+ consecutively to produce a recombinant plasmid (pcTERT-melittin). The recombinant plasmid containing only the enhancer and hTERT promoter was named ‘pcTERT’.
Cell culture
TE1 cells were cultivated in DMEM supplemented with 10% FBS, while Het-1a cells were cultured with bronchial epithelial cell medium (BEGM BulletKit) containing basal medium (BEBM) plus additives (BEGM SingleQuot), and incubated at 37°C with 5% CO2. The cells were passaged every ~2 days. TE1 cells were transfected with recombinant plasmid pcTERT-melittin or pcTERT (2 µg plasmid for 12-well plate and 4 µg plasmid for 6-well plate) at 60% of plate confluence using Lipofectamine 2000, following manufacturer's instructions. Following transfection, the cells were observed using a phase-contrast microscope (Olympus Corporation) (magnification, ×200). For the cell proliferation assay, the measurement of ∆Ψm using tetraethylbenzimidazolylcarbocyanine iodide (JC-1) staining, intracellular ROS quantification assays, Annexin-V-FITC apoptosis assay, cell cycle assay and cell migration assay, the cells were detected immediately after transfection. For the caspase-3 and caspase-9 activity assay and western blot analysis, after the transfection, cells were lysed and proteins were detected immediately. For the RNA extraction and reverse transcription-quantitative (RT-q) PCR assay, after the transfection, RNA was extracted immediately and reverse-transcribed to cDNA, then stored at −80°C for subsequent PCR assays.
Agarose gel electrophoresis
The construction of pcTERT-melittin was identified with agarose gel electrophoresis. In brief, after plasmids digested with restriction endonucleases (HindIII and XbaI) for 8 h at 37°C, samples were analyzed with a 1% agarose gel at 80 V at 37°C. The pcTERT-melittin plasmid was sent to Sangon Biotech Co., Ltd to investigate whether the sequence of melittin had the correct reading frames inserted.
Cell proliferation assay
Determination of the number of viable cells was performed using a TransDetect CCK-8 assay according the manufacturer's instructions. TE1 and Het-1a cells were plated in quadruplicate at a concentration of 5×103 per well in 96-well cell culture plates. Cells were incubated overnight at 37°C with 5% CO2 and then transfected with the recombinant plasmids for 24–72 h. After treatment with 10 µl CCK-8 solution at 37°C for 1 h, the plates were measured using a microplate spectrophotometer at 450 nm. The survival rate was calculated using the formula: Survival rate=(mean absorption of treated group/mean absorption of control group)x100%.
Measurement of ∆Ψm using tetraethylbenzimidazolylcarbocyanine iodide (JC-1) staining. The change in ∆Ψm is a key feature of early stage apoptosis (19). The change in ∆Ψm was monitored using the mitochondrial membrane potential (∆Ψm) assay kit containing the cell-permeable cationic JC-1 dye and fluorescence microscopy according to the manufacturer's instructions. This procedure did not require fixation. In brief, the TE1 cells were plated into 12-well plates at 2×105 cells/well and incubated at 37°C with 5% CO2 overnight. A total of 3×105 TE1 cells were transfected with 2 µg pcTERT-melittin or pcTERT for 24 h, then the medium was replaced with DMEM containing JC-1 and incubated for 20 min at 37°C with 5% CO2. After incubation, the medium was removed and cells were washed with ice-cold JC-1 buffer twice. The cells were observed using fluorescent microscopy, five random fields were observed at a magnification of ×200. Red emission was characteristic of normal polarized mitochondria, while green fluorescence was observed with a depolarized mitochondrial membrane, which is characteristic of early stage apoptosis (20).
Intracellular ROS quantification assays
Intracellular ROS levels were assessed by detecting the oxidative conversion of dichlorodihydrofluorescein diacetate (DCFH-DA) to 2′,7′-dichlorofluorescein in TE1 cells according to the ROS assay kit manufacturer's instructions. TE1 cells were transfected with 2 µg recombinant plasmids for 48 h, then harvested with trypsinization. Then, 1×105 cells were incubated with 5 µl DCFH-DA in 1 ml DMEM for 20 min at 37°C. The cells were then washed three times and re-suspended in PBS, followed via assessment using a fluorescence microplate at excitation and emission wavelengths of 488 and 525 nm, respectively.
Annexin-V-FITC apoptosis assay
The quantification of apoptotic TE1 cells was performed using flow cytometry with Annexin V-FITC and PI dyes, according to the manufacturer's instructions. A total of 7.5×105 TE1 cells in 6-well plates were transfected with 4 µg recombinant plasmids pcTERT-melittin or pcTERT for 24, 48 and 72 h, before being harvested via trypsinization and washed with PBS. Subsequently, 70% cold ethanol was used to fix the TE1 cells for 30 min at 4°C. Pellets were re-suspended and incubated in Annexin V-FITC labelling solution at 15–25°C for 15 min. PI was added at 4°C and stained for 5 min. The apoptotic cells were then analyzed using a flow cytometer (BD Calibur; Becton-Dickinson and Company). Data were analyzed with Cell Quest data acquisition and analysis software (Becton-Dickinson and Company).
Cell cycle assay
TE1 cell cycle distribution was measured using a flow cytometer by quantifying the PI labeled DNA content. After transfection with recombinant plasmids for 48 h, TE1 cells were collected and fixed with 70% cold ethanol at 4°C for 30 min, then washed with PBS. Pellets were re-suspended and incubated in PBS containing RNaseA (50 µg/ml), Triton (0.2%) and PI (20 µg/ml) at 4°C for 30 min in the dark. Cell cycle analysis was performed using a flow cytometer (BD Calibur; Becton-Dickinson and Company). Data were analyzed with Cell Quest data acquisition and analysis software (Becton-Dickinson and Company) after 1 h.
Cell migration assay
Transwell chambers (6.5 mm diameter inserts; 8.0-µm pore size) were used to measure cell migration. After transfection with recombinant plasmids for 48 h, TE1 cells were harvested with trypsinization. Then, 1×105 cells were re-suspended in 200 µl serum-free DMEM medium and cultivated in the upper well of a 24-well Transwell chamber without Matrigel, while 400 µl DMEM containing 10% FBS was plated in the lower chamber. For the upper well with Matrigel (wells precoated with Matrigel for 10 min at 37°C), 1.5×105 cells were seeded. After seeding for 15 h, the cells in the top chamber were removed. Chambers were washed with PBS three times. Migrating cells in the lower chamber were fixed with 4% paraformaldehyde for 15 min at 37°C, washed with PBS and stained with 2% crystal violet at 37°C for 15 min. TE1 cells in the lower chamber were counted using light microscopy (magnification, ×200).
RNA extraction and reverse transcription-quantitative (RT-q)PCR
A total of 3×105 TE1 cells were transfected with 2 µg plasmids for 48 and 72 h in 12-well plates before RNA extraction. Total RNA was extracted using a TRIzol® reagent. Briefly, 500 µl TRIzol reagent was added to the well, before cells were transferred to tubes and then incubated for 5 min at room temperature. The tubes were shaken for 30 sec, then 100 µl chloroform was added and left to stand for 10 min at 37°C. The samples were centrifuged at 12,000 × g at 4°C for 15 min, and the upper layer of supernatant was collected. An equal volume of isopropanol (~100 µl) was added to the supernatant. After standing for 10 min at 37°C, the samples were centrifuged at 12,000 × g for 30 min at 4°C. The RNA pellet was washed with 75% ethanol and dissolved in DEPC treated water. Then, 1 µg total RNA was reverse-transcribed to cDNA using the following temperature protocol, 42°C for 30 min and 85°C for 5 sec using a Primescript™ RT reagent kit. qPCR was performed using a SYBR® premix ex Taq™ II, ROX plus reagent kit, conducted in the Step One plus™ Real-Time PCR system (Thermo Fisher Scientific, Inc.). The primers was as follows: Bax: Forward, primer, 5′-GGCAACTTCAACTGGGGC−3′; reverse, primer, 5′-CCACCCTGGTCTTGGATCC-3′; and Bcl-2 forward, primer, 5′-AGGATTGTGGCCTTCTTTGA-3′; Reverse primer, 5′-TCAGGTACTCAGTCATCCAC-3′. The PCR protocol was as follows: Initial denaturation at 94°C for 10 min, followed by 40 cycles at 90°C for 5 sec and 60°C for 30 sec. Products were verified using melting curve analysis. Each sample was calculated from threshold cycle numbers and GAPDH was monitored as the internal control. Fold-changes in target gene mRNA expression were determined using the 2−ΔΔCq method (21).
Caspase-3 and caspase-9 activity assay
The activities of caspase-3 and caspase-9 in TE1 cells were measured using a caspase activity assay kit following the manufacturer's instructions. Cells were transfected with pcTERT-melittin or pcTERT for 24–72 h, digested with trypsin (0.25%) for 1 min at 37°C, the cells were collected and mixed with lysis buffer (1% Triton X-100, 50 mM HEPES, 50 mM sodium pyrophosphate, 100 mM sodium fluoride, 10 m EDTA and 10 mM sodium vanadate) at 37°C for 5 min. After incubating at 0°C on ice for 20 min, the samples were centrifuged at 12,000 × g at 4°C for 30 min. The supernatant was incubated with reaction buffer at 37°C for 11 h, then the absorbance measured at 450 nm using a microplate reader.
Western blot analysis
For western blot analysis, TE1 cells were lysed on ice with ice-cold lysis buffer (1% Triton X-100; 50 mmol/l HEPES; 50 mmol/l sodium pyrophosphate; 100 mmol/l sodium fluoride; 10 mmol/l EDTA; 10 mmol/l sodium vanadate) containing protease inhibitors cocktail. The lysates were centrifuged for 15 min at 15,000 × g at 4°C. The supernatant was analyzed with a BCA protein assay kit to confirm protein concentration. Subsequently, 50 µg proteins were boiled at 100°C for 5 min, then separated on 8–15% SDS-PAGE, before being transferred onto a PVDF membrane. Membranes were blocked with 5% fat-free milk in TBS-0.1% Tween-20 (TBST) buffer at room temperature for 1 h, and then incubated with the primary antibodies overnight at 4°C. Membranes were washed with TBST buffer three times, followed by incubation with the corresponding secondary antibody at 37°C for 1 h and washing with TBST buffer three times. The protein-antibody bound bands were visualized using an EasySee® western blotting kit and the signal strength of each protein was normalized against the corresponding control (signal strength of tubulin) and analyzed with ImageJ software version 1.52 (National Institutes of Health).
Statistical analysis
Data are presented as the mean ± SEM. Data analysis for comparison between pcTERT group and pcTERT-melittin group was performed using SPSS version 19.0 (IBM Corp). Data were analyzed with two-tailed unpaired Student's t-tests and one-way ANOVA followed by the Least Significant Difference post hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
Construction of recombinant plasmid pcTERT-melittin
The activity of different hTERT promoters was assessed by inserting a green fluorescence protein expressed sequence EGFP after the hTERT promoter. The 378 bp promoter demonstrated the greatest level of transcription compared with the 443 bp, 714 bp promoter and 1,100 bp promoters, and was used to construct the pcTERT-melittin plasmid (Fig. 1). Agarose gel electrophoresis was performed after pcTERT-melittin plasmid was digested with restriction endonucleases (HindIII and XbaI), and the 100 bp fragment of melittin was observed (Fig. S1A). The sequence of melittin was also demonstrated to be inserted with correct reading frames via gene sequencing (Fig. S1B).
Figure 1.

Expression of enhanced green fluorescent protein (EGFP) promoted by different hTERT promoters. The EGFP sequence was placed after hTERT promoters (378, 443, 714 and 1,100 bp) in pcDNA3.1+ plasmids. TE1 cells werse transfected with the recombinant plasmids for 24 h. The fluorescence intensity was detected using a fluorescence microscope with magnification, ×200. TERT, telomerase reverse transcriptase.
Proliferation is inhibited by pcTERT-melittin in TE1 cells
Morphological changes typically occur during cell apoptosis (19). To investigate the effects of pcTERT-melittin on TE1 cells, the morphological changes of transfected TE1 cells were observed using phase-contrast microscopy. Phase-contrast micrographs demonstrated that TE1 cells exposed to pcTERT-melittin exhibited cell shrinkage, apoptotic vacuoles, membrane blebbing and formed floating cells in a time-dependent manner, indicating that pcTERT-melittin induced apoptosis in TE1 cells (Fig. 2A). Cells transfected with pcTERT and untreated cells displayed normal morphology with distinct cell borders (Fig. 2A).
Figure 2.

TE1 cell proliferation inhibited by pcTERT-Mel transfection. (A) TE1 cells were transfected with pcTERT or pcTERT-Mel and cell morphology was observed under a phase-contrast microscope 24–72 h after transfection (original magnification, ×200). (B) TE1 and Het-1a cells were transfected with pcTERT and pcTERT-Mel, and the survival rate (%) of cells was evaluated using a Cell Counting Kit-8 assay. The absorbance was measured at 450 nm using a spectrophotometer. Data are presented as the mean ± SEM, and the results are an average of three independent experiments. *P<0.05 vs. Con group. TERT, telomerase reverse transcriptase; Con, control; Mel, melittin.
A cell proliferation assay was conducted in TE1 cells exposed to recombinant plasmids for 24, 48 and 72 h using a TransDetect CCK-8 assay. pcTERT-melittin induced a significant decrease in cell survival in TE1 cells. The viable cell percentage in pcTERT-melittin treated cell populations was 88.1% of the control after transfection for 24 h, 66% of the control after 48 h and 58.6% of the control after 72 h in TE1 cells (Fig. 2B). However, no significant effects on cell viability were found between pcTERT-melittin treated cells and controls in Het-1a cells. This suggested that pcTERT-melittin treatment inhibited cell proliferation in a time-dependent manner compared with controls in TE1 cells.
Subsequently, pcTERT and pcTERT-melittin plasmids were used to transfect Het-1a and TE1 cells, and the mRNA expression of melittin was measured using RT-qPCR. The mRNA expression of melittin could only be detected in TE1 cells transfected with pcTERT-melittin plasmid for 48 h, but not in Het-1a cells transfected with pcTERT and pcTERT-melittin plasmid (using the same primer pair) or TE1 cells transfected with pcTERT (Fig. S2).
pcTERT-melittin induces apoptosis in TE1 cells associated with damage to mitochondrial membranes and the production of ROS
∆Ψm is a characteristic parameter for monitoring mitochondria-dependent cell apoptosis, since reduction of the ∆Ψm increases the generation of ROS, leading to the release of pro-apoptotic factors such as cytochrome c (22). The current study assessed the influence of recombinant plasmids on ∆Ψm in living cells using a fluorescence microscope with the fluorescent dye JC-1. JC-1 is a cationic dye that accumulates in the lumen of mitochondria, producing red fluorescence in normally polarized mitochondria. As the Ψm deceases, JC-1 becomes monomeric, showing green fluorescence (20). Green fluorescence of JC-1 was observed in TE1 cells treated with pcTERT-melittin, which was reflective of JC-1 existing in a monomeric state, and suggested a reduction in ∆Ψm (Fig. 3A). Moreover, pcTERT treated TE1 cells and untreated cells both exhibited red cell-staining, indicating normal ∆Ψm. The mitochondrial depolarization observed in pcTERT-melittin treated TE1 cells suggested that pcTERT-melittin induces early stage apoptosis.
Figure 3.

Transfection of pcTERT-Mel decreases mitochondrial membrane potential and increases ROS production in TE1 cells, leading to apoptosis. (A) Cells were stained with tetraethylbenzimidazolylcarbocyanine iodide and visualized using a fluorescence microscope at 24 h post-transfection. pcTERT treated cells and untreated cells stained red suggested normal high membrane potentials. pcTERT-Mel treatment caused a significant loss of red fluorescence and an increase of green fluorescence, indicating the loss of mitochondrial membrane potential, which was associated with apoptosis (original magnification, ×200). (B) ROS production was detected with a ROS assay kit. Increased ROS production was observed in pcTERT-melittin treated cells with a fluorescence microplate at excitation and emission wavelengths of 488 and 525 nm, respectively. (C) Quantification of the pcTERT-melittin transfection-induced apoptosis of TE1 cells, as assessed via flow cytometry using Annexin-V and PI staining at 24, 48 and 72 h post-transfection. The percentage of apoptotic cells was presented as the mean ± SEM. Results are an average of three independent experiments. *P<0.05 vs. Con group. TERT, telomerase reverse transcriptase; Con, control; Mel, melittin; ROS, reactive oxygen species.
Reduction in ∆Ψm is typically associated with the opening of mitochondrial permeability transition pores, resulting in the release of ROS (23). It was identified that the production of ROS was significantly increased in pcTERT-melittin treated cells compared with controls (Fig. 3B).
After typical apoptotic morphological changes, low survival rate and mitochondrial depolarization were observed in TE1 cells transfected with pcTERT-melittin, apoptotic cells were counted with the Annexin V-FITC and PI double-staining method using a flow cytometer. TE1 cells transfected with pcTERT-melittin demonstrated a significant increase in Annexin V-positive cells compared with pcTERT treated cells (Fig. 3C). After transfection with pcTERT-melittin for 24 h, apoptotic TE1 cells were significantly higher (14.08±2.53%) compared with the controls (8.15±1.12%). At 48 h post-transfection, the percentage of apoptotic cells that had been transfected with pcTERT-melittin increased to 20.56±0.76% compared with the pcTERT group (10.56±0.86%). After transfection for 72 h, the number of apoptotic TE1 cells was 48.36±8.04% in the pcTERT-melittin group compared with 21.05±1.17% in the pcTERT group.
pcTERT-melittin induces apoptosis in TE1 cells via a mitochondrial pathway
The Annexin V-FITC and PI staining assay results demonstrated that the recombinant plasmid can induce apoptosis in TE1 cells, thus decreasing ∆Ψm, which suggested that pcTERT-melittin induces apoptosis via the mitochondrial pathway. Caspase-9 and caspase-3 serve crucial roles in the apoptotic mitochondrial pathway (24). Therefore, caspase activity assays were conducted to investigate the associated caspase activities in TE1 cells. Caspase-3 activity increased 1.4 fold after pcTERT-melittin transfection for 48 and 72 h, and the activity of caspase-9 was significantly upregulated in pcTERT-melittin treated cells after 24–72 h, indicating the activation of caspase-9 of pcTERT-melittin group appears earlier compared with caspase-3 activation of the pcTRET-melittin group (Fig. 4A).
Figure 4.

pcTERT-Mel triggers apoptosis in TE1 cells via the mitochondrial pathway. (A) Effects of pcTERT-Mel on caspase-3 and caspase-9 activity. TE1 cells were transfected with recombinant plasmids for 24–72 h and the activities of caspases-3 and −9 were analyzed with a caspase activity assay kits. Data are presented as the mean ± SEM of three experiments. *P<0.05 vs. Con group. (B) Relative expression of Bcl-2/Bax ratio was detected using reverse transcription-quantitative PCR. (C) Expression levels of PARP, cleaved caspase-3, caspase-3, cleaved caspase-9 and Bax were analyzed via western blotting in TE1 cells after transfection for 48 h. Data are presented as the mean ± SEM of three experiments, as an average of three independent experiments. *P<0.05 vs. Con group. TERT, telomerase reverse transcriptase; Con, control; Mel, melittin; PARP, poly(ADP-ribose) polymerase 1.
The expression levels of Bax and Bcl-2, which are upstream of caspase-3 and caspase-9 in the apoptotic mitochondrial pathway (25), were investigated using RT-qPCR. The results demonstrated that the relative expression of Bcl-2/Bax ratio in pcTERT-melittin treated TE1 cells was significantly lower compared with controls, in a time-dependent manner (Fig. 4B). Moreover, after 48 h transfection, the expression of pro-apoptotic cleaved caspase-9 was upregulated, followed by an increase in cleaved caspase-3 in pcTERT-melittin treated TE1 cells, compared with controls (Fig. 4C). As the downstream substrate of caspase-3, the expression of cleaved-PARP also increased when caspase-3 was cleaved in pcTERT-melittin treated TE1 cells, compared with controls.
pcTERT-melittin transfection inhibits TE1 cell migration
To identify whether transfection of pcTERT-melittin altered the migration of TE1 cells, a Transwell assay was conducted to assess cell migration. Migration and invasion of TE1 cells to the lower Transwell chamber were significantly decreased in pcTERT-melittin treated TE1 cell compared with pcTERT treated cells after transfection for 48 h (Fig. 5). Thus, transfection of pcTERT-melittin could inhibit TE1 cell migratory and invasive ability.
Figure 5.
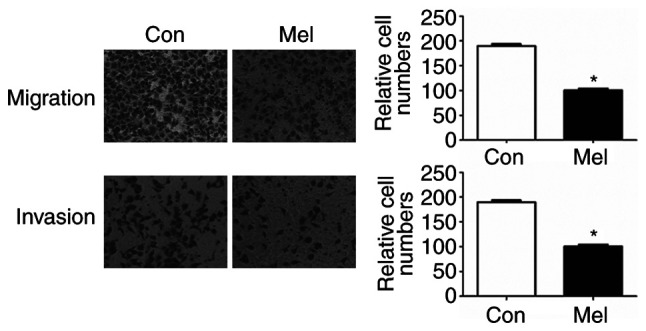
Effect of pc-telomerase reverse transcriptase-Mel transfection on cell migration in TE1 cells. Cell migration and invasion were measured using a Transwell assay after transfection for 48 h. Cells in five random microscope fields (original magnification, ×200) were counted for each group of cells. The results are an average of three independent experiments. *P<0.05 vs. Con group. Con, control; Mel, melittin.
pcTERT-melittin transfection induces cell cycle arrest in TE1 cells
As a highly-ordered and tightly regulated process, normal cell cycle progression is vital to the maintenance of cell structure and integrity (26). Multiple checkpoints determining extracellular growth signals, cell size and DNA integrity are involved in cell cycle progression, and thus dysregulation of the cell cycle is a typical features of cancer development (27). To determine whether cell proliferation inhibition and the pro-apoptotic ability of pcTERT-melittin are associated with an abnormal cell cycle, the distribution of the cell cycle was assessed using flow cytometry in TE1 cells. After transfection for 48 h the percentage of TE1 cells at phase G0/G1 was significantly higher in pcTERT-melittin treated cells (66.73±0.43%) compared with the pcTERT group (50.19±0.74%) (Fig. 6A). As the cells in pcTERT-melittin group were arrested at G0/G1 phase, the percentage of S phase cells of pcTERT-melittin group was significant decreased (Fig. 6A).
Figure 6.

Effects of pcTERT-Mel transfection on cell cycle distribution of TE1 cells. (A-a) Cell cycle distribution of pcTERT-Mel and pcTERT treated TE1 cells and untreated TE1 cells, as measured using flow cytometry after 48 h transfection. (A-b) Representative quantitation of the cell cycle phase, with data presented as the mean ± SEM. (B) Western blot analysis of CDK4, CDK6, cyclin D1 and p53 protein expression levels, performed after TE1 cells were transfected with recombinant plasmids for 48 h. Protein band intensity was quantified using ImageJ, and data are means ± SEM of triplicate experiments normalized to tubulin. *P<0.05 vs. Con group. Con, control; Mel, melittin; TERT, telomerase reverse transcriptase.
The cyclin-dependent kinases, CDK4 and CDK6, are important regulators of retinoblastoma phosphorylation in phase G1/S of the cell cycle for promoting cell proliferation (28). The activation of p53 is also detected as a clear biological outcome of cell cycle arrest (29). It was identified that the expression levels of CDK4 and CDK6 were significantly decreased in pcTERT-melittin treated cells, which was accompanied with a significantly downregulation of cyclin D1. Furthermore, p53 was upregulated in pcTERT-melittin treated cells after transfection for 48 h, compared with the control cells (Fig. 6B).
Discussion
Melittin is an effective anti-inflammatory compound with activity 100 times stronger compared with that of hydrocortisone (30). Previously, the antineoplastic activity of melittin has received increased attention (31,32). As a frequent cancer type, esophagus cancer is a serious threat to human health, and is a form of gastrointestinal tumor with a high incidence rate in China (33). The present study used TE1 cells to investigate the apoptosis inducing capacity of the recombinant plasmid pcTERT-melittin, which contains the melittin coding sequence regulated by the hTERT promoter.
Changes in TE1 cell morphology were observed using phase-contrast microscopy after transfection with the recombinant plasmid for different intervals in the present study, and it was identified that pcTERT-melittin treated cells exhibited distinct apoptosis compared with controls. In addition, pcTERT-melittin transfection induced cell shrinkage, apoptotic vacuole formation and membrane blebbing in a dose and time-dependent manner, and further analysis with a CCK-8 assay demonstrated proliferation inhibition by pcTERT-melittin in TE1 cells.
There are significant changes in mitochondrial structure during the early stage of cell apoptosis, including increasing membrane permeability and decreasing mitochondrial membrane potential (34). Multiple biochemical changes accompany the decrease of ∆Ψm, such as the release of cytochrome c into the cytoplasm and augmented ROS production (35). The present study used JC-1 staining to detect ∆Ψm changes, which indicated the early stage apoptosis of TE1 cells after transfection with pcTERT-melittin. The collapse of ∆Ψm and the increased production of ROS suggested that the intrinsic pathway of apoptosis is the main mechanism via which pcTERT-melittin induces apoptosis.
In the current study, quantifying apoptotic cells using a flow cytometer demonstrated that the percentage of apoptotic cells was significantly increased by pcTERT-melittin. Cytochrome c can bind to the apoptotic protease activating factor-1, recruiting and activating caspase-9 via the caspase recruitment domain, and thus initiating the cascade reaction (36). After transfection with pcTERT-melittin, a significant increase in cleaved caspase-9 expression was identified in TE1 cells. PARP is associated with numerous cell processes, including DNA repair and cell apoptosis (37). Moreover, cleavage of PARP is a sensitive molecular switch in apoptosis (38). In the present study, caspase-3 was significantly activated, which was followed by cleavage of PARP in pcTERT-melittin treated TE1 cells.
The invasion and metastasis of ESCC is not only a common postoperative occurrence, but also causes the loss of potential operation opportunities for numerous patients (39). In the present study, pcTERT-melittin treated TE1 cells demonstrated a significantly reduced ability to migrate and invade compared with controls, indicating the potential inhibitory effects of pcTERT-melittin transfection on cancer cell migration and invasion.
The aberrant activity of various cell cycle proteins such as p53 and ras may result in uncontrolled tumor cell proliferation, which is a characteristic feature of cancer (26). In the current study, TE1 cells were arrested in the G1 phase followed by the downregulation of CDK4, CDK6 and cyclin D1 expression levels. p53 is a pivotal protein associated with apoptosis and cell cycle arrest. When p53 is activated, p21 is highly induced, with p21 binding to the cyclin D/CDK4 complexes that cause G1 arrest (40). The pro-apoptotic protein Bax can also be activated by p53 in the cytosol (41). The present results demonstrated that the expression levels of p53 and Bax were increased in TE1 cells after transfection for 48 h.
In conclusion, the present study constructed a recombinant plasmid pcTERT-melittin, which contained the melittin coding sequence regulated by the hTERT promoter. It was demonstrated that the plasmid pcTERT-melittin plasmid induced apoptosis in TE1 cells via the mitochondrial pathway, and that did not affect healthy esophageal cells. pcTERT-melittin also inhibited TE1 cell migration and invasion, suggesting it may be a potential therapeutic agent in cancer therapy. However, the present study has some limitations, more proteins related to apoptosis, such as proteins related to ER stress or death receptor pathway were not investigated and the effect of pcTERT-melittin in vivo was also not investigated. Future studies will conduct RNA-sequencing and proteomics methods to investigate the detailed underlying molecular mechanisms, and will construct transplantation tumor models in mice to assess the effect of pcTERT-melittin on tumors in vivo.
Supplementary Material
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural Science Foundation of China (grant nos. 81370497 and 81500635) and the Science and Technology Department of Jilin Province (grant no. 20190304067YY).
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding authors on reasonable request.
Authors' contributions
PJ and JM contributed to the study conception and design. CZ, YW, PJ and JM wrote and revised the manuscript. CZ, YL, WZ, BX, JL, YM, YT, WW and QZ performed the experiments. PJ, WW, WY, YL and CZ were responsible for the acquisition and analysis of data. All authors approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Pakzad R, Mohammadian-Hafshejani A, Khosravi B, Soltani S, Pakzed I, Mohammadian M, Salehiniya H, Momenimovahed Z. The incidence and mortality of esophageal cancer and their relationship to development in Asia. Ann Transl Med. 2016;4:29. doi: 10.3978/j.issn.2305-5839.2016.01.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 3.Abnet CC, Arnold M, Wei WQ. Epidemiology of esophageal squamous cell carcinoma. Gastroenterology. 2018;154:360–373. doi: 10.1053/j.gastro.2017.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu X, Zhang M, Ying S, Zhang C, Lin R, Zheng J, Zhang G, Tian D, Guo Y, Du C, et al. Genetic alterations in esophageal tissues from squamous dysplasia to carcinoma. Gastroenterology. 2017;153:166–177. doi: 10.1053/j.gastro.2017.03.033. [DOI] [PubMed] [Google Scholar]
- 5.Testa U, Castelli G, Pelosi E. Esophageal cancer: Genomic and molecular characterization, stem cell compartment and clonal evolution. Medicines (Basel) 2017;4:67. doi: 10.3390/medicines4030067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen M, Shen M, Lin Y, Liu P, Liu X, Li X, Li A, Yang R, Ni W, Zhou X, et al. Adjuvant chemotherapy does not benefit patients with esophageal squamous cell carcinoma treated with definitive chemoradiotherapy. Radiat Oncol. 2018;13:150. doi: 10.1186/s13014-018-1086-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu SX, Wang LH, Luo HL, Xie CY, Zhang XB, Hu W, Zheng AP, Li DJ, Zhang HY, Xie CH, et al. Randomised phase III trial of concurrent chemoradiotherapy with extended nodal irradiation and erlotinib in patients with inoperable oesophageal squamous cell cancer. Eur J Cancer. 2018;93:99–107. doi: 10.1016/j.ejca.2018.01.085. [DOI] [PubMed] [Google Scholar]
- 8.Amer MH. Gene therapy for cancer: Present status and future perspective. Mol Cell Ther. 2014;2:27. doi: 10.1186/2052-8426-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, Liang H, Liu J, Wang Z. Poly (amidoamine) (PAMAM) dendrimer mediated delivery of drug and pDNA/siRNA for cancer therapy. Int J Pharm. 2018;546:215–225. doi: 10.1016/j.ijpharm.2018.05.045. [DOI] [PubMed] [Google Scholar]
- 10.Kim HJ, Kim A, Miyata K, Kataoka K. Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv Drug Deliv Rev. 2016;104:61–77. doi: 10.1016/j.addr.2016.06.011. [DOI] [PubMed] [Google Scholar]
- 11.Zhou Z, Liu X, Zhu D, Wang Y, Zhang Z, Zhou X, Qiu N, Chen X, Shen Y. Nonviral cancer gene therapy: Delivery cascade and vector nanoproperty integration. Adv Drug Deliv Rev. 2017;115:115–154. doi: 10.1016/j.addr.2017.07.021. [DOI] [PubMed] [Google Scholar]
- 12.Orsolic N. Bee venom in cancer therapy. Cancer Metastasis Rev. 2012;31:173–194. doi: 10.1007/s10555-011-9339-3. [DOI] [PubMed] [Google Scholar]
- 13.Rajasekaran G, Dinesh Kumar S, Nam J, Jeon D, Kim Y, Lee CW, Park IS, Shin SY. Antimicrobial and anti-inflammatory activities of chemokine CXCL14-derived antimicrobial peptide and its analogs. Biochim Biophys Acta Biomembr. 2019;1861:256–267. doi: 10.1016/j.bbamem.2018.06.016. [DOI] [PubMed] [Google Scholar]
- 14.Sobral F, Sampaio A, Falcão S, Queiroz MJ, Calhelha RC, Vilas-Boas M, Ferreira IC. Chemical characterization, antioxidant, anti-inflammatory and cytotoxic properties of bee venom collected in Northeast Portugal. Food Chem Toxicol. 2016;94:172–177. doi: 10.1016/j.fct.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 15.Lim HN, Baek SB, Jung HJ. Bee venom and its peptide component melittin suppress growth and migration of melanoma cells via inhibition of PI3K/AKT/mTOR and MAPK Pathways. Molecules. 2019;24:929. doi: 10.3390/molecules24050929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rady I, Siddiqui IA, Rady M, Mukhtar H. Melittin, a major peptide component of bee venom, and its conjugates in cancer therapy. Cancer Lett. 2017;402:16–31. doi: 10.1016/j.canlet.2017.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gajski G, Garaj-Vrhovac V. Melittin: A lytic peptide with anticancer properties. Environ Toxicol Pharmacol. 2013;36:697–705. doi: 10.1016/j.etap.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 18.Leao R, Apolonio JD, Lee D, Figueiredo A, Tabori U, Castelo-Branco P. Mechanisms of human telomerase reverse transcriptase (hTERT) regulation: Clinical impacts in cancer. J Biomed Sci. 2018;25:22. doi: 10.1186/s12929-018-0422-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis. 2003;8:115–128. doi: 10.1023/A:1022945107762. [DOI] [PubMed] [Google Scholar]
- 20.Perelman A, Wachtel C, Cohen M, Haupt S, Shapiro H, Tzur A. JC-1: Alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis. 2012;3:e430. doi: 10.1038/cddis.2012.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rao XY, Huang XL, Zhou XC, Lin X. An improvement of the 2ˆ(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat Bioinforma Biomath. 2013;3:71–85. [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Y, Karakhanova S, Hartwig W, D'Haese JG, Philippov PP, Werner J, Bazhin AV. Mitochondria and mitochondrial ros in cancer: Novel targets for anticancer therapy. J Cell Physiol. 2016;231:2570–2581. doi: 10.1002/jcp.25349. [DOI] [PubMed] [Google Scholar]
- 23.Song SB, Jang SY, Kang HT, Wei B, Jeoun UW, Yoon GS, Hwang ES. Modulation of mitochondrial membrane potential and ROS generation by nicotinamide in a manner independent of SIRT1 and mitophagy. Mol Cells. 2017;40:503–514. doi: 10.14348/molcells.2017.0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin M, Tang S, Zhang C, Chen H, Huang W, Liu Y, Zhang J. Euphorbia factor L2 induces apoptosis in A549 cells through the mitochondrial pathway. Acta Pharm Sin B. 2017;7:59–64. doi: 10.1016/j.apsb.2016.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu R, Tang S, Wang M, Xu X, Yao C, Wang S. MicroRNA-497 induces apoptosis and suppresses proliferation via the Bcl-2/Bax-Caspase9-Caspase3 Pathway and Cyclin D2 Protein in HUVECs. PLoS One. 2016;11:e0167052. doi: 10.1371/journal.pone.0167052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luch A. Cell cycle control and cell division: Implications for chemically induced carcinogenesis. Chembiochem. 2002;3:506–516. doi: 10.1002/1439-7633(20020603)3:6<506::AID-CBIC506>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 27.Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nature reviews. Cancer. 2017;17:93–115. doi: 10.1038/nrc.2016.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov. 2016;6:353–367. doi: 10.1158/2159-8290.CD-15-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med. 2016;6:a026104. doi: 10.1101/cshperspect.a026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee G, Bae H. Anti-inflammatory applications of melittin, a major component of bee venom: Detailed mechanism of action and adverse effects. Molecules. 2016;21:616. doi: 10.3390/molecules21050616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kong GM, Tao WH, Diao YL, Fang PH, Wang JJ, Bo P, Qian F. Melittin induces human gastric cancer cell apoptosis via activation of mitochondrial pathway. World J Gastroenterol. 2016;22:3186–3195. doi: 10.3748/wjg.v22.i11.3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soliman C, Eastwood S, Truong VK, Ramsland PA, Elbourne A. The membrane effects of melittin on gastric and colorectal cancer. PLoS One. 2019;14:e0224028. doi: 10.1371/journal.pone.0224028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 34.Guo LD, Chen XJ, Hu YH, Yu ZJ, Wang D, Liu JZ. Curcumin inhibits proliferation and induces apoptosis of human colorectal cancer cells by activating the mitochondria apoptotic pathway. Phytother Res. 2013;27:422–430. doi: 10.1002/ptr.4731. [DOI] [PubMed] [Google Scholar]
- 35.Bishayee K, Ghosh S, Mukherjee A, Sadhukhan R, Mondal J, Khuda-Bukhsh AR. Quercetin induces cytochrome-c release and ROS accumulation to promote apoptosis and arrest the cell cycle in G2/M, in cervical carcinoma: Signal cascade and drug-DNA interaction. Cell Prolif. 2013;46:153–163. doi: 10.1111/cpr.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu Q, Wu D, Chen W, Yan Z, Yan C, He T, Liang Q, Shi Y. Molecular determinants of caspase-9 activation by the Apaf-1 apoptosome. Proc Natl Acad Sci USA. 2014;111:16254–16261. doi: 10.1073/pnas.1418000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bai P. Biology of poly(ADP-Ribose) polymerases: The factotums of cell maintenance. Mol Cell. 2015;58:947–958. doi: 10.1016/j.molcel.2015.01.034. [DOI] [PubMed] [Google Scholar]
- 38.Los M, Mozoluk M, Ferrari D, Stepczynska A, Stroh C, Renz A, Herceg Z, Wang ZQ, Schulze-Osthoff K. Activation and caspase-mediated inhibition of PARP: A molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell. 2002;13:978–988. doi: 10.1091/mbc.01-05-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang Y, Zhang J, Zhao J, Li Z, Chen H, Qiao Y, Chen X, Liu K, Dong Z. TOPK promotes metastasis of esophageal squamous cell carcinoma by activating the Src/GSK3β/STAT3 signaling pathway via γ-catenin. BMC Cancer. 2019;19:1264. doi: 10.1186/s12885-019-6453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giono LE, Manfredi JJ. The p53 tumor suppressor participates in multiple cell cycle checkpoints. J Cell Physiol. 2006;209:13–20. doi: 10.1002/jcp.20689. [DOI] [PubMed] [Google Scholar]
- 41.McCurrach ME, Connor TM, Knudson CM, Korsmeyer SJ, Lowe SW. Bax-deficiency promotes drug resistance and oncogenic transformation by attenuating p53-dependent apoptosis. Proc Natl Acad Sci USA. 1997;94:2345–2349. doi: 10.1073/pnas.94.6.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding authors on reasonable request.